

SUMMARY
Enzymatic probes of chromatin structure reveal accessible versus inaccessible chromatin states, while super-resolution microscopy reveals a continuum of chromatin compaction states. Characterizing histone H2B movements by Single Molecule Tracking (SMT), we resolved chromatin domains ranging from low to high mobility and displaying different subnuclear localizations patterns. Heterochromatin constituents correlated with the lowest mobility chromatin, whereas transcription factors varied widely with regard to their respective mobility with low or highly mobile chromatin. Pioneer transcription factors, which bind nucleosomes, can access the low mobility chromatin domains, whereas weak or non-nucleosome binding factors are excluded from the domains and enriched in higher mobility domains. Non-specific DNA and nucleosome binding accounted for most of the low mobility of strong nucleosome interactor FOXA1. Our analysis shows how the parameters of the mobility of chromatin-bound factors, but not their diffusion behaviors or SMT-residence times within chromatin, distinguish functional characteristics of different chromatin interacting proteins.
Graphical Abstract
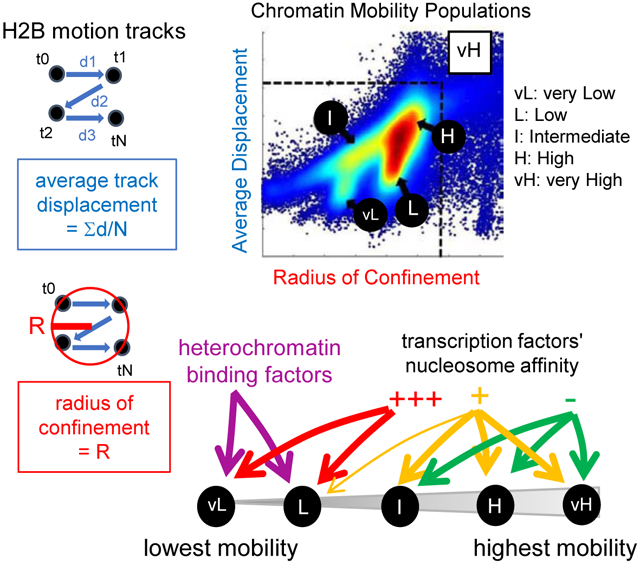
In Brief
A two-parameter assessment of motion tracks of histone H2B in living cells unveils a chromatin mobility landscape that explains differences in single molecule behavior and function of diverse heterochromatin regulators and transcription factors.
INTRODUCTION
During cell fate changes, new genetic networks are established by the interaction of transcription factors with DNA regulatory sites in silent chromatin, including at genes that can be antithetical to the original cell fate. Thus, it is important to understand how transcription factors scan silent chromatin, where DNA accessibility can be restricted by nucleosomes, bound proteins, post-translational histone modifications, remodelers, and physical compaction (Klemm et al., 2019). Rather than the opened or closed view of chromatin accessibility conveyed by enzymatic probes, super-resolution (Boettiger et al., 2016; Ricci et al., 2015) and electron microscopy (Ou et al., 2017) reveal a continuum of chromatin states, from highly compact to much less so. In living cells, these variations in nucleosome densities elicit different mechanical and electrostatic constraints over the Brownian motions of chromatin fibers (Dion and Gasser, 2013). Several observations relate degrees of chromatin movements to functional status, including reduced displacements at the nuclear periphery (Chubb et al., 2002; Heun et al., 2001), where repressed and compact chromatin can be found (van Steensel and Belmont, 2017), versus increased mobility at sites of transcriptional activation (Chuang et al., 2006; Gu et al., 2018; Neumann et al., 2012). In addition, Single-Molecule Tracking (SMT) has revealed a wide range of dynamics for individual histone H2B molecules in the nucleus, with reduced displacements at the nuclear periphery (Nozaki et al., 2017). Nevertheless, the use of a single, normally-distributed parameter is limited in defining discrete chromatin mobility states, which in theory could be scaled to characterize the mobility of chromatin-bound interactors corresponding to their distinct functional capabilities.
In vitro, transcription factors present a spectrum of capacities to interact with target DNA motifs on nucleosomes (Fernandez Garcia et al., 2019; Iwafuchi-Doi and Zaret, 2016; Soufi et al., 2015; Zhu et al., 2018). Accordingly, ChIP-seq (Furey, 2012) and Cut&Run (Meers et al., 2019) show that the genomic occupancy by transcription factor is sensitive to the local chromatin context (Soufi et al., 2015). Nevertheless, cell lysis and chemical treatments inherent to Cut&Run (MNase digestion) and ChIP-seq (formaldehyde fixation) limit the understanding of how different modes of chromatin occupancy relate to different modes of chromatin scanning in living cells, where transcription factors undergo rounds of trial-and-error through nonspecific associations before finding a specific target sequence (von Hippel and Berg, 1989). The process of nonspecific chromatin scanning is integrated into recent models of gene regulation (Cortini and Filion, 2018; Inukai et al., 2017).
In SMT, specific and nonspecific chromatin interactions of transcription factors have been suggested to elicit long-lived and short-lived residence times in chromatin, respectively (Voss and Hager, 2014). Nevertheless, where tested in SMT, transcription factors’ residence times are only moderately affected by point mutations for DNA binding (Callegari et al., 2019; Chen et al., 2014; Paakinaho et al., 2017), and thus how SMT-measured residence time relates to a factor’s specific and nonspecific interactions with chromatin remains unclear. In addition, the SMT-measured residence times of different types of mammalian transcription factors interacting with low-accessibility chromatin are similar (Swinstead et al., 2016), and hence provide little information about the type of chromatin bound by transcription factors. SMT parameters that discriminate transcription factor interactions with low accessibility chromatin thus remain to be found.
Here, we employ SMT with high temporal resolution to define a chromatin mobility landscape by characterizing different aspects of the mobility of histone H2B: the radius of confinement, estimating the area in which the molecule performs its confined motions, and the average displacement, representing the average distance between subsequent steps in a motion track, over time. These two parameters, though generally positively correlated, present systematic deviations allowing us to resolve five chromatin mobility groups, each with distinct subnuclear localizations. The results are recapitulated in several differentiated and transformed cell lines, while induced pluripotent stem cells exhibit chromatin with higher degrees of mobility. In order to further understand the relationship between degrees of mobility and the functional states of chromatin, we used the dual mobility parameters as a benchmark to compare the mobility of diverse proteins while interacting with chromatin. We found that the lowest degree of chromatin mobility at the periphery and throughout the nucleus correlates with that of Lamin-A, revealing its heterochromatic nature. Accordingly, the mobility of heterochromatin factors HP1α, HP1β, HP1γ, SUv39h2, and SUv39h1 correlates with the lower mobility chromatin, but with notable differences in terms of preferential binding and subnuclear localization that relate to functional properties of the proteins and underscores the complex nature of heterochromatin domains (Becker et al., 2017). For transcription factors, we observed a strong correlation between nucleosome binding and the ability to access the lowest mobility chromatin domains. We further use mutations of transcription factor structure to understand the basis for FOXA1 mobility characteristics and show that the main drivers of accessibility to low mobility chromatin domains are nonspecific DNA binding and direct nucleosome contacts. Our two-parameter method helps resolve chromatin interaction types in ways that diffusion and SMT-based residence time assessments do not.
RESULTS
Histone H2B local motions defines a heterogeneous chromatin mobility landscape
Using HiLO-based SMT with a 10 millisecond (ms) exposure (or Fast-SMT, Figure 1A, see STAR-Methods and Chen et al., 2014) in a volume depth of 200 nm in the H2.35 liver cell line (Zaret et al., 1988), we first measured the diffusion coefficients (in μm2/s) (Liu et al., 2014) of histone H2B-Halo, using dCas9-Halo expressed without a guide RNA (dCas9X) to compare for free diffusion track behavior. As seen in the motion tracks, 88% of histone H2B molecules presented diffusion coefficients below the range of free dCas9X (Figure S1A-D), reflecting their association with chromatin.
FIGURE 1: Characterization of H2B single-molecule dynamics allow the definition of a chromatin mobility landscape.
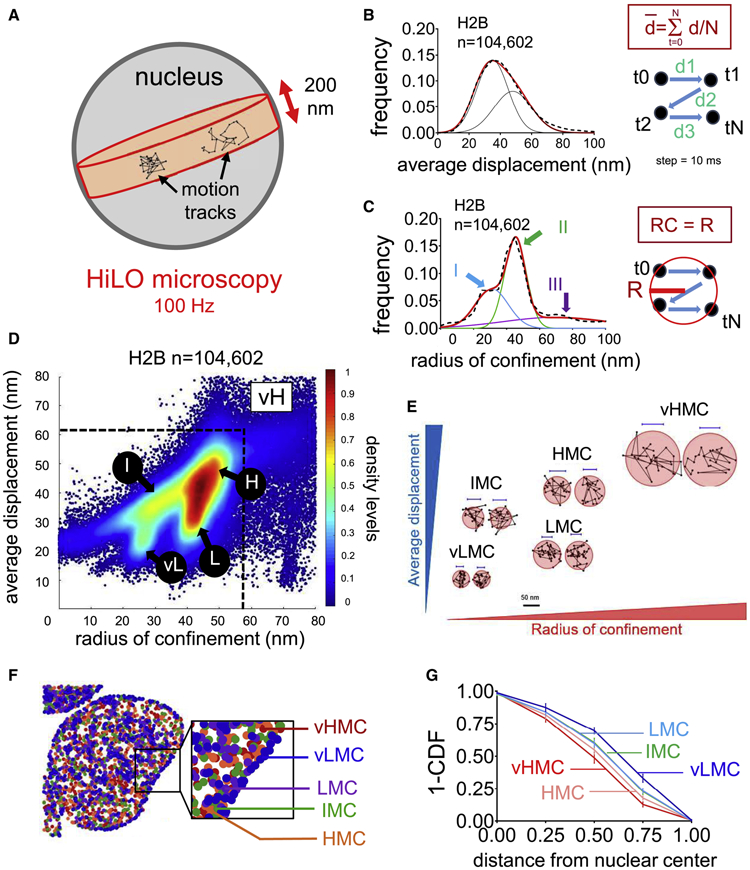
A: Experimental set-up for HiLO microscopy. The framerate for image acquisition is 100 Hz.
