

Abstract
Tafazzin is a mitochondrial enzyme that exchanges fatty acids between phospholipids by phospholipid-lysophospholipid transacylation. The reaction alters the molecular species composition and, as a result, the physical properties of lipids. In vivo, the most important substrate of tafazzin is the mitochondria-specific lipid cardiolipin. Tafazzin mutations cause the human disease Barth syndrome, which presents with cardiomyopathy, skeletal muscle weakness, fatigue, and other symptoms, probably all related to mitochondrial dysfunction. The reason why mitochondria require tafazzin is still not known but recent evidence suggests that tafazzin may lower the energy cost associated with protein crowding in the inner mitochondrial membrane.
Graphical abstract
Introduction
Tafazzin transfers fatty acids from phospholipids to lysophospholipids [1]. As such, it belongs to the category of acyltransferases and to the subcategory of transacylases. The latter is a group of enzymes that move acyl groups between ester bonds, catalyzing what is commonly referred to as “transesterification”. Tafazzin owes its name to the comic character “Tafazzi”, once popular on Italian television, whose masochistic traits resonated with the discoverers of the gene [2]. Since phospholipids are much more abundant than lysophospholipids and tafazzin does not change the phospholipid/lysophospholipid ratio, the overall effect of tafazzin is to redistribute fatty acids among phospholipids. Thus, tafazzin is a membrane-bound protein that affects its own environment by altering the membrane lipid composition.
Historically, the discovery of tafazzin came out of the identification of the genetic cause of Barth syndrome [2], an X-linked disease presenting with cardiomyopathy, skeletal muscle weakness, neutropenia, and abnormal growth [3]. Following a bioinformatics lead that suggested tafazzin might be an acyltransferase [4], Vreken et al demonstrated that tafazzin affects lipids, specifically the lipids of mitochondria [5]. This discovery confirmed the involvement of mitochondria in Barth syndrome, which had already been suspected from the analysis of human biopsies [3], and established the importance of lipids for the disease. Cardiolipin (CL) is the lipid that is most drastically affected by Barth syndrome. Not only does the total abundance of CL decrease [5, 6], there is also a profound change in its molecular species composition [7]. In addition, a new CL derivative, monolyso-cardiolipin (MLCL), is formed, which is usually below the detection limit in healthy tissues [8].
The enzymatic function of tafazzin [1] does not provide a straightforward explanation for its biological effects [5–8]. This is because the tafazzin reaction, as most transesterifications, is reversible. Thus, tafazzin can both produce and consume CL, which does not explain why it increases the CL concentration in vivo. Furthermore, tafazzin can react with a broad spectrum of phospholipid classes and with a large variety of fatty acids [9, 10], which does not explain why tafazzin transfers a select group of fatty acids specifically into CL.
However, we have shown that the physical properties of membranes, more specifically the packing order of membrane lipids provides a potential source of specificity. For instance, the distribution of fatty acids produced by tafazzin is different in the hexagonal phase state than in the micellar phase state [10]. Our most recent data have suggested a new hypothesis according to which the high protein density in mitochondrial membranes may create physical conditions that stimulate the tafazzin reaction and impose substrate specificity on the reaction [11].
Occurrence, localization, and isoforms
First discovered in humans [2], tafazzin homologues have also been identified in yeast [12, 13], fruit flies [14], and zebrafish [15]. The protein database UniProt [16] lists tafazzin homologues in many different species, among them plants, which suggests that tafazzins are conserved throughout the eukaryotic kingdom. Not surprisingly, many mammalian tissues have been shown to express tafazzin, including brain, liver, heart, lung, spleen, kidney, skeletal muscle, gut, monocytes, and endothelial cells [17]. There is also evidence for tafazzin expression in lymphoblasts [18] and germ cells [19] because tafazzin deletion has a large effect on CL in these cells. However, not every tissue is clinically affected in patients with Barth syndrome. For instance, the disease manifests itself in heart and skeletal muscle but not in liver or kidney. The data suggest that most eukaryotic cells express tafazzin but that the importance of tafazzin for cellular function varies considerably.
Tafazzin is localized in mitochondria. This was first hypothesized based on the effects of tafazzin deficiency on mitochondria-specific lipids [5] and then formally proven by subcellular fractionation of yeast [20, 21] and mammalian cells [22]. What turned out to be more difficult was to pinpoint the sub-mitochondrial localization of tafazzin. In part the problem was the protease resistance of endogenous tafazzin in mammalian cells, which could eventually be overcome by careful analysis of tagged tafazzin constructs [22]. Clearly, tafazzin is a membrane-bound protein that is degraded by proteases if the latter get access to the mitochondrial intermembrane space [20–22]. However, the conclusion that tafazzin is solely bound to the inner surface of the outer membrane [20] could not be confirmed [21, 22]. Instead mitochondrial sub-fractionation experiments showed that the distribution of tafazzin is in between that of inner membrane and outer membrane markers [21, 22]. Thus, tafazzin may be associated with both the outer surface of the inner membrane and the inner surface of the outer membrane, essentially lining the intermembrane space [21, 22]. Alternatively, tafazzin may be associated with protein assemblies that form contact sites between the inner and the outer membranes. Since CL is an endogenous substrate of tafazzin, the location of tafazzin implies that CL molecules must have access to the outer leaflet of the inner membrane at least once during their lifetime.
Alternative mRNA splicing creates several tafazzin transcripts in human cells. Apart from the full-length transcript, they include transcripts missing exon 5, exon 7, or both [23]. However, Western blots of human tafazzin show only one dominant protein band with the size of the isoform that lacks exon 5 [22]. Also, mice contain only a single splice variant that is missing exon 5. It is therefore likely that the mRNA without exon 5 is the one that produces native tafazzin in humans and other mammals. In spite of that, the full-length isoform of human tafazzin is active and rescues the tafazzin deletion phenotype in Drosophila [24]. The isoform lacking exon 5 is also active but not the isoforms lacking exon 7 or lacking both exons [12, 24]. Similar to humans, Drosophila expresses several splice variants of tafazzin, all of which carry enzymatic activity, but only the full-length isoform faithfully rescues the phenotype and localizes to mitochondria [24]. Thus, despite the presence of multiple tafazzin mRNAs, only one protein seems to be abundantly expressed and imported into mitochondria both in humans (protein without exon 5) and in Drosophila (full-length protein).
