

Abstract
Background:
MUC16 is a mucin marker that is frequently mutated in melanoma; but whether MUC16 mutations could be useful as a surrogate biomarker for tumor mutation burden (TMB) remains unclear.
Methods:
This study rigorously evaluates the MUC16 mutation as a clinical biomarker in cutaneous melanoma by utilizing genomic and clinical data from patient samples from The Cancer Genome Atlas (TCGA) and two independent validation cohorts. We further extended the analysis to studies with patients treated with immunotherapies.
Results:
Analysis results showed that samples with MUC16 mutations had a higher TMB than the samples of wild-type, with strong statistical significance (P<0.001) in all melanoma cohorts tested. Associations between MUC16 mutations and TMB remained statistically significant after adjusting for potential confounding factors in the TCGA cohort (OR, 9.28 [95% CI, 5.18–17.39]; P<0.001), Moffitt cohort (OR, 31.95 [95% CI, 8.71–163.90]; P<0.001), and Yale cohort (OR, 8.09 [95% CI, 3.12–23.79]; P<0.01). MUC16 mutations were also found associated with overall survival in the TCGA (hazard ratio, 0.62; [95% CI, 0.45–0.85]; P<0.01) and Moffitt cohorts (hazard ratio, 0.49; [95% CI, 0.28–0.87]; P=0.014). Strikingly, MUC16 is the only top frequently mutated gene for which prognostic significance was observed. MUC16 mutations were also found valuable in predicting anti-CTLA-4 and anti-PD-1 therapy responses.
Conclusions:
MUC16 mutation appears to be a useful predictive marker of global TMB and patient survival in melanoma.
Impact:
This is, to the best of our knowledge, the first systematic evaluation of MUC16 mutation as a clinical biomarker and a predictive biomarker for immunotherapy in melanoma.
Keywords: MUC16, Melanoma, prognostic biomarker, tumor mutation burden
Introduction
MUC16 (OMIM 606154, previously known as CA125) is frequently mutated in many cancers. Although the functional role of MUC16 is extensively studied in ovarian cancer, its role as a clinical biomarker remains controversial. In melanoma, MUC16 is one of the top mutated genes according to the Cancer Genome Atlas (TCGA) (Supplementary Figure 1). However, this gene was disregarded in most genomic analyses because it is less likely to be a pathogenic or cancer driver gene. Based on the TCGA gastric cancer data and an independent validation cohort, a recent analysis showed MUC16 mutations are significantly associated with patient overall survival (1). The researchers also explored the potential function of MUC16 in influencing the immune system in gastric cancer.
The functional role of MUC16 in cancer is yet to be elucidated due to several challenges such as its complicated structure and lack of specific antibodies (2). However, one hypothesis is that MUC16 mutations might function through neoantigen presentation and immunoediting (3). Another plausible hypothesis is that MUC16, being large and heavily glycosylated (4), prevents the establishment of a robust immunological synapse between T cells and MHC class I presentation of tumor antigen on the cell surface. The two main questions that are more relevant to melanoma in the clinical setting are (1) how strong are the associations between the mutation status of MUC16 and tumor mutation burden (TMB) and patient prognosis in melanoma--reflecting its potential as a surrogate biomarker for use in clinical practice; and (2) whether the mutation or neoantigen of MUC16 is a predictive biomarker for melanoma patients treated with immunotherapies. To investigate these questions, we analyzed MUC16 mutations and their associations with TMB and prognosis in patients from three independent melanoma cohorts. Further, we investigated how MUC16 mutation is linked with neoantigen presentation and therapy response in melanoma patients treated with anti-CTLA-4 and anti-PD-1 therapies.
Materials and Methods
Genomic and clinical data were acquired from the TCGA, Yale and Moffitt melanoma cohorts (Their clinical characteristics are shown in Supplementary Table 1). Somatic mutation data from whole exome sequencing (WES) and matched gene expression data from RNA sequencing (RNAseq) from the TCGA melanoma cohort (SKCM, n=467 patients) were downloaded from NCI GDC Data Portal (https://portal.gdc.cancer.gov). For the Yale melanoma cohort, the somatic mutation and clinical data were obtained from a previous study (5), which contained data from 132 patients. The Moffitt cohort contains 135 patients with stage 3 and 4 melanoma only, WES and RNAseq data from the Moffitt cohort were included in this study and their deidentified clinical covariates of research interest were obtained from the honest broker for the Moffitt Cutaneous Oncology Program. Tumor-specific mutations were identified with Strelka (6) and MuTect (7), and were annotated to determine genic context using ANNOVAR and summarized using VarSifter (8). To ensure the same sensitivity in calling wild-types and mutants, mutation count in each gene in Moffitt data was adjusted by benchmarking with TCGA data on matched genes. As part of a larger study on computational drug repurposing (MCC# 19147), the study was conducted in accordance with recognized ethical guidelines (e.g., Declaration of Helsinki, CIOMS, Belmont Report, U.S. Common Rule) and was approved by Advarra Institutional Review Board. A Waiver of Consent was granted by Advarra.
