

Abstract
Quiescence is a hallmark of CD4+ T cells latently infected with HIV-1. While reversing this quiescence is an effective approach to reactivate latent HIV from T cells in culture, it can cause deleterious cytokine dysregulation in patients. As a key regulator of T-cell quiescence, FOXO1 promotes latency and suppresses productive HIV infection. We report that in resting T cells, FOXO1 inhibition impaired autophagy and induced ER stress, thereby activating two associated transcription factors: activating transcription factor 4 (ATF4) and nuclear factor of activated T cells (NFAT). Both factors associate with HIV chromatin and were for HIV reactivation. Indeed, inhibition of PKR-like endoplasmic reticulum kinase (PERK), an ER stress sensor that can mediate the induction of ATF4, and calcineurin, a calcium-dependent regulator of NFAT, synergistically suppressed HIV reactivation induced by FOXO1 inhibition. Thus, our studies uncover a link between FOXO1, ER stress, and HIV infection that could be therapeutically exploited to selectively reverse T-cell quiescence and reduce the size of the latent viral reservoir.
The major barrier to eradicating human immunodeficiency virus (HIV-1) from infected patients is the persistence of latently infection, primarily of memory CD4+ T cells. These cells are rare, long-lived, and generally quiescent. Although anti-retroviral therapy (ART) effectively suppresses HIV replication, therapy interruption results in reactivation of the latent reservoir, necessitating life-long treatment, and there is no cure1,2. One strategy to eliminate latently infected cells is to activate virus production with latency reversing agents (LRAs) to trigger cell death through virus-induced cytolysis or immune clearance. Efficient viral reactivation requires a calibrated degree of activation that avoids inducing polyclonal T cell activation or a cytokine storm that would negate the potential benefit. Known LRAs include PKC agonists (e.g. Prostratin), epigenetic modulators (e.g. HDAC or bromodomain inhibitors), and modulators of the PI3K or mTOR signaling pathways3–6. However, clinical trials of existing LRAs have not yet demonstrated successful reduction of the latent reservoir7. Thus, a better understanding of how HIV latency is established and maintained is needed to improve the design of interventions.
The Forkhead box O (FOXO) protein family comprises evolutionarily conserved transcription factors8. FOXO-mediated gene regulation is determined in part by localization; upon phosphorylation by upstream kinases, they exit the nucleus and remain inactive. FOXO isoforms influence multiple pathways, including cell cycle arrest, glucose metabolism, oxidative stress regulation, apoptosis, and the DNA damage response9–11. In the immune system, FOXO proteins regulate a set of genes involved in maintaining quiescence and cell-fate differentiation in CD4+ and CD8+ T cells12–14. Furthermore, inhibition of FOXO1 in naïve and memory CD8+ T cells promotes a more effector-like and cytotoxic phenotype in the context of immune aging and chronic infection15.
In elite controller HIV patients, FOXO3a is downregulated in memory CD4+ T cells, promoting the persistence of these cells16. Furthermore, FOXO proteins negatively regulate HIV transcription through Tat-mediated repression17, and FOXO1 inhibition accelerates productive infection18. Because FOXO1 activity promotes and maintains the quiescent state of memory CD4+ T cells, in which HIV establishes latency, we hypothesized that FOXO1, directly or indirectly, induces HIV latency. Here, we identify a pathway that links HIV latency with FOXO1 activity via ER stress signaling, but without general T cell activation. Having shown that FOXO1 inhibition prevents latency establishment and successfully purges the virus from its latent reservoirs, we propose that the combination of early ART and FOXO1 inhibition may downsize the latent reservoir. In support of our findings, a recent study also showed the potential for FOXO1 inhibition to reactivate HIV latent proviruses19.
Results
FOXO1 is a Specific Regulator of HIV Latency Establishment
To test the potential of FOXO1 inhibition to affect HIV-1 latency, we used the second-generation dual-color reporter virus HIVGKO20 and AS1842856 (Figure 1a), a cell-permeable inhibitor of FOXO1 known to inhibit hepatic glucose production21. In this system, latently infected K562(Cas9) cells and the T cell-based cell line NH7 express the mKO2 fluorescent protein under the control of the EF1α promoter, while cells containing a productive and active virus express both mKO2 and GFP fluorophores, the latter controlled by the HIV-1 LTR. Upon treatment with increasing concentrations of AS1842856, the proportion of latently infected cells decreased, despite no significant changes in the total infection rate or viability (Figure 1a and Extended Data Figure 1a). Concomitantly, the proportion of actively infected cells increased, as expected. The IC50 of the inhibitor observed in our cell culture system was 74 ± 15 nM, near its in vitro IC50 of 33 nM21. Thus, FOXO1 inhibition prevented HIV-1 latency establishment and increased the ratio between actively- and latently-infected cells.
Figure 1. FOXO1 is a Specific Regulator of HIV Latency Establishment.
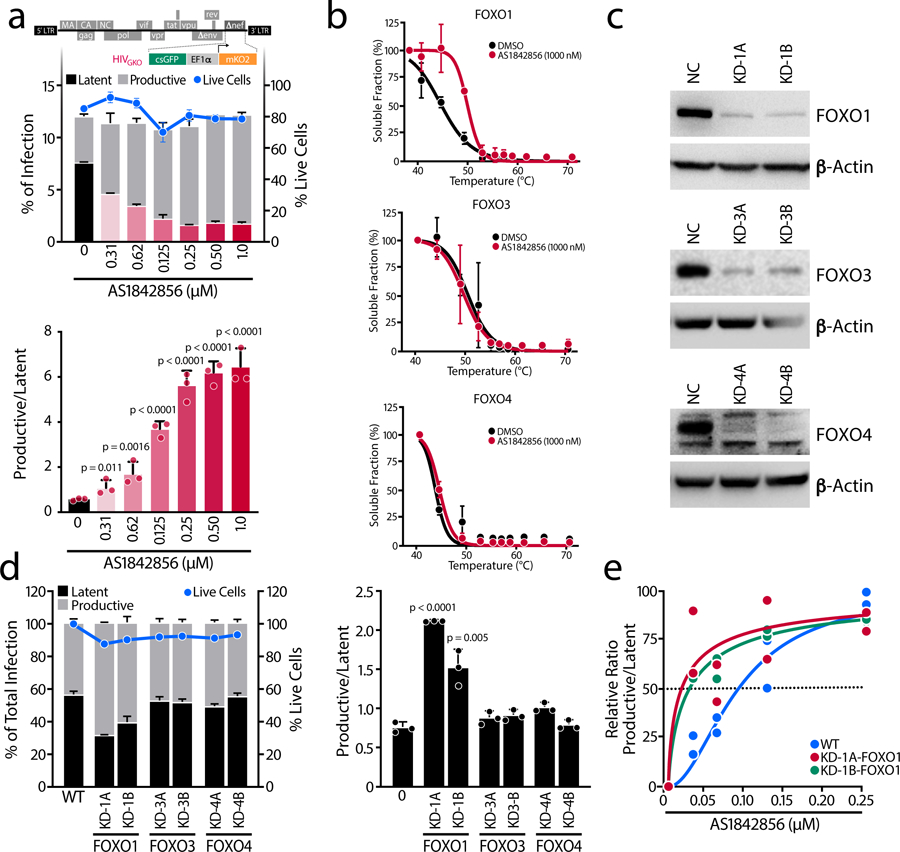
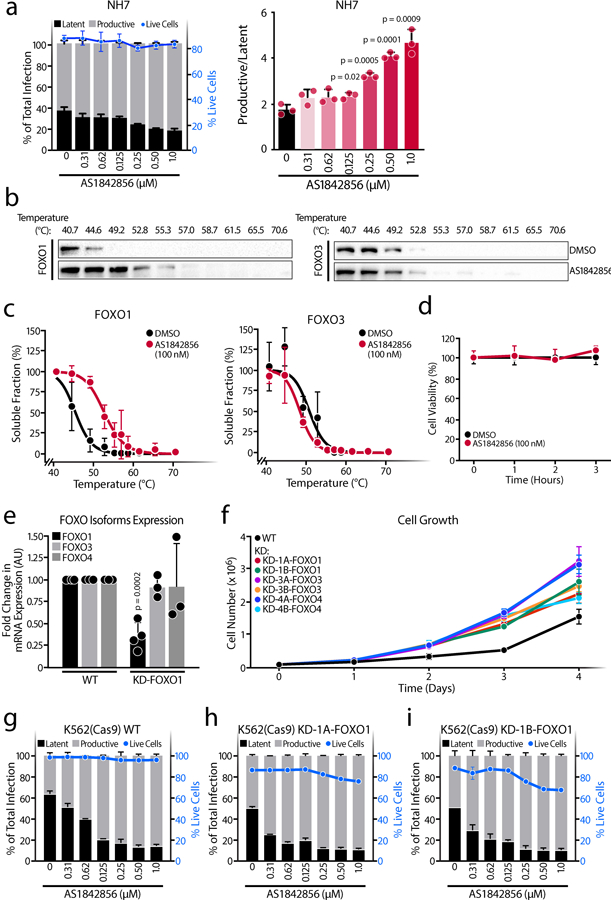
a, Schematic representation of HIVGKO dual-labeled HIV-1 reporter. K562(Cas9) cells were treated with increasing concentrations of the FOXO1 inhibitor AS1842856 just after HIVGKO infection. After 3–4 days, the percentage of latent and productively infected cells were quantified by FACS. In upper panel, amount of productively or latently infected cells (bars) and total cell viability (blue dotted line) of a representative experiment. In the lower panel, ratios of productive versus latent populations relative to the total infection rate upon increasing concentrations of AS1842856 treatments. Data are represented as mean ± SD of triplicate values, representative of n=3 independent experiments. b, FOXO1, FOXO3 and FOXO4 CETSA-melting curve shifts upon the presence or absence of AS1842856 1,000 nM in K562(Cas9) cells. Band intensities obtained from western blot analysis were normalized to the highest western blot signal. Relative FOXO-band intensities were plotted against corresponding incubation temperatures and a nonlinear least-squares regression fit was applied. Data represent the mean ± SD of n=3 independent experiments, except for FOXO4 (n=2). c, Efficiency of FOXO1, FOXO3 and FOXO4 knockdowns with two different sgRNAs determined by western blot. Cells transduced with NC (negative control) sgRNA lentiviruses were used as a control. Representative of n=3 independent experiments, except for FOXO3 (n=2). d, In the left graph, percentage of productive or latent cells relative to the total infection rate and cell viability in the different single knockdown K562(Cas9) cell lines. In the right graph, ratios of productive versus latent populations in K562(Cas9) cell lines with knockdown of FOXO1, FOXO3, or FOXO4. Data are represented as mean ± SD of triplicate values, representative of n=3 independent experiments. e, Ratios of productive versus latent populations upon increasing concentrations of AS1842856 in the WT K562(Cas9) or in the FOXO1-knockdown cell lines. Data are represented as mean of n=2 independent experiments fitted into a model.
We next sought to confirm the specificity of AS1842856 for FOXO1. To that end, we performed cellular thermal shift assays (CETSA) against the FOXO isoforms expressed in K562(Cas9) cells. CETSA evaluates biophysical binding under physiological conditions in living cells. Binding was considered ‘positive’ if there was a significant change in the thermal denaturation profile of the protein upon addition of AS1842856 (Extended Data Figure 1b). We observed a 5.2 °C shift in FOXO1 melting curve upon AS1842856 treatment (1 μM), whereas no changes were found in either FOXO3 or FOXO4 (Figure 1b). Similar results were observed at 100 nM of AS1842856 (Extended Data Figure 1b–d), confirming the specificity of the inhibitor.
To interrogate the role of other FOXO family members in HIV-1 latency establishment, we generated single knockdowns of FOXO1 (KD-1A-FOXO1, KD-1B-FOXO1), FOXO3 (KD-3A-FOXO3, KD-3B-FOXO3) and FOXO4 (KD-4A-FOXO4, KD-4B-FOXO4) by CRISPR interference (CRISPRi) in K562(Cas9) cells (Figure 1c and Extended Data Figure 1e). Because FOXO proteins control the cell cycle and promote a quiescence, each knockdown increased the proliferation rate (Extended Data Figure 1f), in agreement with previous findings on human bladder cancer cells22 and MCF-7 breast adenocarcinoma cells23. However, only FOXO1 downregulation increased productive infection and decreased latent infection (Figure 1d), although the effect was modest. Furthermore, FOXO1 knockdown rendered infected cells more sensitive to AS1842856 treatment, shifting the IC50 (Figure 1e and Extended Data Figure 1g–i). Taken together, these data indicate that pharmacological or genetic inhibition of FOXO1, but not FOXO3 or FOXO4, decreases establishment of HIV-1 latency.
FOXO1 inhibition Reactivates HIV-1 from Latency
The J-Lat clonal cell lines are Jurkat cells that contain a stably integrated, latent HIV-1 (HXB) ∆ env provirus with a GFP reporter gene in place of the nef gene24. In this system, reactivation from established latency can be monitored through GFP mRNA and protein expression. After 48 and 72 hours of treatment, AS1842856 stimulated GFP mRNA expression in J-Lat 5A8 cells to a similar degree as TNFα, a known LRA in J-Lat cells, indicating that FOXO1 inhibition can also reverse established latency (Figure 2a). No major effect on cell viability was observed (Figure 2b). Maximal activation by TNFα was seen after 24 hours, while AS1842856-induced activation progressively increased only after this time point.
Figure 2. FOXO1 Inhibition Reactivates HIV1 from Latency.
