

Abstract
Tyrosine kinase inhibitors have revolutionized the world of cancer treatment in recent years, profoundly improving survival of patients with chronic myeloid leukemia (CML) and beyond. However, off-target toxicities of these inhibitors are well-described, and resistance has become a paramount concern. Novel allosteric inhibitors of the Abelson (ABL) family of tyrosine kinases, including GNF-2, GNF-5, and ABL-001, are equipped to overcome these issues. Several contemporary studies have demonstrated their potential efficacy in three key areas: primary hematologic and solid malignancies, metastasis, and combination with other small molecules. Further, ongoing clinical trials are investigating the efficacy of ABL-001 for the treatment of CML and recurrent solid tumors. This work reviews the current literature of the preclinical testing of GNF-2 and GNF-5 and the preclinical and clinical testing of ABL-001. Future research will continue to evaluate these promising inhibitors as both first-line therapy for solid tumors and salvage therapy when more traditional drugs such as imatinib fail.
Keywords: ABL1, ABL2, kinase, inhibitor, allosteric, cancer
Introduction
The Abelson (ABL) family of tyrosine kinases contains proteins ABL1 and ABL2 that regulate a multitude of cellular processes (1). These kinases garnered major attention in 1982, when de Klein et al. identified their involvement in the Philadelphia chromosome translocation that drives chronic myeloid leukemia (CML) (2). This translocation results in an oncogenic fusion, BCR-ABL1, between the BCR gene (chromosome 22) and the ABL1 gene (chromosome 9). ABL1 and ABL2 (also known as ABL-related gene, or ARG) have since been recognized for their roles in cytoskeletal reorganization, cell survival/proliferation, and disease pathology (1). ABL kinases are now implicated in a variety of disorders outside CML including diabetes, Niemann-Pick type C disease, and chronic pain (3–6). Elevated expression or activation of ABL kinases have also been found in several solid tumors (7). Furthermore, ABL kinase fusions (like BCR-ABL1) have been identified as drivers of non-CML leukemias including B-cell acute lymphoblastic leukemia (B-ALL), T-cell acute lymphoblastic leukemia (T-ALL), and acute myeloid leukemia (AML) (7–10).
Unsurprisingly, ABL kinase inhibition has proven an interesting therapeutic target in oncology. Imatinib mesylate, a first-generation, non-allosteric ABL inhibitor, has transformed the CML treatment landscape by improving ten-year overall survival in CML to over 80%, a nearly 50% reduction in annual age-adjusted mortality (11). However, both imatinib resistance and off-target drug effects for the treatment of CML, chronic eosinophilic leukemia, gastrointestinal stromal tumors, and dermatofibrosarcoma protuberans have been reported (12–16). Imatinib resistance may cause edema, muscle cramps, nausea, diarrhea, rashes, headache/fatigue, and pain (musculoskeletal and abdominal) and manifests as one of two forms (17,18). Primary resistance is intrinsic (patients do not respond at all to treatment), while secondary resistance is acquired (patients lose an achieved response over time) (19). In CML, primary resistance affects 8–13% of patients who fail to achieve major or complete cytogenetic response and 2% of patients who fail to achieve hematologic response (19). Secondary resistance affects 18% of CML patients by five years of treatment (19). Thus, it has become increasingly important to design alternative ABL kinase inhibitors that can overcome such resistance and are more specific to ABL kinases than imatinib for the treatment of leukemias and solid tumors. Aside from imatinib, other non-allosteric ABL kinase inhibitors include nilotinib, dasatinib, and bosutinib. These have all demonstrated some degree of efficacy even in solid tumors that are typically not driven by ABL1 fusions. For example, dasatinib was found to be relatively well tolerated in patients with metastatic solid tumors including gastrointestinal stromal tumor (GIST), melanoma, and multiple sarcomas (20). Bosutinib was found to be well tolerated in patients with advanced solid tumors including colorectal, pancreatic, and ovarian cancers, and resulted in one partial response (breast cancer) and one unconfirmed complete response (pancreatic cancer) (21). Further, Nilotinib was found to be both well tolerated and clinically active in patients with imatinib-resistant GIST; 38 of 53 patients had stable disease and two achieved partial response (22).
