

Abstract
Breast cancer metastasis is a leading cause of cancer-related death of women in the U.S. A hurdle in advancing metastasis-targeted intervention is the phenotypic heterogeneity between primary and secondary lesions. To identify metastasis-specific gene expression profiles we performed RNA sequencing of breast cancer mouse models; analyzing metastases from models of various drivers and routes. We contrasted the models and identified common, targetable signatures. Allograft models exhibited more mesenchymal-like gene expression than genetically engineered mouse models (GEMMs), and primary culturing of GEMM-derived metastatic tissue induced mesenchymal-like gene expression. Additionally, metastasis-specific transcriptomes differed between tail vein and orthotopic injection of the same cell line. Gene expression common to models of spontaneous metastasis included sildenafil response and nicotine degradation pathways. Strikingly, In vivo sildenafil treatment significantly reduced metastasis 54% while nicotine significantly increased metastasis 46%. These data suggest that (i) actionable metastasis-specific pathways can be readily identified, (ii) already-available drugs may have great potential to alleviate metastatic incidence, and (iii) metastasis may be influenced greatly by lifestyle choices such as the choice to consume nicotine products. In summary, while mouse models of breast cancer metastasis vary in in ways that must not be ignored, there are shared features that can be identified and potentially targeted therapeutically.
Graphical Abstract
Introduction
Metastatic breast cancer remains the leading cause of cancer-related death among women world-wide(1). Standard treatment for breast cancer patients diagnosed with stage I-III is surgical resection, indicating that mortality is largely due to recurrence at distant sites(2). Indeed, while non-metastatic breast cancer has a 5-year survival rate of 98.8%, metastatic disease reduces 5-year survival to only 27.4%(1). Therapeutic strategies to treat localized disease, such as molecular profiling and targeted therapy, have been increasingly successful, but patients with disseminated disease continue to face much worse outcomes, as metastases are largely insensitive to current treatments(3). Therefore, to improve outcome for patients with advanced cancer, specific metastasis-targeted strategies will need to be developed, as will a deeper understanding of the unique biological processes that occur during disease progression(4).
Investigations of metastatic biology trail those of primary tumors (PTs) due to difficulty in obtaining appropriate human tissue samples. Metastatic lesions are usually not surgically removed, and the biology of those that are resected are usually confounded by treatment(5), which may enrich for characteristics associated with therapy resistance rather than metastasis. Additionally, early biopsies of metastatic lesions in untreated patients often have low tumor cell content(5) or are not accessible to biopsy, and repeated biopsies of lesions within individuals increases patient morbidity(6). As an added challenge, since metastases within patients exhibit significant heterogeneity at both the biomarker(7) and genomic levels(8), selective biopsies may not capture the full complexity of events associated with tumor progression. Therefore, although human data from smaller genomics projects focused on metastasis are now becoming available(9), our understanding of the somatic events that contribute to metastasis remains far behind the understanding of primary tumorigenesis.
Mouse models overcome many of these limitations and offer additional advantages: (i) a readily available, renewable resource of tissue enables investigation of a genetically equivalent set of tissues over time; (ii) the ability to observe the full natural history of metastatic development, which is not possible in patients due to surgical resection and application of neo-adjuvant and adjuvant therapies; (iii) the existence of syngeneic cell line injection (allograft) models to rapidly test hypotheses generated from genomic characterization; and (iv) genetically engineered models to examine factors that function through somatic microenvironmental effects rather than directly within tumor cells, an aspect often precluded in human studies. Data generated from animal models therefore provide an important vehicle for hypothesis generation and testing that can subsequently be examined and verified in more restrictive human experimental systems.
As mouse models have become integral to the study of metastatic spread, the variety of pre-clinical mouse models has grown to better investigate the steps of the metastatic cascade and capture the many stages of this disease. To identify metastasis-specific gene expression (MSGE) profiles that are integral to the establishment and growth of metastatic lesions, we performed RNA-sequencing (RNA-seq) of breast cancer metastases and paired primary tumors isolated from several mouse models. As the interventions for metastatic cancer are limited to targeting already established secondary tumors(10), we analyzed primary tumors and macrometastases from models of various oncogenic drivers and routes, including orthotopic injection, tail vein injection, intracardiac injection, and genetically engineered mouse models (GEMMs). Here we present data revealing critical differences in MSGE between pre-clinical models of metastatic breast cancer. Additionally, we have identified several core and targetable gene expression pathways common to multiple models, which we further confirmed the significance of in vivo. By combining the analysis of tissues from several models of metastasis we present here a robust strategy for the discovery of clinically targetable pathways that may be integral to metastatic spread.
Materials and Methods
Ethics statement
The research described in this study was performed under the Animal Study Protocol LCBG-004 and LPG-002, approved by the National Cancer Institute (NCI) Animal Use and Care Committee. Animal euthanasia was performed by cervical dislocation after anesthesia by Avertin.
Genetically engineered mouse models
FVB/N-Tg(MMTV-PyVT)634Mul/J and FVB/N-Tg(MMTVneu)202Mul/J male mice were obtained from The Jackson Laboratory. FVB/N-Tg(C3(1)-TAg) and FVB/N-Tg(MMTV-Myc) were a generous gift from Dr. Jeffrey Green (NCI, Bethesda, MD).
Male PyMT mice were crossed with female wild type FVB/NJ, MOLF/EiJ, CAST/EiJ, C57BL/6J, and C57BL/10J mice also obtained from The Jackson Laboratory. Male Her2, Myc, and C3(1)-TAg mice were crossed with female wild type FVB/NJ mice. All female F1 progeny were genotyped by the Laboratory of Cancer Biology and Genetics genotype core for the PyMT or Her2 gene and grown until humane endpoint. Primary tumor, metastatic nodules, and normal (tail) tissue were isolated immediately following euthanasia and snap-frozen in liquid nitrogen. Tissue samples were then stored at −80°C.
Cell culture
The mouse mammary carcinoma cell lines 6DT1, 4T1, Mvt1, and MET1 were a generous gift from Dr. Lalage Wakefield (NCI, Bethesda, MD) whose lab validated and characterized these lines in their 2017 publication (11). The mouse mammary carcinoma cell line M6 was a generous gift from Dr. Jeffrey Green (NCI, Bethesda, MD) whose lab generated the cell line from FVB/N-Tg(C3(1)-TAg) mice, which they generated and published in 2000(12). All cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM), supplemented with 10% Fetal Bovine Serum (FBS), 1% Penicillin and Streptomycin (P/S), and 1% L-Glutamine (Gibco), and maintained at 37°C with 5% CO2. All cell lines were tested for mycoplasma using the MycoAlertTM PLUS Mycoplasma Detection Kit (Lonza) upon receipt from Dr. Wakefield and Dr. Green, before expansion creation of liquid nitrogen stocks. Cells were not cultured beyond 10 passages.
In vivo metastasis
Female virgin FVB/NJ or BALB/cJ mice were obtained from The Jackson Laboratory. Two days prior to in vivo experiments, cells were plated at 1 × 106 cells/condition into T-75 flasks (Corning) in non-selective DMEM. A total of 100,000 cells in 100 μl of PBS were injected per mouse into the fourth mammary fat pad (orthotopic injection), tail vein (tail vein injection), or left ventricle (intracardiac injection). The mice were euthanized between 28–30 days post-injection. Primary tumors were resected, weighed, and lung metastases counted. Primary tumor, metastatic nodules, and normal (tail) tissue were isolated immediately following euthanasia and snap-frozen in liquid nitrogen. Tissue samples were then stored at −80°C. For orthotopic injection resection models, the primary tumors were resected at 2 weeks following initial injection. and mice were euthanized between 42–45 days post-injection. Primary tumors from syngeneic orthotopic injection of r3T, D2A1, E0771, EMT6, F311, HRM-1, and TS/A-E1 were a generous gift from Dr. Lalage Wakefield (NCI, Bethesda, MD).
Syngeneic models:
FVB/NJ - 6DT1, Mvt1, Met1, M6C. BALB/cJ – 4T1, 4T07, 67NR
Sildenafil citrate treatment:
6DT1 orthotopic injection was performed as described above. Dosing with 10 mg/kg sildenafil citrate was initiated on day 7 post-injection and was repeated daily until day 28, the study end-point. Sildenafil citrate salt (Sigma-Aldrich) was resuspended in sterile PBS (Life Technologies) to create a 5 mg/ml stock solution, which was stored at −20°C. Each mouse was weighed daily before dosing and 10 mg/kg dose was calculated and then administered by diluting the sildenafil citrate stock solution with sterile PBS to a final volume of 250 μl. Control mice were weighed and administered 250 μl of PBS. Mice were administered sildenafil citrate by IP injection of 250 μl using a 28-gauge needle (Andwin Scientific).