Tumor mutation burden is defined as log2 transformed total number of nonsynonymous mutations per megabase. We used 38Mb as the estimate of the exome size in TCGA (9). Mutation data from germline samples in TCGA were downloaded from the Genome Data Commons legacy archive (https://portal.gdc.cancer.gov/legacy-archive/). Variant calling from sequence data of germline samples in the Moffitt cohort was based on GATK (10). Somatic mutational signature deconvolution(11) was performed using the method implemented in the R package deconstructSigs (12), which was able to determine the compositions of predefined mutational signatures within a single sample. We used 30 mutational signatures described in the COSMIC database (http://cancer.sanger.ac.uk/cosmic/signatures). Mutational signature analysis confirmed that UV signatures were dominant in melanomas in all studies. RNA sequencing data from the same tumor and survival outcomes are also available.
HLA types and total neoantigen load for each sample from patients treated with immunotherapy were obtained from previously published studies (13–16). Putative MUC16-specific neoantigens were predicted for each patient by defining all novel amino acids—all nonsynonymous somatic mutations in MUC16 identified from the WES analysis were translated into 9-mer or 10-mer peptides flanking the mutant amino acid. The binding affinity of mutant neoepitopes to the patient-specific HLA-matched class I types was then predicted using NetMHCpan 4.0 (17). The thresholds for strong and weak binder were based on the default setting (0.5% rank for a strong binder and 2% for a weak binder).
We used the nonparametric Wilcoxon rank sum test to compare the TMB between MUC16 wild-type and mutant groups, as well as the differences in neoantigen burden. A two-sided P<.05 was considered statistically significant. Multivariable logistic regression was used to assess the association between MUC16 mutations and TMB while adjusting for confounding factors including sex, age, stage and mutational signatures. In the logistic regression, TMB measures were dichotomized by median in the logistic regression. Odds ratios and 95% confidence intervals (CIs) of included covariates were reported. Kaplan-Meier curves and the log-rank test were used to assess the survival differences between patient groups with differing mutation status. Multivariable Cox regression analyses were used to assess the association of MUC16 mutations and prognosis by controlling for potential confounding factors. The association between patient survival and germline variants in the MUC16 region was tested using both single-variable Cox regression and multi-variable Cox regression analysis with backward selection. For this study, we also replicated a recently published trivariate regression analysis by Lee and Ruppin (18) in predicting objective response rate (ORR) to anti-PD-1/PD-L1 therapy across 21 cancer types. We used the same dataset and followed the same statistical procedure provided in the original publication but with the total mutation burden in the model replaced by the MUC16 single-gene mutation burden. The other two variables included in the regression model were the CD8+ T-cell abundance (eCD8T) and fraction of high PD-1 messenger RNA expression (fPD1).
Results
MUC16 association with TMB
Of the 467 patients in the TCGA SKCM cohort, 312 (66.81%) patients carried somatic mutations in MUC16 (Supplementary Figure 1), where BRAF mutations occurring at a frequency of 50%. TCGA SKCM samples with MUC16 mutations had a statistically significantly higher TMB than the samples of wild-type (p<0.0001, Wilcoxon rank sum test; Figure 1A). The difference in the median of the two groups is 2.44 at log2 scale, which is larger than the difference reported in the gastric cancer patients.(1) A significantly higher mutation rate was also observed in the independent Moffitt melanoma samples with MUC16 mutations (p<0.0001, Wilcoxon test; Figure 1B), as well as in a different melanoma dataset from Yale (p<0.0001, Wilcoxon test; Figure 1C). A consistent finding across three studies was that the median TMB (log2 scale) in the MUC16-mutant group was around 9 and the median TMB in the MUC16 wild-type group was around 7. Of the tumors with lower mutation burdens (log2TMB<5), most samples are MUC16 wild-type: 84.2% (101 of 120) in TCGA, 96.2% (25 of 26) in Moffitt data, and 70.0% (21 of 30) in the Yale data. On the basis of the distribution of somatic mutations in the MUC16 region in all three cohorts, no hotspot region was identified (Supplementary Figure 2). As expected, patients with mutations in both of the main regions of MUC16 (N-terminal domain and tandem repeats) showed highest TMB in the entire cohorts. (Supplementary Figure 3).
Figure 1.