Structure, import, and assembly into membranes
Most studies on tafazzin have been carried out in yeast, flies, and mammalian cells. Their full-length sequences are 381, 378, and 292 amino acids long, which correspond to masses of 44 kDa (yeast tafazzin), 43 kDa (full-length Drosophila tafazzin), and 33 kDa (human full-length tafazzin) (Figure 1). As to this date, tafazzin has not been crystallized and therefore little is known about its secondary and tertiary structure, although a speculative structural model has been proposed based on target-template sequence alignment with the glycerol-3-phosphate actyltransferase from squash [25].
Figure 1.

Aligned amino acid sequences of tafazzins from humans, Drosophila, and yeast. Hydrophobic amino acids are marked blue. The red box indicates the conserved catalytic center with the HXXXXD motif characteristic of acyltransferases [31]. The blue box indicates a conserved hydrophobic segment involved in membrane anchorage [21]. The green lines indicate putative segments involved in mitochondrial targeting [26, 27].
Tafazzin does not carry a canonical targeting signal but a conserved hydrophobic segment near the N-terminus was found to be critical for targeting in yeast [26]. In humans, the corresponding region has also been implicated together with another region near the C-terminus [27] (Figure 1). Once imported into mitochondria through the TOM complex, tafazzin interacts with the Tim9-Tim10 complex of the intermembrane space [20, 26]. From there it is inserted into the membrane. It was suggested that tafazzin is inserted first into the outer membrane and then migrates to the inner membrane [26].
The dissociation of tafazzin from mitochondrial membranes requires detergents or strong alkaline conditions [21]. In that sense tafazzin is indistinguishable from transmembrane proteins. However, despite the presence of conserved hydrophobic regions that in theory could form transmembrane segments, the protease accessibility of tagged tafazzin constructs suggested that none of the hydrophobic segments actually traverses the membrane. Thus, it has been suggested that tafazzin binds through hydrophobic regions that insert into the lipid bilayer but do not cross it [21]. In yeast, the amino acid stretch 215–232 has been shown to be critical for membrane association [21] (Figure 1). It is also possible that tafazzin interacts primarily with other membrane proteins rather than with lipids, notwithstanding the obvious necessity that tafazzin comes into contact with lipids in order to carry out catalysis.
The distribution of tafazzin in blue-native gels suggests that it is assembled into multiple protein complexes. Such data were first collected in yeast [21, 28] and then in flies and mammalian cells [29]. The nature of these complexes has not been established but they are probably heterogeneous, have masses between 105-106 Da, and may involve among others the ATP synthase and the ADP/ATP carrier [28]. Cross-linking mass spectrometry in Drosophila mitochondria has also suggested multiple protein interactions (unpublished data from our laboratory). The assembly of tafazzin into these complexes seems to be critical for its stability [30]. Tafazzin has a shorter half-life than other mitochondrial proteins [29], perhaps because it is prone to unfolding and degradation by the i-AAA protease when released from its native environment [30].
In summary, tafazzin is imported into the intermembrane space from where it is passed on to protein complexes of the inner and perhaps also the outer membrane. The tertiary structure and the physical interactions of tafazzin are still not known but current evidence suggests that tafazzin is a transient resident of a heterogeneous group of protein complexes.
Enzymatic mechanism and specificity
Tafazzin belongs to a large family of acyltransferases [4], many of which carry the HX4D sequence motif that has been implicated in the catalytic mechanism [31, 32] (Figure 1). Acyltransferases typically use an activated fatty acid (acyl-CoA) as the acyl donor [33–37] but tafazzin uses a fatty acid linked to a phospholipid [1]. This has two important implications for the tafazzin reaction. First, the reaction is symmetrical, both consuming and producing a phospholipid-lysophospholipid pair, and second, its substrates and products have similar free energies and therefore the reaction is fully reversible. We have shown that tafazzin transfers medium and long chain fatty acids, including saturated and unsaturated ones, and that it reacts with both the sn-1 and the sn-2 position of different phospholipid classes [9, 10]. These include both diacyl-phospholipids and plasmalogens [38]. It seems therefore safe to extrapolate that tafazzin reacts with all phospholipids and lysophospholipids.
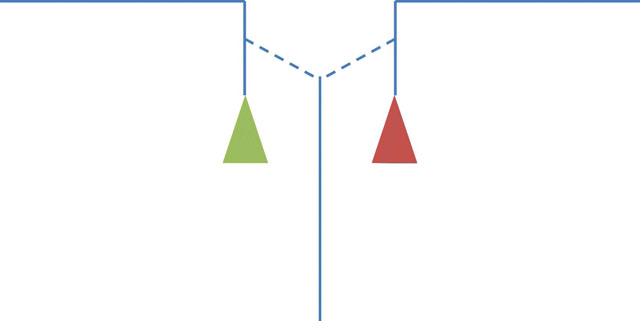
In theory, it is possible that the tafazzin reaction proceeds via a semi-stable acylated enzyme, similar to transacylations catalyzed by phospholipases [39, 40]. However, purified tafazzin does not release free fatty acids to any significant extent, which makes the idea of an acyl-enzyme intermediate unlikely [1]. Instead, we postulate that tafazzin forms an enzyme-substrate complex consisting of two lysophospholipids and one uncommitted fatty acid. This complex can release two alternative phospholipid-lysophospholipid pairs, either the original pair that formed the complex or a new pair (Figure 2). Which of the two will be released depends entirely on their respective free energies. Importantly, free energies are context-dependent, i.e. they are not an intrinsic property of the individual lipid species but are determined by how well these species fit into the packing order of the surrounding lipids. Two important issues arise regarding the reaction, one having to do with directionality and the other with specificity.
Figure 2.
