

Abstract
Genetic variants causing the fast-channel congenital myasthenic syndrome (CMS) have been identified in the α, δ, and ε but not the β subunit of acetylcholine receptor (AChR). A 16-year-old girl with severe myasthenia had low-amplitude and fast-decaying miniature endplate potentials. Mutation analysis revealed two heteroallelic variants in CHRNB1 encoding the AChR β subunit: a novel c.812C>T (p.P248L) variant in M1-M2 linker (p.P271L in HGVS nomenclature), and a ~430 bp deletion causing loss of exon 8 leading to frame-shift and a premature stop codon (p.G251Dfs*21). P248 is conserved in all β subunits of different species, but not in other AChR subunits. Measurements of radio-labeled α-bungarotoxin binding show that βP248L reduces AChR expression to 60% of wild-type. Patch clamp recordings of ACh-elicited single channel currents demonstrate that βP248L shortens channel opening bursts from 3.3 ms to 1.2 ms, and kinetic analyses predict that the decay of the synaptic response is accelerated 2.4-fold due to reduced probability of channel reopening. Substituting βP248 with threonine, alanine or glycine reduces the burst duration to 2.3, 1.7, and 1.5 ms, respectively. In non-β subunits, substituting leucine for residues corresponding to βP248 prolongs the burst duration to 4.5 ms in the α subunit, shortens it to 2.2 ms in the δ subunit, and has no effect in the ε subunit. Conversely, substituting proline for residues corresponding to βP248 prolongs the burst duration to 8.7 ms in the α subunit, to 4.6 ms in the δ subunit, but has no effect in the ε subunit. Thus, this fast channel CMS is caused by the dual defects of βP248L in reducing expression of the mutant receptor and accelerating the decay of the synaptic response. The results also reveal subunit-specific contributions of the M1-M2 linker to the durations of channel opening bursts.
Keywords: Neuromuscular junction, neuromuscular disease, congenital myasthenic syndromes, fast-channel myasthenia, acetylchoine receptor, β subunit, intracellular linker, M1-M2 linker, receptor activation, channel gating
Introduction
Congenital myasthenic syndromes (CMS) are disabling disorders in which the safety margin of neuromuscular transmission is compromised by one or more specific mechanisms. Although no fewer than 33 CMS disease genes have been identified to date (Engel et al. 2015; Thompson et al. 2018; Oury et al. 2019), at least half of the CMS stem from variants of genes encoding subunits of the muscle acetylcholine receptor (AChR).
The AChR is a pentameric ligand-gated cation ion channel composed of homologous subunits with stoichiometry (α1)2β1δε (Fig 1A). Each subunit contains four transmembrane domains: the second transmembrane domain (M2) of each subunit forms the wall of the ion channel, and is flanked by the first (M1), third (M3) and fourth (M4) transmembrane domains (Fig 1B and 1C). The intracellular M1-M2 and M3-M4 linkers connect the M1/M2 and M3/M4 domains, respectively, whereas the extracellular M2-M3 linker connects the M2/M3 domain (Fig 1B). The M2-M3 linker in the α subunit couples to the pre-M1 domain through the β1-β2 (Lee and Sine 2005) and Cys loops (Lee, Free, and Sine 2009), forming a principal pathway that transduces ACh binding into channel gating. The M3-M4 linker constitutes most of the cytoplasmic mass of the receptor (Popot and Changeux, 1984), interacts with rapsyn to cluster AChR at the endplates (Maimone and Merlie 1993; Yu and Hall 1994), is a major determinant of the change from fetal γ to adult ε AChR channel gating kinetics (Bouzat, Bren, and Sine 1994), and contributes to AChR activation in a subunit-specific manner (Shen et al. 2005). However, the functional role of the M1-M2 linker in the activation of muscle AChR is less well understood. In pentameric ligand gated ion channels of non-muscle AChR, the M1-M2 linker contributes to ion selectivity (van Nierop et al. 2005; Cymes and Grosman 2016). In muscle AChR, recordings of macroscopic currents show that only the α subunit M1-M2 linker participates in deactivation and desensitization of the receptor (Papke and Grosman 2014), but the contribution of the M1-M2 linker to the kinetics of activation at the single channel level, and whether its contribution depends on the type of subunit, have not been investigated.
Figure 1.
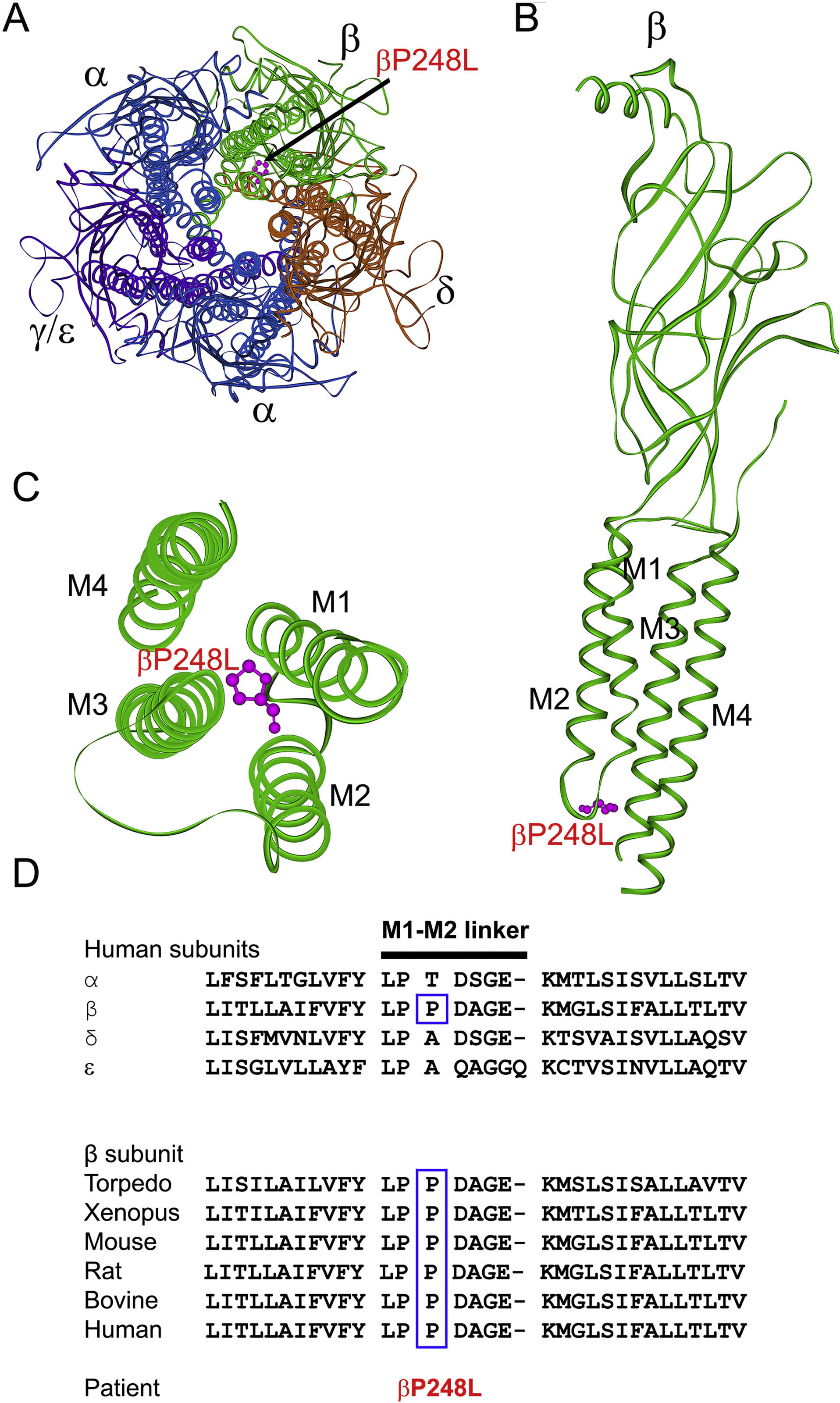
(A) Structure the AChR viewed from the synaptic space indicating the location of the mutant P248 residue (pink) in the M1-M2 linker of the β subunit based on the α-bugarotoxin-bound structure of native Torpedo muscle AChR at a resolution of 2.7 Å (PDB# 6UWZ) (Rahman et al. 2020). (B) Side view of the extracellular and transmembrane domains of the β subunit. The MX helix stemming from the end of M3 is omitted for clarity. (C) Excellular view of the four transmembrane domains of the β subunit indicating the mutant residue βP248L in the M1-M2 linker projecting between the M3 and M4 helices. (D) Multiple sequence alignment of the AChR M1-M2 linkers. Proline at codon 248 in the human β subunit is conserved across all β subunits of different species.