B: Black dashed curve: frequency distribution of the average displacement (, in nm) for n=104,602 histone H2B motion tracks. Red curve: 2 gaussian fitting (R2= 0.9977, mean 39 and 47 nm; s.d. 8 and 12 nm). As indicated in the panel, each step of displacement corresponds to 10 ms.
C: Black dashed curve : frequency distribution of radius of confinement (in nm) for n=104,602 histone H2B motions tracks. Red curve: 3 gaussian fitting (R2= 0.9832). Blue, Green and curves and arrows indicate the 3 populations (I: mean 30 nm, s.d 9 nm, II: mean 46 nm, s.d. 5 nm and III: mean 70 nm, s.d. 31 nm).
D: Scatter density plot of radius of confinement vs. average displacement assigned to n=104,602 histone H2B motion track. vL: vMLC, L: LMC, I: IMC, H: HMC, vH: vHMC.
E: Representative histone H2B motion tracks in each chromatin mobility population
F: Subnuclear localization of the histone H2B mobility groups, randomly downsampled to n=794 tracks. Dark blue: vLMC, purple: LMC, green: IMC, orange: HMC, red: vHMC.
G: For each chromatin mobility group, Cumulative Distribution Function (subtracted to 1, 1-CDF) of the distances between the average position of each individual motion track and the nuclear center (defined as the average coordinates of all motion tracks). The maximal distance was set to 1 arbitrary unit to normalize differences due to nuclear sizes. n=8 cells.
We filtered out the free-diffusing subset of Histone H2B (Figure S1E-H, and see STAR Methods) and used two parameters to characterize the chromatin-bound histone H2B movements: the average displacement in each motion track (in nm, Figure 1B), reflecting the average speed of the molecule over the time of detection at the imaging frame rate (100 Hz); and the radius of confinement (in nm, Figure 1C), estimating the size of the circle best encompassing each motion track (Wieser and Schütz, 2008). As expected (see STAR Methods), neither of the parameters correlated with the duration of the motion tracks (Figure S1I-J). The two parameters positively correlated with each other, but with marked deviations (Figure S1K, black arrows), reflecting that motion tracks with similar average displacements could have different radii of confinement and vice versa (Figure S1L). Consistent with Voronoi segmentation (Figure S1M), the density plot of the two parameters was clustered into five groups: very low mobility chromatin (vLMC; Figure 1D, circle vL, 9% of the molecules); low mobility chromatin (LMC; Figure 1D, circle L 19%); intermediate mobility chromatin (IMC; Figure 1D, circle I, 13%); high mobility chromatin (HMC; Figure 1D, circle H, 43%) and very high mobility chromatin (vHMC; Figure 1D, square vH, 16%). The robustness of our method was evident from observing similar five chromatin mobility groups for H2B in a pancreatic cancer cell line, human fibroblasts, and mouse embryonic fibroblasts (Figure S1N-Q). The main variation was seen in the amount of low mobility chromatin, reflecting biological differences (see Discussion) with the extreme case being human iPS cells, which presented chromatin mobility mostly in the HMC and vHMC range (Figure S1Q, red arrows indicating minimal vLMC and LMC), consistent with previous biophysical studies relating pluripotency with higher chromatin dynamics (Meshorer et al., 2006). Mitotic H2.35 cell chromosomes overall exhibited an increase in vLMC, LMC, and IMC, and a decrease in HMC and vHMC (Figure S1R-S and Table S1). However, each chromosome arm displayed a variety of mobility states (Figure S1T-U), consistent with heterogeneous structures seen in ChromEMT (Ou et al., 2017). In summary, a two-parameter assessment of histone H2B motion tracks reveals distinct chromatin mobility patterns (or fingerprints) that corresponds to different cell states.
Chromatin compaction states found at the nuclear lamina are also present in other nuclear regions
Each H2B-chromatin mobility population was present throughout the nucleus (Figure 1F and Figure S2A-E). vLMC showed an enrichment at the nuclear periphery, as well as did LMC and IMC to a lesser extent (Figure 1G and Figure S2A-C, black arrows), correlating with the location of compact chromatin seen in super-resolution microscopy (Otterstrom et al., 2019; Ricci et al., 2015). HMC and vHMC were localized mostly in central regions (Figure 1F-G and Figure S2D-E). Notably, H2B in vLMC was also observed extensively within the nucleus (Figure 1F and Figure S2A), demonstrating that such compaction states exist independently of the nuclear periphery. We used Delaunay triangulation (Yang and Cui, 2010, Figure S2F and see STAR Methods) to assay the spatial clustering of each chromatin mobility group and observed high degrees of clustering solely for vLMC, confirming its organization in spatially restricted domains (Figure S2G).
The nuclear periphery hosts large heterochromatin domains interacting with the nuclear lamina at Lamin Associated Domains (LADs) (van Steensel and Belmont, 2017). Accordingly, the two-parameter mobility plot of Lamin-A exhibited a clear predominance of vLMC (Figure 2A, red arrow), which we then quantified as a ratio of Lamin-A density over histone H2B in each category, except for vHMC because of data sparsity (Figure 2B and Table S1). Lamin-A motions in the vLMC range were enriched at the nuclear periphery (Figure 2C). In central nuclear regions, Lamin-A was in the higher ranges of chromatin mobility (Figure 2C), which could relate to its recently described roles in euchromatin regulation (Gesson et al., 2016). Notably, both histone H2B and Lamin-A also exhibited higher mobility events close to the nuclear periphery (Figures 1F, 2C), consistent with the heterogeneity in promoter repression at the nuclear lamina, as recently described (Leemans et al., 2019).
FIGURE 2: vLMC corresponds to the mobility of Lamin-A.

A: Scatter density plot of radius of confinement vs. average displacements for Lamin-A motion tracks vL: vMLC, L: LMC, I: IMC, H: HMC, vH: vHMC. The red arrow indicates the predominance of the vLMC range.
B: Relative mean density levels of Lamin-A compared to histone H2B in vLMC, LMC, IMC and HMC. *** indicates p<0.0001, n.s. non-significant differences (p>0.05) as determined by one-way ANOVA, see Table S1). vHMC not included because of high data sparsity.
C: Subnuclear localization of the different mobility groups of Lamin-A.
See also Table S1.
Mapping heterochromatin constituents onto the chromatin mobility landscape
To assess how heterochromatin constituents map onto the chromatin mobility landscape, we assayed the mobility of HALOtag fusions of the heterochromatin epigenetic readers HP1α, HP1β, and HP1γ, and of the heterochromatic writers SUv39h1 and SUv39h2, selecting confined motion tracks (Figure 3A-D, and Figure S3A-C, and Table S1). The mobility pattern of HP1α and SUv39h2 showed enriched levels in the lowest mobility chromatin (vLMC and LMC, Figure 3B-D, red arrows), as well as SUv39h1, to a lesser extent (Figure 3E, F). Concomitantly, HP1α and SUv39h2, but not Suv39h1, were depleted moderately from IMC and strongly from HMC, respectively (Figure 3G, H). Thus, heterochromatin regulators preferentially interact with low mobility chromatin, confirming the latter's repressed nature. Distinctions seen between the behavior of SUv39h1 and SUv39h2 can relate to structural differences among the two isoforms (see Discussion). HP1 isoforms β and γ presented abundant levels in LMC, like HP1α, but not in vLMC (Figure S3D, E) and were depleted in IMC (Figure S3F). HP1γ was less depleted than the other isoforms in HMC (Figure S3G). Variations in the behaviors of HP1 isoforms also correspond to differences in regulatory behaviors and biochemical properties (see Discussion).
FIGURE 3: Heterochromatin constituents map with low mobility chromatin.
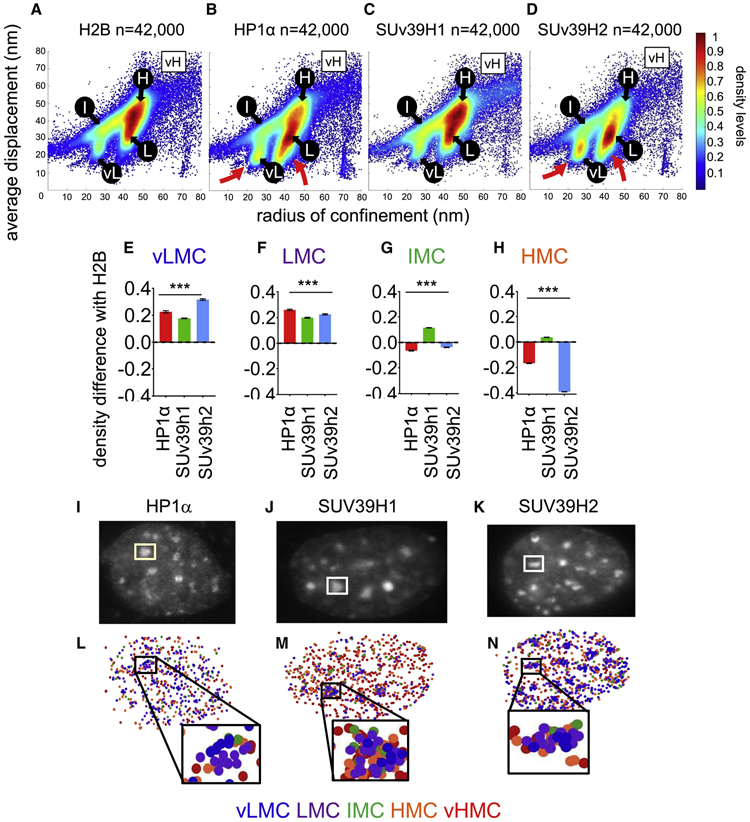
A-D: Scatter density plots of radius of confinement vs average displacements for histone H2B (A) HP1α (B), Suv39h1 (B) and SUV39h2 (C). vL: vMLC, L: LMC, I: IMC, H: HMC, vH: vHMC. The red arrows emphasize strong vLMC and LMC for HP1α and SUv39h2.