Nicotine administration:
6DT1 orthotopic injection was performed as described above. On day 7 following injection mice were given drinking water with nicotine or vehicle alone. (−)-Nicotine (Sigma-Aldrich) was diluted in 100% ethanol to create a stock solution with a final concentration of 100 mg/ml. 250 μl of the nicotine stock solution, or 250 μl of ethanol was added to a 250 ml of mouse drinking water for a final concentration of 100 μg/ml. The water was replaced weekly and the nicotine stock solution was made fresh each week.
Serum isolation
Following mouse anesthesia, 500–1000 μl of blood was removed by cardiac puncture using a 25-gauge needle. Blood was stored on ice with 10 μl of EDTA (0.5M, pH 8) and then centrifuged at 2,000 x g for 15 minutes in a refrigerated centrifuge. The plasma supernatant was then immediately removed and placed into a new tube and stored at −20°C.
Sildenafil citrate trial serum collection:
Serum was collected from tumor bearing mice 1, 4, 6, 8, 18, and 24 hours after the final sildenafil citrate or PBS control IP injection was performed on day 28 (see sildenafil citrate treatment regimen above).
Nicotine trial serum collection:
20 mice were administered 100 μg/ml nicotine in the drinking water, and 5 mice were administered vehicle control drinking water. Serum was collected from 5 mice in the nicotine group once a week for 4 weeks. Serum was collected from the 5 control mice after the final 4th week. Water was replaced weekly.
LC-MS/MS drug-serum level assessment
Nicotine and cotinine plasma concentrations were simultaneously measured using a validated LC-MS/MS assay, with a lower limit of quantitation of 1 ng/mL for both compounds. Briefly, 100 μL of plasma was mixed with 1 ml of dichloromethane containing 2 ng/ml of [2H]4-nicotine as internal standard. The organic layer was isolated, dried under nitrogen, reconstituted with (10/90, v/v) water/methanol, and injected into the LC-MS/MS. The calibration range for both nicotine and cotinine were 1–500 ng/ml.
Sildenafil plasma concentrations in mice dosed orally were measured using a validated LC-MS/MS assay. Briefly, 50 μl of plasma sample was mixed with 5-fold volume of methanol containing 100 ng/ml 2[H]8-sildenafil (as internal standard) to precipitate plasma proteins. The mixture was vortexed and centrifuged, and 200 μl of the resulting supernatant was transferred to a clean 96-well sample plate for measurement. A 10-μL aliquot of sample was injected onto a Kinetex C18 column (50 × 2.1 mm, 1.3 μm; Phenomenex) for chromatographic separation followed by multiple reaction monitoring detection by tandem mass spectrometry (MS/MS) by the mass transition of m/z 475.1 to 283.0 for sildenafil and m/z 482.5 to 283.0 for 2[H]8-sildenafil. Calibration standards of sildenafil were prepared over the range of 2.5 – 2500 ng/ml (in duplicate) with quality control (QC) standards at a low (7.5 ng/ml), mid (400 ng/ml) and high (2000 ng/ml) range, each in quintuplet
RNA isolation
RNA was isolated from flash-frozen tissue. First the tissue was mechanically dissociated using a tissue grinder while submerged in 1 ml of TriPure (Roche). 200 μl of chloroform (Sigma-Aldrich) was then added and the soluble fraction was isolated by centrifugation at 12,000 rpm for 15 minutes at 4°C. RNA was then precipitated with the addition of 500 μl isopropanol and incubation of the sample at −20°C for 2 hours. Pure RNA was then extracted using the RNA: DNA mini-prep kit (Zymogen) and finally samples were eluted in 100 μl (PT) or 50 μl (metastases) of DEPC water (Quality Biological). RNA was isolated from cell lines using TriPure as described above but following isopropanol precipitated RNA was washed with 75% ethanol (Sigma-Aldrich), and then again with 95% ethanol before being resuspended in 100 μl DEPC water.
RNA sequencing
RNA quality was tested using the Agilent 2200 TapeStation electrophoresis system, and samples with an RNA integrity number (RIN) score >7 were sent to the Sequencing Facility at Frederick National Laboratory. Preparation of mRNA libraries and mRNA sequencing was performed by the Sequencing Facility using the HiSeq2500 instrument with Illumina TruSeq v4 chemistry.
RNA sequencing analysis
Differential gene expression analysis:
RNA-seq reads were aligned to the mouse mm9 genome assembly using TopHat Software, and differentially expressed genes were determined using DESeq2. For the comparison of MSGE between orthotopic and tv allograft models The RNA sequences were aligned against mouse genome mm10 by STAR and the RNA expression were calculated by RSEM (RNA-Seq by Expectation Maximization). The differentially expressed genes were found by using DESeq2.
Gene Set Enrichment Analysis (GSEA):
The hierarchical cluster analysis was applied to the quantile normalized RPKM data of the primary tumor and metastatic samples to obtain two groups. One group had 88% allograft samples and another group had 86% GEMM samples. DESeq2 was then applied to find the differentially expressed genes between the two groups and then used GSEA to find significant cancer Hallmark pathways and Gene Ontology (GO) pathways.
Principal Component Analysis:
PCA analysis was performed using Partek Flow software (Kanehisa Laboratories). RNA-seq reads were uploaded into Partek Flow and aligned with the mouse mm9 genome assembly. Gene counts were then determined and normalized before performing unsupervised principal component analysis.
ImmQuant analysis:
DESeq2 fold-change data for those genes differentially regulated in GEMM and allograft models (Supplemental Table 2) were loaded into the ImmQuant Software(13). The following settings were selected for default deconvolution: Cell-type data - ImmGen 207, signature markers, lineage tree pre-compiled based on the reference data, and calculations were performed using all samples. Default color ranges were also selected for the lineage tree output file.
Pathway Analysis:
Pathway analysis was performed using Ingenuity Pathway Analysis (IPA) (Qiagen). Differentially regulated gene lists from sample type comparisons using DESeq2 were uploaded into IPA for Core Expression Analysis of expression data. The Ingenuity Knowledge Base was chosen as the reference set of genes, and both direct and indirect relationships were considered. No other analysis parameters were specified, and the default settings were selected.
qRT-PCR
RNA was isolated from cell lines or flash-frozen tissue as described above was reverse-transcribed using iScript (Bio-Rad). Real-Time PCR was conducted using VeriQuest SYBR Green qPCR Master Mix (Affymetrix). Peptidylprolyl isomerase B (Ppib) was used for normalization of expression levels. Expression of mRNA was defined from the threshold cycle, and relative expression levels were calculated using delta delta Ct after normalization with Ppib.
Primers sequences 5’−3’:
Cdh1 F: AAGTGACCGATGATGATGCC, R: GCGACTCTACCTGTCTCTTC
Epcam F: AACACAAGACGACGTGGACA, R: GCTCTCCGTTCACTCTCAGG
Snai1 F: GTGTGTGGAGTTCACCTTC, R: GGAGAGAGTCCCAGATGAG
Twist1 F: TTCTCCGTCTGGAGGATG, R: TCCTTCTCTGGAAACAATGAC
Ppib F: GGAGATGGCACAGGAGGAAAGAG, R: TGTGAGCCATTGGTGTCTTTGC
Vim F: CTGTACGAGGAGGAGATGCG, R: AATTTCTTCCTGCAAGGATT
Statistics
Statistical significance between groups in in vivo assays was determined using the Mann-Whitney unpaired nonparametric test using Prism (version 5.03, GraphPad Software, La Jolla, CA). Statistical significance between samples in qRT-PCR analysis was determined by an unpaired t test, also using Prism.
Data availability statement
All sequence data that supports the findings of this study can be accessed in the GEO database using the accession number GSE150928.
