

Abstract
Objective:
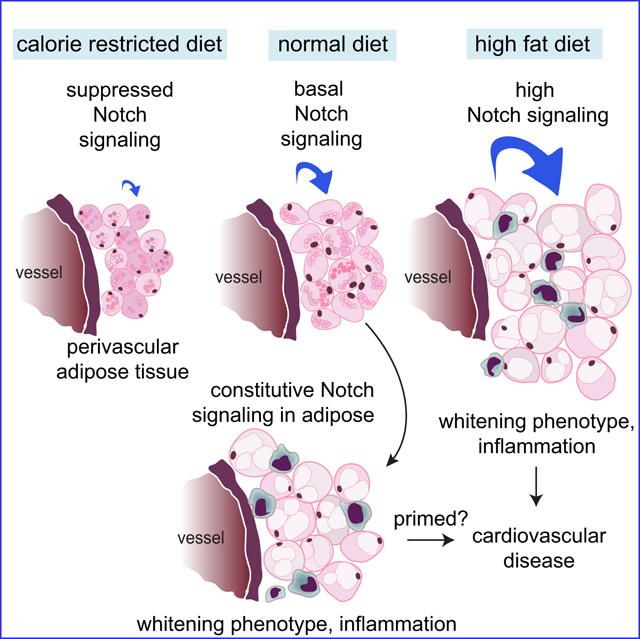
Perivascular adipose tissue (PVAT) surrounding arteries supports healthy vascular function. During obesity, PVAT loses its vasoprotective effect. We study pathological conversion of PVAT, which involves molecular changes in protein profiles and functional changes in adipocytes.
Approach and Results:
C57BL6/J mice were fed a 60% high fat diet (HFD) for 12 weeks or a cardioprotective 30% calorie-restricted diet for 5 weeks. Proteomic analysis identified PVAT as a molecularly distinct adipose depot, and novel markers for thermogenic adipocytes, such as GRP75, were identified. HFD increased the similarity of protein signatures in PVAT and brown adipose, suggesting activation of a conserved whitening pathway. The whitening phenotype was characterized by suppression of UCP1 and increased lipid deposition, leptin, and inflammation, and specifically in PVAT, elevated Notch signaling. Conversely, PVAT from calorie-restricted mice had decreased Notch signaling and less lipid. Using the Adipoq-Cre strain, we constitutively activated Notch1 signaling in adipocytes, which phenocopied the changes in PVAT caused by a HFD, even on a standard diet. Preadipocytes from mouse PVAT expressed Sca1, CD140a, Notch1 and Notch2, but not CD105, showing differences compared to preadipocytes from other depots. Inhibition of Notch signaling during differentiation of PVAT-derived preadipocytes reduced lipid deposition and adipocyte marker expression.
Conclusions:
PVAT shares features with other adipose depots, but has a unique protein signature that is regulated by dietary stress. Increased Notch signaling in PVAT is sufficient to initiate the pathological conversion of PVAT by promoting adipogenesis and lipid accumulation, and may thus prime the microenvironment for vascular disease.
Keywords: perivascular adipose tissue, high fat diet, adipocyte differentiation, Notch signaling, calorie restriction diet, Animal models of human disease, Basic science research, Pathophysiology, Vascular biology, Metabolism
Graphical abstract
Introduction
Obesity is an independent predictor of coronary atherosclerosis, and a direct link between obesity and cardiovascular disease (CVD) was demonstrated by the Framingham Study, which showed a continuous gradient of heart-failure risk with increasing adiposity1. Adipose tissue is an endocrine tissue involved in energy storage and heat generation that is significantly impacted during obesity. There are two major classes of adipose tissue, 1) brown adipose tissue (BAT), which oxidizes lipids to generate heat via the actions of mitochondrial localized uncoupling protein 1 (UCP1), and 2) white adipose tissue (WAT), which stores energy in the form of triglycerides. Beige adipose tissue represents a transient acquisition of thermogenesis by adipocytes within WAT in response to environmental or dietary stimuli.
Perivascular adipose tissue (PVAT) surrounds blood vessels and varies in phenotype in mice from BAT-like around the thoracic aorta (tPVAT) to WAT-like around the abdominal aorta (aPVAT) and mesenteric arteries2, 3. In healthy individuals, PVAT promotes vasodilation and inhibits inflammation through release of adipose-derived relaxing factors and anti-inflammatory cytokines such as nitric oxide and IL-104. Like other adipose depots, PVAT is subject to pathological conversion during obesity5. In humans, imaging to assess PVAT volume or density has been used to correlate with early coronary artery disease6, aortic calcification7, and vasospastic angina8. Potential effects of PVAT on vascular disease have recently been comprehensively reviewed9–13. The mouse is a useful model to study pathological conversion of PVAT in obesity and its impact on CVD. PVAT deficient mice generated by PPARƔ-specific deletion in SM22α positive cells display altered thermogenesis in the vascular microenvironment and enhanced atherogenesis14. In addition, transgenic expression of mitoNEET in BAT/PVAT induced expression of thermogenic genes and was atheroprotective15.
Notch signaling is a highly conserved pathway that regulates cell fate and numerous cardiovascular pathologies16. Adipose tissue specific knockout of Notch1 leads to beiging of WAT and improves metabolic output and insulin sensitivity in mice17. Additionally, HFD-induced fat mass expansion is blunted by adipose specific-Notch1 deletion17. In vitro, Notch signaling inhibits adipogenic differentiation of adipose-derived progenitor cells18, 19. Inhibition of Notch with soluble Jagged-1 increased proliferation of 3T3-L1 preadipocytes20, indicating that Notch regulates adipocyte phenotype. Here, we used sequential window acquisition of all theoretical spectra (SWATH) proteomics to identify adipose tissue-specific proteomic signatures and effects of a HFD. We also characterized Notch signaling and activity on PVAT and show that activation of Notch signaling in adipocytes is sufficient to drive an obesogenic phenotype in PVAT independent of obesity. Successful development of primary preadipocyte cultures also allowed us to characterize progenitor cells in PVAT compared to other adipose tissues, and implicate Notch signaling in accumulation of lipids in PVAT-derived adipocytes. Thus, we have identified a novel pathway by which Notch signaling regulates PVAT function and phenotype.
Materials and Methods
See the Major Resources Table in the Supplemental Material for details on mouse strains, primer sequences and PCR conditions, antibodies, and tissue culture protocols.
Mice
Protocols were approved by the Institutional Animal Care and Use Committee of Maine Medical Center. Singly housed C57BL/6J mice (JAX) were fed ad libitum with a high fat diet (HFD) or sucrose matched control diet for 12 weeks starting at 8 weeks of age. The control diet (Research Diets D12450J) contains 20% kcal protein, 10% kcal fat, 70% kcal carbohydrate with an energy density of 3.82 kcal/g. The HFD (Research Diets D12492) contains 20% kcal protein, 60% kcal fat (soybean oil and lard), 20% kcal carbohydrate with an energy density of 5.21 kcal/g. Since female C57BL/6J do not develop insulin resistance, glucose intolerance or adipose tissue inflammation when fed a 60% fat diet21, obesity studies were restricted to males. For Notch1 activation in vivo, Adiponectin-Cre (Adipoq-cre) mice (JAX) were bred with conditional Notch1 intracellular domain (N1ICD) transgenic mice22. Mice were weaned at 3 weeks of age, single housed at 7 weeks of age, and maintained for 12 weeks on the control diet. For HFD studies, a total of n=10 mice was used per diet for each of 2-independent experiments. Of the 10 mice per group, 3/group were used for qRT-PCR, 4/group for immunoblot and 3/group for immunohistochemistry. For control diet studies using Adipoq-cre mice, a total of n=9 mice per genotype for each of 2 independent experiments with n=3 mice allocated for immunoblot, qRT-PCR and immunohistochemistry. For calorie restriction experiments, 8-week-old, single-housed C57BL/6J mice were fed a control diet or a 30% calorie restricted (CR) diet ad libitum for 4 weeks. The control diet for calorie restriction studies (Research Diets D10012M) contains 15% kcal protein, 9% kcal fat and 76% kcal carbohydrate with an energy density of 3.81 kcal/g. For calorie restriction, food consumption for mice was calculated by measuring normal daily control diet intake for two weeks. CR mice were given 70% of their average food intake daily. A total of 6 mice for each condition were used for CR studies.
Nuclear magnetic resonance (NMR)
Body fat, lean mass and fluid measurements were generated using a Bruker minispec mq NMR. Data were collected using the manufacturer’s software and analyzed in GraphPad PRISM. Calculations were performed as follows: % weight gain=endpoint mass/starting mass, and % body fat=fat mass (g)/endpoint mass (g).
Mouse atherogenic model
PCSK9-AAV containing the D377Y-mPCSK9 construct (Addgene, Watertown, MA) with gain of function mouse PCSK9 mutation D377Y23 under the control of the HCRApoE/hAAT promotor24 was obtained from the Boston Children’s Hospital viral core. Virus was diluted to 5 × 108 genome copies (gc)/μL with sterile 0.9% NaCl (Hospira, Lake Forest, IL) and 100 μL were injected retro-orbitally under isoflurane anesthesia for a 5 × 1010 gc dose per 8 week-old mouse. Mice were maintained on a high fat (21.2%), high cholesterol (0.21%), high sugar (34.9%) western style diet (Research Diets D12079B) for 12 weeks before collection of blood, aortic roots and aortae.
Tissue processing and histology
Tissues were either fixed in formalin for paraffin embedding, or fixed in formalin and infiltrated with 30% sucrose prior to OCT embedding and cryosectioning. For routine histology of paraffin embedded specimens, sections were subjected to routine hematoxylin/eosin stain or further immunostaining protocols. For lipid detection in aortic roots, cryosections were stained with 0.7% oil red O solution (Sigma) in propylene glycol (VWR) and counterstained with hematoxylin. Sections were imaged in 3–4 fields/section for analysis using ImageJ software.
Immunostaining and confocal immunofluorescence
Formalin fixed, paraffin embedded tissues were sectioned. For immunostaining, slides underwent antigen retrieval, quenching and permeabilization, and blocking in 2% BSA and 2% goat serum. Sections were incubated overnight with primary antibodies, which were detected using SignalStain® Boost IHC Detection Reagent and SignalStain® diaminobenzidine substrate kit (Cell Signaling) and counterstained with hematoxylin. For mouse monoclonal antibodies, sections were first incubated with M.O.M. Block (Vector Labs) overnight at 4ºC. Sections were treated as above for immunofluorescence staining. Sections were blocked in PBS with 0.5% Tween-20 (PBS-T) supplemented with 2% BSA and 5% goat serum for 2h at room temperature. Primary antibodies were incubated in PBS supplemented with 2% BSA overnight at 4°C, washed 3 times for 15 minutes in TBS-T and incubated with Alexa Fluor-conjugated secondary antibody in 1% BSA/TBS-T for 1h at room temperature. Sections were washed and incubated for 2 minutes with TrueVIEW Autofluorescence Quenching Kit (Vector Laboratories SP-8400). Sections were washed, incubated with 1μg/ml DAPI for 5 minutes in PBS and coverslipped using Vectashield Hard Set anti-fade mounting medium. Confocal images were captured using a Leica TCS SP8 Laser Scanning Confocal with a 20x/0.75 objective.
