

Abstract
Purpose:
Anti-programmed cell death protein 1 (PD-1) therapy has demonstrated inconsistent therapeutic results in patients with glioblastoma (GBM) including those with profound impairments in CD8 T cell effector responses. We sought to determine the
Experimental Design:
We ablated the CD8alpha gene in BL6 mice and intercrossed them with Ntv-a mice to determine how CD8 T cells affect malignant progression in forming endogenous gliomas. Tumor-bearing mice were treated with PD-1 to determine the efficacy of this treatment in the absence of T cells. The tumor microenvironment of treated and control mice was analyzed by immunohistochemistry and fluorescence activated cell sorting.
Results:
We observed a survival benefit in immunocompetent mice with endogenously-arising intracranial glioblastomas after intravenous administration of anti-PD-1. The therapeutic effect of PD-1 administration persisted in mice even after genetic ablation of the CD8 gene (CD8−/−). CD11b+ and Iba1+ monocytes and macrophages were enriched in the glioma microenvironment of the CD8−/− mice. The macrophages and microglia assumed a proinflammatory M1 response signature in the setting of anti-PD-1 blockade through the elimination of PD-1-expressing macrophages and microglia in the tumor microenvironment. Anti-PD-1 can inhibit the proliferation of and induce apoptosis of microglia through Ab-dependent cellular cytotoxicity (ADCC), as fluorescently-labeled anti-PD-1 was shown to gain direct access to the glioma microenvironment.
Conclusion/Discussion:
Our results show that the therapeutic effect of anti-PD-1 blockade in GBM may be mediated by the innate immune system, rather than by CD8 T cells.
Single sentence summary
Anti-PD-1 immunologically modulates innate immunity in the glioma microenvironment—likely a key mode of activity.
INTRODUCTION
Immunotherapy has revolutionized the treatment of cancer. This has generated interest in harnessing the immune system as a treatment for glioma, the most common primary brain tumor in humans (1–5). However, the effectiveness of immunotherapy against glioma is attenuated by the immunosuppressive tumor microenvironment (6). In the context of metastatic cancer to the brain, immunotherapy has demonstrated significant efficacy, suggesting that this treatment is not impeded by the blood-brain tumor barrier (7). Treatment of GBM patients with immune checkpoint inhibitors may benefit a select patient subset (8, 9). However, these patients are known to be profoundly immunosuppressed (10) and in particular, lymphopenic (11). The number of cytotoxic CD8+ T cells, thought to be critically important to mediate the effects of immunotherapy, is very low in subsets of GBM patients (12), in part related to their sequestration in the bone marrow (13). In GBM patients who demonstrate a response to anti-PD-1 antibody (Ab), it is unclear what immune cell is mediating the antitumor effect because the CD8 T cell is presumed to be completely refractory to immune modulation (10).
The role of the T cell in the process of gliomagenesis is also unclear. GBMs frequently arise de novo but may also originate from a low-grade glioma precursor. Despite an initially indolent course, during which survival time may be many years, low-grade gliomas almost inevitably progress to GBM (14–16). After this malignant transformation, survival rates drop precipitously to 12–15 months. We have previously shown a direct correlation between an immune-suppressive microenvironment and malignant progression (17). As the immune system recognizes and eradicates tumor cells, some tumor cells evade the immune system by avoiding detection or by becoming immune suppressive to diminish the tumoricidal effects of CD8 T cells (18–20). Thus, by the time of diagnosis, GBM has already been subject to immunoediting by T cells and might not be susceptible to this immune cell population, even in the presence of immunotherapies that enhance T cell activity.
Here, we show that PD-1 Ab delivered intravenously significantly increases survival in immunocompetent mice with endogenously-forming tumors (21, 22). To model the lack of CD8 T cell effectors observed in human patients, we genetically modified mice to eliminate the CD8 T cells. We hypothesized that the absence of the CD8 effector response might promote malignant progression. Alternatively, selective pressure of the immune effector response might induce the tumor to become more malignant and immunosuppressive through genetic alterations and instability of the tumor. We show that the CD8 T cell population does not influence glioma formation rates, tumor-free survival times, or malignant progression and that the innate immune system compensates for CD8 T cell loss primarily through the influx of immune-reactive macrophages and microglia. Even in the absence of CD8 T cells, we observed a significant therapeutic effect from the administration of intravenous anti-PD-1 antibodies. Commensurate with this effect was a significant decrease in immune-suppressive PD-1+ macrophages within the tumor microenvironment, possibly allowing for greater tumor clearance by the proinflammatory M1 macrophage population.
MATERIALS AND METHODS
Study Design
Sample size and rules for stopping data collection.
For the binary comparison, we set the control rate at 95%, for a chi-square test with two-sided 5% alpha and 80% power, with 30 mice per group, we could detect a proportion in the experimental group of 67% as being significantly different from the control rate. Because a predefined interim analysis demonstrated marked differences in survival after we used 10 animals per group, further data collection was not needed.
Data inclusion/exclusion criteria.
Prospectively, mice were replaced who died within three weeks after injection of the RCAS vector or that did not receive at least three doses of either anti-PD-1 Ab or IgG.
Outliers.
No outliers were excluded from analysis.
Selection of endpoints.
The primary prospective endpoint of the study was survival.
Replicates.
Experimental replicates are designated in each legend.
Randomization.
Mice were randomly sorted into treatment arms starting on day 21 after the initiation of gliomagenesis when thymic output is at a maximum (23) and after maturation of the immune system (24, 25).
Blinding.
The investigators who assessed, measured, and quantified the results were blinded to the experimental conditions and outcomes. The primary endpoint was death, which was recorded by the animal caretakers and then associated with the treatment group.
Cell lines
The murine microglia cell line EOC-20 was purchased from the American Type Culture Collection (ATCC) and was cultured in Dulbecco’s modified Eagle’s medium (Corning Inc, NY), supplemented with 10% FBS and 1% penicillin/streptomycin at 370 C in a humidified atmosphere of 5% CO2 and 95% air. These cells were maintained by trypsin passage every 2–3 days. The cell line was confirmed to be free of Mycoplasma.
Animals
All mice were housed in the MD Anderson Isolation facility in accordance with Laboratory Animal Resources Commission standards, and all work was supervised by the Institutional Animal Care and Use Committee at MD Anderson (Protocol 00000900-RN01).
Generation of a C57BL/6J congenic strain carrying a null allele of CD8 and the Ntv-a transgene
The Ntv-a transgene (avian cell surface receptor [TVA] for subgroup A avian leukosis virus under the control of a glial progenitor-specific promoter derived from the human nestin [NES] gene) and the CD8α-targeted allele were moved from their respective genetic backgrounds onto the C57BL/6 background by marker-assisted backcrossing (26) to yield Ntv-a/CD8−/− and Ntv-a/CD8+/+ mice.
Vector constructs
The RCAS/Ntv-a model was previously described (21). Both RCAS-PDGFB (27) and RCAS-STAT3 (17) vectors have been described. The vector constructs are propagated in DF-1 chicken fibroblasts. Live virus was produced by transfecting plasmid versions of RCAS vectors into DF-1 cells using FuGene6 (Roche, Nutley, NJ). DF-1 cells senesce 1–2 days after injection. In mice injected with DF-1 cells with non-tumor inducing vectors, no long-term inflammatory response is observed in the brain or the brains appear histologically normal consistent with prior reports using this model (28, 29).
In vivo somatic cell transfer in transgenic mice
To transfer genes via RCAS vectors, 5 × 104 DF-1 producer cells transfected with the RCAS vectors in 1–2 μl of PBS were injected into the frontal lobes of mice using a 10-μl gastight Hamilton syringe (30). Mice were injected within 24–48 h after birth. The mice were killed 90 days after injection or sooner if they demonstrated morbidity. The RCAS-PDGFB and RCAS-STAT3 model has been found to recapitulate many of the key immune features of human gliomas including macrophage infiltration and have been used to study anti-tumor immunity for a variety of immune therapeutic strategies (31, 32).
Quantitative real time-polymerase chain reaction (RT-PCR)
In addition to immunohistochemical methods for detecting expression of CD8α in tumor-bearing tissue, we also performed real time-polymerase chain reaction (RT-PCR) assay on the brains of mice (n = 3). After the mice were killed, their forebrains were removed and frozen in liquid nitrogen. Tissue specimens were homogenized, and RNA was extracted using Qiagen’s RNeasy Mini Kit. Reverse transcription was performed using Bio-Rad’s iScript cDNA Synthesis Kit. Quantitative RT-PCR was performed using FastStart SYBR Green Master reagent (Roche) with the following primers: CD8aF:CAAGCCCAGACCTTCAGAGA CD8aR: TCCCCATCACACCCCTACTA. Data were normalized to internal GAPDH and β2-microglobulin.
Phenotypic characterization of tumor
We dichotomized tumors as either low-grade or high-grade on hematoxylin and eosin (H & E)-stained tumor sections, depending on the presence of histologic features including neovascularization and necrosis. Tumor grading was performed by the study neuropathologist (G.N.F.).
Syngeneic clonotypic intracranial glioma model
To induce intracerebral tumors in C57BL/6J mice, GL261 cells were injected at a dose of 5 × 104 cells/μl as previously described (33).
