

Abstract
G protein-coupled receptors (GPCRs) are privileged structural scaffolds in biology that have the versatility to regulate diverse physiological processes. Interestingly, many GPCR ligands exhibit significant “bias”—the ability to preferentially activate subsets of the many cellular pathways downstream of these receptors. Recently, complementary information from structural and spectroscopic approaches have made significant inroads into understanding the mechanisms of these biased ligands. The consistently emerging theme is that GPCRs are highly dynamic proteins, and ligands with varying pharmacological properties differentially modulate the equilibrium among multiple conformations. Biased signaling and other recently appreciated complexities of GPCR signaling thus appear to be a natural consequence of the conformational heterogeneity of GPCRs and GPCR-transducer complexes.
Keywords: G protein-coupled receptor, arrestin, biased ligand, cellular signaling, pharmacology, biophysics
G protein-coupled receptors
G protein-coupled receptors (GPCRs) comprise a family of over 800 integral membrane proteins in humans that regulate physiological processes ranging from synaptic firing to chemotaxis to metabolism. Binding of extracellular ligands leads to conformational changes that are propagated through the seven-transmembrane (7TM) helix bundle, promoting interactions with and activation of intracellular transducers (see Glossary) (in particular, heterotrimeric G proteins, GPCR kinases [GRKs], and β-arrestins). Impressively, utilizing a highly conserved structural scaffold and these relatively small families of transducers, GPCRs are somehow able to achieve the different spatiotemporal signaling patterns required by the diverse processes they regulate.
Another facet of GPCRs’ versatility is that different agonists of the same receptor can effect qualitatively different patterns of signaling. Classic models of GPCR allostery and activation, describing an equilibrium between a single inactive and a single signaling-competent active conformation, cannot account for the complex pharmacology of these “biased ligands.” This has sparked intense interest in what makes the conformations stabilized by biased ligands “unique.” In this review, we discuss how data from multiple biophysical approaches are revealing that the breadth of pharmacology observed in GPCR ligands inherently follows from the conformational heterogeneity of GPCRs. This conformational heterogeneity also endows GPCRs with the properties needed to serve so universally as privileged structural scaffolds. We focus on Family A GPCRs that are activated by diffusible ligands (unlike rhodopsin, a light-activated Family A GPCR that has been an invaluable model); other GPCR families have large extracellular domains involved in their regulation (reviewed in [1–3]).
Signaling versatility of GPCRs
Most GPCRs couple to one or more heterotrimeric G proteins; coupling specificity is principally determined by the Gα subunits, of which there are 16 in humans [4]. Agonist activation of GPCRs leads to exchange of guanosine-5’-diphosphate (GDP) for guanosine-5’-triphosphate (GTP) in the heterotrimer and dissociation of the Gα and Gβ/Gγ subunits, allowing each subunit to trigger myriad intracellular responses (e.g., Gα-mediated second messenger signaling including 3’,5’-cyclic adenosine monophosphate (cAMP), inositol 1,4,5-phosphate [IP3]). GRKs (GRKs -2, -3, -5, and -6 are broadly expressed) then phosphorylate GPCRs in the C-terminal tail and/or intracellular loop (ICL) 3, facilitating β-arrestin recruitment [5]. The two ubiquitously expressed β-arrestins, β-arrestin1 (also known as arrestin2) and β-arrestin2 (arrestin3), can directly modulate GPCR function by “desensitizing” G protein activation (i.e., by sterically blocking GPCRs’ intracellular transducer-binding pocket) and by promoting receptor endocytosis [5]. In addition, β-arrestins scaffold many signaling proteins and thereby activate receptor-dependent signaling pathways distinct from those initiated by G proteins.
The discovery of so-called “biased ligands” almost two decades ago provided early evidence that GPCR signaling is not simply a series of linear, determinate events. In comparison to a reference ligand, biased ligands vary in their relative efficacy to activate two or more of the multiple responses downstream of GPCRs (Box 1). For example, one of the earliest recognized—and still most pronounced—examples of biased signaling is at the angiotensin II type 1 receptor (AT1R). Certain β-arrestin-biased analogs of the endogenous agonist angiotensin II (AngII) were identified that do not appreciably activate Gq signaling (e.g., increases in IP3 and Ca2+) yet still induce receptor phosphorylation, internalization, and mitogen-activated protein kinase (MAPK) signaling [6, 7]. Other Gq-biased analogs show a gain of function instead of a loss of function, stimulating Gq even more efficaciously than AngII but activating β-arrestin pathways comparably to AngII [8, 9]. These and similar observations at other GPCRs showed that G protein-mediated and GRK/β-arrestin- mediated responses can to some degree be differentially modulated.
Box 1: Ligand efficacy and bias.
Efficacy describes the ability of a ligand to activate a single response (Figure IA, IB). Partial agonists sub-maximally activate a particular response; ligands B, C, and D are all partial agonists for both G protein and β-arrestin activation. Bias describes the relative efficacy of a ligand at multiple pathways as compared to an arbitrary reference ligand, often the endogenous agonist of a GPCR. Importantly, a bias factor is not a unique descriptor of a ligand’s functional profile, as demonstrated in Figure IC. Compared to a reference ligand, ligands A and B are both G protein-biased. Ligand A becomes biased by gaining G protein efficacy relative to the reference ligand, without losing efficacy towards β-arrestin (similar to the AT1R Gq-biased ligands discussed in this review [8, 9]). Ligand B becomes G protein-biased by losing more efficacy towards β-arrestin than towards G protein activation (similar to reported Gi-biased μ-opioid receptor agonists [43]). These two G protein-biased ligands would be expected to stabilize different distributions of receptor conformations. Ligand C is an unbiased partial agonist because it shows a comparable loss of efficacy in both assays. Ligand D is β-arrestin-biased by virtue of losing more efficacy towards G protein than towards β-arrestin (similar to the AT1R β-arrestin-biased ligands discussed in this review [8, 9]).
Figure I shows data for an idealized scenario where all ligands have the same affinity for the receptor, the half maximal effective concentrations (EC50) of the ligand dose response curves accurately reflect the ligands’ binding affinities, and the maximal assay signals are directly proportional to the ligands’ ability to stabilize a signaling-competent receptor conformation and efficacies at each pathway. In practice, many factors in cellular assays can result in the non-linear amplification of responses. Best practices for accurately measuring and calculating ligand bias are still debated and have been discussed elsewhere [66, 67].
Figure I.