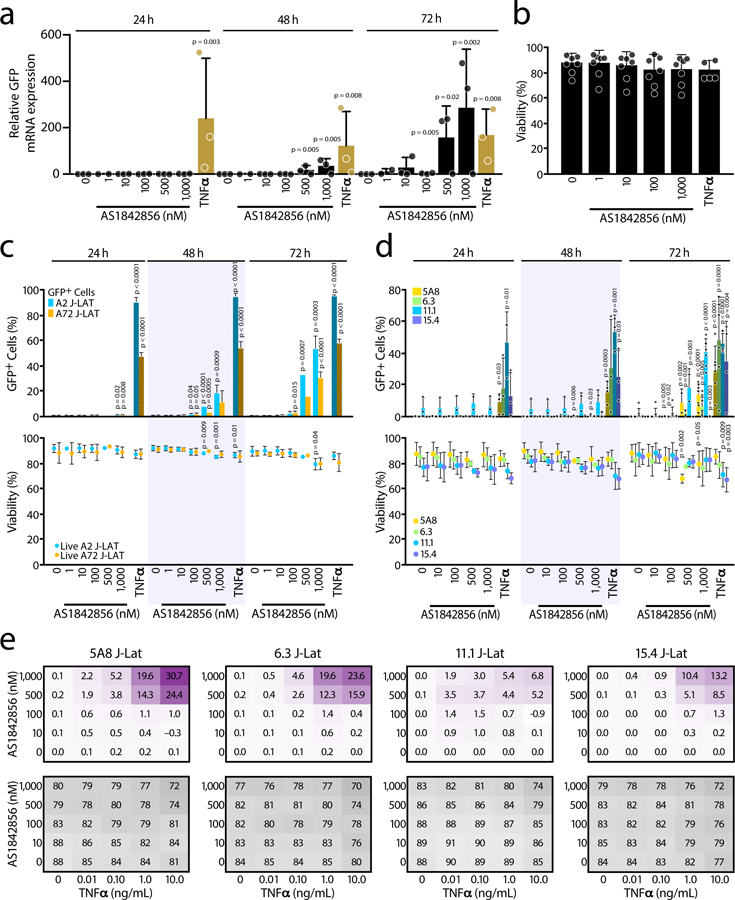
a, J-Lat cell line 5A8 was treated with increasing concentrations (1–1,000 nM) of AS1842856 for 24, 48 and 72 hours and HIV-GFP mRNA reactivation was assessed by RT-qPCR and normalized to RPL13A mRNA. Data represent average ± SD of three independent experiments. p-value relative to the control at each time point. b, Cell viability assessed by flow cytometry at 72 hours of the same experiment as in a. Cell viability was measured by gating on both the live population at the forward scatter (FSC) and side scatter (SSC) plot after staining with the viability dye. Data represent average ± SD of n≥4 independent experiments. c, J-Lat cell lines A2 and A72 were treated with increasing concentrations of AS1842856 for 24, 48 and 72 hours and HIV-GFP reactivation (bars) and cell viability (dots) were analyzed by FACS. HIV-GFP reactivation is reported as a percentage of GFP-expressing cells (% GFP+ cells). Data represent average ± SD of n≥3 independent experiments. d, Same experiments as in Fig. 2c but performed in J-Lat cell lines 5A8, 6.3, 11.1 and 15.4. Data represent average ± SD of n≥3 independent experiments. 10 ng/mL TNFα was used as control. e, J-Lat cell lines 5A8, 6.3, 11.1 and 15.4 were treated for 72 hours with increasing concentrations of both AS1842856 (Y-axis) and TNFα (X-axis) alone or in combination and analyzed by FACS. Calculation of synergy for drug combinations using the Bliss independence model applied to the HIV-GFP reactivation measured as a percentage of GFP-expressing cells (% GFP+ cells). Synergy (in purple) and cell viability (in grey) data points represent the mean effect from three independent experiments.
Treatment with AS1842856 also increased HIV-driven GFP protein expression, similar to mRNA expression, in a dose-dependent manner, as determined by flow cytometry (Figures 2c, d). Because FOXO1 and FOXO4 antagonize transactivation of the HIV-1 promoter through the repression of the viral transactivator Tat17, we tested the ability of AS1842856 to reverse HIV latency in J-Lat cells with and without Tat. AS1842856 increased GFP expression in both J-Lat A2 cells, which do contain Tat, and in A72 cells, which do not24, indicating that the effect of FOXO1 inhibition on HIV reactivation is Tat-independent, although overall levels of reactivation are higher when Tat is present (Figure 2c), in agreement with other reports19. No effect on cell viability was observed in either cell line (Figure 2c).
To rule out effects based on clonal variation or proviral integration sites, we compared AS1842856-induced HIV reactivation in four J-Lat clonal cell lines which harbor latent HIV-1 inserted at distinct genomic sites. After 72 hours of treatment, AS1842856 reactivated HIV in each cell line, as determined by percentage of GFP+ cells (Figure 2d) and the median fluorescence intensity (Extended Data Figure 2a). In all cell lines, viability was unaffected, and treatment with TNFα induced GFP expression as early as 24 hours after treatment, as expected (Figure 2d). Thus, the effect of AS1842856 is independent of the proviral integration site.
Given the different time courses of TNFα- and AS1842856-induced HIV reactivation, we speculated that these LRAs target different mechanisms of reactivation and might synergize. To test this hypothesis, we co-treated multiple J-Lat lines with increasing concentrations of AS1842856 and TNFα (Extended Data Figure 2b, c). We used the Bliss independence model to assess dose combinations that result in greater than additive effects25,26 and observed such synergistic effects across different doses in all clones (Figure 2e, upper panels). There were no major changes in viability in co-treated cell lines (Figure 2e, bottom panels). These data show reversal of established latency by FOXO1 inhibition and indicate a molecular mechanism of activation distinct from TNFα.
FOXO1 inhibition Regulates HIV-1 Latency in Primary CD4+ T cells
To verify these results in primary T-cell models of HIV latency, we measured the capacity of FOXO1 inhibition to impair HIV latency establishment by infecting resting CD4+ T cells from anonymous blood donors with HIVGKO while treating the cells with increasing doses of AS1842856 for 72 hours. As previously observed in K562(Cas9) cells, inhibition of FOXO1 promoted productive infection and decreased latency (Figure 3a). We also tested the ability of AS1842856 to reverse established HIV latency in primary cells by spin-infecting resting CD4+ T cells with a pseudotyped HIV reporter virus containing the firefly luciferase gene27. After 6 days in culture, cells reactivated luciferase expression in response to increasing concentrations of FOXO1 inhibitor (Figure 3b) as well as in response to generalized T cell activation induced with a combination of phytohemagglutinin and interleukin-2 (PHA/IL-2) or with antibodies directed against the CD3 and CD28 receptors (Extended Data Figure 3a). Notably, FOXO1 inhibition did not induce generalized T cell activation, as measured by expression of CD69 and CD25 activation markers on the cell surface (Extended Data Figure 3b). Combining AS1842856 (100 nM) with a low dose of prostratin (250 nM), a natural PKC activator that induces partial T-cell activation25,28, significantly increased the AS1842856 effect in primary T cells, mirroring the synergistic effect of TNFα in J-Lat cells. No change in primary T-cell viability was observed in response to FOXO1 inhibition (Figures 3a–c).
Figure 3. FOXO1 Inhibition Prevents Latency Establishment and Reactivates HIV in Primary CD4+ T cells and HIV-infected CD4+ T cells.
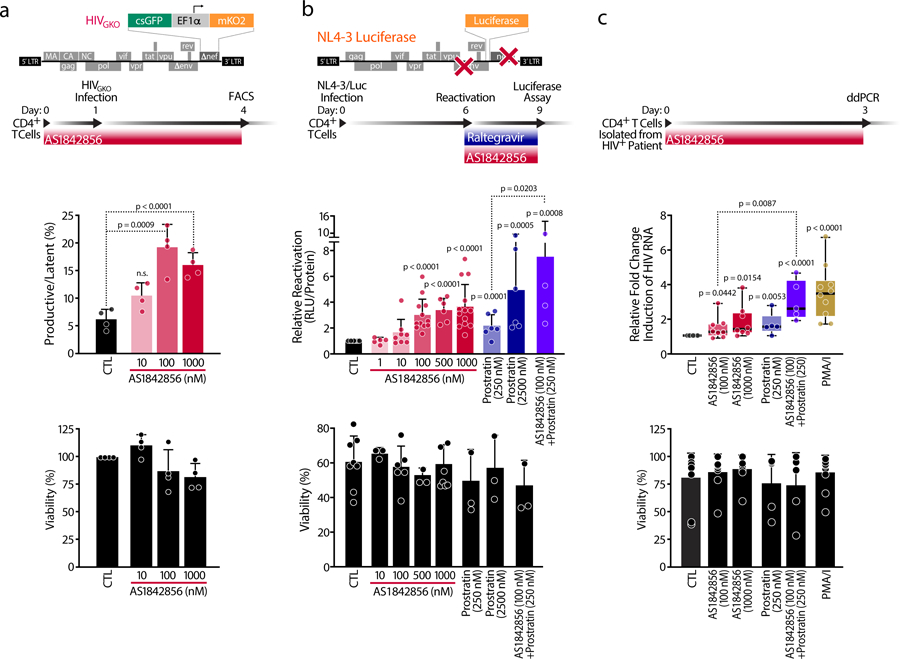
a, Schematic representation of HIVGKO dual-labeled HIV-1 reporter and of strategy used to treat primaryCD4+ T cells purified from blood of healthy donors and pre-treated for 24 hours with increasing concentrations of AS1842856. After infection of HIVGKO, resting cells were treated for 3 days with the same amounts of AS1842856 as in the pre-treatment. Top panel shows ratios of productive versus latent populations of infected CD4+ T cells from four different healthy donors after treatment. Lower panel shows a histogram plot of percent live cells for each drug treatment relative to the control. Data are represented by mean ± SD of n=4 different donors. b, Schematic representation of HIVNL4–3 Luciferase reporter virus and of experimental procedure with primary CD4+ T cells. Briefly, CD4+ T cells were purified from blood of healthy donors, were infected with HIVNL4–3 Luciferase and, after 6 days, the virus was reactivated for 3 days with increasing concentrations of AS1842856 and prostratin alone or in combination, in the presence of raltegravir (30 μM). HIV reactivation was measured by luciferase activity and cell viability by flow cytometry. Data are mean ± SEM of n≥5 individual donors. c, Fold change of cell-associated HIV-1 mRNA expression measured by ddPCR of CD4+ T cells of HIV-infected patients on antiretroviral-therapy with undetectable viral load treated with 50 ng/ml PMA + 1 μM Ionomycin (PMA/I) and with 0.2 % DMSO (CTL). AS1842856- and/or prostratin-treated CD4+ T cells led to an increase in fold change of cell-associated HIV mRNA expression. Data are represented as box plots of n≥5 independent experiments. Cell viability was assessed by Tripan Blue exclusion. Data are represented as mean ± SD of n≥5 independent experiments.
We replicated these observations in CD4+ T cells, isolated from ten HIV-infected patients on antiretroviral therapy for at least 6 months and with undetectable viral loads (< 50 copies/ml). Cells were treated ex vivo with two concentrations of AS1842856 (100 and 1,000 nM), prostratin (250 nM) or combinations thereof for 3 days, and HIV RNA induction was measured by digital droplet PCR (ddPCR). A two-fold induction was observed with AS1842856 treatment alone, which was significantly enhanced with prostratin cotreatment to similar levels as achieved by PMA/I treatment (Figure 3c). These results confirm a role of FOXO1 in the establishment and maintenance of HIV latency in primary resting T cells.
Transcriptional Reprogramming in Response to FOXO1 Inhibition
To determine how FOXO1 inhibition changes the transcriptome of resting CD4+ T cells, we performed RNA sequencing (RNA-Seq) on primary CD4+ T cells treated with AS1842856 or DMSO for 12 hours. AS1842856 treatment resulted in up-regulation of 246 genes and down-regulation of 473 genes (q < 0.05) (Figure 4a). Ingenuity Pathway Analysis of the up-regulated genes revealed significant enrichment of genes involved in tRNA charging (e.g. GARS, MARS, VARS), T and B cell signaling and cytokine production (e.g. IL10, CSF2, SLAMF1), and dysregulation of the endoplasmic reticulum: ER stress pathway (ER stress) and Unfolded Protein Response (UPR) (e.g. CALR, ATF4, HSPA5). Down-regulated genes included those involved in granzyme A signaling (e.g. HIST1), cell junction signaling (e.g. IRS2, ATM), and autophagy (e.g. ATG13, ATG9A, CTSF, NBR1, RB1CC1, ULK1) (Figure 4b). Notably, the autophagy pathway is an established target of FOXO129,30. These results were largely aligned with a recently published microarray analysis in T cells treated with AS1842856 for 7 days19 (Extended Data Figure 4a).
Figure 4. Marked Upregulation of ER Stress in Response to FOXO1 Inhibition in Primary CD4+ T cells.