Analysis of naturally occurring pathogenic AChR variants has provided insights into structure-function relationships and mechanisms governing the postsynaptic response of the neuromuscular junction (Sine and Engel 2006; Engel et al. 2015; Shen et al. 2019). The two agonist binding sites of AChR are formed between the α/δ and the α/ε subunits. The role of the β subunit in activation of AChR has not been investigated as extensively as in other muscle AChR subunits or other β subunits in other AChRs. For example, α4β2-AChR, the most abundant neuronal AChR in the brain, has two subtypes, (α4)2(β2)3 with high agonist sensitivity and (α4)3(β2)2 with low agonist sensitivity (Krashia et al. 2010). Each type contains two nominally equivalent agonist binding sites formed at α4 and β2 interfaces. However, the two binding sites within each isoform and between two isoforms contribute differently to receptor activation due to their different neighboring subunits (Harpsoe et al. 2011; Lucero et al. 2016). Thus, in the (α4)2(β2)3 one of the β2 subunits is a non-agonist binding subunit, and contributes to agonist sensitivity. Unlike the β subunits in neuronal AChRs, the role of the β1 subunit in activation of the muscle AChR is not well documented.
CMS caused by variants in AChR subunits can decrease expression on the cell surface, alter the kinetics of activation, or both (Engel et al. 2015; Thompson et al. 2018). CMS with kinetic defects include the slow- and fast-channel CMS, which are associated with slow- and fast-decaying synaptic currents, respectively. Slow-channel mutations were identified in all four subunits of the adult AChR, whereas fast-channel mutations were identified in the α, δ and ε but not in the β subunit (Benarroch et al. 2019).
Here we trace a fast-channel CMS to a novel proline to leucine variant, P248L (P271L in HGVS nomenclature), in the M1-M2 linker in one allele of the AChR β subunit, and a deletion of exon 8 in the other allele. Expression studies of βP248L and mutations at corresponding positions in non-β subunits reveal that the M1-M2 linker contributes to AChR activation in a subunit-specific manner.
Methods
Human subjects and ethics statement
All human studies described in this paper were approved by the Institutional Review Board of the Mayo Clinic, and the patient’s parents gave informed consent for the patient to participate in the study. The investigation was carried out in accordance with the code of ethics of the World Medical Association (Declaration of Helsinki) for experiments.
Structural studies
Intercostal muscle specimens were obtained from the patient and from control subjects without muscle disease undergoing thoracic surgery. The endplates (EPs) were localized for electron microscopy (EM) (Engel 2004). EM images of individual EP regions were analyzed for the area of the nerve terminal, the area of the postsynaptic region, the postsynaptic membrane length, and the postsynaptic membrane concentration (length per unit postsynaptic area) (Engel 1994).
In vitro electrophysiology studies
Intercostal muscle specimens were obtained from origin to insertion from the propositus and from patients without muscle disease undergoing thoracic surgery. The amplitude of the miniature EP potential (MEPP) and the quantal content of the EP potential (m) were determined as previously described (Engel et al. 1993; Uchitel et al. 1993; Kamenskaya, Elmqvist, and Thesleff 1975; Elmqvist and Quastel 1965). In brief, thin strips of muscle containing a motor nerve were pinned out in a chamber with continuously perfused Tyrode solution containing (in mM): NaCl 135, KCl 5, CaCl2 2, MgCl2 1, NaHCO3 15, Na2HPO4 1.3, Dextrose 11.1, 95% O2 and 5% CO2, at pH of 7.2. MEPP and EP potential (EPP) were recorded by conventional methods. The MEPP amplitude was corrected for resting membrane potential of −80 mV and a fiber diameter of 50 μm in control subjects. The decay time of MEPP was determined by fitting each individual trace with one component exponential equation. The quantal content of the EPP (m) was determined by the variance method using the 6th to 70th EPP in a train of stimulation at 1 Hz, and was corrected for a resting membrane potential of −80 mV, nonlinear summation, and non-Poisson release.
Mutation analysis
We directly sequenced genes encoding the AChR α-, β-, δ-, and ε-subunits using the patient’s genomic DNA as previously described (Ohno et al. 1995). Whole genome sequencing of patient’s genomic DNA was performed by Prevention Genetics (Marshfield, WI) for detection of a large deletion or duplication. End-point quantitative PCR analysis within the deletion breakpoint confirmed the large deletion.
Construction and expression of wild-type and mutant AChR
Sources of human α-, β-, δ-, and ε-subunit cDNAs and subcloning of the cDNAs into the CMV-based mammalian expression vector pRBG4 were as previously described (Ohno et al. 1996). The mutations were engineered into wild-type AChR subunit cDNAs using the QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies, Santa Clara, CA). Presence of each mutation and absence of unwanted mutations were confirmed by sequencing the entire inserts. HEK293 cells (ATTC) were transfected with plasmid DNA encoding α-, β-, δ- and ε-subunits, and pEGFP-N1 at a ratio of 2:1:1:1:1, using the FuGENE 6 transfection reagent (Roche, Basel, Switzerland) as previously described (Shen et al. 2005).
α-Bungarotoxin binding measurements
The total number of 125I-α-bungarotoxin (125I-α-bgt) sites on the surface of transfected HEK cells was determined as described previously (Ohno et al. 1996). Briefly, intact cells were harvested 3 days after transfection by gentle agitation in phosphate buffered saline with 5 mM EDTA. After centrifugation, the cells were resuspended in a high-potassium Ringer’s solution containing (in mM) NaCl 5.4, KCl 140, CaCl2 1.8, MgCl2 1.7, HEPS 25, and 30 mg/L BSA, pH7.4, and divided into aliquots for measurements of α-bgt binding. The total number of α-bgt binding sites was determined by incubation for 1 hour in the presence of 5 nM 125I-α-bgt (Perkin Elmer, Waltham, MA). Unbound toxin was removed by washing with high-potassium Ringer’s solution, followed by filtration using a cell harvester (Brandel Incorporated). Radioactivity retained by glass fiber filters (Whatman GF-B, 1-μm cutoff) was measured with a gamma counter (Wizard 1470, Perkin Elmer). Nonspecific binding was determined by measuring the radioactivity of parallel reaction tubes in presence of 300 μM d-tubocurarine before 125I-α-bgt was added. The difference of radioactivity between reactions without and with d-tubocurarine reflects specific α-bgt binding sites.