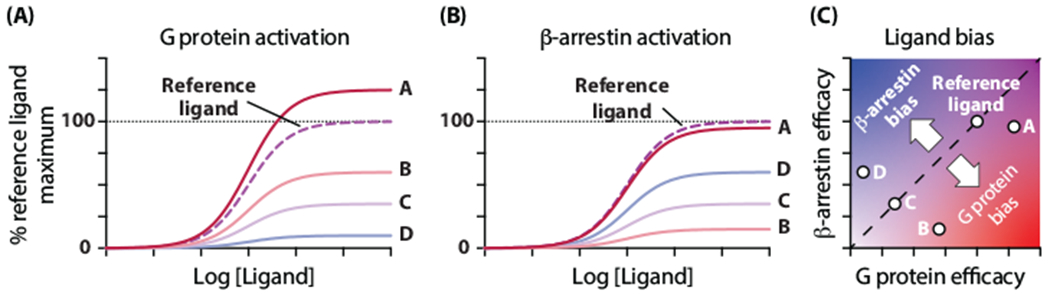
Ligand efficacy and bias. (A), (B) Dose response curves for five different ligands in assays measuring G protein (A) and β-arrestin (B) activation in a hypothetical scenario where the maximal signal is directly proportional to ligand efficacy. (C) Ligand bias is determined based on the relative efficacy of ligands in two (or more) assays. Points falling below the (dashed) line of unity represent G protein-biased ligands, points above the line of unity are β-arrestin-biased ligands, and points lying on the line are unbiased relative to the reference ligand. A reference ligand must be selected to define bias.
GPCR agonists are typically categorized as “balanced,” “G protein-biased,” or “β-arrestin-biased.” However, assays resolving ever-finer aspects of GPCR signaling now suggest that this categorization is too coarse. The same “β-arrestin-biased” ligands that lack the ability to activate the AT1R’s principal cognate G protein, Gq, activate Gi and G12 in bioluminescence resonance energy transfer (BRET) experiments monitoring subunit dissociation [10]—an indicator of bias among G protein subtypes, a pattern which has also been seen in C-C chemokine receptor 5 [11]. Likewise, the manifold functions of β-arrestins are separable. Mutagenesis has shown that β-arrestin desensitization of G protein signaling can be lost without impacting β-arrestin’s recruitment, endocytosis, and MAPK signaling functions [12, 13]. Reports of ligands biased towards different β-arrestin readouts (e.g., recruitment versus β-arrestin-dependent endocytosis [14, 15]) suggest that pharmacological separation of these functions is also possible. Notably, the degree and pattern of GRK phosphorylation can dictate the balance of β-arrestin signaling and trafficking functions [16, 17]. Biased signaling has in many cases been linked not only to whether GRK phosphorylation occurs [18, 19] but also to the selective recruitment of particular GRK isoforms [17, 20].
Integrating multiple views of GPCR activation mechanisms
Just as a single photograph or a video from one angle cannot capture all aspects of an object in movement, no one biophysical method can visualize all aspects of GPCR activation. In general, there is a tradeoff between high-resolution information on the entire protein versus dynamic information on limited regions (Box 2). In the former category, crystal and cryo-electron microscopy (cryoEM) structures have provided comprehensive, atomic-resolution snapshots of scores of GPCRs both in inactive and active conformations, revealing conserved conformational changes associated with activation. Due to their need to bind structurally disparate ligands, different GPCRs vary considerably in the magnitude and nature of the conformational changes in the orthosteric ligand-binding site following agonist binding (Figure 1A) [21]. Yet these changes funnel down through conserved motifs in the interior of the receptor core and induce common conformational changes in the intracellular transducer site—what we refer to as the “canonical active conformation” (Figure 1B) [22]. The largest-scale movement is the rotation and outward displacement of the intracellular end of transmembrane helix 6 (TM6) (~10-14 Å), accompanied by smaller movements and rotations of TMs 5 and 7. These rearrangements “open” the intracellular regions to accommodate transducer binding.
Box 2: Biophysical techniques for analyzing GPCR conformations.
Integrating data from multiple biophysical methods is critical to obtain a full picture of GPCR activation because different techniques deliver complementary information. Advantages and limitations of methods mentioned in this review are outlined below.
Atomic-resolution structures
X-ray crystallography
Atomic-resolution information on the global receptor conformation.
Inherently captures a single, low-energy protein conformation; higher energy activated conformations typically must be stabilized (e.g., by a transducer or conformationally selective antibody).
Has been highly successful for GPCRs bound to ligands or small proteins, but only rarely successful for GPCR-transducer complexes.
Cryo-electron microscopy
Atomic-resolution information on the global receptor conformation.
Potential to resolve multiple conformational states in a single sample.
To date, only used for GPCRs that are either bound to transducers or that have large ectodomains (non-Family A GPCRs); technological improvements may ultimately enable analysis of smaller particles.
Spectroscopic methods
Bioluminescence resonance energy transfer
Detects conformational changes altering the proximity of luciferase and an acceptor fluorescent protein.
Can demonstrate conformational changes inside the cell.
Qualitative indicator of general regions changing conformation.
Requires genetic modification with relatively large tags.
Fluorescence spectroscopy
Detects qualitative changes in the environment of fluorescent labels.
Solvent-accessible residues of interest are chemically labeled.
Single-molecule fluorescence resonance energy transfer
Detects intramolecular conformational changes between a fluorescent donor and acceptor.
Can provide quantitative information on distances between labels and lifetimes of individual conformational transitions.
Solvent-accessible residues of interest are chemically labeled.
Nuclear magnetic resonance
Can detect changes in the environment of isotopically labeled residues and measure exchange rates between different conformations.
Specialized techniques can measure distances between labels.
Solvent-accessible residues are chemically labeled, or residues throughout proteins can be metabolically labeled.
Double electron-electron resonance
Electron paramagnetic resonance technique that provides quantitative information about the distribution of distances between two nitroxide probes (i.e., the fraction of the population in each conformation).
Solvent-accessible residues of interest are chemically labeled.
Mass spectrometry (MS)-based approaches
Hydrogen-deuterium exchange mass spectrometry
Based on exchange of amide hydrogens in protein backbone with deuterium from isotopically labeled water, which is subsequently quantified by MS.
Highlights which solvent-accessible regions undergo conformational changes.
In pulsed formats, can provide kinetic information.
Hydroxyl radical-mediated protein footprinting
Similar to hydrogen-deuterium exchange-MS, but based on X-ray-generated hydroxyl radicals modifying accessible sidechains.
Computational approaches
Molecular dynamics simulations
Computational prediction of how all atoms in a protein change over time.
Limited by quality of input parameters (e.g., the initial structure) and by computational resources that limit run lengths.
Figure 1. Diversity and conservation in GPCR structures.