However, adverse effects such as nausea, rash and fatigue, affected the majority to 100% of patients in all three trials (20–22). Second, only 7% and 16% of patients in the bosutinib and dasatinib studies, respectively, achieved stable disease (20,21). These undesirable toxicities and this moderate efficacy may be attributable to these agents’ nature as non-allosteric inhibitors. Non-allosteric inhibitors may cause off-target drug effects because they do not specifically inhibit ABL kinases. Allosteric inhibitors, by contrast, are entirely specific to ABL kinases. Therefore, cancer treatment with allosteric inhibitors have the potential to be at least as efficacious as non-allosteric inhibitors and may significantly eliminate many off-target effects.
Allosteric ABL inhibitors bind to allosteric sites that are unique to ABL kinases, conferring remarkable specificity for ABL kinase inhibition alone. This review will explore the therapeutic potential of three novel allosteric ABL inhibitors, GNF-2, GNF-5, and ABL-001, in primary malignancies, cancer metastasis, and as combination therapy in leukemias and solid tumors.
Mechanism of action of allosteric ABL inhibitors
ABL kinases are bilobal proteins with large C-terminal domains (23). The two members of the Abelson family in vertebrates, ABL1 and ABL2, share several catalytic and regulatory lobes but differ subtly in function. ABL1 contains nuclear localization motifs, while ABL2 is primarily found in the cytoplasm and at F-actin-rich sites in cells (7). An ATP-binding pocket rests between the N- and C-terminals of ABL1/ABL2 and contains a “gatekeeper” (threonine) residue, which regulates inhibitor binding and specificity. ABL kinase activity is controlled through such intramolecular/intermolecular interactions and through post-translational modifications (7).
A critical intramolecular interaction of ABL kinase activity is myristate binding. ABL kinases are inhibited when a myristoylated residue in the N-lobe binds a hydrophobic pocket in the C-lobe, locking ABL1/ABL2 into a “closed,” or catalytically inactive, conformation (Fig. 1). A major issue with ABL fusion proteins, like BCR-ABL1, is that their chromosomal translocation removes the N-lobe (and its myristate binder) from ABL1/ABL2 (24). This leads to constitutive ABL kinase activity and helps drive leukemias like CML, since the autoinhibitory mechanism of ABL kinases is compromised when the N-terminal cap region is lost (25). The removal of this N-lobe, however, does not affect the myristate binding pocket in the C-lobe of ABL1/ABL2 (24). This observation led researchers to question the utility of allosteric ABL kinase inhibitors, which can mimic the intrinsic myristate of the ABL N-lobe by binding in the C-lobe myristoyl pocket that is retained after chromosomal translocation (26).
Figure 1.

Schematic of the active (open) and inactive (closed) conformations of an ABL kinase. SH2, SH3, and kinase domains are depicted. (Adapted from Hughes et al., 2019 (35).)
Non-allosteric ABL inhibitors, like imatinib and nilotinib, target either the active or inactive conformation of ABL kinases and compete for binding with ATP. In contrast, allosteric inhibitors (GNF-2, GNF-5, and ABL-001) bind specifically to ABL’s myristoyl-binding pocket, abrogating concerns of off-target drug effects. Selectivity profiles of GNF-2, GNF-5, and ABL-001 are displayed in Fig. 2A alongside those of imatinib and nilotinib. Structures of GNF-2, GNF-5, and ABL-001 are shown in Fig. 2B.
Figure 2.
