

Abstract
Immune checkpoint inhibitors (ICI) have the potential to induce durable therapeutic responses, yet response rates in breast cancer are modest and limited to particular subtypes. To expand the applicability of ICI, we examined the role of an essential autophagy gene, FIP200, which has been shown to be important for tumor progression in mammary tumors. Specific disruption of the autophagy function of FIP200 or complete ablation of FIP200 in genetic mouse models revealed that FIP200 autophagy function was required for progression of PyMT-driven mammary tumors. However, a non-canonical autophagy function of FIP200 was responsible for limiting T-cell recruitment and activation of the TBK1-IFN signaling axis. FIP200 also interacted with the TBK1 adaptor protein, AZI2, which was crucial for activation of TBK1 following FIP200 ablation. Accordingly, disrupting the non-canonical autophagy function of FIP200 in combination with ICI therapy led to superior, durable responses in immune-competent models of breast cancer. Collectively, these insights could guide future development of therapeutic agents against FIP200 for combinatorial ICI therapies in nonresponsive breast cancers.
Keywords: FIP200, TBK1, AZI2, Immune checkpoint inhibitor, breast cancer, autophagy
Introduction
Clinical application of immune checkpoint inhibitors (ICIs) such as anti-PD1 and anti-CTLA4 therapy has expanded beyond tumors that have a high burden of non-synonymous mutations (e.g. melanoma and lung cancer) (1,2). Remarkably, robust durable responses and prolonged overall survival can be achieved with these treatment modalities (3,4). However, not all tumors respond and in the case of breast cancer, current clinical trials are largely limited to triple negative breast cancers (TNBC) and Her2+ breast cancers. Several biomarkers have been proposed to predict response to ICIs and these include CD8+ T cell density, PD-L1 expression and the mutational load of tumors (5). TNBCs frequently display higher numbers of tumor-infiltrating lymphocytes (TIL) and are generally more mutagenic, hence the justification for its inclusion in ongoing clinical trials. Even then, objective response rates (ORR) that are reported for advanced TNBC patients are less than 20% (6,7) and in an unselected breast cancer patient cohort, the ORR has been reported to be much lower, at 4.8% (8). Accordingly, it remains a formidable challenge to expand the prominent benefits of immuno-therapy to a large proportion of breast cancer patients.
Macroautophagy (hereafter autophagy) is a process that enables cells to recycle cytoplasmic entities and organelles through the sequestration of cargo within double-membraned structures termed autophagosomes. This self-recycling mechanism allows for sustenance under nutrient limiting conditions and the elimination of defective organelles or proteins (9). FIP200 (RB1CC1) is an essential autophagy gene, that is part of the autophagy initiation complex, which includes ULK1, ATG13 and ATG101 (10). Autophagy has been shown to be important for the secretion of damage-associated molecular patterns (DAMPs) in chemotherapy treated colon carcinoma and melanoma cells (11,12), which leads to efficient recruitment and activation of T cells to elicit anti-tumor immune responses. More recently however, it has been shown in syngeneic transplant models of breast cancer and melanoma that inhibition of autophagy either through genetic depletion (Atg7 or Atg12) or pharmacologically (antimalarial treatment) in tumor cells, does not impair anti-tumor immune responses (13). Nonetheless, a number of studies have shown that depletion of autophagy-related genes in the stromal or immune compartment can heighten anti-microbial or anti-tumor responses, implicating a role for autophagy in these processes (14–21). There are also reports of non-autophagic functions of autophagy-related genes in the regulation of inflammation (22–24), namely LC3-associated phagocytosis (16). Hence, the relative contributions of both the canonical-autophagy and non-autophagic functions of autophagy genes with regards to inflammation is of great interest. As such, there is a need to specifically dissect and distinguish the contribution of autophagy and non-autophagy functions of autophagy genes in the regulation of anti-tumor immunity, both in tumor and stromal cells.
We have shown previously that FIP200 ablation in MMTV-PyMT driven mammary tumors can inhibit tumor growth and progression. In addition FIP200 deletion also increased CD8+ TILs and the expression of interferon (IFN) responsive genes in these tumors (25). This suggests that FIP200 negatively regulates anti-tumor immune responses, but since FIP200 also has non-autophagy related functions (26), it is possible that this negative regulation is due to the non-autophagy functions of FIP200 and may explain the different outcomes observed with anti-tumor immunity when other autophagy related genes are depleted (11–13). Accordingly, we have recently developed a mouse model with a FIP200 knock-in allele that disrupts the association between FIP200 and ATG13, leading to specific impairment of autophagy (27). With this mouse model, we will be able to dissect the role of autophagy or non-autophagy functions of FIP200 in the regulation of tumor growth, progression and anti-tumor immune responses.
In this study, we demonstrated that disruption of FIP200’s autophagy function is sufficient to inhibit the growth and metastatic potential of PyMT driven mammary tumors, to a similar extent as depleting FIP200 completely. However, the non-autophagy function of FIP200 was responsible for increased T cell recruitment and activation of the TBK1-IFN signaling axis. FIP200 was also found to interact with the TBK1 adaptor protein, AZI2, which was crucial for TBK1 activation upon FIP200 ablation. Since tumors with increased CD8+ T cell density and interferon (IFN) signaling have been associated with better responses to ICI therapy in various cancers (5,28,29), we further demonstrated the effects of ablating FIP200 in combination with anti-PD1 and/or anti-CTLA4 therapy to achieve prolonged durable responses in mice bearing PyMT tumors. Ultimately, we have unraveled a superior combinatorial therapeutic strategy for ICIs and this could potentially expand the prominent benefits of ICIs to non-responsive breast cancers.
Material & Methods
Reagents and antibodies
ISG56-Luciferase plasmid was a gift from Dr. Saumendra Sarkar from the University of Pittsburgh (30). pcDNA3.1-hTANK-FLAG construct was obtained from Dr. Ulrich Siebenlist (31), pCMV3Tag8-HA-hSINTBAD-FLAG and pCMV3Tag8-HA-mNAP1-FLAG plasmids were a kind gift from Dr. Shitao Li (32). FIP200 plasmids (Myc-FIP200, Myc-FIP200-N859, Myc-FIP200-CC and Myc-FIP200-CT) were generated as described previously (26) and Myc-FIP200-CT delta 20 amino acid variants were generated using Q5 site directed mutagenesis kit (NEB). For gene silencing experiments, siRNAs used were AZI2 (D-014092-01-0002, D-014092-02-0002), TANK (D-005255-20-0002, D-005255-02-0002), and SINTBAD (D-020406-01-0002, D-020406-02-0002) (Dharmacon). Antibodies used for immunoblotting include b-actin (Sigma A5441), Vinculin (Sigma V4505), GAPDH (CST 2118), FIP200 (CST 12436), p62 (CST 5114), LC3 (CST 2775), phospho-TBK1 (CST 5483), TBK1 (CST 3504), IRF1 (CST 8478), PARP (CST 9532), Azi2 (Proteintech 15042-1-AP), Tank (Proteintech 27065-1-AP), SINTBAD (CST 8605). For immunohistochemistry, antibodies used p62 (Enzo life Sciences BML-PW9860), cleaved caspase3 (CST 9661), Ki67 (Spring Bioscience m3062), CD8 (eBioscience 14-0808-80). For flow cytometry, antibodies used were CD45-Pacific blue (BioLegend 103125), CD3-V500 (BioLegend 100233), CD8-FITC (BioLegend 100706), CD103-APC (BioLegend 121413), CD49b-APC (BioLegend108907).
Cell culture and treatment
Primary tumor cells and their derivatives were cultured in DMEM/F12 supplemented with 10% FBS, 10 ng/mL EGF, 20 mg/mL insulin, and 50 units/mL penicillin–streptomycin. The generation of FIP200f/f;PyMT;CreER cells and FIP200f/KI;PyMT;CreER cells have been described previously (27,33) and deletion of Fip200 was induced by culturing with 100 nmol/L 4-hydroxytamoxifen (4-OHT). Transfection experiments were carried out using Lipofectamine 3000 Reagent (Invitrogen) for cell lines and Amaxa Basic Nucleofector Kit for electroporation for primary cells (Lonza). Production of lentivirus and transduction of cells were carried out as described previously (34). Each sgRNA sequence is as follows: sgRb1cc1 is 5’-caccg CTCCATTGACCACCAGAACC-3’. sgTbk1 is 5’-caccgCATAAGCTTCCTTCGCCCAG-3’. sgAzi2 is 5’-caccgATCTTCTACTAGCGTGTCCA -3’. MDA-MB-231 cells were obtained from ATCC. Cell lines were maintained for less than 20 passages after collection or thawing. Mycoplasma testing was performed on a monthly basis.
Flow cytometry
Single-cell suspensions were prepared from primary tumors as described previously (35). Cells were blocked with TruStain fcX(anti-mouse CD16/32 antibody)(BioLegend 101319) before incubation with antibodies described above for 20 minutes at 4°C. Cells were then rinsed before sorting or analysis by FACSAria or FACSCanto instruments (BD Biosciences, San Jose, CA, USA). Flow cytometry data were analyzed using FlowJo (Ashland, OR, USA) software.
Tumor mice and transplants
Ctrl-MT (Fip200f/f, MMTV-PyMT), and cKO-MT (Fip200f/f, MMTV-PyMT, MMTV-Cre) mice have been described previously (34). Brca1f/f Trp53f/f K14-Cre mice were a kind gift from Dr. Jos Jonkers (Netherlands Cancer Institute) and have been crossed with FIP200f/ki mice as described previously (36). To eliminate potential influence of mouse genetic background on the phenotypes, all strains were backcrossed to FVB/N genetic background (syngeneic to the MMTV-PyMT mice or Brca1f/f Trp53f/f K14-Cre mice used throughout the study) for at least seven times (99.21%). Mice were housed and handled according to local, state, and federal regulations. All experimental procedures were carried out according to protocols approved by the Institutional Animal Care and Use Committee at University of Cincinnati (Cincinnati, OH). For transplantation experiments, 2×106 cells were prepared in PBS:Matrigel at a 1:1 ratio and were injected into the fourth mammary gland fat pad. Mice with transplanted tumors were randomized into respective treatment groups when the diameter of tumors reached to ~5mm. Antibodies for treatment were obtained from BioXCell; anti mouse PD-1 (BP0273), anti mouse CTLA-4 (BE0131), rat IgG2a isotype control (BP0089) and Syrian hamster IgG control (BE0087). Amlexanox was obtained from MedChemExpress (HY-B0713) and administered at 25mg/kg intra-peritoneally daily for 7 days, upon the start of tamoxifen induction in vivo.
