

Abstract
The adenosine nucleotide translocase (ANT) family of proteins are inner mitochondrial membrane proteins involved in energy homeostasis and cell death. The primary function of ANT proteins is to exchange cytosolic ADP with matrix ATP, facilitating the export of newly synthesized ATP to the cell while providing new ADP substrate to the mitochondria. As such, the ANT proteins are central to maintaining energy homeostasis in all eukaryotic cells. Evidence also suggests that the ANTs constitute a pore-forming component of the mitochondrial permeability transition pore (MPTP), a structure that forms in the inner mitochondrial membrane that is thought to underlie regulated necrotic cell death. Additionally, emerging studies suggest that ANT proteins are also critical for mitochondrial uncoupling and for promoting mitophagy. Thus, the ANTs are multifunctional proteins that are poised to participate in several aspects of mitochondrial biology and the greater regulation of cell death, which will be discussed here.
ANT FAMILY
The ANT family belongs to the large mitochondrial solute carrier family of proteins [1, 2]. ANT proteins are encoded in the nuclear DNA and their subsequent production in the cytoplasm is followed by trafficking to the inner mitochondrial membrane where they facilitate the transport of ADP and ATP into or out of the mitochondrial matrix [3]. ANT proteins have a characteristic structure with a tripartite repeat of ~100 amino acids and a total of six transmembrane domains that cluster in a generally cylindrical shape [4–7]. Topologically the ANT proteins have carboxy- and amino-terminals that face the intramembrane space, with 2 exposed cytosolic loops (C1-C2), while the matrix side of ANT is comprised of three hydrophilic loops (M1-M3) that are available for protein interaction and modification (Figure 1) [4, 5, 7]. ANT protein monomers are able to form dimers via interactions in the M1 loop [8, 9], and it is believed that the majority of ANT proteins exist in this dimeric form [10, 11]. However, ANT transport activity is proportional to the number of active ANT monomers, suggesting that regardless of higher order complexes it is the monomeric unit that comprises the translocase activity. [12, 13]. Moreover, structural studies of crystalized ANT monomers demonstrate that substrate binding and transport occur within individual monomers [7]. Experimental evidence shows that engineered covalently-linked ANT homodimers possess normal transport activity [14, 15], although chimeric wildtype-mutant ANT dimers reveal some cooperativity between linked monomers suggesting that dimerization is functionally relevant [16]. Another important structural consideration of ANT proteins is that they tightly bind six molecules of cardiolipin within the mitochondrial inner membrane, which are required for protein function and stability [17–19]. ANT proteins are often the most abundant proteins in the mitochondria inner membrane, comprising ≈10% of all proteins, which may explain why it is often identified as an interacting protein [20].
Figure 1: ANT protein schematic.
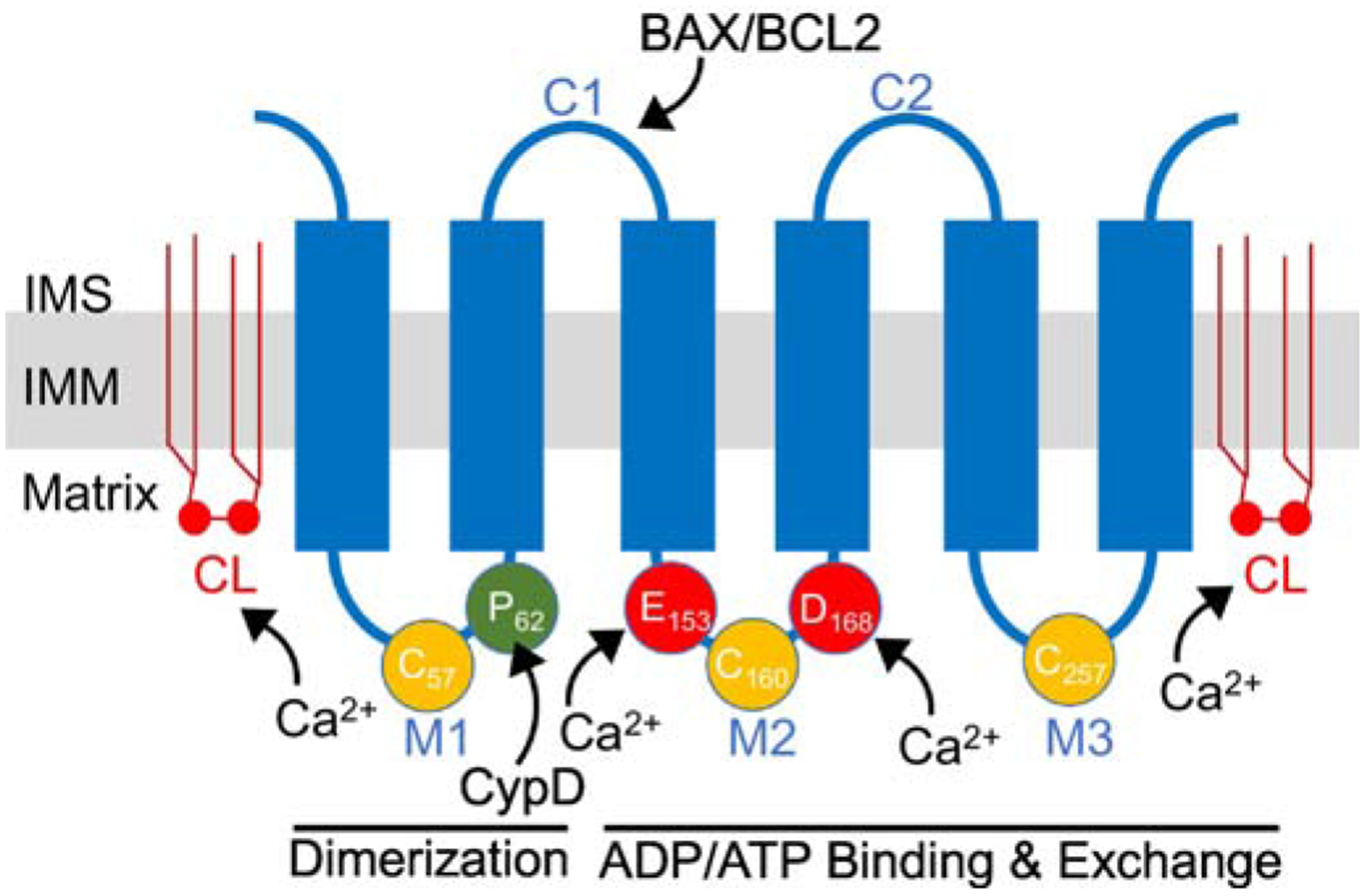
Schematic of ANT protein monomer topology in the inner mitochondrial membrane (IMM). ANT proteins have six transmembrane domains and two mitochondrial intermembrane (IMS) space facing domains (C1-C2). C1 contains a BAX/BCL2 binding motif. In addition, there are three mitochondrial matrix facing loops (M1-M3). The M1 loop is thought to be involved in dimerization and it contains Proline-62 (P62) that is thought to interact with CypD. This M1 loop also contains Cysteine-57 (C57) that is thought to play a role in ANT dimerization. The M2 and M3 loops are involved in ADP/ATP binding and nucleotide exchange. M2 and M3 contain two modifiable cysteines (C160 and C257) that are thought to be sensitive to oxidative stress. In addition, M2 contains two acidic amino acids, Gultamate-153 (E153) and Asparatate-168 (D168), important to ADP binding that may also be involved in Ca2+ sensing and regulation. In addition, ANT proteins are associated with six molecules of cardiolipin (depicted by CL) in the inner membrane that is required for protein folding and sensitivity to Ca2+ regulation. Amino acid residue numbers are taken from human ANT1.
Humans possess four ANT genes that have similar structure and high sequence homology (~80–90%) [21]. ANT1 (SLC25A4) is located on chromosome 4 and is the predominant isoform in terminally differentiated, metabolically active tissues such as heart and skeletal muscle [22–25]. ANT2 (SLC25A5) is encoded on the X-chromosome and it shows expression in many tissues but is enriched in proliferating cell-types [23, 26–28]. ANT3 (SLC25A6) is also located on the pseudoautosomal region of the X and Y chromosomes and it is expressed at low levels in virtually all tissues [23, 24, 29, 30]. The fourth ANT isoform, ANT4 (SLC25A31), is expressed in human brain, liver, and testis [31]. While the differential transcriptional regulation between ANT gene isoforms has been investigated [21], the specific reasons for tissue specificity remain unknown but it is thought to be related to the function of these 4 isoforms in coordinating differing oxidative and glycolytic metabolic requirements [26].
Mice, in contrast to humans, possess only three Ant genes: Ant1, Ant2, and Ant4. Murine Ant1 shows primarily heart and skeletal muscle expression, analogous to ANT1 in humans [32, 33]. Murine Ant2 is expressed in most mouse tissues except skeletal muscle and it appears to function in place of human ANT3, which mice do not possess [33, 34]. Murine Ant4 expression is limited to embryonic stem cells, the testis, and the embryonic ovary [35–38]. Ant4 gene-deleted mice show infertility in males through a defect in spermatogenesis [35, 37]. Interestingly, Ant4 gene expression has been experimentally tied to DNA hypermethylation in somatic cells that tightly restricts its expression to embryonic or germline cells in mice [38, 39]. Ant4 repression can be specifically reversed by treating cells with a CpG-demethylating agent, which induces expression in somatic fibroblasts, suggesting the possibility that Ant4 expression could be induced in certain contexts when Ant1 or Ant2 function are compromised [38, 39].
ATP/ADP EXCHANGE
ANT proteins mediate the movement of one nucleotide across the inner mitochondrial membrane coupled with the movement of another nucleotide in the opposite direction [40, 41]. This transport follows a strict 1:1 ratio, such that no net change in total nucleotide pool levels is achieved [40–43]. Biochemical analysis has demonstrated that ANT proteins bind and transport the free ions of ATP4- and ADP3-, although ANTs have a higher affinity for ADP3- in charged mitochondria [41, 44, 45]. ANTs do not efficiently transport AMP or other nucleotide species [40, 41, 46]. ANTs can participate in homogenous transfers where ATP and ADP are exchanged for themselves or mixed transfer where ATP is exchanged for ADP, as well as vice versa [47]. In uncoupled mitochondria ANTs translocate ATP and ADP equally in either direction without bias [40, 41], however under charged conditions the mitochondrial membrane potential (Δψ) favors importation of ADP into the matrix and exportation of ATP [40, 48], which maximizes [ATP]/[ADP] ratio in the cytosol [48–51]. The 1:1 exchange of ADP3- for ATP4- is electrogenic and this drives transport bias and the production of a net negative charge, which can secondarily erode Δψ and have an energy cost [48, 52].
Recent structural studies have elucidated the mechanism of ADP/ATP exchange in ANT proteins [4, 7, 53]. These studies have validated the hypothesis that ANT transport follows an “induced transition fit” model and involves a single common binding site for nucleotide cargo [3, 54, 55] (FIGURE 2). During a typical transport cycle, cytosolic ADP3- will bind the common binding site while the ANT protein is in the “c-state” in which the protein is open at the intermembrane space but closed to the mitochondrial matrix via a network of salt bridges [4, 7, 53]. ADP3- binding then causes a conformational change in ANT, moving it into the “m-state” in which the protein is open to the mitochondrial matrix and closed to the intermembrane space by a newly formed salt bridge network and a hydrophobic “plug” [7]. This transition involves breaking the c-state matrix salt bridges and causing the ANT protein domains to rotate around the nucleotide binding site like a “fulcrum”, exposing the transported nucleotide to the mitochondrial matrix and forming new cytosolic salt bridges to produce the m-state conformation [7]. Transported ADP3- unbinds the ANT m-state and diffuses into the mitochondrial matrix. ATP4- can then bind the unloaded ANT m-state and induce the reverse conformation change to the c-state, where the nucleotide is then released into the intermembrane space. As discussed above, since Δψ makes the export of ATP4- energetically favorable, this scenario of ADP/ATP exchange dominates and there is net ADP import and ATP export from the mitochondria.
Figure 2: ANT model of ADP/ATP exchange.