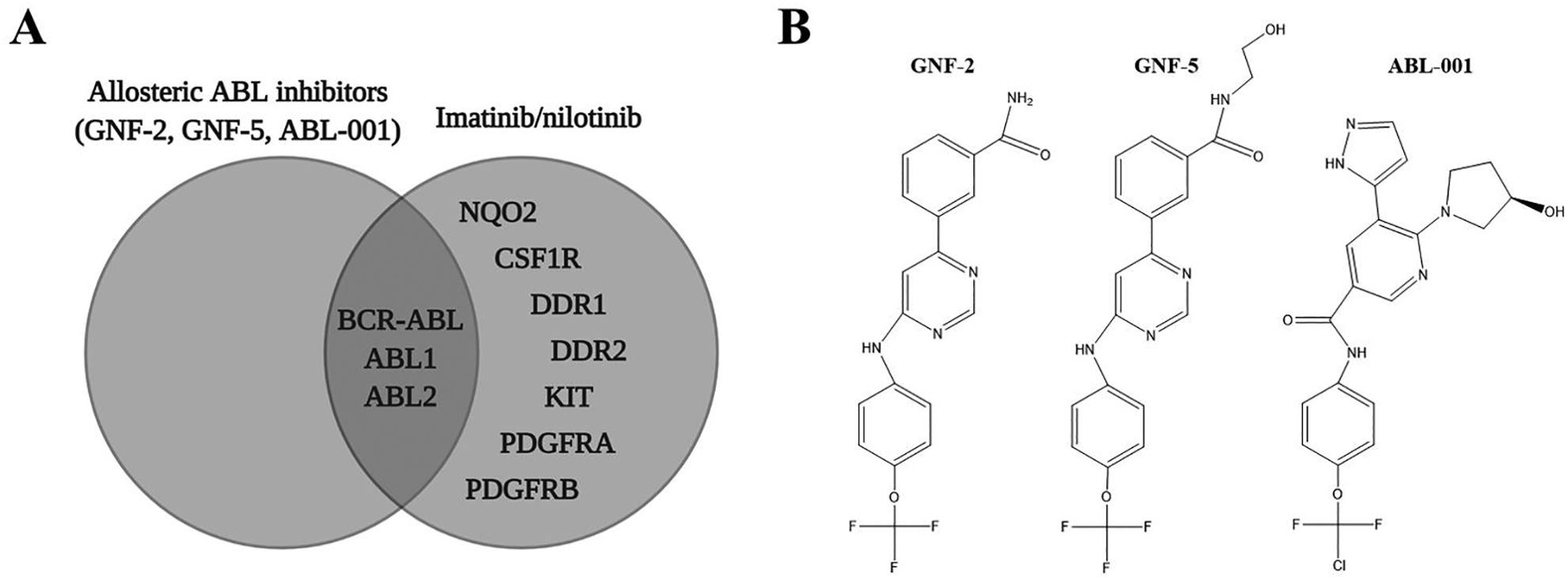
A. Selectivity profiles of GNF-2, GNF-5, and ABL-001 in contrast to two non-allosteric ABL kinase inhibitors, imatinib and nilotinib. (Adapted from Greuber et al., 2013 (7).) B. Chemical structures of GNF-2 (PubChem CID 5311510), GNF-5 (PubChem CID 44129660), and ABL-001 (PubChem CID 72165228).
The selectivity of allosteric inhibitors allows them to be just as effective against hyperactive (unfused) ABL kinases in solid tumors as they are against leukemia-driving ABL fusion proteins (7). Though hyperactive (unfused) ABL kinases retain their intrinsic, N-lobe myristate, allosteric inhibitors can bind the ABL C-lobe pocket with higher affinity and inhibit ABL kinase activity in a more reliable manner than non-allosteric inhibitors. Although the allosteric ABL inhibitors, GNF-2 and GNF-5, have yet to be explored in clinical trials, their potential as such highly specific drugs warrants review alongside that of ABL-001 (asciminib), the first allosteric ABL inhibitor in humans. Since GNF-2 and GNF-5 are so structurally similar to ABL-001, they may demonstrate equally as desirable safety profiles as ABL-001 has thus far.