Frozen sections for immunofluorescence were permeabilized for 5 minutes with 0.1% Tween20 in PBS, then treated with 0.01M sodium citrate buffer at 50°C 15 minutes. Sections were blocked in 1% BSA/TBS-T for 20 minutes before overnight incubation at 4°C with primary antibodies diluted in 1% BSA/TBS-T. Primary antibodies were detected using AlexaFluor conjugated secondary antibodies diluted in 1% BSA/TBS-T for 1h at room temperature. A 2 minute autofluorescent quench with TrueVIEW Autofluorescent Quenching Block (Vector Laboratories) was followed by coverslipping using Vectashield HardSet Antifade Mounting Media (Vector Laboratories). 18 μm z-stacks composed of seven slices interspaced by 3 μm were collected on a Leica SP8 confocal microscope.
RNA isolation and RT-PCR
RNA was isolated using the miRCURY kit (Exiqon 300111) supplemented with lipid lysis additive (Exiqon 300121). Total RNA was quantified using a nanodrop 2000c spectrophotometer (Thermo Scientific) and 250ng were used for reverse transcription using iScript gDNA clear cDNA synthesis kit with DNAase (Biorad 1725035). cDNA was diluted 1:1 with nuclease free water and stored at −20°C. For RT-PCR, 2x PCR master mix (Promega M7502) was mixed with nuclease free water, 12.5ng cDNA, and 300nM primer in a final volume of 20μl and run on a C1000 thermocycler. For qPCR, IQ SYBR Green Supermix (Biorad 1708882) was used with the CFX Connect thermocycler. Gene CT values were normalized to Ppia CT values and fold change calculated by the comparative CT method (2^-ΔΔCT). Relative expression was calculated (1/fold change).
Protein isolation and immunoblotting
Adipose tissues were lysed in RIPA buffer (150mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, and 50mM Tris pH=8.0) supplemented with 1x protease/phosphatase inhibitor cocktail (PPI, Cell Signaling 5872S) on ice. Debris was removed by centrifugation at 14,000g for 20 minutes at 4°C. Proteins were precipitated using 4 volumes ice cold 100% acetone, and incubation at −20°C for 1h. Proteins were pelleted by centrifugation at 10,000g for 10 minutes at 4°C, washed 2x in 70% acetone and air dried. Pellets were resuspended in RIPA and sonicated at 40% output control and duty cycle 3 for 10 pulses on ice using a Branson Sonifier 250, and samples stored at −20°C. Cultured cells were washed 2x in PBS and lysed with RIPA supplemented with 1x PPI. Lysates were centrifuged at 14,000 x g for 20 minutes at 4°C and the supernatant was collected and stored at −20°C. Protein was quantified using the BioRad DC protein assay, put in Laemmli sample buffer with 100mM DTT, and incubated at 95°C for 15 minutes prior to gel electrophoresis. SDS-PAGE was performed using BioRad TGX™ FastCast™ gels between 10% and 12% (1610173 and 1610175) and 20–50μg protein/lane. Gels were transferred to PVDF membranes using Biorad TransBlot Turbo Transfer System and membranes blocked using 5% milk in PBS with 0.1% Tween. Primary antibodies were diluted in 5% milk and incubated overnight at 4°C. Secondary antibodies are detailed in the major resources table. Signal was detecting using enhanced chemiluminescence.
Proteomics
Tryptic digests were obtained using the ProteoExtract kit (Calbiochem) and separated by nanoscale liquid chromatography. SWATH was used via a data independent acquisition method on a Sciex 5600 TripleTOF mass spectrometer. A mouse ion library was constructed using ProteinPilot. For identification, multiple fragment ion chromatograms were retrieved from the spectral library for each peptide. These spectra were compared with the extracted fragment ion traces to identify and quantify the target peptide. SWATH protein responses were determined using PeakView software, and this information was extracted for principle component analysis and T-test comparisons using MarkerView25. Venn diagrams were generated with the Venn Diagram Generator (Whitehead Institute). For PANTHER ontology classification analysis, we used version 13.1 (released 2018–01-27) to functionally classify significantly regulated (p<0.05) proteins. Swath and ion library raw data can be located at the PeptideAtlas repository26, 27, using the following URL: http://www.peptideatlas.org/PASS/PASS01599. PeptideAtlas is a part of the ProteomeXchange Consortium.
Whole mount β-galactosidase detection
Tissues were fixed in 10% formalin for 1h. After fixation, detergent solution (2mM MgCl2, 0.01% sodium deoxycholate, 0.02% NP-40 in PBS) was added to tissues for 2h at room temperature with rocking. Tissues were then incubated in staining solution (5mM C6N6FeK3, 5mM K4Fe(CN)6, 20mM Tris (pH=7.3), 1mg/ml X-gal in detergent solution) overnight at RT in the dark.
Stromal cell isolation and adipocyte differentiation
Adipose tissue was digested at 37°C in 0.225U/ml collagenase B (Sigma), filtered through a tissue strainer, centrifuged, and treated with ACK buffer (Lonza) to lyse red blood cells. Cells were sub-cultured <10 times in DMEM/F12 supplemented with 15% FBS, 100μg/ml primocin (Invitrogen, cat ant-pm-1) and 10ng/ml murine FGF-2 (Peprotech). Once confluent, cells were differentiated for 3 days and incubated in maintenance media for 7 days. 10μM gamma secretase inhibitor (Calbiochem) or DMSO was added after induction at the start of lipid accumulation and replaced with fresh media every 48h. Lipid accumulation was measured by oil red O (ORO) staining28. Cells were fixed in 10% formalin for 20 minutes, washed 2x in PBS and dehydrated in 60% isopropanol. The ORO stock solution (0.35% in isopropanol) was diluted to a final concentration of 0.23% in diH2O and filtered through a 0.2μm filter. The 60% isopropanol was aspirated and cells incubated with the ORO working solution for 10 minutes. Cells were de-stained using 100% isopropanol and absorbance measured at 490nm against ORO standards. Cells were washed and lysed in RIPA and total protein measured.
Flow cytometry
Stromal cells were isolated, washed with PBS, resuspended in FACS buffer (0.5% BSA, 2mM EDTA in PBS), and counted. Cells were treated with TruStain-fcX ™ anti-mouse CD16/32 (Biolegend) to block nonspecific binding, then stained with fluorochrome-conjugated primary antibodies. DAPI was used for live/dead staining. Cells were washed with FACS buffer and signal detected using MacsQuant Analyzer (Miltenyi). Data were analyzed using WinList 5.0.
Statistical analysis
Proteomics studies - For sample comparisons, a t-test was applied using MarkerView software (Sciex) essentially as described25, 29. The calculated t-value was subsequently converted to a P value, yielding the probability that any difference between the two respective sample groups is statistically significant, defined as P<0.05. The P value calculated from the t-test was measured for each possible pair-wise comparison, and each group was also compared to all other samples. Additionally, the first acquired samples were compared to the following fourth sample to help detect potential drift for variables. Samples with a drift of greater than 10% were excluded. Principle component analyses (PCA) utilized Pareto autoscaling, where each value is subtracted from the average and divided by the square root of the standard deviation of the value, as described previously25. PCA was guided using the sample groupings for adipose tissue samples.
Statistical analysis of other data was performed in PRISM software version 7.04. Multiple group comparisons were analyzed using one-way ANOVA with two tailed Student’s t-test and post-hoc Tukey’s Range test. For pair-wise comparisons, Students t-test with post hoc Tukey’s Range test was used. Data are graphed as means +SD. Differences were considered significant at P<0.05 (*), <0.01 (**) or <0.001 (***).
Results
Pathological conversion of tPVAT following HFD.
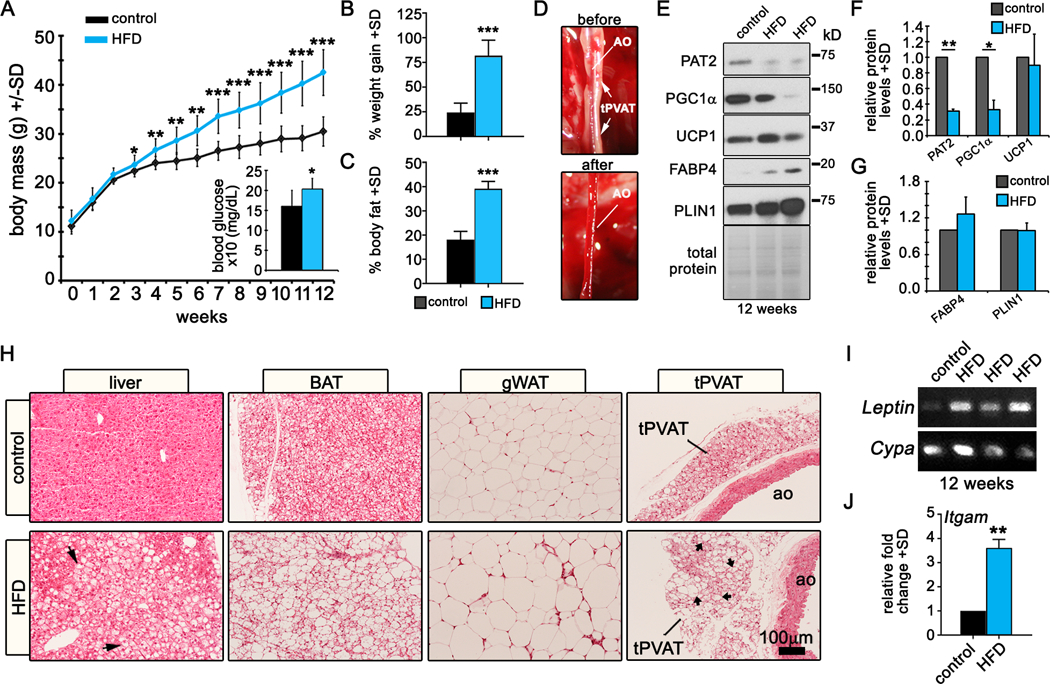
To determine the effects of obesity on tPVAT phenotype, C57BL/6J male mice at 8 weeks of age were fed a 60% HFD or sucrose-matched control for 12 weeks. Mice that consumed the HFD were significantly heavier by week 3, and by week 12, weighed ~40% more than controls (Fig. 1A). At 12 weeks, fasting blood glucose was significantly increased with HFD (inset). Mice fed a HFD gained significantly more weight than control diet mice (Fig. 1B). Further, nuclear magnetic resonance (NMR) showed increased body fat percentage in the HFD-fed mice, suggesting the increased mass and weight gain in HFD-fed group is due to increased fat mass (Fig. 1C). tPVAT was resected from the thoracic aorta (Fig. 1D) and lysed for analysis. Thermogenesis confers a cardioprotective advantage to mice5, 14, 15, and thus we investigated if HFD altered thermogenic proteins in tPVAT. Immunoblot of tPVAT from mice fed HFD for 12 weeks (Fig. 1E-G) showed significant reductions in the levels of the thermogenic markers PAT2 and PGC1α30, while UCP1 varied from unchanged to slightly elevated by HFD, consistent with previous reports31. No significant changes in FABP4 or the lipid droplet-associated protein PLIN1 were observed. Notably, reduction in PAT2, but not PGC1α with HFD was observed in BAT, and none of the thermogenic markers were detected in WAT (Suppl. Fig. IA-B).
Figure 1. Pathological conversion of PVAT in mice fed a HFD.