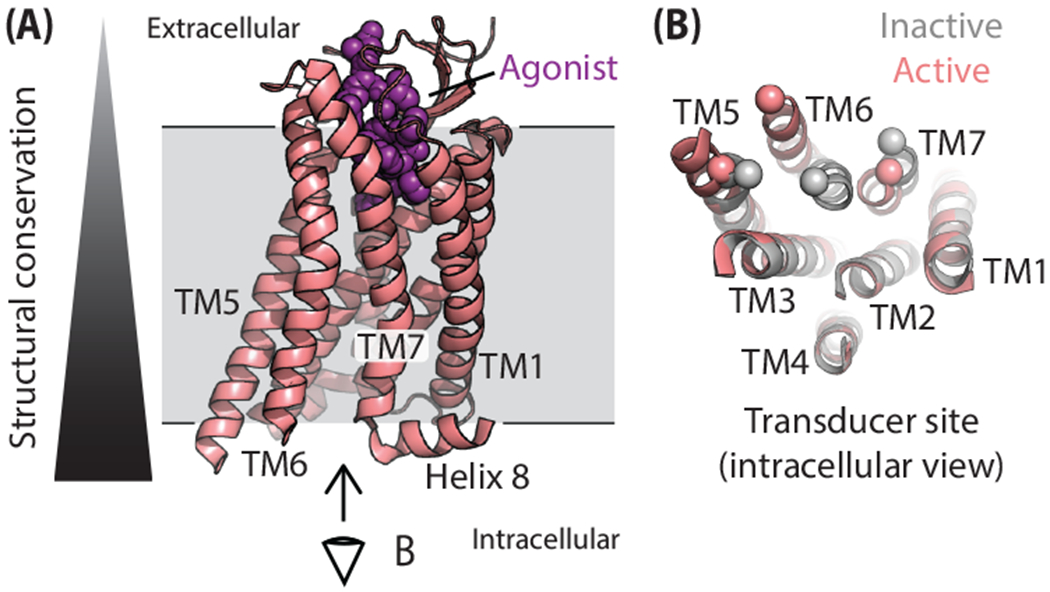
(A) General structure of a GPCR (pink) bound to an agonist (purple). Structural conservation in the family is lowest in the extracellular-facing orthosteric site and highest in the intracellular transducer site. Eye indicates viewing angle in panel B. (B) Conserved conformational changes in the intracellular transducer site of GPCRs in moving from the canonical inactive conformation (gray) to the canonical active conformation (pink). Structures of the AT1R (inactive, PDB: 4YAY; active, PDB: 6OS0), the case study presented in this review, are used. Spheres highlight the movement of Cα atoms of residues at the intracellular ends of TMs 5, 6, and 7. Intracellular loops and helix 8 are not shown for clarity.
Spectroscopic and computational approaches provide complementary information, highlighting the role of conformational dynamics in GPCR activation [23]. In the absence of agonists, the receptor population is typically dominated by conformations closely related to those observed in inactive-state crystal structures [24]. While agonist binding drives the receptor population towards conformations similar to those in active-state structures, a mixture of inactive and active conformations remains, reflecting “loose” or incomplete allosteric coupling between the orthosteric and transducer pockets [24, 25]. Surprisingly, for some GPCRs, and under some experimental conditions, a substantial fraction of unliganded receptors already reside in an active-like conformation, which may be related to their level of basal or constitutive signaling [26, 27].
Conformational heterogeneity induced by functionally diverse ligands
Conformational heterogeneity induced by AT1R biased ligands
The AT1R has been a particularly useful model for mechanistic studies of biased ligands for several reasons. First, the AT1R biased ligands—all analogs of the AngII octapeptide—exhibit well-defined structure-activity relationships (Figure 2A) [6–9]. Mutation of AngII F8 to a residue with a small side chain, or its outright deletion, profoundly impairs Gq but not β-arrestin coupling. Gq-biased ligands, in contrast, retain F8 but have mutations closer to the peptide N terminus.
Figure 2. Mechanism of AT1R activation by AngII and biased agonists.
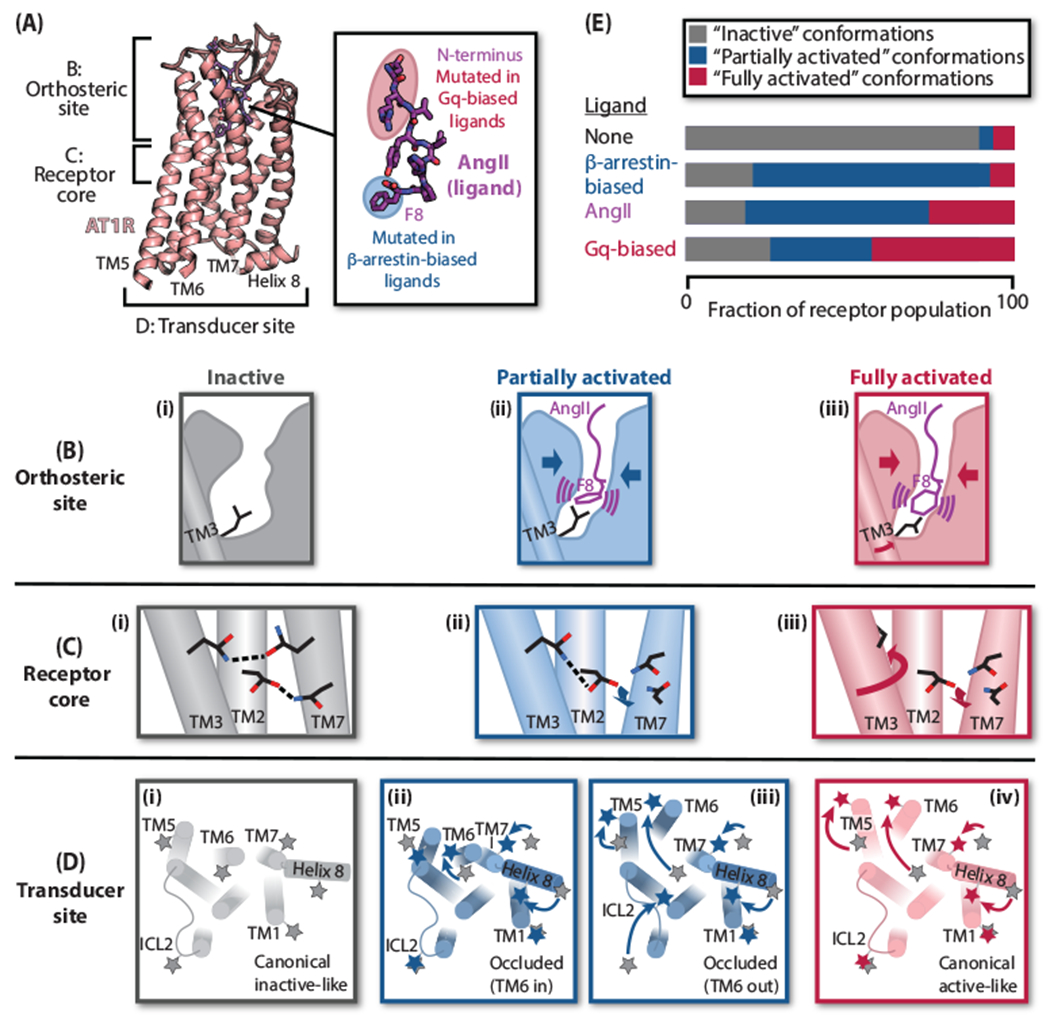
(A) Overview of AT1R-AngII structure (PDB: 6OS0) and structure-activity relationships of biased AngII analogs. (B)-(D) Cartoon representations of the distinct states of the AT1R orthosteric site, receptor core, and transducer site—classified as “inactive” (gray), “partially activated” (blue), and “fully activated” (red)—observed by crystallography, spectroscopy, and MD simulations. (B) Compared to the inactive state (i), binding of either AngII or β-arrestin-biased ligands (lacking F8) causes contraction of the orthosteric site (ii). Certain AngII F8 conformations promote an additional rotation of TM3 (iii). (C) A hydrogen-bonding network involving TMs 2, 3, and 7 in the receptor core stabilizes the inactive state (i). In structures with β-arrestin-biased ligands (ii) and AngII (iii), the movement of TM7 breaks its connection to this network. In the AngII-bound structure, the additional rotation of TM3 removes the key residue from the network (iii). (D) Besides transducer site conformations consistent with the canonical inactive (i) and fully activated (iv) GPCR conformations, DEER spectroscopy delineated two partially activated, “occluded” conformations—one occluded by virtue of an inward TM6 (ii), and the other due to differences in the second intracellular loop (ICL2) and TM5 (iii). Stars represent nitroxide labels; positions of labels from the inactive state (i) are shown in gray in each panel for reference. (E) Model of AT1R biased ligand activation. β-Arrestin-biased ligands stabilize only partially activated conformations; AngII promotes both partially and fully activated conformations; and Gq-biased ligands stabilize fully activated conformations more strongly than AngII.