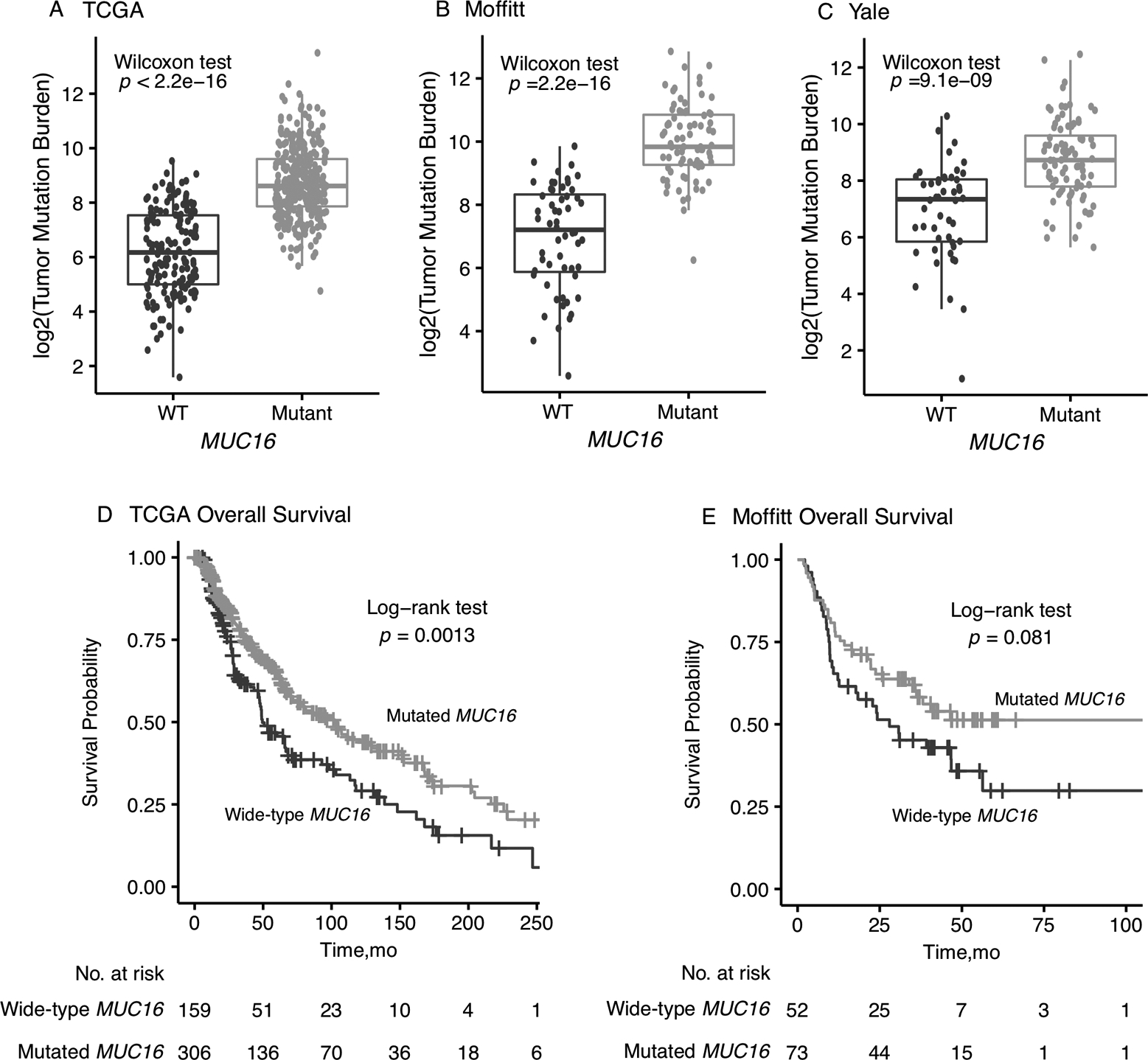
Association of MUC16 mutations with total mutation burden (TMB) in melanoma. (A) TCGA cohort; (B) Moffitt cohort; (C) Yale cohort. Association of MUC16 mutations with overall survival in melanoma in TCGA and Moffitt cohorts (D-E).
To adjust for possible confounding factors, we included covariates including age, sex, stage, POLE status and common mutational signatures in melanoma (Supplementary Figures 4–6) in the logistic regression model. In mutational analysis, as expected, the signatures attributed to UV exposure (Signature 7) and alkylating agents (Signature 11) dominated the mutational signatures in the melanoma mutational landscape. Associations between MUC16 mutations and TMB remained statistically significant in the TCGA cohort (OR, 9.28 [95% CI, 5.18–17.39]; P<0.001) (Figure 2A), as well as in the Moffitt melanoma cohort (OR, 31.95 [95% CI, 8.71–163.90]; P<0.001) (Figure 2C) and Yale cohort (OR, 8.09 [95% CI, 3.12–23.79]; P<0.01) (Supplementary Figure 7A). Beyond melanoma, the strong association between MUC16 and TMB was observed in multiple cancer types in the TCGA data set (Supplementary Figure 8).
Figure 2.

Association of MUC16 mutations with TMB and overall survival in TCGA and Moffitt melanoma datasets by adjusting for confounding factors.