GNF-2, GNF-5, and ABL-001
GNF-2, GNF-5, and ABL-001, are fluorine-containing pyrimidine/pyridine derivatives that specifically target ABL1, ABL2, and BCR-ABL1 (7). ABL-001 is the subject of ten current clinical trials for various diseases (27). GNF-5 has been reported to have better pharmacokinetic properties than GNF-2 including higher bioavailability, longer half-life, and slower clearance, while ABL-001 has been reported to be well tolerated clinically with imatinib-resistant CML (28,29). A comprehensive list of manuscripts exploring GNF-2, GNF-5, and ABL-001 for hematologic and solid malignancies is featured in Table 1.
Table 1.
Selected clinical trials and preclinical studies using GNF-2, GNF-5, and ABL-001.
| Reference | Title | Author | Journal | Year | Summary | Inhibitor(s) | Cell Line |
|---|---|---|---|---|---|---|---|
| Clinical Trials | |||||||
| 35 | Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure | Hughes et al. | New England Journal of Medicine | 2019 | ABL-001 achieved major molecular response (MMR) in 48% of heavily pretreated patients, including those harboring T315I. | ABL-001 | |
| 37 | Phase 1a study results investigating the safety and preliminary efficacy of ABL001 (NOV1501), a bispecific antibody targeting VEGF and DLL4 in metastatic gastrointestinal (GI) cancer | Lee et al. | Journal of Clinical Oncology | 2019 | ABL-001 was well tolerated up to 7.5 mg/kg with no significant treatment-related adverse events in 18 patients pre-treated with at least three lines of chemotherapy. | ABL-001 | |
| 48 | Combination of Asciminib plus Nilotinib or Asciminib plus Dasatinib in Previously Treated Chronic Myeloid Leukemia (CML) Patients: Phase 1 Study Results | DeAngelo et al. | Clinical Lymphoma, Myeloma & Leukemia | 2019 | 9/27 patients treated with ABL-001 combination therapy achieved MMR over the course of treatment; good safety/tolerability of combination treatment despite some drug-related adverse events. | ABL-001 | |
| 49 | Combination of Asciminib, a Novel and Specific BCR-ABL1 Inhibitor, Plus Imatinib in Previously Treated Chronic Myeloid Leukemia (CML) Patients: Phase 1 Study Results | Talpaz et al. | Clinical Lymphoma, Myeloma & Leukemia | 2019 | ABL-001 plus imatinib combination therapy is well tolerated in CML patients resistant/intolerant to at least two other tyrosine kinase inhibitors. | ABL-001 | |
| Preclinical Studies | |||||||
| 3 | Targeting ABL-IRE1alpha Signaling Spares ER-Stressed Pancreatic beta Cells to Reverse Autoimmune Diabetes | Morita et al. | Cell Metabolism | 2017 | ABL kinases rheostatically enhance the enzymatic activity of the ER transmembrane kinase/endoribonuclease IRE1α; GNF-2 interferes with signaling on this axis by disengaging kinase-inhibited endogenous ABL1 from 14-3-3 proteins. | GNF-2 | INS-1 (rat insulinoma), non-diabetic human islets |
| 5 | A Bcr-Abl Inhibitor GNF-2 Attenuates Inflammatory Activation of Glia and Chronic Pain | Song et al. | Frontiers in Pharmacology | 2019 | GNF-2 attenuates markers of inflammatory glial activation and ensuing pain behaviors in an animal model, promoting further research into its use for management of chronic pain. | GNF-2 | BV-2 (immortalized murine microglia), MGCs (mouse primary mixed glial cells) |
| 28 | Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors | Zhang et al. | Nature | 2010 | GNF-2 binds to the myristate pocket of ABL kinases, combinations of established kinase inhibitors with allosteric ABL kinase inhibitors reduce the emergence of resistant mutants, and GNF-5 is effective both in vitro and in vivo against T315I. | GNF-2, GNF-5 | Ba/F3 (fibroblasts) |