Patch-clamp recordings and single-channel kinetic analysis
Recordings were obtained in the cell-attached patch configuration at a membrane potential of −80 mV at 22°C with bath and pipette solutions containing (in mM): 142 KCl, 5.4 NaCl, 1.8 CaCl2, 1.7 MgCl2, 10 HEPES, pH 7.4. Single-channel currents were recorded using an Axopatch 200A amplifier (Molecular Devices, San Jose, CA) at a bandwidth of 50 kHz, digitized at 5-μs intervals using a Digidata 1322A (Molecular Devices) and recorded to a hard disk using the program Clampex 8.2 (Molecular Devices). Recordings obtained with ACh concentrations of 1 μM or lower were analyzed at a uniform bandwidth of 11.7 kHz with an imposed dead time of 15.3 μs using TACx4.0.9 software (Bruxton, Seattle, WA). Open intervals and closed times were fitted to the sum of exponentials using TACFITx4.09 (Bruxton). Burst durations were determined by grouping openings separated by a specified closed time, taken as the intersection between the briefest closed time component and the next longer component. Burst duration histograms were fitted to the sum of exponentials and plotted on logarithmic abscissa (Sigworth and Sine 1987).
In the process of AChR activation, the rate of ACh association with the binding sites depends on the ACh concentration. Thus, to determine the rate constants underpinning activation of wild-type and mutant receptors, we employ a range of intermediate and high concentrations of ACh (10 to 1000 μM) , which have the added benefit that opening and closing events from the same receptor cluster into identifiable activation episodes due to desensitization (Sakmann, Patlak, and Neher 1980; Sine et al. 1995). Recordings were analyzed at a bandwidth of 10 kHz and an imposed dead time of 25 μs. The global fitting of a kinetic scheme to dwell times elicited by different ACh concentrations allows us to simultaneously estimate the dissociation constants of ACh binding and the equilibrium constant for channel gating. Clusters were identified as a series of closely spaced openings preceded and followed by closed intervals greater than a defined critical time. The critical time (tcr), was determined by a method that misclassifies an equal number of events between two adjacent closed-time components (Colquhoun and Sakmann 1985). We calculated the tcr manually by solving equation afexp(−tcr/tf) = as(1-exp(−tcr/ts), here, af and as represent area of fast and slow components of closed times, respectively; and tf and ts represent the time constant of fast and slow components of closed times, respectively. For each receptor, the critical time providing the best fit for the closed-time histogram was chosen for the final analysis. Clusters with fewer than five openings were excluded from analysis. Individual clusters were examined for homogeneity based on the mean open probability and open duration for each cluster, and clusters within two SDs of the means of both were accepted for further analysis (Qin, Auerbach, and Sachs 1997; Shen et al. 2002). We used TAC software to measure the durations of open and close events of each recording, and manually derived the open probability from the sum of open time divided by the sum of open and closed times for each cluster. The resulting global set of open and closed dwell times from wild-type and mutant AChRs were analyzed using the program MIL of QuB suite, which uses an interval-based maximum likelihood method that also corrects for missed events to yield fitted rate constants in a kinetic scheme for receptor activation (Qin, Auerbach, and Sachs 1997). At least 3 patches for each desensitizing concentration of ACh were obtained for both wild-type and mutant AChR.
Statistics
Two tailed Student’s t test was used to compare the surface expression of each mutant AChR with wild-type AChR; P value less than 0.05 was considered significant. Error estimates of the scheme rate constants for wild-type and mutant receptors in Table 3 were computed by the MIL program of QuB suite from the curvature of the likelihood function at its maximum (Qin, Auerbach, and Sachs 1997).
Table 3.
Kinetic parameters of wild-type and mutant AChRs expressed in HEK cells with ACh as agonist
| Rate constants | Wild-type | βP248L |
|---|---|---|
| k+1 (μM−1s−1) | 81± 6 | 127 ± 12 |
| k−1 (s−1) | 1,036 ± 108 | 1,642 ± 230 |
| K1 (μM) | 12 | 13 |
| k+2 (μM−1s−1) | 84±2 | 132 ± 5 |
| k−2 (s−1) | 10,679 ± 167 | 13,682 ± 302 |
| K2 (μM) | 127 | 104 |
| β (s−1) | 57,138 ± 1,219 | 27,054 ± 536 |
| α (s−1) | 2,410 ± 66 | 2,708 ± 39 |
| θ | 24 | 10 |
| Predicted mean burst duration (Tpred) (ms) | 2.6 | 1.1 |
| Low conc mean burst duration (Trec) (ms) | 3.3 | 1.2 |
Values represent mean ± SE. Kinetic parameters and errors estimates are derived from the global fitting of a kinetic scheme to data obtained over a wide range of ACh concentrations (see Methods). The dissociation constants, K1 and K2, are the ratios of k−/k+. The apparent channel gating equilibrium constant θ is the ratio of the apparent opening rate (β) to apparent closing rate (α) constants. The predicted burst mean burst duration is given by (1+ β/k−2)/α.
Results
Clinical findings
A 16-yr-old girl presented with profound weakness, bilateral vocal cord paralysis and club foot at birth. For three months after birth she required G-tube for nutrition, and remained respirator dependent until 13 years old mostly for handling secretions. She learned to walk by the age of 5 years, but still eats only soft foods. She is ambulatory, with decreased muscle bulk, fatigable weakness, gastric dysmotility, ophthalmoplegia, and hypophonic speech. She has no similarly affected family members. The electromyography (EMG) studies showed a 10% to 37% decrement of the fourth compared with the first evoked compound muscle action potential on 2 Hz stimulation in different muscles.
Endplate Studies
Electron microscopy of 55 endplate (EP) regions demonstrated no significant abnormality (data not shown). Table 1 summarizes in vitro microelectrode recordings from patient and control EPs. The mean amplitude of miniature endplate potentials (MEPP) was reduced to 37% and the mean decay time of the MEPP was to 22% of control; the quantal content of the endplate potential fell in the normal range.
Table 1.
Endplate studies
| Patient | Controls | |
|---|---|---|
| MEPP Amplitude (mV)a | 0.37 ± 0.090 (10) | 1.00 ± 0.025 (165) |
| MEPP decay (ms) | 0.83 ± 0.14 (10) | 3.75 ± 0.14 (46) |
| Quantal content (1 Hz)b | 34.39 ± 3.45 (20) | 31 ± 1 (190) |
Values indicate mean ± SEM; values in parentheses represent numbers of muscle fibers. Measurements at 29°C ± 0.5°C for MEPPs and EPPs recordings.
Corrected for resting membrane potential of −80 mV and a fiber diameter of 50 μm;
Corrected for a resting membrane potential of −80 mV, nonlinear summation, and non-Poisson release.