Results
Significant transcriptional differences exist between mouse models of metastatic breast cancer
To investigate the transcriptomic landscape specific to metastatic breast cancer, RNA-seq analysis of the most commonly used mouse models of metastatic breast cancer was performed. Matched primary and metastatic tumor tissues from GEMMs of metastatic breast cancer were collected from four mouse models representing luminal (MMTV-Myc, MMTV-PyMT), basal (C3(1)-TAg), and HER2+ (MMTV-Her2) subtypes of human breast cancer. Since, all of these models are on an FVB/NJ inbred mouse background, we bred MMTV-PyMT mice to two pairs of closely related mouse strains that have significantly different inherited metastatic susceptibilities (C57BL/6J and C57BL/10J or MOLF/EiJ and CAST/EiJ, low and high metastatic, respectively) (14).We hypothesized that relatively close phylogenetic relationships between the pairs (C57BL10/C57BL6 or MOLF/CAST) would reduce the differences in transcription not related to the differences in metastasis between these pairs. This would potential increasing the relative signal-to-noise for identification of important metastasis associated pathways while still attempting to better replicate human population diversity. To control for potential contamination of the pulmonary metastases by adjacent tissues, normal lung and metastasis-free tumor-conditioned lung tissue were also profiled. Additionally, we collected tissues from frequently used syngeneic allograft models of mammary tumor metastases. Specifically, matched primary and metastatic tumors from orthotopic injection, tail vein injection, and intracardiac injection models of metastatic breast cancer (4T1, 6DT1, Mvt1, and M6) were included in the analysis. Together this transcriptional analysis profiled 235 RNA samples isolated from 107 animals (Supplemental Table 1). Unsupervised principle component analysis (PCA) of RNA-seq data revealed that the samples clustered into three groups consisting almost exclusively of either autochthonous GEMM-derived samples or allograft samples, with the third group populated by M6 allograft and C3(1)-TAg samples which model basal-like gene expression (Figure 1A)(11). Metastatic and primary tumors were relatively evenly distributed within each of the clusters, consistent with previous studies that found a high degree of transcriptional similarity between primary and secondary breast cancer samples(15) (Figure 1B).
Figure 1. Model has a stronger influence over the tumor transcriptome than driver or site of origin.
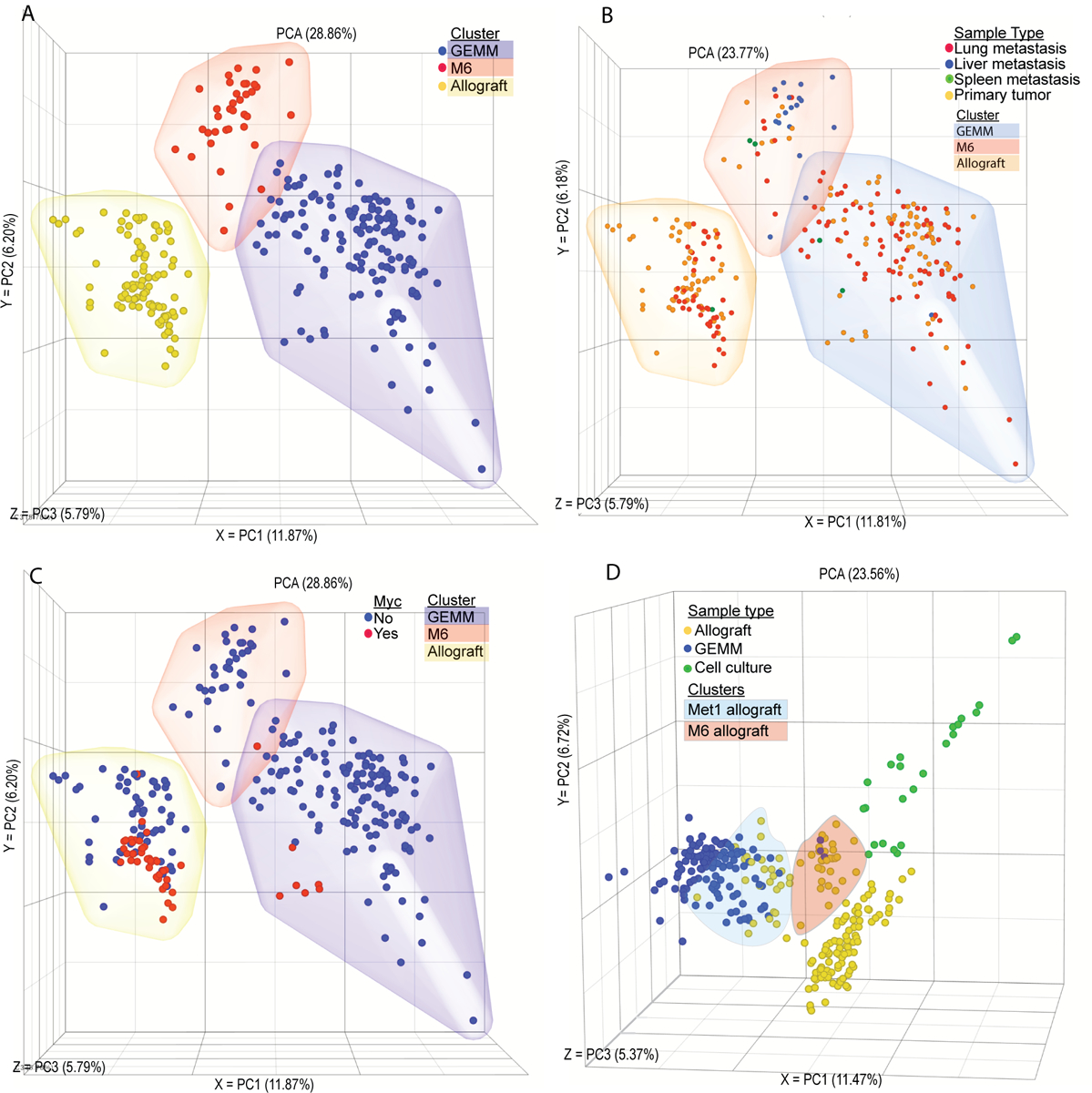
Unsupervised PCA plot showing RNA-seq transcriptomic analysis from matched primary tumor and metastatic nodules. Each point represents one tissue sample from (A) GEMM (blue), M6 (red), or allograft (yellow) clusters. (B) Lung metastasis (red), liver metastasis (blue), spleen metastasis (green), primary tumor (yellow). (C) Samples from a Myc-driven tumor model (red), from a non-Myc-driven tumor model (blue) that is a GEMM model (blue cloud), M6 (red cloud), or allograft (yellow cloud). (D) Allograft samples (blue), GEMM samples (yellow), or cell culture samples (green), with Met1 allograft samples (blue clous) and M6 samples (orange cloud) highlighted.
Allograft models are widely used in the study of metastasis as a model of spontaneous disease, however unsupervised PCA analysis of RNA-seq data revealed that samples clustered by model rather than driver or origin. Despite being derived from the MMTV-Myc model, both primary and metastatic samples from the 6DT1 and Mvt1 cell line allograft models displayed significant separation from the autochthonous MMTV-Myc tumors, suggesting that in vitro growth may result in a permanent transcriptional reprogramming in these cells (Figure 1C). To further explore the potential effect of in vitro culture on allograft transcriptional programming, primary tumors from an additional 11 metastatic and non-metastatic mammary tumor allograft models were profiled, in addition to in vitro cultures from 7 cell lines (Supplemental Table 1). Allograft tumors from the MMTV-PyMT-derived MET1 cell line clustered with the autochthonous MMTV-PyMT samples, and those from the C3(1)-TAg-derived M6 cell line clustered with the C3(1)-TAg samples between the basal and luminal autochthonous tumor samples (Figure 1D). The remaining nine allograft tumors clustered with the original allograft cluster, regardless of oncogenic driver or genetic background. Finally, in vitro samples formed a fourth new cluster adjacent to the allograft samples (Figure 1D). Together these data reveal that models of metastatic breast cancer differ significantly based on the origin of the transformed cells, and that in vitro culturing of metastatic samples permanently alters their transcriptional profile.
To better understand the transcriptional differences between the allograft and GEMM models, variances in the transcriptional profiles were examined. Gene set enrichment analysis of differentially expressed genes between the GEMM and allograft clusters revealed significant differences in immune regulation- and cell motility-related genes (Supplemental Table 2). Analysis of the differentially expressed genes using the ImmQuant software package suggested that the autochthonous GEMM tumors and metastases had decreased levels of infiltrating macrophages, dendritic cells, natural killer cells, and T cells compared to the allograft tumors (Supplemental Figure 1)(13). Moreover, the GEMM cluster had lower expression of genes associated with the mesenchymal phenotype (e.g., Vim, Zeb1), compared to the allograft tumor cluster, and relatively high expression of epithelial markers such as Epcam, Cldn1, and Cldn2 (Figure 2A). Similarly, the MSGE differences between GEMM and allograft models resided mostly in developmental, immune signaling, and EMT pathways (Figure 2B, Supplemental Table 2). This suggests that key differences exist between GEMM and allograft breast cancer metastasis models that involve contributions from both the microenvironment and cell plasticity.
Figure 2. Model is the predominant determinant of EMT signature.