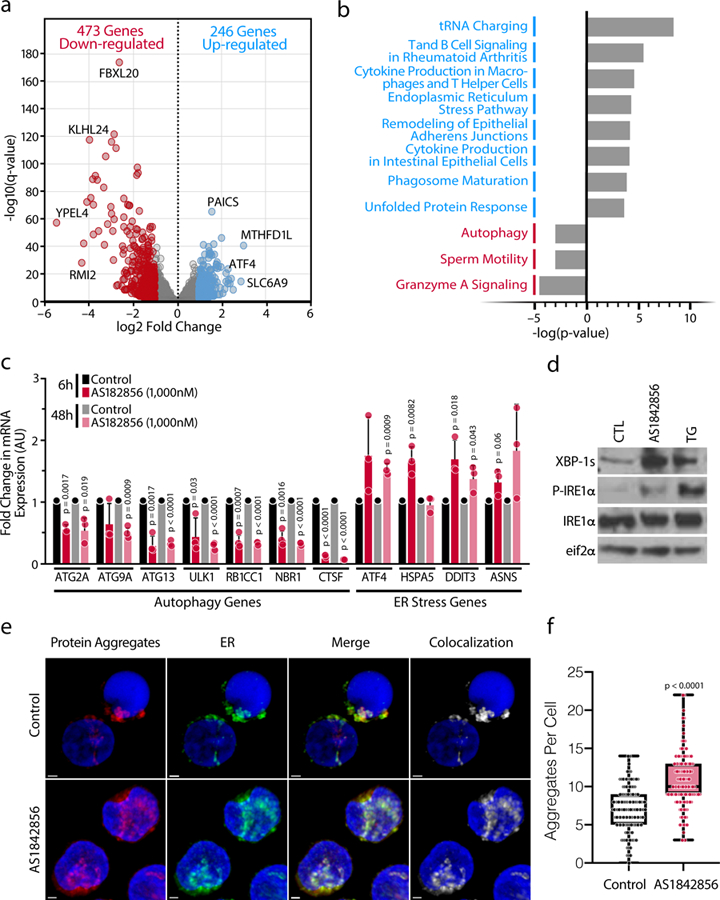
a, Volcano plot from the RNA-Seq data comparing CD4+ T cells treated with AS1842856 (1,000 nM) versus DMSO control for 12 hours. Up-regulated (blue) or down-regulated (red) genes are q-value < 0.05 and log2 fold change ≥ 1 or ≤ −1, respectively. b, IPA pathway analysis of the most dysregulated canonical pathways for the up- and down-regulated genes. c, Confirmation of up- or down-regulated expression after 6 and 48 hours treatment of specific genes from altered pathways by RT-qPCR, normalized to RPL13A mRNA. Data represent mean ± SD of n=3 individual donors. d, Representative blots of protein expression of the ER stress markers XBP-1s, phosphorylated IRE1α and total IRE1α and total eIF2α (control). Representative experiment of n=3 independent experiments. e, Representative confocal microscopy images of primary CD4+ T cells isolated from PBMCs. Cells were treated with AS1842856 (500 nM) for 72 hours and processed for immunostaining after treatment with Proteostat (protein aggregates, red), GRP78 (endoplasmic reticulum, green), and Hoechst (nuclei, blue). The scale bars represent 1μm. Data are representative of n=3 independent experiments. f, The number of protein aggregates per cell. Data are represented as box plots: Control (min: 0, Q1: 5, center: 7, Q3: 9 max: 14), AS1842856 (min: 3, Q1: 9, center: 10, Q3: 13, max: 22). The P value was calculated by a linear mixed model by residual maximum likelihood. p-value < 2-16. n = 135 cells per condition.
We confirmed down-regulation of autophagy markers and up-regulation of ER stress markers, in AS1842856-treated healthy CD4+ T cells (Figure 4c) and in HIV-infected samples (Extended Data Figure 4b) by real-time quantitative PCR (RT-qPCR). Furthermore, in CD4+ T cells FOXO1 inhibition increased the level of the spliced form of the X box-binding protein 1 (XBP-1s), an important transcription factor in the ER stress response31, and the phosphorylation of its upstream activator, inositol-requiring enzyme 1α (IRE1α), whereas eif2α levels remained unchanged (Figure 4d). FOXO1 inhibition also increased protein aggregation in the cytoplasm of CD4+ T cells (Figure 4e, f), especially in the ER (Figure 4e, Extended Data Figure 4c), suggesting that protein accumulation and ER stress may be a result of decreased FOXO1-mediated autophagy.
FOXO1 Inhibition Induces HIV-1 Reactivation via ATF4 and NFAT
Upstream regulator analysis identified ATF4 as the top modulator of AS1842856-induced genes (Figure 5a). ATF4 is a transcription factor specifically upregulated at the protein level as part of the UPR and the integrated stress response (ISR), which involves phosphorylation of the translation initiation factor eIF2α by stress-specific kinases32–34. Consistent with this data, FOXO1 inhibition increased expression of ATF4 protein levels in CD4+ T cells across multiple donors (Figure 5b). Given that the ER is an important storage organelle for calcium35 and that ER stress induces calcium release36, we performed calcium flux measurements in AS1842856-treated CD4+ T cells (Figure 5c) and K562(Cas9) and Jurkat cell lines (Extended Data Figure 5). Basal calcium levels were increased in response to AS1842856 treatment, whereas calcium ER flux levels increased modestly yet significantly after the addition of the Ca2+ ionophore ionomycin in primary CD4+ T cells (Figure 5c) as compared to Jurkat cells, consistent with previous results19.
Figure 5. FOXO1 Inhibition Induces HIV Reactivation in the Absence of NF-kB Recruitment via ATF4 and NFAT.
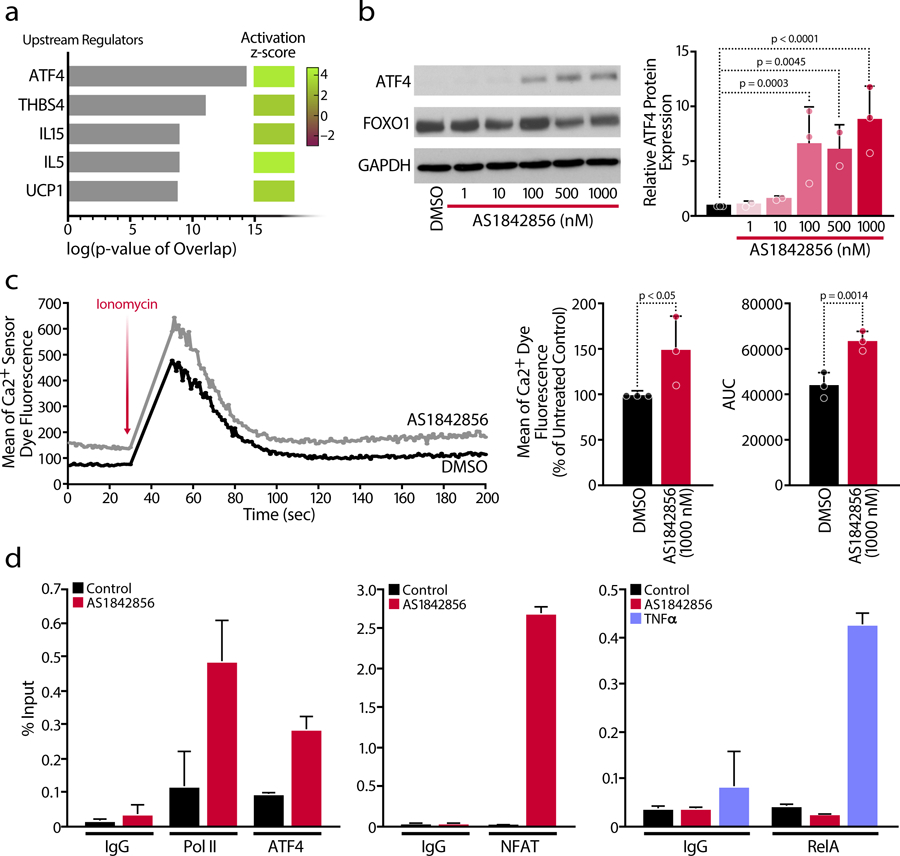
a, Analysis of the five top upstream regulators and table with their activation z-scores according to the RNA-Seq data. b, Representative blot of protein expression of the transcription factors ATF4 and FOXO1 (left) and densitometry analysis of ATF4 protein expression (right) was performed from n=3 individual donors. Data are mean ± SD. c, Representative plot of intracellular calcium-flux kinetics in primary CD4 T cells in the presence or absence of AS1842856 (100 nM). Cells were stained with a membrane permeable calcium sensor dye in PBS and stimulated by adding Ionomycin after 30 seconds resulting in an increase of fluorescence indicating a calcium mobilization from the ER. The mean of the calcium sensor dye fluorescence at basal condition (before Ionomycin stimulation, 0–30 sec) and the parameter Area Under the Curve (AUC) relative to the calcium flux were calculated. Data are represented as mean ± SD of n=3 independent experiments. d, Chromatin immunoprecipitation (ChIP) assays with antibodies against Pol II, ATF4, RelA, NFAT and IgG control at the HIV LTR, followed by qPCR using primers specific for HIV-1 LTR Nuc0 or Nuc1. Chromatin was prepared from J-Lat A2 and 5A8 cells, in which the LTR was stimulated by 1,000 nM AS1842856 treatment, 10 ng/mL TNFα or which were left untreated/DMSO. Representative experiment of n=3 independent biological experiments. Data are represented as mean ± SD of n=3 independent technical replicates.
Because previous research demonstrated that ATF4 binds the simian immunodeficiency virus (SIV) and HIV LTR37 and activates the related human T-cell leukemia virus type 1 (HTLV-1)38, we performed chromatin immunoprecipitation (ChIP) experiments to test whether ATF4 binds the HIV LTR in response to FOXO1 inhibition. While low levels of ATF4 bound HIV LTR chromatin in untreated J-Lat cells, its recruitment was 3-fold enriched after AS1842856 treatment (Figure 5d). We also observed a marked increase in RNA polymerase II recruitment to the HIV promoter after FOXO1 inhibition, consistent with HIV transcription reactivation. Since we observed increased calcium release upon FOXO1 inhibition, we asked whether FOXO1 inhibition also altered HIV promoter occupancy of nuclear factor of activated T cells (NFAT), which drives HIV transcription activated through calcium mobilization after T-cell activation39,40. NFAT was markedly enriched at the HIV LTR after FOXO1 inhibition (Figure 5d). In contrast, the RelA subunit of NF-κB, which regulates HIV transcription in response to T cell activation or TNFα exposure41, was not recruited to the HIV LTR after AS1842856 treatment but was significantly enriched after TNFα treatment, as expected (Figure 5d). Consistent with those results, NFAT can occupy the NF-κB binding sites in the HIV LTR42, indicating that FOXO1 in resting T cells suppresses a (non-canonical) transcriptional program that controls HIV transcription without NF-κB, but with ATF4 and NFAT. This result explains the synergy observed between AS1842856 and TNFα or prostratin, which activate HIV through NF-κB28.
Induction of ER stress promotes HIV reactivation
To investigate the role of the integrated stress response (ISR) in HIV reactivation, we evaluated the effect of AS1842856 in combination with inhibitors of the eIF2α upstream kinases: double-stranded RNA-dependent protein kinase R (PKR), activated by viral infection; PKR-like endoplasmic reticulum kinase (PERK), activated in response to ER stress; and general control nonderepressible 2 (GCN2) kinase, activated by amino acid deficiencies43–48. Treatment with two pharmacological inhibitors of PERK (Figure 6a), but not inhibitors of GCN2 or PKR (Extended Data Figure 6a,b), reduced AS1842856-induced HIV reactivation, consistent with the model that FOXO1 inhibition activates ATF4 through ER stress-driven PERK activation. In contrast, TNFα-mediated HIV reactivation was not affected by PERK inhibition (Figure 6a) but was diminished by GCN2 or PKR inhibitor treatment (Extended Data Figure 6a,b), underscoring the distinct molecular mechanisms underlying latency reversal by the two activators. Similar results were observed in CD4+ T cells from three HIV-infected individuals (Figure 6b). Interestingly, inhibition of IRE1α, which represents a separate branch of the ER Stress pathway, did not affect HIV reactivation induced by FOXO1 inhibition (Extended Data Figure 6d), although the compound successfully suppressed XBP-1s-dependent ER stress induction by thapsigargin (Extended Data Figure 6e). Thus, while FOXO1 inhibition induces protein aggregates and ER stress, HIV latency reactivation appears exclusively dependent on the PERK-ATF4 branch of the UPR.
Figure 6. Induction of ER stress promotes HIV reactivation.