Mutation analysis
Sanger sequencing, whole genomic sequencing and microarray analysis revealed two heteroallelic variants in CHRNB1 encoding the AChR β subunit. One allele harbored a novel c.812C>T variant (p.βP248L in legacy nomenclature, or p.βP271L in HGVS nomenclature). Proline at codon 248 is located in the M1-M2 linker (Fig 1A, B, C), is conserved across all β subunits from different species, but differs from equivalent residues in non-β subunits of human AChR (Fig 1D). The second allele contained a ~430 bp deletion which includes the entire exon 8 with breakpoints in introns 7 and 8, corresponding to a minimum deletion boundary via array comparative genomic hybridization (aCGH) of ChR17:7, 357,605 – 7358,039 (GRCh37/hg19). End-point quantitative PCR (qPCR) analysis confirmed the exon 8 deletion c.821–1?_1044+?del with uncertainty in the breakpoint, predicting deletion of the entire exon 8 and disruption of the open reading frame leading to a premature termination due to a frame-shift, p.G251Dfs*21 variant (p.G274Dfs*21 in HGVS nomenclature).
Of note, in the HGVS nomenclature, the first nucleotide is the A of the ATG initiation codon, whereas in legacy nomenclature the first nucleotide is the first base of a cDNA segment encoding the mature peptide. As the AChR β subunit carries a signal peptide of 23 amino acids, the first codon in legacy nomenclature is codon 24 in HGVS nomenclature. Causative mutations in AChR subunits related to congenital myasthenic syndrome in many previous reports are indicated with location of the mutation in the mature peptide. In this study, we employ the legacy nomenclature (p. βP248L and p.βG251Dfs*21). The indicated codon numbers start with the first codon of the mature peptide (NP_000738.2), and the indicated nucleotide numbers start from the translational start site, with +1 corresponding to the A of the ATG translation initiation codon (NM_000747.3). Hence we term two variants as βP248L and βG251Dfs*21.
Surface expression of βP248L AChR
We engineered the βP248L mutation into the cDNA encoding CHRNB1 within the mammalian expression vector pRBG4, and co-expressed it with cDNAs encoding complementary wild-type AChR subunts in HEK293 cells. Measurements of [125I]α-bungarotoxin binding showed that expression of the βP248L-AChR on the surface of intact cells was reduced to 60% of wild-type (Fig 2).
Figure 2.

Specific [125I]α-bgt binding to surface receptors on intact HEK cells transfected with the indicated wild-type or mutant subunits, together with complementary wild-type subunits. The results are normalized to α-bgt binding for the wild-type AChR and represent the mean and SD of the numbers of experiments indicated in following parenthesis of each receptor, wild-type (10), βP248L (5), αT237L (4), δΑ251L (4), εA246L (5), αT237P (4), δΑ251Ρ (4), εA246P (5), βΡ248Τ (5), βΡ248Α (5), and βP248G (5), respectively. *P<0.05, **P<0.001, ***P<0.0001.
Single-channel currents from the βP248L AChR activated by a low concentration of ACh
To determine the kinetic consequences of the βP248L mutation, we recorded single-channel currents elicited by a limiting low concentration of ACh from HEK293 cells expressing either wild-type or mutant AChRs. We used 50 nM ACh for wild-type AChR and 1 uM ACh for mutant receptor because 50 nM ACh elicited insufficient opening events from the mutant for analysis. Compared to the wild-type AChR, the open intervals and bursts of channel openings from the mutant AChR were noticeably briefer and the number of openings per burst were reduced (Fig 3, left panel; Table 2). After defining bursts as successive openings separated by closings of a defined brief duration, the bursts were compiled into duration histograms, and each histogram was fitted by the sum of exponentials. The fitting analysis revealed 2 exponential components for βP248L, with a mean duration of the longest component of 1.2 ms, and 3 exponential components for the wild-type AChR, with a mean duration of the longest component of 3.3 ms (Fig 3, right panel and Table 2). The mean burst duration provides an independent estimate of the time constant for decay of the synaptic response; the briefer burst duration of the mutant receptor of 1.2 ms is close to the time constant for decay of the MEPP of 0.83 ms (Table 1), which is typical of the fast-channel CMS.
Figure 3.
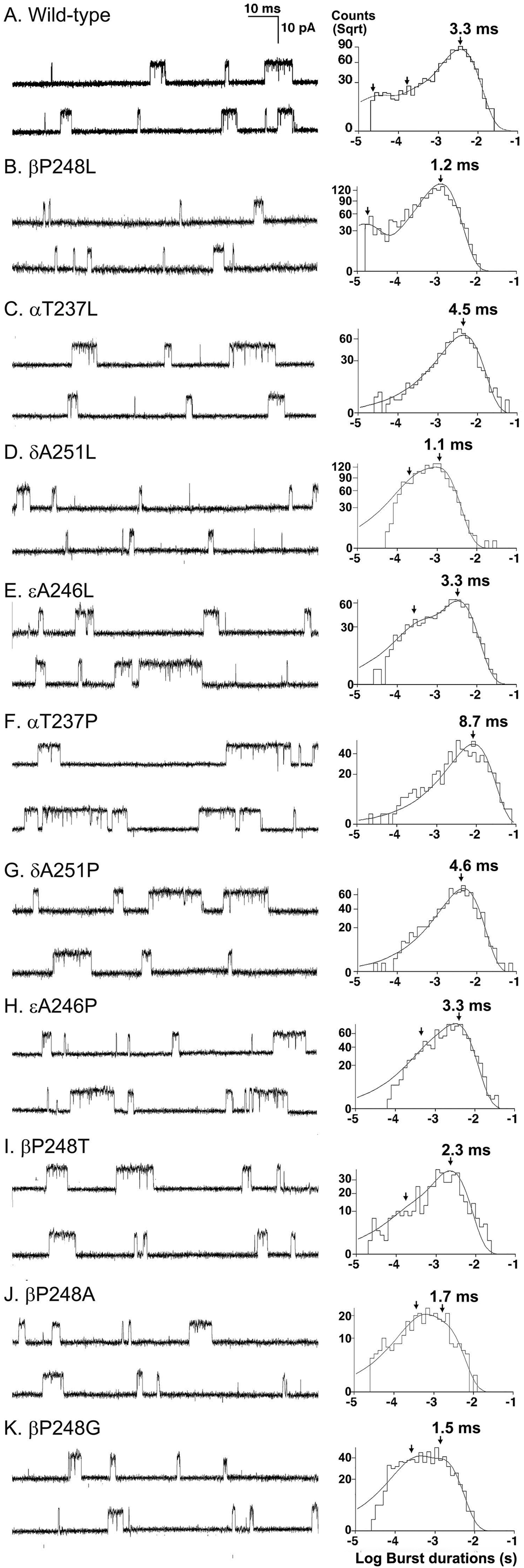
Single channel currents recorded from HEK cells expressing wild-type (A) and the indicated mutant AChRs (B to K). Left panel shows representative traces from the same patch with channel openings depicted as upward deflections. The histograms of burst durations are shown from one patch in wild-type and combined patches in all mutants. The peaks of individual components are indicated by arrows. Bandwidth = 10–12 kHz; [ACh] = 50 nM − 1 μM.
Table 2.