MUC16 is the ONLY prognostic gene in commonly mutated genes in melanoma
In the Kaplan-Meier survival analysis of the TCGA CM cohort (Figure 1D), a significant difference was found in overall survival between the MUC16-mutant and MUC16-WT patient groups (log-rank test, P=0.0013). MUC16-mutant patients have a median overall survival of 103 months [95% CI, 74.7–133 months] compared to 49.5 months [95% CI, 46.9–72 months] in the MUC16-WT patient group. Interestingly, as shown in Figure 3A, the survival difference remained significant (log-rank test, P=0.0096) in patient subgroup of lower than median mutation burden calculated based on genes included in Illumina’s TruSight Tumor 170 (TST170), a panel widely used in clinical setting. As shown in the Cox regression (Figure 2B), the association between MUC16 and overall survival remained statistically significant after adjusting for potential confounding factors (hazard ratio, 0.62; [95% CI, 0.45–0.85]; P<0.01).
Figure 3.
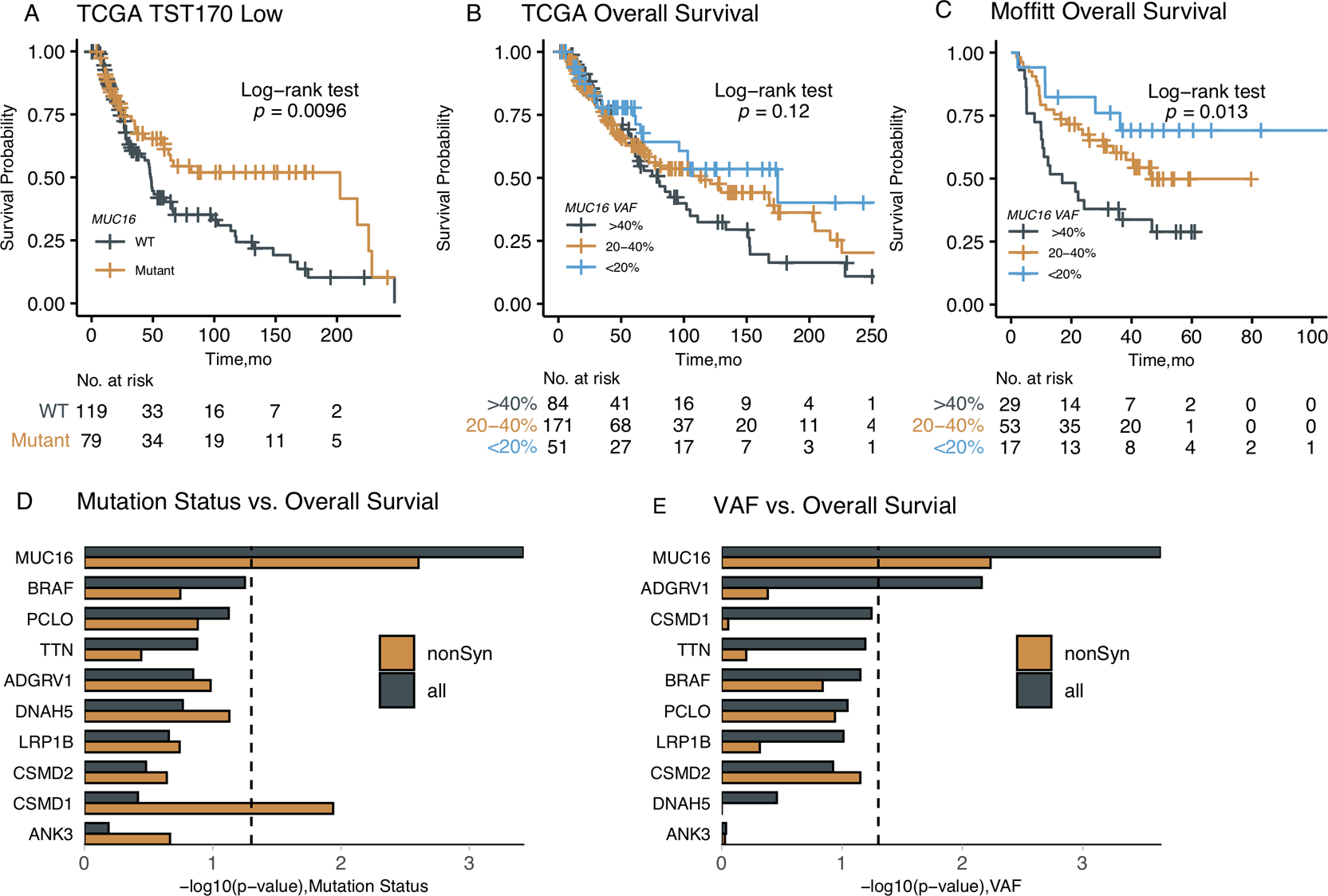
Further analysis of the prognostic value of MUC16 mutations. (A) The MUC16 mutation separates patient survivals in mutation-low subgroup defined by the mutation burden calculated from the TST170 panel. (B) Association of the MUC16 VAF values with overall survival in TCGA SKCM MUC16-Mutant patient group. (C) Association of the MUC16 VAF values with overall survival in Moffitt MUC16-Mutant patient group. (D) The association analysis of mutation status and overall survival in most commonly mutated (top 10) genes in melanoma. (E) The association analysis of VAF values and over survival in most commonly mutated genes in melanoma.