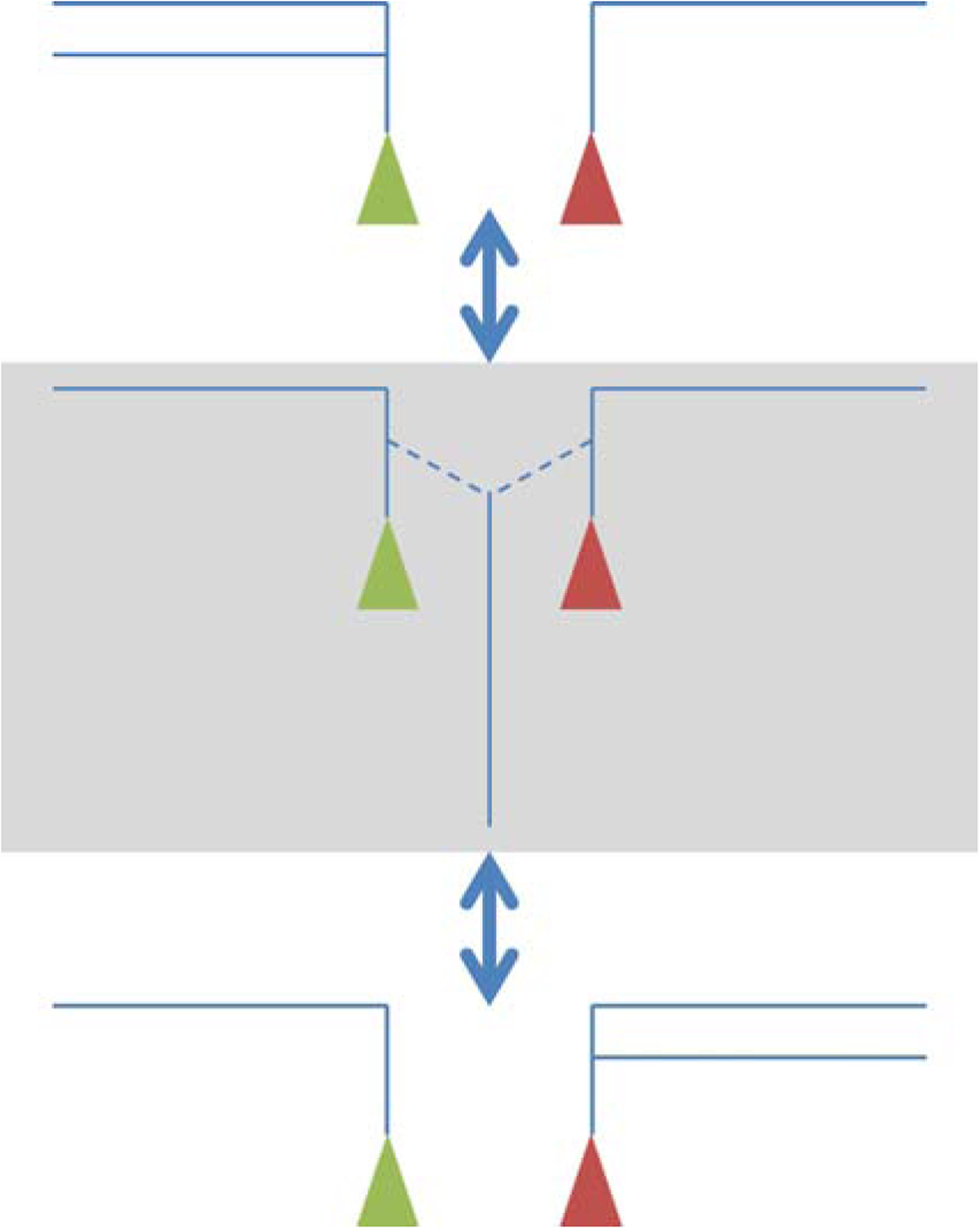
Postulated mechanism of acyl transfer by tafazzin. The enzyme-substrate complex (enzyme-product complex) is shown on a grey background. It contains two lysophospholipids and one uncommitted fatty acid. Lipids are shown as cartoons consisting of fatty acids (long lines), glycerol groups (short lines), and head groups (colored triangles).
(i). Directionality:
Tafazzin is often portrayed as catalyzing a unidirectional process that converts MLCL into CL. However, even for the simplest situation, such as the reaction of purified tafazzin with phosphatidylcholine (PC) and MLCL, there is evidence for bidirectional transfer of fatty acids [41]. This is because the products formed from dioleoyl-PC (PC18:1/18:1) and trilinoleoyl-MLCL (MLCL18:2/18:2/18:2) include not only lyso-PC and CL18:1(18:2)3 but also CL(18:1)2(18:2)2, CL(18:1)318:2, and CL(18:1)4. This is only possible if there is a ping-pong sequence of acyl exchanges between PC and CL (Figure 3). Rather than transferring a specific fatty acid from a specific phospholipid to a specific lysophospholipid, tafazzin shuttles fatty acids back and forth among various lipid species.
Figure 3.