| 30 | C-Abl is not activated in DNA damage-induced and Tap63-mediated oocyte apoptosis in human ovary | Bildik et al. | Cell Death & Disease | 2018 | There is molecular evidence for ovarian toxicity of imatinib, but not GNF-2, in human (imatinib is also a c-KIT inhibitor). | GNF-2 | Normal GV-stage oocytes, HLGCs (human non-mitotic luteinized granulosa cells), mitotic non-luteinized human granulosa cells |
| 31 | N-myristoylated c-Abl tyrosine kinase localizes to the endoplasmic reticulum upon binding to an allosteric inhibitor | Choi et al. | Journal of Biological Chemistry | 2009 | GNF-2 inhibits non-myristoylated ABL1 more potently than myristoylated ABL1, and inhibits enzymatic/cellular kinase activity of ABL2 as well. | GNF-2 | Phoenix™ Ampho (kidney), ABL1/ARG K/O 3T3 (fibroblasts) |
| 32 | The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1 | Wylie et al. | Nature | 2017 | Characterized the resistance profile and kinase activity of ABL-001; ABL-001 plus nilotinib combination therapy eradicated xenografted tumors of CML and led to complete disease control without recurrence. | ABL-001 | Ba/F3 (fibroblasts), KCL-22 (leukemia) |
| 33 | Anti-growth Effects of Imatinib and GNF5 via Regulation of Skp2 in Human Hepatocellular Carcinoma Cells | Kim et al. | Journal of Cancer Prevention | 2018 | GNF-5 inhibits cell growth along with imatinib, inducing caspase-dependent apoptosis and enhancing p27/p21 (however, GNF-2 did not). | GNF-2, GNF-5 | SK-HEP1 (human hepatocarcinoma) |
| 38 | Inactivation of ABL kinases suppresses non-small cell lung cancer metastasis | Gu et al. | JCI Insight | 2016 | ABL1/ABL2 promote metastasis of EGFR- and KRAS-mutated lung cancer cells; cells treated with GNF-5 or knocked down for ABL1/ABL2 display reduced expression of metastasis gene signatures, and GNF-5 can suppress multiple-organ metastasis in vivo. | GNF-5 | H358, PC9 and PC9-derived metastatic PC9M, H460 parental and H460-derived metastatic M4M5 (human NSCLC) |
| 39 | Targeting invadopodia-mediated breast cancer metastasis by using ABL kinase inhibitors | Meirson et al. | Oncotarget | 2018 | GNF-5 (and imatinib/nilotinib) hampers the formation/maturation of invadopodia, spontaneous lung metastasis is impaired in GNF-5-treated mice, and GNF-5 specifically inhibits MMP-dependent invasiveness. | GNF-5 | MDA-MB-231 (human breast adenocarcinoma) |
| 40 | Abl and Arg mediate cysteine cathepsin secretion to facilitate melanoma invasion and metastasis | Tripathi et al. | Science Signaling | 2018 | ABL1/ABL2 activate metastasis-related transcription factors to promote cathepsin protease secretion. | GNF-2 | Melanoma (25 various) |
| 41 | A TAZ-AXL-ABL2 Feed-Forward Signaling Axis Promotes Lung Adenocarcinoma Brain Metastasis | Hoj et al. | Cell Reports | 2019 | ABL-001 treatment significantly decreases brain metastases; GNF-5 affects expression of AXL protein (correlative prognostic value for patients with brain metastases), and ABL-001 and GNF-5 are both blood-brain barrier-penetrant. | GNF-5, ABL-001 | HCC4006, HCC827, H1975, PC9 (NSCLC); H293T (human embryonic kidney) |
| 42 | Allosteric inhibitors of Bcr-abl-dependent cell proliferation | Adrián et al. | Nature Chemical Biology | 2006 | GNF-2 inhibits BCR-ABL1-dependent cell proliferation, enhances imatinib activity on BCR-ABL1-expressing cells, and is subject to resistance with mutations in the myristoyl cleft. | GNF-2 | Ba/F3 (fibroblasts), 32D (murine myeloid precursor) |
| 43 | The new allosteric inhibitor asciminib is susceptible to resistance mediated by ABCB1 and ABCG2 overexpression in vitro | Eadie et al. | Oncotarget | 2018 | ABL-001 resistance can be overcome with ABL-001 plus imatinib/nilotinib combination therapy. | ABL-001 | K562, KU812 (leukemia) |