Histology and Immunohistochemistry
Formalin-fixed paraffin-embedded tumors were sectioned (5μm) and stained for respective antigens as described previously (34). For antigen retrieval, slides were heated in citrate buffer in a pressure cooker. Quantification of positive cells was performed using Image J software.
Immunoblotting
Lysates were prepared from cells using modified RIPA buffer as described previously (34) with the addition of protease and phosphatase inhibitors according to manufacturer’s instructions (Thermo Fisher Scientific). Protein concentrations were then quantified by bicinchoninic acid method, subjected to SDS–PAGE and analyzed by immunoblotting as described previously (34). For phosphatase treatment, cell lysates without phosphatase inhibitors were incubated with FastAP at 37°C for 1hour according to manufacturer’s protocol (Thermo Fisher Scientific).
Quantitative PCR
Total RNA was isolated from cells using GeneGet RNA Purification Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. Equal amounts of RNA were then reverse transcribed using iScript cDNA Synthesis Kit (Bio-rad). qRT-PCR analysis was performed using iTaq Universal SYBR Green Supermix (Bio-rad) with primers used as follows: Actin-Forward; 5’-GGCTGTATTCCCCTCCATCG-3’, Actin-Reverse; 5’-CCAGTTGGTAACAATGCCATGT-3’, Ccl5-Forward; 5’-GCTGCTTTGCCTACCTCTCC-3’, Ccl5-Reverse; 5’-TCGAGTGACAAACACGACTGC-3’, Cxcl10-Forward; 5’-CCAAGTGCTGCCGTCATTTTC-3’, Cxcl10-Reverse; 5’-GGCTCGCAGGGATGATTTCAA-3’, Cxcl9-Forward; 5’-GGAGTTCGAGGAACCCTAGTG-3’, Cxcl9-Reverse; 5’-GGGATTTGTAGTGGATCGTGC
Statistical Analysis
Data were plotted as means and error bars represent standard deviation (SD). One-way comparisons were tested using unpaired t-test with Welch’s correction for figures 2J, 3A–D, 3K–L and 4H. For multiple comparisons, one-way ANOVA with Tukey’s post-hoc test were performed when data sets were all determined to be parametric by Shapiro-Wilk tests, in figures 1G, 2B, 2E, 2G and 4C–F. For multiple comparisons when datasets were non-parametric as determined by Shapiro-Wilk tests, Kruskal-Wallis test followed by Dunn’s post-hoc test were used, in figures 1D, 1E, 2C, 2F, 2H and 3H.For survival curves, log-rank tests were utilized. The threshold for statistical significance was p<0.05. For analyses involving quantification of fields of view from histological samples, triplicate fields of views were averaged and this was plotted to represent each single biological replicate.
Fig. 2.
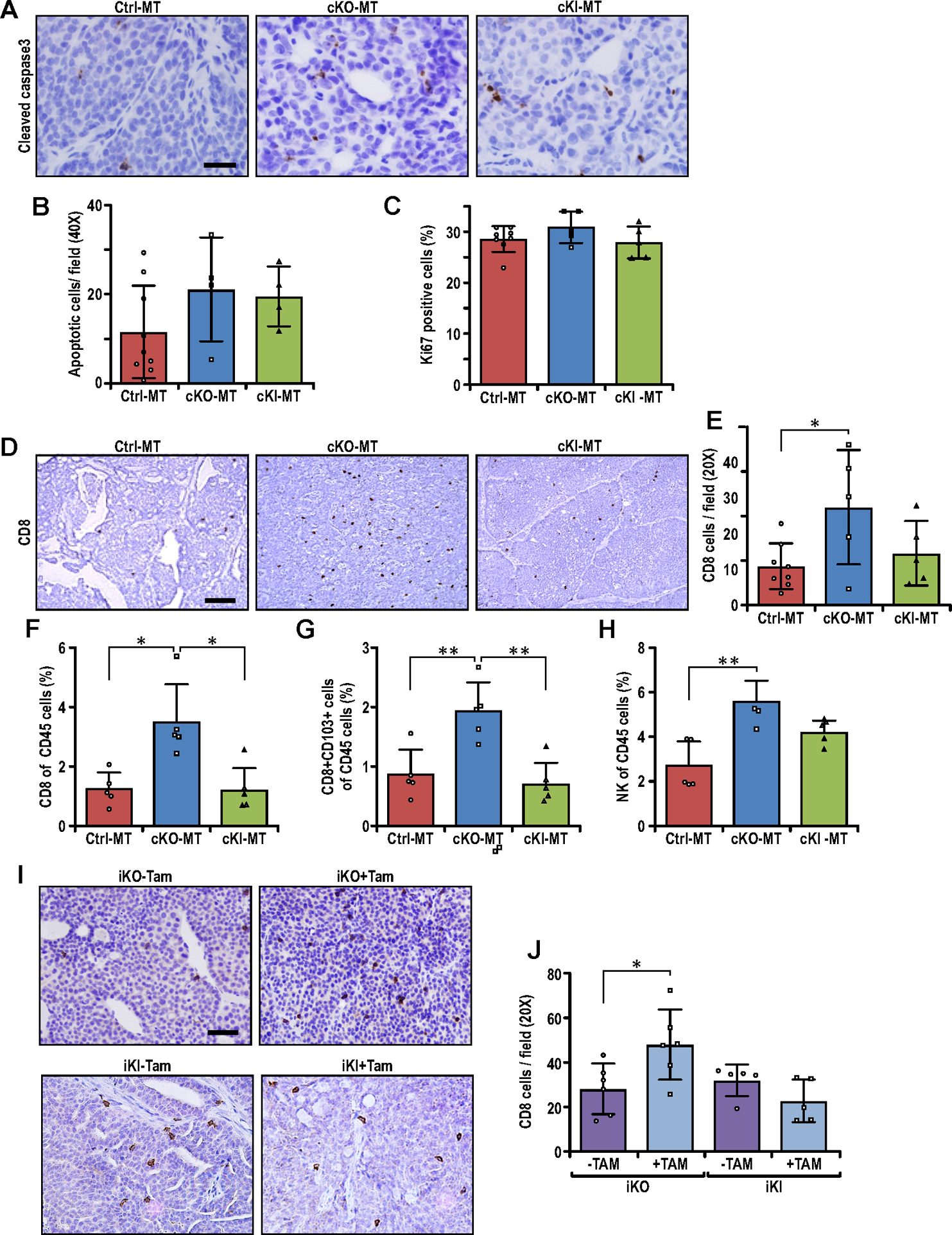
Non-canonical autophagy function of FIP200 is responsible for suppressing CD8+ T-cell infiltration in breast cancer. (A) Representative images of Ctrl-MT, cKO-MT and cKI-MT tumors immuno-stained for cleaved-caspase-3. Scale bar represents 50μm. (B–C) Bar charts showing quantification of (B) number of cleaved-caspase-3 positive cells per field of view (Ctrl-MT; n=9, cKO-MT; n=4, cKI-MT; n=4 mice) and (C) number of Ki67 positive cells per field of view (Ctrl-MT; n=8, cKO-MT; n=5, cKI-MT; n=5 mice). ANOVA with Tukey’s post-hoc test was used for figure 2B whereas Kruskal-Wallis test followed by Dunn’s post-hoc test was used for figure 2C. (D) Representative images of Ctrl-MT, cKO-MT and cKI-MT tumors immuno-stained for CD8. Scale bar represents 100μm. (E) Bar chart showing quantification of CD8 positive cells per field of view (Ctrl-MT; n=11, cKO-MT; n=5, cKI-MT; n=5 mice). ANOVA with Tukey’s post-hoc test was used. (F–H) Flow cytometry analysis of (F) CD8+ T cells, (G) CD8+ CD103+ T cells and (H) CD49B+ NK cells as a percentage of CD45+ cells in Ctrl-MT (n=5), cKO-MT (n=5) and cKI-MT (n=5) tumors. Kruskal-Wallis test followed by Dunn’s post-hoc test was used for figures 2F and 2H whereas ANOVA with Tukey’s post-hoc test was used for figure 2G. (I) Representative images of iKO (−Tam), iKO (+Tam), iKI (−Tam) and iKI (+Tam) tumors immuno-stained for CD8. Scale bar represents 50μm. (J) Bar chart showing quantification of CD8 positive cells per field of view in iKO and iKI tumors (iKO-Tam; n=6, iKO+Tam; n=6, iKI-Tam; n=5, iKI+Tam; n=5 mice). Statistical significance was determined by unpaired t-test with Welch’s correction. * denotes p≤0.05, ** denotes p≤0.01 and *** denotes p≤0.001.
Fig. 3.