Second, data from multiple biophysical techniques have provided complementary perspectives on how these biased ligands exert their effects across the AT1R, framing a relatively comprehensive mechanistic model. A series of crystal structures of AT1R bound to functionally diverse ligands—inverse agonists, AngII, and β-arrestin-biased ligands—and molecular dynamics (MD) simulations have provided information on how biased ligands initiate their effects in the orthosteric site [28–32]. Despite the ligands’ stark functional differences, crystal structures showed that AngII and β-arrestin-biased ligands have essentially identical binding modes and induce a similar remodeling of the orthosteric site [31] (Figure 2B). However, unlike the β-arrestin-biased ligands that lack bulky eighth residues, AngII induces one additional, very localized change at the base of the pocket—a rotation of TM3 that prevents a steric clash with the large aromatic side chain of AngII F8. MD simulations further show that AngII F8 is highly dynamic, and only one of its two main conformations is associated with TM3 rotation [32] (Figure 2B, iii). Notably, simulations with Gq-biased ligands (which have not been crystallized) indicate that their mutations influence the distribution of F8 conformations, driving rotation of TM3 even more effectively than AngII itself [32].
Ligand-specific differences in the AT1R orthosteric site are propagated into the receptor core, where Gq-active ligands promote more extensive changes in conserved interaction networks than β-arrestin-biased ligands. Most notably, a hydrogen-bonding network composed of residues from TMs 2, 3, and 7 stabilizes the inactive state (Figure 2C, i) [28, 29]. In the β-arrestin-biased ligand structures, the inward movement of TM7 rearranges this network (Figure 2C, ii). However, in the AngII structure, the TM3 rotation that began in the orthosteric site pivots the key TM3 residue away from the interior of the TM bundle, severing all stabilizing bridges across the receptor core (Figure 2C, iii) [31].
Crystallizing the AT1R with peptide ligands required the intracellular regions to be stabilized with a conformationally specific nanobody, locking the receptor in the same intracellular conformation [30, 31]. However, double electron-electron resonance (DEER) spectroscopy has demonstrated experimentally that the transducer site is conformationally heterogeneous in the absence of an intracellular binder. DEER provides the distribution of distances between two nitroxide labels installed on a protein. Even though DEER cannot provide the same global atomic-resolution information as crystal or cryoEM structures, analysis of ten labeling pairs allowed the deconvolution and mapping of “silhouettes” of the four major intracellular conformations stabilized by AT1R biased ligands: two conformations consistent with the canonical “inactive” and “active” GPCR conformations, and two “occluded” conformations that appear less accessible to transducer binding (Figure 2D) [33]. One of these occluded conformations lacks the outward displacement of TM6—long considered to be the defining hallmark of GPCR activation—but shows other changes compared to the inactive conformation, particularly in TM7 and helix 8 (Figure 2D, ii). The other occluded conformation displays the outward movement of TM6 and inward movement of TM7 associated with the canonical active conformation, but it shows a smaller rotation of TM5 and a significant rearrangement of ICL2 (Figure 2D, iii).
Each class of biased ligand has distinct effects on the intracellular regions as assessed by DEER (Figure 2E) [33]. AngII stabilizes a mixture of the canonical active and the TM6 outward occluded conformations. Four different β-arrestin-biased ligands stabilize only the two occluded conformations—not the canonical active conformation—but in widely varying ratios. Gq-biased ligands drive the receptor equilibrium in the opposite direction, stabilizing the canonical active conformation even more strongly than AngII. The functional profiles of these ligands suggest that the canonical active conformation is necessary for Gq coupling, while the two occluded conformations and the canonical active conformation all promote β-arrestin coupling.
The crystallographic, simulation, and spectroscopic data all harmonize to suggest a model of biased ligand action that hinges upon (1) the endogenous agonist AngII stabilizing a mixture of “fully” and “partially” activated receptor conformations and (2) Gq having a higher degree of conformational specificity than β-arrestin. Thus, tilting the receptor’s conformational distribution towards “partially activated” states leads to β-arrestin bias; tilting the distribution more towards “fully activated” conformations leads to Gq bias (Figure 2E).
Conservation of biased ligand mechanisms
The AT1R has been an exemplary model for biased signaling precisely because there are orthosteric ligands that show an unusually high degree of bias. This begs the question as to whether the mechanisms seen in the AT1R are broadly conserved and can inform the development of drugs with defined signaling profiles for other GPCRs.
As mentioned above, GPCRs show the most structural divergence in the orthosteric ligand-binding site; therefore, the specific “triggers” that biased ligands use to initiate their effects undoubtedly differ by receptor. In the AT1R, the ligand-receptor interaction that is the key determinant of Gq coupling is at the base of the orthosteric site. In contrast, crystal structures of other GPCRs bound to ligands with varying degrees of bias have highlighted that biased ligands often make more extensive contacts with the receptor near its extracellular face and in the extracellular loops [34, 35]. While no clear predictive rules for how these interactions affect the directionality or degree of bias have emerged, they may play a role analogous to the substitutions in the Gq-biased AT1R biased ligands, modulating the conformational distribution of the orthosteric site [32]. Contacts with the extracellular regions may also influence ligand binding kinetics, which can influence signaling bias in the non-equilibrium conditions of living cells [36] (Box 3).