Flow cytometry analysis
Peripheral blood (50 μL) was collected and placed in 4 ml of mouse RBC lysis buffer (eBioscience). Splenic single-cell suspensions were prepared by mechanical dissociation of the spleen, followed by filtration, and lysis of the RBCs. Cells were stained with anti-mouse CD4, CD8α, CD19, CD3e, NK1.1, λσ-T, FoxP3, and granzyme B antibodies (eBiosciences). For granzyme B measurements, T cell activation was performed before Ab staining. Splenic single-cell suspensions were prepared and cultured in RPMI 1640 (containing 10% FBS) plus IL-2. T cells were activated for 7 days using a Dynabead Mouse T Activator CD3/CD28 kit. Antibodies used for flow cytometry in this study (anti-CD11c, anti-CD11b, anti-CD19, anti-NK1.1, anti-CD3, and anti-MHCII) were from eBioscience, and anti-TNF-α was from Becton-Dickinson. Single-cell suspensions were prepared with a Neural Tissue Dissociation Kit from Miltenyi Biotec. For CD11b and TNF-α analysis, cells were treated with Cell Stimulation Cocktail and a protein transport inhibitor (eBioscience) overnight, stained for surface CD11b, and fixed and permeabilized for TNF-α detection (using flow cytometry). TMEM 119 was used to stain microglia via ex vivo flow cytometry and by immunohistochemistry (34).
Immunohistochemical and immunofluorescence analyses
Mouse brains were fixed in 10% buffered formalin and were paraffin-embedded, with 4-μm sections being used for immunohistochemical analysis. To detect Iba1 (1:1000), the anti-Iba1 Ab from Wako (Richmond, VA) was used. To quantify Iba1-positive cells, we counted the total number of cells and the number of positively-stained cells in the areas of highest tumor-cell density in 10 nonoverlapping microscopic fields (400 X magnification) in tumor-bearing brains taken from mice in each group. To detect CD8+ T cells in brain sections, we performed immunofluorescence using fluor-conjugated antibodies from eBioscience (1:50). PD-1 for immunofluorescence was detected using antibodies from Abcam (ab214421, Cambridge, MA) or from R&D Systems (AF1021, Minneapolis, MN) and TMEM119 using Abcam (ab209064, Cambridge, MA). Alexa Fluor Conjugated secondary antibodies were used from Invitrogen Thermo Scientific (Carlsbad, CA). ProLong Gold Antifade Mountant with 4’-diamidino2-phenylindole (DAPI; Thermo Fisher Scientific, Waltham, MA) was used as the mounting medium. Slides were further processed for imaging and confocal analysis using an Olympus Fluoview FV1000 microscope. We quantified the percentage of positive cells by counting the number of cells that stained for both Iba1 and TMEM119 and PD-1 in at least five non-overlapping microscopic fields (magnification, 400× and/or 600×) from each genotype. The number of positive cells was divided by the total number of DAPI+ cells.
Ex vivo flow cytometry of intracerebral gliomas
When the mice started to show signs of neurological deficit, they were euthanized and the brains were collected after cardiac perfusion with PBS. To isolate immune cells from the brains, the brains were manually dissected, filtered through a 70 μM cell strainer (BD Bioscience) and the myelin was depleted from the single cell suspension with percoll gradient centrifugation or magnetic bead separation (MACS Miltenyi Biotec) according to the manufacturer instructions. Next, cells were incubated with Protein Transport Inhibitor Cocktail 500x (ThermoFisher) for 4 to 5 hours at 37°C. To prevent non-specific binding, cells were incubated with Fc-Block (CD16/32, Biolegend) and then stained with fixable viability dye efluor 780 to exclude dead cells (Thermo Fisher Scientific). To determine the different immune cell subsets, the following antibodies were used: anti-mouse CD45 BV510, anti-mouse CD11b PerCP/Cy5.5, anti-mouse PD-1 BV421,anti-mouse CD3 PerCP/Cy5.5, anti-mouse CD4 FITC, anti-mouse IFN-ɣ PE/Cy7, anti-mouse CD49b – PE/Cy7, anti-mouse CD4 – BV510, (all Biolegend), anti-mouse CD25 BV510 (BD Bioscience), anti-mouse Foxp3 PE (ThermoFisher), anti-mouse TMEM119 (abcam), goat anti-rabbit AlexaFluor 488 (highly cross-absorbed; Life Technologies). For fixation and permeabilization of the cells the eBioscience Foxp3/ Transcription factor Fixation/ Permeablizaion Kit (Thermo Fisher) was used according to manufacturer instructions. The cells were measured using FACS Celesta (BD Bioscience) and the data analysis was done with FlowJo software.
NanoString assay
RNA (200 ng) at a concentration of 40 ng/μl in a total volume of 5 μl was prepared for NanoString assay analysis with the immune-specific gene array kit (NanoString Technologies, Inc.). Sample preparation and hybridization were performed for the assay according to the manufacturer’s instructions. Briefly, RNA samples were prepared by ligating a specific DNA tag (mRNA-tag) onto the 3’ end of each mature mRNA, and excess tags were removed via restriction enzyme digestion at 37°C. After processing with the mRNA sample preparation kit, the entire 10-μl reaction volume containing mRNA and tagged mRNAs was hybridized with a 10-μl reporter CodeSet, 10 μl of hybridization buffer, and a 5-μl capture ProbeSet (for a total reaction volume of 35 μl) at 65°C for 16–20 hours. Excess probes were removed using two-step magnetic bead-based purification with an nCounter Prep Station. The specific target molecules were quantified using an nCounter Digital Analyzer by counting the individual fluorescent bar codes and assessing target molecules. The data were collected using the nCounter Digital Analyzer after obtaining images of the immobilized fluorescent reporters in the sample cartridge using a charge-coupled device camera. These data were then normalized to mRNA gene expression data for the GSE5099 Classical M1 VS Alternative M2 macrophage gene panel (35). The cluster analyses were used to determine deregulated genes between the anti-PD-1 and the IgG isotype control group by multigroup comparison using Qlucore software (Lund, Sweden). Gene counts were loaded into GSEA 4.0.1 as a tab delimited text expression matrix, with each row representing a gene and its expression across the samples. Analyses were run with the default settings and 100 permutations. Dataset labels were created to group the columns that were either anti-PD-1 Ab or IgG treated.
Microglia assays
Cell viability was assessed using the Presto-blue assay according to the manufacturer’s recommendations (Invitrogen, A13261). Briefly, 3000 cells were seeded overnight into a 96-well plate containing 100 μL/well of cell culture medium. Cells were treated with various concentrations of IgG (Bio Xcell; BE0089) and the anti-PD-1 Ab (BioXcell; BE0146) for 24, 48, 72, 96, and 120 hours. Next, 10 μl of cell viability reagent was added at the end of the incubation periods and incubated for 10 minutes in the dark (protected from light). Fluorescence was measured at 560 nm. Cell proliferation was measured using a BrdU incorporation assay according to the manufacturer’s recommendations (Cell Signaling Technology; cat # 6813). Briefly 3000 cells were seeded overnight and treated with various concentrations of IgG or the anti-PD-1 Ab for 24 and 48 hours. Twenty-four hours before the proliferation measurement, the 10X BrdU solution was added at a concentration of 1X and the cells were incubated at 37⁰ C in an atmosphere of 95% air and 5% CO2. The cells were fixed and incubated with the detection Ab for an hour followed by incubation with HRP secondary Ab and the TMB substrate. Absorbance was measured at 450 nm. For assessment of the effects on cell cycle, the EOC-20 cells were harvested, fixed, stained with a PI Ab, and analyzed by flow cytometry after incubating with 125 μg/ml of either the anti-PD-1 Ab or IgG.
Ab-dependent cell cytotoxicity (ADCC) assay
In a typical ADCC assay, an Ab binds on the surface of target cells, the effector cell FC receptor recognizes cell-bound antibodies, and the cross-linking results in apoptosis of target cells. To investigate whether PD-1 signaling was mechanistically required, we used two different blocking antibodies (RPM1–14 [BioXcell, BE0146] and 29F.1A12 [BioXcel, BE0273]) and a non-blocking Ab (RPM1–30 [ebioscience,14–9981-82]). RPM1–14 (anti-PD-1) is produced in rats and as such, the Fc gamma R IV receptor of the mouse doesn’t recognize/react with the rat IgG2a (36). Repeat administration of rat anti-mouse antibodies will trigger in vivo generation of mouse anti-rat responses. To recapitulate the in vivo vaccination response to RPM1–14, mouse anti-rat IgG antibodies were added to experimental arms. The ADCC assay was performed using standard murine FcγRIII ADCC effector cells according to manufacturer’s recommendations (Promega, G7015). Briefly, a day before the assay, target cells (EOC20) were plated in fresh medium in a 96-well plate and incubated in a C02 incubator at 37°C. On the following day, either IgG or anti-PD-1 or secondary antibodies were diluted in medium and added to the target cells, followed by the addition of effector cells. The ratio of Effector cells:Target cells is maintained at 20:1 in triplicate for 24 h. The Bio-Glo luciferase reagent was added and the luminescence (in relative light units, RLU) was determined using a synergyHTX multimode plate reader.
Treatment with anti-PD-1 monoclonal Ab and in vivo depletions
We used RMP1–14, a murine monoclonal Ab against PD-1 (BioXCell, Lebanon, New Hampshire, USA). Mice were injected with RCAS-PDGFB+ RCAS-STAT3 to induce primarily high-grade tumors, consistent with glioblastoma. Mice were randomized between the two treatment arms beginning four weeks after gene transfer in the perinatal period (approximates adolescence in humans) and were treated with either RMP1–14 (200 μg) or an IgG isotype control (BioXcell) intravenously (via tail vein injection) thrice weekly for up to 5 weeks. At three weeks, thymic output is at its maximum (23) and the immune system if full mature (24, 25). Animals that died from tumor progression prior to the initiation of at least three doses of anti-PD-1 Ab were replaced. Mice were monitored as described above and killed when they exhibited neurological morbidity. Their brains were removed and fixed in 4% formalin.