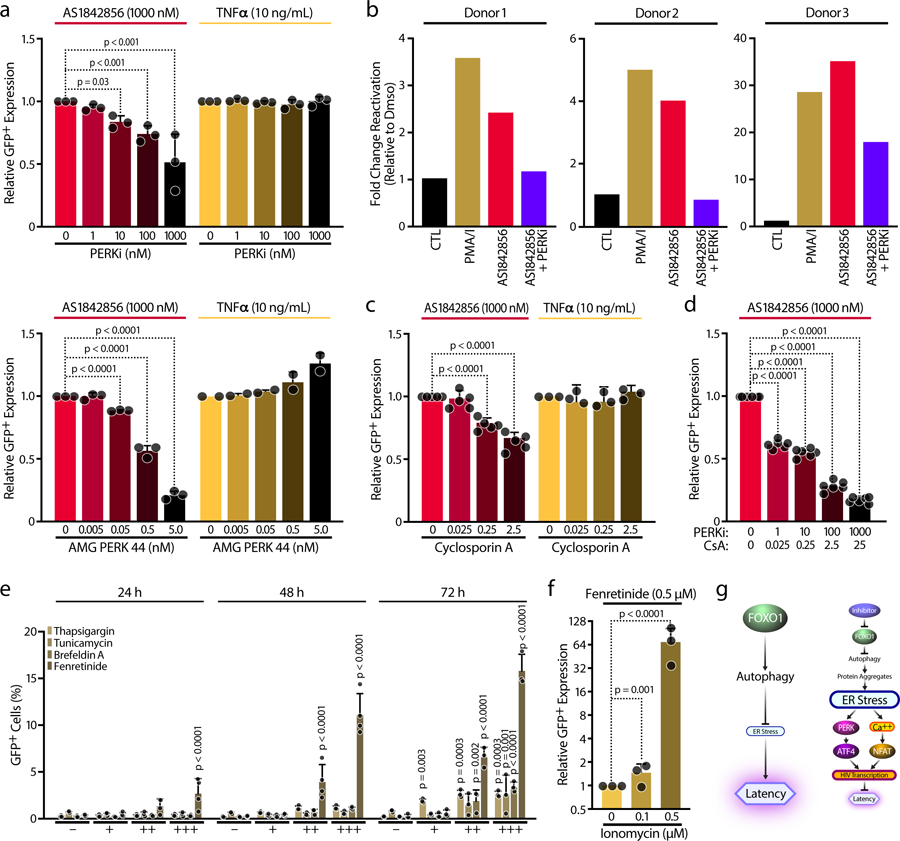
a, J-Lat cell line 5A8 were treated with increasing concentrations of PERKi (GSK2656157 / PERK inhibitor II) (top panels) or the highly specific PERK inhibitor (AMG PERK 44) (lower panels) and co-treated with 1,000 nM AS1842856 (72 hours) or 10 ng/mL TNFα (24 hours). HIV-GFP reactivation was analyzed by FACS and normalized to the control. Data shown are mean ± SD of n=3 independent experiments. b, Effect of 50 ng/ml PMA + 1 μM Ionomycin (PMA/I), AS1842856 (1,000 nM) and AS1842856 (1,000 nM) with 1 μM PERKi (GSK2656157 / PERK inhibitor II) on HIV-1 mRNA expression in CD4+ T cells of n=3 HIV-infected patients on antiretroviral-therapy with undetectable viral load measured by ddPCR. c, Same experiment as in Fig. 6a, but cells were treated with increasing concentrations of Cyclosporin A (CsA). d, Similar experiment that in Fig. 5b or Fig. 5c, but combining increasing concentrations of PERKi (GSK2656157 / PERK inhibitor II) and Cyclosporin A (CsA). Data represent mean ± SD of n=6 independent experiments. e, J-Lat cell line A58 was treated with increasing concentrations of Thapsigargin (0.01, 0.1, 1 µM), Tunicamycin (0.1, 0.5, 1 µg/mL), Brefeldin A (0.01, 0.1, 1 µg/mL) and Fenretinide (0.5, 2, 5 µM) for 24, 48 and 72 hours and HIV-GFP reactivation was analyzed by FACS. Data shown are mean ± SD of n≥3 independent experiments. f, J-Lat cell line A58 was treated with 0.5 µM Fenretinide and increasing concentrations of Ionomycin. HIV-GFP reactivation and cell viability were analyzed by FACS. Data are mean ± SD of n=3 independent experiments. g, Model: FOXO1 inhibition impairs autophagy, thus promoting protein accumulation and leading to ER Stress. Thus, ATF4 activation through PERK and NFAT via cytosolic calcium release will promote HIV transcription and will prevent HIV latency.
Because NFAT was also recruited to the HIV LTR after FOXO1 inhibition but is not regulated by PERK activation, we tested the effect of cyclosporin A (CsA), a potent inhibitor of calcium release and NFAT mobilization known to reduce HIV transcriptional activity49. Increasing concentrations of CsA suppressed HIV reactivation induced by AS1842856, but not TNFα, supporting a specific role of NFAT in FOXO1 inhibitor-mediated latency reversal (Figure 6c). Treatment with the PERK inhibitor or CsA alone suppressed HIV reactivation by ~ 42.5 % (49 ± 22 % (PERKi); 36 ± 7 % (CsA)), whereas the combination almost completely blocked AS1842856-mediated viral reactivation (83 ± 4 %) (Figure 6d), without affecting cell viability (Extended data Figure 6c, f). This result suggests that both ATF4 and NFAT are important for HIV reactivation in response to FOXO1 inhibition.
To validate the effect of ER stress on HIV reactivation, we treated J-Lat cells with known inducers of ER stress50: thapsigargin, tunicamycin, brefeldin A and fenretinide. All resulted in HIV-1 reactivation, with fenretinide exerting the most potent effect (Figure 6e). Each of these agents induced dose-dependent cellular toxicity, as expected51 (Extended Data Figure 6g). To minimize toxicity caused by high concentrations of fenretinide, we combined a nontoxic dose of fenretinide (0.5 μM) with increasing amounts of ionomycin to mobilize additional Ca2+ ions. Ionomycin alone was sufficient to induce HIV reactivation (Extended Data Figure 6h), but the combination with fenretinide significantly increased the effect (~64-fold over fenretinide alone), with minimal toxicity (Figure 6f and Extended Data Figure 6i). These data support a model in which FOXO1 interferes with potentially low-level chronic ER stress in resting T cells, preventing activation of ATF4 and NFAT, thereby promoting HIV latency. In turn, inhibition of FOXO1 activity promotes protein accumulation in the ER leading to ER stress signaling and calcium release, which mobilizes ATF4 and NFAT and activates HIV-1 transcription (Figure 6g).
Discussion
By uncovering a link between FOXO1 and ER stress, this work describes a mechanism through which HIV latency is established and maintained in quiescent CD4+ T cells. Our results are in agreement with work linking ER stress with CD4+ T cell activation43,52, and show that FOXO activity protects quiescent cells from oxidative stress53. We further show that this effect does not involve NF-κB activity, but that FOXO1 activity controls ATF4 and NFAT mobilization. In other systems, ATF4 interacts with FOXO1 and regulates glucose metabolism in osteoblasts54, while FOXO activity suppresses calcineurin/NFAT activation by reducing oxidative stress in cardiac physiology55,56.
Malfunction of the ER, such as excessive secretory activity, Ca2+ depletion, or presence of misfolded proteins, has a pivotal role in establishment of latent viral infections. ER stress activates the UPR, which is necessary for replication of many viruses, such as herpes viruses57. Other viruses are known to hijack the UPR to promote expression of ATF4, including hepatitis C virus58, West Nile virus59, respiratory syncytial virus60, Japanese encephalitis virus61, human cytomegalovirus62, dengue virus (DENV)63, and infectious bronchitis virus (IBV)64. In the context of HIV, our work shows that ER stress promotes active HIV infection, with FOXO1 acting as a critical regulator of this process in resting T cells. These results are in agreement with previous findings that HIV-1 infection is associated with enhanced expression of the ER stress sensors PERK, ATF4, ATF6, and IRE-1 in PBMCs65–67, and that the viral protein Tat induces ER stress in astrocytes68.
While NFAT is not a “classical” ER stress-induced factor, it can be activated by ER stress69. In addition, its close connection with intracellular calcium levels links it to the ER, where the majority of intracellular calcium is stored35. Prolonged ER stress is associated with calcium release, and the ER-resident chaperone binding immunoglobulin protein (BiP), a sensor of unfolded proteins, is itself calcium-regulated70. In the absence of ER stress, BiP is bound to monomeric PERK in the ER membrane. When BiP senses the accumulation of misfolded proteins, it dissociates from PERK, allowing the kinase to dimerize and trans-autophosphorylate31. Phosphorylated PERK promotes phosphorylation of eIF2α, which impairs translation of most mRNAs, with the exception of a few transcripts like ATF471. We hypothesize that this cascade of events explains the late reactivation kinetics of FOXO1 inhibition.
Further experiments are required to better understand the molecular link between FOXO1, ER stress and proteostasis. We demonstrate that in quiescent T cells, FOXO1 inhibition down-regulates autophagy genes and leads to an increased protein accumulation. Other studies have linked FOXO activity to autophagy induction29,30,72, and the loss of dFOXO in Drosophila attenuates autophagy73. While our current data point to inhibition of autophagy, protein accumulation in the ER and a broad activation of ER stress responses as the cause of HIV reactivation in response to FOXO1 inhibition, we cannot exclude a direct effect of FOXO1 on the PERK-ATF4 pathway. PERK is known to phosphorylate FOXO1 to enhance insulin responsiveness74 but no reciprocal effect of FOXO1 on PERK activity has yet been reported. Furthermore, it is unclear whether T cell physiology impacts the phosphorylation state of FOXO1 upon AS1842856 inhibition21,75.
Nevertheless, the discovery of this mechanism opens the door to promising therapeutic options, especially considering the synergistic effects of FOXO1 inhibition in combination with other LRAs and the latency reversing effect of ER stress-inducing agents such as fenretinide, currently in early clinical trials for cancer76. Furthermore, the cytolytic activity of CD8+ T cells, key contributors to a successful “shock and kill” strategy, is strengthened by treatment with the FOXO1 inhibitor15, in contrast to HDAC inhibitors, which suppress CD8+ T cell activity77. A combination of FOXO1 inhibitors with early ART seems especially promising, given the negative effect on latency establishment, potentially prolonging the time window for intervention to prevent maximal reservoir establishment.
Methods
Cell lines and primary cultures.
J-Lat cell lines (clones A2, A72, 6.3, 11.1 14.8 and 5A8) were obtained from the Verdin and Greene Laboratories at Gladstone Institutes, which generated these lines from Jurkat cells24,78. J-Lat and Jurkat cells were cultured at 37°C in RPMI (Corning, VA, USA), supplemented with 10% FBS (Corning, VA, USA), 1% L-glutamine (Corning, VA, USA) and 1% penicillin-streptomycin (Corning, VA, USA). K562(Cas9) cells are a myelocytic cell line derived from a chronic myelogenous leukemia (CML) patient that stably express dCas9-BFP-KRAB necessary for CRISPRi experiments. K562(Cas9) and the T cell-based cell line NH7 cells were a gift from the lab of Johnathan Weissman, at UCSF79 and were maintained in appropriate volume of RPMI medium supplemented with 10% fetal bovine serum (Serum Plus - II, Sigma MO, USA) and 1% penicillin-streptomycin. HEK293T cells (provided and authenticated by ATCC) were cultured in DMEM (Corning, VA, USA), supplemented with 10% FBS, 1% glutamine, and 100 U/mL penicillin-streptomycin at 37°C, 5% CO2. Short-term passages (<15) were used for all experiments. All cells in culture were tested for mycoplasma infection every 4–6 months. Primary CD4+ T cells were isolated from peripheral blood mononuclear cells (PBMCs) and enriched via negative selection with an EasySep Human CD4+ T Cell Isolation Kit (Stemcell Technologies, Canada). Primary CD4+ T cells were cultured at 37°C in RPMI supplemented with 10% FBS, 1% L-glutamine and 1% penicillin-streptomycin.
Chemicals.
Cells were treated with the following compounds: FOXO inhibitor / AS1842856 (344355, Calbiochem); TNFα (300–01A, PeproTech); Raltegravir (CDS023737, Sigma-Aldrich); Prostratin (P4462, LC Laboratories); PHA-M (10576015, Sigma-Aldrich); IL-2 (I2644, Sigma-Aldrich); αCD3 (40–0038, Tonbo Biosciences); αCD28 (70–0289, Tonbo Biosciences); PMA (P8139, Sigma-Aldrich); Ionomycin (I0634, Sigma-Aldrich); PERK inhibitor II / GSK2656157 (504651, Sigma-Aldrich); PERK inhibitor / AMG PERK 44 (5517, Tocris); GCN2 inhibitor / A-92 (2720, Axon Medchem); Imidazolo-oxindole PKR inhibitor C16 (I9785, Sigma-Aldrich); IRE1α inhibitor / MKC8866 (HY-104040, MedChem Express); Cyclosporin A (C3662, Sigma-Aldrich); Thapsigargin (T9033, Sigma-Aldrich); Tunicamycin (T7765, Sigma-Aldrich); Brefeldin A 1,000X Solution (00–4506-51, ThermoFisher Scientific); Fenretinide (17688, Cayman Chemicals).
Virus production.
Pseuodtyped HIVGKO (LTR-HIV-∆-env-nefATG-csGFP-EF1α-mKO2) and HIVNL4–3/Luciferase viral stocks were generated by co-transfecting (standard calcium phosphate method) HEK293T cells with a plasmid encoding HIVGKO or HIVNL4–3/Luciferase, and a plasmid encoding HIV-1 dual-tropic envelope (pSVIII-92HT593.1) or vesicular stomatitis virus G protein (VSVg), respectively. Medium was changed 6–8 h after transfection, and supernatants were collected after 48–72 hours, centrifuged (20 min, 2000 rpm, RT), filtered through a 0.45 μm membrane to clear cell debris, and then concentrated by ultracentrifugation (22,000 g, 2 h, 4 °C). Virus-like particles (VLPs) were similarly generated as described previously80. Here, HEK293T cells were co-transfected with 16 μg of a plasmid encoding Vpr-Vpx (PSIV3+), 16 μg of a plasmid encoding gp160, 40 μg of the packaging vector plasmid 8.91 and 4 μg of pAdvantage DNA (T175 flask). Concentrated virions were resuspended in complete media and stored at −80 °C. Virus concentration was estimated by p24 titration using the FLAQ assay81 (HIVGKO and VLPs) or the Lenti-X™ p24 Rapid Titer Kit (Clontech; HIVNL4–3/Luciferase).