E-H: Relative mean density levels of HP1α (red), SUv39h1 (green) and SUv39h2 (blue) compared to histone H2B in the vLMC (E), LMC (F), IMC (G) and HMC (H). vHMC not included because of high data sparsity. *** indicates p<0.0001, as determined by one-way ANOVA (see Table S1).
I-K: representative nuclei expressing HP1α (I) SUv39h1 (J), and SUv39h2 (K) showing presence of the proteins at the chromocenters.
M-O: Subnuclear localization of HP1α(M) SUv39h1 (N), and SUv39h2 (O). Dark blue: vLMC, purple: LMC, green: IMC, orange: HMC, red: vHMC.
In mice, pericentromeric chromatin is organized in chromocenters, appearing as dense regions and enriched in H3K9me3 marks and associated proteins (Erdel et al., 2020; Guenatri et al., 2004). HP1α, HP1β, SUv39h1, and Suv39h2, but not HP1γ, were found in chromocenters (Figure 3I-K, and Figure S3H, I), mostly in low mobility ranges (vLMC and LMC) (Figure 3M-O, and Figure S3J), consistent with the compacted nature of pericentromeric heterochromatin. At lower levels, we also observed interactions of the heterochromatin regulators with higher chromatin mobility ranges within chromocenters (Figure 3M-O and Figure S3J), potentially reflecting heterogeneities in the structure of chromocenters that have been seen in super-resolution microscopy (Erdel et al., 2020).
In summary, despite variations, chromatin-bound heterochromatin regulators exhibit low mobility states, confirming the relationship between chromatin compaction, gene repression, and lower ranges of mobility.
Nucleosome affinity of transcription factors predicts their chromatin mobility
We sought to understand how the chromatin mobility landscape defines ranges of accessibility for fate-changing transcription factors, such as nucleosome-binding pioneer factors that enable cell fate changes versus factors eliciting terminal differentiation or growth (Iwafuchi-Doi and Zaret, 2016). Accordingly, we selected the liver pioneer factor FOXA1, GATA4, and differentiation factors HNF1A and HNF4A, the ubiquitously expressed cMYC, the pluripotency pioneer factors SOX2, OCT4, and KLF4, and the macrophage pioneer factor PU.1; all were expressed at comparable low levels (Figure S4A-B). Previous diffusion coefficient and residence time measurements studies by SMT (Chen et al., 2014; Liu et al., 2014; Paakinaho et al., 2017; Swinstead et al., 2016), FRAP (Figure S4C), as well as those in our laboratory, did not show a trend matching the different biological functions of the selected transcription factors (Figure S4D-H, and Table S2). For example, the strong nucleosome interactor SOX2 presented diffusion coefficients (Figure S4D) and residence times in chromatin (Figure S4E-H and Table S2) comparable to cMYC or HNF4A, which present weak or virtually no affinity for nucleosomes, respectively. Among strong nucleosome interactors, only FOXA1 exhibited a distinct low-diffusion behavior (Figure S4D), consistent with previous FRAP experiments (Sekiya et al., 2009) and possibly related to its structural similarity with histone H1 (see Discussion).
Heterochromatin constituents showed various diffusion behaviors, with HP1γ presenting a distribution like most transcription factors, suggesting that interaction with compact chromatin is not clearly related to its diffusion coefficient (Figure S4I-M). As an important control, we saw no correlation between the measured SMT parameters and the expression levels of the proteins (Figure S5A-R).
By contrast, the two-parameter mobility plots of chromatin-bound transcription factors exhibited striking differences that correspond to known functions (Figure 4A-J, Figure S6A-E, Table S1). The strong nucleosome binders FOXA1 and SOX2 interacted with vLMC and LMC (Figure 4B-C, red arrows, and Figure 4K-L), but were significantly reduced in HMC (Figure 4N). OCT4, KLF4, and PU.1, other nucleosome interactors, were enriched at slightly lower levels in vLMC and LMC (Figure 4D-F, red arrows, and Figure 4K, L), but still found in HMC (Figure 4N). cMYC and GATA4, weaker nucleosome interactors (Sekiya et al., 2009), were reduced both in vLMC and LMC (Figure 4K-L), mostly binding to HMC (Figure 4N). Finally, HNF1A and HNF4A, differentiation factors, were virtually absent from vLMC and LMC (Figure 4K-L) and present only in the HMC range (Figure 4N). The IMC was poorly discriminating towards transcription factors (Figure 4M) and could be a composite of accessible and inaccessible domains.
FIGURE 4: Nucleosome interaction correlates with enrichment in low mobility groups for chromatin bound transcription factors molecules.

A-J: Scatter density plots of radius of confinement vs. average displacement for histone H2B (A), FOXA1 (B), SOX2 (C), OCT4 (D), KLF4 (E), PU.1 (F), cMYC (G), GATA4 (H), HNF1A (I) and HNF4A (J). vL: vMLC, L: LMC, I: IMC, H: HMC, vH: vHMC. Shades of grey to black in the names of the different mobility ranges indicate increasing proportion of the mobility population. Red arrows indicate the vLMC population of strong nucleosome interacting transcription factors.
K-N: Relative mean density over histone H2B for FOXA1 (light blue), SOX2 (dark blue), OCT4 (orange), KLF4 (pink), PU.1 (grey), GATA4 (brown), cMYC (purple) HNF1A (red) and HNF4A (green) in the vLMC (K), LMC (L), IMC (M), and HMC (N) chromatin mobility ranges. *** indicates p<0.0001, n.s. non-significant differences (p>0.05) as determined by one-way ANOVA, see Table S1).
In summary, the chromatin mobility landscape is differentially accessed by transcription factors with different biological functions, and accession to the most restricted domains correlates with high levels of an intrinsic nucleosome binding ability.
Non-specific DNA interactions can elicit long-lived binding events
Short and long-lived binding events seen in SMT are considered to reflect nonspecific and specific DNA interactions, respectively (Voss and Hager, 2014). Nevertheless, point mutations at the protein-DNA interface had a partial effect on the SMT-measured residence time of various transcription factors (Chen et al., 2014; Paakinaho et al., 2017). Two previously characterized mutants of FOXA1 allow the discrimination of specific and non-specific DNA binding. FOXA1-NHAA disrupts hydrogen bonding to bases in the DNA major groove and has lost DNA binding specificity, but retains non-specific interactions with DNA and nucleosomes in vitro (Sekiya et al., 2009). FOXA1-RRAA disrupts phosphodiester backbone binding and presents an impaired overall DNA binding, but retains specificity for the FOXA1 motif, in vitro (Sekiya et al., 2009) and in vivo (Caravaca et al., 2013; Raccaud et al., 2019). Halo-tagged FOXA1-WT, FOXA1-NHAA, and FOXA1-RRAA were expressed at comparably low levels in H2.35 cells (Figure S7A-B). Using ChIP-qPCR, we observed that loss of specific DNA binding by FOXA1-NHAA-HALO resulted in decreased binding at the albumin enhancer, to a stronger extent than seen upon loss of non-specific DNA binding (FOXA1-RRAA-Halo) (Figure S7C).
We used SlowSMT with an exposure of 500 ms (Chen et al., 2014) to measure the residence times of FOXA1-WT, FOXA1-NHAA, and FOXA1-RRAA over 100 seconds. Fitting the residence time distribution with a double exponential decay, and correcting for photobleaching (see STAR Methods), we measured three parameters: 1) the size of the bound fraction (%); 2) the average duration of short-lived interactions, in seconds; and 3) the average duration of long-lived interactions, in seconds. Consistent with the diminution in residence time distributions (Figure 5A), loss of specific DNA binding (FOXA1-NHAA) resulted in weaker effects on the three parameters than loss of nonspecific DNA binding (FOXA1-RRAA, Figure S7D-F, and Table S2). To extend these findings, we defined a range for the longest measured interactions, defined as the 5th percentile of histone H2B residence times above 20 s (Figure 5A and Figure S7G). In this range of very long-lived interaction events, despite a lower frequency compared to the wildtype protein (Figure 5B), FOXA1-NHAA, mutant for specific DNA binding, exhibited similar residence times as wild type (Figure 5C, and Table S2). These findings indicate that under the conditions of SMT measurements, non-specific binding events can result in long-lived chromatin interactions. Thus, if the FOXA1 interactions observed in SlowSMT are mainly non-specific, it is possible that specific interactions indeed elicit much longer residence times (see Discussion).
FIGURE 5: Nucleosome interaction and non-specific DNA interaction provides the main contribution to the low-diffusion behavior of FOXA1 and to access to low-mobility chromatin.

A: Logarithmic frequency distribution (1-CDF: Cumulated Distribution Function subtracted to 1) of residence times for n=22,000 molecules of FOXA1-WT (blue), NHAA (red) and RRAA (green). The black dashes line indicate the 5th percentile for histone H2B residence times (~20 seconds).
B: fraction (in %) of molecules with residence times above 20 seconds for FOXA1-WT (blue), NHAA (red) and RRAA (green). *** indicates p<0.0001, n.s. non-significant differences (p>0.05) as determined by one-way ANOVA, see Table S2).
C: Average residence times (in seconds) of molecules with residence times above 20 seconds for FOXA1-WT (blue), NHAA (red) and RRAA (green). *** indicates p<0.0001, n.s. non-significant differences (p>0.05) as determined by one-way ANOVA, see Table S2).
D-E: Scatter density plot of the radius of confinement vs. average displacement assigned to every individual motion of FOXA1-NHAA (D) and FOXA1-RRAA (E). vL: vMLC, L: LMC, I: IMC, H: HMC, vH: vHMC. Red arrows indicate the loss of vLMC and LMC range for FOXA1-RRAA.
F-I: Relative mean density levels of FOXA1-NHAA (red), FOXA1-RRAA (green) compared to FOXA1-WT in the vLMC (F), LMC (G), IMC (H), HMC (I) chromatin mobility populations. *** indicates p<0.0001 (one-way ANOVA, see Table S1).