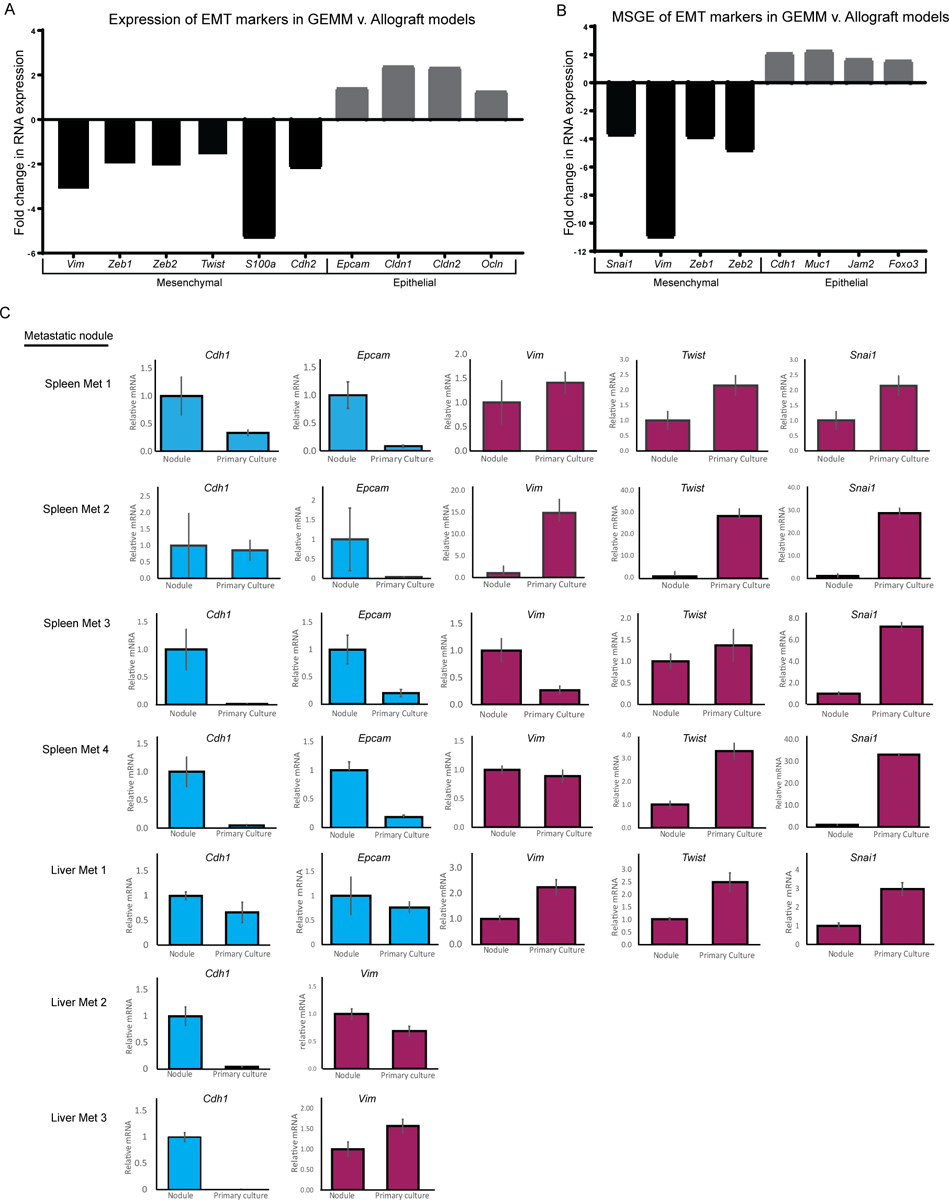
RNA-seq data showing the fold change in expression of EMT markers in (A) All GEMM derived tissues compared to all allograft-derived tissues, or (B) Metastasis-specific gene expression from GEMM models compared to allograft models. (C) qRT-PCR data showing epithelial (blue) or mesenchymal (red) EMT marker expression in spleen metastases and liver metastases that were subsequently grown in primary culture.
To further explore the possibility that in vitro cell culture may promote epithelial to mesenchymal transition (EMT), a gene expression program widely associated with tumor progression and metastatic spread, we measured expression of EMT markers in tissue from an MMTV-Myc mouse harboring lung, liver, and spleen metastases. The spleen and liver metastases collected from this mouse were cut in half and flash frozen, the remaining tissue was then used to create primary cultures which were later injected orthotopically back into ten syngeneic wild type mice per culture. qRT-PCR analysis was performed on RNA isolated from the original metastases, subsequent primary cultures, and two of the ten injected orthotopic primary tumors, which were each sampled three times to account for primary tumor heterogeneity (Supplemental Figure 2A). Pulmonary micro-metastases were observed in 76% of animals (53 out of 70), while only 2.8% (2 or 70) and 1.4% (1 or 70) had liver or spleen metastases, respectively. As observed in our RNA-seq analysis of well-established allograft and GEMM models, culturing of the MMTV-Myc GEMM-derived liver and spleen metastatic cells in vitro for < 5 passages imparted a more mesenchymal-like gene expression pattern and reduced epithelial gene expression (Figure 2C). This change in gene expression was variably maintained once cells were implanted orthotopically in vivo (Supplemental Figure 2B). Taken together, this data suggests that in vitro culturing techniques induce a more mesenchymal and lung-tropic phenotype that variably persists and does not consistently or accurately model the plasticity and tropism of metastatic cells as they originally existed in vivo.
Orthotopically derived and tail vein-injected metastases differ in immune-related gene expression
The preparation of a premetastatic niche at the secondary site is now widely accepted as a key step in the metastatic process (16). Therefore, we examined if metastasis assays performed by tail vein injection, in the absence of a primary tumor and premetastatic niche conditioning, produced metastases with different transcriptional profiles than those arising from orthotopically injected cells. To address this, the MSGE of lung metastases derived from syngeneic tail vein injection or orthotopic injection of three metastatic mouse cell lines (4T1, Mvt1, and 6DT1) was established (Supplemental Figure 3A). The orthotopic injection MSGE was then compared to tail vein injection MSGE for each cell line individually (Figure 3A).
Figure 3. Metastases derived from orthotopic and tail vein injection models differ in a cell line-dependent manner.
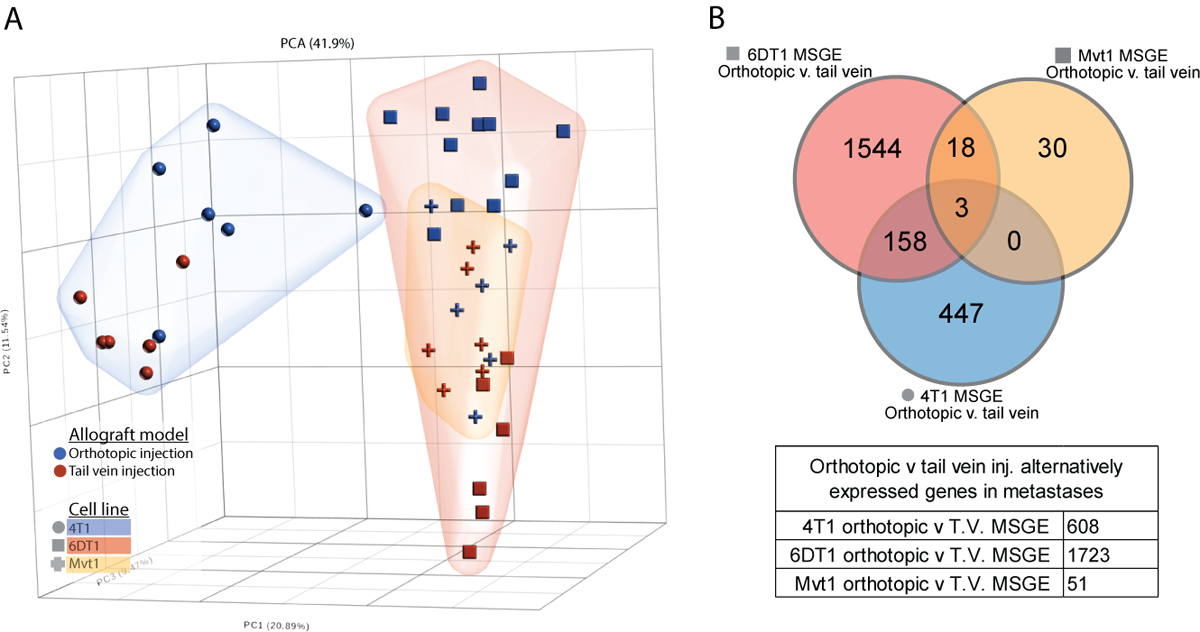
(A) PCA analysis plot showing relative difference between the transcriptomes of tail vein-derived lung metastases (red points) and orthotopic injection model-derived lung metastasis (blue points), with 4T1 cells (blue cloud, circles), 6DT1 cells (red cloud, squares), and Mvt1 cells (yellow cloud, crosses). (B) Venn diagram showing the overlapping orthotopic-specific MSGE from the 4T1 (blue), 6DT1 (red), and Mvt1 (yellow) allograft models.