In the Kaplan-Meier survival analysis of the Moffitt melanoma cohort (Figure 1E), MUC16 mutations were found associated with a better overall survival with a borderline statistical significance (log-rank test, P=0.081). After controlling for covariates including age, sex, stage, POLE mutations and mutational signatures (7 and 11) in the Cox regression, the association between MUC16 and overall survival was found statistically significant (hazard ratio, 0.49; [95% CI, 0.28–0.87]; P=0.014) (Figure 2D). Mutational signature 1 was not included in the Cox regression because very few patients had a non-zero score in this signature. Because of the insufficient reporting of survival outcomes, no association was found between MUC16 and the prognosis defined in the Yale data (Supplementary Figure 7).(5) The association between MUC16 mutation and overall survival was observed in four TCGA cancer types (Supplementary Figure 9).
In addition to the mutation status, we observed that the variant allele frequency (VAF) value (also known as mutant allele burden) of MUC16 was also associated with prognosis in both Moffitt and TCGA cohorts (Figures 3B–C). Both data revealed a previously unreported but consistent phenomena: higher non-zero VAF levels of MUC16 were associated with worse survival in CM patients. We also investigated the mutation status and VAF in other top mutated genes (including TTN and BRAF) in melanoma. Surprisingly, MUC16 is the only gene among the top ten most commonly mutated genes in melanoma for which a clear prognostic significance was observed in both mutation status and VAF (Figure 3D–E and Supplementary Figure 10), indicating that MUC16 is a selective clinical biomarker.
Association with neoantigen load and immunotherapy response
To investigate the predictive value of MUC16 mutations in immunotherapeutic studies, we next examined two sets of melanoma patients treated with anti-CTLA-4(13,14) and two sets of melanoma patients treated with anti-PD-1 therapy,(15,16) in which neoantigen load and patient HLA class data were available. Predicted neoantigens in the MUC16 protein-coding region were identified in 94 of the 174 patients in the anti-CTLA-4 cohorts, as well as in 61 of the 103 patients in the anti-PD-1 cohorts. The numbers of MUC16-specific neoepitopes were found significantly associated with total neoantigen burden across four studies (Wilcoxon rank sum test, P<0.001) (Figure 4A). Associations between MUC16 mutations and TMB were also statistically significant in all four cohorts (Supplementary Figure 11). Of the patients with MUC16-specific neoantigens, the median proportion of neoantigens attributed to MUC16 in these four studies are: 0.20% (Snyder et al.), 0.21% (Van Allen), 1.77% (Hugo), and 0.22% (Riaz). Because only patients in Snyder et al. reported association of mutation and neoantigen load with therapy response, we further examined the association between MUC16-specific neoepitopes and therapy outcomes. As shown in Figure 4B, in the combined patient sets, the long-term benefit patient group has a higher proportion of patients with MUC16-specific neoantigens (18 of 27, 66.67%) than the minimal or no benefit patient group (15 of 37, 40.54%). The discrepancy in such proportion was found larger in the discovery set defined in the original publication. However, the enrichment of patients with neoantigens was not found in patients responsive to anti-PD-1 (Supplementary Figure 12). In terms of mutation status, we observed that MUC16 was associated with overall survival in combined anti-CTLA-4 treated patients with a borderline significance but not in combined anti-PD-1 treated patients (Supplementary Figure 13). This result is expected as TMB itself has not be established as a successfully biomarker for anti-PD-1 response in melanoma.
Figure 4.