Anti-PD-1 fluorescent tagging and in vivo biodistribution analysis
The PD-1 Ab was fluorescently tagged with Alexa fluor 647 using the SAIVI Rapid Ab Labeling Kit according to the manufacturer’s recommendations (S30044, Invitrogen, CA). Briefly, 1 mg of the Ab at a concentration of 2 mg/ml was incubated with the Alexa 647 dye for one hour at room temperature with gentle stirring. A 3-cm column was prepared by using the resin in the kit and washed twice with elution buffer before loading the sample. The labeled Ab was loaded onto the column, and all the eluted fractions were collected. The first-eluted colored bands contained the labeled Ab. Absorbance of the purified conjugated Ab was measured at both A280 and 650 nm and protein concentration was calculated using a Nanodrop 1000 spectrophotometer (Thermo Scientific, CA). To evaluate whether the anti-PD-1 Ab was able to infiltrate brain tumors, C57BL/6 mice with intracerebral GL261 tumors established for 20 days or mice injected with RCAS-PDGFB+RCAS-STAT3 in the Ntv-a+ wild-type or in the homozygous CD8−/− background that were neurologically symptomatic were injected intravenously with 200 μg of Alexa Fluor 647-conjugated anti-PD-1 Ab. The mice were killed, and their organs were harvested after 3 hours and imaged using an IVIS 200 fluorescence imager.
Statistical analysis
The Cochran-Mantel-Haenszel test was used to compare the tumor incidence between different injection sets. Student’s two-sample t test was applied to compare immune cell compositions between different groups. Kaplan–Meier survival curves were used to estimate unadjusted tumor latency. To compare the time-to-event variables between groups, the log-rank test was used to compare distributions. All tests were two-sided, and P < 0.05 was considered statistically significant. Statistical analysis was carried out using R version 3.1.2 software (R Core Team, Vienna, Austria) and Graphpad Prism version 6.01 software (Graphpad Software, Inc., La Jolla, CA, USA).
RESULTS
Enhancement of innate immunity in the glioma microenvironment in the absence of CD8 T cells
To analyze the immune glioma microenvironment in the absence of CD8 T cells, Ntv-a mice were backcrossed into a BL6 background and intercrossed with CD8−/− mice to create Ntv-a/CD8−/− mice. To ascertain the CD8 T cell composition in the CD8-/, heterozygous (CD8−/+), and wild-type mice (CD8+/+), their peripheral blood, bone marrow, and spleens were analyzed. No CD8+ cells were detected in these tissues of CD8−/− mice (Supplementary Fig. 1A). Heterozygous CD8 mice (Supplementary Fig. 1B) displayed a level of CD8 T cells similar to that in wild-type mice (Supplementary Fig. 1C, D; P>0.05). The flow cytometry data were consistent with the genetic analysis of CD8 T cell loss achieved in the CD8 KO background. We identified intratumoral infiltration of CD8+ T cells in the CD8+/+ mice, but as expected, these cells were absent in the CD8−/− mice (Supplementary Fig 1E). To test the hypothesis that the CD8 effector response facilitates evasion of immune detection, thereby decreasing animal survival and promoting malignant progression, gliomas were induced with RCAS-PDGFB+RCAS-STAT3 in Ntv-a mice in either the CD8+/+ or CD8−/− background. CD8+/+ wild-type mice survived longer (49.5 days, 95% CI: 27–69) than the CD8−/− mice (27 days, 95% CI: 21–46) although this was not statistically significant (Fig. 1A, P=0.2281, H.R.1.354). Additionally, there was no difference in the incidence of high-grade gliomas, regardless of CD8 status (Fig. 1B).
Fig. 1. CD8 knockout (CD8−/−) does not impact survival in a genetically-engineered murine model of glioma and demonstrate a compensatory increase in macrophages in the glioma microenvironment.

A, As shown by the Kaplan-Meier curves, survival was not impacted when 5 × 104 DF-1 cells/mouse containing the RCAS-PDGFB+RCAS-STAT3 genes were injected bilaterally into the frontal brain lobes of CD8−/− mice. B, At the time of death or when the animal was moribund, an autopsy was performed, and microscope slides containing sections of the central nervous system (CNS) were stained with hematoxylin and eosin (H & E) for tumor grading. The CD8 status did not influence the glioma grade. C, CD11b+ monocytes and macrophages were found to occur more frequently in the blood and spleens of tumor-bearing CD8−/− mice than in those of CD8+/+ mice (*P=0.0202 for blood; ****P<0.0001 for spleen). The analysis was conducted using ex vivo flow cytometry and the percentage of CD11b+ cells was calculated based on the total alive cells with 30,000 total events analyzed. D, CD11b+ MHC II+ cells were found by ex vivo flow cytometry to be enriched in the CD8−/− group relative to the wild-type CD8+/+ group (****P=0.0001). The percentage of duel expressing CD11b+ MHC II+ cells was calculated based on the total alive cells with 30,000 total events analyzed. E, Dot plot summarizing a significantly higher percentage of Iba1+ cells in gliomas in the CD8−/− mice than in CD8+/+ mice. P=0.001. F, Functional analysis by detecting intracellular TNF-α expression with flow cytometry demonstrated increased numbers of TNF-α+-expressing CD11b+ cells in glioma-bearing CD8−/− mice (***P=0.0001). The analysis was conducted using ex vivo flow cytometry and the percentage of CD11b+ cells that had intracellular expression of TNF-α was calculated based on the total alive cells with 30,000 total events analyzed. G, CD8 knockout mice demonstrate a compensatory increase in the peripheral CD4 compartment. The CD4+ T cell percentage in spleens was increased in CD8−/− mice (***P=0.0002). H, Because there was an increase in the frequency of CD4+ T cells in the CD8−/− mice, the fraction of Tregs within the CD4 compartment was assessed, but no differences were observed in this in either the blood or spleen compartments, regardless of CD8 status. I, The percentage of CD4+ T cells producing granzyme B was found to be increased only in CD8−/− mice harboring intracranial tumors (****P<0.0001).
CD11b is a marker of myeloid cells of the innate immune system including macrophages. In the tumor-bearing mice we found an increase in the CD11b+ cells in the CD8−/− mice relative to CD8+/+ mice in both the spleen and blood (Fig. 1C; P=0.02 for blood and P<0.0001 for spleen). There was also a significant difference in the macrophage and microglia marker Iba1+ in tumors induced in CD8−/− mice compared with CD8+/+ mice (Student’s t test, P=0.0009) (Fig. 1D). MHC+CD11b+ cells demonstrated a >2-fold increase in the gliomas of CD8−/− mice relative to CD8+/+ mice (Fig. 1E). This expansion was not an intrinsic property of the CD8−/− mice, as shown by the lack of an increase in the percentage of CD11b+ cells in the brains of non-tumor-bearing control mice. These macrophages possess features that reflect a pro-inflammatory immune propensity based on TNF-α expression (Fig. 1F; P=0.001) Increases in innate immunity in the glioma microenvironment were also found in a second glioma model in which retrovirus induced PGFR+Pten−/− murine gliomas were depleted of CD8 T cells (37). Cumulatively, these data indicate an association of the innate immune system with gliomagenesis and anti-tumor immune reactivity.
To ascertain if there was a compensatory increase in CD4 T cells in the absence of CD8 T cells in these models, we analyzed blood and spleens for the percentage of CD4 T cells in the CD8−/− and wild-type mice. The frequency of CD4 T cells was elevated in the spleens of CD8−/− mice relative to that in the CD8+/+ mice (P=0.0002). However, there was no difference in the frequency of the CD4 T cells in the blood of the CD8−/− mice relative to the wild-type mice (Fig. 1G). Previously, an increased regulatory T cell fraction was shown to be present within the CD4 compartment in patients with high-grade gliomas (11). To determine if there was an alteration of glioma-induced immune suppression between the CD8−/− and CD8+/+ mice, both their blood and spleens were analyzed for the fraction of CD4+ and FoxP3+ T cells. We found no difference in the fraction of CD4+ FoxP3+ T cells between glioma-bearing CD8−/− and CD8+/+ mice (Fig. 1H), indicating that Tregs are not differentially modulated. Because some CD4 T cells are cytotoxic cells (38, 39), including those directed against malignancies (40, 41) (a perforin-and granzyme B-mediated process (42)) we explored whether the increase in CD4 frequency was tied to these specific functional activities. In mice harboring gliomas, there was a 2.4-fold increase of granzyme β+ CD4 T cells in the spleens of CD8−/− mice relative to that in CD8+/+ mice (Fig. 1I). However, this expansion was not an intrinsic property of the CD8−/− background, as shown by the absence of an increase in the percentage of granzyme β+ CD4 T cells in non-tumor-bearing control mice (not injected with DF-1 cells) possibly related to the immunological recognition of the intracranial tumor. Further analysis of the CD8−/− mice harboring gliomas demonstrated no expansion of this population in the glioma microenvironment (Fig. 2A), indicating that the CD4 cytotoxic T cell does not have a dominant role in the immunological control of gliomagenesis.
Fig. 2. CD8 knockout mice (CD8−/−) demonstrate a compensatory increase in multiple peripheral immune compartments.