Interestingly, this analysis revealed that the differences in metastatic gene expression between models was cell line dependent. Metastases derived from orthotopic or tail vein injection of Mvt1 cells differed only in the expression of 51 genes, but metastases derived from 4T1 cell injections differed in the expression of 608 genes between injection models, and 6DT1 derived metastases differed by 1723 genes (Figure 3B; Supplemental Table 3). There were three genes commonly altered by injection model across all three cell lines Pigr, Sdc1, and Retnla (Figure 3B), all of which were elevated in orthotopically derived metastases. Differentially expressed genes specific to each cell line were assessed by Ingenuity Pathway Analysis (IPA) (Table 1 and Supplemental Table 3), which revealed that the gene expression activated specifically in orthotopically derived metastases comprised T cell activation and immune related-related pathways when compared to tail vein-derived metastases. This suggests that metastatic tumors derived from these two models may undergo different interactions with the immune compartment at the metastatic site and differ significantly in gene expression, the extent of which may be specific to each cell line.
Table 1. Ingenuity canonical pathways of orthotopic allograft model MSGE.
Ingenuity Pathway Analysis results showing the top 10 significantly enriched pathways by orthotopic-specific MSGE of 4T1, 6DT1, and Mvt1. Significance of enrichment for this pathway in each the gene set is indicated by the -log(p-value) where >1.3 is significant.
| 4T1 | −log (p-value) |
6D1 | −log (p-value) |
Mvt1 | −log (p-value) |
|---|---|---|---|---|---|
| Granulocyte Adhesion and Diapedesis | 9.25 | Th1 and Th2 Activation Pathway | 17.3 | Antigen Presentation Pathway | 7.19 |
| Agranulocyte Adhesion and Diapedesis | 7.16 | Th1 Pathway | 14.9 | Allograft Rejection Signaling | 5.79 |
| Th1 and Th2 Activation Pathway | 7.14 | Th2 Pathway | 14.2 | OX40 Signaling Pathway | 4 |
| VDR/RXR Activation | 6.89 | iCOS-iCOSL Signaling in T Helper Cells | 11.8 | Cytotoxic T Lymphocyte-mediated Apoptosis of Target Cells | 3.27 |
| Th2 Pathway | 6.83 | Altered T Cell and B Cell Signaling in Rheumatoid Arthritis | 11.5 | Cdc42 Signaling | 3.21 |
| T Helper Cell Differentiation | 6.2 | Granulocyte Adhesion and Diapedesis | 11.3 | Graft-versus-Host Disease Signaling | 2.97 |
| LXR/RXR Activation | 4.97 | Type I Diabetes Mellitus Signaling | 11 | Autoimmune Thyroid Disease Signaling | 2.95 |
| Th1 Pathway | 4.97 | CD28 Signaling in T Helper Cells | 10.9 | Protein Ubiquitination Pathway | 2.59 |
| iCOS-iCOSL Signaling in T Helper Cells | 4.54 | T Helper Cell Differentiation | 10.8 | Crosstalk between Dendritic Cells and Natural Killer Cells | 2.44 |
| Nur77 Signaling in T Lymphocytes | 4.09 | Dendritic Cell Maturation | 10.4 | Systemic Lupus Erythematosus In T Cell Signaling Pathway | 2.35 |
Nicotine and Sildenafil processing are targetable metastasis-specific transcriptional programs
As metastatic nodules in human patients are often resistant to treatments that the primary tumor was sensitive to, there is a clear need for metastasis-specific therapies. We therefore examined targetable transcriptional programs common to spontaneous metastases in both allograft and GEMM models. First, the combined MSGE for spontaneous allograft models was determined for each cell line. MSGE common across 6DT1, 4T1, and Mvt1 orthotopically injected cell lines was then compared to MSGE of GEMMs (Supplemental Figure 3B). 360 genes were commonly regulated in metastases from both allograft and autochthonous models of spontaneous metastasis. IPA of this gene set analysis revealed several signaling, motility, and metabolic pathways in addition to several targetable pathways including “Cellular Effects of Sildenafil Citrate (Viagra)” and “Nicotine Degradation” (Table 2, Figure 4A and 4B, Supplemental Table 4). Interrogation of these pathways as regulators of breast cancer metastasis was then performed by spontaneous metastasis assays using orthotopic injection of metastatic breast cancer 6DT1 cells in the presence of sildenafil citrate or nicotine.
Table 2. Ingenuity canonical pathways of MSGE common to spontaneous models of metastasis.
Ingenuity Pathway Analysis results showing the pathways populated by MSGE common to both spontaneous allograft and GEMM models. Significance of enrichment for this pathway in each the gene set is indicated by the -log(p-value) where >1.3 is significant.
| -log(p-value) | |
|---|---|
| Nicotine Degradation II | 6.98 |
| Estrogen Biosynthesis | 5.23 |
| Acetone Degradation I (to Methylglyoxal) | 5.13 |
| Bupropion Degradation | 4.4 |
| Agranulocyte Adhesion and Diapedesis | 3.94 |
| Nicotine Degradation III | 3.66 |
| Hepatic Fibrosis / Hepatic Stellate Cell Activation | 3.35 |
| Atherosclerosis Signaling | 3.23 |
| Intrinsic Prothrombin Activation Pathway | 3.04 |
| Cellular Effects of Sildenafil (Viagra) | 2.91 |
| Granulocyte Adhesion and Diapedesis | 2.88 |
Figure 4. Common MSGE of several models of spontaneous metastasis present targetable metastasis-specific pathways.
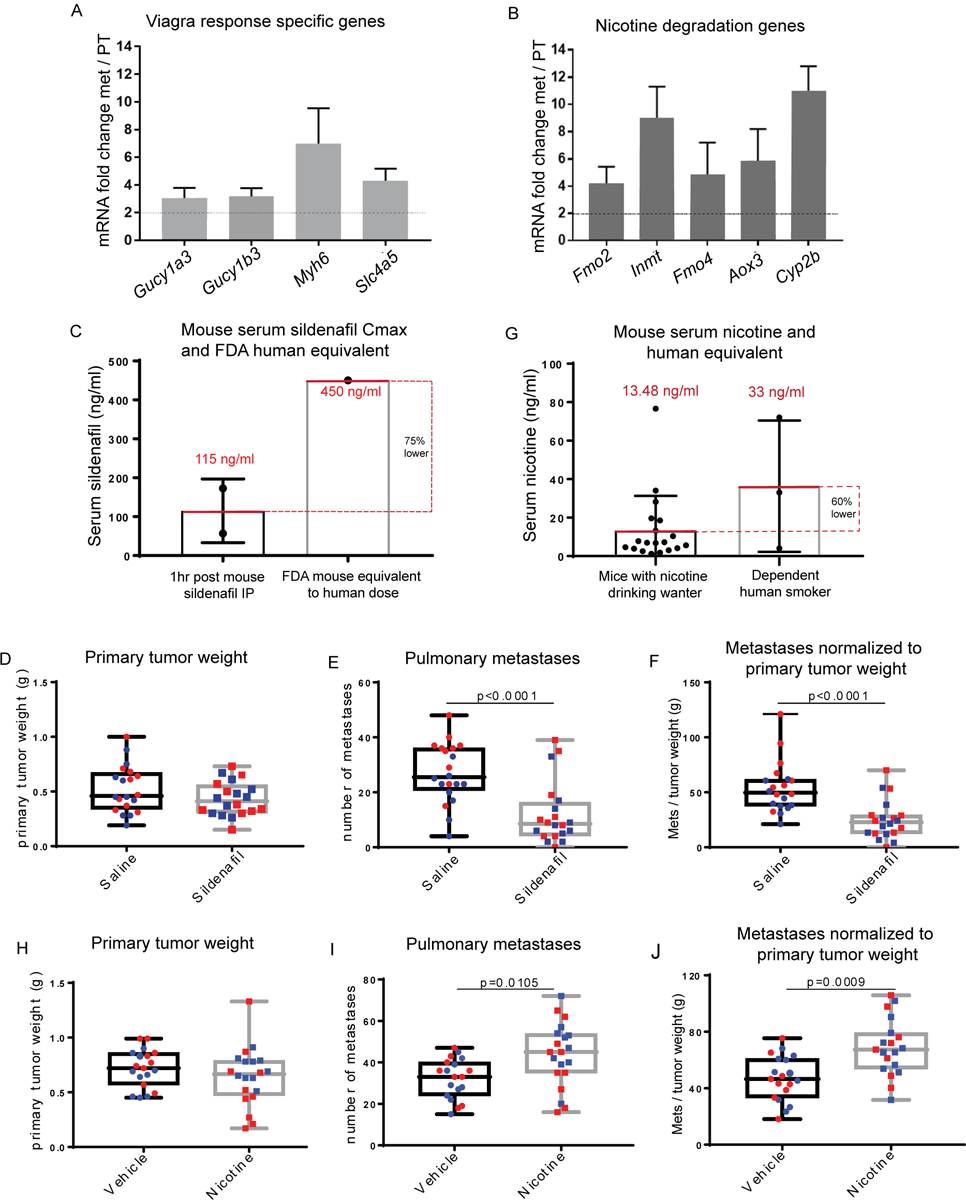
RNA-seq data showing expression of factors from the IPA sildenafil response pathway (A) and Nicotine degradation pathways (B) in metastatic v. primary tumor tissue. (C) Max Serum (Cmax) of sildenafil 1hr post IP injection of 10 mg/kg sildenafil (each point corresponds to one mouse) compared to the FDA mouse equivalent sildenafil serum levels of a human dose (D) Primary tumor weight (g), (E) lung nodule counts, and (F) lung nodule counts normalized to paired primary tumor weight at end point following syngeneic orthotopic injection of 1×105 6DT1 cells and treatment with saline (circles) or sildenafil (10 mg/kg) (squares). (G) Serum levels of nicotine achieved in this study following constant access to nicotine 100 μg/ml drinking water (each point corresponds to one mouse) compared to the average nicotine serum levels of dependent human smokers (range 4 – 72 ng/ul, average 33 ng/μl(19)). (H) Primary tumor weight (g), (I) lung nodule counts, and (J) lung nodule counts normalized to paired primary tumor weight at end point following syngeneic orthotopic injection of 1×105 6DT1 cells and access to nicotine or vehicle water (E-J). Two independent experiments shown (red and blue).