A) Body weights of male C57BL/6J male mice fed a control or HFD over a 12 week period. Inset shows blood glucose readings after a 12h fast from control or HFD fed mice after 12 weeks (n=10/group). Average weight gain (B) and percentage of body fat (C) for each group after 12 weeks are shown. D) Mouse thoracic aorta before and after tPVAT dissection. E) Representative immunoblot of two independent experiments of 5 mice/group for the indicated proteins from control or HFD fed mice, and quantification of proteins (F-G). H) Hematoxylin and eosin (H&E) staining of tissues from mice fed a control or HFD for 12 weeks. Arrows show steatosis in the liver and arrowheads indicate unilocular white-like adipocytes in tPVAT. I) RT-PCR to detect Leptin mRNA from control or HFD-fed mice. J) Quantitative RT-PCR analysis of the inflammatory cell marker Itgam from mice fed a control or HFD (n=4/group). Graphed are means +SD. Data were analyzed using Student’s t-test with post-hoc Tukey’s range test. *P≤0.05, **P≤0.01, ***P≤0.001
As expected, UCP1 was significantly upregulated in BAT by HFD32 (Suppl. Fig. IC) while PLIN1 was unchanged in BAT and slightly increased in gWAT by HFD. Next, we histologically analyzed liver, BAT, gWAT and tPVAT. Mice fed a HFD had widespread liver steatosis, marked hypertrophy of gWAT and a phenotypic conversion of BAT and tPVAT from multilocular to unilocular, with increased lipid accumulation (Fig. 1H), indicating that tPVAT is subject to expansion and pathological conversion similar to other adipose depots. In PVAT, transcript levels of the hormone Leptin (Fig. 1I) and the inflammatory marker Itgam (Fig. 1J) were elevated in mice fed a HFD. Overall, 12 weeks of HFD led to a pathological phenotype in PVAT characterized by morphological adipocyte whitening, suppression of thermogenic proteins, and increased inflammatory characteristics consistent with symptoms of metabolic dysfunction.
The PVAT proteome is unique and affected by HFD.
We performed unbiased protein analysis using SWATH mass spectrometry. Proteomic data were used for a principle component analysis (PCA, Fig. 2A). We observed strong aggregation of group replicates, and clustering of each adipose tissue, regardless of diet. Because of the phenotypic similarity of PVAT with BAT, we compared those two depots without WAT (Fig. 2B). In mice fed a HFD, protein profiles in PVAT and BAT were significantly more concordant, suggesting a similar response to HFD. Considering all means at p<0.05 to be significantly different, we identified shared and unique proteins comparing PVAT to WAT (Fig. 2C, Suppl. Tables Ia-b, IIa-b), PVAT to BAT (Fig. 2D, Suppl. Tables IIIa-b, IVa-b), and WAT to BAT (Fig. 2E, Suppl. Tables Va-b, VIa-b). We quantified the overlap to understand protein diversity: PVAT-WAT were 44% similar on standard diet and 48% similar on HFD, and WAT-BAT were 53% similar on standard diet and 46% similar on HFD. Strikingly, while PVAT-BAT were 43% similar on a standard diet, this overlap increased to 76% similar on a HFD, suggesting conserved pathways in BAT and PVAT during pathological conversion associated with obesity. Comparison of mice under control diets validated proteins known to be shared between BAT and tPVAT (UCP1), but also identified novel proteins associated with thermogenic adipocytes, such as electron transferring flavoprotein alpha (ETFA) and GRP75 (Fig. 2F). Interestingly, human mutations in ETF family members lead to metabolic disorders involving fatty acid metabolism33, although these proteins have not been studied in adipocytes. GRP75, encoded by hspa9, is also unexplored in adipocytes, although it is suppressed in the liver following a HFD34.
Figure 2. PVAT is a molecularly distinct adipose tissue.
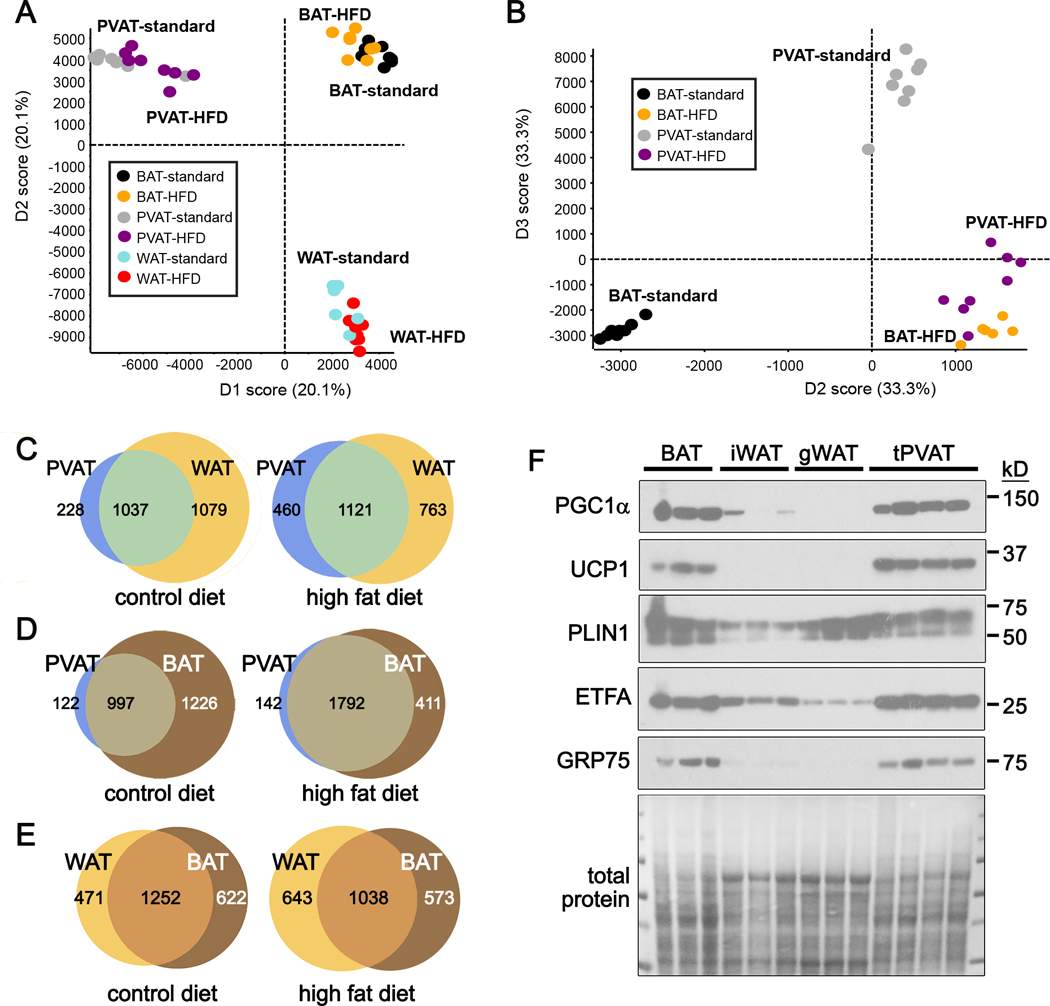
A) Principle component analysis (PCA) along axes of abundance variation from SWATH-MS analysis of tPVAT, BAT and gWAT from mice fed a control or HFD. B) PCA of tPVAT and BAT from control or HFD fed mice. Each circle represents independent mice. C-E) Venn diagram representation of shared and unique proteins identified from SWATH mass spectrometry between tPVAT and WAT (C), tPVAT and BAT (D) and WAT and BAT (E) from mice fed a control or HFD. F) Immunoblot of proteins that distinguish WAT from thermogenic BAT and PVAT from mice on a standard diet.
The increased overlap of proteins shared between PVAT and BAT following HFD prompted us to seek potential “whitening” proteins, or those associated with loss of thermogenesis. We stratified our SWATH dataset to identify proteins significantly higher in WAT compared to PVAT or BAT (1,336 proteins). We identified those that were significantly increased in PVAT on a HFD compared to standard diet (269 proteins), and with those significantly increased in BAT on a HFD compared to standard diet (23 proteins). We identified 12 proteins represented in all groups (Suppl. Table VII) as proteins associated with a whitening phenotype. We also identified 61 PVAT-enriched proteins related to lipid metabolism, energy generation, and mitochondrial function (Suppl. Table VIII). Some of these PVAT-enriched proteins were also present in BAT, such as GRP75 and ETFA (Fig. 2F). To further understand changes specifically in PVAT in response to a HFD, we analyzed proteins that were significantly different between diets. Suppl. Table IXa shows the 94 proteins higher in PVAT from mice on a standard diet, and Suppl. Table IXb shows the 714 proteins higher in PVAT from mice fed a HFD. Of the 714 proteins elevated in PVAT from mice fed a HFD, 150 (21%) are potentially secreted, based on the presence of a signal sequence. We predict that some of these proteins produced by PVAT, could have paracrine effects on the underlying blood vessel. We also found an interesting trend in the levels of the signaling protein, Notch2, which was increased 8-fold in PVAT from mice fed a HFD, compared to mice on a control diet, although this was not statistically significant due to variability in expression between individual mice. However, due to the intriguing role of Notch signaling in the regulation of adipose tissue phenotype, we focused further studies on Notch signaling.
Notch signaling is upregulated in tPVAT by HFD.
Notch signaling regulates adipogenesis in vitro18, 19, 35 and suppresses the brown fat phenotype in vivo17, 36. Notch signaling in PVAT has not been characterized. We hypothesized that dysfunctional PVAT from mice fed an obesogenic diet would exhibit elevated Notch signaling. RT-PCR of tPVAT isolated from mice fed a HFD for 12 weeks showed marked upregulation of mRNA (Fig. 3A-B) and protein (Fig. 3C-D) for NOTCH1 and NOTCH2, and downstream targets HES1 and HEY1 compared to tPVAT from mice fed a control diet. Immunostaining of tPVAT confirmed elevated NOTCH1, NOTCH2 and HES1 localized to adipocytes in HFD mice (Fig. 3E, arrows show nuclear HES1). Sections stained with secondary antibody only or isotype-matched IgG control were negative (Fig. 3F). Interestingly, BAT from mice fed a HFD for 12 weeks showed reduced Notch signaling, while gWAT had relatively robust NOTCH1 and HEY1 protein, regardless of diet (Suppl. Fig. IIA-C). Thus, Notch seems selectively activated in PVAT in mice fed a HFD.
Figure 3. Notch signaling is upregulated in PVAT by HFD.
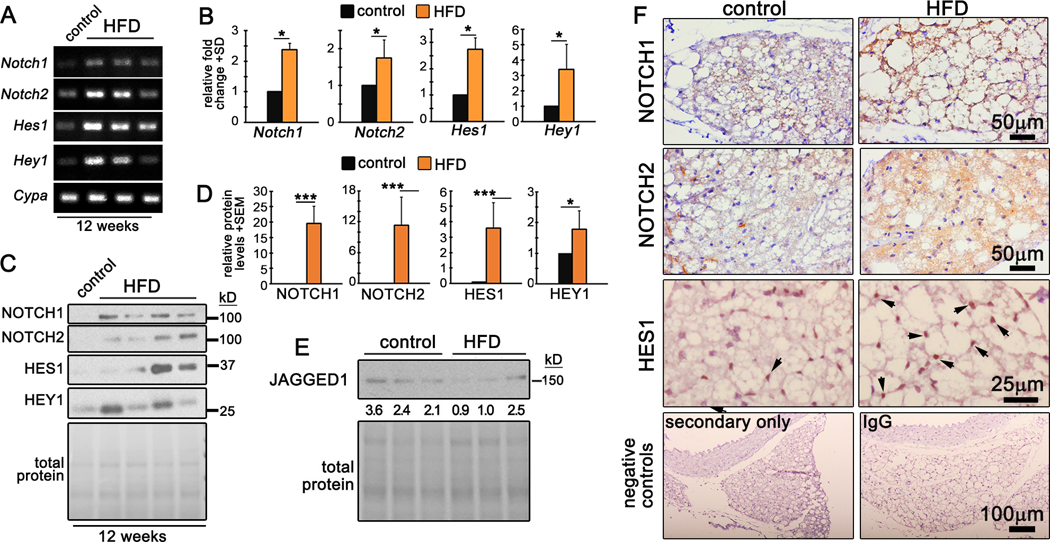
A-B) RT-PCR (A) and quantification (B) of tPVAT from control or HFD fed mice. Shown is a representative from 2 independent experiments, n=3 mice each. C-D) Immunoblot (C) and quantification (D) from tPVAT of control or HFD fed mice. Shown is a representative from 2 independent experiments, n=3–4 mice each. E) Immunoblot to detect Notch ligand JAGGED1. F) Immunohistochemistry of tPVAT for the indicated proteins from control or HFD fed mice or negative controls stained with secondary antibody only, or isotype matched IgG control. Arrowheads indicate nuclear HES1 protein. Data were analyzed with one-way ANOVA with pairwise comparison and Tukey’s post-hoc analysis. *P≤0.05, **P≤0.01, ***P≤0.001
We observed elevated Notch signaling in tPVAT as early as 8 weeks of HFD as demonstrated by increased Notch1 and Hey1 transcripts, and NOTCH1 protein (Suppl. Fig. IID). In contrast, Notch1 was suppressed and Hey1 unchanged by HFD in BAT (Suppl. Fig. IIE), and both transcripts were suppressed by 8 weeks HFD in gWAT (Suppl. Fig. IIF). Thus, despite the similarities between BAT and tPVAT phenotype and function, discrete temporal mechanisms may regulate their whitening in response to HFD. Consistently, Notch signaling was upregulated in tPVAT by 8- and 12-weeks of HFD feeding.