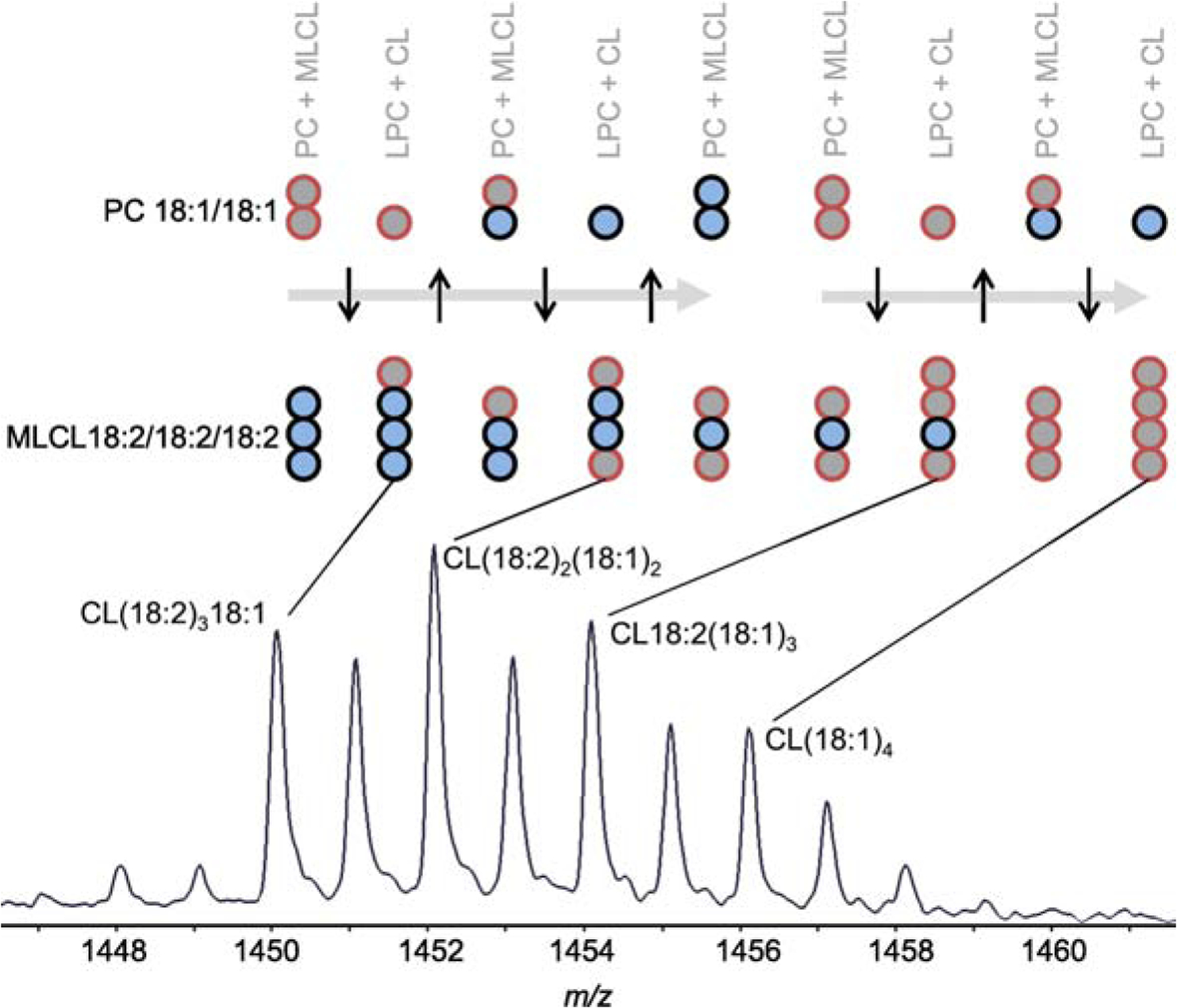
PC-CL acyl exchange. The mass spectrum shows the molecular composition of CL formed from dioleoyl-PC (PC18:1/18:1) and trilinoleoyl-MLCL (MLCL18:2/18:2/18:2) by purified tafazzin [41]. The fatty acids are presented as circles with different colors. In order to achieve the CL composition in the spectrum, multiple acyl exchange reactions between PC and CL have to occur. They involve the transfer of both oleoyl (18:1) and linoleoyl (18:2) groups. PC, phosphatidylcholine; LPC, lysophosphatidylcholine; CL, cardiolipin; MLCL monolyso-cardiolipin.
(ii). Specificity:
In vivo, tafazzin produces a specific composition of molecular species, most notably in CL [5–7]. At first glance that seems to be at odds with the promiscuous substrate usage of tafazzin [9, 10], but a brief review of basic chemical principles reveals that specificity can arise in different ways and does not necessarily require a specific enzyme. In essence, specificity can arise either when alternative substrates cause different reaction velocities or when alternative substrates cause different reaction outcomes. The former is the kinetic specificity, i.e. the fact that one type of fatty acid is transferred faster than another. The latter is the thermodynamic specificity, i.e. the fact that one type of fatty acid shows a different distribution between phospholipid classes than another in the equilibrium state. Kinetic specificity is a property of the enzyme whereas thermodynamic specificity is a property of the lipids. Although previous discussions have mostly focused on kinetics, it is the thermodynamic specificity that determines the reaction pathway in the setting of multiple competing transacylations. This is because thermodynamics determines which lipid composition is produced whereas kinetics merely determines how long it takes to achieve that result.
These theoretical considerations are fully supported by experimental data. Transgenic experiments have confirmed that the enzyme is responsible for remodeling the fatty acids but is not actually conferring the specificity. For instance, human tafazzin faithfully replicates the characteristic species pattern of CL in Drosophila even though Drosophila CL and human CL are very different [24]. The same applies to human tafazzin in yeast [12]. These data demonstrate that the specificity of CL remodeling is controlled by the host, that is the environment of tafazzin, but not by the enzyme. This is consistent with the idea of a non-specific catalyst that enables the self-organization of the lipids. Kinetic specificity of tafazzin has been described [42] but the results are confounded by the fact that the rates of reversible chemical reactions do not only depend on the catalyst but also on the thermodynamic driving force [43, 44]. When we separated the contributions of the two, we found that the kinetic contribution of the enzyme was small compared to the thermodynamic contribution of the reaction [41].
In summary, tafazzin catalyzes bidirectional and non-specific acyl transfer from phospholipids to lysophospholipids, which results in a global inter-lipid acyl exchange. This process “finds” the acyl distribution with the lowest free energy and thus enables lipids to self-organize into their most stable state.
Biological function
The role of tafazzin in cellular physiology is to improve mitochondrial performance. Exactly how this is accomplished has remained an open question. Tafazzin is not essential for survival. Deletion models in yeast [13], flies [14], and mice [19] are viable even though they show evidence of mitochondrial dysfunction particularly in tissues that are dependent on high rates of oxidative phosphorylation. Patients with genetic mutations in the tafazzin gene may be asymptomatic at times but may also experience episodes of acute deterioration [3].