A, CD4+ T cells were not detected in the gliomas of either CD8−/− mice (CD8−/−) or wild-type mice (CD8+/+) by immunohistochemical staining. B, CD19+ cells and C, NK1.1+ populations in the blood and spleen of glioma-bearing mice were found to be significantly higher in the CD8−/− group relative to the CD8+/+ group (***P=0.0004 and ****P <0.0001, respectively). However, these cell populations were not detected within the brain tumors. Spleen staining is the positive control. Representative immunohistochemically-stained images at 200x magnification for A and B left panels, bar = 100 μM; A and B middle and right panels at 400x magnification, bar = 50 μM. D, Kaplan-Meier estimates of tumor-free survival in glioblastoma-bearing Ntv-a mice in the wild type (CD8+/+) or CD8−/− background treated with anti-PD-1 or IgG isotype control (n = 11–13 per group). The median overall survival time was 68 days in the anti-PD-1 Ab-treated wild type mice and 40 days in the control group (log rank test, P=0.0002) (left panel). The median overall survival time was 61 days in the anti-PD-1 Ab-treated CD8−/− mice and 39 days in the control group (log rank test, P<0.0001).
Compensatory immune cell expansion occurs peripherally in the absence of CD8 T cells
Because we observed expansion of the CD4 T cell population in the spleens of mice with the CD8−/− background (Fig 2A), we investigated whether this occurred with other immune cell populations such as CD19+ B cells (Fig. 2B) and NK1.1+ cells (Fig. 2C). In the CD8−/− glioma-bearing mice, there was a compensatory increase in both B cells and NK cells, in the blood and spleen. However, we saw no CD19+ B cells in gliomas arising in either the CD8−/− (n = 8) or CD8+/+ wild-type mice (n = 9). Similar findings were obtained for the NK1.1 population in the CD8−/− (n = 8) and wild-type backgrounds (n = 9), again indicating that although there may be expansion of these populations in the periphery as a reaction to the tumor but there was no evidence of their involvement in trafficking to and exerting an effector response in the tumor microenvironment.
Anti-PD-1 exerts a therapeutic effect in the absence of CD8 T cells
To investigate the role of anti-PD-1 in the absence of CD8 T cells, we induced high-grade gliomas in Ntv-a+/BL6 mice of the CD8+/+ background by coinjecting RCAS-PDGFB+RCAS-STAT3, as has been previously described (17). Mice were then observed for 4 weeks and treated with anti-PD-1 intravenously through tail-vein injection. Animals were treated with 200 μg of either rat IgG (control) or anti-PD-1 administered intravenously every week for up to 5 weeks. Mice are weaned at 3–4 weeks after birth and they reach maturity (adulthood) by 8–10 weeks (43). Based on this report, the mice used in these experiments started treatment as teenagers with the vast majority of treatments occurring in adulthood. The median survival time for the anti-PD-1-treated group was 68 days, and in the IgG control group it was 40 days; (log-rank test, P=0.0002) (Fig. 2D), demonstrating therapeutic activity of anti-PD-1 in this model and similar to the GL261 model (44–46). To investigate whether anti-PD-1 would exert a therapeutic effect in the CD8−/− mice, we treated a cohort of these mice. Compared with the anti-PD-1-treated groups, median survival times for the IgG treated controls were 40 days (relative to the PD-1 Ab-treated group of 68 days; log-rank test, P=0.0002) and 39 days (relative to the PD-1 Ab-treated control group of 61 days; log-rank test, P<0.0001) in the CD8+/+ and the CD8−/− mice, respectively (Fig. 2D). The difference in the survival of the CD8+/+ mice and the CD8−/− mice treated with anti-PD-1 (Fig. 2D) was not statistically significantly different (68 vs 61 days respectively, log rank test p=0.0727). These results demonstrate that a similar therapeutic effect was obtained for anti-PD-1 regardless of the presence of CD8 T cells.
CD11b+ myeloid cells are the predominant immune cells population in the brains of tumor-bearing Ntv-a/CD8−/− and Ntv-a/CD8+/+ mice
To investigate which immune cells subsets could mediate the anti-PD-1 mediated survival benefit in the absence of CD8 T cells, we isolated immune cells from the whole brains of tumor-bearing Ntv-a/CD8−/− and Ntv-a/CD8+/+ mice treated with PD-1 antibody or IgG control for multicolor flow cytometry (Supplementary Fig. 2). First, we quantified the percentage of myeloid cells (defined as CD11b+ cells), CD4 T cells (defined as CD3+ CD4+ cells), and NK cells (defined as CD3- CD49+ cells) since those immune cells have already been reported to express PD-1 (Fig. 3A). With an average of around 70%, CD11b+ myeloid cells (Fig. 3A, left) represented the majority of immune cells in all groups, whereas the contribution of CD4 T cells (middle), was less than 5%, and NK cells (right), were less than 1%. There was no statistical significant difference in the percentage of the different immune cells subsets between any of treatment groups within their genotype. Next, we determined the PD-1 expression levels on each of these cell subsets. The CD11b+ myeloid cell subset showed an average PD-1 expression between 10 and 20% (Fig. 3B, left) with no significant differences between the treatment groups. CD11b+ myeloid cells, but not CD4 T cells, are present throughout the brain parenchyma. Notably, myeloid cells only showed PD-1 expression in the presence of glioma (Supplementary Fig. 3). Thus, the flow cytometry analysis, which analyzed the whole brain including the glioma, underestimates the modulation of this population in the setting of anti-PD-1 treatment within the glioma microenvironment. In the CD4 T cell subset (Fig. 3B, middle panel), there was much more variability in the percentage of PD-1 expression within the different groups (Fig. 3B, middle panel) with expression levels ranging from 11.5 to 89.4%. There was a statistically significant difference detected between the anti-IgG and the anti-PD-1 in the Ntv-a CD8+/+ mice (p=0.0425) but not in the Ntv-a/CD8−/− mice. In the NK cell subset, only 2 to 4% showed PD-1 expression and there was no difference between the different groups (Fig. 3B, right). Because the CD4 T cells showed quite high PD-1 expression, even though they are not very frequent and contribute only to very small extent to the immune cell composition, we examined if the anti-PD-1 treatment influences their functional status. Therefore we stained the CD4 T cells for IFN-ɣ (Fig. 3C, left), TNF-α (Fig. 3C, middle), and for regulatory T cells (Fig. 3C, right), but we could not detect any significant changes, indicating the CD4 T cells are not the main contributors of our observed effects.
Fig. 3. Flow cyotmetric analysis of immune cell subsets isolated from the whole brain of glioblastoma-bearing Ntv-a mice in the wild type (CD8+/+) or CD8−/− background treated with PD-1 antibody or IgG isotype control.
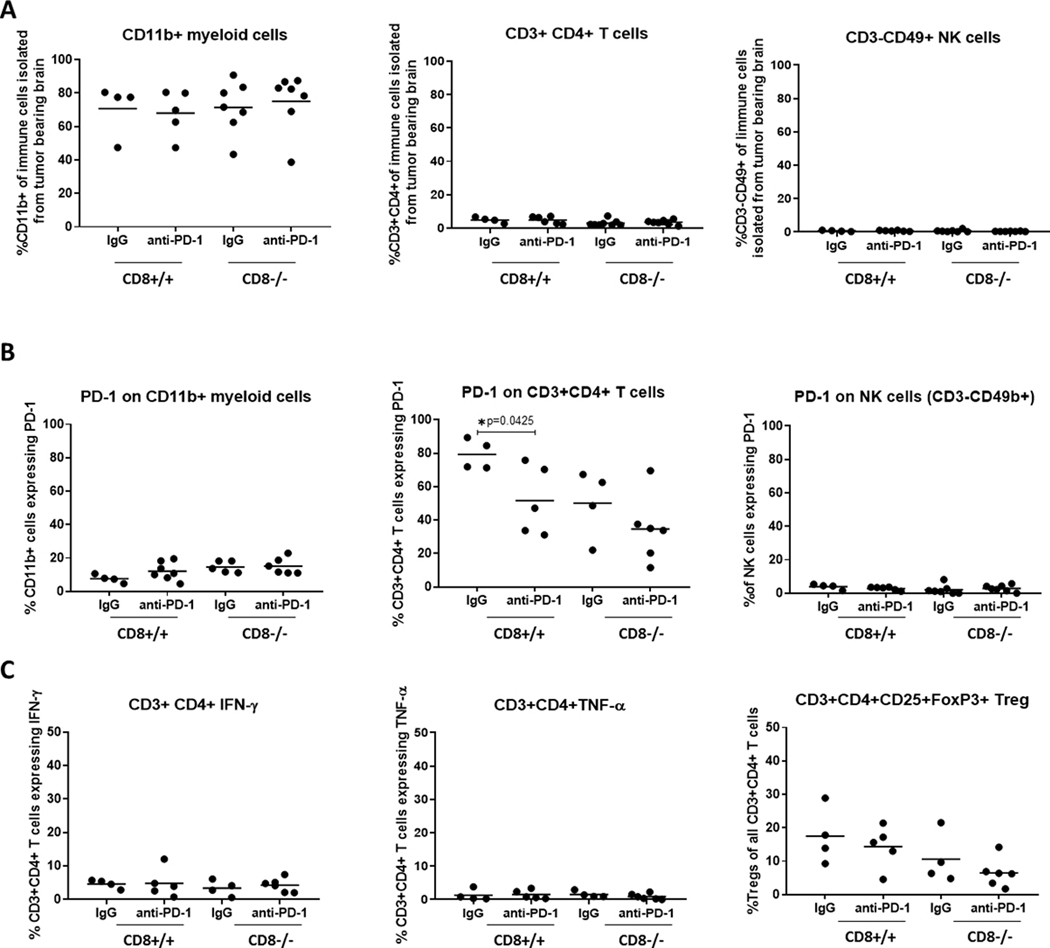
A, Percentage of CD11b+ myeloid (left) [CD8+/+ IgG vs anti-PD-1 p=0.7933; CD8−/− IgG vs anti-PD-1 p=0.6872], CD3+ CD4+ T cells (middle) [CD8+/+ IgG vs anti-PD-1 p=0.9975; CD8−/− IgG vs anti-PD-1 p=0.5572], and cells NK cells (right) [CD8+/+ IgG vs anti-PD-1 p=0.7146; CD8−/− IgG vs anti-PD-1 p=0.3732 ] of all immune cells isolated from the whole brains of tumor bearing Ntv-a mice. B, PD-1 expression on CD11b+ myeloid cells (left) [CD8+/+ IgG vs anti-PD-1 p=0.1551; CD8−/− IgG vs anti-PD-1 p= 0.8539], CD3+ CD4+ T cells (middle) [CD8+/+ IgG vs anti-PD-1 p= 0.0425; CD8−/− IgG vs anti-PD-1 p= 0.2647], and NK cells (right) [CD8+/+ IgG vs anti-PD-1 p= 0.2294; CD8−/− IgG vs anti-PD-1 p= 0.5275]. C, Quantification of functional CD3+ CD4+ T cell subsets in the brains of tumor-bearing mice, percentage of IFN-ɣ (left) [CD8+/+ IgG vs anti-PD-1 p= 0.9441; CD8−/− IgG vs anti-PD-1 p= 0.5492] and TNF-α (middle) [CD8+/+ IgG vs anti-PD-1 p= 0.8415; CD8−/− IgG vs anti-PD-1 p= 0.2869] expressing cytotoxic CD3+ CD4+ T cells, and percentage of regulatory T cells (Treg; right) [CD8+/+ IgG vs anti-PD-1 p=0.5431; CD8−/− IgG vs anti-PD-1 p=0.2994]. Two-sided unpaired t-test was performed to compare the treatment groups within the genotypes, only the statistical significant values (p≤ 0.05) are indicated in the figure.