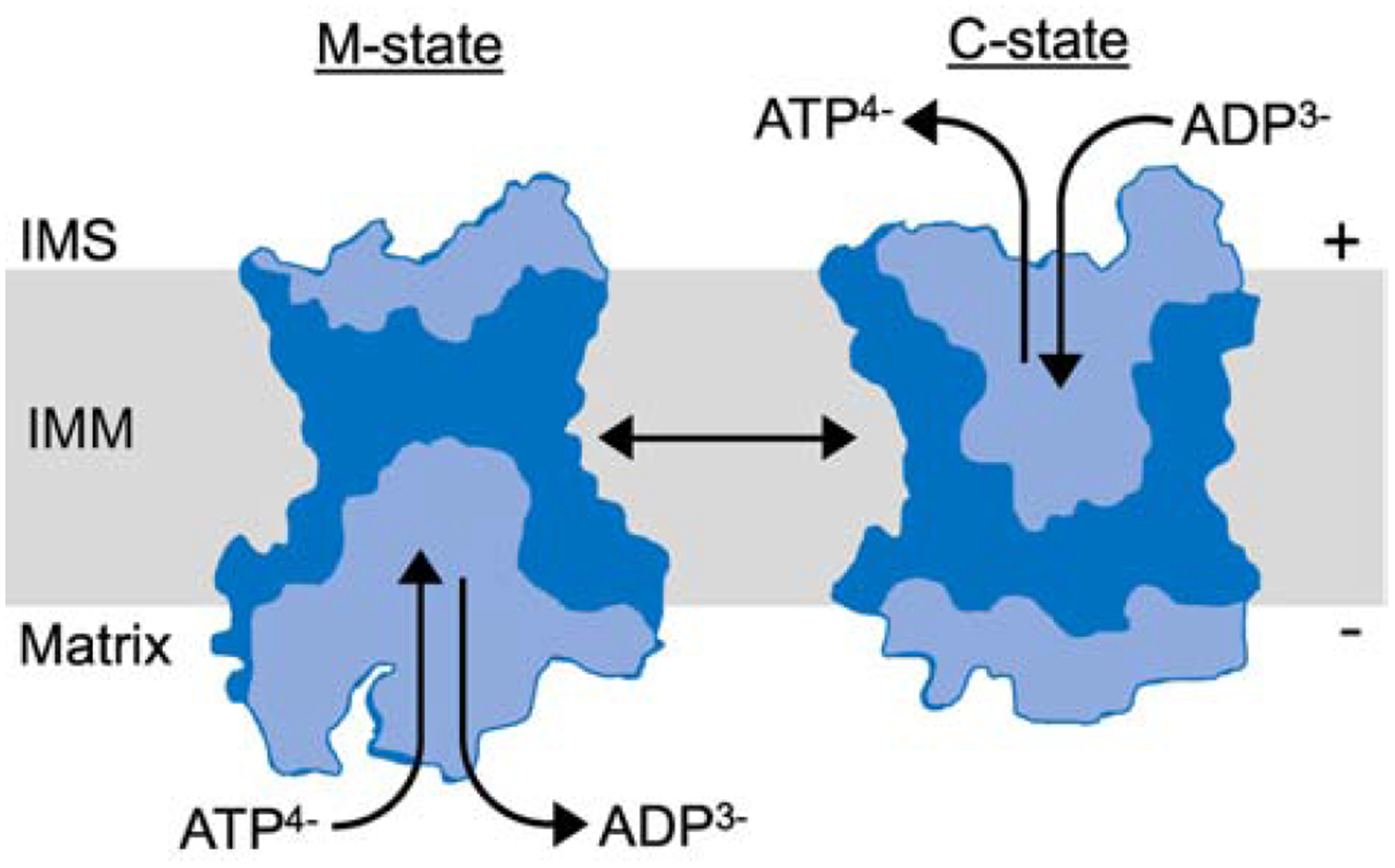
ANT interconverts between a mitochondrial matrix facing m-state conformation and intermembrane space (IMS) facing c-state. In charged mitochondria ATP is exported and ADP is imported, causing the conformational change. The ANT m-state and c-state diagrams were adapted from the ANT crystal structures [7]. Light blue areas depict ANT solute accessible cavities.
ANT research has been highly informed by two unique classes of ANT inhibitors. The first is atractyloside (ATR), a toxic molecule from the Atractylis gummifera thistle plant [56]. ATR is a potent inhibitor of ATP/ADP transfer that binds ANT proteins in the nucleotide binding site, but exclusively when ANT is in the c-state conformation [4, 57–59]. As such, ATR is only able to inhibit ANT function when applied to the exterior of mitochondria and is useful for trapping ANT proteins in the c-state [3, 4, 59]. Notably, this inhibition can be overcome via competition with ADP, which drives ANT proteins back into the m-state [59, 60]. Carboxyatractyloside (CATR), a higher affinity analogue of ATR, locks ANT-proteins in the c-state irreversibly [59, 61]. The second class of ANT inhibitor, bongkrekic acid (BKA), is a respiratory toxin produced by the bacteria Burkholderia gladioli pathovar cocovenenans [62–64]. BKA is another potent inhibitor of ATP/ADP exchange, but unlike ATR it binds exclusively to the m-state conformation from inside the mitochondrial lumen [7, 59, 65–67]. BKA can be used to lock ANT proteins in the m-state [7, 68]. The differential targeting of these two inhibitors has proved invaluable in determining the kinetic and structural features of ANT proteins.
Mitochondrial oxidative metabolism requires diverse enzymatic processes working in concert towards the goal of producing ATP. Evidence exists that one way the mitochondria coordinates this activity is by grouping proteins into supercomplexes to coordinate their activity or increase efficiency. One mitochondrial supercomplex, the so-called “ATP synthasome”, contains the F1F0-ATP synthase as well as ANT and phosphate carrier (PiC) proteins in a 1:1:1 stoichiometric ratio so that the fundamental ATP metabolite transporters are proximal to the site of ATP production [69, 70]. It is thought that this structure ensures a robust supply of inorganic phosphate and ADP to the F1F0-ATP synthase as well as rapid export of produced ATP. While an appreciable amount of the ATP synthasome species can be observed in mitochondria from several tissues, it should be noted that only a small subset of ANT and PiC proteins are found in ATP synthasome structures, while the majority of these proteins are found elsewhere in the mitochondria [70]. ANT proteins have also been shown to associate with the respiratory chain supercomplexes (RCS), including the respirasome, the main RCS species comprised of electron chain complexes I, III, and IV [71, 72]. While ANT association with RCS is not required for the individual function of electron chain complexes or ANT proteins themselves, it has been noted that overall ATP/ADP exchange rates are decreased when RCS assembly is inhibited [71]. This suggests that RSC integrity is important to ANT function, although it is unclear if this is due to direct ANT-RSC interactions or due to secondary effects from changes in Δψ from altered electron transport chain function [71]. Conversely, ANT proteins have also been implicated in RSC and respirasome assembly or stability, since genetic knockdown of ANT1 in H9C2 cardiomyoblasts causes respirasome disintegration [72]. It is currently unclear whether association with mitochondrial supercomplexes affects the non-ADP/ATP transport functions of ANT proteins (as discussed below).
ANT FAMILY AS THE MITOCHONDRIAL PERMEABILITY TRANSITION PORE
The mitochondrial permeability transition pore (MPTP) is a mitochondrial inner membrane channel or group of channels that open in response to acute elevations in matrix Ca2+ concentration, causing the organelle to swell and rupture, precipitating necrotic cell death [73, 74]. Specifically, elevated matrix Ca2+, alone or in combination with oxidative stress, facilitates the formation of a non-specific megachannel with a conductance of 1.3 nS (although smaller conductance states down to 600 pS have been reported) and a diameter of 2.1 nm [74–77]. Opening of the MPTP allows solutes of up to 1.5 kDa to freely diffuse across the inner mitochondrial membrane [74, 77]. MPTP opening also causes the mitochondrial H+ gradient to dissipate, which uncouples the mitochondria and collapses Δψ. This in turn abolishes the mitochondrial capacity to produce ATP and even causes the F1F0 ATP synthase to hydrolyze ATP to pump H+ in an attempt to re-establish Δψ, further consuming ATP reserves [75, 78, 79]. This energetic catastrophe along with mitochondrial swelling and rupture initiates necrotic cell death. Substantial evidence exists that MPTP has physiological roles beyond cell death [80]. It has been noted that MPTP may exhibit low conductance or transient activation events that may contribute to cellular processes such as oxidative energy metabolism [81, 82], mitochondrial Ca2+ homeostasis [81, 83–85], or reactive oxygen species signaling [86, 87]. The exact molecular identity of the MPTP remains controversial [88], but proposed models with ANT will be discussed below.
One universally agreed upon and well-defined molecular component of the MPTP is cyclophilin-D, a matrix peptidyl-prolyl cis-trans isomerase (CypD) that resides in the mitochondrial matrix [89]. A primary chemical inhibitor of MPTP activation is the immunosuppressant drug cyclosporine A (CsA) [90], which directly inhibits CypD activity and its ability to facilitate MPTP opening [91]. Liver mitochondria isolated from mice lacking the Ppif gene (which encodes CypD) demonstrate desensitized MPTP opening, confirming the role of CypD in this process [92–95]. However, while the Ca2+ sensitivity of MPTP activation is reduced in Ppif−/− mitochondria, much higher matrix Ca2+ levels are still able to activate MPTP opening in mitochondria lacking CypD [92–95]. This suggests that CypD is a critical regulator of MPTP but at the same time it is not required for MPTP activation. CsA sensitive, CypD regulated mitochondrial swelling is used as a diagnostic hallmark of MPTP activation experimentally.
In 1990 Halestrap and Davidson proposed that the ANT family of proteins could be the MPTP forming species [96]. This concept stems from the fact that several ANT ligands and inhibitors modulate MPTP opening activity. Adenosine nucleotides, particularly ADP, are potent desensitizers of MPTP opening [96–100]. Depletion of matrix nucleotides through the exogenous treatment of the mitochondria with pyrophosphate, which can be exchanged by ANT proteins albeit with a much lower affinity than ATP or ADP, has the opposite effect and promotes MPTP opening [101]. The ANT inhibitors ATR and CATR sensitize the pore to matrix Ca2+ and increase MPTP activation [96, 98]. Conversely, BKA is a potent desensitizer of MPTP opening and in the presence of this agent substantially higher increases in Ca2+ are required to induce mitochondrial swelling [96, 98]. Mitochondrial uncoupling and oxidative stress are also potent inducers of MPTP [102–107]. ANT proteins, due to their interaction with Δψ and associated ATP/ADP exchange, possess the ability to sense and be regulated by uncoupling [48, 108]. Moreover, ANT proteins possess exposed cysteine residues that previous studies have shown can be chemically modified, thereby altering ATP/ADP exchange [109–111]. Specifically, N-ethylmaleimide (NEM), an inhibitor of ATP/ADP exchange and desensitizer of MPTP opening, binds the m-state of ANT and is similar to BKA in mediating its effect [110, 112]. Conversely, phenylarsine oxide (PAO), a potent inducer of MPTP opening, oxidizes critical cysteine residues that reduces ADP binding and promotes the ANT c-state, analogous to ATR and CATR [107, 112, 113]. ANT proteins clearly have a highly specific association with MPTP opening and remain attractive candidates to be direct molecular components of this pore.
To function as the MPTP-forming species, ANT would need to bind and be regulated by CypD in a CsA dependent manner [96]. Early studies demonstrated that oxidative stress caused by t-butyl hydroperoxide or diamide treatment caused increased in MPTP opening, as well as increased CypD binding to the inner mitochondrial membrane [114, 115]. Some studies have suggested that Ca2+ may enhance this association and ADP treatment may prevent CypD binding in intact mitochondria [116, 117]. The key experimental evidence linking CypD to the ANT family was collected by Woodfield et al., 1998, who demonstrated that ANT was a key protein from a mitochondrial membrane preparation that remained bound to a CypD affinity column following extensive washing [118]. Critically, this tight ANT binding to the column was abolished with CsA pretreatment [118]. A caveat of this study is that ANT is the most abundant inner mitochondrial membrane protein, meaning that less abundant or weaker CypD interacting proteins could have been missed. A similar study showed that isolated ANT could also form a complex with the voltage dependent anion channel (VDAC) from the outer mitochondrial membrane in conjunction with CypD [119]. The isolated ANT/VDAC/CypD-fusion protein complex was reconstituted in proteoliposomes and shown to be permeabilized (opened) by Ca2+ in a CsA-dependent manner [119]. Collectively these studies suggested that ANT proteins can interact with CypD in a CsA dependent manner, which is a critical prerequisite of ANT serving as a potential direct component of the MPTP.
Another key feature of MPTP activity exhibited by ANT proteins is regulation by mitochondrial matrix Ca2+. Studies have demonstrated that increasing matrix Ca2+ in isolated, energized cardiac mitochondria can induce conformational changes in ANT as measured by a characteristic change in light scattering [96, 120]. This conformational change is reversed by BKA or ADP treatment, and this inhibition was reversible with application of CATR, suggesting that matrix Ca2+ specifically promotes the ANT c-state [96]. This change in conformation was not caused by Sr2+ application, which is consistent with the observed specificity of MPTP opening by only Ca2+ [96]. Consistent with these findings, Ca2+ treatment of purified ANT in reconstituted vesicles induced a non-selective pore without CypD; however this system required millimolar concentrations of Ca2+, which is in excess of the MPTP activation threshold in intact mitochondria [121]. While the exact mechanism or binding site for Ca2+ regulation is still unclear, these studies suggests that ANT can interact with matrix Ca2+ in a manner consistent with MPTP regulation.