J-K: Scatter density plot of the average displacement vs radius confinement assigned to every individual motion of FOXA1-WT (D) and FOXA1-EKQ/AAA (E). vL: vMLC, L: LMC, I: IMC, H: HMC, vH: vHMC. Shades of grey to black indicate increasing proportion of the mobility population.
L: Relative mean density levels of FOXA1-EKQ compared to FOXA1-WT in vLMC, LMC, IMC, HMC and vHMC. *** indicates p<0.0001 (one-way ANOVA, see Table S1).
Association of FOXA1 with low mobility chromatin is largely dependent on non-specific nucleosome binding
In FastSMT, consistent with previous FRAP results (Sekiya et al., 2009), loss of nonspecific DNA binding (FOXA1-RRAA) impairs the diffusion coefficients of FOXA1 more markedly than loss of specific DNA binding (FOXA1-NHAA, Figure S7H). Thus, non-specific DNA binding, which correlates with non-specific nucleosome binding (Sekiya et al., 2009), provides the strongest contribution to the low diffusion of FOXA1, consistent with most interactions seen in FastSMT being nonspecific chromatin binding events.
We then measured the radius of confinement and average displacements for the two FOXA1 mutants (Figure S7I-J). The two-dimensional density plots after loss of loss of non-specific DNA binding (FOXA1-RRAA) showed greater changes than upon loss of specific DNA binding (FOXA1-NHAA) (Figure 5D, E, red arrows). Indeed, loss of non-specific DNA binding (FOXA1-RRAA) causes a strong skew away from vLMC and LMC, compared to FOXA1-WT, whereas loss of specific DNA binding (FOXA1-NHAA) leads to a more modest depletion (Figure 5F, G and Table S1). The other mobility ranges were less affected by the mutations (Figure 5H, I and Table S1). Thus, nonspecific DNA interactions are the main driver of interactions with the most compact chromatin domains.
Recent studies found that FOXA1 makes direct contact with core histones at two sites, including via lysine 270, outside of the DNA binding domain (Iwafuchi et al., 2020). Point mutations at lysine 270 and its adjacent residues (FOXA1-EKQ/AAA) did not impair the free DNA binding of FOXA1 in vitro (Figure S7K, L). FOXA1-EKQ/AAA presented a similar diffusion profile as FOXA1-WT (Figure S7M). Nevertheless, the two-parameter mobility plot of FOXA1-EKQ/AAA showed a strong decrease in the vLMC range (Figure 5J, L, red arrow), along with an increase in the HMC range (Figure 5L). In conclusion, nonspecific nucleosome interactions are crucial for the pioneer factor FOXA1 to access the most restricted chromatin domains.
DISCUSSION
The hypothesis that chromatin functional states are reflected by local mobility was founded on the observation of low mobility chromatin at the nuclear periphery (Chubb et al., 2002; Heun et al., 2001; Marshall et al., 1997; Nozaki et al., 2017), as well as of mobility changes during the transcriptional activation of individual loci (Chuang et al., 2006; Gu et al., 2018; Neumann et al., 2012). To test the hypothesis at the scale of the entire nucleus, we developed a two-parameter SMT approach, measuring the radius of confinement and average displacement of the motion tracks of individual histone H2B molecules, with a time resolution of 10 ms. By classifying histone H2B motion tracks and comparing them to a dCAS9 control (see STAR Methods), we were able to exclude free-diffusing molecules for the measurements. We thus defined a chromatin mobility landscape composed of five ranges, with distinct subnuclear localizations, that were recapitulated in other cell lineage and biological contexts. In iPS cells, the absence of vLMC and LMC is consistent with less defined chromatin compaction states (Meshorer et al., 2006; Schlesinger and Meshorer, 2019). In mitotic chromosome arms, the generally decreased chromatin mobility agrees with higher levels of compaction. Yet we still observed heterogeneity within chromosome arms, consistent with the spectrum of accessibilities seen for transcription factors in mitosis (Raccaud et al., 2019). Such heterogeneities could also reflect local domains that are inherited epigenetically, in the absence of a higher-order TAD structure (Naumova et al., 2013).
Very low mobility chromatin (vLMC) was enriched at the nuclear periphery, where it presented mobility features like Lamin-A, and thus likely represents low-accessible chromatin domains with compaction states corresponding to those found in LADs. Yet finding vLMC throughout the nucleus indicates that such compaction states can be built independently of association with the nuclear lamina. vLMC and LMC, which had slightly more accessible features, were explored by heterochromatin regulators, confirming its compacted nature. Stronger enrichment of SUv39h2 in vLMC compared to SUv39h1 may relate to an additional basic, amino-terminal domain in SUv39h2, which confers an enhanced interaction with major satellite repeat-associated RNA (Velazquez Camacho et al., 2017). These distinctions illustrate how the two-parameter measurements of chromatin mobility can distinguish functions of epigenetic proteins. Similarly, stronger enrichment of HP1α compared to HP1β and HP1γ correlates with a stronger affinity for free DNA in vitro (Nishibuchi et al., 2014) and enhanced binding stability in vivo (Bryan et al., 2017). Interestingly, HP1α presents lower affinity for the H3K9me3 modification compared to HP1β and HP1γ (Canzio et al., 2014), which suggests that DNA binding may drive a stronger accession of vLMC more than recognition of repressive histone marks.
vLMC and LMC were explored by transcription factors with a strong nucleosome affinity in vitro, including FOXA1, SOX2, and to a lesser extent OCT4, KLF4 and PU.1, consistent with recent in vitro studies showing a spectrum of nucleosome affinities among transcription factors, rather than a binary behavior (Fernandez Garcia et al., 2019; Zhu et al., 2018). The importance of nucleosome interaction in accessing vLMC was confirmed by a mutant of FOXA1 (FOXA1-EKQ/AAA), which presents an impaired interaction with histones, but normal interaction with DNA. An exception is HNF1A, which shows significant nucleosome interactions in vitro (Garcia et al., 2019). However, in vivo, HNF1A dimers are in a large complex with a dimer of DCoH (Mendel et al., 1991), which could sterically limit its interactions with compact chromatin (Ou et al., 2017).
At the other end of the chromatin mobility landscape, HMC and vHMC are more homogeneously distributed in the nucleus. Although heterochromatin regulators were generally depleted from HMC, SUv39h1, and HP1γ show higher levels in HMC than other isoforms, consistent with H3K9me3 domains occurring across the spectrum of heterochromatin to euchromatin (Becker et al., 2017; Vakoc et al., 2005). HMC is explored by all transcription factors, except for FOXA1 and SOX2, which could reflect the dominance of low nucleosome off-rates compared to that for free DNA (Cirillo and Zaret, 1999; Donovan et al., 2019) and would be consistent with a lateral chromatin scanning mechanism, instead of free diffusion in the nucleoplasm (Cortini and Filion, 2018; von Hippel and Berg, 1989).
The high proportion of chromatin-interacting FOXA1 molecules, as seen by diffusion coefficient measurements, may not be a general characteristic of nucleosome interactors reflecting access to vLMC, since SOX2, OCT4 or KLF4 exhibit higher rates of free diffusion, but rather could be linked to the structural similarity of FOXA1 with linker histone H1 (Cirillo et al., 1998; Clark et al., 1993; Ramakrishnan et al., 1993). We speculate that FOXA1 explores local chromatin regions through repetitive interaction events, including additional interactions with histones (Cirillo et al., 2002; Iwafuchi et al., 2020). Differences in chromatin exploration have been seen for cMYC and PTEF-b (Izeddin et al., 2014). The main contribution of nonspecific DNA binding to the model of FOXA1 chromatin scanning is consistent with what is seen for interaction of transcription factors with mitotic chromatin (Caravaca et al., 2013; Raccaud et al., 2019), and local folding of chromatin polymers could increase occupancy of transcription factors by a local increase of their concentration, independently of specific chromatin binding (Cortini and Filion, 2018).
As seen previously (Swinstead et al., 2016), we find no correlation between SMT-measured residence times of transcription factors and their interaction with different functional types of chromatin, as elicited by affinity for nucleosomes. As with many other SMT studies (Callegari et al., 2019; Chen et al., 2014; Paakinaho et al., 2017; Raccaud et al., 2019; Swinstead et al., 2016), the residence times of transcription factors we measured here are on the order of 10-20 seconds. By contrast, residence times measured in vitro can go up to hundreds of seconds (Donovan et al., 2019; Luo et al., 2014). Also, residence times measured in vivo by SMT are poorly compatible with the measured transcriptional bursting dynamics on the scale of several minutes (Otto, 2019). Here, we show that nonspecific binding by FOXA1 can still give rise to long-lived chromatin binding events, by SMT. Hence, due to noisy measures and dependence on experimental conditions, SMT could fail to identify the longest-lived binding events. This failure could possibly arise from the fact that the size of specific consensus motifs is only a small fraction of the entire genome. Hence, the probability to observe a specific binding event by SMT could be low, especially as only a subset of the nuclear volume is imaged. The high rate of photobleaching in SMT might also prevent the identification of very long-lived binding events, despite mathematical corrections, which could be improved by the introduction of long dark times between imaging acquisition, as previously shown (Liu et al., 2018). Regardless, since we found that FOXA1 interactions observed in SlowSMT are mainly nonspecific, we suggest that specific interactions in vivo may indeed involve much longer residence times. Previous studies showed that transcription factors mutations impairing DNA binding (Callegari et al., 2019; Chen et al., 2014; Paakinaho et al., 2017) had little effects on their residence time, but rather caused a strong decrease in the frequency of binding events, consistent for what we observe for the very long residence times of FOXA1. This suggests that in live cell SMT, the size of the bound fraction might reflect more accurately binding affinity than the residence time per se.