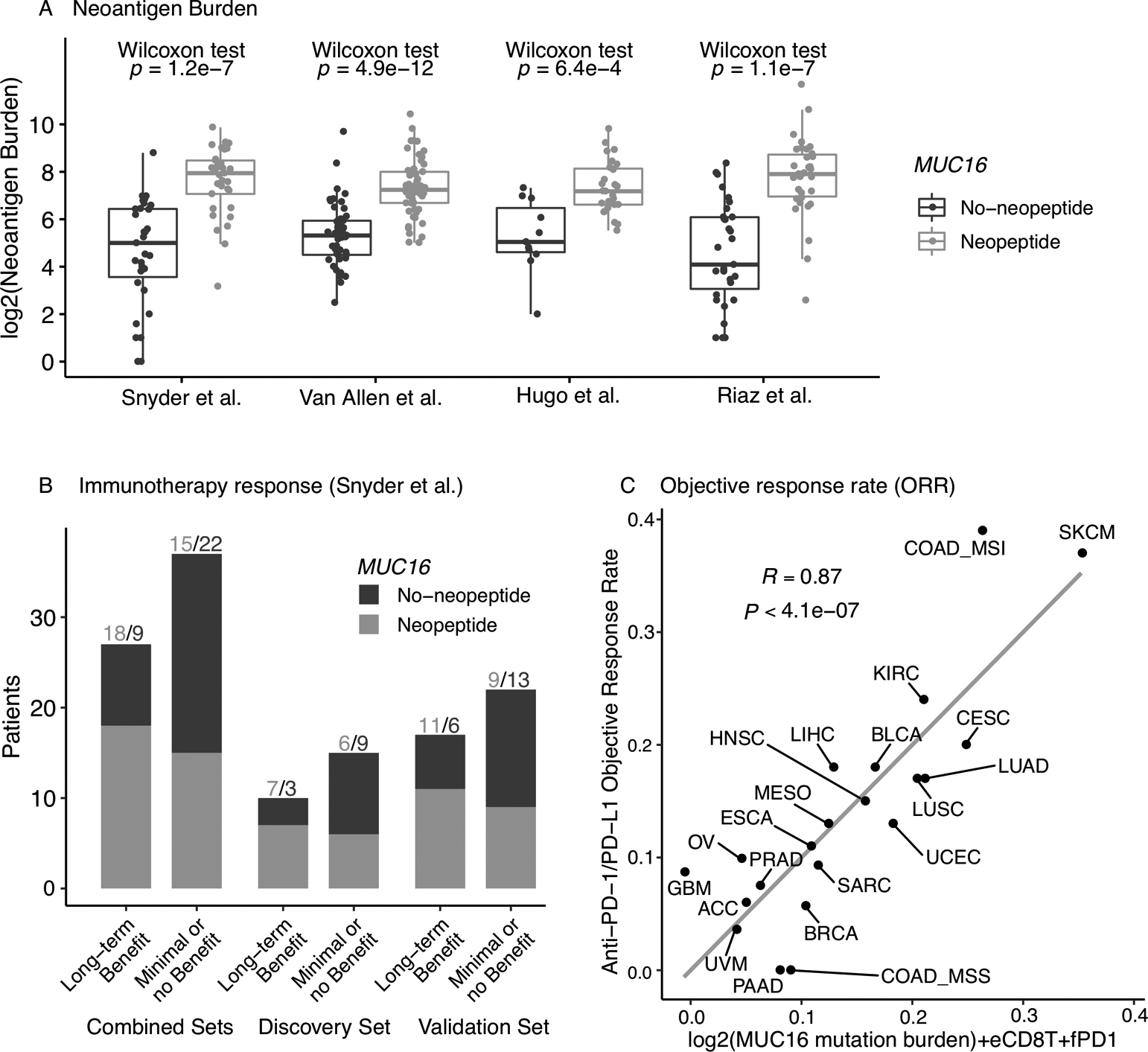
Association of MUC16 with neoantigen load and immunotherapy response. (A) Association of MUC16 neoepitopes with total neoantigen burden in patients treated in immunotherapies. (B) Association of MUC16 neoepitopes with treatment response in patients treated in immunotherapies. (C) Combined MUC16-eCD8T-fDP1 regression model for predicting anti-PD-1/PD-L1 therapy response across 21 cancer types (refer to Lee and Ruppin 18 for details on the original model).
The result from the replicated analysis of the trivariate regression model (18) for predicting the ORR to anti-PD-1/PD-L1 therapy across 21 cancer types is presented in Figure 4C. The combined predictive model of MUC16 mutation burden, eCD8T and fPD1 levels achieved a comparably high prediction accuracy (R=0.87, P<4.1×10−7) compared to the original analysis where total mutation burden was used. Notably, the response rate of SKCM was found closer to the predicted value from the MUC16-based trivariate model (comparing with Figure 2B in Lee and Ruppin (18)), providing additional evidence that MUC16 mutation is a promising candidate when designing a minimalist biomarker panel for melanoma.
Prognostic potential of germline variants in MUC16
The functional roles of germline SNPs in the MUC16 region remain under-investigated. Currently, only 9 variants in MUC16 are annotated in ClinVar and most of them are classified as benign. Through multivariable Cox regression and controlling for clinical covariates, we found 34 germline SNPs in the MUC16 region that were associated with overall survival (Supplementary Table 2) in the Moffitt melanoma cohort. Three common SNPs (rs1609459, rs10402812, and rs7245949) that are in high linkage disequilibrium (LD) were associated with overall survival in the Moffitt data (Cox regression, P=0.01) and were found associated with survival with a borderline significance in TCGA cohort (P=0.07) while other SNPs showed a similar trend of effects (Supplementary Figures 14 and 15). Genetic ancestries were not considered in this explorative analysis because of the limited sample size and the fact that the majority of melanoma patients were Caucasians.
Discussion
Due to its large genome size and as yet understudied gene function, MUC16 is an often-overlooked gene in cancer genomic analysis. In this report, we analyzed mutational data from cutaneous melanoma samples in multiple independent datasets. Results showed that the somatic MUC16 mutation was a reliable surrogate biomarker for global TMB across all datasets. Because the discrepancy of mutation rate between mutation groups was larger in melanoma compared to other cancer types, MUC16 can be a more effective biomarker in clinical use for melanoma. One appealing benefit in using MUC16 mutation status as a stable barometer of TMB in melanoma is that MUC16 is lowly expressed (Supplementary Figure 16) and thus it is less likely to be subject to transcription-coupled repair. MUC16 also has important prognostic implications as MUC16-mutant and lower VAF were found associated with improved survival (but TTN-mutant was not).