Notch1 activation promotes pathological conversion of tPVAT.
Adiponectin (Adipoq) is a chemokine whose expression is restricted to adipose tissue. We bred adiponectin-Cre (Adipoq-Cre, B6.FVB-Tg(Adipoq-cre)1Evdr/J37) mice to ROSA26 Cre reporter mice to assess Cre recombinase activity. Adipoq-Cre;ROSA26 transgenic mice had beta-galactosidase (β-gal) activity in BAT, gWAT and tPVAT, but not lung and heart tissue, as indicated by whole mount staining (Suppl. Fig. IIIA). ROSA26 mice lacking the Adipoq-Cre transgene were devoid of β-gal activity in all tissues. Immunoblot confirmed expression of β-gal in adipose tissues from double transgenic mice, but not lung and heart tissue, or ROSA26 single transgenic mice (Suppl. Fig. IIIB). To determine if Notch1 activation promotes PVAT whitening, we constitutively expressed Notch1 intracellular domain (N1ICD) in adipose tissue by breeding conditional N1ICD transgenic mice (Suppl. Fig. IIIC) to Adipoq-Cre mice (Adipoq-Cre;N1ICD). At 12 weeks of age, PVAT from Adipoq-Cre;N1ICD mice was harvested and stained using antibodies to detect the myc epitope tag (N1ICD transgene) and Notch1. Confocal immunofluorescence showed robust nuclear myc epitope in tPVAT from Adipoq-Cre;N1ICD mice (+Cre) that co-localized with NOTCH1 (Fig. 4A, arrows), while N1ICD mice (-Cre) were negative for the myc epitope and exhibited low levels of NOTCH1. RNA and protein were isolated from tPVAT to assess Notch activation. Compared to –Cre, +Cre mice had significantly higher transcripts for Notch1 and downstream targets Hes1 and Hey1 (Suppl. Fig. IIID-F). Likewise, NOTCH1 and its downstream target HES5 were significantly upregulated in +Cre mice compared to control (Fig. 4B-D), demonstrating activation of Notch signaling in +Cre mice.
Figure 4. Notch1 activation promotes pathological conversion of tPVAT in mice fed a control diet.
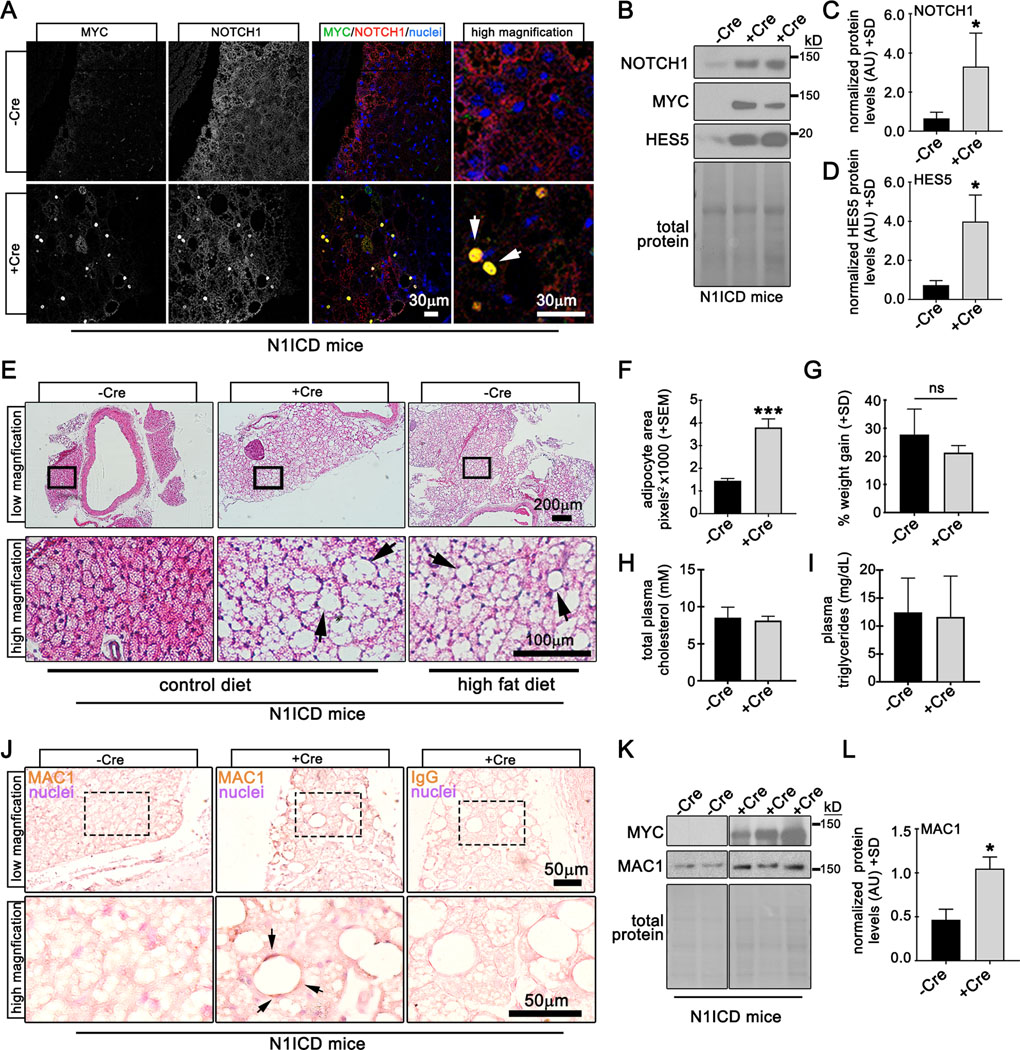
A) Verification of myc-tagged N1ICD transgene was performed by confocal immunofluorescence of tPVAT from N1ICD (-Cre) or Adipoq-Cre;N1ICD (+Cre) mice for indicated proteins. Arrows show nuclear colocalization of the myc epitope and N1ICD. B) Immunoblot of tPVAT from -Cre or +Cre N1ICD mice, with quantification of protein levels (C-D). E) H&E staining of aorta with tPVAT from -Cre or +Cre mice on the indicated diets (arrows, lipid accumulation). F) Quantification of adipocyte area from iWAT from mice fed a control diet for 12 weeks, indicating adipocyte hypertrophy. G) Weight gain for each group was not different. Total plasma cholesterol (H) and triglycerides (I) from -Cre or +Cre N1ICD mice are shown. J) PVAT sections were immunostained to detect macrophages (MAC1), which were prominent in +Cre mice compared to -Cre controls. Negative control with IgG only is shown. K-L) Immunoblot to detect MAC1 in PVAT verifies an increase in +Cre N1ICD mice. Graphed are means +SD. Data were analyzed using Student’s t-test with post-hoc Tukey’s range test. *P≤0.05 ***P≤0.001, ns = not significant
Because Notch signaling is upregulated in tPVAT in mice fed a HFD, we considered that Notch signaling may induce pathological conversion of tPVAT in vivo. To test this, +Cre and –Cre male mice were placed on a control for 12 weeks and tPVAT compared to –Cre male mice on a HFD. Morphological analysis of tPVAT by H&E revealed expansion of PVAT mass surrounding the thoracic aorta (Fig. 4E, top row), coinciding with increased lipid storage (Fig. 4E, bottom row) and adipocyte area (Fig. 4F) in +Cre mice on a control diet compared to –Cre mice. tPVAT from +Cre control fed mice was morphologically indistinguishable from –Cre mice fed a HFD, suggesting Notch activation drives expansion and pathological conversion of tPVAT during obesity. Similar morphological changes in tPVAT were also apparent in +Cre female mice compared to controls (data not shown). Because Adipoq-Cre is also expressed in BAT and WAT, we analyzed those adipose depots, and found that activation of Notch was sufficient to increase lipid accumulation (Suppl. Fig. IVA) and adipocyte area (Suppl. Fig. IVB-C). Despite increased lipid accumulation in adipose tissue of Adipoq:N1ICD mice, +Cre were found to have slightly lower body fat (Suppl. Fig. IVD) and reduced overall bodyweight (Suppl. Fig. IVE) compared to –Cre mice, however normalized weight gain between the two groups was unchanged (Fig. 4G). Food intake measurements revealed that food consumption was unchanged between –Cre and +Cre mice when normalized to lean mass (Suppl. Fig. IVF), further emphasizing the pathological effect of Notch activation on adipose tissue independent of diet. Further, total cholesterol and triglycerides were measured from plasma of –Cre and +Cre male and female mice to test if physiological metabolism was altered by Notch activation in +Cre mice. We observed no differences in free cholesterol (Fig. 4H) or circulating triglycerides (Fig. 4I) in the plasma of –Cre and +Cre mice. Baseline non-fasting blood glucose was also unchanged between groups (data not shown). In summary, Notch activation in tPVAT exerts a local cellular effect on adipose resulting in increased lipid storage and adipocyte size, independent of metabolic parameters (i.e. obesity, dyslipidemia).
Increased inflammatory cell infiltration during obesity contributes to adipose tissue inflammation and is associated with insulin resistance and metabolic dysfunction38. Crown-like structures comprised of proinflammatory macrophages encompassing dying adipocytes form during obesity and signify pathological conversion of adipose tissue. Given that mice fed an obesogenic diet for 12 weeks exhibited increased lipid deposition and inflammatory marker expression (Fig. 1), and that Notch1 activation causes increased lipid deposition independent of obesity (Fig. 4E-F and Suppl. Fig. IVA-E), we asked if inflammation was present in tPVAT of Notch1-activated control diet mice. Compared to -Cre mice, tPVAT from +Cre mice had significantly elevated Itgam and Leptin (Suppl. Fig. IVG-H). Immunohistochemistry for MAC1 in tPVAT of +Cre mice revealed increased MAC1 staining that was organized into crown-like structures in tPVAT (Fig. 4J, arrows). To quantify MAC1 protein in tPVAT, we performed Immunoblot of whole tissue lysates and confirmed a significant upregulation of MAC1 in +Cre mice compared to controls (Fig. 4K-L) indicating that Notch1 activation promotes inflammation in tPVAT of non-obese mice. Collectively, these observations are consistent with an obesity-like phenotype39, 40, and demonstrate that activation of Notch signaling in adipose tissue is sufficient to phenocopy key pathologies associated with obesity and adipose tissue dysfunction.