In summary, we developed a two-parameter method to define a chromatin mobility landscape that is differentially explored by transcription factors and epigenetic chromatin binding proteins. Mapping of the chromatin binding factors onto the mobility landscape provided insights that relate to the factors' inherent biochemical capacities and biological functions. Further work is needed to understand how the dynamic binding of transcription factors to any domain of chromatin relates to the remarkable specificity for factors detected at target motifs in the genome.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
For other reagents generated in this study or questions about the reagents, please contact Ken Zaret (zaret@pennmedicine.upenn.edu).
Materials availability
All the materials generated in this study are accessible upon request.
Data and Code availability
The data and code used in this study are accessible upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Lines and Tissue Culture
H2.35 cells (Zaret et al., 1988) were grown in Low Glucose Medium (ThermoFisher 10567 low glucose DMEM , GlutaMAX™ Supplement, pyruvate, supplemented with GlutaMAX™, characterized fetal bovine serum (Hyclone SH300071.03) , 0.2 μM dexamethasone (Sigma D4902), 100 units/ml penicillin and 100 μg/ml streptomycin(Thermo Fischer 15140122) at 33C with 5% CO2. Three days before imaging, H2.35 cells were incubated at 37C with 5% CO2. Imaging experiments were carried out in Phenol red-free Low Glucose Medium (ThermoFisher 11054020 DMEM, pyruvate, supplemented with 1X GlutaMAX™ Supplement, 4% tetracycline-free fetal bovine serum and 0.2 μM M dexamethasone) in an imaging chamber heated at 37°C (more details in the Single Molecule Live Cell Imaging section).
For stable clone generation, one million H2.35 cells were seeded in a 10cm dish. After 24h, 1mL of non-concentrated corresponding retrovirus were added directly to the cell culture medium. The medium was changed after 24h. After another 24h, cells were stained for 20 min with HaloTag-TMR (Promega G8251) at 5 μM, before, and cells were observed under a Nikon Eclipse ti 2000-D epifluorescence microscope to assay the efficiency of infection. Typically, ~50% of the cells were expressing the Halotag fusion proteins. Cells were sorted with a BD FACSJazz™ by fluorescence intensity of HaloTag-TMR (emission 575 nm). The lowest expressing cells was replated in a 10 cm dish with fresh Low Glucose Medium. After 48h, the cells were seeded at limit dilution in a 96 well plate. Wells containing 1 clone were then amplified, and expression levels assayed by western blot.
METHOD DETAILS
Plasmid Construction and Genome Editing
pCMV plasmids:
FOXA1-HaloTag : The FOXA1-Wasabi vector (source :laboratory) was digested with KpnI and BsrGI, in order to replace the Wasabi protein at the C-terminal end of FOXA1 by the HaloTag from pENTR4-HaloTag (source Addgene #29644), amplified with primers JL1 and JL2 (see Table S3), allowing the addition of a long polyglycine linker (SGGGGSGGGGSGGGGSGGGGS) between FOXA1 and the HaloTag.
Other HaloTag constructs: pCMV-FOXA1-HaloTag was digested by EcoRI and KpnI. The gene of interest was amplified with the adequate primers (see Table S3, primers JL3 to JL22) and either ligated with vector using T4 DNA ligase (NEB M0202T, HNF1A, HNF4A), either assembled with the vector using Gibson Assembly® Master Mix kit (NEB E2611L, All other proteins).
pGCDNsam plasmids (retroviral vectors):
pGCDNsam-FOXA1-HALO, pGCDNsam-FOXA1-NHAA-HALOm and pGCDNsam-FOXA1-RRAA-HALO.
FOXA1-HaloTag from pCMV-FOXA1-HaloTag was amplified from the correct position using JL23 and JL24 (see Table S3) and ligated into BamHI digested pGCDNsam-FOXA1 (Addgene plasmid #33003). FOXA1-NHAA-HALO and FOXA1-RRAA-HALO from pCMV-FOXA1-NHAA/RRAA-HALO were amplified using JL25 and JL26 and ligated into XbaI + HindIII digested pGCDNsam-FOXA1-RRAA-HALO.
TETO-FUW plasmids (lentiviral vectors):
TETO-FUW-HNF1A/TETO-FUW-HNF4A/TETO-FUW-MYC/TETO-FUW-GATA4/TETO-FUW-OCT4/TETO-FUW-KLF4/TETO-FUW-SOX2/TETO-FUW-PU.1/TETO-FUW-FOXA1EKQAAA
ORF of interest (see Key resource table) from the corresponding pCMV-ORF-HaloTag were PCR amplified with the adequate primers (see Table S3, JL27 to JL35) and assembled using Gibson Assembly® Master Mix kit (NEB E2611L) with EcoRI (NEB) digested TETO-FUW-OCT4 (Addgene plasmid #20323).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-FOXA1 | ABCAM | 55178 |
| Mouse anti-POL2 | COVANCE | 8WG16 |
| Bacterial and Virus Strains | ||
| pENTR4/HALO | Gift from Eric Campeau | Addgene W876-1 |
| H2B-Cherry | Gift from Robert Benezra | Addgene 20972 |
| pmWasabi-CT | Allele Biotech | ABP-FP-WCNCSP |
| Tet-O-FUW-OCT4 | Gift from Rudolf Jaenisch | Addgene 20323 |
| FOXA1-HALO | This paper | FOXA1-HALO |
| FOXA1-NHAA-HALO | This paper | FOXA1-NHAA-HALO |
| FOXA1-RRAA-HALO | This paper | FOXA1-RRAA-HALO |
| FOXA1-EKQAAA-HALO | This paper | FOXA1-EKQAAA-HALO |
| GATA4-HALO | This paper | GATA4-HALO |
| HNF1A-HALO | This paper | HNF1A-HALO |
| HNF4A-HALO | This paper | HNF4A-HALO |
| cMYC-HALO | This paper | cMYC-HALO |
| SOX2-HALO | This paper | SOX2-HALO |
| OCT4-HALO | This paper | OCT4-HALO |
| KLF4-HALO | This paper | KLF4-HALO |
| PU.1-HALO | This paper | PU.1-HALO |
| SUv39h1-HALO | This paper | SUv39h1-HALO |
| SUv39h2-HALO | This paper | SUv39h2-HALO |
| HP1α-HALO | This paper | HP1α-HALO |
| HP1β-HALO | This paper | HP1β-HALO |
| HP1γ-HALO | This paper | HP1γ-HALO |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Critical Commercial Assays | ||
| MinElute PCR purification kit | QIAGEN | 28004 |
| MiniPrep | QIAGEN | 27106 |
| MAXIPREP | QIAGEN | 12263 |
| Gel extraction kit | QIAGEN | 28706 |
| Gibson Assembly® kit | NEB | E5510 |
| T4 DNA Ligase high concentration | NEB | M0202T |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| DH5A competent cells | Invitrogen | 18265017 |
| H2.35 | Lab stock | H2.35 |
| H2.35 FOXA1 | This paper | H2.35_FOXA1-HALO |
| H2.35 FOXA1-NHAA | This paper | H2.35_NHAA-HALO |
| H2.35 FOXA1-RRAA | This paper | H2.35_RRAA-HALO |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| Table S3 | This paper | Table S3 |
| Recombinant DNA | ||
| Software and Algorithms | ||
| Serial Cloner | SerialBasics | Serial Cloner 2.6 |
| PRISM 7 | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Fiji is Just ImageJ (Fiji) version 2.0.0-rc/1.51f | ImageJ | https://imagej.net/ |
| Oni acquisition software | Oxford Nanoimaging | https://oni.bio |
| MATLAB | Mathworks | www.mathworks.com |
| Microsoft Office | Microsoft | www.office.com |
| MATLAB script - track classification | MATLAB script | SMT_Motion_Classifier.m |
| MATLAB script - MSD comparison | MATLAB script | Compare_MSD_Results |
| Other | ||
| Oni Nanoimager S (HiLO microscopy) | Oxford Nanoimaging | https://oni.bio |
Lentiviral and retroviral production and concentration
Retrovirus were produced as described in Sekiya and Suzuki, 2011. In brief, 293GPG cells were co-transfected with the retroviral expression vector (pGCDN) and pCMV-VSV-G. After 24h, the medium containing the retrovirus was centrifuged at 2,000 rpm for 10 minutes, the supernatant passed through a 0.45 μm filter and used directly to infect the cells.
Lentivirus were produced as described in (Becker et al., 2017). In brief, 293T cells were co-transfected with lentiviral expression vector, psPAX2 and PMDG. Fresh medium was added after 24h. After another 72h, the medium containing the lentivirus was centrifuges at 2,000 rpm for 10 min, passed through a 0.45 μm filter, pelleted by ultracentrifugation (24,000 rpm 3 hours) and resuspended at high concentration in 200 μL DMEM high glucose. Lentivirus were titered in H2.35 cells. Suboptimal M.O.I. (Multiplicity of Infection) was used (<1), in order to obtain low expression levels.
Western blotting
H2.35 cells were incubated for 3 days at 37C. Nuclear extracts were performed as previously described (Schreiber et al., 1989), and run on 10 % Bis-Tris gels (Life technologies), followed by standard western blotting procedures. FOXA1 was detected with a primary antibody (ABCAM 55178 1:1000) and a anti mouse secondary antibody (Santa Cruz SC-2005, 1:10,000). Detection was performed with ECL Prime reagent (SuperSignal™ West Pico PLUS Chemiluminescent Substrate, ThermoFisher 34580) and the Amersham 600 imager.
Electro Mobility Shift Assay (EMSA)
EMSA was performed on mouse recombinant FOXA1-WT and EKQAAA using the same methodology than in (Fernandez Garcia et al., 2019; Iwafuchi et al., 2020). In brief, end-labeled oligonucleotides were incubated with recombinant proteins in DNA-binding buffer (10 mM Tris-HCl pH7.5, 1 mM MgCl2, 10 mM ZnCl2, 1 mM DTT, 50 mM KCl, 3 mg/ml BSA, 5% Glycerol) at room temperature for 30 min. Free and bound DNA were separated on 4% non-denaturing polyacrylamide gels run in 0.5X Tris–borate–EDTA. EMSA gels were run at 90 V at room temperature nd visualized using with an Amersham Typhoon RGB Biomolecular Imager using Cy5 fluorescence setting (excitation at 633 nm and emission filter 670 BP 30) and a high sensitivity setting.