Anti-PD-1 recalibrates the glioma-infiltrating macrophages/microglia to an M1 phenotype
Because PD-1 expression has been previously described for tumor-associated macrophages (47), we evaluated whether PD-1 expression was present within gliomas and coexpressed with macrophages within the CNS. Using dual immunofluorescence, clusters of PD-1- and Iba1-positive cells were detected in both the CD8+/+ and CD8−/− mice (Supplementary Figs.4, 5). Throughout the glioma microenvironment there was heterogeneous expression of Iba1+PD-1-, Iba1-PD-1+, and Iba1+PD-1+ cells. Given that the tumor-associated macrophages express PD-1, and that there are scant CD8 T cells in gliomas at diagnosis, we hypothesized that PD1 blockade might act independently of CD8 T cell-based immune responses. To assess whether PD-1 blockade has any role in the regulation of macrophages/microglia in the tumor microenvironment we performed multi-immunofluorescent staining for the microglia-specific marker TMEM119, Iba1 and PD-1 (Fig. 4A). We noticed that in the anti-PD-1 treated group relative to the IgG control during the therapeutic window, the microglia/macrophage expression was decreased in both the CD8+/+ and the CD8−/− backgrounds. Notably, relative to the IgG control group, we detected a marked decrease in the populations expressing Iba1+ TMEM119+ PD1+cells within the tumor in both CD8+/+ and CD8−/− mice (p<0.0001) (Fig. 4B). The decreased expression of macrophage/microglia markers Iba1 and TMEM119 (Supplementary Fig. 6) suggest that the innate immune system may be mediating the effects of the anti-PD-1 Ab.
Fig. 4. Anti-PD-1 recalibrates the glioma-infiltrating macrophages/microglia to an M1 phenotype.

A, Representative immunofluorescent images of Iba1+TMEM119+PD-1+ microglia infiltration in a glioblastoma-bearing mouse (CD8+/+ background, with similar staining in the CD8−/− background) after IgG or anti-PD-1 Ab treatment, 400x magnification, scale bar = 50 μM. B, Scatter plot demonstrating difference in PD-1 expressing Iba1+TMEM119+ expression in the wild-type CD8+/+ and KO CD8−/− background mice demonstrating a significant reduction in the number of Iba1+TMEM119+ microglia after anti-PD-1 Ab treatment (P<0.0001, t test, n=5 per group). Quantification was done by as described in methods. C, The genetic signature of a proinflammatory M1 phenotype in macrophages/microglia compared between gliomas treated with an isotype control IgG and anti-PD-1 Ab. A GSEA waterfall plot shows that only a small subset of the total M1 genes were found to be preferentially expressed in the IgG-treated group (usually in the wild-type CD8+/+ background). Overall, the M1 profile was strongly associated with the use of the anti-PD-1 Ab.
Given that the PD-1-expressing macrophage/microglial population was reduced in the setting of anti-PD-1 therapy, we evaluated whether the remaining macrophage/microglia population was exerting a proinflammatory or M1 classical skewed phenotype, as these cells can exist either in a nonpolarized state or along a continuum that is either pro- or anti-inflammatory (48). Gliomas in either the CD8+/+ or the CD8−/− backgrounds treated with anti-PD-1 demonstrated a significant skewing in genes associated with the proinflammatory M1 phenotype relative to the IgG isotype control treated mice based on nanostring profiling (Fig. 4C). The up regulated genes were related to class I MHC mediated antigen processing and presentation (e.g. TRIM69), skewing to or maintaining the M1 phenotype (e.g., LP1N1, EPST1 ZNF229, HDAC3), macrophage differentiation (e.g., ERMAP), toll-like receptor transcription (e.g., HCFC2), phagocytosis (e.g., ATP10A), and survival (e.g., E4F1) (Supplementary Fig. 7). Together, these results indicate that anti-PD-1 enriches for a classical M1 skewed macrophage phenotype in the glioma microenvironment.
Anti-PD-1 has direct effects on PD-1-expressing macrophage/microglia expressing cells but also triggers ADCC
To define the direct effects of the anti-PD-1 Ab on macrophages/microglia, we used EOC-20, a murine microglia cell line that expresses PD-1 (Fig. 5A) to a similar extent as tumor infiltrating myeloid cells (defined as CD45+ CD11b+ cells, Fig 5B), to assay cell viability, proliferation, and cell cycle. At a concentration of 125 μg/ml of anti-PD-1, there was direct loss of cell viability after one day of coincubation that was further enhanced by extended exposure (Fig. 5C). The effect of the anti-PD-1 Ab was partially mediated by direct inhibition of cellular proliferation (Fig. 5D). After treatment with the anti-PD-1 Ab for 48 hours, EOC20 cells were analyzed for cell cycle distribution with flow cytometry, and there was no difference upon exposure to anti-PD-1 (Fig. 5E). Given that the maximum circulating concentration of anti-PD-1 was approximately 3 μg/ml (200 μg infusion in an 60 mL blood volume), we tested if the anti-PD-1 Ab could be further potentiated secondary to the involvement of ADCC. There was no substantial increase in cytotoxicity as detected by luminescence released from the microglia target cells in the presence of the isotype IgG control and effector cells, but the presence of anti-PD-1 increased this by 113% (Fig. 5F). Because repeated administration of the anti-PD-1 rat anti-mouse Ab in vivo would trigger an anti-rat humoral response, a secondary Ab (anti-rat IgG) was added to the ADCC assay, which further enhanced luminescence release by 233%. To further investigate if PD-1 signaling is mechanistically required, both blocking antibodies (RMP1–14 and 29F.1A12) and a non-blocking Ab (RMP1–30) were evaluated in the ADCC assay. All three antibodies triggered ADCC elimination of PD-1-expressing microglia, indicating that anti-PD-1 triggers the elimination of PD-1-expressing immune-suppressive innate immune cells in the CNS through an ADCC-mediated mechanism independent of PD-1 blockade.
Fig. 5. Direct and Ab-dependent cellular cytotoxicity (ADCC) activity of anti-PD-1 Ab on PD-1 expressing macrophage/microglia.

A, PD-1 expression of EOC-20 cells, gated on CD45+ CD11b+ cells. B, PD-1 expression on myeloid cells (CD45+, CD11b+) isolated from tumor bearing Ntv-a mice. C, Coincubation of only the anti-PD-1 Ab at the designated concentrations with PD-1 expressing EOC-20 microglia resulted in diminished cellular viability starting one day after exposure, which was further enhanced with increased exposure time. D, Coincubation of BrdU-labeled EOC-20 microglia with increasing concentrations of anti-PD-1 relative to the IgG control demonstrated decreased proliferative capacity. E, Cell cycle analysis of EOC-20 cells exposed to IgG control or anti-PD-1 demonstrating that these antibodies do not impact cellular proliferation. F, Ab-dependent cellular cytotoxicity (ADCC) assay detecting lactic dehydrogenase leakage (luminescence; relative light units [RLU]) from target EOC-20 microglia cells upon exposure to anti-PD-1 and in the presence of effector cells capable of mediating ADCC. Mouse microglial target cells, EOC20, were incubated with control Ab or anti-PD-1 Ab at a concentration of 125 μg/ml or 12.5 μg/ml, followed by the addition of effector cells. The E:T ratio was 20:1. After 8h of induction at 37o C, Bio-Glo luciferase reagent was added, and luminescence (RLU) was determined. The ADCC was further potentiated by the presence of a secondary Ab (mouse anti-rat) that could be generated by repeat administration of a rat anti-mouse Ab (anti-PD-1) in vivo. M = Media; T = Target; E = Effector Cell. When the anti-PD-1 Ab was decreased to 12.5 μg/ml, similar results were obtained.