To determine if sildenafil citrate treatment could modify breast cancer metastasis, FVB/NJ mice bearing 6DT1 orthografts were treated with a daily low dose of sildenafil citrate by intraperitoneal injection starting 7 days post-injection of cells and lasting 21 days until endpoint. Pharmacokinetic/pharmacodynamic (PK/PD) analysis of serum collected at several time points following sildenafil citrate injection indicated that a Cmax of 115 ng/ml was achieved at 1hr post sildenafil IP, a serum level equivalent to 25% of typical human dose according to FDA guidelines for human and mouse equivalent dosing (Figure 4C, Supplemental Table 4) (17,18). While treatment with sildenafil did not significantly alter primary tumor growth it did significantly reduce the number of surface metastases on the lungs (p < 0.0001) (Figure 4D–F), indicating that sildenafil processing is an integral, metastasis-specific pathway necessary for efficient metastatic spread.
To determine if cellular nicotine degradation pathway activity is a modifier of breast cancer metastasis, FVB/NJ mice bearing 6DT1 orthografts were administered nicotine in their drinking water starting 7 days post- injection of cells and lasting 21 days until endpoint. PK/PD analysis of nicotine and its metabolite cotinine in serum collected throughout a 4-week course of nicotine treatment indicated that mouse nicotine serum levels averaged approximately 13.48 ng/ml, which is 60% lower than that of a dependent smoker (Figure 4G, Supplemental Figure 4, Supplemental Table 4)(19). Treatment with nicotine did not alter primary tumor growth but did significantly increase the number of surface metastases on the lung (p < 0.001) (Figure 4H–J). Again, this data suggests that the activation of nicotine degradation gene expression pathways is a metastasis-specific function resulting in more efficient metastatic spread.
Discussion
A major barrier to progress in the development of metastasis-specific drugs is a lack of available human tissue for study, forcing researchers to rely heavily on pre-clinical mouse models to delineate the metastatic process and advance therapeutics. However, 95% of agents that enter phase 1 of clinical development fail to make it into the clinic (20). This failure can be partly attributed to the inadequate predictive value of conventional preclinical models across the entire human population (4,21). Therefore, in addition to on-going efforts to develop models that better represent human disease (22), a greater understanding of our current models is also necessary (23). Currently, a variety of mouse models of metastatic breast cancer exist to interrogate oncogenic drivers, genetic predisposition, and stages of the metastatic cascade (24). However, no single model likely captures the full complexity of metastatic breast cancer observed in humans. We therefore hypothesized that gene expression observed in metastases from each model may exhibit both model specific as well as common and essential metastatic programs. Here we have presented transcriptomic data from a wide variety of pre-clinical mouse models of metastatic breast cancer, to better inform the use of these tools and advance the development of metastasis-specific interventions.
Several studies published in recent years have compared genetically engineered breast cancer mouse model tumor transcriptomes to human disease (25–27), delineated disease progression observed in the mouse (28), and providing characterization of murine cell line orthotopic injection models (11). Here we have expanded these efforts to interrogate metastasis-specific gene expression in 18 different models of metastatic breast cancer, including cells in culture, intravenous injection and orthotopic implantation of cells, and genetically engineered mouse models. While this is by no means an exhaustive list of mouse models used to study breast cancer metastasis, they represent many of the most widely used models (23). This study does not, however, include human cell lines, patient derived xenograft models, organoid implantation, or syngeneic transplantation of murine tumor fragments. These models may be informative tools for interrogating breast cancer etiology (29–31), however due to the low rate of metastasis and/or the need for immune-deficient mice they were not included in this study. In addition to limiting our study to immune-competent models, we have excluded the use of labeled cells and instead examined metastatic gene expression of bulk tumor isolates to characterize a more complete tumor environment with minimal experimental manipulation of the different models.
In agreement with the findings of other studies, we have identified a number of differences between the primary tumor and metastatic transcriptomes (29,30,32,33). However, by combining data from several models we show that more significant differences exist in the gene expression between models than between primary tumors and matched metastases, and in fact that model may play a larger role in shaping the metastatic transcriptome than tissue type or oncogenic driver. Perhaps the most striking observation from our data was the association of the allograft models with the EMT gene expression signature. EMT is postulated to be a key mechanism by which tumor cells acquire an invasive and metastatic phenotype (34). However, most of the evidence for this signature as metastasis-driving is derived from in vitro experimental systems and cancer cell lines (35). Work by Trimboli et al. on EMT in PyMT, Neu, and Myc GEMMs shows that EMT-like gene expression signatures are only observed for the Myc GEMM, despite the high metastatic propensity of the PyMT model (36). Similarly, our data suggest that the EMT phenomenon is not predominant in primary tumor or metastatic tissues collected from breast cancer GEMM models, as the samples analyzed in this study overwhelmingly exhibit more epithelial-like gene expression profile compared to cell-line-derived allograft tissue samples. Some studies have reported the ability of cells to undergo mesenchymal to epithelial transition (MET) upon outgrowth at the metastatic site (37). Therefore, as we only included macro-metastases in transcriptomic analysis for this study, we cannot rule out that EMT followed by MET may have occurred and was not captured here. Despite this caveat, and in agreement with the findings of Trimboli et al (36), we see by PCA analysis that MMTV-Myc tissue samples cluster with the remaining GEMM samples but are located on the periphery of this cluster closest to the allograft cluster, potentially signifying this model’s propensity for a more EMT-like phenotype. Additionally, the higher EMT signature expression in allograft models, as well as our analysis of metastatic tissue grown in primary culture supports the observation that in vitro culturing of mouse mammary cancer cells may impart a more mesenchymal-like gene expression signature, although we cannot discount the confounding variable of tumor cell enrichment through primary culturing. Together our data suggest that EMT may be a marker of in vitro culturing of tumor cells rather than metastatic potential when working with mouse models of metastatic breast cancer.
In addition to the variable EMT signature expression observed across our models, we also focused on the metastasis-specific differences that exist in the transcriptomes of both spontaneously generated nodules in GEMMs and orthotopic injection models. In this study, we identified a decrease in immune cell infiltration of GEMM derived metastases, suggesting key differences in tumor cell – stroma interactions. There are several factors that may contribute to this observation when looking at our cohorts overall. Firstly, this study includes a higher diversity of mouse strains within the GEMM cohort sampled (5 strains), than the allograft cohort (2 strains). Different mouse strains have previously shown to possess differing levels of tumor-immunogenicity (BALB/cJ and FVN/nJ: high, C57BL/6: Low) (38), therefore potentially biasing most allograft experiments towards higher immune-compartment interactions. Secondly, oncogenic driver has been shown to impact the immunogenicity of tumor cells and directly modify the stromal gene expression and immune response (39).While the majority of GEMs sampled in this study are MMTV-PyMT, this oncogenic driver only comprises one third of the allograft models sampled. Despite these factors, on a case by case basis our data reveals variability in gene expression between metastases generated from allograft models and the GEMMs from which they were derived. Some of this variability is undoubtedly due to divergent somatic evolution of the tumor cells in in vitro culture. However, the rapid conversion of the gene expression program observed with our explanted metastasis tissues suggest that in vitro cultures impart a rapid, strong selective pressure for a more mesenchymal phenotype, potentially mediated by epigenetic reprogramming. Further studies will be necessary to investigate this possibility.