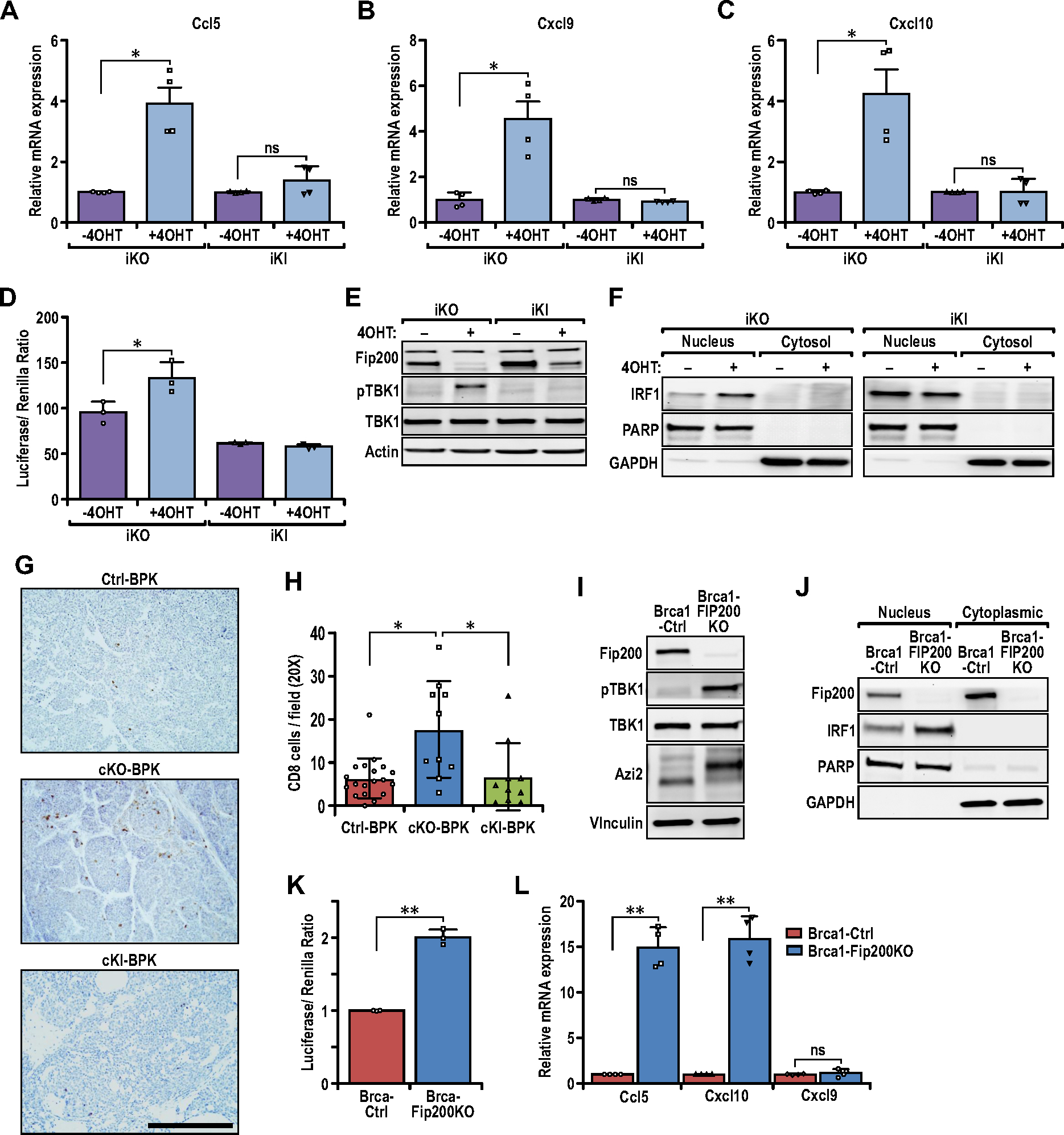
Loss of FIP200’s non-autophagy function activates the TBK1-IRF-IFN signaling axis for pro-inflammatory chemokine expression. (A–C) Bar charts showing the relative transcript levels of (A) Ccl5, (B) Cxcl9, and (C) Cxcl10 in iKO (−4OHT), iKO (+4OHT), iKI (−4OHT) and iKI (+4OHT) tumor cells, quantified via qRT-PCR (n=4 for each sample). Statistical significance was determined by unpaired t-test with Welch’s correction. (D) Bar charts showing the luciferase/Renilla luminescence ratio for iKO and iKI cells transfected with the ISG56-reporter plasmid (n=3 for each sample). Statistical significance was determined by unpaired t-test with Welch’s correction. (E) Immunoblots showing the levels of FIP200, p-TBK1, TBK1 and Actin in iKO (−4OHT), iKO (+4OHT), iKI (−4OHT) and iKI (+4OHT) tumor cells. (F) Immunoblots showing the levels of IRF1, PARP and GAPDH in nuclear and cytoplasmic extracts of iKO and iKI cells. (G) Representative images of Ctrl-BPK, cKO-BPK and cKI-BPK tumors immuno-stained for CD8. Scale bar represents 200μm. (H) Bar chart showing quantification of CD8 positive cells per field of view (Ctrl-BPK; n=19, cKO-BPK; n=10, cKI-BPK; n=10 mice). Kruskal-Wallis test followed by Dunn’s post-hoc test was used. (I) Immunoblots showing the levels of FIP200, p-TBK1, TBK1, AZI2 and Vinculin in BRCA1-Ctrl and BRCA1-FIP200KO tumor cells. (J) Immunoblots showing the levels of FIP200, IRF1, PARP and GAPDH in nuclear and cytoplasmic extracts of BRCA1-Ctrl and BRCA1-FIP200KO tumor cells. (K) Bar charts showing the luciferase/Renilla luminescence ratio for BRCA1-Ctrl and BRCA1-FIP200KO cells transfected with the ISG56-reporter plasmid (n= 3 for each sample). Statistical significance was determined by unpaired t-test with Welch’s correction. (L) Bar charts showing the relative transcript levels of Ccl5, Cxcl9 and Cxcl10 in BRCA1-Ctrl and BRCA1-FIP200KO tumor cells, quantified via qRT-PCR (n=4 for each sample). Statistical significance was determined by unpaired t-test with Welch’s correction.
Fig. 4.
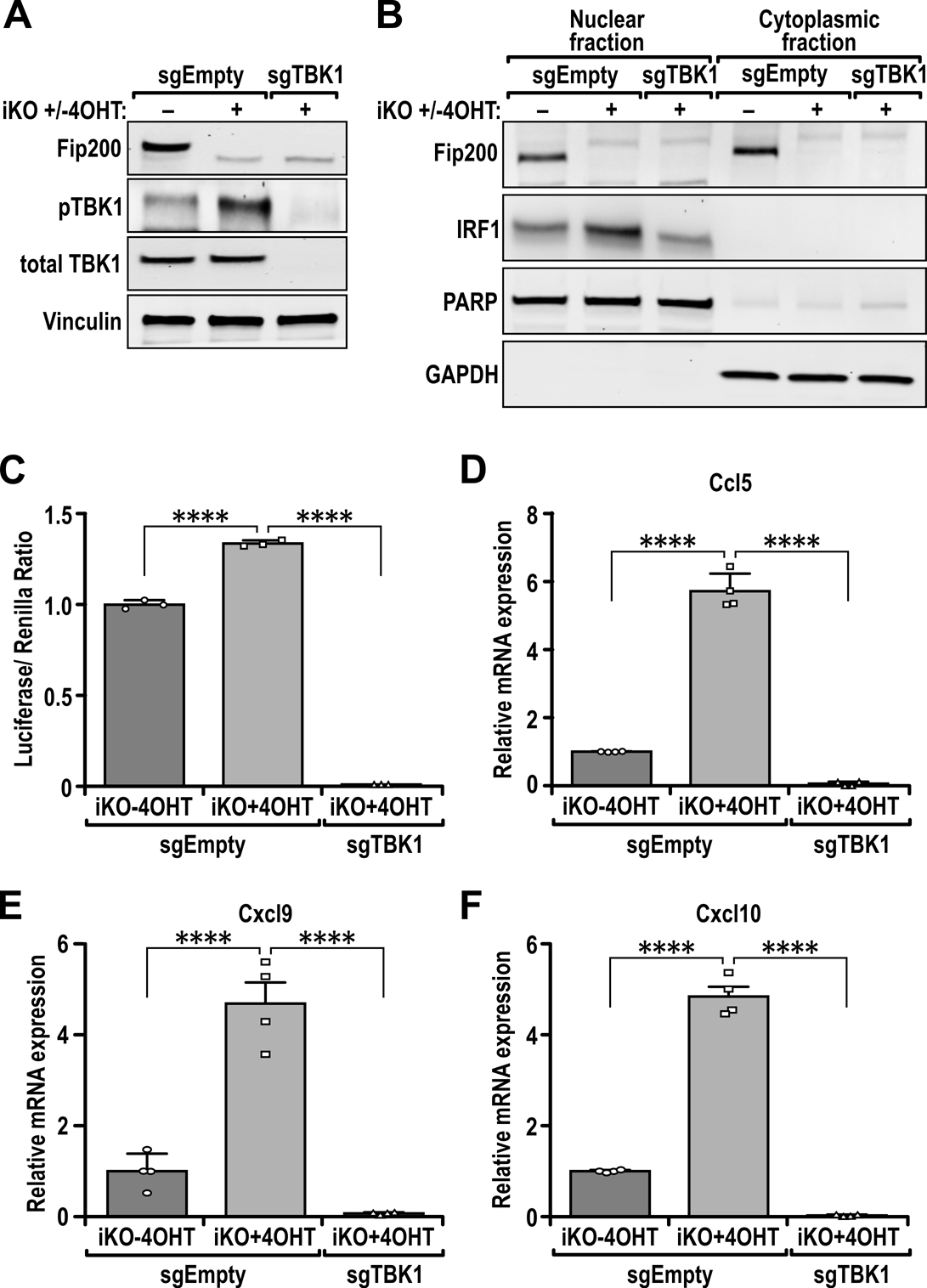
TBK1 is required for increased IRF1 nuclear localization, ISG56-reporter activity and expression of chemokines. (A) Immunoblots showing the levels of FIP200, p-TBK1, TBK1 and Vinculin in iKO (−4OHT), iKO (+4OHT) [FIP200 KO] and iKO (+4OHT +sgTBK1) [FIP200/TBK1 2KO] cells. (B) Immunoblots showing the levels of FIP200, IRF1, PARP and GAPDH in nuclear and cytoplasmic extracts of iKO (−4OHT), iKO (+4OHT) [FIP200 KO] and iKO (+4OHT +sgTBK1) [FIP200/TBK1 2KO] cells. (C) Bar charts showing the luciferase/Renilla luminescence ratio for iKO (−4OHT), iKO (+4OHT) [FIP200 KO] and iKO (+4OHT +sgTBK1) [FIP200/TBK1 2KO] cells transfected with the ISG56-reporter plasmid (n= 3 for each sample). (D–F) Bar charts showing the relative transcript levels of (D) Ccl5, (E) Cxcl9 and (F) Cxcl10 in iKO (−4OHT), iKO (+4OHT) [FIP200 KO] and iKO (+4OHT +sgTBK1) [FIP200/TBK1 2KO] tumor cells, quantified via qRT-PCR (n=4 for each sample). Statistical significance was determined by ANOVA with Tukey’s post-hoc test for figures 4C-F , *** denotes p≤0.001 and **** denotes p≤0.0001.
Fig. 1.