To investigate whether MUC16 mutations and MUC16-specific neoantigens can be used as predictive biomarkers for immunotherapy outcomes, we analyzed patient mutation data from four studies with immunotherapies. One noteworthy finding is that the MUC16-specific neoepitopes were significantly associated with total neoantigen burden in tumors in all studies. However, the association of MUC16 mutations with therapy outcome was only observed in one study with anti-CTLA-4 treated patients, where MUC16-mutant patients were found enriched in the group of long-term benefit. In the pooled sample analysis, it was found that the MUC16 mutation status was associated with better survival outcomes in anti-CTLA-4 treated cohorts, but not in anti-PD-1 cohorts. The lack of evidence for prognostic associations in these studies may be due to limited patient sample size and varying definitions of response to immunotherapy.
We also sought to explore germline genetic variants in MUC16, which may alter the gene’s function and activity in both cancer and immune cells, and the prognosis of melanoma. Although preliminary, we observed wild-type genotypes in multiple SNPs that appeared to be associated with worse prognosis. A very interesting SNP is rs7245949, which is in strong LD with two other SNPs; having homozygous wild-type genotype was clearly associated with lower expression in markers of T cell responses (Supplementary Figure 17) and accordingly worse survival. This missense SNP changes the codon from T (threonine) at position 2891 to I (isoleucine), which is located within the heavily O-glycosylated N-terminal region.(4) This makes the hypothesis that MUC16 acts through modulating the synapse between T cells and MHC presentation more plausible. It would encourage further research on how these missense SNPs, discovered mainly in the N-terminus of MUC16, impact upon cancer immunogenicity and prognosis. As a way to further explore whether a gene is functional, we calculated the VAF of all genes in the TCGA melanoma cohort. Interestingly, it was found that the VAF of MUC16 was increased to 50% (ranked 4th in all genes) when we only look at tumors with high purity (Supplementary Figure 18) —which provides additional suggestive evidence that MUC16 might be functional and clonal despite its relatively low expression in melanoma.
Overall, the findings presented support the hypothesis that MUC16 mutations were associated with TMB and prognosis. In addition, we observed that MUC16-specific neoantigens were associated with total neoantigen load in tumors. These results support the use of MUC16 mutation as a potential surrogate biomarker for global TMB and neoantigen load analysis. Although it was hypothesized that MUC16 mutations might act through neoantigen presentation and immunoediting,(3) we did not observe a direct relation between its mutation and therapy response based on existing data collected from anti-PD-1 treatments. However, we were able to replicate a predictive model for predicting the response rate to anti-PD-1/PD-L1 therapy across tumor types based on the MUC16 mutation burden, further supporting the proposal that MUC16 provides a potential gauge biomarker for global tumor neoantigens. The findings from this study encourage further studies in MUC16 and other non-driver hotspot mutations—which may contain special secondary structures such as hairpins and loops (19). Mutations in those genes regions are not necessarily associated with oncogenic driver; rather, these are mere passenger mutations, but should be better measurements of overall mutational burden than other regions of the genome.
We argue that the inter-tumor heterogeneity will have a negligible impact on the mutational analysis based on TCGA, Yale and Moffitt cohorts, especially for somatic results on the TMB association. However, study heterogeneity and other unknown sources of patient heterogeneity should be carefully investigated in future large-sample analysis for the immunotherapy response prediction.
Conclusions
Collectively, our analysis revealed that MUC16 mutation is a reliable and independent biomarker for TMB in melanoma. Importantly, we identified that MUC16 is associated with survival outcomes across different data sets. It is also, to the best of our knowledge, the first analysis of its kind showing that MUC16 is the only gene among top mutated genes that are associated with survival outcomes in CM. Although TMB is not a successful biomarker for predicting anti-PD-1/PD-L1 response in CM, we demonstrated that MUC16 could still be useful as a predictor of neoantigen load and anti-CTLA-4 responses. When combined with CD8+ T-cell abundance and PD-1 expression, MUC16 can be used to generate a minimalist predictive panel for predicting the ORR to anti-PD-1/PD-L1 therapy across cancer types. Therefore, we conclude that MUC16 is a valid surrogate biomarker for TMB as well as a promising prognostic biomarker in melanoma. Its role in predicting response to immunotherapy should be further studied when more data have been collected.
Supplementary Material
Acknowledgements
This study was supported in part by Moffitt Skin Cancer SPORE grant P50 CA168536 (to V.K. Sondak); Moffitt Skin Cancer SPORE Career Enhancement Program (to X. WANG); Institutional Research Grant 14-189-19 from the American Cancer Society (to X. WANG); as well as National Institute of Health grants R01CA157664, R01CA124515, R01CA178687, and R01CA211913 (to J.R. Conejo-Garcia). Support for shared resources was provided by Cancer Center Support Grant (CCSG) CA076292 to H. Lee Moffitt Cancer Center.