Elegant reductionist studies have demonstrated that isolated ANT proteins can recapitulate several other aspects of MPTP biology. Specifically it has been shown that purified ANT from heart mitochondria can form 300 – 600 pS non-specific pores when reconstituted in vesicles in response to millimolar Ca2+ [121, 122]. The single channels were gated at > 150mV and closed at pH < 5.5, which is consistent with MPTP opening activity [102, 104, 121]. Formation of this pore was desensitized by ADP or BKA treatment, but was not prevented by treatment with CATR, showing that this channel activity was ANT dependent and that only the ANT m-state was protected from opening [121, 122]. This system lacked CypD, which accounts for a lack of CsA inhibition and potentially for the high Ca2+ levels needed to generate the opening event, consistent with observations of Ppif−/− mice [92–94, 121]. Subsequent and more definitive studies using a neurospora crassa ANT recombinant reconstitution system that included CypD in the preparation demonstrated Ca2+ dependent pore activity with a conductance of 600 pS that was inhibited by the addition of CsA, as well as ADP and BKA [123]. However, it should be noted that this system also required millimolar Ca2+ for ANT-pore formation. Collectively these results demonstrate that isolated ANT can generate an MPTP-like activity and that pore formation is regulated by CypD interaction.
ANT MODEL OF MPTP FORMATION
An early proposed model of the MPTP involved the regulator CypD, VDAC within the outer mitochondrial membrane, and ANT proteins in the inner membrane, all interacting to form the MPTP [79, 108, 119, 124]. In this model the ANT effectively has three states, the m-state and the c-state, which are involved in adenine nucleotide exchange as discussed above, and a third permeable state, the p-state, which occurs when ANT exhibits non-selective pore activity (FIGURE 3). Since MPTP desensitizers such as ADP, BKA, and NEM function to promote the ANT m-state, this conformation can be considered protected and unlikely to directly transition to the p-state [96, 98, 110]. Conversely, ATR, CATR, and PAO sensitize MPTP by stabilizing the c-state of ANT, which can be considered the ‘sensitized state’, since it appears to be the form that transitions into the ANT p-state in response to elevated matrix Ca2+ [96, 98, 110]. Consistent with this concept, nucleotide depletion or mitochondrial uncoupling would equalize the distribution of ANT conformations, increasing the proportion of ANT proteins in the sensitized c-state and therefore facilitating the transition into the p-state [3, 101]. Ca2+lcium itself is also able to promote the shift of ANT from the protected m-state into the c-state, which may be mechanistically important for the eventual transition into the p-state [96, 120]. Recent structural studies have elucidated that the protected m-state is sealed by a combination of “tyrosine braced” intermembrane space facing salt bridges as well as by a hydrophobic “plug” structure, while the sensitized c-state is closed only by a salt bridge network on the matrix side [4, 7, 53]. Moreover, these structural studies have demonstrated that the ANT c-state has an extensive water-filled cavity that is significantly larger than in the m-state cavity [4, 7, 53]. The width of these binding cavities in the c-state and m-state are each ~2.1 nm suggesting that ANT monomers could possess a channel of this width if ANT is simultaneously open to both the matrix and intermembrane space, which would be consistent with MPTP conductance measurements [7]. While subsequent studies have ruled out the requirement of VDAC as a component of the MPTP [125], other work has shown that the ANT c-state can specifically associate with BAX [126], which is consistent with our current understanding of BAX and BAK and their putative role in MPTP-dependent cell death [127]. Collectively, the proposed working model suggests that ANT in the c-state conformation experiences a regulated triggering event that causes a conformational shift into the non-selective p-state.
Figure 3: ANT model of MPTP.
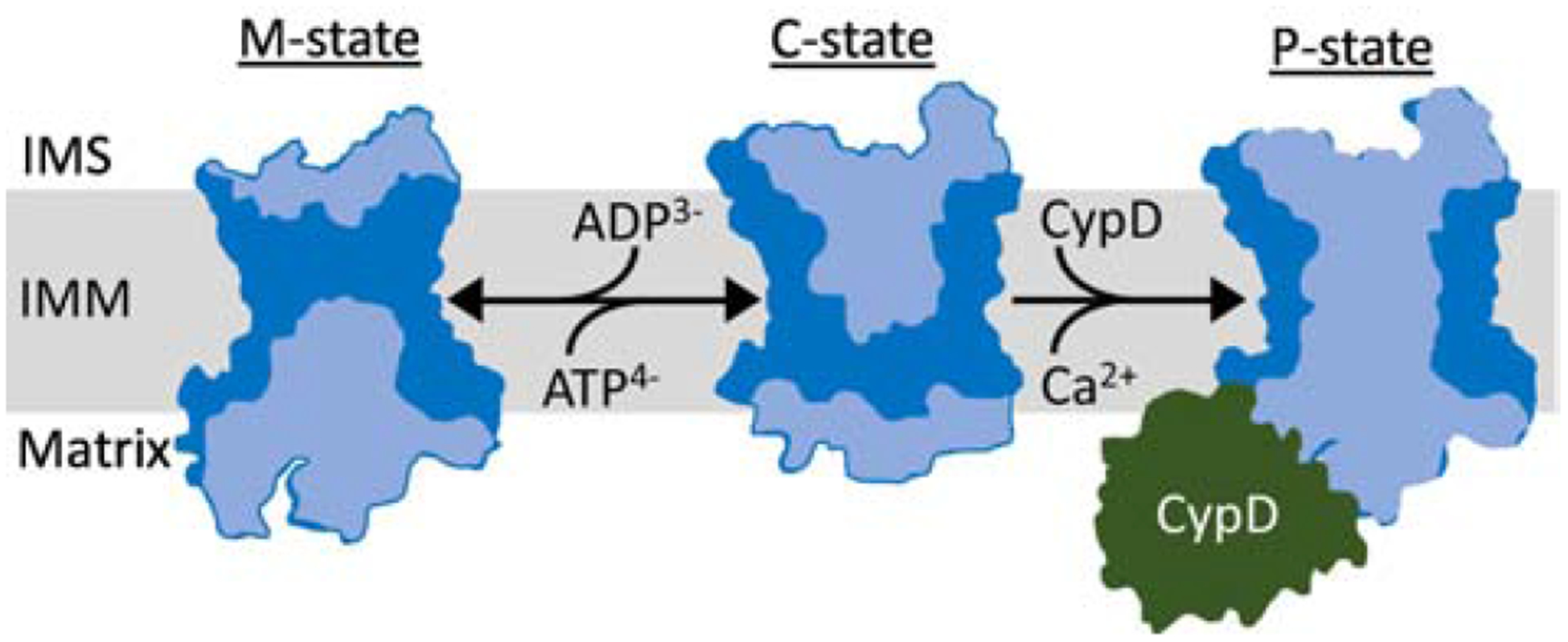
ANT is hypothesized to form a non-selective pore with properties similar to the MPTP. The ANT interconverts between the m- and c-states during ADP/ATP exchange. The m-state is protected and cannot transition directly into the MPTP-like pore, the ‘p-state’. The ANT c-state can convert into the p-state in response to a variety of activating stresses including binding CypD and high matrix Ca2+ levels. According to this model the MPTP is produced when ANT monomers become simultaneously open to both the matrix and intermembrane space (IMS), forming a pore. The ANT m-state and c-state diagrams were adapted from ANT crystal structures [7], while the hypothetical p-state diagram was generated by superimposing the m- and c-states crystal structures to create an ANT-species open to both the matrix and IMS side of the inner mitochondrial membrane (IMM). Light blue areas depict solute accessible ANT cavities. CypD is not drawn to scale and is included for illustration only.
Given the variety of ANT-associated MPTP-modifying compounds and a structural understanding of ANT proteins, some predictions can be made (FIGURE 1). The first prediction is that ANT Proline-62 (Pro62, Human ANT1) is the primary site of CypD binding and activation of ANT [108]. Pro62 is highly conserved in ANT isoforms that demonstrate CsA-sensitive MPTP opening but is absent in yeast ANT that does not possess CsA regulated MPTP opening [108, 118, 128, 129]. Moreover, Pro62 is positioned on the M1 loop that undergoes conformational changes during the transition from the ANT m-state to c-state, suggesting that this loop could be important for ANT channel gating [3, 108]. The second prediction is that conserved cysteine residues located in loop M1 (Cys57), loop M2 (Cys160), and loop M3 (Cys257), are important sites targeted by oxidative stress that also facilitate the pore open state [108, 112, 113]. PAO has been shown to crosslink Cys160 and Cys257, which substantially reduces ANT affinity for ADP, promoting the ANT c-state and MPTP opening [113]. Cys57 has been implicated in ANT dimerization and when alkylated by NEM it promotes the ANT m-state, thus inhibiting MPTP opening [8, 9, 112, 130, 131]. The position of Cys57 in proximity to Pro62 may be mechanistically related to CypD regulation of the pore [108].
The exact nature of the Ca2+ activated trigger remains unclear, although one hypothesis states that cardiolipin in the inner mitochondrial membrane serves as the ANT Ca2+ sensor [3, 121]. As mentioned above, ANT requires cardiolipin for function and stability and 6 molecules of cardiolipin are isolated per molecule of ANT [17–19]. In this situation matrix Ca2+ or oxidative stress can disrupt the association of cardiolipin with a charged collar of lysine residues in the matrix side of ANT, which would then open the matrix gate of the ANT c-state [3, 121]. Consistent with this idea, recent structural studies have revealed that cardiolipin forms an interdomain bridge that may be important in stabilizing ANT conformations [7, 53]. Furthermore, it has been observed that mitochondria isolated from tafazzin knockdown mice, which contain significantly reduced mature cardiolipin levels, display increased basal mitochondrial swelling as well as reduced rates of Ca2+ induced swelling, which suggests a functional link between cardiolipin and MPTP function [132]. An alternate model suggests that Ca2+ directly interacts with acidic residues in the core of the ANT c-state. According to this view the acidic residues in the M2 and M3 ANT loops may orient in such a way as to create a Ca2+ binding pocket [108]. Specifically, glutamate-153 (Glu153) and aspartate-168 (Asp168) in M2 have been suggested as potential Ca2+ sensors [108]. These residues are involved in ADP binding that perhaps offers a mechanistic rationale for ADP desensitization of MPTP opening [3, 108, 133]. Consistent with a role for Glu153 in regulating MPTP opening through Ca2+, treatment with high concentrations of NEM can modify the adjacent Cys160 in loop M2, which can also decrease Ca2+ sensitivity [112]. It is further possible that treatment with arginine-modifying reagents phenylglyoxal and 2,3-butanedione, which results in decreased Ca2+ sensitivity, chemically modifies Arg152, disrupting Glu153 Ca2+ sensing [108, 134]. Finally, Ca2+ could interact with or directly disrupt the ANT c-state matrix salt bridge network, which would cause ANT to open into a pore-like structure [4, 7]. However, to date the experiments needed to distinguish between these scenarios as the Ca2+ sensing mechanism of ANT have not been reported.
EVIDENCE AGAINST THE ANT MODEL
Kokoszka et al suggested that the MPTP could still form in mitochondria isolated from livers of Ant1/2 double deleted mice [135]. More specifically, Kokoszka et al generated mice lacking both ANT1 and ANT2 protein expression in the liver by crossing Ant1 (Slc25a4) gene-deleted mice (Ant1−/−)[32] with mice harboring conditional Ant2 loxP (f)-site targeted alleles (Ant2f/f) with a liver specific Cre recombinase transgenic line driven by the albumin promotor (AlbCre). The resulting Ant1−/−Ant2f/fAlbCre mice were viable and displayed virtually complete loss of ANT1 and ANT2 proteins in the liver. Ant1/2-deficient liver mitochondria displayed similar levels of baseline oxygen consumption to control mitochondria but were unable to be metabolically stimulated with 125 nmols of ADP, indicating compromised ANT activity. The key finding of this study was that Ant1/2 –deficient liver mitochondria retained functional MPTP activity that was activated by oxidative stress or matrix Ca2+ [32]. This Ant1/2-independent MPTP activity was also inhibited by CsA, a hallmark of the process, but was no longer sensitized by the ANT inhibitor ATR. However, loss of Ant1/2 in liver mitochondria did cause a significant change in MPTP sensitivity to Ca2+, requiring 3-fold more Ca2+ to produce swelling, which is approximately equivalent to the protection generated from CsA treatment [32]. Moreover, treating Ant1/2-deficient mitochondria with CsA had a profound synergistic effect where 10-fold more matrix Ca2+ was required to activate MPTP opening compared with control mitochondria. Therefore, while MPTP activation occurred in Ant1/2-deficient mitochondria, the behavior of this process was dramatically altered. Despite obvious alterations in MPTP activity, this study was adopted as initial proof that ANT proteins were not a pore-forming species of the MPTP.