Several recent studies have compared the gene expression of metastatic nodules produced from orthotopic implantation and intravenous injection. Nakayama et al compared gene expression of xenograft models using the human breast cancer cell line MDA-MB-231 in immune-deficient mice. They identified chemotaxis and cell adhesion gene expression enriched in pulmonary metastases generated by the orthotopic implantation model, and antigen recognition and cell adhesion in metastases from TV derived metastases (40). Similarly, Pillar et al has found a significant difference between the microRNA expression profiles of metastases generated by orthotopic or intravenous injection of 4T1 cells in a syngeneic model (41). Here we analyzed the metastasis specific gene expression differences between orthotopic and intravenous injection models using three metastatic mouse mammary cancer cell lines and syngeneic mouse strains. Interestingly, we have not only confirmed the significant divergence in gene expression signatures between routes of injection model, but also we show that the extent of this variation is unique to each cell line. Of note, 6DT1 and Mvt1 cells are FVB/NJ derived and therefore injected into the same syngeneic strain for this analysis. However, tumor cell intrinsic factors unique to each cell line generate highly variable metastatic gene expression for 6DT1 cells, and minimally different expression in Mvt1 metastases, depending on injection site. Furthermore, for all three cell lines examined, immune-related gene expression pathways were highly enriched in orthotopically-derived compared to tail vein-derived metastases, suggesting altered tumor cell – immune compartment interactions between injection models. This data reveals potentially significant changes in tumor cell – stroma interactions between injection model that are reflected in metastatic gene expression, and with a more informed selection of cell line researchers may potentially minimize or capitalize on this difference.
Despite the significant transcriptional differences between models, we have also identified common and targetable gene expression patterns in metastases derived from several models of spontaneous metastasis. Comparison of MSGE between allograft and genetically engineered models identified pathways common to metastases produced from seven oncogenic drivers and six mouse strains. Of note, our data revealed an upregulation of factors responsible for the cellular effects of sildenafil citrate (Viagra) and nicotine degradation. These pathways were subsequently validated as key metastasis modifying pathways in allograft transplantation models, using therapeutically relevant dosing schemas. These gene expression pathways are also enriched in lung metastases from three breast cancer PDX models generated by Alzubi et al (29,42), providing further support of their relevance to human breast cancer metastasis. The molecular target of sildenafil, phosphodiesterase type 5 (PDE5) has been identified as a tumor biomarker in several human cancers (43,44) and work with human cancer cell lines has revealed that expression and activation of PDE5 stimulates invasive, migratory, and cancer stem cell phenotypes (44). Several studies have also reported the pro-metastatic effects of nicotine on tumor cells in culture, including promotion of an invasive phenotype and EMT (45). Additional work using colorectal cancer cell lines suggests that the pro-metastatic effects of nicotine can be activated through MAP kinase signaling (46). While our transcriptomics data and research by others would suggest that modified metastatic potential by sildenafil and nicotine is likely tumor cell-intrinsic, we cannot rule out the possibility that administration of either compound in the in vivo experiments presented here may induce systemic contributions to this phenotype.
Physiologically, sildenafil and nicotine can have opposing effect on nitric oxide (NO)-induced vasodilation, and blood pressure. PDE5 is highly expressed in the vasculature and by inhibiting PDE5 sildenafil positively regulates cellular cGMP stability, mimicking NO-induction of vascular cGMP. cGMP-induced smooth muscle relaxation in blood vessels leads to vasodilation and increased blood flow (47). In contrast, nicotine has been reported to both increase and decrease NO depending on the tissue type (48,49), as nicotine has been found to constrict skin blood vessels and coronary blood vessels, but dilate blood vessels in the skeletal muscle (48,49). Due to the opposing effects of sildenafil and nicotine on dilation of the coronary blood vessels these compounds produce opposing effects on blood pressure. Therefore, the resulting changes to vascular wall stiffness, blood pressure, and tissue oxygenation may be contributing factors to the metastasis-modifying effects of these compounds, potentially impacting extravasation and colony formation at the secondary site. Due to its low toxicity, a dedicated study of sildenafil as an adjuvant or neo-adjuvant therapy may be warranted to determine if this compound may be effective in reducing metastasis of breast and other cancers. Clinically, sildenafil is administered almost exclusively to men, and therefore no data is available to interrogate the effects of sildenafil on breast cancer patient outcomes. Conversely, nicotine is consumed by both men and women, and the data is clear that breast cancer patients who are current or former smokers have a higher incidence of disease progression and mortality (50). While current human data sets cannot distinguish the effect of nicotine from tobacco, our data suggest that exposure to nicotine alone may be pro-metastatic, implicating tobacco-free nicotine products as potentially increasing the risk of metastatic progression in breast cancer patients.
In summary, by performing a survey of the MSGE profiles of pulmonary macrometastases from several allograft models and GEMMs, we have discovered key differences between models as well as common metastasis-specific programs that can be readily targeted. By using the strategy outlined here, the continued characterization of preclinical mouse models of metastasis will provide further insight into the associations between clinical subtypes, primary tumor drivers, and MSGE programs, ultimately creating new opportunities for the generation of metastasis-targeted therapies.
Supplementary Material
Implication Statement.
The data we present here exposes critical variances between pre-clinical models of metastatic breast cancer and identifies targetable pathways integral to metastatic spread.
Acknowledgements
This work utilized the computational resources of the NIH HPC Biowulf cluster. (http://hpc.nih.gov).
This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research (K. Hunter and W. Figg).
Funding: National Cancer Institute Project ZIA BC 011255
Footnotes
Conflict of Interest Statement: The author(s) declare(s) that there is no conflict of interest.
References
- 1.Female Breast Cancer - Cancer Stat Facts [Internet]. [cited 2018 Apr 11]. Available from: https://seer.cancer.gov/statfacts/html/breast.html
- 2.Treatment of Breast Cancer Stages I-III [Internet]. [cited 2019 Dec 18]. Available from: https://www.cancer.org/cancer/breast-cancer/treatment/treatment-of-breast-cancer-by-stage/treatment-of-breast-cancer-stages-i-iii.html
- 3.Mattson J, Huovinen R. [Treatment of disseminated breast cancer]. Duodecim. 2015;131:1033–1040. [PubMed] [Google Scholar]
- 4.Steeg PS. Targeting metastasis. Nat Rev Cancer. 2016;16:201–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vakiani E, Janakiraman M, Shen R, Sinha R, Zeng Z, Shia J, et al. Comparative genomic analysis of primary versus metastatic colorectal carcinomas. J Clin Oncol. 2012;30:2956–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shachar SS, Fried G, Drumea K, Shafran N, Bar-Sela G. Physicians’ considerations for repeat biopsy in patients with recurrent metastatic breast cancer. Clin Breast Cancer. 2016;16:e43–8. [DOI] [PubMed] [Google Scholar]
- 7.Hoefnagel LDC, van der Groep P, van de Vijver MJ, Boers JE, Wesseling P, Wesseling J, et al. Discordance in ERα, PR and HER2 receptor status across different distant breast cancer metastases within the same patient. Ann Oncol. 2013;24:3017–3023. [DOI] [PubMed] [Google Scholar]
- 8.Kuukasjärvi T, Karhu R, Tanner M, Kähkönen M, Schäffer A, Nupponen N, et al. Genetic heterogeneity and clonal evolution underlying development of asynchronous metastasis in human breast cancer. Cancer Res. 1997;57:1597–1604. [PubMed] [Google Scholar]
- 9.Wagle N, Painter C, Anastasio E, Dunphy M, McGillicuddy M, Kim D, et al. The Metastatic Breast Cancer (MBC) project: Accelerating translational research through direct patient engagement. J Clin Oncol. 2017;35:1076–1076. [Google Scholar]
- 10.Diagnostic tests for metastatic breast cancer - InformedHealth.org - NCBI Bookshelf [Internet]. 2016. [cited 2019 Oct 7]. Available from: https://www.ncbi.nlm.nih.gov/books/NBK361015/