Notch activation suppresses thermogenesis and promotes whitening of tPVAT.
The brown adipose marker UCP1 regulates non-shivering thermogenesis and promotes cardiometabolic health41, 42. Obesity is linked to UCP1 activity43, and UCP1 null mice have an increased propensity to become obese with age44. PLIN1 regulates lipid deposition45 and PLIN1 is upregulated during obesity. Since tPVAT of +Cre mice develops a pathological morphology independent of obesity, we asked whether UCP1 and/or PLIN1 levels where changed. tPVAT was stained to detect UCP1 and PLIN1 from 15-week old +Cre and -Cre male mice. Confocal immunofluorescence revealed robust UCP1 staining in -Cre tPVAT that was reduced by Notch activation (+Cre) (Fig. 5A-B). Consistent with increased adipocyte size, PLIN1 was increased and more uniform in +Cre mice compared to controls, which exhibited mosaic expression levels (Figs. 5A and 5C). Isotype controls were negative while internal controls showed no UCP1 or PLIN1 staining in the aortic wall adjacent to the tPVAT (Fig. 5A, right column). Parallel qPCR analysis showed significantly decreased Ucp1 and increased Plin1 in +Cre mice compared to controls (Fig. 5D-E). Collectively, Notch1 activation in adipose tissues induces an obesity-like phenotype in tPVAT independent of weight gain.
Figure 5. The effects of Notch activation on thermogenic markers in tPVAT.
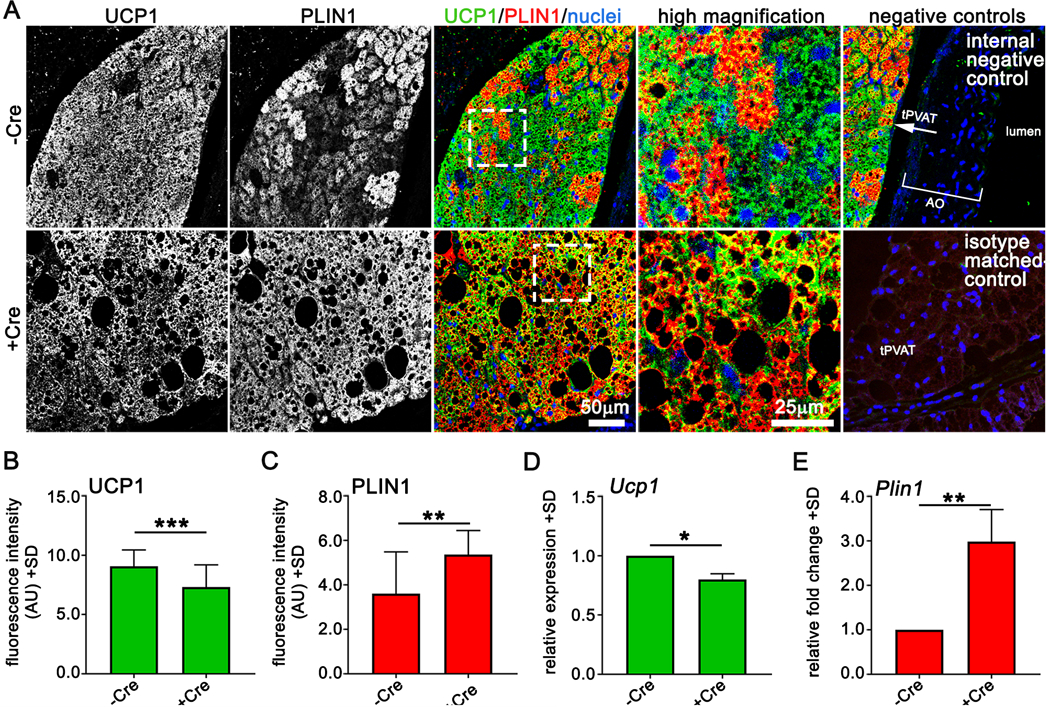
A) Confocal immunofluorescence of tPVAT from -Cre or +Cre mice for indicated proteins. B-C) Fluorescence intensity of UCP1 (B) and PLIN1 (C) from tPVAT of indicated mice, n=3 mice per group. D-E) qPCR of Ucp1 and Plin1 from tPVAT of indicated mice, n=3 mice per group. Graphed are means +SD. Data were analyzed using Student’s t-test with post-hoc Tukey’s range test. *P≤0.05, **P≤0.01, ***P≤0.001.
Notch-mediated whitening of PVAT alone is not sufficient to alter atherogenesis in the aortic root.
To start to test effects of constitutive activation of Notch in PVAT on the underlying blood vessel, we tested a model of atherogenesis. We initiated plaque formation using a gain of function AAV-PCSK9DY model. After 12 weeks, mice had plaque development in the aortic root, but not in the aorta or brachiocephalic artery. Thus, we quantified plaque size, lipid accumulation, and inflammation in the aortic root in -Cre versus +Cre mice (Suppl. Fig. V). The addition of constitutive Notch signaling in PVAT did not alter features of the atherosclerotic plaque. Because we showed that Notch signaling is already induced on a high fat diet (Fig. 3), this is a possible reason that constitutive activation of Notch did not further alter the phenotype.
PVAT-derived progenitors are Sca1low/CD140a-/CD105-.
Progenitor cells reside within a stromal vascular niche46. During obesity, adipose tissue expands via hypertrophy of adipocytes and de novo differentiation of progenitor cells within the stromal vascular fraction (SVF). Stem cell antigen1 (Sca1) identifies progenitor cells in hematopoietic tissue such as bone marrow47 and non-hematopoietic tissue such as mammary glands and adipose tissue48. Sca1 positive cells with adipogenic potential have been reported in brown and white adipose depots, but it is unclear if they are present in tPVAT. We isolated the SVF from BAT, iWAT, retroperitoneal (RP) adipose, tPVAT and abdominal aortic PVAT (aPVAT) and performed flow cytometry on live cells for Sca1 and CD31. Gating on the CD45 negative population, we detected a CD31+/Sca1+ endothelial cell population in all adipose depots49 (Fig. 6A-E), with BAT, iWAT and aPVAT having the highest proportion of endothelial cells (Fig. 6F). In all depots, we detected a CD31-/Sca1+ population, which we further gated into CD31-/Sca1high and CD31-/Sca1low subpopulations. We noted significantly fewer Sca1high cells/mg tissue in tPVAT and aPVAT (Fig. 6G), with no significant differences in Sca1low cells (Fig. 6H). Adipose progenitor cells also reportedly express CD10550 and/or CD140a51, which we analyzed within the Sca1low/CD31-/CD45- population (Suppl. Fig. VIA-E) and the Sca1high/CD31-/CD45- population (Suppl. Fig. VIF-J). The majority of Sca1+ cells were devoid of CD105. Sca1high cell populations were positive for CD140a in all depots (Suppl. Fig. VF-J). In contrast, the Sca1low population was CD140a negative (Suppl. Fig. VID-E), suggesting they represent a more committed precursor population. Our findings identify a difference in cell surface marker expression in PVAT-derived progenitor compared to those derived from brown or white adipose tissues.
Figure 6. PVAT-derived progenitors are Sca1low/CD140a+/CD105-.
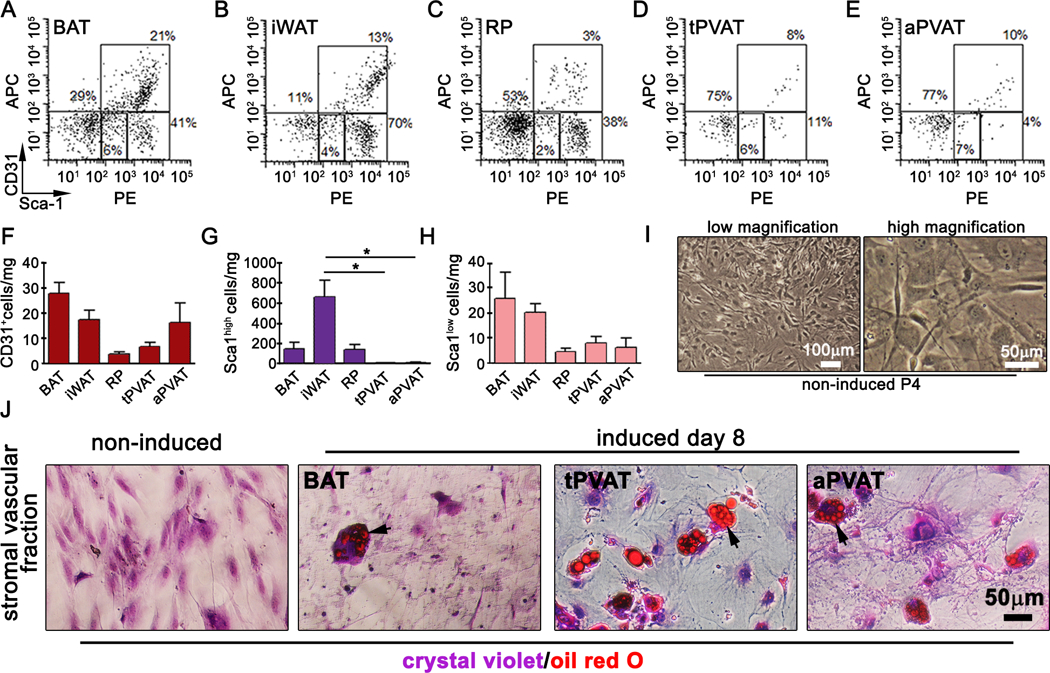
Flow cytometry of Sca1+ and CD31+ cells within the CD45- population of live/freshly isolated progenitors from BAT (A), inguinal WAT (B), retroperitoneal (C), tPVAT (D) and abdominal PVAT (E). Scatter plots are representative of two independent experiments, n=4. Quantification of CD31+ (F), Sca1high (G) and Sca1low (H) cells/mg tissue. I) Phase contrast images of passage 4 tPVAT-derived progenitors. J) Non-induced or induced progenitor cells were grown for 8d before staining with oil red O to detect lipids. Cells counterstained with crystal violet. Graphed are means + SD. Data were analyzed using one-way ANOVA and student’s t-test with post-hoc Tukey’s range test. *P≤0.05
We further analyzed the adipogenic potential of tPVAT-derived progenitor cells in vitro. SVF from tPVAT was isolated and expanded 4 passages in culture (Fig. 6I) before inducing adipogenic differentiation28. SVF from BAT and iWAT were run in parallel. We observed accumulation of lipids in PVAT-derived progenitors by 8-day post adipogenic induction as indicated by oil red O (ORO) staining (Fig. 6J). Non-induced tPVAT-derived progenitor cells were included as a negative control. Our data show that PVAT-derived progenitor cells are CD45-/CD31-/CD105-/CD140a+/Sca1low and have adipogenic potential in vitro.
Notch signaling regulates lipid accumulation in mouse tPVAT-derived progenitor cells.