RE-EVALUATING THE ROLE OF ANT IN MPTP FORMATION
In the years following the publication of Kokoszka et al it was recognized that mammals possess an additional ANT isoform, and in the mouse this represents ANT4 (Slc25a31) [31]. In rodents ANT4 expression is typically restricted to gamete producing reproductive cells, primarily in the testicles, while in humans ANT4 mRNA has also been detected in the brain and liver [31, 37]. In light of this finding, we reevaluated the role of ANT proteins in MPTP with the hypothesis that Ant4 may provide compensation in Ant1/2-deficient liver as originally reported by Kokoszka et al [135, 136]. We first examined the expression profile of the three murine ANT isoforms using specific antibodies and confirmed that ANT4 expression is induced in the liver following loss of ANT1 and ANT2 protein from the same gene-targeted mice [136]. To examine whether ANT4 could account for the remaining MPTP activity in Ant1/2-defcieint livers we crossed Ant1/2 liver-deleted mice (Ant1−/−Ant2f/fAlbCre)[135] with Ant4 (Slc25a31) deleted mice [35], to generate mice lacking expression of all three rodent ANT isoforms in the liver (ANT-null mice). We found that livers from ANT-null mice still exhibited MPTP opening, however substantially higher levels of matrix Ca2+ were required to induce pore opening. This MPTP-desensitization to Ca2+ was significantly beyond the protection conveyed by Ant1/2 deletion alone indicating that the ANT4 protein contributes to some of the residual MPTP activity observed in Ant1/2-deficient liver mitochondria reported by Kokoszka et al. [136]. However, despite a total loss of all known ANT proteins in the liver, MPTP was still observed, meaning that ANT proteins cannot account for total MPTP activity in this tissue. However, when ANT-null liver mitochondria were treated with CsA, MPTP opening was completely prevented, despite the mitochondria taking up >1.6 mM CaCl2/mg mitochondria, which was the limit of the assay (when Ca2+ absorption by the mitochondria could no longer be measured) [136]. The converse experiment was then performed by treating isolated CypD-null (Ppif−/−) liver mitochondria with ADP, an ANT substrate that promotes the ANT m-state conformation and inhibits ANT-pore formation in isolated systems [96–100]. This experiment showed that inhibiting ANT-pore formation with ADP essentially silenced MPTP opening to supraphysiologic Ca2+ in mitochondria lacking CypD. We then combined these two mouse models to generate quadruple null mice lacking both CypD and all known isoforms of ANT (CypD-ANT-null mice). Liver mitochondria isolated from these mice failed to undergo Ca2+ stimulated MPTP at Ca2+ levels that saturated the assay. This represents the first genetic model in which MPTP opening was completely abrogated. Collectively these findings indicate that MPTP opening in liver mitochondria requires the presence of at least one isoform of ANT protein and CypD.
Another surprising aspect of ANT-null and CypD-ANT-null liver mitochondria is that they were still able to transport and metabolically respond to exogenous ADP despite a lack of ANT proteins [136]. However, when we generated mouse embryonic fibroblasts (MEFs) lacking all three Ant genes from our gene-targeted mice, mitochondria from them did not transport or metabolically respond to exogenous ADP [136]. Mitoplasts isolated from wildtype or ANT2-only MEFs showed channel opening and conductance consistent with the MPTP, and these channels could be inhibited by ADP, an MPTP opening suppressor. Conversely, ANT-null MEF mitochondria showed a much lower prevalence of large conductance pores, and all the observed rare pores were unresponsive to ADP, suggesting that no MPTP-like channels were present in the absence of ANT proteins in MEFs. These results suggest that ANT proteins are the primary and perhaps only MPTP-forming species in MEFs [136].
THE MULTIPLE PORE MODEL OF MPTP
We believe that the data discussed above demonstrates that the ANT family of proteins can function as MPTP-forming components, although a more comprehensive model that explains all aspects of the data in the current literature as of the year 2020 is that of a “multiple pore” model (FIGURE 4) [136]. The first species consists of ANT proteins that can form MPTP pores in response to high matrix Ca2+ or oxidative stress and can be modulated with ADP and ANT inhibitors. ANT proteins, while regulated by CypD, can activate independent of it in response to very high matrix Ca2+ levels. However, the unknown second pore-forming species absolutely requires CypD activity and cannot activate in its absence. If the “multiple pore” model of MPTP is correct and there are at least two pore-forming components involved in the process, this may explain the difficulty in defining the molecular identity of the pore for so many years. Hence, future investigation is needed to identify the other potential proteins that can additionally generate the pore-forming aspect of the MPTP. Several leading hypotheses have been proposed for additional candidates that can form the MPTP.
Figure 4: The “Multiple Pore” model of MPTP.
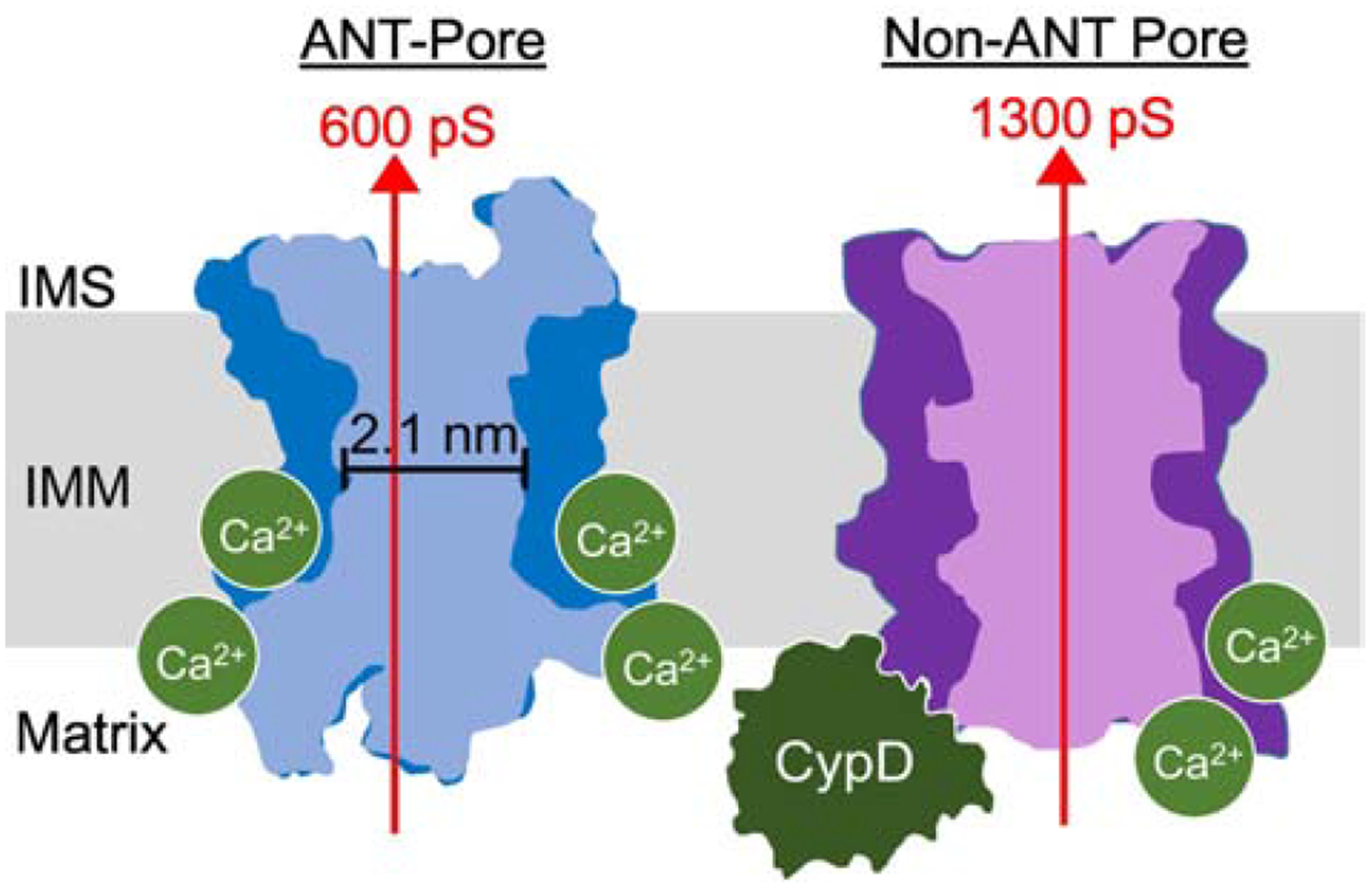
The MPTP is hypothesized to be comprised of multiple protein species, one of which is the ANT. In our model, ANT generates a pore with a conductance of up to 300–600 pS [123, 151] and with a theoretical diameter of 2.1 nm (based on the narrowest channel opening in ANT crystal structures [7]). ANT-pore activity is regulated by CypD but can also activate in the absence of CypD in response to very high levels of matrix Ca2+. It is currently unclear if Ca2+ binds to ANT directly to regulate its MPTP-activity, however we favor a model where Ca2+ directly activates the channel since Ca2+ treatment can activate channel behavior in vesicles containing purified ANT [121–123]. There is at least one non-ANT pore forming species. This non-ANT pore responds to matrix Ca2+ but absolutely requires CypD to activate. This non-ANT pore may generate a conductance of 1300 pS [151]. The ANT pore diagram was generated from superimposing the m-state and c-state crystal structures as in Figure 3 [7].
One hypothesis is that of the “non-selective pore”, in which other select SLC25A family members of translocases or exchangers present within the mitochondrial inner membrane also adopt a CypD regulated activity like ANT. For example, it has been observed that several mitochondrial carrier family proteins including uncoupling protein 1 (UCP1) and the glutamate/aspartate exchanger, can form non-selective protein channels with similar properties to the MPTP in response to thiol reagent treatment or lipotoxicity [137–139]. However, these specific channels do not show CsA or Ca2+ regulation, although they do serve as an example that this general class of SLC25A proteins have such pore generating properties. Furthermore, not all mitochondrial SLC25A carrier proteins display this activity, potentially suggesting that only a subset of these type of carrier proteins would be relevant [140]. The phosphate carrier (Slc25a3; PiC) is a likely candidate for this alternate MPTP model [141]. Phosphate, the substrate of PiC, is a strong inducer of MPTP opening [106], and one study showed that PAO can activate MPTP in the absence of ANT, where it was suggested that PiC was the critical remaining MPTP effector [141]. Moreover, CypD is able to bind PiC and this binding can be modulated by CsA treatment or oxidative stress [141]. However, heart-specific Slc25a3 (PiC) gene-deleted mice retained MPTP activity, which was originally interpreted to exclude PiC from being the pore-forming species in MPTP activity, although this interpretation is potentially incorrect if ANT fully compensated as part of the multiple pore model proposed here [142]. Another potential MPTP candidate is one or more proteins that account for the unknown source of ADP transport in ANT-null liver mitochondria that we identified [136]. It is conceivable that since ANT family members function as pore-forming units, one or more proteins with unrecognized adenosine nucleotide translocator activity might also possess MPTP activity.
A second hypothesis that some groups have proposed is that the F1F0 ATP synthase may be the pore-forming species in MPTP activity [143–147], although this remains controversial [88]. Recently, three studies targeted key subunits of the F1F0ATP synthase that are predicted to directly impact MPTP activity in this complex, but the results instead showed that MPTP opening occurred in the absence of every theoretically important F1F0ATP synthase subunit suggesting that neither monomers or dimers of this complex could comprise the MPTP [148–150]. Moreover, a recent study by Neginskaya et al., 2019 showed that mitoplasts lacking the F1F0ATP synthase c-subunit, which was predicted to be important for F1F0 ATP synthase-pore formation [144, 145], had a pore conductance of only ~300 pS, lower than the 1.3 nS channel recorded in wildtype mitoplasts with bonafide MPTP activity [151]. This 300 pS channel has a conductance consistent with some measurements of isolated ANT channels (which typically range from 300–600 pS) and could be inhibited by BKA suggesting it is generated by ANT [121, 151]. The results of this study are consistent with the multiple pore model of MPTP and a role for ANT pore formation in this process, as well as a potential role for the F1F0 ATP synthase.
A third hypothesis that fits a multiple protein species MPTP paradigm is the model put forward by He and Lemasters whereby mitochondrial permeability transition may be the result of misfolded protein aggregates forming pores in the inner mitochondrial membrane [152]. In this model there are two distinct modes of MPTP activation, a “regulated” mode that is activated by CypD and Ca2+ and an “unregulated” mode that is insensitive to CsA and is induced by elevated stresses. In this context Ca2+ and CypD isomerase activity drive protein misfolding that increases aggregation and causes MPTP-formation. Our research into the role of ANT in MPTP is interesting when viewed through this lens. Since mitochondria lacking all ANT isoforms lack “unregulated” CypD-independent MPTP activation, it directly contradicts the “misfolded protein” hypothesis [136]. Hence, the “unregulated” MPTP activation model has a specific genetic identity and is not due to a general misfolded protein effect. However, given the sheer abundance of ANT in the inner mitochondrial membrane and its association with mitochondrial protein supercomplexes it is possible that loss of ANT prevents “unregulated” opening by reducing the total amount of SLC25a protein in themitochondrial inner membrane below a permissive concentration threshold [69, 71, 72, 152]. Despite these considerations, the current experimental evidence cannot distinguish between a “multiple-pore” model and “misfolded protein” model of MPTP.