Not surprisingly, the most prominent effect of tafazzin deficiency is an alteration in the mitochondrial phospholipid composition. Mitochondria contain less CL, the fatty acids of CL become more saturated, and MLCL accumulates [5–8, 13]. MLCL accumulation is a specific sign of tafazzin deficiency and a diagnostic marker of Barth syndrome [45] as it is not present in normal tissues except for testis [19]. However, the reason why MLCL accumulates has been controversial. Initially, we and others have postulated that MLCL is the intermediate of a deacylation-reacylation cycle that remodels CL. According to this idea, MLCL accumulates because the lack of tafazzin prevents its reacylation [1, 13]. But the idea of such a cycle has been contradicted by evidence showing that tafazzin prolongs the lifetime of fatty acids attached to CL whereas CL deacylation, catalyzed by Cld1, shortens the lifetime [11]. Since tafazzin and Cld1 have opposite effects on the turnover of fatty acids, they cannot be parts of a mechanistically linked reaction cycle. Furthermore, the deacylation-reacylation cycle is inconsistent with the bidirectional nature of the tafazzin reaction.
We have shown that MLCL accumulates because it is an intermediate of CL degradation and that tafazzin deficiency triggers CL degradation by weakening the association of CL with proteins [11, 46]. The lack of CL provides an explanation for the functional effects of tafazzin deficiency including the dissociation of respiratory supercomplexes [20, 46, 47] and the decrease in the maximal respiratory capacity [48, 49] because the respiratory chain is strongly affected by CL. It has been shown that the lack of CL, rather than its altered species composition, is the main reason for impaired bioenergetics in tafazzin-deficient yeast [49, 50].
The question then arises as to why tafazzin is necessary to maintain normal CL levels. One important clue is that the outcome of the tafazzin reaction, in other words the lipid composition that the reaction is creating, critically depends on the phase state of lipids [10]. This result can be conceptualized on the basis of the shape of lipid molecules and the associated packing properties [51]. For instance, if the phase state requires wedge-shaped molecules because it involves negatively curved lipid layers, such as the hexagonal state, wedge-shaped species, such as tetralinoleoyl-CL, naturally accumulate in a free-for-all exchange of fatty acids. Therefore, we propose that tafazzin stabilizes CL in vivo by creating a more stable physical state of mitochondrial lipids.
The second clue is that the stabilizing effect of tafazzin has to do with the interaction between CL and proteins. Specifically, we observed synergy between tafazzin and the complexes of oxidative phosphorylation. Increasing the expression of respiratory supercomplexes in the inner membrane partially reverses the CL instability induced by the lack of tafazzin [46]. Conversely, a global decrease in supercomplexes destabilizes CL. Our preliminary data indicate that the effects on CL correlate with the global expression of complexes of oxidative phosphorylation, not just with the abundance of supercomplexes. Genetic manipulations that cause a substantial reduction in the protein expression levels have the same effect on CL as deletion of tafazzin, including a reduced CL concentration, an altered CL species composition, and an elevated MLCL/CL ratio [11]. In contrast, other disturbances of mitochondria, even if they inhibit respiratory function, do not affect CL as long as they do not prevent the accumulation of complexes of oxidative phosphorylation in the inner membrane [11].
On the basis of these data, we have postulated that the high protein concentration in the inner membrane is the main reason why tafazzin is needed in mitochondria. Mitochondrial cristae are among the protein-richest membranes in the cell [52]. High surface density of membrane proteins has been shown to disturb the packing order of lipids, which increases the energy cost of membrane assembly [51]. According to the flexible surface model [51] this disturbance induces curvature elastic stress, which can be mitigated by creating intrinsic negative curvature in both leaflets of the membrane (Figure 4). Thus, we propose that tafazzin by creating lipids with intrinsic negative curvature such as unsaturated CL, alters the fluid-mechanical properties of mitochondrial membrane lipids, and by doing so reduces the energy cost of protein crowding in the inner membrane. As a result of tafazzin’s action, more proteins can be accommodated in the inner membrane without compromising membrane stability and mitochondria are able to sustain higher rates of ATP production. While this hypothesis is still to be tested, it provides answers to questions, such as why is tafazzin present in mitochondria and why are tissues with high-energy expenditure most susceptible to tafazzin deficiency.
Figure 4.