Box 3: From receptor conformations to cellular context.
In cellular contexts, orthosteric ligands are just one of many factors that can alter a GPCR’s ability to activate particular signaling pathways. A few of the cellular mechanisms that have been demonstrated or proposed to further modify GPCR signaling are outlined below. Importantly, many of these mechanisms can modify GPCR activity in a cell-type-specific manner.
Post-translational modifications, including phosphorylation, may directly modulate receptor conformations [68].
Endogenous allosteric ligands such as ions [69] and lipids [70] can directly modulate the conformational distributions of GPCRs. Membrane lipids further regulate coupling of G proteins [71, 72], GRKs [70], and β-arrestins [53, 73] to GPCRs.
Different alternative splicing patterns of GPCRs can bias their signaling through different pathways [74].
Protein expression levels can determine whether or not certain pathways are activated in particular cell types [19].
Interactions between GPCRs—homo- and hetero-oligomerization—can bias receptors’ ligand and transducer specificities [75].
- Interactions among different transducers allows G protein and β-arrestin mediated pathways to influence each other. This likely accounts for recent controversy in the field as to whether β-arrestin-mediated signaling can occur in the absence of G proteins [76, 77]. Some of the known ways crosstalk can occur among various GPCR-activated pathways include:
The conserved interaction networks in the receptor core are likely key regions where biased ligand mechanisms converge. In most Family A GPCRs, the TM2-TM3-TM7 network that stabilizes the inactive AT1R inactive conformation is part of the “sodium-binding site,” a conserved polar network that binds sodium in the inactive state and collapses in active-state structures [37]. The AT1R deviates from the GPCR consensus in one residue in this network and does not bind sodium in the inactive conformation [28]. It is not yet understood whether such receptor-specific features contribute to the ability of certain GPCRs to readily achieve a dramatic range of signaling bias. However, mutation of this region can bias signaling of other GPCRs, suggesting that directly manipulating this network with allosteric ligands could be a general means to alter the balance of transducer activation [37, 38].
Consistent with the fact that GPCRs show the most structural conservation in the transducer site, various biophysical approaches applied to multiple GPCRs have implicated an intracellular conformation with changes in TM7 and helix 8 but not TM6—features similar to one of the occluded AT1R conformations (Figure 2D, ii)—in selective coupling to β-arrestin. Fluorescence probes detected qualitative conformational changes in TM7 or helix 8, but not TM6, for both the V2 vasopressin receptor bound to an orthosteric β-arrestin-biased ligand (deficient in Gs signaling) [39] and the cannabinoid type 1 receptor bound to an allosteric β-arrestin-biased ligand (which blocks orthosteric agonist-induced Gi activation but activates β-arrestin) [40]. Likewise, nuclear magnetic resonance (NMR) studies using probes on the intracellular ends of TMs 6 and 7 of the β2-adrenergic receptor (β2AR) indicated that an orthosteric β-arrestin-biased ligand (deficient in Gs signaling) selectively causes changes in TM7 [41]. Single-molecule fluorescence of the β2AR has further indicated that the lifetime of conformational changes in TM7 is associated with ligand bias [42]. This suggests some ligands may effect bias by altering GPCRs’ conformational kinetics, not just the equilibrium conformational distribution (a thermodynamic effect).
Importantly, the model for biased ligand action at the AT1R—which rests on the promiscuity of β-arrestin—cannot account for the loss of β-arrestin activation seen in biased ligands of other GPCRs. In several cases, G protein bias involving the loss of β-arrestin activity is known to be a downstream consequence of impaired GRK phosphorylation [18, 43]. Like G proteins and β-arrestins, GRKs interact with GPCRs in a conformationally dependent manner, and mutation data at the β2AR suggest that altering GPCRs’ conformational distribution can drive differential phosphorylation by GRKs [18]. NMR studies of the μ-opioid receptor bound to a Gi-biased ligand (impaired in phosphorylation and β-arrestin activation) show that the receptor favors open, accessible intracellular conformations [44]. Additional studies are needed to understand what distinguishes these conformations from the canonical active conformations which effectively promote GRK phosphorylation and β-arrestin coupling.
Conformational heterogeneity induced by partial agonists
There are also interesting parallels between the biophysical mechanisms of AT1R β-arrestin-biased ligands and those of partial agonists of other GPCRs that do not show appreciable signaling bias (Box 1). There is a long-standing debate as to whether the submaximal efficacy of partial agonists is due to stabilization of a lower fraction of receptors in a fully active conformation, or to stabilization of a conformation that is less efficacious in transducer coupling. Multiple spectroscopic and computational studies have now found that partial agonists can stabilize intracellular conformations that are distinguishable from the canonical, fully activated conformation but nevertheless show significant outward movement of TM6 [25, 27, 45–48]. It is intriguing to speculate that these conformations may be similar to the AT1R occluded conformation in which TM6 has moved outward (Figure 2D, iii), and that for some GPCRs stabilization of this conformation leads to low-efficacy G protein activation.
β-Arrestin-biased AT1R ligands stabilize widely varying ratios of two very different “occluded” intracellular conformations (Figure 2D, ii and iii, 2E) [33], even though these ligands have indistinguishable pharmacological profiles [8, 9]. These ligands may promote more similar conformational distributions in the context of the cell membrane, or more nuanced assays may ultimately resolve differences in the cellular pathways they activate. However, these data raise the possibility that different receptor conformations can be functionally redundant. That is, there are multiple conformational solutions that achieve the same level and balance of transducer activation. Here again, there is evidence for similar phenomena in partial agonist mechanisms. In NMR studies of the M2 muscarinic receptor (M2R), which used 13CH3-ε-methionine metabolic labeling to analyze residues throughout the receptor, each member of a structurally diverse panel of agonists stabilized a distinct receptor conformation, even though several agonists showed similar efficacy and bias towards Gq activation and β-arrestin recruitment [49]. Likewise, pharmacological analyses have found that β2AR partial agonists with similar efficacies towards Gs can differentially stabilize defined receptor states and fall into several distinct mechanistic categories [50].