ANT IN MITOCHONDRIAL UNCOUPLING
Mitochondrial uncoupling occurs when H+ leaks across the inner mitochondrial membrane in a manner that bypasses utilization by the F1F0 ATP synthase. In brown and beige adipose tissue this transfer is facilitated by UCP1, which co-transports H+ with short chain fatty acid (FA) anions into the mitochondrial matrix [153–156]. However, several tissues exhibit H+ leak that do not express UCP1, particularly heart and skeletal muscle [157], which can be inhibited by CATR treatment, implicating ANT proteins in this process [158–161]. Bertholet et al., provided compelling evidence that ANT proteins are the major mediator of H+ leak in a variety of tissues [162]. Utilizing a series of mitoplast-based electrophysiological experiments the authors demonstrate that skeletal muscle, heart, and liver mitochondria exhibit FA-activated H+ leak that can be inhibited by CATR treatment [162]. This contrasts with brown adipose tissue that showed much larger proton leak that was inhibited by guanosine diphosphate treatment, which is a strong inhibitor of UCP1 [162]. More definitively, the authors showed that this CATR-sensitive, FA-activated H+ leak was not observed in heart or skeletal muscle mitoplasts lacking ANT2 (isolated from Ant2 hypomorphic mice [163]) providing direct genetic evidence for involvement of ANT2 in this process. Moreover, the authors also observed that FA-activated H+ leak persists in heart or skeletal muscle mitoplasts lacking UCP1, UCP2, or UCP3, demonstrating this phenomenon is independent of canonical uncoupling proteins. In contrast, brown adipose mitoplasts lost the majority of H+ leak in the absence of UCP1 expression [162]. The authors suggest that FA-activated H+ leak can occur in either the ANT c-state or m-state. This study provides conclusive evidence that ANT proteins are a primary source of FA-activated mitochondrial uncoupling in a majority of tissues and likely underlies important physiological processes such as muscle thermogenesis [3].
ANT IN MITOPHAGY
A recent paper by Hoshino et al., proposed a novel role for ANT proteins in mitophagy [164]. This group conducted CRISPR-Cas9 genome-wide screen to identify novel mitophagy genes. The authors found that genetic deletion of Ant1 (Slc25a4) or Ant2 (Slc25a5) suppressed mitophagy in a variety of cell-types in response to uncoupling or treatment with metabolic toxins. Strikingly, reduced mitophagy was observed in the context of compromised energetics since Ant1 and Ant2 deletion displayed greater depolarization than control cells. Paradoxically, pharmacologically inhibiting ANT proteins with BKA or ibipinabant [165] in wildtype cells promoted mitophagy, as would be expected from the metabolic effects of inhibiting ADP/ATP transport. This suggests that ANT proteins regulate mitophagy via a mechanism independent from their role as ADP/ATP translocators. Consistent with this, mitophagy could be rescued in cells lacking ANT1 by expressing the ATP-binding mutant Ant1K43E/R244E [166], which lacks ADP/ATP transport. Conversely, Ant1 mutants identified from cardiomyopathy patients (Ant1A123D) [167] and external opthalmoplegia patients (Ant1A90D) [168], which retain ADP/ATP translocase activity [164, 169], were unable to rescue mitophagy in cells lacking ANT1. The authors interpret these data to suggest that ANT regulates mitophagy through physical interactions with the mitophagic machinery.
A key observation was that PTEN-induced kinase 1 (PINK1) fails to accumulate on the outer mitochondrial membrane in cells lacking ANT1 or ANT2 [164]. PINK1 is a key component of the mitophagic machinery that marks dysfunctional mitochondria for degradation [170]. In healthy mitochondria, PINK1 is consistently translocated via the TOM and TIM23 complex into the mitochondrial matrix and degraded [170]. During mitochondrial dysfunction, loss of Δψ prevents PINK1 translocation into the mitochondrial matrix via TIM23 since this process requires Δψ [170]. Since PINK1 cannot enter the matrix, it instead accumulates on the outer mitochondrial membrane where it recruits the E3 ubiquitin ligase Parkin, which initiates the mitophagic process [170]. The fact that cells lacking ANT1 and ANT2 do not accumulate PINK1 despite mitochondrial uncoupling is surprising and challenges the widely accepted model of PINK1 processing. The authors demonstrate that ANT proteins physically interact with the TIM23 complex via the protein TIM44, an important subunit involved in peptide import, and that disrupting this interaction via mutations on ANT or TIM44 prevents PINK1 accumulation and disrupts mitophagy [164]. The authors propose that ANT proteins act as bioenergetic sensors that suppress TIM23 translocation of PINK1, thus facilitating the stabilization of PINK1 on the outer mitochondrial membrane and activating mitophagy. The fact that PINK1 translocation continues to occur in uncoupled mitochondria lacking ANT proteins represents a substantial challenge not only to the understanding of mitophagy, but also to the current model of how proteins are transported into the mitochondrial matrix [171]. As with any novel concept, a number of unresolved questions remain to be addressed, such as testing whether ANT is required for TIM23-dependent mitochondrial protein import in general, as would be predicted by the authors’ model. Another question is if the observed uncoupling behavior of ANT proteins in mitophagy underlie the known cardiac pathology in Ant1 null mice and humans. Given that uncoupling mitochondria is a necessary step in PINK1 dependent mitophagy in vivo, it would not be outlandish to suggest that either FA-dependent H+ leak or MPTP-opening could be upstream of mitophagy. Future studies using knock-in mouse models of the various ANT-mutants utilized in Hoshino et al., 2019 could be valuable tools for understanding the significance of the proposed model in pathophysiology in vivo [164], especially since PINK1 null mice lack a mitophagy phenotype in metabolically active tissues in vivo [172].
CONCLUSION AND FUTURE DIRETIONS
ANT proteins are critical metabolic effectors of mitochondrial energy homeostasis through ADP/ATP exchange. However, a large body of experimental evidence demonstrates that ANT proteins are also involved in MPTP-dependent cell death and recent work by our group has suggested that the ANT family of proteins represent part of the pore-forming molecular machinery of the MPTP. Additionally, recent studies have provided evidence that ANT proteins can cause FA-dependent H+ leak and uncoupling, and that ANT proteins serve as the mitophagy receptor. An interesting facet of this collective work is that ANT can sense and initiate cellular responses to abnormal mitochondrial conditions such as hyperpolarization, mitochondrial damage, or energetic catastrophe. As such, ANT proteins are likely key integration sites for mitochondrial health, function and cell death. In moving forward beyond 2020, additional studies will be needed to define the regulatory mechanisms of these processes and to better characterize the different modes of ANT action.
With the current state of genetic tools and advances in mitochondrial experimental approaches the field has an opportunity to delve into the complex biology of ANT proteins. With recently published [7, 13, 53], high quality ANT protein structure data, experiments can be designed targeting individual amino acid residues thought to be critical in these divergent processes. For example, the generation of ANT-mutants that can participate in normal ADP/ATP exchange, but which disrupt an individual alternate ANT function (MPTP, H+ leak, mitophagy) will be valuable in isolating alternate ANT effects from metabolism, as well as determine the role of each individual ANT gene product. From an MPTP perspective, generating knock-in mutants of ANT that lack CypD regulation or that target the putative calcium binding effects will provide further evidence for the role of ANT in generating the MPTP. Similarly, generating ANT mutants lacking FA-dependent H+ leak while retaining ADP/ATP transport will allow the field to explore the metabolic and whole body physiology of this process in vivo.
Another open question is which of these processes are specific to the ANT family of proteins and which are more general features of mitochondrial carriers. Given the high homology of the SLC25a mitochondrial carrier family it has been proposed that additional carriers could share common structural and functional features with ANT and play a role in MPTP, mitophagy, or the H+ leak FA transport mechanism [7]. However, with the sheer abundance of ANT proteins in the inner mitochondrial membrane, it is possible that they dominate based on mass action instead of specificity. As such, past studies do not rule out the possibility that other SLC25A mitochondrial carrier proteins possess the same activities that could also become important in the absence of ANT. In particular we are interested in which aspects of ANT biology are shared by the phosphate carrier, PiC, which is also a highly abundant SLC25A family member in the inner membrane. As discussed above, PiC has been implicated as a MPTP-forming species and shares many structural properties with ANT proteins [141]. Genetic deletion of Slc25a3 (PiC), particularly in the context of cells with reduced ANT expression, could be a method to test the specific role of PiC in MPTP, H+ leak, and mitophagy. The role of other SLC25A proteins could also be similarly addressed using genetics in mice or cell lines.
Within this review we favored a definition of MPTP that includes all protein channels and electrophysiological currents that contribute since we believe this approach best reflects the current state of the field. However with the emerging knowledge that ANT proteins comprise part of the complement of MPTP proteins it is now possible to design research that assigns specific conductance and regulatory characteristics to both the ANT and non-ANT contributions to the overall MPTP. As our understanding of the protein species involved in MPTP improves, we expect that MPTP will be defined in a genetic way in the future (such as MPTPANT1, MPTPANT2, MPTPf1/f0ATPase, etc).
Highlights.