| 44 | ABL kinase inhibition sensitizes primary lung adenocarcinomas to chemotherapy by promoting tumor cell differentiation | Khatri et al. | Oncotarget | 2019 | Treatment of tumor-bearing mice with GNF-5 sensitizes lung adenocarcinomas to chemotherapy. | GNF-5 | Kras(G12D/+);p53(−/−) mouse |
Hematologic and solid malignancies
Preclinically, GNF-2, GNF-5, and ABL-001 have all been effective against solid tumors and/or on tumor cell lines expressing high levels of ABL1/ABL2 (30–33). In 2009, GNF-2 was shown to inhibit the enzymatic and cellular kinase activities of ABL1, ABL2, and recombinant ABL and to inhibit the proliferation of BCR-ABL1-expressing Ba/F3 fibroblast cells at a potency comparable to that of imatinib (IC50=0.24 μM) (31). These findings were confirmed by evaluating expression of phosphorylated CRKII, a downstream ABL substrate, which decreased in a time- and dose-dependent manner following allosteric ABL inhibition (31). Notably, this same study confirmed that GNF-2 binds to ABL kinases allosterically. GNF-2 inhibited non-myristoylated ABL1 more effectively than myristoylated ABL1, and mutations in the myristate binding pocket of this kinase severely compromised the inhibitor’s capacity to bind (31). Additionally, Bildik et al. compared the off-target effects of GNF-2 with imatinib in a study of ovarian cancer. While imatinib promoted “bizarre shaped follicles lacking oocytes,” increased follicular atresia, and histomorphological abnormalities in human ovarian graft follicles, GNF-2 treatment did not cause any such toxic effects (30). Taken together, these results suggest that allosteric ABL inhibitors selectively kill tissues expressing high levels of ABL kinases, while desirably sparing surrounding healthy cells.
In 2018, Kim et al. found that the more pharmacokinetically active derivative of GNF-2, GNF-5, can inhibit the growth of human hepatocellular carcinoma (SK-HEP1) cells – increasing the expression of tumor suppressors (p27 and p21) alongside markers of apoptosis such as caspases and poly (ADP-ribose) polymerase (33). Further, Malki et al. designed several alloxazine derivatives of imatinib that also bind to ABL kinases’ allosteric sites (34). Ultimately, these derivatives improved on-target cytotoxicity for various ovarian, colon carcinoma, and breast cancer cell lines between 10- and 50-fold.
Wylie et al. demonstrated that ABL-001 can lead to complete tumor regression in mice implanted with CML and ALL patient-derived cell lines (32). Remarkably, this inhibitor was also active at relatively low nanomolar concentrations against every known mutation in the catalytic site of ABL kinases, including T315I. The T315I mutation, also known as the “gatekeeper mutation,” commonly emerges after treatment with traditional kinase inhibitors such as imatinib and is thought to decrease ABL kinases’ affinity for inhibitor binding while increasing their oncogenic potential. ABL-001 has not resulted in T315I emergence thus far. In Hughes et al.’s study with 141 CML patients, only 4 out of 86 screened patients developed mutations – and all mutations were found within the myristoyl-binding pocket (35). However, the doses of ABL-001 required to achieve “complete cytogenic and major molecular responses” in patients with a T315I mutation were higher than those without this mutation (35). This relative lack of sensitivity was also found preclinically (32) and the mechanisms for this are not currently clear. Encouragingly, ABL-001 was the recent subject of a Phase 1 dose escalation and expansion study for patients with advanced (resistant) solid tumors (NCT03292783), closed on December 27, 2019 (36). Preliminary results demonstrate that ABL-001 displays anti-tumor activity including modulations to VEGF/VEGFR and DLL4/Notch1 pathways and is well tolerated up to 7.5 mg/kg without significant adverse events (37).