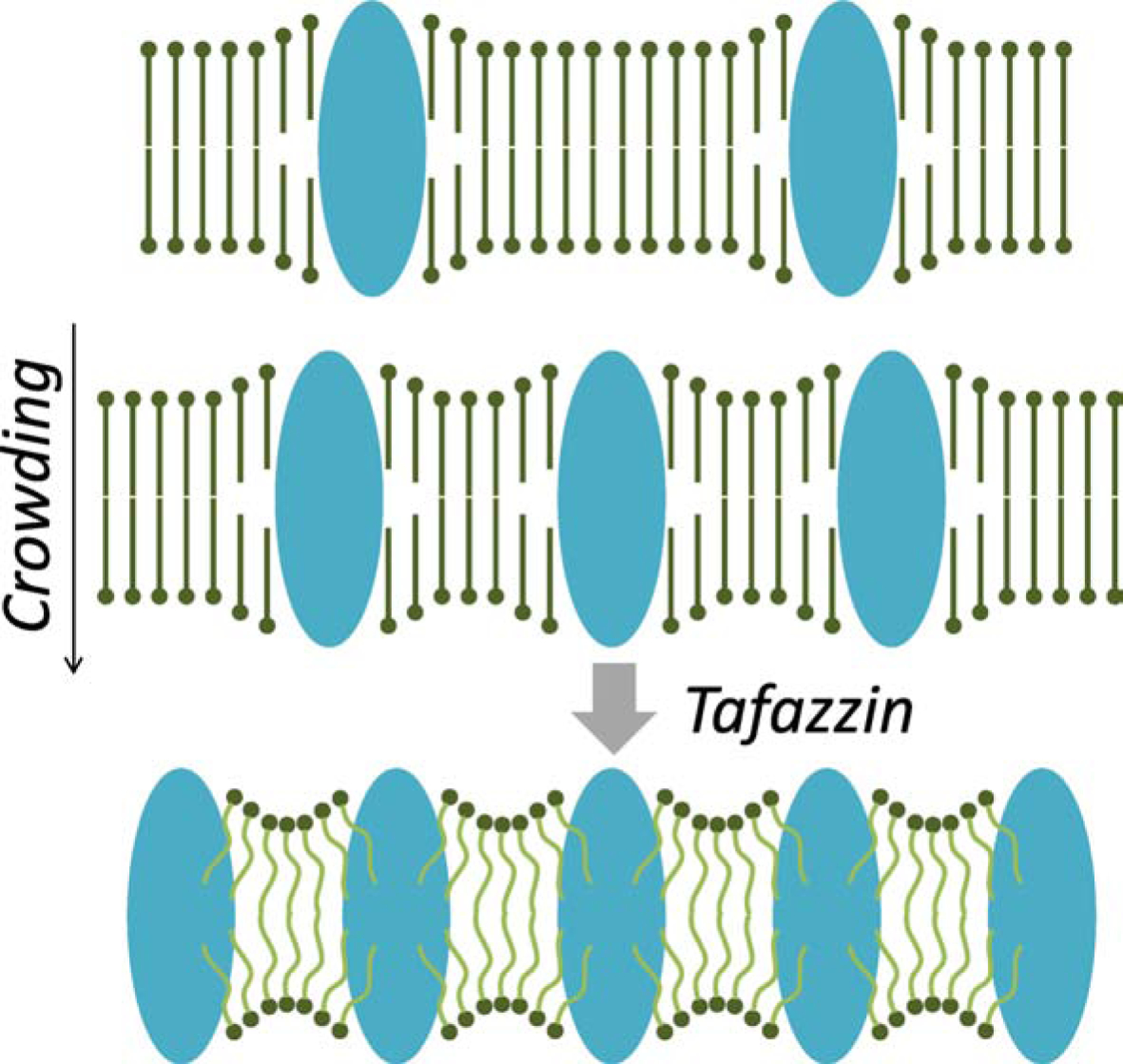
Postulated function of tafazzin. Top: Membrane proteins disturb the packing order of bilayer lipids due to hydrophobic mismatch and other distortions at the lipid-protein interface. Middle: The more proteins are present per surface area the more lipids are affected. This creates curvature elastic stress, i.e. lipids cannot establish a smooth packing order because their intrinsic curvature is at odds with the imposed curvature [51]. Bottom: By forming lipids with intrinsic negative curvature, such as unsaturated CLs, tafazzin relieves the packing stress. This creates a stable membrane despite the high protein density.
Perspective
Although tafazzin was discovered >20 years ago, the enzymatic reaction of tafazzin was identified >10 years ago, and the relevance to Barth syndrome has encouraged funding of tafazzin research, some of the most fundamental aspects of the enzyme have remained elusive. In that sense, the discoverers of tafazzin have shown remarkable foresight by naming the enzyme after a masochistic clown [2]. Among the many unresolved issues, the following four are the most pressing in our opinion. (i) It has not been clarified why several splice variants of tafazzin are transcribed, whether more than one splice variant is translated, and if so what the biological significance is. (ii) The crystal structure of tafazzin is not known. This is a barrier to a better understanding of the catalytic mechanism and the membrane association of tafazzin. (iii) The precise localization, or the spectrum of localizations of tafazzin within mitochondria have remained ambiguous. This pertains in particular to identifying the spectrum of protein interactions tafazzin is engaged in. (iv) Finally and perhaps most importantly, the biological function of tafazzin remains to be defined. Our proposal that tafazzin supports protein crowding in the inner membrane, is an idea that still requires rigorous proof.
Highlights.
Tafazzin is a mitochondrial enzyme
It exchanges fatty acids between phospholipids and lysophospholipids
The reaction alters the molecular species composition of membrane lipids
The most important substrate of tafazzin cardiolipin
Tafazzin mutations cause the human disease Barth syndrome
Acknowledgements
The author’s work is supported by grant R01GM115593 from the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Xu Y, Malhotra A, Ren M, Schlame M, The enzymatic function of tafazzin, J. Biol. Chem 281 (2006) 39217–39224. [DOI] [PubMed] [Google Scholar]
- [2].Bione S, D’Adamo P, Maestrini E, Gedeon AK, Bolhuis PA PA, Toniolo D, A novel X-linked gene, G4.5. is responsible for Barth syndrome, Nat. Genet 12 (1996) 385–389. [DOI] [PubMed] [Google Scholar]
- [3].Barth PG, Scholte HR, Berden JA, Van JM der Klei-Van Moorsel, I.E. Luyt-Houwen, E.T. Van ‘t Veer-Korthof, J.J. Van der Harten, M.A. Sobotka-Plojhar, An X-linked mitochondrial disease affecting cardiac muscle, skeletal muscle and neutrophil leucocytes, J. Neurol. Sci 62 (1983) 327–355. [DOI] [PubMed] [Google Scholar]
- [4].Neuwald AF, Barth syndrome may be due to an acyltransferase deficiency, Curr. Biol 7 (1997) R465–466. [DOI] [PubMed] [Google Scholar]
- [5].Vreken P, Valianpour F, Nijtmans LG, Grivell LA, Plecko B, Wanders RJA, Barth PG, Defective remodeling of cardiolipin and phosphatidylglycerol in Barth syndrome, Ciochem. Biophys. Res. Comm 279 (2000) 378–382. [DOI] [PubMed] [Google Scholar]
- [6].Schlame M, Kelley RI, Feigenbaum A, Towbin JA, Heerdt PM, Schieble T, Wanders RJA, DiMauro S, Blanck TJJ, Phospholipid abnormalities in children with Barth syndrome, J. Am. Coll. Cardiol 42 (2003) 1994–1999. [DOI] [PubMed] [Google Scholar]
- [7].Schlame M, Towbin JA, Heerdt PM, Jehle R, DiMauro S, Blanck TJJ, Deficiency of tetralinoleoyl-cardiolipin in Barth syndrome, Ann. Neurol 51 (2001) 634–737. [DOI] [PubMed] [Google Scholar]
- [8].Valianpour F, Mitsakos V, Schlemmer D, Towbin JA, Taylor JM, Ekert PG, Thorburn DR, Munnich A, Wanders RJ, Barth PG PG, Vaz FM, Monolysocardiolipins accumulate in Barth syndrome but do not lead to enhanced apoptosis, J. Lipid Res 46 (2005) 1182–1195. [DOI] [PubMed] [Google Scholar]
- [9].Malhotra A, Xu Y, Ren M, Schlame M, Formation of molecular species of mitochondrial cardiolipin. 1. A novel transacylation mechanism to shuttle fatty acids between sn-1 and sn-2 positions of multiple phospholipid species, Biochim Biophys. Acta 1791 (2009) 314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Schlame M, Acehan D, Berno B, Xu Y, Valvo S, Ren M, Stokes DL, Epand RM, The physical state of lipid substrates provides transacylation specificity for tafazzin, Nat. Chem. Biol 8 (2012) 862–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Xu Y, Anjaneyulu M, Donelian A, Yu W, Greenberg ML, Ren M, Owusu-Ansah E, Schlame M, Assembly of the complexes of oxidative phosphorylation triggers the remodeling of cardiolipin, Proc. Natl. Acad. Sci. USA 116 (2019) 11235–11240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Vaz FM, Houtkooper RH, Valianpour F, Barth PG, Wanders RJ, Only one splice variant of the human TAZ gene encodes a functional protein with a role in cardiolipin metabolism, J. Biol. Chem 278 (2003) 43089–43094. [DOI] [PubMed] [Google Scholar]