Adenosine Nucleotide Transporters are multi-functional mitochondrial proteins
ANT proteins exchange ATP/ADP across the inner-mitochondrial membrane
ANTs can generate a mitochondrial permeability transition pore (MPTP) activity
MPTP is comprised of multiple proteins and has both ANT and non-ANT pores
ANT proteins are also involved in mitochondrial H+ leak and mitophagy
Sources of funding
This work was supported by grants from the National Heart Lung and Blood Institute of the National Institutes of Health (R01-HL132831 to JDM and DMB, R01-HL132831 JDM, R01-HL030077 to DMB), the Howard Hughes Medical Institute (JDM), and by a grant from the Fondation Leducq (JDM). MJB was supported by a grant from American Heart Association (18POST33960216).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest:
All authors confirm no conflict of interest with the current study
REFERENCES
- [1].Aquila H, Link TA, Klingenberg M. Solute carriers involved in energy transfer of mitochondria form a homologous protein family. FEBS Lett 1987; 212: 1–9. [DOI] [PubMed] [Google Scholar]
- [2].Monne M, Palmieri F. Antiporters of the mitochondrial carrier family. Curr Top Membr 2014; 73: 289–320. [DOI] [PubMed] [Google Scholar]
- [3].Klingenberg M. The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta 2008; 1778: 1978–2021. [DOI] [PubMed] [Google Scholar]
- [4].Pebay-Peyroula E, Dahout-Gonzalez C, Kahn R, Trezeguet V, Lauquin GJ, Brandolin G. Structure of mitochondrial ADP/ATP carrier in complex with carboxyatractyloside. Nature 2003; 426: 39–44. [DOI] [PubMed] [Google Scholar]
- [5].Aquila H, Misra D, Eulitz M, Klingenberg M. Complete amino acid sequence of the ADP/ATP carrier from beef heart mitochondria. Hoppe Seylers Z Physiol Chem 1982; 363: 345–9. [PubMed] [Google Scholar]
- [6].Saraste M, Walker JE. Internal sequence repeats and the path of polypeptide in mitochondrial ADP/ATP translocase. FEBS Lett 1982; 144: 250–4. [DOI] [PubMed] [Google Scholar]
- [7].Ruprecht JJ, King MS, Zogg T, Aleksandrova AA, Pardon E, Crichton PG, et al. The Molecular Mechanism of Transport by the Mitochondrial ADP/ATP Carrier. Cell 2019; 176: 435–447 e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hashimoto M, Majima E, Goto S, Shinohara Y, Terada H. Fluctuation of the first loop facing the matrix of the mitochondrial ADP/ATP carrier deduced from intermolecular cross-linking of Cys56 residues by bifunctional dimaleimides. Biochemistry 1999; 38: 1050–6. [DOI] [PubMed] [Google Scholar]
- [9].Majima E, Ikawa K, Takeda M, Hashimoto M, Shinohara Y, Terada H. Translocation of loops regulates transport activity of mitochondrial ADP/ATP carrier deduced from formation of a specific intermolecular disulfide bridge catalyzed by copper-o-phenanthroline. J Biol Chem 1995; 270: 29548–54. [DOI] [PubMed] [Google Scholar]
- [10].Hackenberg H, Klingenberg M. Molecular weight and hydrodynamic parameters of the adenosine 5’-diphosphate--adenosine 5’-triphosphate carrier in Triton X-100. Biochemistry 1980; 19: 548–55. [DOI] [PubMed] [Google Scholar]
- [11].Ryan MT, Muller H, Pfanner N. Functional staging of ADP/ATP carrier translocation across the outer mitochondrial membrane. J Biol Chem 1999; 274: 20619–27. [DOI] [PubMed] [Google Scholar]
- [12].Bamber L, Harding M, Monne M, Slotboom DJ, Kunji ER. The yeast mitochondrial ADP/ATP carrier functions as a monomer in mitochondrial membranes. Proc Natl Acad Sci U S A 2007; 104: 10830–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Kunji ER, Harding M. Projection structure of the atractyloside-inhibited mitochondrial ADP/ATP carrier of Saccharomyces cerevisiae. J Biol Chem 2003; 278: 36985–8. [DOI] [PubMed] [Google Scholar]
- [14].Hatanaka T, Hashimoto M, Majima E, Shinohara Y, Terada H. Functional expression of the tandem-repeated homodimer of the mitochondrial ADP/ATP carrier in Saccharomyces cerevisiae, Biochem Biophys Res Commun 1999; 262: 726–30. [DOI] [PubMed] [Google Scholar]
- [15].Trezeguet V, Le Saux A, David C, Gourdet C, Fiore C, Dianoux A, et al. A covalent tandem dimer of the mitochondrial ADP/ATP carrier is functional in vivo. Biochim Biophys Acta 2000; 1457: 81–93. [DOI] [PubMed] [Google Scholar]
- [16].Huang SG, Odoy S, Klingenberg M. Chimers of two fused ADP/ATP carrier monomers indicate a single channel for ADP/ATP transport. Arch Biochem Biophys 2001; 394: 67–75. [DOI] [PubMed] [Google Scholar]
- [17].Beyer K, Klingenberg M. ADP/ATP carrier protein from beef heart mitochondria has high amounts of tightly bound cardiolipin, as revealed by 31P nuclear magnetic resonance. Biochemistry 1985; 24: 3821–6. [DOI] [PubMed] [Google Scholar]
- [18].Beyer K, Nuscher B. Specific cardiolipin binding interferes with labeling of sulfhydryl residues in the adenosine diphosphate/adenosine triphosphate carrier protein from beef heart mitochondria. Biochemistry 1996; 35: 15784–90. [DOI] [PubMed] [Google Scholar]
- [19].Hoffmann B, Stockl A, Schlame M, Beyer K, Klingenberg M. The reconstituted ADP/ATP carrier activity has an absolute requirement for cardiolipin as shown in cysteine mutants. J Biol Chem 1994; 269: 1940–4. [PubMed] [Google Scholar]
- [20].Boxer DH, Feckl J, Klingenberg M. Identity between the major protein located at the outer face of the inner mitochondrial membrane and carboxyatractylate binding protein. FEBS Lett 1977; 73: 43–6. [PubMed] [Google Scholar]
- [21].Brenner C, Subramaniam K, Pertuiset C, Pervaiz S. Adenine nucleotide translocase family: four isoforms for apoptosis modulation in cancer. Oncogene 2011; 30: 883–95. [DOI] [PubMed] [Google Scholar]
- [22].Li K, Warner CK, Hodge JA, Minoshima S, Kudoh J, Fukuyama R, et al. A human muscle adenine nucleotide translocator gene has four exons, is located on chromosome 4, and is differentially expressed. J Biol Chem 1989; 264: 13998–4004. [PubMed] [Google Scholar]
- [23].Stepien G, Torroni A, Chung AB, Hodge JA, Wallace DC. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J Biol Chem 1992; 267: 14592–7. [PubMed] [Google Scholar]
- [24].Cozens AL, Runswick MJ, Walker JE. DNA sequences of two expressed nuclear genes for human mitochondrial ADP/ATP translocase. J Mol Biol 1989; 206: 261–80. [DOI] [PubMed] [Google Scholar]
- [25].Wijmenga C, Winokur ST, Padberg GW, Skraastad MI, Altherr MR, Wasmuth JJ, et al. The human skeletal muscle adenine nucleotide translocator gene maps to chromosome 4q35 in the region of the facioscapulohumeral muscular dystrophy locus. Hum Genet 1993; 92: 198–203. [DOI] [PubMed] [Google Scholar]
- [26].Giraud S, Bonod-Bidaud C, Wesolowski-Louvel M, Stepien G. Expression of human ANT2 gene in highly proliferative cells: GRBOX, a new transcriptional element, is involved in the regulation of glycolytic ATP import into mitochondria. J Mol Biol 1998; 281: 409–18. [DOI] [PubMed] [Google Scholar]
- [27].Ku DH, Kagan J, Chen ST, Chang CD, Baserga R, Wurzel J. The human fibroblast adenine nucleotide translocator gene. Molecular cloning and sequence. J Biol Chem 1990; 265: 16060–3. [PubMed] [Google Scholar]
- [28].Schiebel K, Mertz A, Winkelmann M, Nagaraja R, Rappold G. Localization of the adenine nucleotide translocase gene ANT2 to chromosome Xq24-q25 with tight linkage to DXS425. Genomics 1994; 24: 605–6. [DOI] [PubMed] [Google Scholar]
- [29].Slim R, Levilliers J, Ludecke HJ, Claussen U, Nguyen VC, Gough NM, et al. A human pseudoautosomal gene encodes the ANT3 ADP/ATP translocase and escapes X-inactivation. Genomics 1993; 16: 26–33. [DOI] [PubMed] [Google Scholar]
- [30].Schiebel K, Weiss B, Wohrle D, Rappold G. A human pseudoautosomal gene, ADP/ATP translocase, escapes X-inactivation whereas a homologue on Xq is subject to X-inactivation. Nat Genet 1993; 3: 82–7. [DOI] [PubMed] [Google Scholar]
- [31].Dolce V, Scarcia P, Iacopetta D, Palmieri F. A fourth ADP/ATP carrier isoform in man: identification, bacterial expression, functional characterization and tissue distribution. FEBS Lett 2005; 579: 633–7. [DOI] [PubMed] [Google Scholar]
- [32].Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, Wallace DC. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat Genet 1997; 16: 226–34. [DOI] [PubMed] [Google Scholar]
- [33].Levy SE, Chen YS, Graham BH, Wallace DC. Expression and sequence analysis of the mouse adenine nucleotide translocase 1 and 2 genes. Gene 2000; 254: 57–66. [DOI] [PubMed] [Google Scholar]
- [34].Ellison JW, Salido EC, Shapiro LJ. Genetic mapping of the adenine nucleotide translocase-2 gene (Ant2) to the mouse proximal X chromosome. Genomics 1996; 36: 369–71. [DOI] [PubMed] [Google Scholar]
- [35].Brower JV, Lim CH, Jorgensen M, Oh SP, Terada N. Adenine nucleotide translocase 4 deficiency leads to early meiotic arrest of murine male germ cells. Reproduction 2009; 138: 463–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lim CH, Brower JV, Resnick JL, Oh SP, Terada N. Adenine nucleotide translocase 4 is expressed within embryonic ovaries and dispensable during oogenesis. Reprod Sci 2015; 22: 250–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Brower JV, Rodic N, Seki T, Jorgensen M, Fliess N, Yachnis AT, et al. Evolutionarily conserved mammalian adenine nucleotide translocase 4 is essential for spermatogenesis. J Biol Chem 2007; 282: 29658–66. [DOI] [PubMed] [Google Scholar]
- [38].Rodic N, Oka M, Hamazaki T, Murawski MR, Jorgensen M, Maatouk DM, et al. DNA methylation is required for silencing of ant4, an adenine nucleotide translocase selectively expressed in mouse embryonic stem cells and germ cells. Stem Cells 2005; 23: 1314–23. [DOI] [PubMed] [Google Scholar]
- [39].Brower JV, Lim CH, Han C, Hankowski KE, Hamazaki T, Terada N. Differential CpG island methylation of murine adenine nucleotide translocase genes. Biochim Biophys Acta 2009; 1789: 198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Pfaff E, Klingenberg M. Adenine nucleotide translocation of mitochondria. 1. Specificity and control, Eur J Biochem 1968; 6: 66–79. [DOI] [PubMed] [Google Scholar]
- [41].Pfaff E, Heldt HW, Klingenberg M. Adenine nucleotide translocation of mitochondria. Kinetics of the adenine nucleotide exchange. Eur J Biochem 1969; 10: 484–93. [DOI] [PubMed] [Google Scholar]
- [42].Pfaff E, Klingenberg M, Heldt HW. Unspecific permeation and specific exchange of adenine nucleotides in liver mitochondria. Biochim Biophys Acta 1965; 104: 312–5. [DOI] [PubMed] [Google Scholar]
- [43].Klingenberg M. The adenine-nucleotide exchange in submitochondrial (sonic) particles. Eur J Biochem 1977; 76: 553–65. [DOI] [PubMed] [Google Scholar]
- [44].Chan SH, Barbour RL. Adenine nucleotide transport in hepatoma mitochondria. Characterization of factors influencing the kinetics of ADP and ATP uptake. Biochim Biophys Acta 1983; 723: 104–13. [DOI] [PubMed] [Google Scholar]
- [45].Duyckaerts C, Sluse-Goffart CM, Fux JP, Sluse FE, Liebecq C. Kinetic mechanism of the exchanges catalysed by the adenine-nucleotide carrier. Eur J Biochem 1980; 106: 1–6. [DOI] [PubMed] [Google Scholar]
- [46].Duee ED, Vignais PV. Kinetics and specificity of the adenine nucleotide translocation in rat liver mitochondria. J Biol Chem 1969; 244: 3920–31. [PubMed] [Google Scholar]
- [47].Kramer R, Klingenberg M. Electrophoretic control of reconstituted adenine nucleotide translocation. Biochemistry 1982; 21: 1082–9. [DOI] [PubMed] [Google Scholar]
- [48].Klingenberg M. The ADP-ATP translocation in mitochondria, a membrane potential controlled transport. J Membr Biol 1980; 56: 97–105. [DOI] [PubMed] [Google Scholar]
- [49].Soboll S, Scholz R, Heldt HW. Subcellular metabolite concentrations. Dependence of mitochondrial and cytosolic ATP systems on the metabolic state of perfused rat liver. Eur J Biochem 1978; 87: 377–90. [DOI] [PubMed] [Google Scholar]
- [50].Kauppinen RA, Hiltunen JK, Hassinen IE. Subcellular distribution of phosphagens in isolated perfused rat heart. FEBS Lett 1980; 112: 273–6. [DOI] [PubMed] [Google Scholar]
- [51].Heldt HW, Klingenberg M, Milovancev M. Differences between the ATP-ADP ratios in the mitochondrial matrix and in the extramitochondrial space. Eur J Biochem 1972; 30: 434–40. [DOI] [PubMed] [Google Scholar]
- [52].Villiers C, Michejda JW, Block M, Lauquin GJ, Vignais PV. The electrogenic nature of ADP/ATP transport in inside-out submitochondrial particles. Biochim Biophys Acta 1979; 546: 157–70. [DOI] [PubMed] [Google Scholar]
- [53].Ruprecht JJ, Hellawell AM, Harding M, Crichton PG, McCoy AJ, Kunji ER . Structures of yeast mitochondrial ADP/ATP carriers support a domain-based alternating-access transport mechanism. Proc Natl Acad Sci U S A 2014; 111: E426–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Klingenberg M. Ligand-protein interaction in biomembrane carriers. The induced transition fit of transport catalysis. Biochemistry 2005; 44: 8563–70. [DOI] [PubMed] [Google Scholar]
- [55].Klingenberg M. The ADP, ATP shuttle of the mitochondrion, Trends in Biochemical Sciences 1979; 4: 249–252. [Google Scholar]
- [56].Daniele C, Dahamna S, Firuzi O, Sekfali N, Saso L, Mazzanti G. Atractylis gummifera L. poisoning: an ethnopharmacological review. J Ethnopharmacol 2005; 97: 175–81. [DOI] [PubMed] [Google Scholar]