Metastatic malignancies
Four seminal papers have indicated that allosteric ABL inhibitors may be of utility in cancer metastasis (38–41). ABL kinase activity is required for cell invasion, migration, and motility, and ABL1/ABL2 are involved in actin polymerization and the formation of invadopodia (7). Accordingly, work by Gu et al. demonstrated that ABL kinases are required for non-small cell lung carcinoma (NSCLC) metastasis (38). This group showed that genetic knockdown and pharmacological allosteric inhibition of ABL1/ABL2 via GNF-5 can reduce expression of metastasis gene signatures and decrease metastasis in vivo, thereby increasing survival in multiple lung cancer cell lines.
More recently, GNF-5 was found to significantly reduce the formation and maturation of invadopodium precursors in breast cancer cells, along with their actin polymerization, matrix degradation, and 3D invasion (39). Tripathi et al. showed that treatment of at least one human melanoma cell line, WM3248, with GNF-2 can decrease secretion of the cathepsin proteases B and L, which are involved in matrix degradation and cancer cell invasion (40). Lastly, Hoj et al. identified a TAZ-AXL-ABL2 autocrine signaling axis that is required for brain metastases of lung adenocarcinoma – and that ABL-001, which can penetrate the blood-brain barrier, impairs such metastasis (41). ABL-001 treatment also increased the median overall survival of tumor-bearing mice in their study by approximately 20 days (41).
Combination therapy
Recent studies suggest that allosteric ABL inhibitors may synergize with more traditional therapies to improve outcomes for various cancers (28,32,42–45). In 2006, treatment with both GNF-2 and imatinib was shown to inhibit the growth of BCR-ABL1-expressing Ba/F3 cells more effectively than treatment with either inhibitor alone (42). Moreover, Zhang et al. demonstrated that combining GNF-2 and imatinib reduced the number of resistant Ba/F3 cell clones by over 90% in their study (28). This same group discovered that combining GNF-5 with nilotinib resulted in additive inhibition of both wild-type and T315I-mutated BCR-ABL1. After combination treatment, mice transplanted with T315I-mutated, BCR-ABL1-transduced bone marrow also survived significantly longer than those treated with GNF-5 or nilotinib as single agents alone (28). Similarly promising results with GNF-5 plus docetaxel combination therapy were observed in a lung cancer model (44). This combination treatment significantly decreased lung tumor progression compared to vehicle or singly treated mice and significantly reduced high-level expression of nuclear YAP1, a transcriptional regulatory factor associated with pro-proliferative pathways in tumorigenesis (44).
Finally, at least three established publications offer insight into the potential utility of ABL-001 in combination therapy (32,43,45). Eadie et. al. generated a line of ABL-001-resistant K562 (human CML) cells (43). Intriguingly, treating these cells with ABL-001 in combination with clinically achievable doses of either imatinib or nilotinib reversed the group’s engineered resistance phenotype (43). Inhibiting ABCB1 and ABCG2, two drug efflux transporters, with cyclosporine (ABCB1) and Ko143 (ABCG2) likewise reversed this ABL-001-resistant phenotype in vitro (43). Wylie et al. demonstrated that ABL-001 and nilotinib have complementary resistance profiles, suggesting that their combination may be effective in delaying resistance in human patients (32). To this point, combination treatment with ABL-001 and nilotinib evoked durable complete regressions of CML in their study in vivo. Treatment with either ABL-001 or nilotinib as single agents also led to tumor regression, but resistance due to T315I mutations after nilotinib treatment and resistance due to A337V and P223S mutations after ABL-001 treatment ultimately emerged (32). Lastly, Elrashedy et al. demonstrated that allosteric binding causes structural modifications of ABL kinases, allowing for increased interactions upon nilotinib binding (45). Co-bound BCR-ABL1 induced more stable and compact protein structures than when singly bound (45).