FRAP
All FRAP experiments were carried out on Leica TCS SP8 Confocal microscope, equipped with 405, 488, 552, 638 nm laser lines, an environmental chamber to control humidity and temperature, three spectral detection channels (2 PMTs and 1 HyD detector), a 40x water immersion objective (Nikon, NA = 1.49), at a resolution of 512*512 and a scanning speed of 1400. Cells were cultured in μ-Slide 8 Well from IBIDI (Ref: 80826), transfected with the appropriate pCMV plasmid and then incubated at 37°C 5% CO2 for 3 days. Ten prebleach frames were taken to estimate fluorescence intensity every 0.371 second. 4 rounds of bleaching of 0.371 second each were applied, followed by the acquisition of ten post-bleaching frames every 0.371 s and 60 frames every 2 seconds. Analysis was performed as in (Sekiya et al., 2009).
Single Molecule Live Cell Imaging
All single molecule live cell imaging was carried out on a Nanoimager S from Oxford Nanoimaging Limited (ONI), comporting a temperature and humidity controlled chamber, a scientific Complementary metal–oxide–semiconductor (sCMOS) camera with a 2.3 electrons rms read noise at standard scan, a 100X, 1.49 NA oil immersion objective and a 561 nm green laser. Images were acquired with the included Nanoimager software. 30,000 H2.35 cells were cultured in LabTek-II chambered 8 well plates (Lab-Tek 155049), transfected with the appropriate tetracycline-inducible lentivirus when necessary (without rtTA or doxycycline to keep low levels of expression) and then incubated at 37C 5% CO2 for 3 days. Before imaging, cells were treated with 5nM of Janelia Fluor 549 (JF549) HaloTag ligand (a kind gift from Luke Lavis, HHMI) for 15 minutes. Cells were subsequently washed three times in PBS at 37C, and Phenol Red-free Low Glucose medium was added to each well. All imaging was carried out under HILO conditions (Tokunaga et al., 2008). For imaging experiments, one frame was acquired with 100ms of exposure time (10 Hz) to measure the intensity of fluorescence of the nuclei. For FastSPT experiments, 5000 frames were acquired with an exposure of 10ms (100 Hz). For SlowSPT experiments, 200 frames were acquired with an exposure time of 500ms (2 Hz). Acquisition were realized over a field of 50 x 80 μm with a depth of 0.2 μm.
QUANTIFICATION AND STATISTICAL ANALYSIS
Two Parameters Single Molecule Tracking Analysis - Tracking algorithm
Imaging was performed over a depth of 200 nm, hence molecules motions are tracked in 3D but projected on a plane. For each individual imaged, a movie is generated as a TIF stack, and analyzed by the Matlab-based SLIMfast script (Teves et al., 2016), a modified version of MTT (Sergé et al., 2008). Frame-to-frame motions are defined by the distance between consecutive positions of the particle, and can be potentially related to (a) Brownian (random walk) or confined motions of the molecule (b) other artifactual effects such as the support or imperceptible movements of the nucleus. It is thus necessary, as described previously (Chen et al., 2014) to define a Maximal expected Diffusion Coefficient (DMax), that defines the maximal distance (dm) between two consecutive frames for a particle to be considered as the same object. As in the original publication (Chen et al., 2014), a cutoff was set to 3dm, to ensure a 99% confidence level. In FastSMT, higher time resolution (10ms) allows to visualize and track free-diffusing molecules with high diffusion coefficients, hence DMax was of 3 μm2/s−1. In SlowSMT, due to longer exposure times (500ms) free diffusing molecules are blurred in the background. In order to measure residence times of confined molecules only (corresponding to the binding at a single locus), the DMax setting was lower (0.1 μm2/s−1), excluding potential molecules with higher ranges of motion.
For each imaged nucleus, SLIMfast gives an output in the form of a .txt file consisting of a series of successive (x,y) coordinates and times of detection, corresponding to the displacement of each individual molecule in the nucleus. For analysis purposes, the output SLIMfast .txt files was reorganized by a homemade Matlab script in .csv format with the following data order : iteration (1-n, with n number of rows of the csv table), frame (the frame on which each single molecule was detected), time (t=frame*exposure), trajectory (ID number of the trajectory), x and y (2D coordinates of the molecule in μm). Each imaged individual nucleus thus has its corresponding .csv file.
Two Parameters Single Molecule Tracking Analysis - Classification of the tracks:
This section describes how single molecules motion trajectories (or tracks) are classified in order to exclude free-diffusing motions from the two-parameters analysis. The single molecule tracking .csv files (see previous section) were first analyzed by the homemade Matlab script“SMT_Motion_Classifier.m". Single molecule trajectories (or tracks) with a track duration shorter than 5 frames were discarded from the analysis. The T-MSD (or MSD=f(tlag), where tlag is the delay) curves of each motion track was fitted with a power law distribution (MSD = 4·D·tα, where D is the diffusion coefficient and α a coefficient that indicates the motion type) (Ernst and Köhler, 2012). Motion tracks were then classified in different groups: tracks with α≤0.7 were considered as Confined; motion tracks with 0.7<α<1 as Brownian; and motion tracks with α≥1 as Directed. In addition, the motion tracks showing a behavior similar to a levy-flight (presenting mixed Confined and Directed/Brownian behavior) were detected by the presence of a jump superior to the average jump among the track + a jump threshold of 1.5, and classified as “Butterfly”. Butterfly motion tracks were segmented into their corresponding Confined and Directed/Brownian sub-trajectories for posterior analysis. As an additional filtering step of Confined motions (including confined segments of Butterfly tracks), we defined a jump threshold of 100nm, to filter out motion tracks with an average frame-to-frame jump size bigger than 100nm. For H2B, the discarded tracks represented only 4% of all the confined tracks.
For the two-parameters analysis of chromatin, heterochromatin factors, andtranscription factors, we defined the bound state as being the pool of Confined motion tracks and of the Confined segments of the Butterfly motion tracks.
Two Parameters Single Molecule Tracking Analysis - Analysis of trajectories
After the track classification, the trajectories were analyzed by a homemade Matlab script “Compare_MSD_Results.m” to quantify diffusion coefficients, residence times, radius of confinement and average frame-to-frame displacement.
Two Parameters Single Molecule Tracking Analysis - Radius of Confinement
The T-MSD curves of each track were fitted using least squares with a circle confined diffusion model (Equation 1, Wieser and Schütz, 2008):
| (1) |
The fitting provides the radius of confinement R, the diffusion coefficient at short time scales D, and a constant offset due to the localization precision limit inherent to all the localization-based microscopy methods. In our case, we estimate a localization precision of 13 nm. To discard fitting errors related to effects of the number of steps (track duration) or to artifacts such as erroneously connected jumps, we have discarded the trajectories with squared norm of the residual (or RSS) higher than 10−5, and radius of confinement (R) higher than 300nm. In this step, less than 5% of the total data was discarded. The radius of confinement thus obtained represents the circle best encompassing the motion track, rather than encompassing it strictly. Thus, as seen in Figure S1I, the measurement of the radius of confinement is independent of the track duration. In fact, the measure of the radius of confinement is performed only on selected confined motions (see classification of the tracks, and Figure S1E-H), which T-MSD curves are defined by a plateau aspect. Thus, the T-MSD curves exhibit limited evolution over time, and the measure is independent of the track duration.
Two Parameters Single Molecule Tracking Analysis - Average displacement
The average displacement was computed by measuring the average Euclidean distance between two consecutive positions of the molecule in each individual motion track, or frame-to-frame jump.
Two Parameters Single Molecule Tracking Analysis - Radius of Confinement vs. Average displacement
For the joint representation, we have built scatter density plots using the same number of tracks for each condition (randomly downsampling, if necessary). For this purpose, as well as for building Voronoi diagrams of the data, we used the freely available Scatplot.m Matlab function. Apart from the plotting, we measured the local density (given by Scatplot.m) in the different chromatin mobility ranges to quantitatively compare the all the studied proteins with histone H2B . Density values were first normalized by dividing the local density of each point by the maximum density for each studied protein, and were then iterated point by point on the H2B density data, to find the 5 closest points in the density plot of each transcription factor. The density of these 5 points was then averaged the difference between each studied protein and H2B was computed for each individual motion track. For more intuitiveness, the whole scale was multiplied by −1. Then, positive 1 means that there is a difference of 100% where the studied protein is denser than H2B and negative values correspond to regions where H2B is denser.
Two Parameters Single Molecule Tracking Analysis - Subnuclear localization of the different mobility populations
For each individual nucleus, we defined the position of each motion track as a point corresponding to the average coordinates of the successive position of its corresponding tracked single molecule. To measure the radial distribution of each chromatin mobility group, the center of the cell was defined as the average coordinates of all identified motion tracks positions. To prevent any shape related biases, we used we selected cells with rounded shaped nuclei, and used Euclidean transformation on the motion tracks position coordinates, so that their distribution forms an object as round as possible around the nucleus center. The Euclidean distance of each motion track position to the cell center was then measured, and the cumulative distribution function of distance plotted on a horizontal axis. To normalize for differences in nuclear size, we set the maximal distance from the nucleus center as equal to 1 for all cells.
Two Parameters Single Molecule Tracking Analysis - Delaunay triangulation
The motion track position (as defined in the previous section) for each chromatin group was down-sampled to study the same number of objects. The Delaunay function of Matlab was used to triangulate the motion tracks position and get the sizes of the different Delaunay areas, which logarithmic frequency was subsequently plot on a horizontal axis.