Cell infection.
K562(Cas9) cell lines expressing dCas9-BFP-KRAB were spinoculated for 2 h at 2000 rpm at 32°C with the HIVGKO VSV-G pseudotyped dual-reporter virus encoding for GFP and mKO2 (MOI = 0.01), followed by in vivo culture with complete RPMI medium. Increasing concentrations of AS1842856 were added onto cells immediately after HIVGKO infection and were not washed away until analysis. Purified CD4+ T cells isolated from healthy peripheral blood cells were also spinoculated with HIVNL4–3 VSV-G pseudotyped virus encoding a luciferase reporter at a concentration of 0.25–1 μg of p24 per 1×106 cells. For Figure 3a, purified resting CD4+ T cells were infected with VLPs at day 0. Cells were spin-infected in a 96 well V-bottom plates (1×106 cells/well) with 100 ng of p24 VLPs per 1×106 cells (final volume 100 μl/well) for 2 h at 2,000 rpm at 4 °C and then incubated at 37°C. The following day, 100 μL of fresh complete 1640 RPMI with or without the different drugs was added to each well. Finally, 24 hours later, treated and untreated cells were spin-infected with HIVGKO as described previously82. Briefly, 1×106 cell were spin-infected with serial dilutions of HIVGKO (1/2 dilutions), starting with 300 ng of p24 for 2 h at 2,000 rpm at 32 °C. After spin-infection, fresh drugs were added to the media, and cells were cultured for an additional 4 days. Infected cells were analyzed by flow cytometry 4 days post-infection. Resting CD4+ T cells were treated 24 hours pre-HIVGKO infection with 0–10-100–1,000 nM AS1842856. The same drug concentrations were added onto cells right after HIVGKO spin-infection and were not washed away until data analysis.
T-cell activation analysis and Flow cytometry.
Cells were seeded in drug-supplemented media in triplicates onto 96-well plates with a target cell density of 0.125–0.25–0.5 × 106 cells/mL, and then incubated at 37 °C for 72–48-24 hours, respectively. In preparation for flow cytometry, cells were washed in V-shaped 96-well plates (500 x g, 5 min, 4 °C), and stained with Fixable Viability Dye eFluor 780 (65–0865-14; ThermoFisher Scientific). For T cell activation analysis, CD69 and CD25 expression was measured by flow cytometry gating on CD3+CD4+ T cells using FITC-labeled antibodies for CD3 (11–0048-42, eBioscience), APC-conjugated CD25 antibodies (17–0259-42, eBioscience), PerCP-labeled antibodies for CD4 (300528, Biolegend), and CD69-V450 (560740, BD Horizon). Staining was performed for 30 min on ice in FACS buffer (PBS, 2% FBS) before analysis. 5A8 J-Lat cells were measured on a BD FACSCalibur system (BD Biosciences) in an uncompensated setting. Infected or uninfected K562(Cas9) and CD4+ T cell data was collected using the BD LSRII flow cytometer (BD Biosciences). Data were analyzed using FlowJo V10.1 software (Tree Star).
Viability.
Cell viability (% survival) was measured by flow cytometry gating on forward and side scatter analysis and using Fixable Viability Dye eFluor 780 (65–0865-14; ThermoFisher Scientific). Trypan blue dye exclusion was used to evaluate the cell viability in the CETSA assays in K562(Cas9) cells and in the reactivation studies in HIV+ donors. A 10 μL cell aliquot from each sample was briefly mixed with an equal volume of 0.4% (w/v) trypan blue dye solution (Gibco, NY, USA) and counted. Cells with the ability to exclude trypan blue were considered viable.
Cellular Thermal Shift Assay (CETSA).
For CETSA, K562(Cas9) cell were freshly seeded one day before the experiment. On the day of the experiment, equal numbers of cells were counted by Moxi Mini automated cell counter (Orflo) and 0.6×10^6 cells per data point were seeded in T-25 cell culture flasks (VWR, PA, USA) in appropriate volume of culture medium. Cells were exposed to 100 nM AS1842856 or equal DMSO volume for 3 hours in an incubator with 5% CO2 and 37°C. Following the incubation, cells were harvested, washed with PBS and diluted in PBS supplemented with EDTA-free complete protease inhibitor cocktail (ROCHE). Then, the cells were divided into 100 μl aliquots and heated individually at different temperatures for 3 minutes (Thermal cycler, BIO-RAD) followed by cooling for 2 minutes at room temperature. Cell suspensions were freeze-thawed three times with liquid nitrogen. The soluble fraction was separated from the cell debris by centrifugation at 20000 g for 20 minutes at 4°C. The supernatants were transferred to new microcentrifuge tubes and analyzed by SDS-PAGE followed by immunoblotting analysis. CETSA experiments were performed as triplicates on different days. Immunoblotting results were subjected to densitometric analysis (Image J) and melting temperatures of FOXO1, FOXO3 and FOXO4 were determined by Prism 7 (GraphPad) using nonlinear least-squares regression fit to Y = 100/ (1 + 10 ^ (Log IC50 – X) *H), where H = Hill slope (variable). The viability of the cells was assessed in triplicate by trypan blue exclusion.
Generation of stable CRISPRi Knockdown.
Individual sgRNAs from human v2 library83 were cloned into lentiviral vectors expressing either a Puromycin resistance cassette (60955, Addgene) or into a modified vector expressing a Blasticidin resistance cassette (Gift from Nevan Krogan, UCSF). Lentiviruses were prepared in HEK293T. For single knockdowns, K562(Cas9) cell lines expressing dCas9-BFP-KRAB were transduced with a single construct and pooled selected with either Puromycin (1 μg/mL, Sigma-Aldrich) or Blasticidin (10 μg/mL, Sigma-Aldrich). See Table 1.
Table 1.
sgRNA list.
| Name | sgRNA | Resistance |
|---|---|---|
| KD-1A-FOXO1 | GCCGCAGGAGAGCCAAGAGG | Puromycin |
| KD-1B-FOXO1 | GTGCTGCCTGTTGAATGTGG | Blasticidin |
| KD-3A-FOXO3 | GGAGGGGAAAGGGAAGCGCC | Puromycin |
| KD-3B-FOXO3 | GTGGCGGCGCGCGAGCTGAC | Blasticidin |
| KD-4A-FOXO4 | GAAACAGAGACGTCAGGAGG | Puromycin |
| KD-4B-FOXO4 | GGAGCAGGAAGCTGAGTGAG | Blasticidin |
Immunofluorescence.
Primary naïve CD4+ T cells were isolated from PBMCs through negative selection (19555, STEMCELL Technologies) and treated with AS1842856 for 72 hours. Cells were collected after treatment and plated onto poly-l-lysine (P4707, Sigma-Aldrich) coated, 22- by 22-mm no. 1.5 coverslips. Cells were fixed in 4% paraformaldehyde, permeabilized with 0.1% Triton X-100, and blocked in 3% bovine serum albumin. Cells were then immunostained with the indicated antibodies: Anti-GRP78-Alexa Fluor 647 (PA1–014A-A647, Invitrogen), Proteostat Aggregate Stain (enz-51035-K100, Enzo Life Sciences), and Hoescht 33258 (H3569, Invitrogen). Coverslips were mounted onto glass slides using ProLong Gold Antifade Mountant (P36934, Invitrogen) and analyzed by confocal microscopy (Zeiss LSM 880). High-resolution images were acquired on the Zeiss LSM 880 with Airyscan using a 63×/1.4 M27 oil immersion objective. The resulting Z-stack was reconstructed and rendered in 3D using Imaris software (Bitplane). Images were formatted for publication with ImageJ. Protein aggregate foci were reconstructed via the Imaris spot detection function, which provided an analysis of total number of aggregates within a cell. The Imaris spot detection function identifies foci based on fluorescence intensity. The Imaris colocalization function was used to determine overlap of fluorescence. Thresholding for background fluorescence was determined by the Imaris automatic thresholding tool, which utilizes the Costes approach. The thresholded Mander’s correlation coefficient (MCC) measures the fraction of voxels with fluorescence positive for one channel that also contains fluorescence from another channel.
Western blot.
Cells were lysed in radioimmunoprecipitation assay buffer (50 mM Tris-HCl, pH 8, 150 mM NaCl, 1 % NP-40, 0.5 % sodium deoxycholate, and 0.1 % SDS, supplemented with protease inhibitor cocktail; Sigma-Aldrich) for 30 min at 4°C and the protein content was measured by DC Protein Assay (Bio-Rad). Protein samples (20 μg) were resuspended in Laemmli buffer and separated by SDS-PAGE. Next, samples were transferred to PVDF membranes (Immobilon-P, Millipore) and blocked with 5 % dry milk supplemented with 0.05% Tween 20 in PBS. The membranes were then immunoblotted by specific antibodies: FOXO1 (2880; Cell Signaling Technology), FOXO3 (D19A7; Cell Signaling Technology), FOXO4 (9472; Cell Signaling Technology), XBP-1s (83418; Cell Signaling Technology), IRE1α (3294; Cell Signaling Technology), IRE1α [pSer724] (NB100–2323SS; Novus Biologicals), eIF2α (9722; Cell Signaling Technology), ATF4 (11815; Cell Signaling), GAPDH (2118; Cell Signaling Technology) and β-actin (A5316; Sigma-Aldrich). For chemiluminescent detection, we used enhanced luminol-based chemiluminescent substrate (ECL) and ECL Hyperfilm (Amersham). Immunoblotting results were analyzed by densitometry (Image J).
RNA extraction, RT, and quantitative RT-PCR.
Total RNA from samples was extracted using the Direct-zol RNA kit (R2060; Zymogen). cDNA was generated using 500 ng for CD4+ T cells or 1,000 ng of total RNA for J-Lats with Superscript III reverse transcription (18080–044; ThermoFisher) and oligo(dT)12–18 (18418–012; ThermoFisher). Quantitative RT-PCR was done using Maxima SYBR Green qPCR Master Mix (Thermo Scientific) on SDS 2.4 software (Applied Biosystems) in a total volume of 12 μL. The SYBR green qPCR reactions contained 5 µl of 2× Maxima SYBR green/Rox qPCR Master Mix (K0221; ThermoFisher), 5 µl of diluted cDNA, and 1 nmol of both forward and reverse primers. The reactions were run as follows: 50°C for 2 min and 95°C for 10 min, followed by 40 cycles of 95°C for 5 s and 62°C for 30 s. See Table S2 for a qRT-PCR primer list. Primer efficiencies were around 100%. Dissociation curve analysis after the end of the PCR confirmed the presence of a single and specific product. All qPCRs were independently repeated at least three times, averaged and compared using standard deviation (±SD).
Measurement of intracellular HIV-1 mRNA by digital droplet PCR from isolated CD4+ T cells from ART-suppressed individuals.
This study sampled HIV-infected participants from the Zuckerberg San Francisco General Hospital clinic SCOPE cohorts. These individuals met the criteria of having had started ART during chronic infection and being under suppressive ART, with undetectable plasma HIV-1 RNA levels (<50 copies/mL) for a minimum of one year. The UCSF Committee on Human Research approved this study (IRB #10–01330), and the participants gave informed, written consent before enrollment. Peripheral blood mononuclear cells (PBMCs) were isolated from fresh blood (100 mL) using Lymphocyte Separation Medium (25–072-CI, Corning) and CD4+ T cells were isolated using negative selection by EasySep kit (19052, STEMCELL) according to the manufacturer’s instructions. Isolated CD4+ T cells were aliquoted at a density of 1×10^6 cells per well in 5 mL RPMI medium supplemented with 10% FBS and corresponding drugs. All drugs were prepared in the culture medium from stock solutions dissolved in DMSO. After the 72 hours treatment, total RNAs from the cells were extracted miRNeasy kit (217004, QIAGEN) with the optional on-column DNase treatment step, followed by a TURBO™ DNase (AM2238, Invitrogen) treatment post isolation. RNA was quantified using a NanoDrop Spectrophotometer ND-1,000 (NanoDrop Technologies). The SuperScript III RT First-Strand Synthesis system (18080051, Invitrogen) was used to reverse-transcribe 450–1,000 ng of RNA with random hexamers (48190–011, Invitrogen). Following cDNA synthesis, absolute quantification of the HIV Long Terminal Repeat (LTR) was performed in a duplex-digital droplet PCR reaction using the Raindrop System (Raindrop Technologies / Bio-Rad). The Raindrop Source instrument was used to generate uniform aqueous droplets (5 picoliter) for each sample on a 8 well microfluidic chip (Raindrop Source Chip). Cell-associated HIV-1 RNA was detected using LTR-specific primers F522–43 (5’ GCC TCA ATA AAG CTT GCC TTG A 3’; HXB2 522–543) and R626–43 (5’ GGG CGC CAC TGC TAG AGA 3’; 626–643). Reactions were carried out in 50 μl volumes with 2μl 25x Droplet Stabilizer (Raindance Technologies), 25μl 2x Taqman Genotyping Master Mix (Thermo Fisher Scientific), 900nm primers, 250nm probes and 5μl cDNA. Droplets were thermocycled at 95°C for 10 minutes, 45 cycles of 95°C for 15 second, 59°C for 1 minutes, followed by 98°C for 10 minutes. The thermal-cycled 8-tube strip was placed into the deck of the RainDrop Sense instrument with a second microfluidic chip (RainDrop Sense chip) used for single droplet fluorescence measurements. Data were analyzed using the Raindrop Analyst Software on a two dimensional histogram with FAM intensity on the X-axis and Vic intensity on the Y-axis, normalized to RNA input. Each drug combination was normalized to the DMSO (0.2%) control to calculate the fold induction.