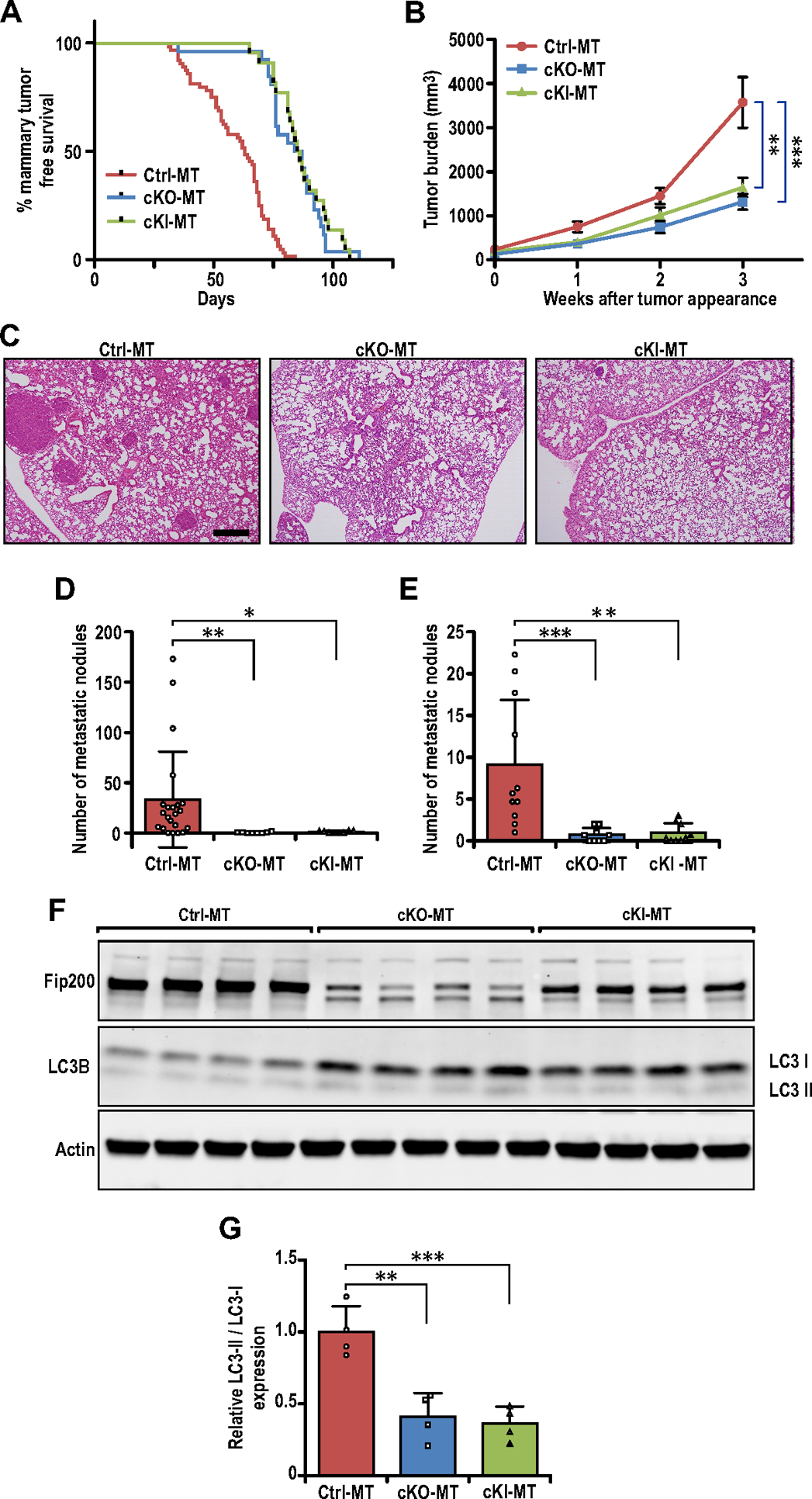
Specific targeting of FIP200’s autophagy function inhibits breast cancer development and metastasis. (A) Tumor free survival curves of Ctrl-MT (n=64 mice), cKO-MT (n=22 mice) and cKI-MT (n=26 mice) cohorts. (B) Tumor growth curves of Ctrl-MT (n=30), cKO-MT (n=12) and cKI-MT (n=14) mice. (C) Representative images for lung sections from Ctrl-MT, cKO-MT and cKI-MT mice. Scale bar represents 500μm. (D) Bar chart showing quantification for number of metastatic nodules per field of view (Ctrl-MT; n=22 mice, cKO-MT; n= 8 mice, cKI-MT n=8), when compared at the same final timepoint. Kruskal-Wallis test followed by Dunn’s post-hoc test was used. (E) Bar chart showing quantification for number of metastatic nodules per field of view (Ctrl-MT; n=11mice, cKO-MT; n=9 mice, cKI-MT; n=9 mice), when lung sections from mice bearing similar tumor loads were compared. Kruskal-Wallis test followed by Dunn’s post-hoc test was used. (F) Immuno-blots showing levels of FIP200, LC3B and β-Actin in Ctrl-MT, cKO-MT and cKI-MT tumors (n=4 for each sample). (G) Bar charts showing quantification of relative LC3-II/LC3-I expression levels. Statistical significance was determined by ANOVA with Tukey’s post-hoc test. * denotes p≤0.05, ** denotes p≤0.01 and *** denotes p≤0.001.
Results
Specific targeting of FIP200’s autophagy function inhibits breast cancer development and metastasis
To investigate the specific role of FIP200’s autophagy function in breast cancer in vivo, we first created mammary epithelial-specific FIP200 knock-in mutant mice in the PyMT breast cancer model (FIP200f/KI;MMTV-Cre;MMTV-PyMT, designated as cKI-MT mice) by crossing our recently developed FIP200+/KI and FIP200f/+;MMTV-Cre;MMTV-PyMT mice (25,27). Mammary epithelial cells in cKI-MT mice express only the FIP200-4A mutant from the KI allele (floxed allele deleted by MMTV-Cre), which specifically abrogate the autophagy function of FIP200 in these cells by disrupting its interaction with Atg13 (27). Mammary tumor development in these mice was monitored by physical palpation as described previously (25,37). cKI-MT mice showed significantly suppressed tumor development compared to littermate controls (Ctrl-MT mice) and to a similar extent as that of cKO-MT mice described previously (25)(Fig. 1A), suggesting that specific blockade of FIP200’s autophagy function is sufficient to inhibit mammary tumor formation in vivo. Blocking FIP200’s autophagy function specifically also inhibited mammary tumor growth (Fig. 1B), as well as metastasis to the lungs (Figs. 1C and 1D) in cKI-MT mice to a similar extent as cKO-MT mice, when mice were analyzed 3 weeks after tumor appearance. We also examined the number of metastatic nodules for the different mice with comparable primary tumor size (tumor burden of ~1500mm3) and found significantly reduced number of nodules in both cKI-MT and cKO-MT mice (Fig. 1E), indicating that decreased metastasis upon ablating autophagy function of FIP200 was not simply due to the reduced tumor growth in these mice vs Ctrl-MT mice.
We next prepared lysates from multiple primary tumors in different mice and analyzed by Western blotting for FIP200 expression and autophagy in tumor cells. Fig. 1F shows significantly reduced expression of FIP200 in the tumors from cKO-MT mice, as expected, and a slightly decreased level of FIP200 in tumors from cKI-MT mice (i.e. FIP200-4A mutant protein encoded by the KI allele), possibly caused by the loss of the floxed allele of FIP200. Measurements of LC3-II/LC3-I ratios provided validation for autophagy blockage upon deletion of FIP200 in cKO-MT or loss of its autophagy function in cKI-MT tumors (Figs. 1F and 1G), as expected. Together, these results suggest that the decreased breast cancer development and metastasis upon FIP200 deletion is due to specific loss of its autophagy function rather than its non-autophagy functions.
Non-autophagy function of FIP200 is responsible for suppressing CD8+ T-cell infiltration in breast cancer
Our previous studies suggested that FIP200 deletion inhibited mammary tumorigenesis through both tumor cell-intrinsic mechanisms and increased infiltration of CD8+ effector T cells to enhance immune surveillance in cKO-MT mice (25). To evaluate whether either of these activities are mediated by autophagy and non-autophagy functions of FIP200, we examined tumor sections from cKI-MT mice in comparison with those from Ctrl-MT and cKO-MT mice. A trend of increased apoptosis was observed for both cKI-MT and cKO-MT tumors (Figs. 2A–2B), but proliferation rates were comparable to Ctrl-MT as measured by Ki67 staining (Fig. 2C). In contrast to these similar tumor cell intrinsic activities, we found that loss of the autophagy function of FIP200 in cKI-MT tumors was not sufficient to increase CD8+ TILs, but only total ablation of FIP200 in cKO-MT mice led to increased CD8+ TILs (Figs. 2D and 2E). Furthermore, analysis of immune cell populations by flow cytometry verified that cKO-MT, but not cKI-MT tumors, had increased levels of CD8+ T cells (Fig. 2F), and also revealed more specifically increased CD8+ CD103+ tissue resident memory T-cells (Fig. 2G), relative to Ctrl-MT tumors. The increase in cytotoxic T cells were also accompanied by higher levels of CD49b+ natural killer (NK) cells in cKO-MT but not cKI-MT tumors (Fig. 2H). Overall, these results indicated that elevated anti-tumor-immune responses was a consequence of tumors that lack FIP200.
To further elucidate the mechanisms of increased CD8+ T-cell infiltration upon the loss of FIP200’s non-autophagy functions, we prepared mammary tumor cells from FIP200f/KI;MMTV-PyMT mice (lacking MMTV-Cre) and infected them with MSCV.CreERT2, to allow for inducible deletion of FIP200, as described previously (38). The resulting FIP200f/KI;MMTV-PyMT:CreER (designated as iKI) and the previously described FIP200f/f;MMTV-PyMT:CreER (designated as iKO)(38) tumor cells were transplanted into syngeneic FVB recipient mice, and the recipient mice were then treated with or without tamoxifen (TAM) to induce Cre expression and consequent deletion of the floxed FIP200 allele in these cells. FIP200 deletion in the established tumors (i.e. mice with iKO cell transplant + TAM) led to increased CD8+ T cell infiltration (compared to the same mice - TAM), whereas blocking FIP200 autophagy function (i.e. mice with iKI cell transplant + TAM) did not show significant increase in CD8+ T cell infiltration (Figs. 2I and 2J). These results are consistent with our observations in cKO-MT and cKI-MT mice, and together, they suggest that FIP200’s non-canonical autophagy functions (lost in FIP200 deletion but not FIP200-4A mutation), but not its canonical autophagy function (lost in both), is required for suppressing CD8+ T-cell infiltration in breast cancer.