Analyses of open and burst durations for wild-type and mutant AChRs in HEK cells
| AChR | Open Intervals | Bursts | ||||
|---|---|---|---|---|---|---|
| T1,ms (a1) |
T2, ms (a2) |
T3, ms (a3) |
T1,ms (a1) |
T2, ms (a2) |
T3, ms (a3) |
|
| Wild type | 0.037 ± 0.033a | 0.31 ± 0.050 | 1.35 ± 0.051 | 0.036 ± 0.0017b | 0.47 ± 0.059 | 3.31 ± 0.12 |
| (0.17 ± 0.022) | (0.27 ± 0.038) | (0.67 ± 0.042) | (0.24 ± 0.021) | (0.21 ± 0.027) | (0.58 ± 0.038) | |
| βP248L | 0.12 ± 0.014 | 0.88 ± 0.014 | 0.14 ± 0.019c | 1.24 ± 0.026 | ||
| (0.23 ± 0.035) | (0.77 ± 0.035) | (0.21 ± 0.013) | (0.86 ± 0.069) | |||
| αT237L | 1.59 ± 0.12 | 4.50 ± 0.27 | ||||
| (1.00± 0.00) | (1.00± 0.00) | |||||
| δA251L | 0.21 ± 0.065c | 0.85 ± 0.17 | 0.29 ± 0.091c | 1.11 ± 0.15 | ||
| (0.24 ± 0.066) | (0.84 ± 0.088) | (0.30 ± 0.083) | (0.80 ± 0.11) | |||
| εA246L | 1.35 ± 0.066 | 0.30 ± 0.00d | 3.27 ± 0.18 | |||
| (1.00± 0.00) | (0.26± 0.00) | (0.91 ± 0.86) | ||||
| αT237P | 2.02 ± 0.050 | 8.70 ± 1.20 | ||||
| (1.00± 0.00) | (1.00± 0.00) | |||||
| δA251P | 1.67 ± 0.048 | 4.55 ± 0.50 | ||||
| (1.00 ± 0.00) | (1.00 ± 0.00) | |||||
| εA246P | 1.26 ± 0.11 | 0.30 ± 0.024 | 3.33 ± 0.11 | |||
| (1.00 ± 0..00) | (0.15± 0.036) | (0.86 ± 0.036) | ||||
| βP248T | 1.05 ± 0.10 | 0.15 ± 0.084 | 2.26 ± 0.11 | |||
| (1.00 ± 0.00) | (0.17 ± 0.035) | (0.83 ± 0.035) | ||||
| βP248A | 0.051 ± 0.0012c | 0.68 ± 0.079 | 0.38 ± 0.10 | 1.68 ± 0.21 | ||
| (0.18 ± 0.098) | (0.88 ± 0.081) | (0.52 ± 0.15) | (0.48 ± 0.15) | |||
| βP248G | 0.61 ± 0.088 | 0.28 ± 0.067 | 1.54 ± 0.098 | |||
| (1.00 ± 0.00) | (0.44 ± 0.14) | (0.56 ± 0.14) | ||||
Values indicate mean ± SE from 21 patches for wild-type, 3 patches for all mutants except for αT237L with 5 patches. [ACh] = 50 nM for wild-type, δA251L, εA246L, βP248A, and βP248G; 1 μM for others. Final bandwidth 10–12 kHz. MP = −80 mV. τn and an are time constants and fractional areas.
corresponding component of open and burst durations were not detected for 12, 3, 1, and 2 patches, respectively.
Single-channel recordings from βP248L AChRs at increased ACh concentrations
To determine the kinetic consequences of βP248L, we recorded single-channel currents activated by a range of intermediate and high ACh concentrations (10 to 1000 μM) (Fig 4). At increased ACh concentrations, channel openings appear in clusters of many successive openings, all from the same AChR. The temporal distributions of openings and closings within clusters provide kinetic information on state transitions between closed and open states. Fitting a kinetic scheme to the sequences of intra-cluster dwell times yields estimates of rate constants for state transitions in the scheme. For both the wild-type and mutant AChRs, the intra-cluster closings become progressively briefer as the ACh concentration increases, and the closed duration histograms mirror the concentration dependent change from long to brief intra-cluster closings observed in the single channel current traces (Fig 4). In addition, at intermediate ACh concentrations, the ratio of brief to long closings is reduced for the mutant compared to the wild-type AChR. These qualitative changes in intra-cluster closings suggest the mutation affects one or more rate constants in a kinetic scheme for receptor activation.
Figure 4.
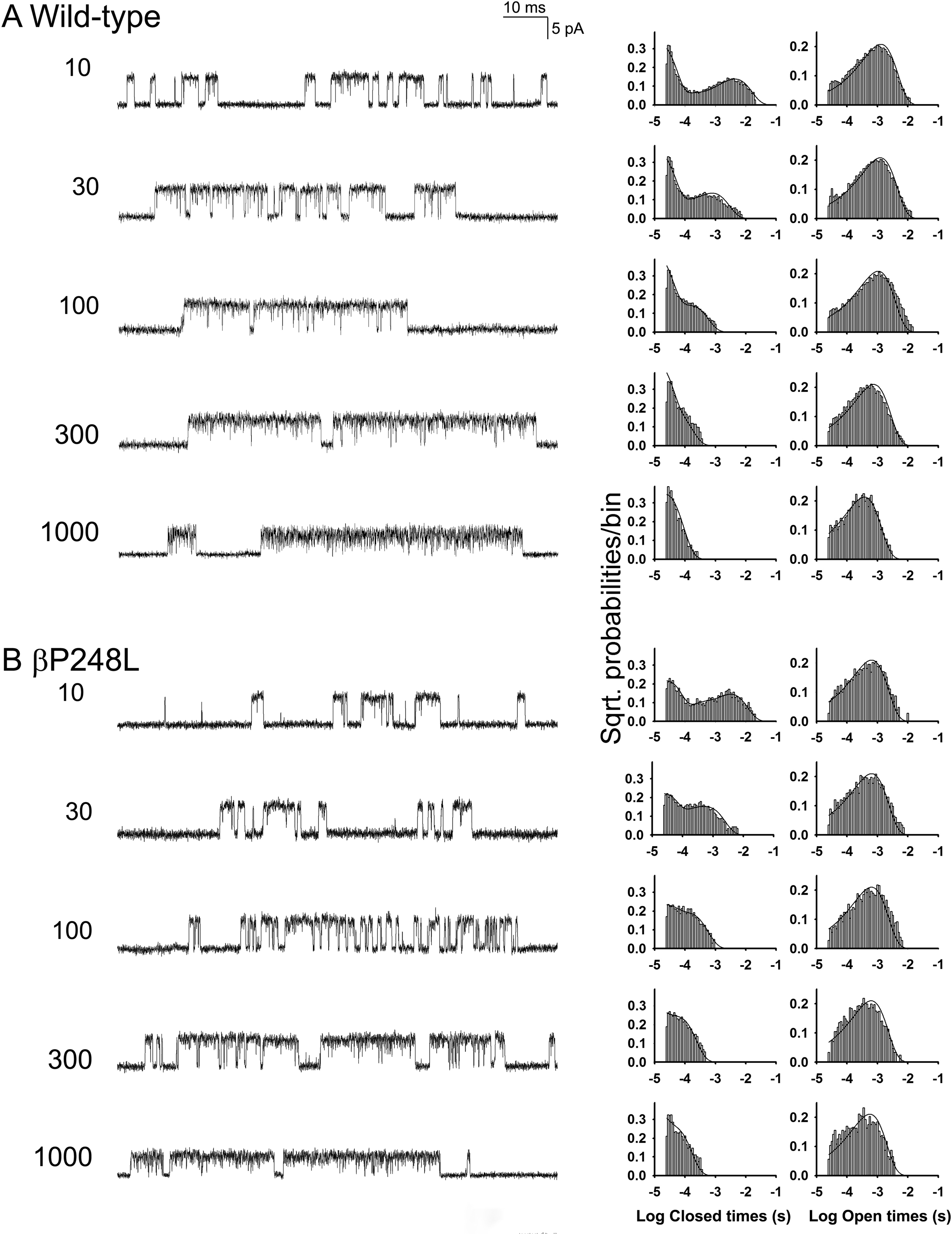
Single channel recordings from wild-type and βP248L AChRs elicited by the indicated concentrations of ACh (μM). The left columns show representative single channel currents recorded from HEK cells. The center and right columns show corresponding histograms of closed and open dwell times with the global fits for Scheme 1 superimposed. Fitted rate constants are shown in Table 3.