- [13].Gu Z, Valianpour F, Chen S, Vaz FM, Hakkaart GA, Wanders RJ, Greenberg ML, Aberrant cardiolipin metabolism in the yeast taz1 mutant: a model for Barth syndrome, Mol. Microbiol 51 (2004) 149–158. [DOI] [PubMed] [Google Scholar]
- [14].Xu Y, Condell M, Plesken H, Edelman-Novemsky I, Ma J, Ren M, Schlame M, A Drosophila model of Barth syndrome, Proc. Natl. Acad. Sci. U S A 103 (2006) 11584–11588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Khuchua Z, Yue Z, Batts L, Strauss AW, A zebrafish model of human Barth syndrome reveals the essential role of tafazzin in cardiac development and function, Circ. Res 99 (2006) 201–208. [DOI] [PubMed] [Google Scholar]
- [16].The UniProt Consortium, UniProt: the universal protein knowledgebase, Nucleic Acids Research 45 (2017) D158–D159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Lu B, Kelher MR, Lee DP, Lewin TM, Coleman RA, Choy PC, Hatch GM GM, Complex expression pattern of the Barth syndrome gene product tafazzin in human cell lines and murine tissues, Biochem. Cell. Biol 82 (2004) 569–576. [DOI] [PubMed] [Google Scholar]
- [18].Xu Y, Sutachan JJ, Plesken H, Kelley RI, Schlame M, Characterization of lymphoblast mitochondria from patients with Barth syndrome, Lab. Invest 85 (2005) 823–830. [DOI] [PubMed] [Google Scholar]
- [19].Ren M, Xu Y, Erdjument-Bromage H, Donelian A, Phoon CKL, Terada N, Strathdee D, Neubert TA, Schlame M, Extramitochondrial cardiolipin suggests a novel function of mitochondria in spermatogenesis, J. Cell Biol 218 (2019) 1491–1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Brandner K, Mick DU, Frazier AE, Taylor RD, Meisinger C, Rehling P, Taz1, an outer mitochondrial membrane protein, affects stability and assembly of inner membrane protein complexes: Implications for Barth syndrome, Mol. Biol. Cell 16 (2005) 5202–5214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Claypool SM, McCaffery M, Koehler CM, Mitochondrial mislocalization and altered assembly of a cluster of Barth syndrome mutant tafazzins, J. Cell Biol 174 (2006) 379–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lu YW, Galbraith L, Herndon JD, Lu YL, Pras-Raves M, Vervaart M, Van Kampen A, Luyf A, Koehler CM, McCaffery JM, Gottlieb E, Vaz FM, Claypool SM, Defining functional classes of Barth syndrome mutation in humans, Hum. Mol. Gen 25 (2016) 1754–1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Kirwin SM, Manolakos A, Swain Barnett S, Gonzalez IL, Tafazzin splice variants and mutations in Barth syndrome, Molecular Genetics and Metabolism 111 (2014) 26–32. [DOI] [PubMed] [Google Scholar]
- [24].Xu Y, Zhang S, Malhotra A, Edelman-Novemsky I, Ma J, Kruppa A, Cernicica C, Blais S, Neubert TA, Ren M, Schlame M, Characterization of tafazzin splice variants from humans and fruit flies, J. Biol. Chem 284 (2009) 29230–29239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hijikata A, Yura K, Ohara O, Go M, Structural and functional analyses of Barth syndrome-causing mutations and alternative splicing in the tafazzin acyltransferase domain, Meta Gene 4 (2015) 92–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Herndon JD, Claypool SM, Koehler CM, The Taz1p transacylase is imported and sorted into the outer mitochondrial membrane via a membrane anchor domain, Eukaryotic Cell 12 (2013) 1600–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dinca AA, Chien W-M, Chin MT, Identification of novel mitochondrial localization signals in human tafazzin, the cause of the inherited cardiomyopathic disorder Barth syndrome, Journal of Molecular and Cellular Cardiology 114 (2018) 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Claypool SM, Boontheung P, McCaffery JM, Loo JA, Koehler CM, The cardiolipin transacylase, tafazzin, associates with two distinct respiratory components providing insight into Barth syndrome, Molecular Biology of the Cell 19 (2008) 5143–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Xu Y, Malhotra A, Claypool SM, Ren M, Schlame M, Tafazzins from Drosophila and mammalian cells assemble in large protein complexes with a short half-life, Mitochondrion 21 (2015) 27–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Claypool SM, Whited K, Srijumnong S, Han X, Koehler CM, Barth syndrome mutations that cause tafazzin complex lability, J. Cell Biol 192 (2011) 447–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Lewin TM, Wang P, Coleman RA, Analysis of amino acid motifs diagnostic for the snglycerol-3-phosphate acyltransferase reaction, Biochemistry 38 (1999) 5764–5771. [DOI] [PubMed] [Google Scholar]