Loss of the non-autophagy function of FIP200 activates the TBK1-IRF-IFN signaling axis for pro-inflammatory chemokine expression
Our previous studies showed that ablation of FIP200 in cKO-MT tumors can lead to increased expression of IFN-responsive genes, including several chemokines such as Ccl5, Cxcl9, and Cxcl10 (25). Such changes are likely responsible for the increased CD8+ TILs in FIP200-null tumor cells in both cKO-MT mice ((25), Figs. 2D and 2E) as well as recipient mice with iKO transplant following Tam (see Figs. 2I and 2J), as these chemokines have been associated with increased recruitment of TILs (39). To evaluate this notion further, we examined their expression in iKO and iKI tumor cells with or without 4OHT, the active metabolite of tamoxifen, for inducing deletion in these cells in vitro. Similar to tumor cells in cKO-MT mice, we found that FIP200 deletion increased expression of Ccl5, Cxcl9, and Cxcl10 in iKO cells (Figs. 3A–3C). In contrast, FIP200-4A mutation (i.e. iKI cells + 4OHT) led to a much smaller increase for Ccl5, and Cxcl10, and no difference for Cxcl9, despite similar levels of autophagy inhibition in both iKI and iKO cells after 4OHT treatment (Fig. S1). Moreover, 4OHT treatment of iKO cells to delete FIP200, but not iKI cells for FIP200-4A mutation, increased IRF transcriptional activity relative to their respective controls (i.e. vehicle treated cells) in ISG56 promoter-reporter assays (Fig. 3D). These results suggest that up-regulation of these chemokines after the loss of non-canonical autophagy functions of FIP200 (but not loss of its canonical autophagy function) is responsible for the increased CD8+ TILs.
We next examined the levels of phosphorylated TBK1 in iKO and iKI cells with or without 4OHT treatment, as FIP200 has been implicated in the regulation of TBK1, a key node in IFN-signaling (40). TBK1 phosphorylation increased specifically in iKO cells, but not iKI cells, following 4OHT treatment relative to the vehicle-treated, control cells (Fig. 3E). Moreover, we observed increased nuclear localization of interferon regulatory factor 1 (IRF1) in iKO cells, but not iKI cells, after 4OHT treatment (Fig. 3F). Immunohistochemical analysis of PyMT tumors also revealed increased levels of nuclear IRF1 in cKO-MT but not cKI-MT tumors relative to Ctrl-MT tumors (Figs. S2A–S2B). Importantly, the regulation of CD8+ TILs by FIP200 can be recapitulated in a mouse model of BRCA1-null basal-like breast cancer; Brca1F/F;Trp53F/F;K14-Cre driven tumors (Ctrl-BPK tumors) (Fig. 3G). We observed that conditional deletion of FIP200 (cKO) increased CD8+ TIL infiltration in cKO-BPK mammary tumors, whereas mammary tumors expressing an autophagy-deficient mutant knock-in allele (cKI-BPK), did not exhibit any significant increase in CD8+ TILs (Fig. 3G–3H). Ablation of FIP200 by CRISPR-CAS9 mediated targeting in BPK tumor cells also led to increased phosphorylation of TBK1 (Fig. 3I) and increased nuclear localization of IRF1 (Fig. 3J) in BRCA1-FIP200KO (transduced with sgFIP200) cells relative to BRCA1-Ctrl cells (transduced with sgEmpty). Concomitantly, increased ISG56-luciferase reporter activity (Fig.3K) and increased expression of Ccl5 and Cxcl10 was also observed in these cells (Fig. 3L). This indicates that the negative regulation of TBK1-IRF signaling and tumor immune responses by FIP200 occurs in more than one breast cancer subtype.
To further determine whether the increased IRF activity and expression of pro-inflammatory cytokines upon ablation of FIP200 were a consequence of increased TBK1 activation, we genetically ablated TBK1 in 4OHT-treated iKO cells (i.e. FIP200 KO tumor cells) by CRISPR-CAS9 to generate FIP200/TBK1 2KO cells. We found that TBK1 KO in FIP200 KO tumor cells (Fig. 4A) reversed the increase in nuclear IRF1 levels by FIP200 ablation to a similar level as in vehicle-treated iKO cells (Fig. 4B). Accordingly, there was also less IRF transcription factor activity in FIP200/TBK1 2KO cells, as determined by ISG56-reporter assays (Fig. 4C). Further, the increased expression of Ccl5, Cxcl9, and Cxcl10 in FIP200 KO tumor cells relative to Ctrl tumor cells were also diminished in FIP200/TBK1 2KO cells (Figs. 4D–4F). We also attempted to determine the effects of TBK1 KO on CD8+ TIL infiltration but iKO cells depleted of TBK1 did not form any tumors. It is possible that TBK1 is required for the survival of cells, especially in vivo (41,42) and we observed increased cleaved caspase-3 levels upon TBK1 knockout (Fig. S3A) but the effects on cell growth in vitro were not as prominent (Fig. S3B). Alternatively, we have transplanted iKO (+Tam) tumors and treated them with the TBK1 inhibitor, amlexanox, and this led to a trend in decreased CD8 TILs in iKO (+Tam) tumors (Figs. S3C–D). Together, these results suggest that FIP200 suppresses TBK1 phosphorylation and consequent up-regulation of IFN-signaling through its non-canonical autophagy functions, and loss of which triggers the TBK1-IRF-IFN signaling for CD8+ TIL recruitment in mammary tumors.
FIP200 interacts with and regulates the phosphorylation of AZI2
We next sought to determine the potential molecular mechanisms for the increased TBK1 phosphorylation upon FIP200 deletion in tumor cells. Although FIP200 has been previously reported to co-immunoprecipitate with TBK1 (43), we were unable to confirm this interaction under the conditions we tested. Nonetheless, another previous study identified FIP200 as an interactor of SINTBAD/TBKBP1 (32), which is a known adaptor protein important in the regulation of TBK1 (44,45). Hence, we set out to evaluate the potential interactions between FIP200 and several different TBK1 adaptor proteins, including SINTBAD, AZI2/NAP1 and TANK. Indeed, we could verify the interaction between HA/FLAG-SINTBAD and MYC-FIP200, but interestingly, we also found that FIP200 could interact with AZI2 (Fig. 5A). This is also true for the reciprocal IP with immuno-precipitation of FLAG tag (Fig. S4A). Despite the ability of FIP200 to interact with AZI2 and SINTBAD, we were not able to co-IP FIP200 with TANK (Fig. 5A, Fig. S4A), suggesting that there is specificity in the regulation of TBK1 adaptors by FIP200. To delineate the contribution of TBK1 adaptor proteins in the activation of TBK1 upon FIP200 ablation, we utilized siRNAs against TBK1 adaptor proteins to silent AZI2, TANK and SINTBAD independently in MDA-MB-231 cells that were transduced with sgEmpty or sgFIP200 (Fig. 5B). Similar to iKO cells following 4OHT treatment (see Fig. 3E), FIP200 KO increased TBK1 phosphorylation in MDA-MB-231 (Fig.5B) and MCF7 (Fig. S4B) human breast cancer cells. Importantly, silencing of AZI2 was most effective in diminishing p-TBK1 (Fig. 5B), indicating that FIP200 regulates TBK1 activation largely in part through AZI2. The ablation of AZI2 in iKO (+4OHT) cells could also abolish TBK1 activation upon loss of FIP200 (Fig. 5C) and reduced IRF transcriptional activity, measured through ISG56-reporter assays (Fig. S5A). However, the interaction between AZI2 and TBK1 was not increased in cells that were depleted of FIP200. This suggests that FIP200 does not compete with TBK1 for binding to AZI2, as a mechanism of inhibition (Fig. S5B).
Fig. 5.
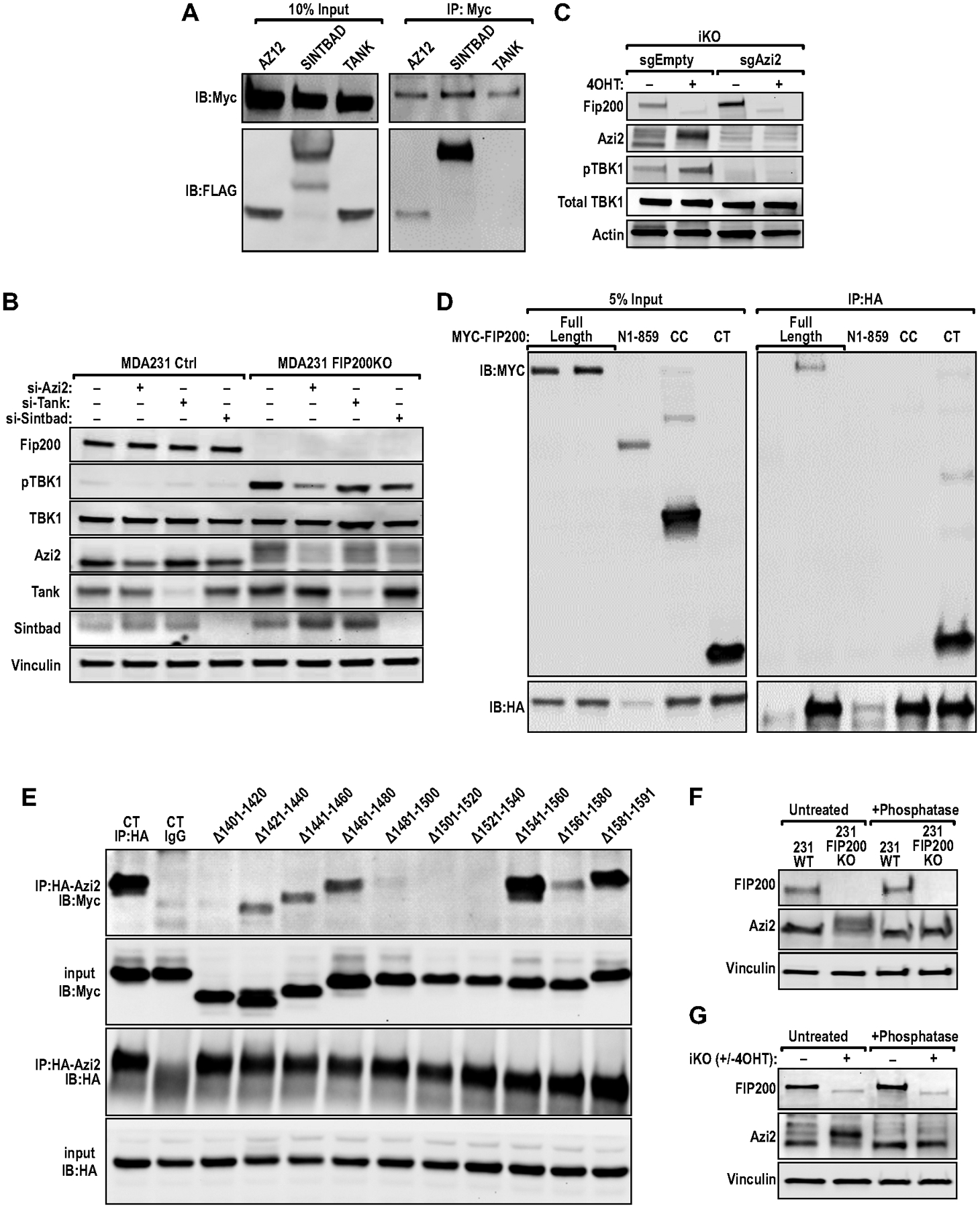
FIP200 interacts with and regulates the phosphorylation of AZI2. (A) Immunoblots of co-IP experiments with MYC antibody, in HEK293 cells transfected with either HA/FLAG-AZI2, HA/FLAG-SINTBAD or FLAG-TANK along with MYC-FIP200. (B) Immunoblots showing the levels of FIP200, p-TBK1, TBK1, AZI2, TANK, SINTBAD and Vinculin in MDA-MB-231 Ctrl (sgEmpty) and MDA-MB-231 FIP200KO (sgFIP200) cells transfected with indicated siRNAs against AZI2, TANK or SINTBAD. (C) Immunoblots showing the levels of FIP200, AZI2, p-TBK1, TBK1 and Actin in iKO sgEmpty and iKO sgAZI2 cells, ±4OHT treatment. (D–E) Immunoblots of co-IP experiments with HA antibody, in HEK293 cells transfected with MYC-FIP200 (D) fragment or (E) deletion mutant constructs and HA/FLAG-AZI2. (F–G) Immunoblots showing levels of FIP200, AZI2 and Vinculin in (F) MDA-MB-231 or (G) iKO cells ±phosphatase treatment.