Luciferase Reporter Assay.
For reactivation of latent HIV-1 provirus, cells were counted and collected as pellets by centrifugation at 1500 rpm for 10 min. Cells were then plated in 96-well U-bottom plates at 1×106 per 200 μl in the presence of 30 μM Raltegravir (Santa Cruz Biotechnology) and the indicated activator. Cells were harvested 72 hours after stimulation, washed one time with PBS, and lysed in 60 μl of Passive Lysis Buffer (Promega). After 15 min of lysis, the luciferase activity in cell extracts was quantified with a SpectraMax i3x Multi-Mode plate reader (Molecular Devices, USA) after mixing 20 μl of lysate with 100 μl of substrate (Luciferase Assay System-Promega). Relative light units (RLU) were normalized to protein content determined by DC Protein assay (BIO-RAD).
Synergy Bliss model.
To analyze drug combinatorial effects, we used the Bliss independence model from the SynergyFinder web application (https://synergyfinder.fimm.fi)84. The Bliss score (∆faxy) is the difference between the calculated reactivation value if the two drugs act independently and the observed combined reactivation values. Synergy is defined as ∆faxy > 0, while ∆faxy < 0 indicates antagonism. Positive Bliss scores represent dose combinations which have greater than additive effect26.
RNA-Seq and Ingenuity Pathway Analysis.
RNA was prepared from CD4+ T cells using the QIAgen RNeasy Plus Kit. The Gladstone Institutes Genomics Core carried out the downstream processing of the RNA samples. Strand-specific cDNA libraries were prepared using the Nugen Ovation kit (Nugen) and the libraries were deep sequenced on NextSeq 500 using single-end 75 bp sequencing. RNA-seq analysis was done using the Illumina RNAexpress application v 1.1.0. Briefly, alignment of RNA-Seq reads was performed with the STAR aligner and, after alignment of aligned reads to genes, differential gene expression was analyzed with DESeq2. Pathway analysis was carried out using Ingenuity Pathway Analysis (QIAGEN) and Gene Ontology AmiGO Term Enrichment tool (Biological Processes and Cellular Components). For the Ingenuity analysis, the entire dataset was uploaded, and a two-direction analysis carried using filters to restrict the analysis to differentially expressed genes as above. Refer to the manufacturer’s website (http://www.ingenuity.com) for further details. For Gene Ontology, individual lists of genes were uploaded and analyzed separately. Venn diagrams were generated using the online tool: http://genevenn.sourceforge.net/.
Calcium flux analysis.
For calcium flux measurement, cells were seeded in complete RPMI medium in presence or absence of AS1842856 (100 nM) at a density of 2.5 × 105 cells/mL, and then incubated at 37 °C for 72 hours. In preparation for flow cytometry, equal numbers of cells were washed and incubated with the membrane-permeable calcium sensor dye eFluor™ 514 (65–0859, eBioscience) in PBS for 15 min at room temperature. Changes in calcium intracellular free concentration were measured over 200 sec by flow cytometric analysis on a BD LSRII flow cytometer (BD Biosciences). Ionomycin (1μg/ml, Thermo Fisher Scientific) was added after 30 sec. related fluorescence was measured on a BD FACSCalibur system (BD Biosciences) in an uncompensated setting). Data were analyzed using FlowJo V10.1 software (Tree Star).
Chromatin immunoprecipitation (ChIP).
J-Lat A2, A72 and 5A8 cells were treated with TNFα (10 ng/ml) or AS1852856 (1,000 nM) for 72 hours. Cells were fixed with 1% formaldehyde (v/v) in fixation buffer (1 mM EDTA, 0.5 mM EGTA, 50 mM HEPES, pH 8.0, 100 mM NaCl), and fixation was stopped after 30 min by addition of glycine to 125 mM. The cell membrane was lysed for 15 min on ice (5 mM Pipes, pH 8.0, 85 mM KCl, 0.5% NP40, protease inhibitors). After washing with nuclear swell buffer (25 mM HEPES, pH 7.5, 4 mM KCl, 1 Mm DTT, 0.5% NP-40, 0.5 mM PMSF) and micrococcal nuclease (MNase) digestion buffer (20 mM Tris pH 7.5, 2.5 mM CaCl2, 5 mM NaCl, 1 mM DTT, 0.5 % NP-40), the pellet was resuspended in MNase buffer (15 mM Tris-HCl, pH 7.5, 5 mM MgCl2, 1 mM CaCl2, 25 mM NaCl). Subsequently, samples were incubated with MNase (New England Biolabs) for 30 min at 37°C. The reaction was quenched with 0.5 M EDTA and incubated on ice for 5 min. Nuclei were lysed (1% SDS, 10 mM EDTA, 50 mM Tris-HCl, pH 8.1, protease inhibitors), and chromatin DNA was sheared to 200–1,000 bp average size through sonication (Ultrasonic Processor CP-130, Cole Parmer). Cellular debris was pelleted, and the supernatant was recovered. Lysates were incubated overnight at 4°C with 5 μg antibody against RNA Pol II (39097, Active Motif), RelA (A301–824A, Bethyl), ATF4 (11815, Cell Signaling), NFAT1 (ab2722, Abcam), or IgG control (P120–101, Bethyl). After incubation with protein A/G Dynabeads for 2 h and washing three times with low salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl), one time with high salt buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl) and twice with TE-buffer (1 mM EDTA, 10 mM Tris-HCl, pH 8.1), chromatin was eluted (1% SDS, 0.1M NaHCO3)and recovered with Agencourt AMPure XP beads (Beckman Coulter). Bound chromatin and input DNA were treated with RNase H (New England Biolabs) and Proteinase K (Sigma-Aldrich) at 37 °C for 30 min. Immunoprecipitated chromatin was quantified by real-time PCR using the Maxima SYBR Green qPCR Master Mix (Thermo Scientific) and the ABI 7700 Sequence Detection System (Applied Biosystems). SDS 2.4 software (Applied Biosystems) was used for analysis. The specificity of each PCR reaction was confirmed by melting curve analysis using the Dissociation Curve software (Applied Biosystems). All chromatin immunoprecipitations and qPCRs were repeated at least three times, and representative results are shown. Primer sequences were HIV LTR Nuc0 forward: 5′ ATCTACCACACACAAGGCTAC 3′, HIV LTR Nuc0 reverse: 5′ GTACTAACTTGAAGCACCATCC 3′. HIV LTR Nuc1 forward: 5′ AGTGTGTGCCCGTCTGTTGT 3′, HIV LTR Nuc1 reverse: 5′ TTCGCTTTCAGGTCCCTGTT 3′.
Statistical analysis.
Two groups were compared using a two-tailed Student’s t-test. Multiple comparisons to a control were calculated using two-way ANOVA, followed by Dunnett’s test. Data are presented as mean ± SD for independent biological replicates. Statistical significance is indicated in all figures by p-values.
Extended Data
Extended Data Fig 1.
a, Jurkat-derived NH7(Cas9) cells were treated with increasing concentrations of the FOXO1 inhibitor AS1842856 just after HIVGKO infection. After 3–4 days, the percentage of latent and productively infected cells were quantified by FACS. In left panel, percentage of productively or latently cells relative to the total infection rate (bars) and total cell viability (blue dotted line) of a representative experiment. In the right panel, ratios of productive versus latent populations relative to the total infection rate upon increasing concentrations of AS1842856 treatments. Data are represented as mean ± SD of triplicate values, representative of n = 3 independent experiments. b, Representative western blots of CETSA assays in FOXO1 and FOXO3 in the presence or absence of 100 nM AS1842856. c, FOXO1 and FOXO3 CETSA-melting curves upon the presence or absence of AS1842856 100 nM in K562(Cas9) cells. Band intensities obtained from western blot analysis were normalized to the highest western blot signal which has been set to 100 %. Relative FOXO-band intensities were plotted against corresponding incubation temperatures and a nonlinear least-squares regression fit was applied. Data represent the mean ± SD of n = 3 individual experiments. d, Cell viability of CETSA assay in FOXO1 experiments assessed by Trypan Blue exclusion. Data represent the mean ± SD of three individual experiments. e, FOXO1, FOXO3 and FOXO4 gene expression in KD-1A-FOXO1 K562(Cas9) cell line analyzed by RT-qPCR, normalized to RPL13A mRNA. Data represent mean ± SD of n = 3 individual experiments, except for FOXO1 (n = 4). f, Cell growth analysis of WT, FOXO1, FOXO3 and FOXO4 knockdowns K562(Cas9) cell lines. Data are represented as mean ± SD of triplicate values. g-i, Percentage of productive or latent cells relative to the total infection rate and cell viability upon increasing concentrations of AS1842856 treatment in the WT K562(Cas9) (g) or in the FOXO1-knockdown cell lines KD-1A (h) and KD-1B (i). Data are represented as mean ± SD of n = 2 independent experiments.
Extended Data Fig 2.
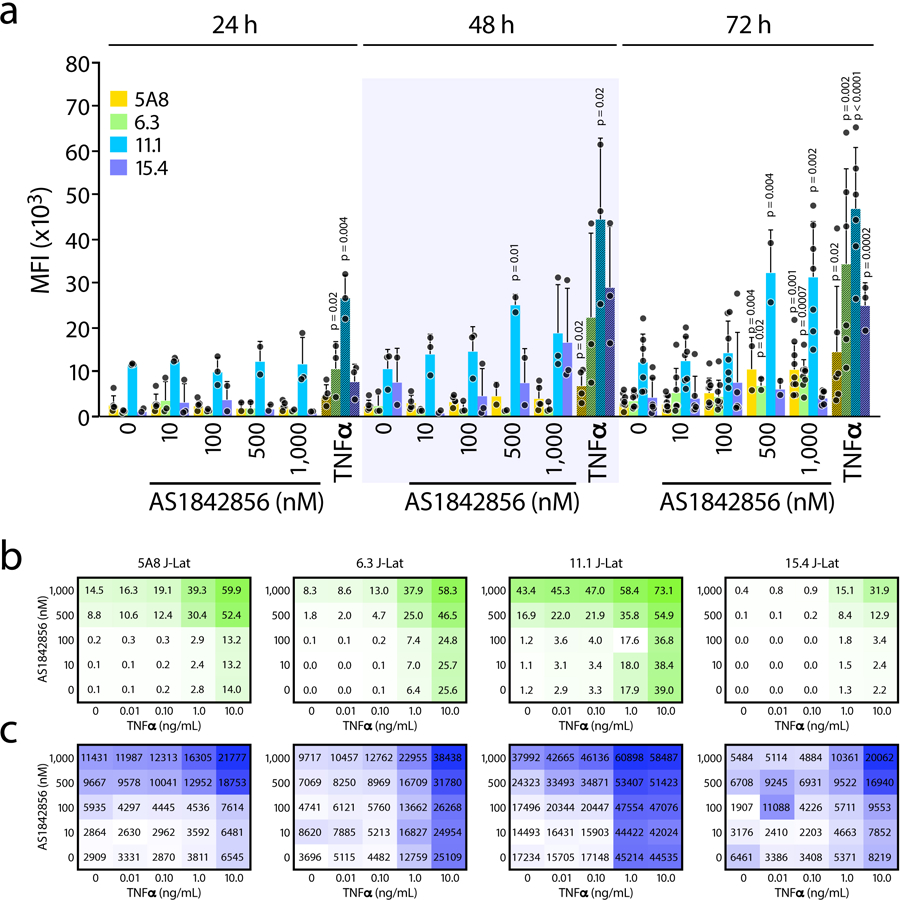
a, J-Lat cell lines 5A8, 6.3, 11.1 and 15.4 were treated with increasing concentrations of AS1842856 for 24, 48 and 72 h and HIV-GFP reactivation was analyzed by flow cytometry. HIV-GFP reactivation is reported as a Mean Intensity Fluorescence (MFI) of GFP-expressing cells. Data represent mean ± SD of n ≥ 3 independent experiments. 10 ng/mL TNFα was used as control. b, J-Lat cell lines 5A8, 6.3, 11.1 and 15.4 were treated for 72 h with increasing concentrations of both AS1842856 (Y-axis) and TNFα (X-axis) alone or in combination and analyzed by FACS. HIV-GFP reactivation is reported as a percentage of GFP-expressing cells (% GFP + cells) or c, Mean Intensity Fluorescence (MFI). Data represent mean of n = 3 independent experiments.
Extended Data Fig 3.