To identify rate constants altered by the mutation, we used the program MIL within QuB suite to fit the following kinetic scheme (Scheme 1) to sequences of open and closed dwell times obtained over a range of increased ACh concentrations:
| Scheme 1 |
In Scheme 1, two agonists (A) bind to the receptor (R) with association rate constants k+1 and k+2 and dissociate with rate constants k−1 and k−2. The doubly occupied receptor opens with the rate constant β and closes with the rate constant α; ACh blocks the open channel with the rate constant k+b, and unblocks from the open channel with the rate constant k−b. Owing to bandwidth limitations, the previously identified closed state between A2R and A2R*, known as either flip or primed (Lape, Colquhoun, and Sivilotti 2008; Mukhtasimova et al. 2009), is not included; thus the fitted parameters β, α, and k−2 are apparent rate constants. Also, a desensitized state is not included because each cluster begins following recovery from desensitization and ends upon return to the desensitized state, so that closings within clusters represent transitions between activatable states. We fittted Scheme 1 to the same set of global sequences of dwell times, with and without assuming that the association and dissociation rate constants are equivalent at each binding site. We found that fitting without constraining the association and dissociation rates gave better loglikelihood values than fitting with the constrained rate constants. Fitting Scheme 1 to the global sequences of open and closed dwell times obtained over a range of ACh concentrations showed that βP248L decreases the channel opening (β) rate constant 2.12-fold, and increases closing (α) rate constant 1.12-fold, resulting in a 2.37-fold decrease in the equilibrium constant (θ) for channel gating, defined by β/α. (Fig. 4 and Table 3). In addition, the mutation increases the rate constant for ACh dissociation from the diliganded receptor k−2 by 1.28-fold. The fitted rate constants predict that the probability the diliganded receptor-channel will open is 0.83 for wild-type and 0.66 for the mutant estimated from the ratio β/(β + k−2), or a 1.3-fold reduction. Of note, the extent of reduction of opening probability of diliganded channel is less than the reduction in the amplitude of the MEPP recorded from patient endplates (Table 1), suggesting the severity of disease of the patient is attributed to dual defects of reduced expression of AChR at the endplates and the impaired channel gating kinetics.
To validate the fitted rate constants, we computed the mean burst durations for wild-type and mutant AChRs from the relationship τ =(β/k−2+1)/α and the values in Table 3, and compared the predicted and measured burst durations (Table 2). The predicted burst durations are 2.6 ms for wild-type AChR and 1.1 ms for βP248L AChR, in good agreement with the measured burst durations of 3.3 and 1.2 ms, respectively.
Substitution of leucine for residues equivalent to βΡ248 in non-β subunits
To determine whether the effects of the βP248L mutation are subunit specific, we substituted leucine at equivalent positions of the α (αT237L), δ (δA251L), and ε (εA246L) subunits (Figs. 2 and 3, Table 2), measured cell-surface expression, and recorded single channel currents elicited by a limiting low concentration of ACh. The αT237L mutation reduces cell-surface expression to 25% of wild-type and prolongs the mean burst duration to 4.5 ms, whereas δA251L does not alter surface expression but shortens the burst duration to 1.1 ms, and εA246L does not affect surface expression or the mean burst duration.
Substitution of proline for residues equivalent to βP248 in non-β subunits
Given that βP248 is conserved in all β subunits but not in non-β subunits, we substituted proline for the corresponding residues in non-β subunits (Figs 2 and 3, Table 2), measured cell-surface expression, and recorded single channel currents elicited by a limiting low concentration of ACh. Substituting proline in either the α (αT237P) or δ (δA251P) subunits does not affect cell-surface expression, but prolongs the mean burst duration to 8.7 and 4.6 ms, respectively. Substituting proline in the ε subunit (εA246P) increases cell-surface expression to 144% of wild-type but does not affect the mean burst duration.
Substitution of βΡ248 by equivalent residues in non-β subunits
To determine whether the deleterious consequences of βP248L depend on the side chain of the substituting residue, we replaced βP248 with threonine (the corresponding residue in the α subunit), alanine (the corresponding residue in the δ and ε subunits), and glycine (Figs 2 and 3, Table 2). The mutants βP248T, βP248A and βP248G do not affect cell surface expression, but reduce the mean burst duration to 2.3 ms, 1.7 ms, and 1.6 ms, respectively. Thus the kinetic consequences of mutations of βP248 do not depend systematically on size or conformational mobility by the side chain of the substituting residue.
Discussion
By clinical, ultrastructural, electrophysiologic, biochemical and molecular genetics approaches, we traced a severe fast-channel CMS to novel variants in CHRNB1: a missense variant βP248L in the M1-M2 linker in one allele, and a large deletion in the other allele resulting in loss of exon 8, frame shift and a premature stop codon. Thus the impaired synaptic response at the patient endplate arises from AChRs containing the βP248L variant, which causes a 2.7-fold decrease of the MEPP decay time and also decrease the opening burst duration of the βP248L receptor. The low amplitude and fast decay of MEPPs reduces the post-synaptic depolarization after each nerve stimulus, resulting in activation of fewer postsynaptic voltage-dependent sodium channels, and a reduced safety margin of neuromuscular transmission. To our knowledge, this is the first report of a fast-channel CMS caused by a mutation in the AChR β subunit. In this patient, the safety margin of neuromuscular transmission is compromised by the reduced amplitude and accelerated decay of the synaptic response due to the combined effects of reduced expression and impaired channel gating of AChR.
Our single channel analysis of the pathogenic βP248L variant reveals that the structure of the M1-M2 linker contributes to elementary state transitions that open and close the AChR channel. Furthermore, studies of mutations at corresponding positions in the non-β subunits reveal that the contribution of the M1-M2 linker to channel gating is subunit specific. While most studies of the M1-M2 linker focused on its contribution to ion selectivity (Corringer et al. 1999; Cymes and Grosman 2016), studies of the M1-M2 linker in the inhibitory glycine receptor revealed that mutations in the linker reduced maximal glycine activated current, increased the EC50 for glycine, and converted partial agonists to antagonists (Lynch et al. 1997; Saul et al. 1999). The collective studies at the level of macroscopic currents suggested that the structure of the M1-M2 linker is crucial for efficient channel opening. However, our study shows that the M1-M2 linker contributes to channel gating at the single channel level.