To characterize the interaction between FIP200 andAZI2 with more precision, we analyzed a series of FIP200 segments for interactions with AZI2. We found that the FIP200 C-terminal region (CT: residues 1401–1591) as well as full length FIP200, but not the N-terminal half (N859: residues 1–859) or the coiled coil domain (CC: residues 860–1373), bound to AZI2 (Fig. 5D). We then prepared another series of Myc-tagged FIP200 CT, each deleting 20 residues within 1401–1591 (i.e. 1401–1420, 1421–1440, etc). These constructs were co-transfected with HA-AZI2 into HEK-293T cells to detect association by co-immunoprecipitation. We found that mutations lacking residues between 1401–1420, 1481–1540 or 1561–1580 showed significantly reduced binding to AZI2, relative to the control construct FIP200 CT (Fig. 5E), suggesting residues within these regions may mediate FIP200 interaction with AZI2. Interestingly, we also observed that deletion of FIP200 resulted in a band-shift for AZI2 (slower migrating band) in MDA-MB-231 cells (Fig. 5F), and this band-shift could be eliminated by phosphatase treatment. The same shift was also found in iKO and BPK cells upon FIP200 deletion (Fig. 5G, 3I) suggesting that the regulation of AZI2 phosphorylation by FIP200 is conserved between mouse and human breast cancer cells. Together, our results suggest the interesting possibility that non-canonical autophagy functions of FIP200 negatively regulates TBK1 through its interaction with AZI2.
Ablation of FIP200 enhanced the efficacy of immune checkpoint inhibitor therapy in non-responsive mammary tumors
As increased CD8+ TIL density and activation of IFN-signaling have been reported to be associated with better ICI responses in various cancers (5,28,46,47), hence we posit that specifically targeting the non-canonical autophagy function of FIP200 will improve ICI efficacy. In order to dissect more specifically the canonical autophagy and non-canonical autophagy roles of FIP200 in regulating enhanced responses to ICI therapy, we leveraged upon the iKI and iKO cells (Figs. 2J–K) that can be induced by tamoxifen in vivo. These cells were respectively transplanted into syngeneic FvB/N hosts and when tumors reached about 5mm in diameter (Day 0), they were randomized into groups treated with 3 consecutive daily doses of tamoxifen to induce deletion of the floxed FIP200 allele or treated with corn oil as vehicle controls (Fig. 6A). Consequently, vehicle treated mice (−Tam) and tamoxifen treated mice (+Tam) were further randomized into cohorts that were treated either with isotype control antibody (IgG), anti-PD1 antibody (PD1), anti-CTLA4 antibody (CTLA4) or the combination of both anti-PD1 and anti-CTLA4 (ICI-Combo) (Fig. 6A). By comparing the growth curves of iKI (−Tam) and iKI (+Tam) tumors, it is apparent that loss of the autophagy function of FIP200 retards tumor growth (Fig. 6B). However, the progression of iKI (+Tam) tumors were not affected by anti-PD1 therapy relative to IgG treated controls and the same is true for iKI (−Tam) tumors (Fig. 6B–C). Due to the lack of treatment response upon anti-PD1 therapy in iKI (−Tam) and iKI (+Tam) tumors, anti-CTLA4 and ICI-Combo regiments were not tested further in these cohorts. These results suggest that the canonical autophagy function of FIP200 is not responsible for suppressing effective responses to anti-PD1 therapy.
Fig. 6.
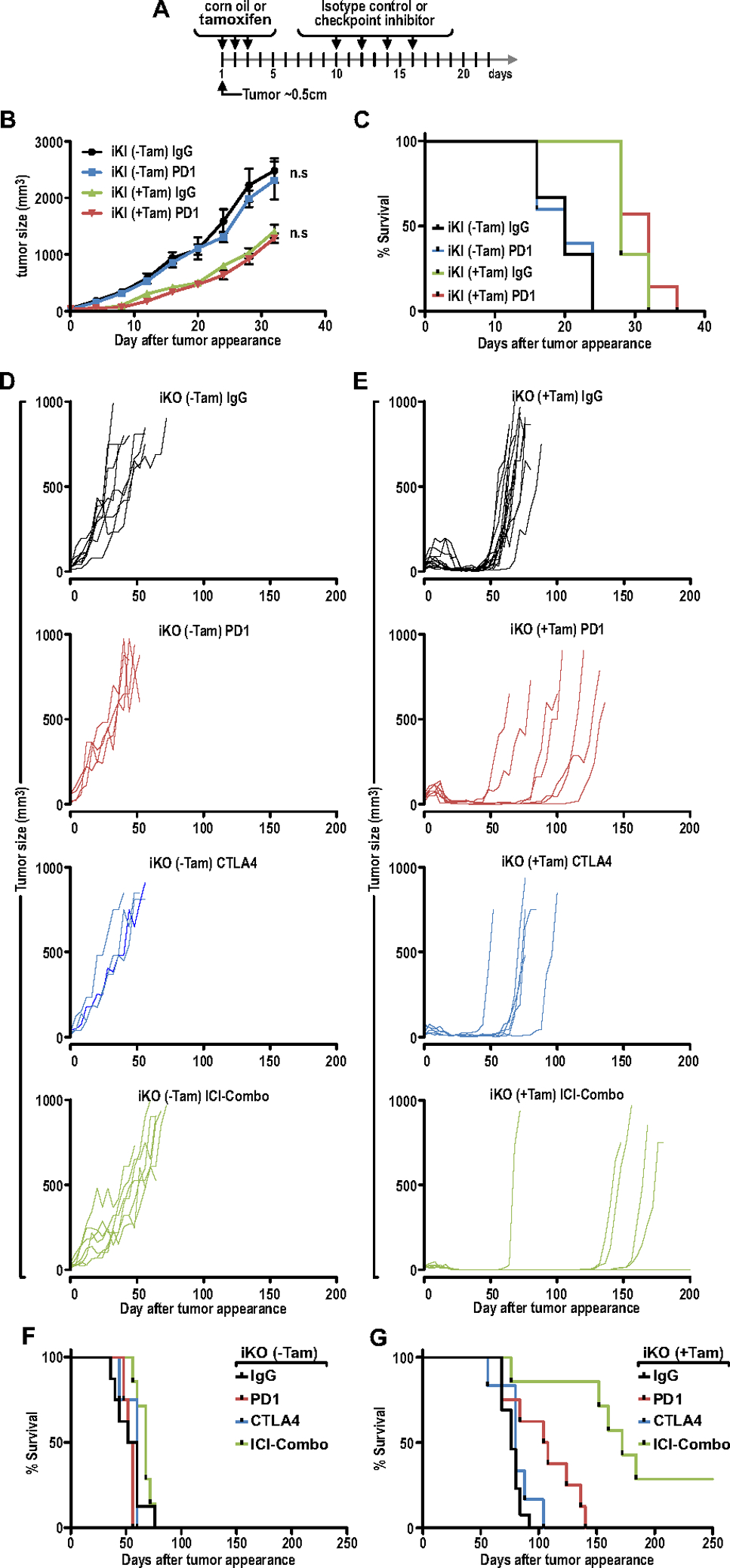
Ablation of FIP200 enhanced the efficacy of immune checkpoint inhibitor therapy in non-responsive mammary tumors. (A) Treatment regimen for transplanted iKO or iKI cells into syngeneic FVB/N mice. Mice cohorts were treated with vehicle or tamoxifen to induce deletion when tumors reached ~50mm3. A week later, IgG control, anti-PD1, anti-CTLA4 or ICI-Combo therapy was administered (8mg/kg I.P every other day; 4 doses). (B) Tumor growth curves for iKI (±Tam) tumors treated with IgG or anti-PD1. (C) Survival plots for iKI (±Tam) tumors treated with IgG or anti-PD1. (iKI-Tam IgG; n=6, iKI-Tam PD1; n=5, iKI+Tam IgG; n=6, iKI+Tam PD1; n=7) (D) Tumor growth curves for iKO (−Tam) tumors treated with IgG, anti-PD1, anti-CTLA4 or ICI-Combo therapy (IgG; n=8, PD1; n=4, CTLA4; n=4, ICI-Combo; n=7). (E) Tumor growth and relapse curves for iKO (+Tam) tumors treated with IgG, anti-PD1, anti-CTLA4 or ICI-Combo therapy (IgG; n=14, PD1; n=7, CTLA4; n=6, ICI-Combo; n=7). (F–G) Survival plots for (F) iKO (−Tam) tumors and (G) iKO (+Tam) tumors treated with IgG, anti-PD1, anti-CTLA4 or ICI-Combo therapy.
Consistent with a lack of response to ICI therapy and similar to iKI (−Tam) tumors, PD1, CTLA4 or ICI-Combo treatment had negligible effects on tumor growth relative to IgG treated tumors for iKO (−Tam) tumors (Figs. 6D, 6F). In contrast, all IgG, single ICI and combination ICI treated iKO (+Tam) tumors regressed upon administration of tamoxifen (Fig. 6E), indicating that FIP200 contributes to tumor growth, as observed previously (38). However, iKO (+Tam) tumors that were treated with IgG relapsed and reached tumor endpoints after a median of about 76 days (Figs. 6E and 6G). On the other hand, iKO (+Tam) tumors treated with anti-PD1 or anti-CTLA4 had an extended median survival of 106 and 80 days respectively (Figs. 6E and 6G). Intriguingly, although the combination of anti-PD1 and anti-CTLA4 therapy had no significant effect on the progression of iKO (−Tam) tumors (Fig. 6F), 2/7 (28.8%) of iKO (+Tam) tumors that were treated with the ICI combination regressed and remained progression-free for up to 250 days after tumor appearance (Figs. 6E and 6G). Together, these results demonstrate that blocking FIP200’s non-autophagy functions, but not the loss of its autophagy function, led to activation of TBK1-IRF-IFN signaling, CD8+ TIL infiltration and enhanced the responses of ICI therapy in mammary tumors.