- [32].Heath RJ, Rock CO, A conserved histidine is essential for glycerolipid acyltransferase catalysis, J. Bacteriol 180 (1998) 1425–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Gijon MA, Riekhof WR, Zarini S, Murphy RC, Voelker DR, Lysophospholipid acyltransferases and arachidonate recycling in human neutrophils, J. Biol. Chem 283 (2008) 30235–30245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Shindou H, Hishikawa D, Harayama T, Endo M, Shimizu T, Generation of membrane diversity by lysophospholipid acyltransferases, J. Biochem 154 (2013) 21–28. [DOI] [PubMed] [Google Scholar]
- [35].Yamashita A, Hayashi Y, Nemoto-Sasaki Y, Ito M, Oka S, Tanikawa T, Waku K, Sugiura T, Acyltransferases and transacylases that determine the fatty acid composition of glycerolipids and the metabolism of bioactive lipid mediators in mammalian cells and model organisms, Progr. Lipid Res 53 (2014) 18–81. [DOI] [PubMed] [Google Scholar]
- [36].Gonzalez-Baro MR, Coleman RA, Mitochondrial acyltransferases and glycerophospholipid metabolism, Biochim. Biophys. Acta Mol. Cell. Biol. Lipids 1862 (2017) 49–55. [DOI] [PubMed] [Google Scholar]
- [37].Bradley RM, Duncan RE, The lysophosphatidic acid acyltransferases (acylglycerophosphate acyltransferases) family: one reaction, five enzymes, many roles, Curr. Opin. Lipidol 29 (2018) 110–115. [DOI] [PubMed] [Google Scholar]
- [38].Kimura T, Kimura AK, Ren M, Berno B, Xu Y, Schlame M, Epand RM, Substantial decrease in plasmalogen in the heart associated with tafazzin deficiency, Biochemistry 57 (2018) 2162–2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ghosh M, Tucker DE, Burchett SA, Leslie CC, Properties of the Group IV phospholipase A2 family, Prog. Lipid Res 45 (2006) 487–510. [DOI] [PubMed] [Google Scholar]
- [40].Ma Z, Turk J, The molecular biology of the group VIA Ca2+-independent phospholipase A2, Prog. Nucleic Acid Res. Mol. Biol 67 (2001) 1–33. [DOI] [PubMed] [Google Scholar]
- [41].Schlame M, Xu Y, Ren M, The basis for acyl specificity in the tafazzin reaction, J. Biol. Chem 292 (2017) 5499–5506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Abe M, Hasegawa Y, Oku M, Sawada Y, Tanaka E, Sakai Y, Miyoshi H, Mechanism for remodeling of the acyl chain composition of cardiolipin catalyzed by Saccharomyces cerevisiae tafazzin, J. Biol. Chem 291 (2016) 15491–15502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Alberty RA, Relations between biochemical thermodynamics and biochemical kinetics, Biophys. Chem 124 (2006) 11–17. [DOI] [PubMed] [Google Scholar]
- [44].Noor E, Flamholz A, Liebermeister W, Bar-Even A, Milo R, A note on the kinetics of enzyme action: a decomposition that highlights thermodynamic effects, FEBS Lett. 587 (2013) 2772–2777. [DOI] [PubMed] [Google Scholar]
- [45].Kulik W, van Lenthe H, Stet FS, Houtkooper RH, Kemp H, Stone JE, Steward CG, Wanders RJ, Vaz FM, Bloodspot Assay Using HPLC–Tandem Mass Spectrometry for Detection of Barth Syndrome, Clin. Chem 54 (2008) 371–378. [DOI] [PubMed] [Google Scholar]
- [46].Xu Y, Phoon CKL, Berno B, D’Souza K, Hoedt E, Zhang G, Neubert TA, Epand RM, Ren M, Schlame M, Loss of protein association causes cardiolipin degradation in Barth syndrome, Nature Chem. Biol 12 (2016) 641–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].McKenzie M, Lazarou M, Thorburn DR, Ryan MT, Mitochondrial respiratory chain supercomplexes are destabilized in Barth syndrome patients, J. Mol. Biol 361 (2006) 462–469. [DOI] [PubMed] [Google Scholar]
- [48].Wang G et al. , Modeling the mitochondrial cardiomyopathy of Barth syndrome with induced pluripotent stem cell and heart-on-chip technologies, Nat. Med 20 (2014) 616–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Baile MG, Sathappa M, Lu Y-W, Pryce E, Whited K, McCaffery JM, Han X, Alder NN, Claypool SM, Unremodeled and remodeled cardiolipin are functionally indistinguishable in yeast, J. Biol. Chem 289 (2014) 1768–1778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Ye C, Lou W, Li Y, Chatzispyrou IA, Hüttemann M, Lee I, Houtkooper RH, Vaz FM, Chen S, Greenberg ML, Deletion of the cardiolipin-specific phospholipase Cld1 rescues growth and life span defects in the tafazzin mutant. Implications for Barth syndrome, J. Biol. Chem 289 (2014) 3114–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Brown MF, Soft matter in lipid-protein interactions, Annu. Rev. Biophys 46 (2017) 379–410. [DOI] [PubMed] [Google Scholar]
- [52].Lotan R, Nicolson GL, Plasma membranes of eukaryotes, in Advanced Cell Biology, Schwartz LM, Azar MM, eds, Van Nostrand-Reinhold, Princeton, NJ, (1981) 129–154. [Google Scholar]