The residue βP248 targeted by the pathogenic mutation is conserved among muscle β subunits of different species, but is not present in any other type of muscle or neuronal AChR subunit. The unique presence of proline in the β subunit, together with its contribution to channel gating, suggest positions equivalent to βP248 in other subunits may also contribute to channel gating of the muscle AChR. To test this notion, we replaced the residue equivalent to βP248 in non-β subunits with proline and measured mean burst durations, but observed no effect in the ε subunit, and prolonged rather than shortened bursts in the α and δ subunits. Conversely, substituting the pathogenic residue leucine at corresponding positions in non-β subunits showed no effect in the ε subunit, prolonged bursts in the α subunit, and shortened bursts in the δ subunit. Thus, proline at position 248 of the β subunit is unique in contributing to the mean burst duration.
The M1-M2 linker flanks the M2 domain that lines the channel pore, and is structurally suited to serve as a hinge about which the lower part of the M2 domain moves (Fig. 1B). The structure of the M1-M2 linker may thus contribute to this movement. Adjacent to βP248 is a second proline that is conserved in all AChR subunits as shown in Fig 1D, and the resulting vicinal pair of prolines in the β subunit may provide additional restriction of the protein backbone required for proper channel gating. To test whether proline at position 248 of the β subunit is unique in conferring proper channel gating, we substituted Thr, Ala and Gly for βP248, and found that each of these substitutions reduced the mean burst duration to a different extent. Thus, given the range of sizes of the substituted side chains, from Gly to Leu, we conclude that the size of the side chain at position 248 of the β subunit does not correlate with the change in mean burst duration. Thus we suggest that the additional restriction of the protein backbone provided by a second proline at position 248 of the β subunit is crucial for the proper hinge motion of the M1-M2 linker. Alternatively, articulation of βP248 with side chains stemming from the M3 and M4 domains (Fig. 1C) may also contribute to the proper hinge motion.
Finally, our expression studies show that (1) replacement of Pro248 by Leu in the β subunit reduces the surface expression but substitutions of the same residue by Thr, Ala, and Gly have no effect; (2) replacement of Thr237 in the α subunit by Leu reduces the surface expression but substitution by Pro has no effect; (3) replacement of Ala in the δ or ε subunit by Leu have no effect; and (4) substitution of Ala in the δ subunit by Pro has no effect, but that in ε subunit increases the surface expression. The overall results suggest that (1) the residue in the middle of M1-M2 linker of each subunit contributes to surface expression of the receptor in a subunit specific way; (2) the branched structure of leucine is harmful to receptor assembly in the α and β subunits but not in δ and ε subunits; and (3) the additional five-membered nitrogen-containing ring of Pro next to the highly conserved Pro in all subunits is not harmful to the receptor expression.
In summary, we characterize a fast-channel CMS (CMS2B) caused by a novel variant in the AChR β subunit. Compared to our previously reported cases of fast-channel CMS, the extent of kinetic defects of the βP248L variant is relatively mild. The severity of phenotype may be due to additional unidentified factors. Nonetheless, our study reveals that the M1-M2 linker is a structural determinant of AChR channel gating, and that its contribution is subunit specific.
Highlights.
First report of an AChR β subunit variant causing a fast-channel myasthenia
Role of the M1-M2 linker in activating AChR examined at the single channel level
Subunit-specific contributions of the M1-M2 linkers to channel gating
Acknowledgement
This work was supported by NIH grants NS109491 (AGE, DS, XMS) and NS031744 (SMS), and a Myasthenia Gravis Foundation of America grant FP00094864 (AGE).
Abbreviations:
- CMS
congenital myasthenic syndrome
- ACh
acetylcholine
- AChR
acetylcholine receptor
- M1, M2, M3, M4
first, second, third and fourth transmembrane domain
- EP
endplate
- EMG
electromyography
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none.
References
- Benarroch L, Bonne G, Rivier F, and Hamroun D. 2019. ‘The 2020 version of the gene table of neuromuscular disorders (nuclear genome)’, Neuromuscul Disord, 29: 980–1018. [DOI] [PubMed] [Google Scholar]
- Bouzat C, Bren N, and Sine SM. 1994. ‘Structural basis of the different gating kinetics of fetal and adult acetylcholine receptors’, Neuron, 13: 1395–402. [DOI] [PubMed] [Google Scholar]
- Colquhoun D, and Sakmann B. 1985. ‘Fast events in single-channel currents activated by acetylcholine and its analogues at the frog muscle end-plate’, J. Physiol, 369: 501–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corringer PJ, Bertrand S, Galzi JL, Devillers-Thiery A, Changeux JP, and Bertrand D. 1999. ‘Mutational analysis of the charge selectivity filter of the alpha7 nicotinic acetylcholine receptor’, Neuron, 22: 831–43. [DOI] [PubMed] [Google Scholar]
- Cymes GD, and Grosman C. 2016. ‘Identifying the elusive link between amino acid sequence and charge selectivity in pentameric ligand-gated ion channels’, Proc Natl Acad Sci U S A, 113: E7106–E15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmqvist D, and Quastel DM. 1965. ‘A quantitative study of end-plate potentials in isolated human muscle’, J. Physiol, 178: 505–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engel AG 1994. “Quantitative morphological studies of muscle” In Myology, edited by Engel AG, Franzini-Armstrong C, 1018–45. New York, NY: McGraw-Hill. [Google Scholar]
- Engel AG 2004. ‘The muscle biopsy’ in Engel AG, Franzini-Armstrong C (ed.), Myology (McGraw-Hill: New York, NY: ). [Google Scholar]
- Engel AG, Nagel A, Walls TJ, Harper CM, and Waisburg HA. 1993. ‘Congenital myasthenic syndromes: I. Deficiency and short open-time of the acetylcholine receptor’, Muscle Nerve, 16: 1284–92. [DOI] [PubMed] [Google Scholar]
- Engel AG, Shen XM, Selcen D, and Sine SM. 2015. ‘Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment’, Lancet Neurol, 14: 420–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harpsoe K, Ahring PK, Christensen JK, Jensen ML, Peters D, and Balle T. 2011. ‘Unraveling the high- and low-sensitivity agonist responses of nicotinic acetylcholine receptors’, J Neurosci, 31: 10759–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hassaine G, Deluz C, Grasso L, Wyss R, Tol MB, Hovius R, Graff A, Stahlberg H, Tomizaki T, Desmyter A, Moreau C, Li XD, Poitevin F, Vogel H, and Nury H. 2014. ‘X ray structure of the mouse serotonin 5-HT3 receptor’, Nature, 512: 276–81. [DOI] [PubMed] [Google Scholar]