Diffusion coefficients
The first 4 points of each T-MSD curve corresponding to each trajectory were fitted with a linear distribution to estimate the diffusion coefficient (Equation 2, Michalet, 2010):
| (2) |
Where D is the diffusion coefficient, tlag is the time between the two positions of the molecule used to calculate the displacement. The offset is due to the limited localization precision inherent to localization-based microscopy methods (~14 nm for our experiments). We set a coefficient of determination R2≥ 0.8 to ensure the good quality of the fitting performed to estimate D. Since the distribution of D follows a log-normal distribution (Nandi et al., 2012), the Log10(D) was used for a proper visualization and fitting of the Gaussian Bi-modal distribution.
Residence times:
We measured the residence times as performed previously (Chen et al., 2014; Mazza et al., 2013). In brief, the “residence_time.m” Matlab script extracts the duration of every detected track and converted it in a residence time (Res.Time = Track_Duration·Exposure_Time).
The 1-cumulative distribution function (1-CDF) of the residence time of every detected track was fitted with a two-exponential decay equation on GraphPad Prism 8, to separate the 1-CDF in a short-lived and a long-lived population (Equation 3).
| (3) |
k1 and k2 are the unbinding constant rates in seconds−1, t1=1/k1 and t2=1/k2 the residence times in seconds, and f a number from 0 to 1 measuring the fraction belonging to each population. As photobleaching highly affects the measure of residence times, the measured k1,2 can be separated into their two contributions:
| (4) |
where koff is the corrected unbinding rate and kb the rate due to photobleaching. In order to measure kb, we measured the fluorescence decay issued by constant illumination at the laser power used to perform SlowSMT, and fit the function by a single exponential decay equation to measure kb, (0.05 s-1) which was subtracted to the measured k1 and k2 to obtain the koff1 and koff2.
Supplementary Material
TABLE S1 (related to Figure 3): Radius of confinement and average displacement density differences scatter plot multiple comparison by one-way ANOVA
Values of mean density difference between the two factors, in each range of mobility (see Figure 1H) are indicated. A one-way ANOVA was performed to test the hypothesis H0: the mean of protein 1 is different from the mean of protein 2, and p-values are indicated. For each condition, n=25,000.
TABLE S2 (related to Figure 5): Residence times comparison
Values of mean long-lived fraction and short/long-lived residence time or size of long-lived fraction of two protein are indicated. A one-way ANOVA was performed to test the hypothesis H0: the mean of protein 1 is different from the mean of protein 2, and p-values are indicated. For each condition, n>100
TABLE S3 (related to STAR METHODS): oligonucleotides used for cloning
Highlights.
Dual-motion parameter analysis of histone H2B defines a mobility landscape
The chromatin mobility landscape, not residence times, reflects functional states
Heterochromatin constituents are variably present in low mobility chromatin
Low mobility chromatin exploration is enabled by nucleosome interactions
ACKNOWLEDGEMENTS
We thank Robert Tjian for promoting our collaboration with HHMI. We thank Luke Lavis for kindly providing the Halo-ligand coupled to JF549 fluorophore and Jingchao Zhang for providing iPS cells. We thank Penn Cell and Developmental Biology microscopy core. J.L. was supported by a fellowship from Fondation pour la Recherche Medicale code 40334. M.L was supported by a Linda Pechenik Montague Investigator Award. The research was supported by NIH grant R01GM36477 to K.S.Z.
Footnotes
DECLARATION OF INTERESTS
The authors have no conflicts of interest to declare.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Becker JS, McCarthy RL, Sidoli S, Donahue G, Kaeding KE, He Z, Lin S, Garcia BA, and Zaret KS (2017). Genomic and Proteomic Resolution of Heterochromatin and Its Restriction of Alternate Fate Genes. Mol. Cell 68, 1023–1037.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boettiger AN, Bintu B, Moffitt JR, Wang S, Beliveau BJ, Fudenberg G, Imakaev M, Mirny LA, Wu C, and Zhuang X (2016). Super-resolution imaging reveals distinct chromatin folding for different epigenetic states. Nature 529, 418–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan LC, Weilandt DR, Bachmann AL, Kilic S, Lechner CC, Odermatt PD, Fantner GE, Georgeon S, Hantschel O, Hatzimanikatis V, et al. (2017). Single-molecule kinetic analysis of HP1-chromatin binding reveals a dynamic network of histone modification and DNA interactions. Nucleic Acids Res. 45, 10504–10517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callegari A, Sieben C, Benke A, Suter DM, Fierz B, Mazza D, and Manley S (2019). Single-molecule dynamics and genome-wide transcriptomics reveal that NF-kB (p65)-DNA binding times can be decoupled from transcriptional activation. PLOS Genet. 15, e1007891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Canzio D, Larson A, and Narlikar GJ (2014). Mechanisms of functional promiscuity by HP1 proteins. Trends Cell Biol. 24, 377–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caravaca JM, Donahue G, Becker JS, He X, Vinson C, and Zaret KS (2013). Bookmarking by specific and nonspecific binding of FoxA1 pioneer factor to mitotic chromosomes. Genes Dev. 27, 251–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Zhang Z, Li L, Chen B-C, Revyakin A, Hajj B, Legant W, Dahan M, Lionnet T, Betzig E, et al. (2014). Single-molecule dynamics of enhanceosome assembly in embryonic stem cells. Cell 156, 1274–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chuang C-H, Carpenter AE, Fuchsova B, Johnson T, Lanerolle P. de, and Belmont AS (2006). Long-Range Directional Movement of an Interphase Chromosome Site. Curr. Biol 16, 825–831. [DOI] [PubMed] [Google Scholar]
- Chubb JR, Boyle S, Perry P, and Bickmore WA (2002). Chromatin motion is constrained by association with nuclear compartments in human cells. Curr. Biol. CB 12, 439–445. [DOI] [PubMed] [Google Scholar]
- Cirillo LA, and Zaret KS (1999). An early developmental transcription factor complex that is more stable on nucleosome core particles than on free DNA. Mol. Cell 4, 961–969. [DOI] [PubMed] [Google Scholar]
- Cirillo LA, McPherson CE, Bossard P, Stevens K, Cherian S, Shim EY, Clark KL, Burley SK, and Zaret KS (1998). Binding of the winged-helix transcription factor HNF3 to a linker histone site on the nucleosome. EMBO J. 17, 244–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, and Zaret KS (2002). Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 9, 279–289. [DOI] [PubMed] [Google Scholar]
- Clark KL, Halay ED, Lai E, and Burley SK (1993). Co-crystal structure of the HNF-3/fork head DNA-recognition motif resembles histone H5. Nature 364, 412–420. [DOI] [PubMed] [Google Scholar]
- Cortini R, and Filion GJ (2018). Theoretical principles of transcription factor traffic on folded chromatin. Nat. Commun 9, 1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dion V, and Gasser SM (2013). Chromatin Movement in the Maintenance of Genome Stability. Cell 152, 1355–1364. [DOI] [PubMed] [Google Scholar]
- Donovan BT, Chen H, Jipa C, Bai L, and Poirier MG (2019). Dissociation rate compensation mechanism for budding yeast pioneer transcription factors. ELife 8, e43008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erdel F, Rademacher A, Vlijm R, Tünnermann J, Frank L, Weinmann R, Schweigert E, Yserentant K, Hummert J, Bauer C, et al. (2020). Mouse Heterochromatin Adopts Digital Compaction States without Showing Hallmarks of HP1-Driven Liquid-Liquid Phase Separation. Mol. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst D, and Köhler J (2012). Measuring a diffusion coefficient by single-particle tracking: statistical analysis of experimental mean squared displacement curves. Phys. Chem. Chem. Phys 15, 845–849. [DOI] [PubMed] [Google Scholar]
- Fernandez Garcia M, Moore CD, Schulz KN, Alberto O, Donague G, Harrison MM, Zhu H, and Zaret KS (2019). Structural Features of Transcription Factors Associating with Nucleosome Binding. Mol. Cell 75, 921–932.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furey TS (2012). ChIP-seq and beyond: new and improved methodologies to detect and characterize protein–DNA interactions. Nat. Rev. Genet 13, 840–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia MF, Moore CD, Schulz KN, Alberto O, Donague G, Harrison MM, Zhu H, and Zaret KS (2019). Structural Features of Transcription Factors Associating with Nucleosome Binding. Mol. Cell 0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gesson K, Rescheneder P, Skoruppa MP, von Haeseler A, Dechat T, and Foisner R (2016). A-type lamins bind both hetero- and euchromatin, the latter being regulated by lamina-associated polypeptide 2 alpha. Genome Res. 26, 462–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu B, Swigut T, Spencley A, Bauer MR, Chung M, Meyer T, and Wysocka J (2018). Transcription-coupled changes in nuclear mobility of mammalian cis-regulatory elements. Science 359, 1050–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenatri M, Bailly D, Maison C, and Almouzni G (2004). Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J. Cell Biol 166, 493–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heun P, Laroche T, Shimada K, Furrer P, and Gasser SM (2001). Chromosome dynamics in the yeast interphase nucleus. Science 294, 2181–2186. [DOI] [PubMed] [Google Scholar]
- von Hippel PH, and Berg OG (1989). Facilitated target location in biological systems. J. Biol. Chem 264, 675–678. [PubMed] [Google Scholar]