- [57].Kemp A Jr., Slater EC. The Site of Action of Atractyloside. Biochim Biophys Acta 1964; 92: 178–80. [DOI] [PubMed] [Google Scholar]
- [58].Weidemann MJ, Erdelt H, Klingenberg M. Adenine nucleotide translocation of mitochondria. Identification of carrier sites. Eur J Biochem 1970; 16: 313–35. [DOI] [PubMed] [Google Scholar]
- [59].Klingenberg M, Grebe K, Scherer B. The binding of atractylate and carboxy-atractylate to mitochondria. Eur J Biochem 1975; 52: 351–63. [DOI] [PubMed] [Google Scholar]
- [60].Vignais PV, Duee ED, Vignais PM, Huet J. Effects of atractyligenin and its structural analogues on oxidative phosphorylation and on the translocation of adenine nucleotides in mitochondria. Biochim Biophys Acta 1966; 118: 465–83. [DOI] [PubMed] [Google Scholar]
- [61].Luciani S, Martini N, Santi R. Effects of carboxyatractyloside a structural analogue of atractyloside on mitochondrial oxidative phosphorylation. Life Sci II 1971; 10: 961–8. [DOI] [PubMed] [Google Scholar]
- [62].Van Veen AG, Mertens WK. Die Giftstoffe der sogenannten Bongkrek‐Vergiftungen auf Java. Recueil des Travaux Chimiques des Pays-Bas 1934; 53: 257–266. [Google Scholar]
- [63].Lumbach G, Cox H, Berends WJ. Elucidation of the chemical structure of bongkrekic acid—I: Isolation, purification and properties of bongkrekic acid. Tetrahedron 1970; 26: 5993–5999. [Google Scholar]
- [64].Welling W, Cohen J, Berends WJ. Disturbance of oxidative phosphorylation by an antibioticum produced by Pseudomonas cocovenenans. Biochem Pharmacol 1960; 3: 122–135. [DOI] [PubMed] [Google Scholar]
- [65].Henderson PJ, Lardy HA. Bongkrekic acid an inhibitor of the adenine nucleotide translocase of mitochondria. J Biol Chem 1970; 245: 1319–1326. [PubMed] [Google Scholar]
- [66].Weidemann MJ, Erdelt H, Klingenberg M. Effect of bongkrekic acid on the adenine nucleotide carrier in mitochondria: tightening of adenine nucleotide binding and differentiation between inner and outer sites . Biochem Biophys Res Commun 1970; 39: 363–70. [DOI] [PubMed] [Google Scholar]
- [67].Klingenberg M, Appel M, Babel W, Aquila H. The binding of bongkrekate to mitochondria. Eur J Biochem 1983; 131: 647–54. [DOI] [PubMed] [Google Scholar]
- [68].Kramer R, Klingenberg M. Reconstitution of inhibitor binding properties of the isolated adenosine 5’-diphosphate, adenosine 5’-triphosphate carrier-linked binding protein. Biochemistry 1977; 16: 4954–61. [DOI] [PubMed] [Google Scholar]
- [69].Chen C, Ko Y, Delannoy M, Ludtke SJ, Chiu W, Pedersen PL. Mitochondrial ATP synthasome: three-dimensional structure by electron microscopy of the ATP synthase in complex formation with carriers for Pi and ADP/ATP. J Biol Chem 2004; 279: 31761–8. [DOI] [PubMed] [Google Scholar]
- [70].Nuskova H, Mracek T, Mikulova T, Vrbacky M, Kovarova N, Kovalcikova J, et al. Mitochondrial ATP synthasome: Expression and structural interaction of its components. Biochem Biophys Res Commun 2015; 464: 787–93. [DOI] [PubMed] [Google Scholar]
- [71].Lu YW, Acoba MG, Selvaraju K, Huang TC, Nirujogi RS, Sathe G, et al. Human adenine nucleotide translocases physically and functionally interact with respirasomes. Mol Biol Cell 2017; 28: 1489–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Parodi-Rullan RM, Chapa-Dubocq X, Guzman-Hernandez R, Jang S, Ayala-Pena S, Javadov S. The Role of Adenine Nucleotide Translocase in the Assembly of Respiratory Supercomplexes in Cardiac Cells. Cells 2019; 8: E1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Raaflaub J. Swelling of isolated mitochondria of the liver and their susceptibility to physicochemical influences. Helvetica physiologica et pharmacologica acta 1953; 11: 142. [PubMed] [Google Scholar]
- [74].Zoratti M, Szabò I. The mitochondrial permeability transition. Biochimica et Biophysica Acta -Reviews on Biomembranes 1995; 1241: 139–176. [DOI] [PubMed] [Google Scholar]
- [75].Szabo I, Zoratti M. The mitochondrial megachannel is the permeability transition pore. J Bioenerg Biomembr 1992; 24: 111–7. [DOI] [PubMed] [Google Scholar]
- [76].Petronilli V, Szabo I, Zoratti M. The inner mitochondrial membrane contains ion-conducting channels similar to those found in bacteria. FEBS Lett 1989; 259: 137–43. [DOI] [PubMed] [Google Scholar]
- [77].Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys 1979; 195: 460–7. [DOI] [PubMed] [Google Scholar]
- [78].Halestrap AP, Kerr PM, Javadov S, Woodfield KY. Elucidating the molecular mechanism of the permeability transition pore and its role in reperfusion injury of the heart. Biochim Biophys Acta 1998; 1366: 79–94. [DOI] [PubMed] [Google Scholar]
- [79].Halestrap AP, Clarke SJ, Javadov SA. Mitochondrial permeability transition pore opening during myocardial reperfusion--a target for cardioprotection. Cardiovasc Res 2004; 61: 372–85. [DOI] [PubMed] [Google Scholar]
- [80].Kwong JQ, Molkentin JD. Physiological and pathological roles of the mitochondrial permeability transition pore in the heart. Cell Metab 2015; 21: 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Elrod JW, Wong R, Mishra S, Vagnozzi RJ, Sakthievel B, Goonasekera SA, et al. Cyclophilin D controls mitochondrial pore-dependent Ca(2+) exchange, metabolic flexibility, and propensity for heart failure in mice. J Clin Invest 2010; 120: 3680–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Menazza S, Wong R, Nguyen T, Wang G, Gucek M, Murphy E. CypD(−/−) hearts have altered levels of proteins involved in Krebs cycle, branch chain amino acid degradation and pyruvate metabolism. J Mol Cell Cardiol 2013; 56: 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Korge P, Yang L, Yang JH, Wang Y, Qu Z, Weiss JN. Protective role of transient pore openings in calcium handling by cardiac mitochondria. J Biol Chem 2011; 286: 34851–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Bernardi P, von Stockum S. The permeability transition pore as a Ca(2+) release channel: new answers to an old question. Cell Calcium 2012; 52: 22–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ichas F, Mazat JP. From calcium signaling to cell death: two conformations for the mitochondrial permeability transition pore. Switching from low- to high-conductance state. Biochim Biophys Acta 1998; 1366: 33–50. [DOI] [PubMed] [Google Scholar]
- [86].Wang W, Fang H, Groom L, Cheng A, Zhang W, Liu J, et al. Superoxide flashes in single mitochondria, Cell 2008; 134: 279–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Li K, Zhang W, Fang H, Xie W, Liu J, Zheng M, et al. Superoxide flashes reveal novel properties of mitochondrial reactive oxygen species excitability in cardiomyocytes. Biophys J 2012; 102: 1011–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Karch J, Molkentin JD. Identity of the elusive mitochondrial permeability transition pore: What it might be, what it was, and what it still could be. Curr Opinion Physiol 2018; 3: 57–62. [Google Scholar]
- [89].Elrod JW, Molkentin JD. Physiologic functions of cyclophilin D and the mitochondrial permeability transition pore. Circ J 2013; 77: 1111–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Broekemeier K, Dempsey M, Pfeiffer DR. Cyclosporin A is a potent inhibitor of the inner membrane permeability transition in liver mitochondria. J Biol Chem 1989; 264: 7826–7830. [PubMed] [Google Scholar]
- [91].Woodfield KY, Price NT, Halestrap AP. cDNA cloning of rat mitochondrial cyclophilin. Biochim Biophys Acta 1997; 1351: 27–30. [DOI] [PubMed] [Google Scholar]
- [92].Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005; 434: 652. [DOI] [PubMed] [Google Scholar]
- [93].Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005; 434: 658. [DOI] [PubMed] [Google Scholar]
- [94].Basso E, Fante L, Fowlkes J, Petronilli V, Forte MA, Bernardi P. Properties of the permeability transition pore in mitochondria devoid of Cyclophilin D. J Biol Chem 2005; 280: 18558–18561. [DOI] [PubMed] [Google Scholar]
- [95].Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, et al. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci U S A 2005; 102: 12005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Halestrap AP, Davidson AM. Inhibition of Ca2(+)-induced large-amplitude swelling of liver and heart mitochondria by cyclosporin is probably caused by the inhibitor binding to mitochondrial -matrix peptidyl-prolyl cis-trans isomerase and preventing it interacting with the adenine nucleotide translocase. Biochem J 1990; 268: 153–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Chappell JB, Crofts AR. Calcium Ion Accumulation and Volume Changes of Isolated Liver Mitochondria. Calcium Ion-Induced Swelling. Biochem J 1965; 95: 378–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Le Quoc K, Le Quoc D. Involvement of the ADP/ATP carrier in calcium-induced perturbations of the mitochondrial inner membrane permeability: importance of the orientation of the nucleotide binding site. Arch Biochem Biophys 1988; 265: 249–57. [DOI] [PubMed] [Google Scholar]
- [99].Haworth RA, Hunter DR. Control of the mitochondrial permeability transition pore by high-affinity ADP binding at the ADP/ATP translocase in permeabilized mitochondria. J Bioenerg Biomembr 2000; 32: 91–6. [DOI] [PubMed] [Google Scholar]
- [100].Novgorodov SA, Gudz TI, Milgrom YM, Brierley GP. The permeability transition in heart mitochondria is regulated synergistically by ADP and cyclosporin A. J Biol Chem 1992; 267: 16274–82. [PubMed] [Google Scholar]
- [101].Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J 1995; 307: 93–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Bernardi P. Modulation of the mitochondrial cyclosporin A-sensitive permeability transition pore by the proton electrochemical gradient. Evidence that the pore can be opened by membrane depolarization. J Biol Chem 1992; 267: 8834–9. [PubMed] [Google Scholar]
- [103].Petronilli V, Nicolli A, Costantini P, Colonna R, Bernardi P. Regulation of the permeability transition pore, a voltage-dependent mitochondrial channel inhibited by cyclosporin A. Biochim Biophys Acta 1994; 1187: 255–9. [DOI] [PubMed] [Google Scholar]
- [104].Scorrano L, Petronilli V, Bernardi P. On the voltage dependence of the mitochondrial permeability transition pore. A critical appraisal. J Biol Chem 1997; 272: 12295–9. [DOI] [PubMed] [Google Scholar]
- [105].Beatrice MC, Stiers DL, Pfeiffer DR. The role of glutathione in the retention of Ca2+ by liver mitochondria. J Biol Chem 1984; 259: 1279–87. [PubMed] [Google Scholar]
- [106].Crompton M, Costi A. Kinetic evidence for a heart mitochondrial pore activated by Ca2+, inorganic phosphate and oxidative stress. A potential mechanism for mitochondrial dysfunction during cellular Ca2+ overload. Eur J Biochem 1988; 178: 489–501. [DOI] [PubMed] [Google Scholar]
- [107].Lenartowicz E, Bernardi P, Azzone GF. Phenylarsine oxide induces the cyclosporin A-sensitive membrane permeability transition in rat liver mitochondria. J Bioenerg Biomembr 1991; 23: 679–88. [DOI] [PubMed] [Google Scholar]
- [108].Halestrap AP, Brenner C. The adenine nucleotide translocase: a central component of the mitochondrial permeability transition pore and key player in cell death. Curr Med Chem 2003; 10: 1507–25. [DOI] [PubMed] [Google Scholar]
- [109].Leblanc P, Clauser H. ADP-dependent inhibition of sarcosomal adenine nucleotide translocase by N-ethylmaleimide. FEBS Lett 1972; 23: 107–113. [DOI] [PubMed] [Google Scholar]
- [110].Aquila H, Klingenberg M. The reactivity of-SH groups in the ADP/ATP carrier isolated from beef heart mitochondria. Eur J of Biochem 1982; 122: 141–145. [DOI] [PubMed] [Google Scholar]
- [111].Vignais PV, Vignais PM. Effect of SH reagents on atractyloside binding to mitochondria and ADP translocation. Potentiation by ADP and its prevention by uncoupler FCCP. FEBS Lett 1972; 26: 27–31. [DOI] [PubMed] [Google Scholar]
- [112].McStay GP, Clarke SJ, Halestrap AP. Role of critical thiol groups on the matrix surface of the adenine nucleotide translocase in the mechanism of the mitochondrial permeability transition pore . Biochem J 2002; 367: 541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Halestrap AP, Woodfield KY, Connern CP. Oxidative stress, thiol reagents, and membrane potential modulate the mitochondrial permeability transition by affecting nucleotide binding to the adenine nucleotide translocase. J Biol Chem 1997; 272: 3346–54. [DOI] [PubMed] [Google Scholar]
- [114].Connern CP, Halestrap AP. Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel. Biochem J 1994; 302: 321–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Connern CP, Halestrap AP. Chaotropic agents and increased matrix volume enhance binding of mitochondrial cyclophilin to the inner mitochondrial membrane and sensitize the mitochondrial permeability transition to [Ca2+]. Biochemistry 1996; 35: 8172–80. [DOI] [PubMed] [Google Scholar]
- [116].Andreeva L, Crompton M. An ADP-sensitive cyclosporin-A-binding protein in rat liver mitochondria. Eur J Biochem 1994; 221: 261–8. [DOI] [PubMed] [Google Scholar]