Since Notch activation in adipose tissue induces tPVAT whitening, we hypothesized that Notch regulates adipocyte maturation in vitro. Progenitor cells were pooled from tPVAT of 8-week-old C57BL/6J mice and plated for expansion in vitro. After 9 passages, we evaluated progenitor markers and Notch receptor expression by flow cytometry. In agreement with freshly isolated tPVAT-derived progenitors, cultured progenitors expressed Sca1 and CD140a and were negative for CD105 (Suppl. Fig. VIIA-C), but expressed the mesenchymal marker CD73 (Suppl. Fig. VIID). As expected, progenitors were negative for immune and endothelial markers CD45 and CD31 (Suppl. Fig. VIIE-F). Notch signaling has not been described in tPVAT-derived progenitors, and we identified cell surface expression of both NOTCH1 and NOTCH2 (Suppl. Fig. VIIG-H). To determine if Notch signaling regulates adipocyte maturation, we differentiated the cells to adipocytes for 3 days28, followed by a 5-day lipid accumulation phase in the presence or absence of the Notch inhibitor DAPT. ORO staining at day 5 revealed multilocular lipid droplets in vehicle (DMSO) and DAPT (gamma secretase inhibitor) treated cells (Fig. 7A, arrows), however DAPT cultures had a significantly lower percentage of ORO-positive cells (Fig. 7B). Of the cells that were ORO positive in DAPT-treated cultures, there was significantly less lipid accumulation compared to DMSO (Fig. 7C). We also evaluated changes in protein expression in undifferentiated and differentiated cultures. Immunoblot confirmed expression of NOTCH1 and NOTCH2 and robust activation of HES1 upon adipogenic differentiation that was blocked by inclusion of DAPT (Fig. 7D). Next, we analyzed levels of the thermogenesis markers PGC1α and PLIN1. Compared to control, adipogenic induction caused strong upregulation of PGC1α and PLIN1 that was significantly inhibited by DAPT (Fig. 7E-G). FABP4 marks mature adipocytes52, and levels remained consistent in control and DAPT treated cultures, suggesting that lack of lipid accumulation in DAPT cultures is not due to de-differentiation of the cells. Thus, Notch signaling regulates maturation and lipid accumulation of differentiated tPVAT-derived progenitors.
Figure 7. Notch signaling regulates lipid accumulation in mouse tPVAT-derived adipocyte progenitor cells.
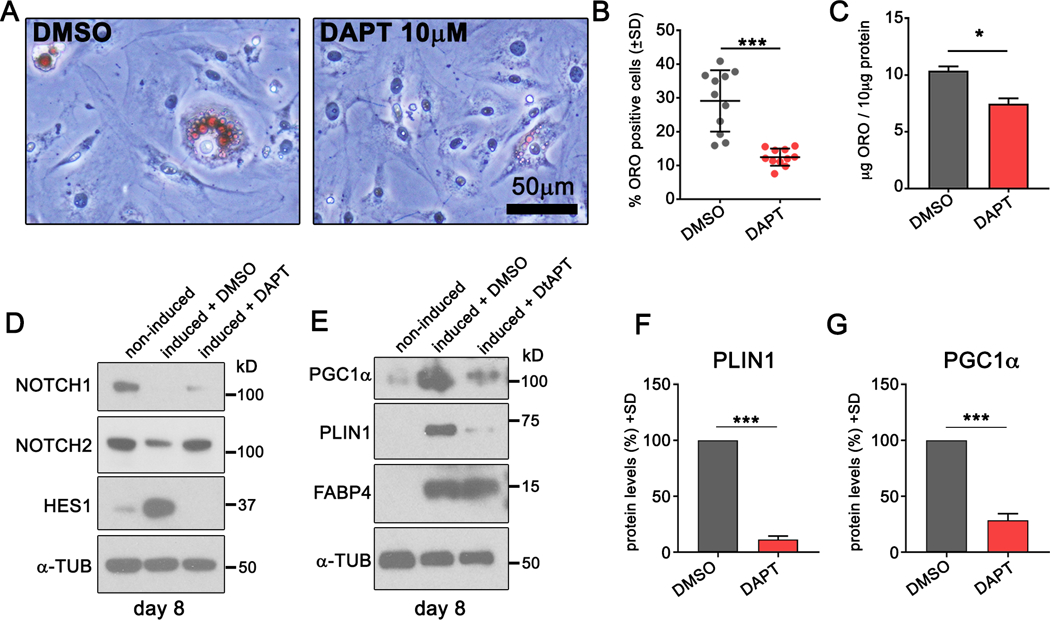
A) PVAT-derived progenitor cells were differentiated for 3 days, and 10μM DAPT or control DMSO was added at days 3–8 during the lipid accumulation stage. Cells were stained with ORO to detect lipid deposition. B-C) quantification of ORO positive cells (B) or ORO uptake (C) in control or DAPT treated cultures. D-E) Immunoblot from non-induced and induced cells treated with DMSO control or DAPT. F-G) Relative protein levels of perilipin1 (F) and PGC1α (G) in DMSO or DAPT treated cultures. Shown are representative data from two independent experiments. Graphed are means +SD. Data were analyzed using Student’s t-test with post-hoc Tukey’s range test. *P≤0.05, ***P≤0.001.
Caloric restriction down regulates Notch signaling in tPVAT.
merging evidence suggests that a CR diet can improve metabolic health and expand lifespan in humans and mice53, 54. We investigated whether a CR diet regulates Notch signaling in tPVAT. Male C57BL/6J mice were fed a control diet ad libitum or a 30% CR diet formulated to reduce carbohydrate and fat intake without inducing nutritional deficiency for five weeks. Weight measurements showed a stark loss in body weight in the CR group during the first week, a slight loss during week two and eventual plateau in weight by week three (Fig. 8A). qRT-PCR of tPVAT revealed reduced inflammation in the CR diet group as indicated by reduced Itgam (Fig. 8B). Histological analysis of tPVAT showed a reduction in lipid droplet size in PVAT and gWAT from mice on the CR diet, while changes in BAT were subtler (Fig. 8C). Consistent with published reports, a CR diet caused shrinkage of adipocytes and acquisition of a multilocular phenotype within WAT55. qRT-PCR of tPVAT for Notch pathway components indicated significant downregulation of Notch1, Notch2, Hes1 and Hey2 in tPVAT of CR mice compared to controls (Fig. 8D). Furthermore, NOTCH2, its ligand JAG1, and downstream effectors HES1 and HES5 were all significantly down regulated while NOTCH1 levels remained unchanged (Fig. 8E-F). We also tested whether Notch signaling is altered in tPVAT in female mice on a CR diet. Histological analysis of tPVAT from female mice kept for 8 weeks on a CR diet (Suppl. Fig. VIIIA) indicated that the phenotype was comparable to tPVAT from males, marked by reduced adipocyte size. Adipoq was unchanged by the CR diet, but Leptin was significantly downregulated, consistent with improved metabolic parameters in the tPVAT (Suppl. Fig. VIIIB-C). In agreement with our observations of PVAT from male mice on the CR diet for 4 weeks, immunoblot of tPVAT from female mice on the CR diet showed drastically reduced Notch signaling (Suppl. Fig. VIIID). These results suggest that Notch signaling in PVAT is sensitive to nutritional stress and is suppressed following a CR diet.
Figure 8. Caloric restriction suppresses Notch signaling in tPVAT.
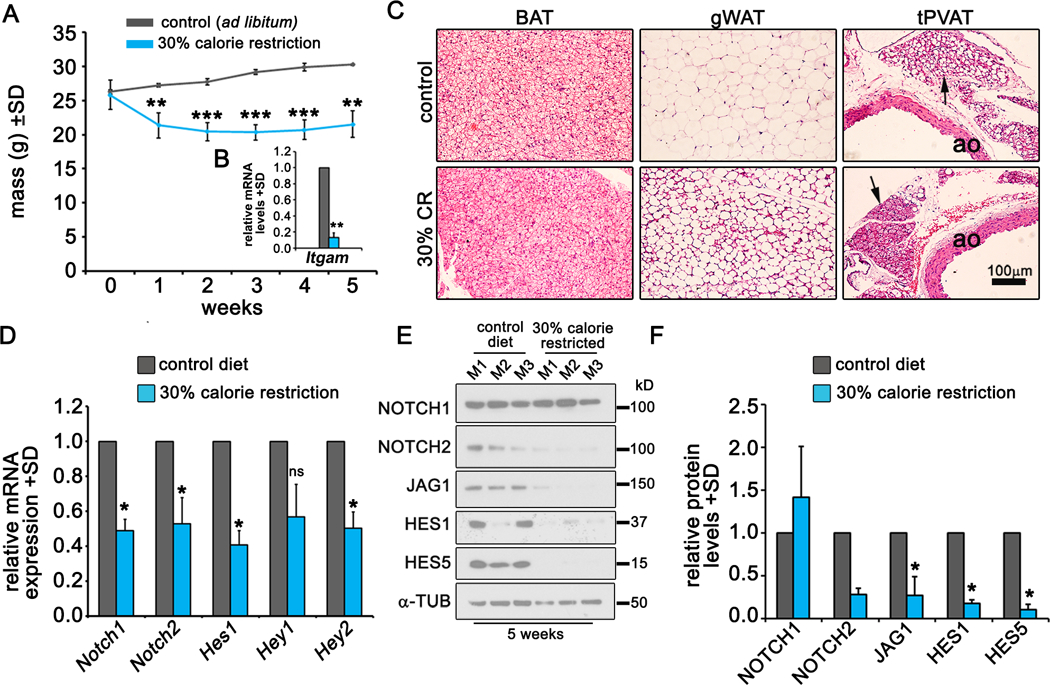
A) Weight measurements of control and 30% CR mice. B) qPCR for Itgam expression in tPVAT from control or CR mice at week 5. C) H&E staining of the indicated adipose tissues from control mice or those fed a CR diet at week 5. Arrows, tPVAT. D) qPCR from tPVAT of control mice or those fed a CR diet at week 5. E-F) Immunoblot (E) and quantification (F) of the indicated proteins from control or CR mice at week 5. N=3 mice per group. Graphed are means + SD. Data were analyzed using A) Pairwise comparison and Tukey’s post-hoc analysis; D-F) One-way ANOVA with pairwise comparison and Tukey’s post-hoc analysis. *P≤0.05, **P≤0.01, ***P≤0.001, ns = not significant
Discussion
PVAT is a thermogenic adipose depot whose dysfunction during metabolic disease exacerbates vascular inflammation and disease progression56. In our study, Notch signaling in tPVAT was found to be elevated during obesity and suppressed with caloric restriction, thus associating Notch activation with loss of thermogenesis. Previously, Bi et al.17 reported that an adipose-specific Notch1 null strain exhibited a browning phenotype in WAT, and conversely, expression of a constitutively active N1ICD in adipocytes decreased thermogenic genes in WAT and had reduced cold-induced browning in inguinal WAT. However, the previous study did not examine tPVAT, which is the focus of our current report. In addition, we characterize the proteomic profiles of WAT, BAT, and tPVAT on a control diet versus HFD, and validate PVAT as a unique adipose depot based on its basal molecular profile. We also defined a high similarity in protein signatures in BAT and tPVAT after HFD feeding, suggesting that they undergo similar transitions corresponding to loss of thermogenesis, or a whitening phenotype. This is of translational importance, given the association of adipose whitening with obesity, which includes adipocyte hypertrophy, increased lipid storage, and metabolic imbalance. Further, recent concepts of inducing beige/thermogenic adipocytes to address metabolic dysfunction requires understanding both thermogenic and whitening pathways.
We demonstrate for the first time that Notch1 activation phenocopies the pathological effects of obesity (i.e. lipid accumulation and inflammation) in tPVAT independent of dietary input. Support for a causal role for Notch activation comes from our observation that activation of Notch1 signaling in PVAT increased Leptin, Plin1 and Itgam expression, and suppressed UCP1, even in mice on a control diet. These data suggest that Notch over-activation in PVAT may prime the vascular microenvironment for disease susceptibility by creating a pro-inflammatory, lipid storage phenotype. In one atherogenesis model that includes HFD feeding after administration of a dominant active PCSK9, we did not find differences in atherogenesis between Notch activated or control PVAT, despite a higher basal level of inflammation and lipid content in the adipose tissue surrounding the vessel. One possible reason for this is our initial observation that HFD itself activates Notch signaling in PVAT. It will be of future interest to utilize both activation and suppression of Notch signaling in PVAT to further define effects on diverse vascular disease paradigms.