Discussion
In this study, we have provided genetic proof of principle that targeting FIP200 can improve the efficacy of combination ICI therapy. Such a strategy is vital and imperative in breast cancers because the majority of breast cancers (especially luminal subtypes) do not respond to ICI therapy (6). This was evidently illustrated in our study, where anti-PD1 in combination with anti-CTLA4 had no significant effect on the growth of Ctrl-MT tumors, which are classified as luminal B tumors. Despite the ineffectiveness of combination ICI therapy on Ctrl-MT tumors, ablation of FIP200 was able to induce tumor regression and remarkably, the administration of combination ICI therapy in tumors lacking FIP200 could prevent relapse and elicit prolonged durable responses. Notably, the median tumor-free survival for cKO-MT tumors treated with combination ICI therapy were improved significantly and 28.8% of mice in this cohort remained tumor-free for up to 250 days. It is worth noting that this is a relatively long duration of follow-up and highlights the superior durable responses that can be achieved with this combination. These results illustrate the promising potential of targeting FIP200 as a means to sensitize non-responsive luminal breast tumors to ICI therapy and this may allow a larger breast cancer patient cohort to reap the benefits of immunotherapy.
As FIP200 is a large protein (~200kDa), it has multiple domains that can function independently to coordinate various cellular pathways (48). Among these, FIP200 has been identified as a key regulator of autophagy (10) that is required in the initiation complex, along with ULK1, ATG13 and ATG101. In order to specifically dissect its autophagy functions, we have recently developed mice with a knock-in allele of FIP200 that is unable to bind ATG13 and initiate autophagy (27). Through the use of this canonical autophagy-deficient allele of FIP200, we were able to dissect specifically for the first time in a mouse model of breast cancer that FIP200’s autophagy function promoted the initiation, growth and metastasis of PyMT driven mammary tumors. These observations are consistent with our previous studies demonstrating a pro-tumorigenic role of FIP200 in breast cancer cells (27,34,35,49), but more specifically, provides justification for the inhibition of autophagy as a therapeutic strategy in breast cancer. Importantly, these observations have clinical implications, since autophagy inhibitors such as hydroxychloroquine are being tested in a number of clinical trials (50).
In addition to the role of FIP200 in canonical autophagy, our findings also demonstrated that FIP200 can regulate the activation of TBK1, a key player in the induction of IFN signaling (32). The negative regulation of TBK1 by FIP200 was not dependent on the ability of FIP200 to interact with ATG13, suggesting that this regulation does not involve the canonical autophagy function of FIP200. This is in line with a study that showed TBK1 activation was increased upon ablation of FIP200, and that the activation of TBK1 in these cells were important for ferritin turnover that was autophagy-independent (40). However, Goodwin et al. also observed TBK1 activation upon ULK1/2 or ATG13 ablation, whereas disrupting FIP200-ATG13 binding in our iKI (+4OHT) cells did not induce pronounced TBK1 activation. These findings may suggest that the negative regulation of TBK1 occurs through members of the ULK1 complex but does not require the interaction between FIP200 and ATG13 (i.e. distinct complex conformation despite the same complex members being involved). It is also worth noting that TBK1 itself is involved in autophagy and phosphorylates cargo receptors such as p62, NDP52 and TAX1BP1 to promote cargo binding (51). More recently, TBK1 has also been implicated in the initiation of autophagy, by phosphorylating Syntaxin 17 to promote the formation of the initiation complex (52). Although autophagy has been shown to negatively regulate TBK1 activation by degrading upstream activators such as MAVS (53) and STING (51), in our studies, the regulation of TBK1 by FIP200 occurs through a canonical autophagy-independent manner. It is possible that TBK1 regulation will depend on the sum of signaling inputs that are present within each cellular context, but TBK1 activation was observed upon FIP200 ablation in a wide range of breast cancer cells in this study (i.e. PyMT, BRCA-1 null, MDA-MB-231 and MCF-7 cells).
Activation of the TBK1-IFN signaling axis has emerged as an effective strategy to improve the efficacy of ICIs. Various modalities such as STING agonists (54) or poly(I:C) treatment (28), which converge on TBK1, have proven to be effective in making ICIs more efficacious. Moreover, the loss of IFN pathway genes has been described as a mechanism of resistance to anti-CTLA4 therapy (47). Importantly, we observed a consistent association between improved ICI efficacy, increased CD8+ TILs and TBK1 activity, where these parameters were increased upon ablation of FIP200 but not when the interaction between FIP200 and ATG13 was disrupted. The lack of responses to anti-PD1 therapy and CD8+ T cell infiltration in the cKI model is also consistent with a study which showed that autophagy inhibition either by genetic or pharmacological means did not affect anti-tumor T cell recruitment and responses (13). Hence, our study highlights the need to target the non-canonical autophagy functions of FIP200 to improve ICI efficacy. Furthermore, we also demonstrated that the increased IRF activity and elevated expression of pro-inflammatory cytokines upon FIP200 ablation were dependent on TBK1 activation. As for the recruitment of CD8+ TILs, treatment with amlexanox, an inhibitor of TBK1 and IKKε, could diminish the number of CD8+ TILs (Figs. S3C and S3D). This suggests that the increased TBK1 activation upon FIP200 depletion contributes to this process but the lack of specificity of amlexanox is a limitation of this particular experiment. In addition, we have excluded the involvement of IFNAR1 signaling as a dominant upstream regulator of TBK1 and AZI2 (Fig. S6). Mechanistically, we have discovered that FIP200 can interact with and regulate the phosphorylation of AZI2, a TBK1 adaptor protein. Our independent observations (Figs. 5A–C) are consistent with a recently published study describing AZI2/SINTBAD binding regions within the C-terminus of FIP200 (55). Furthermore, genetic ablation of AZI2 in FIP200-null tumor cells indicated that AZI2 was a major contributor to TBK1 activation in these cells. To our knowledge, there have been no prior reports on the regulation of post-translational modifications of AZI2. Hence, we have unraveled an additional layer of TBK1 regulation that involves FIP200. The specific phosphorylation site(s) of AZI2 that were negatively regulated by FIP200 and its significance for downstream signaling pathways is of great interest and could be exploited for pharmaceutical intervention in future studies.
Importantly, the regulation of pro-inflammatory chemokines that are important for CD8+ TIL infiltration was not limited to mouse mammary tumor cells. In fact, we also observed increased expression of CXCL10 in MDA-MB-231 cells upon FIP200 deletion (Fig. S7). Accordingly, we then examined the TCGA breast cancer cohort for correlations between FIP200 (RB1CC1) expression with TBK1, CCL5, CXCL9 and CXCL10 (Fig. S8A). For the association between FIP200 and TBK1, there was a significant positive correlation (Fig. S8B). However, since our results indicated a change in phospho-TBK1 levels rather than its expression upon FIP200 ablation, the significance of this correlation will need to be investigated in future studies. Coherently, there was an inverse correlation between FIP200 and CCL5 expression (Fig. S8C). This would be in line with our observations that FIP200 limits the expression of chemokines. On the other hand, no significant associations were found between FIP200 levels with CXCL9 or CXCL10 (Fig. S8D–E). It is possible that the lack of association between FIP200 and CXCL9/CXCL10 may be due to confounding factors such as stromal/immune cells expression of these genes in these whole tumor samples.
In summary, we have found that FIP200 plays multiple pro-tumorigenic roles in mammary tumors. This includes a role in autophagy that is important for tumor initiation, growth and metastasis. On the other hand, FIP200 also negatively regulates the AZI2/TBK1/IFN signaling axis (non-autophagy function) that impinges on the expression of pro-inflammatory cytokines, CD8+ T cell infiltration and ICI therapeutic efficacy. These observations provide rationale for targeting these separate functions of FIP200 in concert. Importantly, we have also demonstrated that FIP200 ablation in combination with ICIs can achieve prolonged durable responses in luminal breast tumors, that are otherwise non-responsive to ICI therapy.
Supplementary Material
Significance:
Findings show that deletion of FIP200 enhances immune checkpoint inhibitor efficacy in nonresponsive breast cancer.
Acknowledgements
We would like to thank Nyiva Muyanga for help with genotyping and the University of Cincinnati LAMS staff for their support with regards to mouse colony maintenance and husbandry. We appreciate the help from Glenn Doerman for graphics support and preparation of figures. We are grateful to members of the Guan lab for critical appraisal and suggestions in the preparation of this manuscript. J-L Guan received NIH R01 CA211066, R01 HL073394 and R01 NS094144. S. Chen was supported by NSFC grants (81572877,81773132).
Funding
This research was supported by NIH grants R01 CA211066, R01 HL073394 and R01 NS094144 to J.-L. Guan. S. Chen was supported by NSFC grants (81572877,81773132).
Footnotes
Competing interests
The authors declare no competing interests.
Data Availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data available from authors upon request.