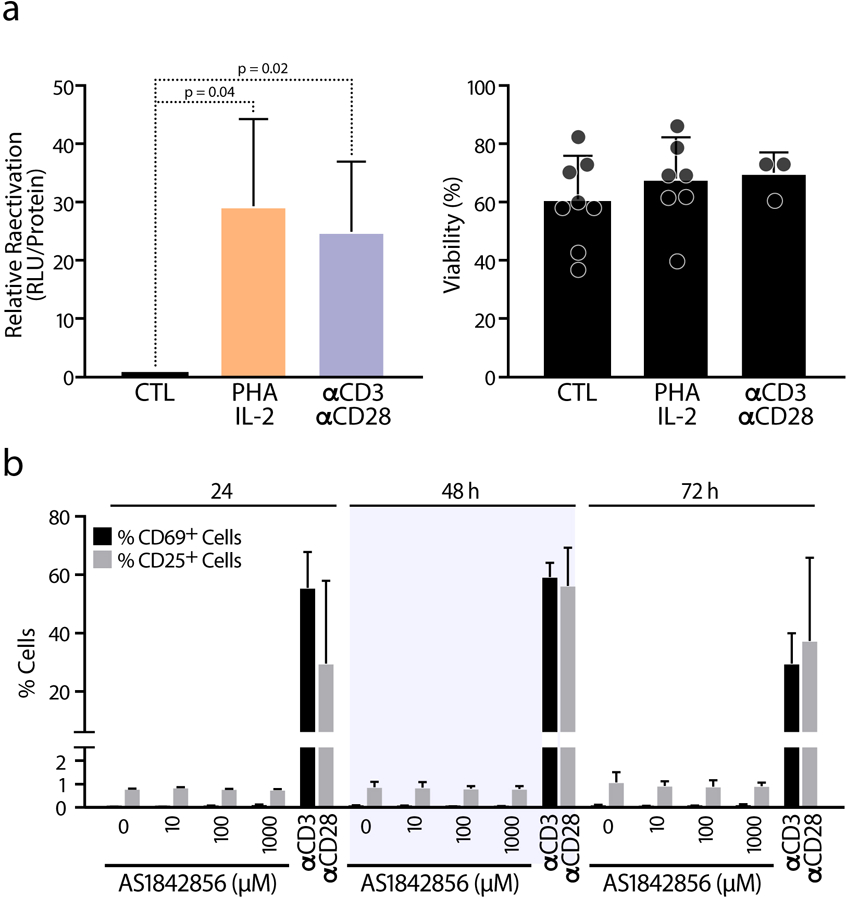
a, HIV reactivation was measured by luciferase activity and cell viability by flow cytometry assessed in CD4 T cells purified from blood of healthy donors and infected with HIV and letting them rest for 6 days before reactivation was induced with 10 μg/mL PHA + 100 U/mL IL-2 and 10 μg/mL αCD3 + 1 μg/mL αCD28 for 72 h, in the presence of raltegravir (30 μM). Data represent average ± SD of n ≥ 3 independent experiments. b, The cell surface CD69 and CD25 T cell activation markers were measured by FACS in CD4 T cells upon AS1842856 treatment for 24, 48 and 72 h. 10 μg/mL αCD3 and 1 μg/mL αCD28 was used as control. Data is shown as mean of percentage of positive cells and as mean ± SD of n = 2 biological replicates.
Extended Data Fig 4.
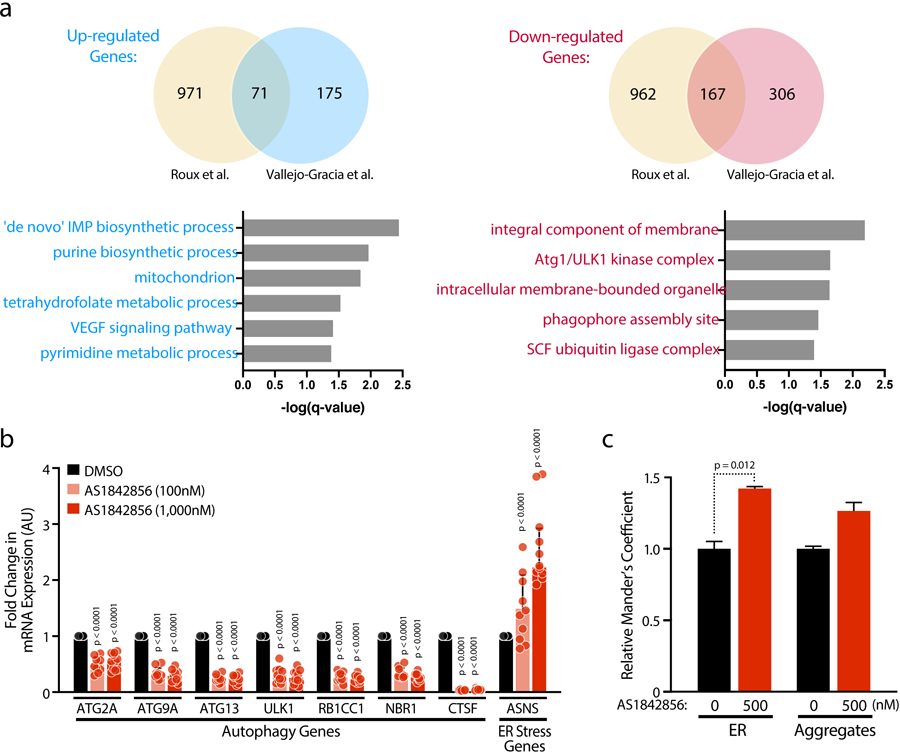
a, Venn diagrams (top panels) comparing the up- and down-regulated genes from a recently published Affimmetrix microarray[19] and RNA-Seq data presented here (Vallejo-Gracia et al.). GO Enrichment Analysis (Biological Processes and Cellular Components) from overlapped dysregulated genes from the two datasets. b, Confirmation of selected up- or down-regulated genes and pathways after 72 h AS1842856 treatment in CD4 T cells of HIV-infected patients on antiretroviral therapy with undetectable viral load by RT-qPCR, normalized to RPL13A mRNA. Data represent mean ± SD of n = 10 individual donors. c, The thresholded Mander’s correlation coefficients were determined and P value was calculated by unpaired Student’s t test. n = 90 cells per condition. Data are represented as mean ± SD.
Extended Data Fig 5.
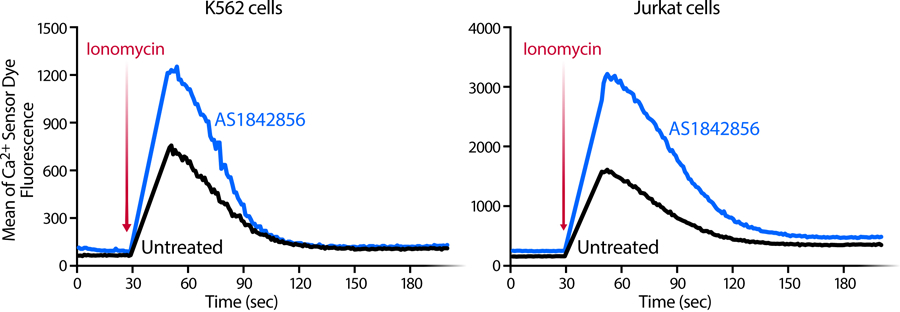
Representative plots of intracellular calcium-flux kinetics in K562(Cas9) and Jurkat cell lines in the presence or absence of AS1842856 (100 nM). Cells were stained with a membrane permeable calcium sensor dye in PBS and stimulated by adding Ionomycin after 30 seconds resulting in an increase of fluorescence indicating a calcium mobilization from the ER. Representative experiments of n = 3 independent experiments.
Extended Data Fig 6.

a, J-Lat cell line A58 was treated with increasing concentrations of GCN2i (A-92) in combination with 1,000 nM AS1842856 (72 h) or 10 ng/mL TNFα (24 h). In the upper panel, HIV-GFP reactivation was analyzed by FACS and relativized to the control. In the lower panel, histogram plots of percent live cells for each drug treatment are shown. Data are represented by mean ± SD of n = 3 different experiments. b, Same experiment as in Extended Data Fig. 6a but treating cells with increasing concentrations of PKRi (Imidazolo-oxindole PKR inhibitor C16). Data are represented by mean ± SD of three different experiments. c, Same experiment as in Extended Data Fig. 6a but treating cells with increasing concentrations of PERKi (GSK2656157 / PERK inhibitor II) (top panels) or the highly specific PERK inhibitor (AMG PERK 44) (lower panels). Histogram plots of percent live cells for each drug treatment are shown. Data represent mean ± SD of n = 3 independent experiments. d, Same experiment as in Extended Data Fig. 6a, but cells were treated with increasing concentrations of IRE1αi (MKC8866). Data are represented by mean ± SD of n = 3 different experiments. e, J-Lat cell line A58 was treated with increasing concentrations of IRE1αi (MKC8866) in combination with 1 μM Thapsigargin (6 h). Data are represented by mean ± SD of n = 3 different experiments. f, Same experiment as in Extended Data Fig. 6a but treating cells with increasing concentrations of CsA (Cyclosporin A) (left panels) or the combined concentrations of PERKi and Cyclosporin A (right panels). Histogram plots of percent live cells for each drug treatment are shown. Data represent mean ± SD of n ≥ 3 independent experiments. g, J-Lat cell line A58 was treated with increasing concentrations of Thapsigargin (0.01, 0.1, 1 μM), Brefeldin A (0.01, 0.1, 1 μg/mL) and Fenretinide (0.5, 2, 5 μM) for 24, 48 and 72 h (bottom right). Histogram plots of percent live cells for each drug treatment are shown. Data represent mean ± SD of n = 3 independent experiments. h, J-Lat cell line A58 was treated with increasing concentrations of Ionomycin (0.01, 0.1, 0.5, 1 μM) for 24, 48 and 72 h and HIV-GFP reactivation (upper panel) and cell viability (lower panel) were analyzed by FACS. Data represent mean ± SD of n = 3 independent experiments. i, J-Lat cell line 5A8 was treated for 72 h with increasing concentrations of both Fenretinide (Y-axis) and Ionomycin (X-axis) alone or in combination and analyzed by FACS. HIV-GFP reactivation is reported as a percentage of GFP-expressing cells (% GFP + cells) (upper panel) and viability was measured by FACS (bottom panel). Data represent average of n = 3 independent experiments.
Supplementary Material
Acknowledgements
We thank all members of the Ott, Verdin and Pillai laboratories for helpful discussions, reagents and expertise. We thank Dante Lacuadra and Austin Patel for assistance, Marielle Cavrois, Nandhini Raman, and the Gladstone Flow Cytometry Core for assistance with FACS, Natasha Carli, Jim McGuire and the Gladstone Genomics Core for assistance with RNA-Seq, John Carroll for graphics, Kathryn Claiborn and Brett Mensh for editorial assistance, and Veronica Fonseca and Lauren Weiser for administrative assistance. This work was supported by NIH/NIAID R01939 and NIH/NIAID R01985 to M.O., NIH/NIDA R01DA041742 and NIH/NIAID R01AI117864 to E.V., NIH/NIGMS R01GM117901 to S.K.P., and NIH P30 AI027763 to Flow Cytometry Core. Research was also supported as part of the amfAR Institute for HIV Cure Research, with funding from amfAR grant number 109301. D.B. was also funded by the Gilead HIV Cure Mentored Scientist Award from the amfAR Institute for HIV Cure Research at the UCSF AIDS Research Institute (ARI).
Footnotes
Data availability
The complete array datasets of the RNA sequencing data associated with Figure 4a, b and Extended Data Figure 4a are available at the Gene Expression Omnibus Omnibus with the accession number GSE129522. The data that support the findings of this study are available from the corresponding author upon request.
Competing interests
The authors declare that there is no conflict of interest.