Abbreviations:
- CM
Cutaneous melanoma
- COSMIC
Catalogue of Somatic Mutations in Cancer
- eCD8T
CD8+ T-cell abundance
- fPD1
PD-1 messenger RNA expression
- ORR
Objective response rate
- OR
Odds ratio
- TMB
Tumor mutation burden
- TCGA
The Cancer Genome Atlas
- UV
Ultra violet
- VAF
Variant allele frequency
- WES
Whole exome sequencing
Footnotes
Competing interests
C. Duan is a research investigator at Bristol-Myers Squibb. J.K. Teer has ownership interest (including patents) and is a consultant/advisory board member at Interpares Biomedicine. V.K. Sondak is a consultant at Bristol Myers-Squibb, Novartis, Poly- noma, Regeneron, and Merck. Jose R. Conejo-Garcia is a consultant at Compass Therapeutics, Anixa Biosciences, and Leidos, reports receiving a commercial research grant from Compass Therapeutics and Anixa Biosciences, and has ownership interest (including patents) in Compass Therapeutics and Anixa Bisociences. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Li X, Pasche B, Zhang W, Chen K. Association of muc16 mutation with tumor mutation load and outcomes in patients with gastric cancer. JAMA Oncology 2018;4:1691–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Haridas D, Ponnusamy MP, Chugh S, Lakshmanan I, Seshacharyulu P, Batra SK. MUC16: molecular analysis and its functional implications in benign and malignant conditions. FASEB J 2014;28:4183–99 [DOI] [PubMed] [Google Scholar]
- 3.Smyth EC, Fitzgerald RC. Muc16 mutations and prognosis in gastric cancer: A little goes a long way. JAMA Oncology 2018;4:1698–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Das S, Batra SK. Understanding the Unique Attributes of MUC16 (CA125): Potential Implications in Targeted Therapy. Cancer research 2015;75:4669–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Krauthammer M, Kong Y, Bacchiocchi A, Evans P, Pornputtapong N, Wu C, et al. Exome sequencing identifies recurrent mutations in NF1 and RASopathy genes in sun-exposed melanomas. Nature genetics 2015;47:996–1002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumor–normal sample pairs. Bioinformatics 2012;28:1811–7 [DOI] [PubMed] [Google Scholar]
- 7.Cibulskis K, Lawrence MS, Carter SL, Sivachenko A, Jaffe D, Sougnez C, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nature biotechnology 2013;31:213–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teer JK, Green ED, Mullikin JC, Biesecker LG. VarSifter: visualizing and analyzing exome-scale sequence variation data on a desktop computer. Bioinformatics (Oxford, England) 2012;28:599–600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome medicine 2017;9:34- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, et al. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome research 2010;20:1297–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Alexandrov LB, Nik-Zainal S, Wedge DC, Aparicio SAJR, Behjati S, Biankin AV, et al. Signatures of mutational processes in human cancer. Nature 2013;500:415–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rosenthal R, McGranahan N, Herrero J, Taylor BS, Swanton C. DeconstructSigs: delineating mutational processes in single tumors distinguishes DNA repair deficiencies and patterns of carcinoma evolution. Genome biology 2016;17:31- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Snyder A, Makarov V, Merghoub T, Yuan J, Zaretsky JM, Desrichard A, et al. Genetic Basis for Clinical Response to CTLA-4 Blockade in Melanoma. New England Journal of Medicine 2014;371:2189–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Van Allen EM, Miao D, Schilling B, Shukla SA, Blank C, Zimmer L, et al. Genomic correlates of response to CTLA-4 blockade in metastatic melanoma Science (New York, NY: ) 2015;350:207–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hugo W, Zaretsky JM, Sun L, Song C, Moreno BH, Hu-Lieskovan S, et al. Genomic and Transcriptomic Features of Response to Anti-PD-1 Therapy in Metastatic Melanoma. Cell 2016;165:35–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, et al. Tumor and Microenvironment Evolution during Immunotherapy with Nivolumab. Cell 2017;171:934–49.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jurtz V, Paul S, Andreatta M, Marcatili P, Peters B, Nielsen M. NetMHCpan-4.0: Improved Peptide-MHC Class I Interaction Predictions Integrating Eluted Ligand and Peptide Binding Affinity Data. Journal of immunology (Baltimore, Md : 1950) 2017;199:3360–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee JS, Ruppin E. Multiomics Prediction of Response Rates to Therapies to Inhibit Programmed Cell Death 1 and Programmed Cell Death 1 Ligand 1. JAMA Oncol 2019 [DOI] [PMC free article] [PubMed]
- 19.Buisson R, Langenbucher A, Bowen D, Kwan EE, Benes CH, Zou L, et al. Passenger hotspot mutations in cancer driven by APOBEC3A and mesoscale genomic features. Science 2019;364:eaaw2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.