Anti-PD-1 antibodies can be detected in CNS gliomas
To ascertain whether anti-PD-1 was able to penetrate CNS gliomas, the anti-PD-1 Ab was fluorescently labeled and subsequently injected intravenously into mice. Non-glioma-bearing, untreated C57BL/6 mice showed no baseline fluorescence, but mice treated with the labeled anti-PD-1 Ab demonstrated fluorescence in the brain and systemic organs (Fig. 6A). In tumor-bearing mice treated with labeled anti-PD-1 Ab, ex vivo analysis of the entire brain demonstrated elevated fluorescence in the right frontal lobe where the GL261 glioma cells were implanted, but no focal fluorescence was detected in non-tumor-bearing mice (Fig. 6B). Increased fluorescence was detected in the cerebellum (i.e., posterior fossa) including in non-glioma-bearing mice. Subsequent sequential coronal sections from anterior to posterior directly confirmed the presence of tumor in the right frontal lobe that correlated with this increased fluorescence (Fig. 6C). Because GL261 is surgically implanted and grows in a focal manner, this increased fluorescence could be an artifact of the model. As such, we evaluated the penetration of fluorescently-labeled anti-PD-1 in the Ntv-a model in which gliomagenesis is triggered in the neonatal period. Additionally, gliomas generated in the Ntv-a model are diffusely infiltrative and can be multifocal, more closely recapitulating human gliomas. In the Ntv-a CD8−/− mice bearing gliomas treated with labeled anti-PD-1, increased fluorescence was detected in multiple areas of the CNS relative to mice without gliomas (Fig. 6D). These areas were confirmed to histologically contain glioma (Fig. 6E). Cumulatively, these data indicate that the anti-PD-1 can pass into the CNS, especially within gliomas in which there is breakdown of the blood-brain barrier (BBB) (such as GL261) and in gliomas that have evolved intrinsically within the CNS that are more diffusely infiltrating, such as those induced in Ntv-a mice.
Fig. 6. Anti-PD-1 in vivo biodistribution analysis.

A, C57BL/6 mice were either untreated (control) or injected with 200 μg of fluorescently-labeled anti-PD-1. After 3 hours, their organs were harvested, rinsed in PBS, positioned on a petri dish, and then imaged using the IVIS 200 Fluorescence Imager. The organs were then photographed. B, The brain from a non-tumor-bearing C57BL/6 mouse treated with fluorescently-labeled anti-PD-1 (top panel) or bearing intracerebral GL261 (bottom panel). The arrow denotes the location of the GL261 implantation. C, The brains from B were then sequentially coronally sectioned, with the non-tumor-bearing brain on the left and the GL261-implanted brain on the right. The sections were positioned anterior to posterior on the petri dish and imaged. The horizontal arrow denotes the location of the implanted GL261 cells in the right frontal lobe. Increased fluorescent intensity is detected in the posterior cerebellum. All brain sections were imaged for the same exposure time. D, Non-glioma-bearing (control) or glioma-bearing Ntv-a mice in the knockout (CD8−/−) background were treated with 200 μg of fluorescently-labeled anti-PD-1. After 3 hours, their brains were harvested, rinsed in PBS, coronally sectioned, positioned on a petri dish, and then imaged using the IVIS 200 Fluorescence Imager. The brains were then photographed. Increasing fluorescence intensity is seen to correlate with increasing concentration of anti-PD-1. E, Hematoxylin and eosin (H & E) stained coronal sections of GL261 (top) and Ntv-a CD8−/− (bottom) from C and D panels. 20x magnification, scale bar = 100μM. Arrows indicate the region of the tumor.
DISCUSSION
In our previous clinical trial of recurrent glioblastoma patients that were treated with anti-PD-1 prior to surgery, immune profiling of the tumor microenvironment revealed a marked paucity of effector T cells but a profound predominance of macrophages displaying heterogeneous immune-stimulatory and immune-suppressive phenotypes. In our clinical trial (49) and in another (8), glioblastoma patients treated in an adjuvant setting had better than expected outcomes. Thus, there arises the paradox of an anti-PD-1 agent having therapeutic activity in an oncological setting that is notable for T cells that are sequestered in the bone marrow and completely refractory to being reinvigorated with immune checkpoint inhibitors (10). It is likely that anti-PD-1 can switch its therapeutic effect between various immune populations given their relative frequencies. In malignancies enriched in T cell infiltration, anti-PD-1 likely exerts most of its therapeutic activity through direct T cell-ligand interactions. In contrast, in malignancies such as glioblastoma that are devoid of T cells, anti-PD-1 activity may exert a therapeutic effect through alternative immune populations such as macrophages and microglia. In this latter scenario, the therapeutic activity is mediated through the elimination of an immune-suppressive, tumor-supportive PD-1+ macrophage/microglia population by myeloid to myeloid ADCC-mediated fratricide mechanisms and/or to M1 macrophage polarization. PD-1 has previously been shown to be expressed by macrophages, which limits their phagocytic capacity (47) and as such, treatment with PD-1 may be enhancing this activity in vivo. Proinflammatory M1 macrophages mediate direct tumor killing through secreted products like nitric oxide or tumor necrosis factor (50). This alternative mechanism of anti-PD-1 activity also provides an explanation for the failure of biomarkers to predict clinical responses in cancers such as glioblastoma, as these markers are focused on the immune functional features of the adaptive immune system such as the abundance of antigens (i.e., mutational burden) and the presence of T cell infiltration and ligand frequency (i.e., PD-1+ TILs, PD-L1), which are unlikely to be the dominant mechanism of therapeutic activity of anti-PD-1 in the setting of myeloid-enriched malignancies. Because PD-1 is not expressed in gliomas, it is unlikely that the anti-PD-1 is exerting a direct effect on the tumor cell. We are unable to exclude the possibility that other immune populations are also contributing to the therapeutic effect of the anti-PD-1 and it is likely that a combination of immune populations play a role in activity. Cumulatively, this is the first study to demonstrate that the anti-PD-1 Ab may promote a pro-inflammatory M1 signature within the glioma microenvironment.
The purpose of this study was not to suggest that monotherapy with anti-PD-1 in glioblastoma is effective for GBM since our own clinical trial studies fail to support even the more optimistic strategy of use of anti-PD-1 in the adjuvant setting (51). Rather, the purpose of our study was to more fully understand the mechanisms of action of this agent in GBM so that we might more appropriately select rational combinations and identify potential response biomarkers. To date, immune checkpoint inhibitor response biomarkers are surrogates for anti-tumor T cell responses which include T cell counts in the tumor microenvironment; tumor mutational burden, microsatellite instability, and POLE mutations as T cell targets; interferon signatures; and the inhibitory ligand for PD-1 - PD-L1. We are also not attempting to refute the contribution of CD4 cytotoxic T cells to these responses (52–54) but are rather expanding consideration to PD-1 expressing macrophages and microglia as also being involved in the anti-tumor immune responses with these agents. As such, future clinical trials using immune checkpoint inhibitors should also consider including the immune phenotype and function of macrophages which may be all the more relevant in glioblastoma given the predominance of this immune population. Combinatorial strategies, such as macrophage polarization therapeutics, are being actively considered with anti-PD-1. However, our data indicates this may not be necessary since anti-PD-1 is already eliminating PD-1 expressing microglia and driving M1 polarization and as such should be deprioritized.
Because CNS macrophages arise from peripherally-derived monocytes, the anti-PD-1 Ab need not have penetration into the CNS in order to eliminate PD-1+ immune-suppressive macrophages in the glioma microenvironment. Most immune cells, including the monocyte-derived macrophages in the tumor microenvironment, arise from the periphery where they can interact with the anti-PD-1 Ab. However, the elimination of PD-1-expressing microglia, which originate in the CNS during embryogenesis, implies that the anti-PD-1 Ab is capable of some degree of CNS penetration, especially in tumor regions in which there is breakdown of the BBB. Fluorescently-tagged anti-PD-1 accumulated in the glioma relative to the surrounding brain, confirming that the anti-PD-1 Ab can gain access to the glioma microenvironment. Future studies will be directed at assessing the concentrations of the anti-PD-1 Ab in infiltrating non-contrast-enhancing regions of the brain that do not have appreciable breakdown of the BBB.