References
- 1.Barré-Sinoussi F, Ross AL & Delfraissy J-F Past, present and future: 30 years of HIV research. Nat. Rev. Microbiol 11, 877–883 (2013). [DOI] [PubMed] [Google Scholar]
- 2.Archin NM, Sung JM, Garrido C, Soriano-Sarabia N & Margolis DM Eradicating HIV-1 infection: seeking to clear a persistent pathogen. Nat. Rev. Microbiol 12, 750–764 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ruelas DS & Greene WC An Integrated Overview of HIV-1 Latency. Cell 155, 519–529 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Besnard E et al. The mTOR Complex Controls HIV Latency. Cell Host Microbe 20, 785–797 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dahabieh MS, Battivelli E & Verdin E Understanding HIV Latency: The Road to an HIV Cure. Annu. Rev. Med 66, 407–421 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spivak AM & Planelles V Novel Latency Reversal Agents for HIV-1 Cure. Annu. Rev. Med 69, 421–436 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rasmussen TA & Lewin SR Shocking HIV out of hiding: where are we with clinical trials of latency reversing agents? Curr. Opin. HIV AIDS 11, 394–401 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Manning BD & Toker A AKT/PKB Signaling: Navigating the Network. Cell 169, 381–405 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hedrick SM, Michelini RH, Doedens AL, Goldrath AW & Stone EL FOXO transcription factors throughout T cell biology. Nat. Rev. Immunol 12, 649–661 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Webb AE, Kundaje A & Brunet A Characterization of the direct targets of FOXO transcription factors throughout evolution. Aging Cell 15, 673–685 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Accili D & Arden KC FoxOs at the crossroads of cellular metabolism, differentiation, and transformation. Cell 117, 421–426 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Newton RH et al. Maintenance of CD4 T cell fitness through regulation of Foxo1. Nat. Immunol 19, 838–848 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gray SM, Amezquita RA, Guan T, Kleinstein SH & Kaech SM Polycomb Repressive Complex 2-Mediated Chromatin Repression Guides Effector CD8 + T Cell Terminal Differentiation and Loss of Multipotency. Immunity 1–13 (2017). doi: 10.1016/j.immuni.2017.03.012 [DOI] [PMC free article] [PubMed]
- 14.Delpoux A et al. Continuous activity of Foxo1 is required to prevent anergy and maintain the memory state of CD8+ T cells. J. Exp. Med 215, 575–594 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jeng MY et al. Metabolic reprogramming of human CD8+ memory T cells through loss of SIRT1. J. Exp. Med 215, 51–62 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Grevenynghe J et al. Transcription factor FOXO3a controls the persistence of memory CD4+ T cells during HIV infection. Nat. Med 14, 266–274 (2008). [DOI] [PubMed] [Google Scholar]
- 17.Oteiza A & Mechti N FoxO4 negatively controls Tat-mediated HIV-1 transcription through the post-transcriptional suppression of Tat encoding mRNA. J. Gen. Virol 98, 1864–78 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Trinité B et al. Suppression of Foxo1 activity and down-modulation of CD62L (L-selectin) in HIV-1 infected resting CD4 T cells. PLoS One 9, (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roux A et al. FOXO1 transcription factor plays a key role in T cell — HIV-1 interaction. PLoS Pathog 15, 1–23 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Battivelli E et al. Distinct chromatin functional states correlate with HIV latency reactivation in infected primary CD4 + T cells. Elife 7, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nagashima T et al. Discovery of Novel Forkhead Box O1 Inhibitors for Treating Type 2 Diabetes: Improvement of Fasting Glycemia in Diabetic db/db Mice. Mol. Pharmacol 78, 961–70 (2010). [DOI] [PubMed] [Google Scholar]
- 22.Shiota M et al. Foxo3a Suppression of Urothelial Cancer Invasiveness through Twist1, Y-Box-Binding Protein 1, and E-Cadherin Regulation. Clin. Cancer Res 16, 5654–5663 (2010). [DOI] [PubMed] [Google Scholar]
- 23.Tezil T, Bodur C, Kutuk O & Basaga H IKK-β mediates chemoresistance by sequestering FOXO3; A critical factor for cell survival and death. Cell. Signal 24, 1361–1368 (2012). [DOI] [PubMed] [Google Scholar]
- 24.Jordan A, Bisgrove D & Verdin E HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. EMBO J 22, 1868–1877 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Laird GM et al. Ex vivo analysis identifies effective HIV-1 latency–reversing drug combinations. J. Clin. Invest 125, 1901–1912 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.BLISS CI The Toxicity of Poisons Applied Jointly. Ann. Appl. Biol 26, 585–615 (1939). [Google Scholar]
- 27.Lassen KG, Hebbeler AM, Bhattacharyya D, Lobritz MA & Greene WC A flexible model of HIV-1 latency permitting evaluation of many primary CD4 T-cell reservoirs. PLoS One 7, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reuse S et al. Synergistic activation of HIV-1 expression by deacetylase inhibitors and prostratin: Implications for treatment of latent infection. PLoS One 4, (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Y et al. Cytosolic FoxO1 is essential for the induction of autophagy and tumour suppressor activity. Nat. Cell Biol 12, 665–675 (2010). [DOI] [PubMed] [Google Scholar]
- 30.Webb AE & Brunnet A FOXO transcription factors: key regulators of cellular quality control. Trends Biochem. Sci 39, 159–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ron D & Walter P Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol 8, 519–529 (2007). [DOI] [PubMed] [Google Scholar]
- 32.Pakos-Zebrucka K et al. The integrated stress response. EMBO Rep 17, 1374–1395 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kilberg MS, Shan J & Su N ATF4-dependent transcription mediates signaling of amino acid limitation. Trends Endocrinol. Metab 20, 436–43 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Castilho BA et al. Keeping the eIF2 alpha kinase Gcn2 in check. Biochim. Biophys. Acta - Mol. Cell Res 1843, 1948–1968 (2014). [DOI] [PubMed] [Google Scholar]
- 35.Carreras-Sureda A, Pihán P & Hetz C Calcium signaling at the endoplasmic reticulum: fine-tuning stress responses. Cell Calcium 70, 24–31 (2018). [DOI] [PubMed] [Google Scholar]
- 36.Deniaud A et al. Endoplasmic reticulum stress induces calcium-dependent permeability transition, mitochondrial outer membrane permeabilization and apoptosis. Oncogene 27, 285–299 (2008). [DOI] [PubMed] [Google Scholar]
- 37.Jiang G et al. HIV Exploits Antiviral Host Innate GCN2-ATF4 Signaling for Establishing Viral Replication Early in Infection Downloaded from. MBio 8, 1518–1534 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Reddy TR, Tang H, Li X & Wong-Staal F Functional interaction of the HTLV-1 transactivator Tax with activating transcription factor-4 (ATF4). Oncogene 14, 2785–2792 (1997). [DOI] [PubMed] [Google Scholar]
- 39.Flanagan WF, Corthesy B, Bram RJ & Crabtree GR. Nuclear association of a T-cell transcription factor blocked by FK-506 and cyclosporin A. Nature 352, 803–807 (1991). [DOI] [PubMed] [Google Scholar]
- 40.Clipstone NA & Crabtree GR. Identification of calcineurin as a key signalling enzyme in T lymphocyte activation. Nature 356, 695–697 (1992). [DOI] [PubMed] [Google Scholar]
- 41.Schütze S, Wiegmann K, Machleidt T & Krönke M TNF-Induced Activation of NF-κB. Immunobiology 193, 193–203 (1995). [DOI] [PubMed] [Google Scholar]
- 42.Kinoshita S, Chen BK, Kaneshima H & Nolan GP Host control of HIV-1 parasitism in T cells by the nuclear factor of activated T cells. Cell 95, 595–604 (1998). [DOI] [PubMed] [Google Scholar]
- 43.So J-S Roles of Endoplasmic Reticulum Stress in Immune Responses. Mol. Cells 41, 705–716 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Walter P & Ron D The Unfolded Protein Response: From Stress Pathway to Homeostatic Regulation. Mol. Biotechnol 34, 279–290 (2006). [DOI] [PubMed] [Google Scholar]
- 45.Harding HP et al. Regulated Translation Initiation Controls Stress-Induced Gene Expression in Mammalian Cells. Mol. Cell 6, 1099–1108 (2000). [DOI] [PubMed] [Google Scholar]
- 46.Jammi NV, Whitby LR & Beal PA Small molecule inhibitors of the RNA-dependent protein kinase. Biochem. Biophys. Res. Commun 308, 50–57 (2003). [DOI] [PubMed] [Google Scholar]
- 47.Axten M et al. Discovery of GSK2656157: An Optimized PERK Inhibitor Selected for Preclinical Development. J. Med. Chem 4, 964–968 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brazeau J-F & Rosse G Triazolo[4,5-d]pyrimidine Derivatives as Inhibitors of GCN2. ACS Med. Chem. Lett 5, 282–283 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cron RQ et al. NFAT1 Enhances HIV-1 Gene Expression in Primary Human CD4 T Cells. Clin. Immunol 94, 179–191 (2000). [DOI] [PubMed] [Google Scholar]
- 50.Corazzari M et al. Targeting homeostatic mechanisms of endoplasmic reticulum stress to increase susceptibility of cancer cells to fenretinide-induced apoptosis: the role of stress proteins ERdj5 and ERp57. Br. J. Cancer 96, 1062–1071 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hail N, Kim HJ & Lotan R Mechanisms of fenretinide-induced apoptosis. Apoptosis 11, 1677–1694 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Thaxton JE et al. Modulation of Endoplasmic Reticulum Stress Controls CD4+ T-cell Activation and Antitumor Function. Cancer Immunol. Res 5, 666–675 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kops GJPL et al. Forkhead transcription factor FOXO3a protects quiescent cells from oxidative stress. Nature 419, 316–321 (2002). [DOI] [PubMed] [Google Scholar]
- 54.Kode A et al. FoxO1 Protein Cooperates with ATF4 Protein in Osteoblasts to Control Glucose Homeostasis. J. Biol. Chem 287, 8757–8768 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sundaresan NR et al. Sirt3 blocks the cardiac hypertrophic response by augmenting Foxo3a-dependent antioxidant defense mechanisms in mice. J. Clin. Invest 119, 2758–2771 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ni YG et al. Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation 114, 1159–1168 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Leo A, Chen HS, Hu CCA & Lieberman PM Deregulation of KSHV latency conformation by ER-stress and caspase-dependent RAD21-cleavage. PLoS Pathog 13, 1–22 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chan S & Egan PA Hepatitis C virus envelope proteins regulate CHOP via induction of the Unfolded Protein Response. FASEB J 1, 1–22 (2005). [DOI] [PubMed] [Google Scholar]
- 59.Medigeshi GR et al. West Nile Virus Infection Activates the Unfolded Protein Response, Leading to CHOP Induction and Apoptosis. J. Virol 81, 10849–10860 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bitko V & Barik S An Endoplasmic Reticulum-Specifc Stress-Activated Caspase (Caspase-12) is Implicated in the Apoptosis of A549 Epithelial Cells by Respiratory Syncytial Virus. J. Cell. Biochem 80, 441–454 (2001). [DOI] [PubMed] [Google Scholar]
- 61.Su H, Liao C & Lin Y Japanese Encephalitis Virus Infection Initiates Endoplasmic Reticulum Stress and an Unfolded Protein Response. J. Virol 76, 4162–4171 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Isler JA, Skalet AH & Alwine JC Human Cytomegalovirus Infection Activates and Regulates the Unfolded Protein Response. J. Virol 79, 6890–6899 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Fraser JE, Wang C, Chan KWK, Vasudevan SG & Jans DA Novel dengue virus inhibitor 4-HPR activates ATF4 independent of protein kinase R e like Endoplasmic Reticulum Kinase and elevates levels of eIF2 a phosphorylation in virus infected cells. Antiviral Res 130, 1–6 (2016). [DOI] [PubMed] [Google Scholar]
- 64.Liao Y et al. Upregulation of CHOP / GADD153 during Coronavirus Infectious Bronchitis Virus Infection Modulates Apoptosis by Restricting Activation of the Extracellular Signal-Regulated Kinase Pathway. J. Virol 87, 8124–8134 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Borsa M et al. HIV infection and antiretroviral therapy lead to unfolded protein response activation Retroviruses. Virol. J 12, 1–11 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Caselli E, Benedetti S, Gentili V, Grigolato J & Di Luca D Short communication: Activating transcription factor 4 (ATF4) promotes HIV type 1 activation. AIDS Res. Hum. Retroviruses 28, 907–912 (2012). [DOI] [PubMed] [Google Scholar]
- 67.Lee SD et al. Understanding of the functional role(s) of the Activating Transcription Factor 4(ATF4) in HIV regulation and production. BMB Rep 51, 388–393 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fan Y & He JJ HIV-1 tat induces unfolded protein response and endoplasmic reticulum stress in astrocytes and causes neurotoxicity through glial fibrillary acidic protein (GFAP) activation and aggregation. J. Biol. Chem 291, 22819–22829 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jiang S, Zhang E, Zhang R & Li X Altered activity patterns of transcription factors induced by endoplasmic reticulum stress. BMC Biochem 17, 2–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Rizzuto R, Hendershot L, Meldolesi J & Lievremont J-P BiP, a Major Chaperone Protein of the Endoplasmic Reticulum Lumen, Plays a Direct and Important Role in the Storage of the Rapidly Exchanging Pool of Ca2+. J. Biol. Chem 272, 30873–30879 (1997). [DOI] [PubMed] [Google Scholar]
- 71.Vattem KM & Wek RC Reinitiation involving upstream ORFs regulates ATF4 mRNA translation in mammalian cells. Proc. Natl. Acad. Sci 101, 11269–11274 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mammucari C et al. FoxO3 Controls Autophagy in Skeletal Muscle In Vivo. Cell Metab 6, 458–471 (2007). [DOI] [PubMed] [Google Scholar]
- 73.Juhász G et al. Gene expression profiling identifies FKBP39 as an inhibitor of autophagy in larval Drosophila fat body. Cell Death Differ 14, 1181–1190 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang W et al. ER stress potentiates insulin resistance through PERK-mediated FOXO phosphorylation. Genes Dev 27, 441–449 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zou P et al. Targeting FoxO1 with AS1842856 suppresses adipogenesis. Cell Cycle 13, 3759–3767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.[https://clinicaltrials.gov/ct2/show/NCT02495415], C. go. ClinicalTrials.gov [Internet] Identifier: NCT02495415, Primary ID: FEN T-14 , Secondary ID: NCI-2015–01195, Trial of Intravenous Fenretinide Emulsion for Patients With Relapsed/Refractory Peripheral T-cell Lymphomas Available from: https://clinicaltrials.
- 77.Tsuji G, Okiyama N, Villarroel VA & Katz SI Histone deacetylase 6 inhibition impairs effector CD8 T-cell functions during skin inflammation. J. Allergy Clin. Immunol 135, 1228–1239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chan JK, Bhattacharyya D, Lassen KG, Ruelas D & Greene WC Calcium/calcineurin synergizes with prostratin to promote NF-κB dependent activation of latent HIV. PLoS One 8, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gilbert LA et al. Resource Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Geng X, Doitsh G, Yang Z, Galloway NLK & Greene WC Efficient delivery of lentiviral vectors into resting human CD4 T cells. Gene Ther 21, 444–449 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Goadsby PJ, Kurth T & Pressman A Fluorescence-linked Antigen Quantification (FLAQ) Assay for Fast Quantification of HIV-1 p24Gag. Bio-Protocol 35, 1252–1260 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Battivelli E & Verdin E HIV GKO: A Tool to Assess HIV-1 Latency Reversal Agents in Human Primary CD4+ T Cells. Bio-Protocol 8, 1–14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Horlbeck MA et al. Compact and highly active next- generation libraries for CRISPR-mediated gene repression and activation. Elife 9, 1–20 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Ianevski A, He L, Aittokallio T & Tang J SynergyFinder: A web application for analyzing drug combination dose-response matrix data. Bioinformatics 33, 2413–2415 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.