- Hibbs RE, and Gouaux E. 2011. ‘Principles of activation and permeation in an anion-selective Cys-loop receptor’, Nature, 474: 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenskaya MA, Elmqvist D, and Thesleff S. 1975. ‘Guanidine and neuromuscular transmission. II. Effect on transmitter release in response to repetitive nerve stimulation’, Arch. Neurol, 32: 510–18. [DOI] [PubMed] [Google Scholar]
- Krashia P, Moroni M, Broadbent S, Hofmann G, Kracun S, Beato M, Groot-Kormelink PJ, and Sivilotti LG. 2010. ‘Human alpha3beta4 neuronal nicotinic receptors show different stoichiometry if they are expressed in Xenopus oocytes or mammalian HEK293 cells’, PLoS One, 5: e13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lape R, Colquhoun D, and Sivilotti LG. 2008. ‘On the nature of partial agonism in the nicotinic receptor superfamily’, Nature, 454: 722–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WY, Free CR, and Sine SM. 2009. ‘Binding to gating transduction in nicotinic receptors: Cys-loop energetically couples to pre-M1 and M2-M3 regions’, J. Neurosci, 29: 3189–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WY, and Sine SM. 2005. ‘Principal pathway coupling agonist binding to channel gating in nicotinic receptors’, Nature, 438: 243–47. [DOI] [PubMed] [Google Scholar]
- Li SX, Huang S, Bren N, Noridomi K, Dellisanti CD, Sine SM, and Chen L. 2011. ‘Ligand- binding domain of an alpha7-nicotinic receptor chimera and its complex with agonist’, Nat. Neurosci, 14: 1253–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucero LM, Weltzin MM, Eaton JB, Cooper JF, Lindstrom JM, Lukas RJ, and Whiteaker P. 2016. ‘Differential alpha4(+)/(−)beta2 Agonist-binding Site Contributions to alpha4beta2 Nicotinic Acetylcholine Receptor Function within and between Isoforms’, J Biol Chem, 291: 2444–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch JW, Rajendra S, Pierce KD, Handford CA, Barry PH, and Schofield PR. 1997. ‘Identification of intracellular and extracellular domains mediating signal transduction in the inhibitory glycine receptor chloride channel’, EMBO J, 16: 110–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maimone MM, and Merlie JP. 1993. ‘Interaction of the 43 kd postsynaptic protein with all subunits of the muscle nicotinic acetylcholine receptor’, Neuron, 11: 53–66. [DOI] [PubMed] [Google Scholar]
- Mukhtasimova N, Lee WY, Wang HL, and Sine SM. 2009. ‘Detection and trapping of intermediate states priming nicotinic receptor channel opening’, Nature, 459: 451–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Hutchinson DO, Milone M, Brengman JM, Bouzat C, Sine SM, and Engel AG. 1995. ‘Congenital myasthenic syndrome caused by prolonged acetylcholine receptor channel openings due to a mutation in the M2 domain of the epsilon subunit’, Proc. Natl. Acad. Sci. U. S. A, 92: 758–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno K, Wang HL, Milone M, Bren N, Brengman JM, Nakano S, Quiram P, Pruitt JN, Sine SM, and Engel AG. 1996. ‘Congenital myasthenic syndrome caused by decreased agonist binding affinity due to a mutation in the acetylcholine receptor epsilon subunit’, Neuron, 17: 157–70. [DOI] [PubMed] [Google Scholar]
- Oury J, Liu Y, Topf A, Todorovic S, Hoedt E, Preethish-Kumar V, Neubert TA, Lin W, Lochmuller H, and Burden SJ. 2019. ‘MACF1 links Rapsyn to microtubule- and actin-binding proteins to maintain neuromuscular synapses’, J Cell Biol, 218: 1686–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke D, and Grosman C. 2014. ‘The role of intracellular linkers in gating and desensitization of human pentameric ligand-gated ion channels’, J Neurosci, 34: 7238–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin F, Auerbach A, and Sachs F. 1997. ‘Maximum likelihood estimation of aggregated Markov processes’, Proc. Biol. Sci, 264: 375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman MM, Teng J, Worrell BT, Noviello CM, Lee M, Karlin A, Stowell MHB, and Hibbs RE. 2020. ‘Structure of the Native Muscle-type Nicotinic Receptor and Inhibition by Snake Venom Toxins’, Neuron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sali A, and Blundell TL. 1993. ‘Comparative protein modelling by satisfaction of spatial restraints’, J Mol Biol, 234: 779–815. [DOI] [PubMed] [Google Scholar]
- Saul B, Kuner T, Sobetzko D, Brune W, Hanefeld F, Meinck HM, and Becker CM. 1999. ‘Novel GLRA1 missense mutation (P250T) in dominant hyperekplexia defines an intracellular determinant of glycine receptor channel gating’, J Neurosci, 19: 869–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen XM, Milone M, Wang HL, Banwell B, Selcen D, Sine SM, and Engel AG. 2019. ‘Slow-channel myasthenia due to novel mutation in M2 domain of AChR delta subunit’, Ann Clin Transl Neurol, 6: 2066–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen XM, Ohno K, Fukudome T, Tsujino A, Brengman JM, De Vivo DC, Packer RJ, and Engel AG. 2002. ‘Congenital myasthenic syndrome caused by low-expressor fast-channel AChR delta subunit mutation’, Neurology, 59: 1881–88. [DOI] [PubMed] [Google Scholar]
- Shen XM, Ohno K, Sine SM, and Engel AG. 2005. ‘Subunit-specific contribution to agonist binding and channel gating revealed by inherited mutation in muscle acetylcholine receptor M3-M4 linker’, Brain, 128: 345–55. [DOI] [PubMed] [Google Scholar]
- Sigworth FJ, and Sine SM. 1987. ‘Data transformations for improved display and fitting of single-channel dwell time histograms’, Biophys. J, 52: 1047–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sine SM, and Engel AG. 2006. ‘Recent advances in Cys-loop receptor structure and function’, Nature, 440: 448–55. [DOI] [PubMed] [Google Scholar]
- Sine SM, Ohno K, Bouzat C, Auerbach A, Milone M, Pruitt JN, and Engel AG. 1995. ‘Mutation of the acetylcholine receptor alpha subunit causes a slow-channel myasthenic syndrome by enhancing agonist binding affinity’, Neuron, 15: 229–39. [DOI] [PubMed] [Google Scholar]
- Thompson R, Abicht A, Beeson D, Engel AG, Eymard B, Maxime E, and Lochmuller H. 2018. ‘A nomenclature and classification for the congenital myasthenic syndromes: preparing for FAIR data in the genomic era’, Orphanet J Rare Dis, 13: 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchitel O, Engel AG, Walls TJ, Nagel A, Atassi MZ, and Bril V. 1993. ‘Congenital myasthenic syndromes: II. Syndrome attributed to abnormal interaction of acetylcholine with its receptor’, Muscle Nerve, 16: 1293–301. [DOI] [PubMed] [Google Scholar]
- van Nierop P, Keramidas A, Bertrand S, van Minnen J, Gouwenberg Y, Bertrand D, and Smit AB. 2005. ‘Identification of molluscan nicotinic acetylcholine receptor (nAChR) subunits involved in formation of cation- and anion-selective nAChRs’, J Neurosci, 25: 10617–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu XM, and Hall ZW. 1994. ‘The role of the cytoplasmic domains of individual subunits of the acetylcholine receptor in 43 kDa protein-induced clustering in COS cells’, J Neurosci, 14: 785–95. [DOI] [PMC free article] [PubMed] [Google Scholar]