Conformational heterogeneity in GPCR-transducer complexes
The binding of ligands with varying degrees of bias clearly alters the conformational distribution of GPCRs, but it is less apparent why these conformational differences result in differential coupling of transducers. Two “matched pairs” of GPCR-G protein and GPCR-β-arrestin structures are now available (neurotensin receptor 1 [NTSR1]-Gi, NTSR1-β-arrestin1, M2R-Gi, M2R-β-arrestin1) [51–54]. The receptor conformations are surprisingly similar once fully engaged with each transducer, showing only minor differences in the very intracellular ends of the TMs. At face value, this observation seems at odds with the clear differences in intracellular receptor conformations stabilized by biased ligands in the absence of transducers. However, mounting evidence suggests that transducer coupling specificity is determined primarily during the multi-step process of engagement, and methods to probe dynamic aspects of complex assembly will be required to elucidate this.
While the overwhelming majority of GPCR-G protein structures display highly similar quaternary structures, a growing body of structural, spectroscopic, and computational data have provided evidence for alternative conformations that may represent intermediates of complex formation. Both single-molecule fluorescence resonance energy transfer and mass spectrometry labeling approaches have detected intermediate configurations of β2AR-Gs complexes that precede GDP release and formation of the stable, nucleotide-free conformations seen in the overwhelming majority of structures [47, 55]. Two structures have now captured high-resolution views of what such intermediates may look like [52, 56]. Both experimental and MD simulation data suggest these initial interactions establish the specificity of GPCRs’ coupling to particular G protein isoforms [55, 57].
GPCR-β-arrestin complexes are characterized by even more dramatic conformational heterogeneity, which has been observed by negative stain electron microscopy [58]. Due to the large differential in β-arrestin’s relatively high affinity for GRK-phosphorylated motifs and much lower affinity for the receptor transducer-binding site, GPCR-β-arrestin complexes appear to be in equilibrium between conformations in which β-arrestin engages only phosphorylated intracellular regions and conformations in which it engages both the phosphorylated regions and the transducer site [58]. Receptor phosphorylation thus increases the effective concentration of β-arrestin in the vicinity of the transducer site; this “proximity effect” may contribute to β-arrestin’s ability to engage the sorts of occluded receptor conformations stabilized by AT1R biased ligands [33]. Interestingly, β-arrestin’s affinity for the transducer site varies substantially among receptors, and these differences correlate with β-arrestin’s ability to block receptor-mediated G protein activation [59]. This conformational heterogeneity thus provides a biophysical explanation for why desensitization is functionally separable from the internalization and signaling functions associated with β-arrestin’s scaffolding role [12, 13]. This conformational equilibrium also allows the coincident engagement of G proteins and β-arrestins, which has been visualized by electron microscopy [60, 61]. Such an arrangement may facilitate G protein-mediated signaling from internalized receptors that has unique downstream effects [62] (Box 3).
GPCR-GRK interactions have been particularly challenging to study since they are transient enzyme-substrate interactions. A host of techniques, including chemical crosslinking, hydrogen-deuterium exchange mass spectrometry, electron microscopy, and MD simulations, was used to show that GRK5 undergoes major conformational changes upon interacting with β2AR, allosterically activating its kinase activity [63]. The ICLs of β2AR appear to be particularly important for GRK5 binding, but additional work is needed to understand the more general principles of how GRKs recognize agonist-activated receptors.
Concluding Remarks
The synthesis of structural and dynamic information on GPCRs has shown that conformational heterogeneity is not an extraneous property of these receptors. Rather, it is the fundamental reason why they are versatile enough to stand at the crux of diverse physiological processes. The ability of GPCRs—and many other classes of proteins [64, 65]—to explore multiple conformations broadens the pharmacology that their ligands can achieve. If we can continue to elucidate the mechanisms underlying the intricate signaling mechanisms of GPCRs (see Outstanding Questions), we may ultimately be able exploit this complexity to rationally hone the properties of therapeutics.
Outstanding questions.
How generally conserved are the mechanisms that biased ligands use to modulate GPCR conformations?
Do thermodynamic effects (the equilibrium population of receptor conformations) or kinetic effects (the on- and off-rates of ligands, and the lifetimes of receptor conformations) principally govern biased signaling? Do the relative contributions of thermodynamics and kinetics vary among GPCRs?
Are there receptor-specific features that determine a GPCR’s propensity to exhibit biased signaling?
How are the conformational distributions of GPCRs influenced by their cellular context (e.g., endogenous allosteric modulators, membrane environment)? There is a need to better recapitulate these variables in in vitro biophysical studies, and to develop tools capable of analyzing GPCRs’ conformations in cellular systems.
What are the steps required for G proteins, GRKs, and β-arrestins to productively engage GPCRs, and how do the conformational distributions of GPCRs influence these processes?
What can information about the conformations stabilized by ligands’ and transducers’ conformations teach us about the most effective strategies to selectively modulate GPCR signaling? For example, if many GPCRs lack features required for orthosteric ligands to initiate conformational changes leading to biased signaling, allosteric ligands that directly engage the intracellular regions of GPCRs could provide a more general solution to this problem.
Highlights.
G protein-coupled receptors (GPCRs) activate multiple pathways in the cell, and certain “biased” GPCR ligands are able to preferentially stimulate particular pathways.
A full picture of how different ligands produce different signaling patterns requires data from multiple biophysical techniques—providing high-resolution structural details and dynamic information on GPCR conformations.
GPCRs sample more than just a single “inactive” and a single “active” conformation, and the endogenous agonists of most receptors stabilize a mixture of multiple conformations.
Biased ligands can achieve their distinctive signaling profiles by modulating this distribution of receptor conformations.
This conformational heterogeneity is likely a major reason why GPCRs are privileged structural scaffolds in biology and provides a biophysical basis for their versatility.
Acknowledgments
Funding was provided by NIH grant R01HL16037 (R.J.L.). R.J.L. is an investigator with the Howard Hughes Medical Institute.