- Inukai S, Kock KH, and Bulyk ML (2017). Transcription factor–DNA binding: beyond binding site motifs. Curr. Opin. Genet. Dev 43, 110–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwafuchi M, Cuesta I, Donahue G, Takenaka N, Osipovich AB, Magnuson MA, Roder H, Seeholzer SH, Santisteban P, and Zaret KS (2020). Gene network transitions in embryos depend upon interactions between a pioneer transcription factor and core histones. Nat. Genet 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwafuchi-Doi M, and Zaret KS (2016). Cell fate control by pioneer transcription factors. Dev. Camb. Engl 143, 1833–1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izeddin I, Récamier V, Bosanac L, Cissé II, Boudarene L, Dugast-Darzacq C, Proux F, Bénichou O, Voituriez R, Bensaude O, et al. (2014). Single-molecule tracking in live cells reveals distinct target-search strategies of transcription factors in the nucleus. ELife 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klemm SL, Shipony Z, and Greenleaf WJ (2019). Chromatin accessibility and the regulatory epigenome. Nat. Rev. Genet 20, 207–220. [DOI] [PubMed] [Google Scholar]
- Leemans C, van der Zwalm MCH, Brueckner L, Comoglio F, van Schaik T, Pagie L, van Arensbergen J, and van Steensel B (2019). Promoter-Intrinsic and Local Chromatin Features Determine Gene Repression in LADs. Cell 177, 852–864.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Dong P, Ioannou MS, Li L, Shea J, Pasolli HA, Grimm JB, Rivlin PK, Lavis LD, Koyama M, et al. (2018). Visualizing long-term single-molecule dynamics in vivo by stochastic protein labeling. Proc. Natl. Acad. Sci 115, 343–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Z, Legant WR, Chen B-C, Li L, Grimm JB, Lavis LD, Betzig E, and Tjian R (2014). 3D imaging of Sox2 enhancer clusters in embryonic stem cells. ELife 3, e04236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Y, North JA, Rose SD, and Poirier MG (2014). Nucleosomes accelerate transcription factor dissociation. Nucleic Acids Res. 42, 3017–3027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall WF, Straight A, Marko JF, Swedlow J, Dernburg A, Belmont A, Murray AW, Agard DA, and Sedat JW (1997). Interphase chromosomes undergo constrained diffusional motion in living cells. Curr. Biol. CB 7, 930–939. [DOI] [PubMed] [Google Scholar]
- Mazza D, Ganguly S, and McNally JG (2013). Monitoring dynamic binding of chromatin proteins in vivo by single-molecule tracking. Methods Mol. Biol. Clifton NJ 1042, 117–137. [DOI] [PubMed] [Google Scholar]
- Meers MP, Janssens DH, and Henikoff S (2019). Pioneer Factor-Nucleosome Binding Events during Differentiation Are Motif Encoded. Mol. Cell [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendel DB, Khavari PA, Conley PB, Graves MK, Hansen LP, Admon A, and Crabtree GR (1991). Characterization of a cofactor that regulates dimerization of a mammalian homeodomain protein. Science 254, 1762–1767. [DOI] [PubMed] [Google Scholar]
- Meshorer E, Yellajoshula D, George E, Scambler PJ, Brown DT, and Misteli T (2006). Hyperdynamic Plasticity of Chromatin Proteins in Pluripotent Embryonic Stem Cells. Dev. Cell 10, 105–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalet X (2010). Mean square displacement analysis of single-particle trajectories with localization error: Brownian motion in an isotropic medium. Phys. Rev. E 82, 041914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi A, Heinrich D, and Lindner B (2012). Distributions of diffusion measures from a local mean-square displacement analysis. Phys. Rev. E 86, 021926. [DOI] [PubMed] [Google Scholar]
- Naumova N, Imakaev M, Fudenberg G, Zhan Y, Lajoie BR, Mirny LA, and Dekker J (2013). Organization of the Mitotic Chromosome. Science 342, 948–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann FR, Dion V, Gehlen LR, Tsai-Pflugfelder M, Schmid R, Taddei A, and Gasser SM (2012). Targeted INO80 enhances subnuclear chromatin movement and ectopic homologous recombination. Genes Dev. 26, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishibuchi G, Machida S, Osakabe A, Murakoshi H, Hiragami-Hamada K, Nakagawa R, Fischle W, Nishimura Y, Kurumizaka H, Tagami H, et al. (2014). N-terminal phosphorylation of HP1α increases its nucleosome-binding specificity. Nucleic Acids Res. 42, 12498–12511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nozaki T, Imai R, Tanbo M, Nagashima R, Tamura S, Tani T, Joti Y, Tomita M, Hibino K, Kanemaki MT, et al. (2017). Dynamic Organization of Chromatin Domains Revealed by Super-Resolution Live-Cell Imaging. Mol. Cell 67, 282–293.e7. [DOI] [PubMed] [Google Scholar]
- Otterstrom J, Castells-Garcia A, Vicario C, Gomez-Garcia PA, Cosma MP, and Lakadamyali M (2019). Super-resolution microscopy reveals how histone tail acetylation affects DNA compaction within nucleosomes in vivo. Nucleic Acids Res. 47, 8470–8484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otto G (2019). Enhancers and promoters regulate burst kinetics. Nat. Rev. Mol. Cell Biol 20, 134. [DOI] [PubMed] [Google Scholar]
- Ou HD, Phan S, Deerinck TJ, Thor A, Ellisman MH, and O’Shea CC (2017). ChromEMT: Visualizing 3D chromatin structure and compaction n interphase and mitotic cells. Science 357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paakinaho V, Presman DM, Ball DA, Johnson TA, Schiltz RL, Levitt P, Mazza D, Morisaki T, Karpova TS, and Hager GL (2017). Single-molecule analysis of steroid receptor and cofactor action in living cells. Nat. Commun 8, 15896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raccaud M, Friman ET, Alber AB, Agarwal H, Deluz C, Kuhn T, Gebhardt JCM, and Suter DM (2019). Mitotic chromosome binding predicts transcription factor properties in interphase. Nat. Commun 10, 487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramakrishnan V, Finch JT, Graziano V, Lee PL, and Sweet RM (1993). Crystal structure of globular domain of histone H5 and its implications for nucleosome binding. Nature 362, 219–223. [DOI] [PubMed] [Google Scholar]
- Ricci MA, Manzo C, García-Parajo MF, Lakadamyali M, and Cosma MP (2015). Chromatin Fibers Are Formed by Heterogeneous Groups of Nucleosomes In Vivo. Cell 160, 1145–1158. [DOI] [PubMed] [Google Scholar]
- Schlesinger S, and Meshorer E (2019). Open Chromatin, Epigenetic Plasticity, and Nuclear Organization in Pluripotency. Dev. Cell 48, 135–150. [DOI] [PubMed] [Google Scholar]
- Schreiber E, Matthias P, Müller MM, and Schaffner W (1989). Rapid detection of octamer binding proteins with “mini-extracts”, prepared from a small number of cells. Nucleic Acids Res. 17, 6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekiya S, and Suzuki A (2011). Direct conversion of mouse fibroblasts to hepatocyte-like cells by defined factors. Nature 475, 390–393. [DOI] [PubMed] [Google Scholar]
- Sekiya T, Muthurajan UM, Luger K, Tulin AV, and Zaret KS (2009). Nucleosome-binding affinity as a primary determinant of the nuclear mobility of the pioneer transcription factor FoxA. Genes Dev. 23, 804–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergé A, Bertaux N, Rigneault H, and Marguet D (2008). Dynamic multiple-target tracing to probe spatiotemporal cartography of cell membranes. Nat. Methods 5, 687–694. [DOI] [PubMed] [Google Scholar]
- Soufi A, Garcia MF, Jaroszewicz A, Osman N, Pellegrini M, and Zaret KS (2015). Pioneer transcription factors target partial DNA motifs on nucleosomes to initiate reprogramming. Cell 161, 555–568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Steensel B, and Belmont AS (2017). Lamina-Associated Domains: Links with Chromosome Architecture, Heterochromatin, and Gene Repression. Cell 169, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swinstead EE, Miranda TB, Paakinaho V, Baek S, Goldstein I, Hawkins M, Karpova TS, Ball D, Mazza D, Lavis LD, et al. (2016). Steroid Receptors Reprogram FoxA1 Occupancy through Dynamic Chromatin Transitions. Cell 165, 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teves SS, An L, Hansen AS, Xie L, Darzacq X, and Tjian R (2016). A dynamic mode of mitotic bookmarking by transcription factors. ELife 5, e22280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vakoc CR, Mandat SA, Olenchock BA, and Blobel GA (2005). Histone H3 Lysine 9 Methylation and HP1γ Are Associated with Transcription Elongation through Mammalian Chromatin. Mol. Cell 19, 381–391. [DOI] [PubMed] [Google Scholar]
- Velazquez Camacho O, Galan C, Swist-Rosowska K, Ching R, Gamalinda M, Karabiber F, De La Rosa-Velazquez I, Engist B, Koschorz B, Shukeir N, et al. (2017). Major satellite repeat RNA stabilize heterochromatin retention of Suv39h enzymes by RNA-nucleosome association and RNA:DNA hybrid formation. ELife 6, e25293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voss TC, and Hager GL (2014). Dynamic regulation of transcriptional states by chromatin and transcription factors. Nat. Rev. Genet 15, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieser S, and Schütz GJ (2008). Tracking single molecules in the live cell plasma membrane-Do’s and Don’t’s. Methods San Diego Calif 46, 131–140. [DOI] [PubMed] [Google Scholar]
- Yang X, and Cui W (2010). A Novel Spatial Clustering Algorithm Based on Delaunay Triangulation. J. Softw. Eng. Appl 03, 141–149. [Google Scholar]
- Zaret KS, DiPersio CM, Jackson DA, Montigny WJ, and Weinstat DL (1988). Conditional enhancement of liver-specific gene transcription. Proc. Natl. Acad. Sci. U. S. A 85, 9076–9080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu F, Farnung L, Kaasinen E, Sahu B, Yin Y, Wei B, Dodonova SO, Nitta KR, Morgunova E, Taipale M, et al. (2018). The interaction landscape between transcription factors and the nucleosome. Nature 562, 76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 (related to Figure 3): Radius of confinement and average displacement density differences scatter plot multiple comparison by one-way ANOVA
Values of mean density difference between the two factors, in each range of mobility (see Figure 1H) are indicated. A one-way ANOVA was performed to test the hypothesis H0: the mean of protein 1 is different from the mean of protein 2, and p-values are indicated. For each condition, n=25,000.
TABLE S2 (related to Figure 5): Residence times comparison
Values of mean long-lived fraction and short/long-lived residence time or size of long-lived fraction of two protein are indicated. A one-way ANOVA was performed to test the hypothesis H0: the mean of protein 1 is different from the mean of protein 2, and p-values are indicated. For each condition, n>100
TABLE S3 (related to STAR METHODS): oligonucleotides used for cloning
Data Availability Statement
The data and code used in this study are accessible upon request.