References
- 1.Blankenstein T, Coulie PG, Gilboa E, Jaffee EM. The determinants of tumour immunogenicity. Nat Rev Cancer 2012;12:307–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bedognetti D, Maccalli C, Bader SB, Marincola FM, Seliger B. Checkpoint Inhibitors and Their Application in Breast Cancer . Breast Care (Basel) 2016;11:108–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Topalian SL, Hodi FS, Brahmer JR, Gettinger SN, Smith DC, McDermott DF, et al. Safety, activity, and immune correlates of anti-PD-1 antibody in cancer. N Engl J Med 2012;366:2443–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, et al. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014;32:1020–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Topalian SL, Taube JM, Anders RA, Pardoll DM. Mechanism-driven biomarkers to guide immune checkpoint blockade in cancer therapy. Nat Rev Cancer 2016;16:275–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Emens LA, Kok M, Ojalvo LS. Targeting the programmed cell death-1 pathway in breast and ovarian cancer. Curr Opin Obstet Gynecol 2016;28:142–7 [DOI] [PubMed] [Google Scholar]
- 7.Nanda R, Chow LQ, Dees EC, Berger R, Gupta S, Geva R, et al. Pembrolizumab in Patients With Advanced Triple-Negative Breast Cancer: Phase Ib KEYNOTE-012 Study. J Clin Oncol 2016;34:2460–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dirix L, Takacs I, Nikolinakos P, Jerusalem G, Arkenau H-T, Hamilton E, et al. Abstract S1–04: Avelumab (MSB0010718C), an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: A phase Ib JAVELIN solid tumor trial. Cancer Research 2016;76:S1-04-S1- [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mizushima N, Komatsu M. Autophagy: renovation of cells and tissues. Cell 2011;147:728–41 [DOI] [PubMed] [Google Scholar]
- 10.Hara T, Takamura A, Kishi C, Iemura S, Natsume T, Guan JL, et al. FIP200, a ULK-interacting protein, is required for autophagosome formation in mammalian cells. J Cell Biol 2008;181:497–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Michaud M, Martins I, Sukkurwala AQ, Adjemian S, Ma Y, Pellegatti P, et al. Autophagy-dependent anticancer immune responses induced by chemotherapeutic agents in mice. Science 2011;334:1573–7 [DOI] [PubMed] [Google Scholar]
- 12.Michaud M, Xie X, Bravo-San Pedro JM, Zitvogel L, White E, Kroemer G. An autophagy-dependent anticancer immune response determines the efficacy of melanoma chemotherapy. Oncoimmunology 2014;3:e944047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Starobinets H, Ye J, Broz M, Barry K, Goldsmith J, Marsh T, et al. Antitumor adaptive immunity remains intact following inhibition of autophagy and antimalarial treatment. J Clin Invest 2016;126:4417–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.DeVorkin L, Pavey N, Carleton G, Comber A, Ho C, Lim J, et al. Autophagy Regulation of Metabolism Is Required for CD8. Cell Rep 2019;27:502–13.e5 [DOI] [PubMed] [Google Scholar]
- 15.Lim J, Park H, Heisler J, Maculins T, Roose-Girma M, Xu M, et al. Autophagy regulates inflammatory programmed cell death via turnover of RHIM-domain proteins. Elife 2019;8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cunha LD, Yang M, Carter R, Guy C, Harris L, Crawford JC, et al. LC3-Associated Phagocytosis in Myeloid Cells Promotes Tumor Immune Tolerance. Cell 2018;175:429–41.e16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin PK, Marchiando A, Xu R, Rudensky E, Yeung F, Schuster SL, et al. Autophagy proteins suppress protective type I interferon signalling in response to the murine gut microbiota. Nat Microbiol 2018;3:1131–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Samie M, Lim J, Verschueren E, Baughman JM, Peng I, Wong A, et al. Selective autophagy of the adaptor TRIF regulates innate inflammatory signaling. Nat Immunol 2018;19:246–54 [DOI] [PubMed] [Google Scholar]
- 19.Yang A, Herter-Sprie G, Zhang H, Lin EY, Biancur D, Wang X, et al. Autophagy Sustains Pancreatic Cancer Growth through Both Cell-Autonomous and Nonautonomous Mechanisms. Cancer Discov 2018;8:276–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Poillet-Perez L, Xie X, Zhan L, Yang Y, Sharp DW, Hu ZS, et al. Autophagy maintains tumour growth through circulating arginine. Nature 2018;563:569–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Karsli-Uzunbas G, Guo JY, Price S, Teng X, Laddha SV, Khor S, et al. Autophagy is required for glucose homeostasis and lung tumor maintenance. Cancer Discov 2014;4:914–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Jung H, Leal-Ekman JS, Lu Q, Stappenbeck TS. Atg14 protects the intestinal epithelium from TNF-triggered villus atrophy. Autophagy 2019;15:1990–2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui J, Jin S, Wang RF. The BECN1-USP19 axis plays a role in the crosstalk between autophagy and antiviral immune responses. Autophagy 2016;12:1210–1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang YT, Zaitsev K, Lu Q, Li S, Schaiff WT, Kim KW, et al. Select autophagy genes maintain quiescence of tissue-resident macrophages and increase susceptibility to Listeria monocytogenes. Nat Microbiol 2020 [DOI] [PMC free article] [PubMed]
- 25.Wei H, Wei S, Gan B, Peng X, Zou W, Guan JL. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 2011;25:1510–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gan B, Peng X, Nagy T, Alcaraz A, Gu H, Guan JL. Role of FIP200 in cardiac and liver development and its regulation of TNFalpha and TSC-mTOR signaling pathways. J Cell Biol 2006;175:121–33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen S, Wang C, Yeo S, Liang CC, Okamoto T, Sun S, et al. Distinct roles of autophagy-dependent and -independent functions of FIP200 revealed by generation and analysis of a mutant knock-in mouse model. Genes Dev 2016;30:856–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Brockwell NK, Owen KL, Zanker D, Spurling A, Rautela J, Duivenvoorden HM, et al. Neoadjuvant Interferons: Critical for Effective PD-1-Based Immunotherapy in TNBC. Cancer Immunol Res 2017;5:871–84 [DOI] [PubMed] [Google Scholar]
- 29.Minn AJ, Wherry EJ. Combination Cancer Therapies with Immune Checkpoint Blockade: Convergence on Interferon Signaling. Cell 2016;165:272–5 [DOI] [PubMed] [Google Scholar]
- 30.Zhu J, Smith K, Hsieh PN, Mburu YK, Chattopadhyay S, Sen GC, et al. High-throughput screening for TLR3-IFN regulatory factor 3 signaling pathway modulators identifies several antipsychotic drugs as TLR inhibitors. J Immunol 2010;184:5768–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chariot A, Leonardi A, Muller J, Bonif M, Brown K, Siebenlist U. Association of the adaptor TANK with the I kappa B kinase (IKK) regulator NEMO connects IKK complexes with IKK epsilon and TBK1 kinases. J Biol Chem 2002;277:37029–36 [DOI] [PubMed] [Google Scholar]
- 32.Li S, Wang L, Berman M, Kong YY, Dorf ME. Mapping a dynamic innate immunity protein interaction network regulating type I interferon production. Immunity 2011;35:426–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei H, Wang C, Groce C, Guan J. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev 2014;28:1204–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wei H, Wei S, Gan B, Peng X, Zou W, Guan J. Suppression of autophagy by FIP200 deletion inhibits mammary tumorigenesis. Genes Dev 2011;25:1510–27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yeo SK, Wen J, Chen S, Guan JL. Autophagy Differentially Regulates Distinct Breast Cancer Stem-like Cells in Murine Models via EGFR/Stat3 and Tgfβ/Smad Signaling. Cancer Res 2016;76:3397–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yeo SK, Paul R, Haas M, Wang C, Guan JL. Improved efficacy of mitochondrial disrupting agents upon inhibition of autophagy in a mouse model of BRCA1-deficient breast cancer. Autophagy 2018;14:1214–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Luo M, Fan H, Nagy T, Wei H, Wang C, Liu S, et al. Mammary epithelial-specific ablation of the focal adhesion kinase suppresses mammary tumorigenesis by affecting mammary cancer stem/progenitor cells. Cancer Res 2009;69:466–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wei H, Wang C, Croce CM, Guan JL. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev 2014;28:1204–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oelkrug C, Ramage JM. Enhancement of T cell recruitment and infiltration into tumours. Clin Exp Immunol 2014;178:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goodwin JM, Dowdle WE, DeJesus R, Wang Z, Bergman P, Kobylarz M, et al. Autophagy-Independent Lysosomal Targeting Regulated by ULK1/2-FIP200 and ATG9. Cell Rep 2017;20:2341–56 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu D, Jin T, Zhu H, Chen H, Ofengeim D, Zou C, et al. TBK1 Suppresses RIPK1-Driven Apoptosis and Inflammation during Development and in Aging. Cell 2018;174:1477–91.e19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lafont E, Draber P, Rieser E, Reichert M, Kupka S, de Miguel D, et al. TBK1 and IKKε prevent TNF-induced cell death by RIPK1 phosphorylation. Nat Cell Biol 2018;20:1389–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Behrends C, Sowa ME, Gygi SP, Harper JW. Network organization of the human autophagy system. Nature 2010;466:68–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Goncalves A, Bürckstümmer T, Dixit E, Scheicher R, Górna MW, Karayel E, et al. Functional dissection of the TBK1 molecular network. PLoS One 2011;6:e23971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ryzhakov G, Randow F. SINTBAD, a novel component of innate antiviral immunity, shares a TBK1-binding domain with NAP1 and TANK. EMBO J 2007;26:3180–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-γ-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127:2930–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN-γ Pathway Genes in Tumor Cells as a Mechanism of Resistance to Anti-CTLA-4 Therapy. Cell 2016;167:397–404.e9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gan B, Guan JL. FIP200, a key signaling node to coordinately regulate various cellular processes. Cell Signal 2008;20:787–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wei H, Gan B, Wu X, Guan JL. Inactivation of FIP200 leads to inflammatory skin disorder, but not tumorigenesis, in conditional knock-out mouse models. J Biol Chem 2009;284:6004–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chude CI, Amaravadi RK. Targeting Autophagy in Cancer: Update on Clinical Trials and Novel Inhibitors. Int J Mol Sci 2017;18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prabakaran T, Bodda C, Krapp C, Zhang BC, Christensen MH, Sun C, et al. Attenuation of cGAS-STING signaling is mediated by a p62/SQSTM1-dependent autophagy pathway activated by TBK1. EMBO J 2018;37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kumar S, Gu Y, Abudu YP, Bruun JA, Jain A, Farzam F, et al. Phosphorylation of Syntaxin 17 by TBK1 Controls Autophagy Initiation. Dev Cell 2019 [DOI] [PMC free article] [PubMed]
- 53.Jin S, Tian S, Luo M, Xie W, Liu T, Duan T, et al. Tetherin Suppresses Type I Interferon Signaling by Targeting MAVS for NDP52-Mediated Selective Autophagic Degradation in Human Cells. Mol Cell 2017;68:308–22.e4 [DOI] [PubMed] [Google Scholar]
- 54.Ohkuri T, Ghosh A, Kosaka A, Zhu J, Ikeura M, David M, et al. STING contributes to antiglioma immunity via triggering type I IFN signals in the tumor microenvironment. Cancer Immunol Res 2014;2:1199–208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ravenhill BJ, Boyle KB, von Muhlinen N, Ellison CJ, Masson GR, Otten EG, et al. The Cargo Receptor NDP52 Initiates Selective Autophagy by Recruiting the ULK Complex to Cytosol-Invading Bacteria. Mol Cell 2019 [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.