Glossary
- Allosteric ligand
A ligand that binds anywhere outside the orthosteric site
- Allostery
Transmission of conformational changes in one region of a protein to another region; e.g., transmission of conformational changes in the orthosteric site of GPCRs (due to agonist binding) to the transducer site
- Biased ligand
Also known as a functionally selective ligand; a ligand that preferentially activates a subset of downstream responses compared to a reference ligand (typically the endogenous agonist)
- Efficacy
The relative ability of an agonist to activate a particular response
- Inverse agonist
A ligand that reduces basal activation of downstream responses, also referred to as a negative antagonist
- Orthosteric site
The extracellular-facing pocket occupied by most endogenous GPCR agonists
- Partial agonist
A ligand that activates a particular response with sub-maximal efficacy
- Transducer
A protein that recognizes and is activated by an activated GPCR, allowing it to initiate further cellular responses; principally refers to heterotrimeric G proteins, GRKs, and β-arrestins
- Transducer site
The cytoplasmic-facing cleft in the center of the 7-transmembrane helix bundle of GPCRs where both G proteins and β-arrestins interact. This region undergoes the largest-scale conformational changes upon activation of Family A GPCRs
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Wootten D and Miller LJ (2020) Structural basis for allosteric modulation of class B G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol 60, 89–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kozielewicz P, et al. (2020) Molecular pharmacology of class F receptor activation. Mol. Pharmacol 97, 62–71 [DOI] [PubMed] [Google Scholar]
- 3.Leach K and Gregory KJ (2017) Molecular insights into allosteric modulation of Class C G protein-coupled receptors. Pharmacol. Res 116, 105–118 [DOI] [PubMed] [Google Scholar]
- 4.Gilman AG (1995) Nobel Lecture. G proteins and regulation of adenylyl cyclase. Biosci. Rep 15, 65–97 [DOI] [PubMed] [Google Scholar]
- 5.Gurevich VV and Gurevich EV (2019) GPCR signaling regulation: The role of GRKs and Arrestins. Front. Pharmacol 10, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Holloway AC, et al. (2002) Side-chain substitutions within angiotensin II reveal different requirements for signaling, internalization, and phosphorylation of type 1A angiotensin receptors. Mol. Pharmacol 61, 768–777 [DOI] [PubMed] [Google Scholar]
- 7.Wei H, et al. (2003) Independent beta-arrestin 2 and G protein-mediated pathways for angiotensin II activation of extracellular signal-regulated kinases 1 and 2. Proc. Natl. Acad. Sci. USA 100, 10782–10787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Strachan RT, et al. (2014) Divergent transducer-specific molecular efficacies generate biased agonism at a G protein-coupled receptor (GPCR). J. Biol. Chem 289, 14211–14224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rajagopal S, et al. (2011) Quantifying ligand bias at seven-transmembrane receptors. Mol. Pharmacol 80, 367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Namkung Y, et al. (2018) Functional selectivity profiling of the angiotensin II type 1 receptor using pathway-wide BRET signaling sensors. Sci. Signal 11, eaat1631. [DOI] [PubMed] [Google Scholar]
- 11.Lorenzen E, et al. (2018) G protein subtype-specific signaling bias in a series of CCR5 chemokine analogs. Sci. Signal 11, eaao6152. [DOI] [PubMed] [Google Scholar]
- 12.Cahill TJ 3rd, et al. (2017) Distinct conformations of GPCR-beta-arrestin complexes mediate desensitization, signaling, and endocytosis. Proc. Natl. Acad. Sci. USA 114, 2562–2567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumari P, et al. (2017) Core engagement with beta-arrestin is dispensable for agonist-induced vasopressin receptor endocytosis and ERK activation. Mol. Biol. Cell 28, 1003–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.De Vries L, et al. (2019) Innovative bioluminescence resonance energy transfer assay reveals differential agonist-induced D2 receptor intracellular trafficking and arrestin-3 recruitment. Mol. Pharmacol 96, 308–319 [DOI] [PubMed] [Google Scholar]
- 15.Kurko D, et al. (2014) Analysis of functional selectivity through G protein-dependent and -independent signaling pathways at the adrenergic alpha(2C) receptor. Brain Res. Bull 107, 89–101 [DOI] [PubMed] [Google Scholar]
- 16.Kim J, et al. (2005) Functional antagonism of different G protein-coupled receptor kinases for beta-arrestin-mediated angiotensin II receptor signaling. Proc. Natl. Acad. Sci. USA 102, 1442–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nobles KN, et al. (2011) Distinct phosphorylation sites on the beta(2)-adrenergic receptor establish a barcode that encodes differential functions of beta-arrestin. Sci. Signal 4, ra51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Choi M, et al. (2018) G protein-coupled receptor kinases (GRKs) orchestrate biased agonism at the beta2-adrenergic receptor. Sci. Signal 11, eaar7084. [DOI] [PubMed] [Google Scholar]
- 19.Urs NMV et al. (2016) Distinct cortical and striatal actions of a beta-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties. Proc. Natl. Acad. Sci. USA 113, E8178–E8186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zidar DAV et al. (2009) Selective engagement of G protein coupled receptor kinases (GRKs) encodes distinct functions of biased ligands. Proc. Natl. Acad. Sci. USA 106, 9649–9654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Venkatakrishnan AJV et al. (2016) Diverse activation pathways in class A GPCRs converge near the G-protein-coupling region. Nature 536, 484–487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Erlandson SCV et al. (2018) Structural basis for G protein-coupled receptor signaling. Annu Rev Biophys 47, 1–18 [DOI] [PubMed] [Google Scholar]
- 23.Latorraca NRV et al. (2017) GPCR dynamics: Structures in motion. Chem. Rev 117, 139–155 [DOI] [PubMed] [Google Scholar]
- 24.Manglik A, et al. (2015) Structural insights into the dynamic process of beta2-adrenergic receptor signaling. Cell 161, 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dror RO, et al. (2011) Activation mechanism of the beta2-adrenergic receptor. Proc. Natl. Acad. Sci. USA 108, 18684–18689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Staus DP, et al. (2019) Detergent- and phospholipid-based reconstitution systems have differential effects on constitutive activity of G-protein-coupled receptors. J. Biol. Chem 294, 13218–13223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ye L, et al. (2016) Activation of the A2A adenosine G-protein-coupled receptor by conformational selection. Nature 533, 265–268 [DOI] [PubMed] [Google Scholar]
- 28.Zhang H, et al. (2015) Structural basis for ligand recognition and functional selectivity at angiotensin receptor. J. Biol. Chem 290, 29127–29139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhang Hv et al. (2015) Structure of the Angiotensin receptor revealed by serial femtosecond crystallography. Cell 161, 833–844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wingler LM, et al. (2019) Distinctive activation mechanism for angiotensin receptor revealed by a synthetic nanobody. Cell 176, 479–490 e412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wingler LM, et al. (2020) Angiotensin and biased analogs induce structurally distinct active conformations within a GPCR. Science 367, 888–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Suomivuori CM, et al. (2020) Molecular mechanism of biased signaling in a prototypical G protein-coupled receptor. Science 367, 881–887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wingler LM, et al. (2019) Angiotensin analogs with divergent bias stabilize distinct receptor conformations. Cell 176, 468–478 e411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee Yv et al. (2020) Molecular determinants of β-arrestin coupling to formoterol-bound β1-adrenoceptor. bioRxiv, DOI: 10.1101/2020.1103.1127.011585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wacker Dv et al. (2017) Crystal structure of an LSD-bound human serotonin receptor. Cell 168, 377–389 e312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Klein Herenbrink, Cv et al. (2016) The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun 7,10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Katritch V, et al. (2014) Allosteric sodium in class A GPCR signaling. Trends Biochem. Sci 39, 233–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zarzycka Bv et al. (2019) Harnessing ion-binding sites for GPCR pharmacology. Pharmacol. Rev 71, 571–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rahmeh Rv et al. (2012) Structural insights into biased G protein-coupled receptor signaling revealed by fluorescence spectroscopy. Proc. Natl. Acad. Sci. USA 109, 6733–6738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fay JF and Farrens DL (2015) Structural dynamics and energetics underlying allosteric inactivation of the cannabinoid receptor CB1. Proc. Natl. Acad. Sci. USA 112, 8469–8474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu JJV et al. (2012) Biased signaling pathways in beta2-adrenergic receptor characterized by 19F-NMR. Science 335, 1106–1110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lamichhane Rv et al. (2020) Biased signaling of the G-protein-coupled receptor beta2AR is governed by conformational exchange kinetics. Structure 28, 371–377 e373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.DeWire SMV et al. (2013) A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther 344, 708–717 [DOI] [PubMed] [Google Scholar]
- 44.Okude J, et al. (2015) Identification of a conformational equilibrium that determines the efficacy and functional selectivity of the mu-opioid receptor. Angew. Chem. Int. Ed. Engl 54, 15771–15776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Solt AS, et al. (2017) Insight into partial agonism by observing multiple equilibria for ligand-bound and Gs-mimetic nanobody-bound beta1-adrenergic receptor. Nat. Commun 8, 1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vogel R, et al. (2006) Agonists and partial agonists of rhodopsin: retinal polyene methylation affects receptor activation. Biochemistry 45, 1640–1652 [DOI] [PubMed] [Google Scholar]
- 47.Gregorio GG, et al. (2017) Single-molecule analysis of ligand efficacy in beta2AR-G-protein activation. Nature 547, 68–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Masureel M, et al. (2018) Structural insights into binding specificity, efficacy and bias of a beta2AR partial agonist. Nat. Chem. Biol 14, 1059–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu J, et al. (2019) Conformational complexity and dynamics in a muscarinic receptor revealed by NMR spectroscopy. Mol. Cell 75, 53–65 e57 [DOI] [PubMed] [Google Scholar]
- 50.Staus DP, et al. (2016) Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation. Nature 535, 448–452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Huang W, et al. (2020) Structure of the neurotensin receptor 1 in complex with beta-arrestin 1. Nature 579, 303–308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kato HE, et al. (2019) Conformational transitions of a neurotensin receptor 1-Gi1 complex. Nature 572, 80–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Staus DP, et al. (2020) Structure of the M2 muscarinic receptor-beta-arrestin complex in a lipid nanodisc. Nature 579, 297–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Maeda S, et al. (2019) Structures of the M1 and M2 muscarinic acetylcholine receptor/G-protein complexes. Science 364, 552–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Du Y, et al. (2019) Assembly of a GPCR-G protein complex. Cell 177, 1232–1242 e1211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Liu X, et al. (2019) Structural insights into the process of GPCR-G protein complex formation. Cell 177, 1243–1251 e1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sandhu M, et al. (2019) Conformational plasticity of the intracellular cavity of GPCR-G-protein complexes leads to G-protein promiscuity and selectivity. Proc. Natl. Acad. Sci. USA 116, 11956–11965 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Shukla AK, et al. (2014) Visualization of arrestin recruitment by a G-protein-coupled receptor. Nature 512, 218–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Staus DP, et al. (2018) Sortase ligation enables homogeneous GPCR phosphorylation to reveal diversity in beta-arrestin coupling. Proc. Natl. Acad. Sci. USA 115, 3834–3839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thomsen ARB, et al. (2016) GPCR-G protein-beta-arrestin super-complex mediates sustained G protein signaling. Cell 166, 907–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nguyen AH, et al. (2019) Structure of an endosomal signaling GPCR-G protein-beta-arrestin megacomplex. Nat. Struct. Mol. Biol 26, 1123–1131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Tsvetanova NG and von Zastrow M (2014) Spatial encoding of cyclic AMP signaling specificity by GPCR endocytosis. Nat. Chem. Biol 10, 1061–1065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Komolov KE, et al. (2017) Structural and functional analysis of a beta2-adrenergic receptor complex with GRK5. Cell 169, 407–421 e416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Moraga I, et al. (2015) Tuning cytokine receptor signaling by re-orienting dimer geometry with surrogate ligands. Cell 160, 1196–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McDonnell DP and Wardell SE (2010) The molecular mechanisms underlying the pharmacological actions of ER modulators: implications for new drug discovery in breast cancer. Curr. Opin. Pharm 10, 620–628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Onaran HO, et al. (2017) Systematic errors in detecting biased agonism: Analysis of current methods and development of a new model-free approach. Sci. Rep 7, 44247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kenakin T (2016) Measurement of receptor signaling bias. Curr. Protoc. Pharmacol 74, 2.15.1–2.15.15 [DOI] [PubMed] [Google Scholar]
- 68.Shiraishi Y, et al. (2018) Phosphorylation-induced conformation of beta2-adrenoceptor related to arrestin recruitment revealed by NMR. Nat. Commun 9, 194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ye L, et al. (2018) Mechanistic insights into allosteric regulation of the A2A adenosine G protein-coupled receptor by physiological cations. Nat. Commun 9, 1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dawaliby Rv et al. (2016) Allosteric regulation of G protein-coupled receptor activity by phospholipids. Nat. Chem. Biol 12, 35–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Strohman M et al. (2019) Local membrane charge regulates beta2 adrenergic receptor coupling to Gi3. Nat. Commun 10, 2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Yen HY, et al. (2018) Ptdlns(4,5)P2 stabilizes active states of GPCRs and enhances selectivity of G-protein coupling. Nature 559, 423–427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mayer Dv et al. (2019) Distinct G protein-coupled receptor phosphorylation motifs modulate arrestin affinity and activation and global conformation. Nat. Commun 10, 1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Xu J, et al. (2017) Alternatively spliced mu opioid receptor C termini impact the diverse actions of morphine. J. Clin. Invest 127, 1561–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fuxe K, et al. (2014) Diversity and bias through receptor-receptor interactions in GPCR heteroreceptor complexes. Focus on examples from dopamine D2 receptor heteromerization. Front. Endocrinol. (Lausanne) 5, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Luttrell LM, et al. (2018) Manifold roles of beta-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci. Signal 11, eaat7650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Grundmann Mv et al. (2018) Lack of beta-arrestin signaling in the absence of active G proteins. Nat. Commun 9, 341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Daaka Yv et al. (1997) Receptor and G betagamma isoform-specific interactions with G protein-coupled receptor kinases. Proc. Natl. Acad. Sci. USA 94, 2180–2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Smith JS, et al. (2019) Noncanonical scaffolding of Gαi and β-arrestin by G protein-coupled receptors. bioRxiv, DOI: 10.1101/629576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Gupte TMV et al. (2017) Priming GPCR signaling through the synergistic effect of two G proteins. Proc. Natl. Acad. Sci. USA 114, 3756–3761 [DOI] [PMC free article] [PubMed] [Google Scholar]