Eliminating the CD8 T cell from the microenvironment of an evolving glioblastoma enabled us to isolate the macrophage as the cell mediating the therapeutic effect of checkpoint inhibition. We were also able to study how the absence of CD8+ T cells affects glioma development, malignancy, and survival. A previous study showed that the CD8+ T cell is defective and nonoperational in murine models of spontaneously-arising astrocytomas, even at early stages of tumor development (55). However, this contrasts with recent reports that levels of CD8+ tumor-infiltrating lymphocytes are inversely correlated with glioma grade and are associated with long-term survival (56). Alternatively, the CD8+ T cell population may only have a role during the early stages of tumor development (57, 58). Our primary hypothesis was that the elimination of the CD8 immune effector population would produce an increased incidence of high-grade gliomas resulting from the lack of immunological recognition and control. Because the presumptive antitumor immune effector population is not present in the CD8−/− genetic background, relative to wild-type mice, the tumor-bearing CD8−/− mice should have had an environment favoring growth of tumor cells. Alternatively, without the selective pressure of CD8 T cells culling tumor cells sensitive to elimination, the more resistant cells escaping immune surveillance might not proliferate, resulting in some degree of quiescence. If this hypothesis were correct, then there would be increased survival in mice with the CD8−/− genetic background associated with less malignant progression. Surprisingly, we found that there was no difference in survival time and no difference in the incidence of glioma grade between the two genotypes. These results show that CD8 T cells do not influence survival of mice that undergo de novo glioma formation. Yet, Kane et al. report that while CD8 T cells do not influence survival similar to our glioma model, the presence of CD8 T cells do influence the tumor genome, phenotype (oncogenes/tumor suppressors, MAPK activation), and composition of the immunological microenvironment (37).
Another observation from our study was that there was elevated MHC-II and Iba1 expression associated with the monocyte/macrophage population present in the glioma in the absence of CD8 T cells. These cells demonstrated a proinflammatory activated profile. Activated macrophages have been shown to be capable of tumoricidal activity (59). Similar compensatory findings have been reported in a GL261 glioblastoma model treated with a tumor lysate vaccine with OX40L-Fc stimulant in which the vaccine efficacy was independent of the CD8+ T cell population, but dependent on CD4+ T cells, NK cells, and B cells (60); however, this study did not specifically delve into the role of the proinflammatory macrophage. The macrophages also demonstrated immune-suppressive features, including PD-1 expression and elaboration of arginase. Consistent with our findings from human GBM specimens (48), the glioma-infiltrating macrophages in our models demonstrated marked immunological heterogeneity of phenotype and function.
A confounder of our study is the possibility that the CD19+ and NK1.1+ populations play a role in the earlier stages of gliomagenesis and the CD8 KO model may impact the development of other immune cells. However, given the sustained survival in this model and the fact that gliomagenesis is induced in the postnatal period, it is not technically feasible to perform in vivo depletions in neonatal mice. This study also underscores the importance of genetically-engineered mouse models in which gliomas are formed de novo in the brain for the study of the immune system, as there was a significant difference in the infiltration of T cells and macrophages between endogenously forming tumors and orthotopic xenografts. These results and the studies by others (37) indicate that CD8 T cells might influence features of glioma development such as genotype, phenotype, immunogenicity, and the microenvironment through immunoediting, and that these effector cells might not be responsible for responses to anti-PD-1 blockade. This may be related to an unappreciated role of the innate immune system in modulating malignant degeneration, as has been previously suggested (61).
Supplementary Material
TRANSLATIONAL RELEVANCE.
Immune checkpoint inhibition (ICI) has largely been ineffective against glioblastoma, likely due to the uniquely immunosuppressed microenvironment of this primary brain tumor. The paucity of CD8+ T cells in GBM has long been considered the reason for the failure of ICI. However, the population of PD1+ macrophages is very robust in GBM and may be targeted by ICI. We show that ICI targeting PD-1 results in significant survival gains in glioma-bearing immunocompetent mice even when CD8 T cells are absent. Treatment with anti PD-1 Ab shifts the polarization of remaining macrophages to the inflammatory (and anti-tumor) M1 phenotype. In humans, this strategy may not have direct anti-tumor effect, but may be useful to reverse immunosuppression by the resident macrophage population. Thus, combinatorial immune strategies which include ICI, may be a rational next step in the treatment of glioblastoma.
Acknowledgments
We thank David M. Wildrick, Ph.D., for scientific editing of the manuscript and Audria Patrick for administrative support.
Funding: Grant support was provided by the Brockman Foundation, the Dr. Marnie Rose Foundation, The University of Texas MD Anderson Cancer Center GBM Moonshot program, and the National Institutes of Health CA1208113, P50 CA127001, and NS094615. This study made use of the Research Animal Support Facility-Smithville (Laboratory Animal Genetic Services), which is supported by P30 CA016672 DHHS/NCI Cancer Center Support Grant to The University of Texas MD Anderson Cancer Center.
Footnotes
Competing interests: the authors have no competing interests.
Data and materials availability: All data associated with this study are available in the main text or the supplementary materials.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Liau LM, Prins RM, Kiertscher SM, Odesa SK, Kremen TJ, Giovannone AJ, et al. Dendritic cell vaccination in glioblastoma patients induces systemic and intracranial T-cell responses modulated by the local central nervous system tumor microenvironment. Clin Cancer Res. 2005;11(15):5515–25. [DOI] [PubMed] [Google Scholar]
- 2.Prins RM, Soto H, Konkankit V, Odesa SK, Eskin A, Yong WH, et al. Gene expression profile correlates with T-cell infiltration and relative survival in glioblastoma patients vaccinated with dendritic cell immunotherapy. Clin Cancer Res. 2011;17(6):1603–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sampson JH, Aldape KD, Archer GE, Coan A, Desjardins A, Friedman AH, et al. Greater chemotherapy-induced lymphopenia enhances tumor-specific immune responses that eliminate EGFRvIII-expressing tumor cells in patients with glioblastoma. Neuro Oncol. 2011;13(3):324–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sampson JH, Heimberger AB, Archer GE, Aldape KD, Friedman AH, Friedman HS, et al. Immunologic escape after prolonged progression-free survival with epidermal growth factor receptor variant III peptide vaccination in patients with newly diagnosed glioblastoma. J Clin Oncol. 2010;28(31):4722–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yu J, Liu G, Ying H, Yong W, Black K, Wheeler CJ. Vaccination with tumor lysate-pulsed dendritic cells elicits antigen-specific, cytotoxic T-cells in patients with malignant glioma. Cancer Res. 2004;64(14):4973–9. [DOI] [PubMed] [Google Scholar]
- 6.Nduom EK, Weller M, Heimberger AB. Immunosuppressive mechanisms in glioblastoma. Neuro-oncology. 2015;17 Suppl 7:vii9–vii14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tawbi HA, Forsyth PA, Algazi A, Hamid O, Hodi FS, Moschos SJ, et al. Combined Nivolumab and Ipilimumab in Melanoma Metastatic to the Brain. N Engl J Med. 2018;379(8):722–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25(3):477–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhao J, Chen AX, Gartrell RD, Silverman AM, Aparicio L, Chu T, et al. Immune and genomic correlates of response to anti-PD-1 immunotherapy in glioblastoma. Nat Med. 2019;25(3):462–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-Cell Exhaustion Signatures Vary with Tumor Type and Are Severe in Glioblastoma. Clin Cancer Res. 2018;24(17):4175–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fecci PE, Mitchell DA, Whitesides JF, Xie W, Friedman AH, Archer GE, et al. Increased regulatory T-cell fraction amidst a diminished CD4 compartment explains cellular immune defects in patients with malignant glioma. Cancer Res. 2006;66(6):3294–302. [DOI] [PubMed] [Google Scholar]
- 12.Hussain SF, Yang D, Suki D, Aldape K, Grimm E, Heimberger AB. The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro-oncology. 2006;8(3):261–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med. 2018;24(9):1459–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kreth FW, Faist M, Grau S, Ostertag CB. Interstitial 125I radiosurgery of supratentorial de novo WHO Grade 2 astrocytoma and oligoastrocytoma in adults: long-term results and prognostic factors. Cancer. 2006;106(6):1372–81. [DOI] [PubMed] [Google Scholar]
- 15.Jaeckle KA, Decker PA, Ballman KV, Flynn PJ, Giannini C, Scheithauer BW, et al. Transformation of low grade glioma and correlation with outcome: an NCCTG database analysis. Journal of neuro-oncology. 2011;104(1):253–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jakola AS, Myrmel KS, Kloster R, Torp SH, Lindal S, Unsgard G, et al. Comparison of a strategy favoring early surgical resection vs a strategy favoring watchful waiting in low-grade gliomas. JAMA. 2012;308(18):1881–8. [DOI] [PubMed] [Google Scholar]
- 17.Doucette TA, Kong LY, Yang Y, Ferguson SD, Yang J, Wei J, et al. Signal transducer and activator of transcription 3 promotes angiogenesis and drives malignant progression in glioma. Neuro Oncol. 2012;14(9):1136–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lohr J, Ratliff T, Huppertz A, Ge Y, Dictus C, Ahmadi R, et al. Effector T-cell infiltration positively impacts survival of glioblastoma patients and is impaired by tumor-derived TGF-beta. Clin Cancer Res. 2011;17(13):4296–308. [DOI] [PubMed] [Google Scholar]