- [117].Andreeva L, Tanveer A, Crompton M. Evidence for the involvement of a membrane-associated cyclosporin-A-binding protein in the Ca(2+)-activated inner membrane pore of heart mitochondria. Eur J Biochem 1995; 230: 1125–32. [DOI] [PubMed] [Google Scholar]
- [118].Woodfield K, Ruck A, Brdiczka D, Halestrap AP. Direct demonstration of a specific interaction between cyclophilin-D and the adenine nucleotide translocase confirms their role in the mitochondrial permeability transition. Biochem J 1998; 336: 287–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Crompton M, Virji S, Ward JM. Cyclophilin-D binds strongly to complexes of the voltage-dependent anion channel and the adenine nucleotide translocase to form the permeability transition pore . Eur J Biochem 1998; 258: 729–35. [DOI] [PubMed] [Google Scholar]
- [120].Halestrap AP. The regulation of the oxidation of fatty acids and other substrates in rat heart mitochondria by changes in the matrix volume induced by osmotic strength, valinomycin and Ca2+. Biochem J 1987; 244: 159–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Brustovetsky N, Klingenberg M. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+. Biochemistry 1996; 35: 8483–8. [DOI] [PubMed] [Google Scholar]
- [122].Ruck A, Dolder M, Wallimann T, Brdiczka D. Reconstituted adenine nucleotide translocase forms a channel for small molecules comparable to the mitochondrial permeability transition pore. FEBS Lett 1998; 426: 97–101. [DOI] [PubMed] [Google Scholar]
- [123].Brustovetsky N, Tropschug M, Heimpel S, Heidkamper D, Klingenberg M. A large Ca2+-dependent channel formed by recombinant ADP/ATP carrier from Neurospora crassa resembles the mitochondrial permeability transition pore. Biochemistry 2002; 41: 11804–11. [DOI] [PubMed] [Google Scholar]
- [124].Kroemer G, Galluzzi L, Brenner C. Mitochondrial membrane permeabilization in cell death. Physiol Rev 2007; 87: 99–163. [DOI] [PubMed] [Google Scholar]
- [125].Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 2007; 9: 550–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Brenner C, Cadiou H, Vieira HL, Zamzami N, Marzo I, Xie Z, et al. Bcl-2 and Bax regulate the channel activity of the mitochondrial adenine nucleotide translocator. Oncogene 2000; 19: 329–36. [DOI] [PubMed] [Google Scholar]
- [127].Karch J, Kwong JQ, Burr AR, Sargent MA, Elrod JW, Peixoto PM, et al. Bax and Bak function as the outer membrane component of the mitochondrial permeability pore in regulating necrotic cell death in mice. Elife 2013; 2: e00772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Jung DW, Bradshaw PC, Pfeiffer DR. Properties of a cyclosporin-insensitive permeability transition pore in yeast mitochondria. J Biol Chem 1997; 272: 21104–12. [DOI] [PubMed] [Google Scholar]
- [129].Manon S, Roucou X, Guerin M, Rigoulet M, Guerin B. Characterization of the yeast mitochondria unselective channel: a counterpart to the mammalian permeability transition pore? J Bioenerg Biomembr 1998; 30: 419–29. [DOI] [PubMed] [Google Scholar]
- [130].Aquila H, Eiermann W, Klingenberg M. Incorporation of N-ethylmaleimide into the membrane-bound ADP/ATP translocator. Isolation of the protein labeled with N-[3H]ethylmaleimide. Eur J Biochem 1982; 122: 133–9. [DOI] [PubMed] [Google Scholar]
- [131].Boulay F, Vignais PV. Localization of the N-ethylmaleimide reactive cysteine in the beef heart mitochondrial ADP/ATP carrier protein. Biochemistry 1984; 23: 4807–12. [DOI] [PubMed] [Google Scholar]
- [132].Jang S, Lewis TS, Powers C, Khuchua Z, Baines CP, Wipf P, et al. Elucidating Mitochondrial Electron Transport Chain Supercomplexes in the Heart During Ischemia-Reperfusion. Antioxid Redox Signal 2017; 27: 57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Goto S, Chuman H, Majima E, Terada H. How does the mitochondrial ADP/ATP carrier distinguish transportable ATP and ADP from untransportable AMP and GTP?Dynamic modeling of the recognition/translocation process in the major substrate binding region. Biochim Biophys Acta 2002; 1589: 203–18. [DOI] [PubMed] [Google Scholar]
- [134].Linder MD, Morkunaite-Haimi S, Kinnunen PK, Bernardi P, Eriksson O. Ligand-selective modulation of the permeability transition pore by arginine modification. Opposing effects of p-hydroxyphenylglyoxal and phenylglyoxal. J Biol Chem 2002; 277: 937–42. [DOI] [PubMed] [Google Scholar]
- [135].Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004; 427: 461–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Karch J, Bround MJ, Khalil H, Sargent MA, Latchman N, Terada N, et al. Inhibition of mitochondrial permeability transition by deletion of the ANT family and CypD. Sci Adv 2019; 5: eaaw4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Rial E, Rodriguez-Sanchez L, Gallardo-Vara E, Zaragoza P, Moyano E, Gonzalez-Barroso MM. Lipotoxicity, fatty acid uncoupling and mitochondrial carrier function. Biochim Biophys Acta 2010; 1797: 800–6. [DOI] [PubMed] [Google Scholar]
- [138].Dierks T, Salentin A, Heberger C, Kramer R. The mitochondrial aspartate/glutamate and ADP/ATP carrier switch from obligate counterexchange to unidirectional transport after modification by SH-reagents. Biochim Biophys Acta 1990; 1028: 268–80. [DOI] [PubMed] [Google Scholar]
- [139].Dierks T, Salentin A, Kramer R. Pore-like and carrier-like properties of the mitochondrial aspartate/glutamate carrier after modification by SH-reagents: evidence for a performed channel as a structural requirement of carrier-mediated transport. Biochim Biophys Acta 1990; 1028: 281–8. [DOI] [PubMed] [Google Scholar]
- [140].Crichton PG, Parker N, Vidal-Puig AJ, Brand MD. Not all mitochondrial carrier proteins support permeability transition pore formation: no involvement of uncoupling protein 1. Biosci Rep 2009; 30: 187–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Leung AW, Varanyuwatana P, Halestrap AP. The mitochondrial phosphate carrier interacts with cyclophilin D and may play a key role in the permeability transition. J Biol Chem 2008; 283: 26312–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Kwong JQ, Davis J, Baines CP, Sargent MA, Karch J, Wang X, et al. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ 2014; 21: 1209–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Bernardi P. Why F-ATP Synthase Remains a Strong Candidate as the Mitochondrial Permeability Transition Pore. Front Physiol 2018; 9: 1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Bernardi P, Di Lisa F, Fogolari F, Lippe G. From ATP to PTP and Back: A Dual Function for the Mitochondrial ATP Synthase. Circ Res 2015; 116: 1850–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Alavian KN, Beutner G, Lazrove E, Sacchetti S, Park HA, Licznerski P, et al. An uncoupling channel within the c-subunit ring of the F1FO ATP synthase is the mitochondrial permeability transition pore. Proc Natl Acad Sci U S A 2014; 111: 10580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Mnatsakanyan N, Llaguno MC, Yang Y, Yan Y, Weber J, Sigworth FJ, et al. A mitochondrial megachannel resides in monomeric F1FO ATP synthase. Nat Commun 2019; 10: 5823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Giorgio V, von Stockum S, Antoniel M, Fabbro A, Fogolari F, Forte M, et al. Dimers of mitochondrial ATP synthase form the permeability transition pore. Proc Natl Acad Sci U S A 2013; 110: 5887–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Carroll J, He J, Ding S, Fearnley IM, Walker JE. Persistence of the permeability transition pore in human mitochondria devoid of an assembled ATP synthase. Proc Natl Acad Sci U S A 2019; 116: 12816–12821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].He J, Carroll J, Ding S, Fearnley IM, Walker JE. Permeability transition in human mitochondria persists in the absence of peripheral stalk subunits of ATP synthase. Proc Natl Acad Sci U S A 2017; 114: 9086–9091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].He J, Ford HC, Carroll J, Ding S, Fearnley IM, Walker JE. Persistence of the mitochondrial permeability transition in the absence of subunit c of human ATP synthase. Proc Natl Acad Sci U S A 2017; 114: 3409–3414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Neginskaya MA, Solesio ME, Berezhnaya EV, Amodeo GF, Mnatsakanyan N, Jonas EA, et al. ATP Synthase C-Subunit-Deficient Mitochondria Have a Small Cyclosporine A-Sensitive Channel, but Lack the Permeability Transition Pore. Cell Rep 2019; 26: 11–17 e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].He L, Lemasters JJ. Regulated and unregulated mitochondrial permeability transition pores: a new paradigm of pore structure and function? FEBS Lett 2002; 512: 1–7. [DOI] [PubMed] [Google Scholar]
- [153].Bouillaud F, Weissenbach J, Ricquier D. Complete cDNA-derived amino acid sequence of rat brown fat uncoupling protein. J Biol Chem 1986; 261: 1487–90. [PubMed] [Google Scholar]
- [154].Aquila H, Link TA, Klingenberg M. The uncoupling protein from brown fat mitochondria is related to the mitochondrial ADP/ATP carrier. Analysis of sequence homologies and of folding of the protein in the membrane . EMBO J 1985; 4: 2369–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Bertholet AM, Kazak L, Chouchani ET, Bogaczynska MG, Paranjpe I, Wainwright GL, et al. Mitochondrial Patch Clamp of Beige Adipocytes Reveals UCP1-Positive and UCP1-Negative Cells Both Exhibiting Futile Creatine Cycling. Cell Metab 2017; 25: 811–822 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Fedorenko A, Lishko PV, Kirichok Y. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 2012; 151: 400–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Divakaruni AS, Brand MD. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 2011; 26: 192–205. [DOI] [PubMed] [Google Scholar]
- [158].Wojtczak L, Schonfeld P. Effect of fatty acids on energy coupling processes in mitochondria. Biochim Biophys Acta 1993; 1183: 41–57. [DOI] [PubMed] [Google Scholar]
- [159].Andreyev A, Bondareva TO, Dedukhova VI, Mokhova EN, Skulachev VP, Volkov NI . Carboxyatractylate inhibits the uncoupling effect of free fatty acids. FEBS Lett 1988; 226: 265–9. [DOI] [PubMed] [Google Scholar]
- [160].Andreyev A, Bondareva TO, Dedukhova VI, Mokhova EN, Skulachev VP, Tsofina LM, et al. The ATP/ADP-antiporter is involved in the uncoupling effect of fatty acids on mitochondria. Eur J Biochem 1989; 182: 585–92. [DOI] [PubMed] [Google Scholar]
- [161].Brustovetsky N, Klingenberg M. The reconstituted ADP/ATP carrier can mediate H+ transport by free fatty acids, which is further stimulated by mersalyl. J Biol Chem 1994; 269: 27329–36. [PubMed] [Google Scholar]
- [162].Bertholet AM, Chouchani ET, Kazak L, Angelin A, Fedorenko A, Long JZ, et al. H(+) transport is an integral function of the mitochondrial ADP/ATP carrier. Nature 2019; 571: 515–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Cho J, Seo J, Lim CH, Yang L, Shiratsuchi T, Lee MH, et al. Mitochondrial ATP transporter Ant2 depletion impairs erythropoiesis and B lymphopoiesis. Cell Death Differ 2015; 22: 1437–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Hoshino A, Wang WJ, Wada S, McDermott-Roe C, Evans CS, Gosis B, et al. The ADP/ATP translocase drives mitophagy independent of nucleotide exchange. Nature 2019; 575: 375–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Schirris TJ, Ritschel T, Herma Renkema G, Willems PH, Smeitink JA, Russel FG . Mitochondrial ADP/ATP exchange inhibition: a novel off-target mechanism underlying ibipinabant-induced myotoxicity. Sci Rep 2015; 5: 4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Di Marino D, Oteri F, della Rocca BM, D’Annessa I, Falconi M. Mapping multiple potential ATP binding sites on the matrix side of the bovine ADP/ATP carrier by the combined use of MD simulation and docking. J Mol Model 2012; 18: 2377–86. [DOI] [PubMed] [Google Scholar]
- [167].Palmieri L, Alberio S, Pisano I, Lodi T, Meznaric-Petrusa M, Zidar J, et al. Complete loss-of-function of the heart/muscle-specific adenine nucleotide translocator is associated with mitochondrial myopathy and cardiomyopathy. Hum Mol Genet 2005; 14: 3079–88. [DOI] [PubMed] [Google Scholar]
- [168].Clemencon B, Babot M, Trezeguet V. The mitochondrial ADP/ATP carrier (SLC25 family): pathological implications of its dysfunction. Mol Aspects Med 2013; 34: 485–93. [DOI] [PubMed] [Google Scholar]
- [169].Kawamata H, Tiranti V, Magrane J, Chinopoulos C, Manfredi G. adPEO mutations in ANT1 impair ADP-ATP translocation in muscle mitochondria. Hum Mol Genet 2011; 20: 2964–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Jin SM, Youle RJ. PINK1- and Parkin-mediated mitophagy at a glance. J Cell Sci 2012; 125: 795–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Mokranjac D, Neupert W. Energetics of protein translocation into mitochondria. Biochim Biophys Acta 2008; 1777: 758–62. [DOI] [PubMed] [Google Scholar]
- [172].McWilliams TG, Prescott AR, Montava-Garriga L, Ball G, Singh F, Barini E, et al. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab 2018; 27: 439–449 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]