- 11.Yang Y, Yang HH, Hu Y, Watson PH, Liu H, Geiger TR, et al. Immunocompetent mouse allograft models for development of therapies to target breast cancer metastasis. Oncotarget. 2017;8:30621–30643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Green JE, Shibata MA, Yoshidome K, Liu ML, Jorcyk C, Anver MR, et al. The C3(1)/SV40 T-antigen transgenic mouse model of mammary cancer: ductal epithelial cell targeting with multistage progression to carcinoma. Oncogene. 2000;19:1020–1027. [DOI] [PubMed] [Google Scholar]
- 13.Frishberg A, Brodt A, Steuerman Y, Gat-Viks I. ImmQuant: a user-friendly tool for inferring immune cell-type composition from gene-expression data. Bioinformatics. 2016;32:3842–3843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lifsted T, Le Voyer T, Williams M, Muller W, Klein-Szanto A, Buetow KH, et al. Identification of inbred mouse strains harboring genetic modifiers of mammary tumor age of onset and metastatic progression. Int J Cancer. 1998;77:640–644. [DOI] [PubMed] [Google Scholar]
- 15.Weigelt B, Glas AM, Wessels LFA, Witteveen AT, Peterse JL, van’t Veer LJ. Gene expression profiles of primary breast tumors maintained in distant metastases. Proc Natl Acad Sci USA. 2003;100:15901–15905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doglioni G, Parik S, Fendt S-M. Interactions in the (pre)metastatic niche support metastasis formation. Front Oncol. 2019;9:219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nichols DJ, Muirhead GJ, Harness JA. Pharmacokinetics of sildenafil after single oral doses in healthy male subjects: absolute bioavailability, food effects and dose proportionality. Br J Clin Pharmacol. 2002;53 Suppl 1:5S–12S. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm. 2016;7:27–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Russell MA, Jarvis M, Iyer R, Feyerabend C. Relation of nicotine yield of cigarettes to blood nicotine concentrations in smokers. Br Med J. 1980;280:972–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Moreno L, Pearson AD. How can attrition rates be reduced in cancer drug discovery? Expert Opin Drug Discov. 2013;8:363–368. [DOI] [PubMed] [Google Scholar]
- 21.Ireson CR, Alavijeh MS, Palmer AM, Fowler ER, Jones HJ. The role of mouse tumour models in the discovery and development of anticancer drugs. Br J Cancer. 2019;121:101–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Liu Z, Wang Y, Kabraji S, Xie S, Pan P, Liu Z, et al. Improving orthotopic mouse models of patient-derived breast cancer brain metastases by a modified intracarotid injection method. Sci Rep. 2019;9:622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gómez-Cuadrado L, Tracey N, Ma R, Qian B, Brunton VG. Mouse models of metastasis: progress and prospects. Dis Model Mech. 2017;10:1061–1074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khanna C, Hunter K. Modeling metastasis in vivo. Carcinogenesis. 2005;26:513–523. [DOI] [PubMed] [Google Scholar]
- 25.Rennhack J, To B, Wermuth H, Andrechek ER. Mouse Models of Breast Cancer Share Amplification and Deletion Events with Human Breast Cancer. J Mammary Gland Biol Neoplasia. 2017;22:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rennhack JP, To B, Swiatnicki M, Dulak C, Ogrodzinski MP, Zhang Y, et al. Integrated analyses of murine breast cancer models reveal critical parallels with human disease. Nat Commun. 2019;10:3261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hollern DP, Swiatnicki MR, Andrechek ER. Histological subtypes of mouse mammary tumors reveal conserved relationships to human cancers. PLoS Genet. 2018;14:e1007135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cai Y, Nogales-Cadenas R, Zhang Q, Lin J-R, Zhang W, O’Brien K, et al. Transcriptomic dynamics of breast cancer progression in the MMTV-PyMT mouse model. BMC Genomics. 2017;18:185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alzubi MA, Turner TH, Olex AL, Sohal SS, Tobin NP, Recio SG, et al. Separation of breast cancer and organ microenvironment transcriptomes in metastases. Breast Cancer Res. 2019;21:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bradford JR, Wappett M, Beran G, Logie A, Delpuech O, Brown H, et al. Whole transcriptome profiling of patient-derived xenograft models as a tool to identify both tumor and stromal specific biomarkers. Oncotarget. 2016;7:20773–20787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan H, Demirci U, Chen P. Emerging organoid models: leaping forward in cancer research. J Hematol Oncol. 2019;12:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iwamoto T, Niikura N, Ogiya R, Yasojima H, Watanabe K-I, Kanbayashi C, et al. Distinct gene expression profiles between primary breast cancers and brain metastases from pair-matched samples. Sci Rep. 2019;9:13343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Varešlija D, Priedigkeit N, Fagan A, Purcell S, Cosgrove N, O’Halloran PJ, et al. Transcriptome characterization of matched primary breast and brain metastatic tumors to detect novel actionable targets. J Natl Cancer Inst. 2019;111:388–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pastushenko I, Blanpain C. EMT Transition States during Tumor Progression and Metastasis. Trends Cell Biol. 2019;29:212–226. [DOI] [PubMed] [Google Scholar]
- 35.Mittal V Epithelial mesenchymal transition in tumor metastasis. Annu Rev Pathol. 2018;13:395–412. [DOI] [PubMed] [Google Scholar]
- 36.Trimboli AJ, Fukino K, de Bruin A, Wei G, Shen L, Tanner SM, et al. Direct evidence for epithelial-mesenchymal transitions in breast cancer. Cancer Res. 2008;68:937–945. [DOI] [PubMed] [Google Scholar]
- 37.Banyard J, Bielenberg DR. The role of EMT and MET in cancer dissemination. Connect Tissue Res. 2015;56:403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lechner MG, Karimi SS, Barry-Holson K, Angell TE, Murphy KA, Church CH, et al. Immunogenicity of murine solid tumor models as a defining feature of in vivo behavior and response to immunotherapy. J Immunother. 2013;36:477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen KB, Spranger S. Modulation of the immune microenvironment by tumor-intrinsic oncogenic signaling. J Cell Biol. 2020;219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nakayama J, Ito E, Fujimoto J, Watanabe S, Semba K. Comparative analysis of gene regulatory networks of highly metastatic breast cancer cells established by orthotopic transplantation and intra-circulation injection. Int J Oncol. 2017;50:497–504. [DOI] [PubMed] [Google Scholar]
- 41.Pillar N, Polsky AL, Weissglas-Volkov D, Shomron N. Comparison of breast cancer metastasis models reveals a possible mechanism of tumor aggressiveness. Cell Death Dis. 2018;9:1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ross C, Szczepanek K, Lee M, Yang H, Qiu T, Sanford J, et al. The genomic landscape of metastasis in treatment-naive breast cancer models. BioRxiv. 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Piazza GA, Thompson WJ, Pamukcu R, Alila HW, Whitehead CM, Liu L, et al. Exisulind, a novel proapoptotic drug, inhibits rat urinary bladder tumorigenesis. Cancer Res. 2001;61:3961–3968. [PubMed] [Google Scholar]
- 44.Catalano S, Campana A, Giordano C, Győrffy B, Tarallo R, Rinaldi A, et al. Expression and function of phosphodiesterase type 5 in human breast cancer cell lines and tissues: implications for targeted therapy. Clin Cancer Res. 2016;22:2271–2282. [DOI] [PubMed] [Google Scholar]
- 45.Dasgupta P, Rizwani W, Pillai S, Kinkade R, Kovacs M, Rastogi S, et al. Nicotine induces cell proliferation, invasion and epithelial-mesenchymal transition in a variety of human cancer cell lines. Int J Cancer. 2009;124:36–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiang T, Fei R, Wang Z, Shen Z, Qian J, Chen W. Nicotine enhances invasion and metastasis of human colorectal cancer cells through the nicotinic acetylcholine receptor downstream p38 MAPK signaling pathway. Oncol Rep. 2016;35:205–210. [DOI] [PubMed] [Google Scholar]
- 47.Dishy V, Sofowora G, Harris PA, Kandcer M, Zhan F, Wood AJ, et al. The effect of sildenafil on nitric oxide-mediated vasodilation in healthy men. Clin Pharmacol Ther. 2001;70:270–279. [DOI] [PubMed] [Google Scholar]
- 48.Rejali AR, Rejali M, Zhang L, Yang S. Effect of nicotine on intracellular Ca2+ concentrations and nitric oxide release in coronary artery endothelial cells | The FASEB Journal. FASEB J. 2008;21:A523–A523. [Google Scholar]
- 49.Toda N, Toda H. Nitric oxide-mediated blood flow regulation as affected by smoking and nicotine. Eur J Pharmacol. 2010;649:1–13. [DOI] [PubMed] [Google Scholar]
- 50.Goldvaser H, Gal O, Rizel S, Hendler D, Neiman V, Shochat T, et al. The association between smoking and breast cancer characteristics and outcome. BMC Cancer. 2017;17:624. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All sequence data that supports the findings of this study can be accessed in the GEO database using the accession number GSE150928.