- 19.Arrieta VA, Cacho-Diaz B, Zhao J, Rabadan R, Chen L, Sonabend AM. The possibility of cancer immune editing in gliomas. A critical review. Oncoimmunology. 2018;7(7):e1445458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dunn GP, Fecci PE, Curry WT. Cancer immunoediting in malignant glioma. Neurosurgery. 2012;71(2):201–22; discussion 22–3. [DOI] [PubMed] [Google Scholar]
- 21.Holland EC, Varmus HE. Basic fibroblast growth factor induces cell migration and proliferation after glia-specific gene transfer in mice. Proceedings of the National Academy of Sciences of the United States of America. 1998;95(3):1218–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kong LY, Wu AS, Doucette T, Wei J, Priebe W, Fuller GN, et al. Intratumoral mediated immunosuppression is prognostic in genetically engineered murine models of glioma and correlates to immunotherapeutic responses. Clinical cancer research : an official journal of the American Association for Cancer Research. 2010;16(23):5722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hale JS, Boursalian TE, Turk GL, Fink PJ. Thymic output in aged mice. Proc Natl Acad Sci U S A. 2006;103(22):8447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Simon AK, Hollander GA, McMichael A. Evolution of the immune system in humans from infancy to old age. Proc Biol Sci. 2015;282(1821):20143085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zlotoff DA, Schwarz BA, Bhandoola A. The long road to the thymus: the generation, mobilization, and circulation of T-cell progenitors in mouse and man. Semin Immunopathol. 2008;30(4):371–82. [DOI] [PubMed] [Google Scholar]
- 26.Wakeland E, Morel L, Achey K, Yui M, Longmate J. Speed congenics: a classic technique in the fast lane (relatively speaking). Immunol Today. 1997;18(10):472–7. [DOI] [PubMed] [Google Scholar]
- 27.Dai C, Celestino JC, Okada Y, Louis DN, Fuller GN, Holland EC. PDGF autocrine stimulation dedifferentiates cultured astrocytes and induces oligodendrogliomas and oligoastrocytomas from neural progenitors and astrocytes in vivo. Genes & development. 2001;15(15):1913–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holland EC, Hively WP, DePinho RA, Varmus HE. A constitutively active epidermal growth factor receptor cooperates with disruption of G1 cell-cycle arrest pathways to induce glioma-like lesions in mice. Genes Dev. 1998;12(23):3675–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fults D, Pedone C, Dai C, Holland EC. MYC expression promotes the proliferation of neural progenitor cells in culture and in vivo. Neoplasia. 2002;4(1):32–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shih AH, Dai C, Hu X, Rosenblum MK, Koutcher JA, Holland EC. Dose-dependent effects of platelet-derived growth factor-B on glial tumorigenesis. Cancer Res. 2004;64(14):4783–9. [DOI] [PubMed] [Google Scholar]
- 31.Wei J, Wang F, Kong LY, Xu S, Doucette T, Ferguson SD, et al. miR-124 inhibits STAT3 signaling to enhance T cell-mediated immune clearance of glioma. Cancer Res. 2013;73(13):3913–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xu S, Wei J, Wang F, Kong L-Y, Ling X- Y, Doucette TA, et al. Effect of miR-142–3p on the M2 macrophage and therapeutic efficacy against murine glioblastoma. J Natl Cancer Inst. 2014;106(8):pii: dju162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heimberger AB, Crotty LE, Archer GE, Hess KR, Wikstrand CJ, Friedman AH, et al. Epidermal growth factor receptor VIII peptide vaccination is efficacious against established intracerebral tumors. Clin Cancer Res. 2003;9(11):4247–54. [PubMed] [Google Scholar]
- 34.Bennett ML, Bennett FC, Liddelow SA, Ajami B, Zamanian JL, Fernhoff NB, et al. New tools for studying microglia in the mouse and human CNS. Proc Natl Acad Sci U S A. 2016;113(12):E1738–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Martinez FO, Gordon S, Locati M, Mantovani A. Transcriptional profiling of the human monocyte-to-macrophage differentiation and polarization: new molecules and patterns of gene expression. J Immunol. 2006;177(10):7303–11. [DOI] [PubMed] [Google Scholar]
- 36.Dahan R, Sega E, Engelhardt J, Selby M, Korman AJ, Ravetch JV. FcgammaRs Modulate the Anti-tumor Activity of Antibodies Targeting the PD-1/PD-L1 Axis. Cancer Cell. 2015;28(3):285–95. [DOI] [PubMed] [Google Scholar]
- 37.Kane JR, Zhao J, Tsujiuchi T, Laffleur B, Mahajan A, Rao G, et al. CD8+ T-cell-mediated immunoediting influences genomic evolution and immune evasion in murine gliomas. bioRxiv preprint for Biology. In-Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fleischer B Acquisition of specific cytotoxic activity by human T4+ T lymphocytes in culture. Nature. 1984;308(5957):365–7. [DOI] [PubMed] [Google Scholar]
- 39.Appay V The physiological role of cytotoxic CD4(+) T-cells: the holy grail? Clin Exp Immunol. 2004;138(1):10–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Porakishvili N, Kardava L, Jewell AP, Yong K, Glennie MJ, Akbar A, et al. Cytotoxic CD4+ T cells in patients with B cell chronic lymphocytic leukemia kill via a perforin-mediated pathway. Haematologica. 2004;89(4):435–43. [PubMed] [Google Scholar]
- 41.Echchakir H, Bagot M, Dorothee G, Martinvalet D, Le Gouvello S, Boumsell L, et al. Cutaneous T cell lymphoma reactive CD4+ cytotoxic T lymphocyte clones display a Th1 cytokine profile and use a fas-independent pathway for specific tumor cell lysis. J Invest Dermatol. 2000;115(1):74–80. [DOI] [PubMed] [Google Scholar]
- 42.Yasukawa M, Ohminami H, Arai J, Kasahara Y, Ishida Y, Fujita S. Granule exocytosis, and not the fas/fas ligand system, is the main pathway of cytotoxicity mediated by alloantigen-specific CD4(+) as well as CD8(+) cytotoxic T lymphocytes in humans. Blood. 2000;95(7):2352–5. [PubMed] [Google Scholar]
- 43.Dutta S, Sengupta P. Men and mice: Relating their ages. Life Sci. 2016;152:244–8. [DOI] [PubMed] [Google Scholar]
- 44.Hung AL, Maxwell R, Theodros D, Belcaid Z, Mathios D, Luksik AS, et al. TIGIT and PD-1 dual checkpoint blockade enhances antitumor immunity and survival in GBM. Oncoimmunology. 2018;7(8):e1466769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim JE, Patel MA, Mangraviti A, Kim ES, Theodros D, Velarde E, et al. Combination Therapy with Anti-PD-1, Anti-TIM-3, and Focal Radiation Results in Regression of Murine Gliomas. Clin Cancer Res. 2017;23(1):124–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86(2):343–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gordon SR, Maute RL, Dulken BW, Hutter G, George BM, McCracken MN, et al. PD-1 expression by tumour-associated macrophages inhibits phagocytosis and tumour immunity. Nature. 2017;545(7655):495–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, et al. Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI insight. 2016;1(2):pii: e85841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.de Groot J, Penas-Prado M, Alfaro-Munoz K, Hunter K, Pei B-L, O’Brien B, et al. Window-of-opportunity clinical trial of Pembrolizumab in Patients with Recurrent GBM reveals a predominance of immune suppressive macrophages. Neuro Oncol. In-Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carswell EA, Old LJ, Kassel RL, Green S, Fiore N, Williamson B. An endotoxin-induced serum factor that causes necrosis of tumors. Proc Natl Acad Sci U S A. 1975;72(9):3666–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Groot J, Penas-Prado M, Alfaro-Munoz KD, Hunter K, Pei B, O’Brien B, et al. Window-of-opportunity clinical trial of pembrolizumab in patients with recurrent glioblastoma reveals predominance of immune-suppressive macrophages. Neuro-oncology. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Yan H, Hou X, Li T, Zhao L, Yuan X, Fu H, et al. CD4+ T cell-mediated cytotoxicity eliminates primary tumor cells in metastatic melanoma through high MHC class II expression and can be enhanced by inhibitory receptor blockade. Tumour Biol. 2016. [DOI] [PubMed] [Google Scholar]
- 53.Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4(+) T Cell Immunity. Cell. 2019;177(3):556–71 e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ahrends T, Spanjaard A, Pilzecker B, Babala N, Bovens A, Xiao Y, et al. CD4(+) T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity. 2017;47(5):848–61 e5. [DOI] [PubMed] [Google Scholar]
- 55.Tran Thang NN, Derouazi M, Philippin G, Arcidiaco S, Di Berardino-Besson W, Masson F, et al. Immune infiltration of spontaneous mouse astrocytomas is dominated by immunosuppressive cells from early stages of tumor development. Cancer Res. 2010;70(12):4829–39. [DOI] [PubMed] [Google Scholar]
- 56.Kmiecik J, Poli A, Brons NH, Waha A, Eide GE, Enger PO, et al. Elevated CD3+ and CD8+ tumor-infiltrating immune cells correlate with prolonged survival in glioblastoma patients despite integrated immunosuppressive mechanisms in the tumor microenvironment and at the systemic level. J Neuroimmunol. 2013;264(1–2):71–83. [DOI] [PubMed] [Google Scholar]
- 57.Marijt KA, Blijleven L, Verdegaal EME, Kester MG, Kowalewski DJ, Rammensee HG, et al. Identification of non-mutated neoantigens presented by TAP-deficient tumors. J Exp Med. 2018;215(9):2325–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marijt KA, Doorduijn EM, van Hall T. TEIPP antigens for T-cell based immunotherapy of immune-edited HLA class I(low) cancers. Mol Immunol. 2018. [DOI] [PubMed] [Google Scholar]
- 59.Beatty GL, Chiorean EG, Fishman MP, Saboury B, Teitelbaum UR, Sun W, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Murphy KA, Erickson JR, Johnson CS, Seiler CE, Bedi J, Hu P, et al. CD8+ T cell-independent tumor regression induced by Fc-OX40L and therapeutic vaccination in a mouse model of glioma. Journal of immunology. 2014;192(1):224–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gajewski TF, Schreiber H, Fu YX. Innate and adaptive immune cells in the tumor microenvironment. Nature immunology. 2013;14(10):1014–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.