One possible target of Notch activity is the preadipocyte in the stromal compartment of PVAT. We were successful in isolating and characterizing adipocyte progenitors from mouse PVAT, a methodology that has not been widely performed in the mouse, where PVAT tissue is limiting. Markers used to define committed pre-adipocytes from multipotent adipose stem cells are still under debate. Expression of the progenitor marker Sca1 marks committed pre-adipocytes57. In inguinal fat, Sca1high progenitor populations have much greater propensity for adipogenic differentiation than Sca1low progenitor cells from the same tissue58. Our observation that progenitors from PVAT exhibit lower Sca1 expression than from other depots may imply that PVAT-derived progenitors have reduced capacity to undergo adipogenic differentiation in vivo than Sca1+ cells from other adipose depots. However, other reports demonstrate that adipose stem cells express CD105 and CD140a and loss of both markers indicates a committed pre-adipocyte59. Side population analysis of Sca1low cells showed that PVAT-derived cells were devoid of CD105 and CD140a. This provides additional support that adipose progenitors from tPVAT have a phenotype consistent with more committed progenitors. These PVAT-derived Sca1low/CD140a+/CD105- progenitor cells express NOTCH1 and NOTCH2 and have adipogenic capacity. Blockade of Notch during adipocyte maturation of PVAT-derived progenitor cells inhibits lipid accumulation and blunts adipogenic markers PGC1α and PLIN1, suggesting a primary role for Notch in promoting lipid accumulation. In support of this observation, CR mice have significantly reduced Notch signaling in their tPVAT. These findings provide the first evidence that Notch signaling drives pathological conversion of tPVAT in response to energy intake.
Our results that Notch regulates tPVAT phenotype support increasing evidence of Notch’s involvement in metabolism and adipose tissue phenotype36. Mouse models in which Notch has been inhibited result in improved glucose uptake, increased insulin sensitivity, and resistance to obesity when fed an obesogenic diet17. Mechanistically, suppression of Notch signaling in WAT activates the thermogenic gene program. Our results are consistent with this work in that Notch1 activation in PVAT increases inflammatory marker expression and promotes aberrant lipid deposition independent of weight gain. Intriguingly, in healthy weight patients, PVAT volume is increased at spastic regions of the coronary artery8, implying that obesity is not required for PVAT expansion. Conversely, it may be possible to suppress PVAT dysfunction in the presence of obesity by specifically targeting Notch signaling. Our findings reinforce Notch as potential target of interest in mitigating PVAT inflammation and dysfunction during obesity. It is also of translational interest to understand that therapies that utilize inhibitors of Notch signaling (e.g. gamma secretase inhibitors in cancer patients60) may also have effects on adipose tissue in the vasculature.
Obesity increases the risk for cardiometabolic disorders such as atherosclerosis, hypertension, hyperlipidemia and diabetes; and greatly increases the risk of developing CVD61, 62. PVAT is of particular importance because it encompasses athero-prone and small resistance arteries, and exerts direct paracrine effects on the vasculature63. Randomized longitudinal studies comparing the patency and atherosclerotic burden of bypass vessels used in coronary artery bypass grafting have demonstrated that leaving the PVAT on the saphenous vein (no-touch procedure), improves left ventricular ejection fraction compared to removal of PVAT64. In our studies, we induced atherogenesis using a gain-of-function PCSK9 mutant. On the background of our Adipoq-Cre;N1ICD mice (C57BL/6;FVB), plaques were limited to the aortic root after 12 weeks on a western diet. In the aortic roots, we did not detect changes in plaque size or composition. We were unable to evaluate atherosclerosis in the aorta, where there is direct and abundant contact with PVAT, as plaques did not develop during the course of the experiment, regardless of the expression of N1ICD in the PVAT. Future studies of aortic atherogenesis will require the use of models (e.g. ApoE null with HFD) where plaque formation is high in the aorta.
Obesity abrogates the conferred vasoprotective effects of PVAT on the cardiovasculature. Obesity-associated PVAT dysfunction alters the secreted factors released from a vasoprotective profile (adiponectin, nitric oxide, adipose derived relaxing factors) to proatherogenic profile (TNF-α, IL-6, resistin). The mechanisms that drive pathological phenotype conversion of PVAT are poorly understood. Our results support the hypothesis that obesity causes upregulation of Notch signaling in PVAT, which drives tissue whitening. In summary, our work identifies Notch signaling as a novel regulator of tPVAT phenotype in vivo and in vitro and identifies an initiating mechanism involved in regulating the pathological conversion of PVAT. Future studies will characterize Notch1-induced pathological phenotype to understand the adverse effects of elevated Notch signaling in PVAT on vascular tone and inflammation.
Supplementary Material
Highlights.
Mouse PVAT is a unique adipose tissue with altered proteomic profiles with nutritional stress; an obesogenic diet induces a pathological whitening phenotype in PVAT
The whitening phenotype in PVAT is associated a decreased thermogenic profile, increased lipid accumulation, and increased inflammation
Notch signaling is activated in PVAT from mice fed a high fat diet and suppressed in mice fed a calorie restricted diet
Activation of Notch signaling in adipose tissue is sufficient to promote the whitening phenotype in PVAT, even in mice fed a control diet
Suppression of Notch in PVAT-derived preadipocytes decreases their ability to mature into adipocytes and accumulate lipid
Acknowledgements
a) The authors acknowledge the expert tissue processing and histology support provided by Graziana Mangoba and Mayasah Al Hashimi of our Histopathology and Histomorphometry Core Facility. We are also grateful to our colleague Dr. Robert Koza for providing scientific feedback and advice on this research.
b) Sources of funding. This research was supported by NIH grants R01HL070865 and R01HL141149 (LL) and American Heart Association grants 17GRNT33670972 and 19TPA34850041 (LL). JB was partially supported by a pilot project from NIH grant 5P30GM106391, which also supported the Progenitor Cell Analysis Core, which was used for flow cytometry (R. Friesel PI). The following additional core facilities were used: Histopathology and Histomorphometry and the Proteomics and Lipidomics (supported by NIH grant P20GM121301, LL PI), and the Mouse Transgenic and In Vivo Imaging Core Facility (partially supported by U54GM115516, CJR PI).
List of abbreviations
- aPVAT
abdominal aorta PVAT
- BAT
brown adipose tissue
- CR
calorie restricted
- CVD
cardiovascular disease
- DAPT
(2S)-N-[(3,5-Difluorophenyl)acetyl]-L-alanyl-2-phenyl]glycine 1,1-dimethylethyl ester
- DMSO
dimethyl sulfoxide
- ETFA
electron transferring flavoprotein alpha
- gWAT
gonadal white adipose tissue
- HFD
high fat diet
- iWAT
inguinal white adipose tissue
- NotchICD
Notch intracellular domain
- ORO
oil red O
- PCSK9
proprotein convertase subtilisin/kexin type 9
- PVAT
perivascular adipose tissue
- RP
retroperitoneal
- Sca1
stem cell antigen 1
- SVF
stromal vascular fraction
- SWATH
sequential window acquisition of all theoretical spectra
- tPVAT
thoracic aorta PVAT
- UCP1
uncoupling protein 1
- WAT
white adipose tissue
Footnotes
c) Disclosures. The authors have nothing to disclose.
References
- 1.Hubert HB, Feinleib M, McNamara PM and Castelli WP. Obesity as an independent risk factor for cardiovascular disease: a 26-year follow-up of participants in the Framingham Heart Study. Circulation. 1983;67:968–77. [DOI] [PubMed] [Google Scholar]
- 2.Police SB, Thatcher SE, Charnigo R, Daugherty A and Cassis LA. Obesity promotes inflammation in periaortic adipose tissue and angiotensin II-induced abdominal aortic aneurysm formation. Arterioscler Thromb Vasc Biol. 2009;29:1458–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Fitzgibbons TP, Kogan S, Aouadi M, Hendricks GM, Straubhaar J and Czech MP. Similarity of mouse perivascular and brown adipose tissues and their resistance to diet-induced inflammation. Am J Physiol Heart Circ Physiol. 2011;301:H1425–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernandez-Alfonso MS, Gil-Ortega M, Garcia-Prieto CF, Aranguez I, Ruiz-Gayo M and Somoza B. Mechanisms of perivascular adipose tissue dysfunction in obesity. Int J Endocrinol. 2013;2013:402053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Brown NK, Zhou Z, Zhang J, Zeng R, Wu J, Eitzman DT, Chen YE and Chang L. Perivascular adipose tissue in vascular function and disease: a review of current research and animal models. Arterioscler Thromb Vasc Biol. 2014;34:1621–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Antonopoulos AS, Sanna F, Sabharwal N, Thomas S, Oikonomou EK, Herdman L, Margaritis M, Shirodaria C, Kampoli AM, Akoumianakis I, Petrou M, Sayeed R, Krasopoulos G, Psarros C, Ciccone P, Brophy CM, Digby J, Kelion A, Uberoi R, Anthony S, Alexopoulos N, Tousoulis D, Achenbach S, Neubauer S, Channon KM and Antoniades C. Detecting human coronary inflammation by imaging perivascular fat. Sci Transl Med. 2017;9. [DOI] [PubMed] [Google Scholar]
- 7.Shields KJ, El Khoudary SR, Ahearn JM and Manzi S. Association of aortic perivascular adipose tissue density with aortic calcification in women with systemic lupus erythematosus. Atherosclerosis. 2017;262:55–61. [DOI] [PubMed] [Google Scholar]
- 8.Ohyama K, Matsumoto Y, Nishimiya K, Hao K, Tsuburaya R, Ota H, Amamizu H, Uzuka H, Takahashi J, Ito K and Shimokawa H. Increased Coronary Perivascular Adipose Tissue Volume in Patients With Vasospastic Angina. Circ J. 2016;80:1653–6. [DOI] [PubMed] [Google Scholar]
- 9.Ahmadieh S, Kim HW and Weintraub NL. Potential role of perivascular adipose tissue in modulating atherosclerosis. Clin Sci (Lond). 2020;134:3–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang X, Qi Y, Dai F, Gu J and Yao W. PVAT: an important guardian of the cardiovascular system. Histol Histopathol. 2020:18211. [DOI] [PubMed] [Google Scholar]
- 11.Queiroz M and Sena CM. Perivascular adipose tissue in age-related vascular disease. Ageing Res Rev. 2020;59:101040. [DOI] [PubMed] [Google Scholar]
- 12.Rafeh R, Viveiros A, Oudit GY and El-Yazbi AF. Targeting perivascular and epicardial adipose tissue inflammation: therapeutic opportunities for cardiovascular disease. Clin Sci (Lond). 2020;134:827–851. [DOI] [PubMed] [Google Scholar]
- 13.Tinajero MG and Gotlieb AI. Recent Developments in Vascular Adventitial Pathobiology: The Dynamic Adventitia as a Complex Regulator of Vascular Disease. Am J Pathol. 2020;190:520–534. [DOI] [PubMed] [Google Scholar]
- 14.Chang L, Villacorta L, Li R, Hamblin M, Xu W, Dou C, Zhang J, Wu J, Zeng R and Chen YE. Loss of perivascular adipose tissue on peroxisome proliferator-activated receptor-gamma deletion in smooth muscle cells impairs intravascular thermoregulation and enhances atherosclerosis. Circulation. 2012;126:1067–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiong W, Zhao X, Garcia-Barrio MT, Zhang J, Lin J, Chen YE, Jiang Z and Chang L. MitoNEET in Perivascular Adipose Tissue Blunts Atherosclerosis under Mild Cold Condition in Mice. Front Physiol. 2017;8:1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boucher J, Gridley T and Liaw L. Molecular pathways of Notch signaling in vascular smooth muscle cells. Front Physiol. 2012;3:81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bi P, Shan T, Liu W, Yue F, Yang X, Liang XR, Wang J, Li J, Carlesso N, Liu X and Kuang S. Inhibition of Notch signaling promotes browning of white adipose tissue and ameliorates obesity. Nat Med. 2014;20:911–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vujovic S, Henderson SR, Flanagan AM and Clements MO. Inhibition of gamma-secretases alters both proliferation and differentiation of mesenchymal stem cells. Cell Prolif. 2007;40:185–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osathanon T, Subbalekha K, Sastravaha P and Pavasant P. Notch signalling inhibits the adipogenic differentiation of single-cell-derived mesenchymal stem cell clones isolated from human adipose tissue. Cell Biol Int. 2012;36:1161–70. [DOI] [PubMed] [Google Scholar]
- 20.Urs S, Turner B, Tang Y, Rostama B, Small D and Liaw L. Effect of soluble Jagged1-mediated inhibition of Notch signaling on proliferation and differentiation of an adipocyte progenitor cell model. Adipocyte. 2012;1:46–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pettersson US, Walden TB, Carlsson PO, Jansson L and Phillipson M. Female mice are protected against high-fat diet induced metabolic syndrome and increase the regulatory T cell population in adipose tissue. PloS one. 2012;7:e46057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Venkatesh DA, Park KS, Harrington A, Miceli-Libby L, Yoon JK and Liaw L. Cardiovascular and hematopoietic defects associated with Notch1 activation in embryonic Tie2-expressing populations. Circ Res. 2008;103:423–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bjorklund MM, Hollensen AK, Hagensen MK, Dagnaes-Hansen F, Christoffersen C, Mikkelsen JG and Bentzon JF. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114:1684–9. [DOI] [PubMed] [Google Scholar]
- 24.Miao CH, Ohashi K, Patijn GA, Meuse L, Ye X, Thompson AR and Kay MA. Inclusion of the hepatic locus control region, an intron, and untranslated region increases and stabilizes hepatic factor IX gene expression in vivo but not in vitro. Mol Ther. 2000;1:522–32. [DOI] [PubMed] [Google Scholar]
- 25.Ivosev G, Burton L and Bonner R. Dimensionality reduction and visualization in principal component analysis. Anal Chem. 2008;80:4933–44. [DOI] [PubMed] [Google Scholar]
- 26.Deutsch EW. The PeptideAtlas Project. Methods Mol Biol. 2010;604:285–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Desiere F, Deutsch EW, King NL, Nesvizhskii AI, Mallick P, Eng J, Chen S, Eddes J, Loevenich SN and Aebersold R. The PeptideAtlas project. Nucleic Acids Res. 2006;34:D655–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rim JS, Mynatt RL and Gawronska-Kozak B. Mesenchymal stem cells from the outer ear: a novel adult stem cell model system for the study of adipogenesis. FASEB journal : official publication of the Federation of American Societies for Experimental Biology. 2005;19:1205–7. [DOI] [PubMed] [Google Scholar]
- 29.Liaw L, Prudovsky I, Koza RA, Anunciado-Koza RV, Siviski ME, Lindner V, Friesel RE, Rosen CJ, Baker PR, Simons B and Vary CP. Lipid Profiling of In Vitro Cell Models of Adipogenic Differentiation: Relationships with Mouse Adipose Tissues. J Cell Biochem. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nascimento EB, Boon MR and van Marken Lichtenbelt WD. Fat cells gain new identities. Sci Transl Med. 2014;6:247fs29. [DOI] [PubMed] [Google Scholar]
- 31.Fromme T and Klingenspor M. Uncoupling protein 1 expression and high-fat diets. Am J Physiol Regul Integr Comp Physiol. 2011;300:R1–8. [DOI] [PubMed] [Google Scholar]
- 32.Surwit RS, Wang S, Petro AE, Sanchis D, Raimbault S, Ricquier D and Collins S. Diet-induced changes in uncoupling proteins in obesity-prone and obesity-resistant strains of mice. Proc Natl Acad Sci U S A. 1998;95:4061–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schiff M, Froissart R, Olsen RK, Acquaviva C and Vianey-Saban C. Electron transfer flavoprotein deficiency: functional and molecular aspects. Mol Genet Metab. 2006;88:153–8. [DOI] [PubMed] [Google Scholar]
- 34.Bondia-Pons I, Boque N, Paternain L, Santamaria E, Fernandez J, Campion J, Milagro F, Corrales F and Martinez JA. Liver proteome changes induced by a short-term high-fat sucrose diet in wistar rats. J Nutrigenet Nutrigenomics. 2011;4:344–53. [DOI] [PubMed] [Google Scholar]
- 35.Ugarte F, Ryser M, Thieme S, Fierro FA, Navratiel K, Bornhauser M and Brenner S. Notch signaling enhances osteogenic differentiation while inhibiting adipogenesis in primary human bone marrow stromal cells. Exp Hematol. 2009;37:867–875 e1. [DOI] [PubMed] [Google Scholar]
- 36.Bi P and Kuang S. Notch signaling as a novel regulator of metabolism. Trends Endocrinol Metab. 2015;26:248–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J, Estall JL, Klein U, Maratos-Flier E and Rosen ED. Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 2011;13:249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Boutens L and Stienstra R. Adipose tissue macrophages: going off track during obesity. Diabetologia. 2016;59:879–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang R and Barouch LA. Leptin signaling and obesity: cardiovascular consequences. Circ Res. 2007;101:545–59. [DOI] [PubMed] [Google Scholar]
- 40.Boschmann M, Engeli S, Adams F, Gorzelniak K, Franke G, Klaua S, Kreuzberg U, Luedtke S, Kettritz R, Sharma AM, Luft FC and Jordan J. Adipose tissue metabolism and CD11b expression on monocytes in obese hypertensives. Hypertension. 2005;46:130–6. [DOI] [PubMed] [Google Scholar]
- 41.Nedergaard J, Golozoubova V, Matthias A, Asadi A, Jacobsson A and Cannon B. UCP1: the only protein able to mediate adaptive non-shivering thermogenesis and metabolic inefficiency. Biochimica et biophysica acta. 2001;1504:82–106. [DOI] [PubMed] [Google Scholar]
- 42.Aldiss P, Davies G, Woods R, Budge H, Sacks HS and Symonds ME. ‘Browning’ the cardiac and peri-vascular adipose tissues to modulate cardiovascular risk. Int J Cardiol. 2017;228:265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kozak LP and Anunciado-Koza R. UCP1: its involvement and utility in obesity. International journal of obesity. 2008;32 Suppl 7:S32–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kontani Y, Wang Y, Kimura K, Inokuma KI, Saito M, Suzuki-Miura T, Wang Z, Sato Y, Mori N and Yamashita H. UCP1 deficiency increases susceptibility to diet-induced obesity with age. Aging cell. 2005;4:147–55. [DOI] [PubMed] [Google Scholar]
- 45.Itabe H, Yamaguchi T, Nimura S and Sasabe N. Perilipins: a diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017;16:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zwick RK, Guerrero-Juarez CF, Horsley V and Plikus MV. Anatomical, Physiological, and Functional Diversity of Adipose Tissue. Cell Metab. 2018;27:68–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Holmes C and Stanford WL. Concise review: stem cell antigen-1: expression, function, and enigma. Stem Cells. 2007;25:1339–47. [DOI] [PubMed] [Google Scholar]
- 48.Rodeheffer MS, Birsoy K and Friedman JM. Identification of white adipocyte progenitor cells in vivo. Cell. 2008;135:240–9. [DOI] [PubMed] [Google Scholar]
- 49.Pratumvinit B, Reesukumal K, Janebodin K, Ieronimakis N and Reyes M. Isolation, characterization, and transplantation of cardiac endothelial cells. Biomed Res Int. 2013;2013:359412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Klimczak A and Kozlowska U. Mesenchymal Stromal Cells and Tissue-Specific Progenitor Cells: Their Role in Tissue Homeostasis. Stem cells international. 2016;2016:4285215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Rostovskaya M and Anastassiadis K. Differential expression of surface markers in mouse bone marrow mesenchymal stromal cell subpopulations with distinct lineage commitment. PLoS One. 2012;7:e51221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moseti D, Regassa A and Kim WK. Molecular Regulation of Adipogenesis and Potential Anti-Adipogenic Bioactive Molecules. International journal of molecular sciences. 2016;17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fontana L. Calorie restriction and cardiometabolic health. Eur J Cardiovasc Prev Rehabil. 2008;15:3–9. [DOI] [PubMed] [Google Scholar]
- 54.Omodei D and Fontana L. Calorie restriction and prevention of age-associated chronic disease. FEBS Lett. 2011;585:1537–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Fabbiano S, Suarez-Zamorano N, Rigo D, Veyrat-Durebex C, Stevanovic Dokic A, Colin DJ and Trajkovski M. Caloric Restriction Leads to Browning of White Adipose Tissue through Type 2 Immune Signaling. Cell Metab. 2016;24:434–446. [DOI] [PubMed] [Google Scholar]
- 56.Xia N and Li H. The role of perivascular adipose tissue in obesity-induced vascular dysfunction. British journal of pharmacology. 2017;174:3425–3442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ussar S, Lee KY, Dankel SN, Boucher J, Haering MF, Kleinridders A, Thomou T, Xue R, Macotela Y, Cypess AM, Tseng YH, Mellgren G and Kahn CR. ASC-1, PAT2, and P2RX5 are cell surface markers for white, beige, and brown adipocytes. Sci Transl Med. 2014;6:247ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Tokunaga M, Inoue M, Jiang Y, Barnes RH 2nd, Buchner DA and Chun TH. Fat depot-specific gene signature and ECM remodeling of Sca1(high) adipose-derived stem cells. Matrix Biol. 2014;36:28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cawthorn WP, Scheller EL and MacDougald OA. Adipose tissue stem cells meet preadipocyte commitment: going back to the future. J Lipid Res. 2012;53:227–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ran Y, Hossain F, Pannuti A, Lessard CB, Ladd GZ, Jung JI, Minter LM, Osborne BA, Miele L and Golde TE. gamma-Secretase inhibitors in cancer clinical trials are pharmacologically and functionally distinct. EMBO Mol Med. 2017;9:950–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hall JE, do Carmo JM, da Silva AA, Wang Z and Hall ME. Obesity-induced hypertension: interaction of neurohumoral and renal mechanisms. Circ Res. 2015;116:991–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lovren F, Teoh H and Verma S. Obesity and atherosclerosis: mechanistic insights. The Canadian journal of cardiology. 2015;31:177–83. [DOI] [PubMed] [Google Scholar]
- 63.Fernandez-Alfonso MS, Somoza B, Tsvetkov D, Kuczmanski A, Dashwood M and Gil-Ortega M. Role of Perivascular Adipose Tissue in Health and Disease. Compr Physiol. 2017;8:23–59. [DOI] [PubMed] [Google Scholar]
- 64.Johansson B, Samano N, Souza D, Bodin L, Filbey D, Mannion JD and Bojo L. The no-touch vein graft for coronary artery bypass surgery preserves the left ventricular ejection fraction at 16 years postoperatively: long-term data from a longitudinal randomised trial. Open heart. 2015;2:e000204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.