Two clinical trials are now testing the effects of ABL-001 in combination for cancer patients. NCT02081378 is evaluating ABL-001 vs. ABL-001 plus nilotinib, imatinib, or dasatinib, and NCT03578367 is assessing ABL-001 plus imatinib vs. imatinib/nilotinib alone (46,47). Preliminary results for NCT02081378 suggest first that ABL-001 is well tolerated as monotherapy, either achieving or maintaining a major molecular response (MMR) in 48% of heavily pretreated CML patients (including some harboring the T315I mutation) (35). Second, these results suggest that all combinations of ABL-001 with nilotinib, imatinib, or dasatinib are promisingly efficacious (48,49). Though adverse effects have been common (affecting >80% of the patient population), 8/19 patients have achieved MMR and 3/20 a molecular response (MR) when administered ABL-001 plus imatinib. Approximately 35% of patients treated with ABL-001 plus either nilotinib or dasatinib achieved MMR by 48 weeks (48,49). No preliminary findings are yet available for NCT03578367, which is estimated to be completed by June 2022 (47).
Conclusions
ABL kinase inhibitors including imatinib have transformed the world of cancer treatment, improving overall survival with CML, in particular, by decades. However, off-target, adjuvant toxicities of treatment with these inhibitors are serious issues, and resistance has become a major problem in recent years. The allosteric ABL inhibitors GNF-2, GNF-5, and ABL-001 are novel, mostly preclinical drugs that may overcome these concerns. Many contemporary studies have demonstrated their potential efficacy for primary malignancies, in metastasis, and as a key player in combination therapy with other small molecules. Given these encouraging data, further research with these inhibitors is warranted. Future studies will investigate the efficacy of allosteric ABL kinase inhibitors simultaneously as first-line therapy for solid tumors and as salvage therapy when more established tyrosine kinase inhibitors such imatinib fail. The results of ongoing clinical trials with ABL-001 will be especially elucidating, as will the continued preclinical evaluation of GNF-2 and GNF-5.
Supplementary Material
Acknowledgements:
Funding (all to EMT): NIH (R03 NS114629–01), NIH (P50 CA190991), NIH (P30 CA014236), Chetna & Meena Trust. The authors wish to thank Anne Marie Pendergast, PhD, and Jacob Hoj for their helpful insights.
Footnotes
Conflicts of Interest:
The authors declare no potential conflicts of interest.
References
- 1.Khatri A, Wang J, Pendergast AM. Multifunctional Abl kinases in health and disease. J Cell Sci 2016;129(1):9–16 doi 10.1242/jcs.175521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Klein A, van Kessel AG, Grosveld G, Bartram CR, Hagemeijer A, Bootsma D, et al. A cellular oncogene is translocated to the Philadelphia chromosome in chronic myelocytic leukaemia. Nature 1982;300(5894):765–7 doi 10.1038/300765a0. [DOI] [PubMed] [Google Scholar]
- 3.Morita S, Villalta SA, Feldman HC, Register AC, Rosenthal W, Hoffmann-Petersen IT, et al. Targeting ABL-IRE1alpha Signaling Spares ER-Stressed Pancreatic beta Cells to Reverse Autoimmune Diabetes. Cell Metab 2017;25(5):1207 doi 10.1016/j.cmet.2017.04.026. [DOI] [PubMed] [Google Scholar]
- 4.Contreras PS, Gonzalez-Zuniga M, Gonzalez-Hodar L, Yanez MJ, Dulcey A, Marugan J, et al. Neuronal gene repression in Niemann-Pick type C models is mediated by the c-Abl/HDAC2 signaling pathway. Biochim Biophys Acta 2016;1859(2):269–79 doi 10.1016/j.bbagrm.2015.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Song GJ, Rahman MH, Jha MK, Gupta DP, Park SH, Kim JH, et al. A Bcr-Abl Inhibitor GNF-2 Attenuates Inflammatory Activation of Glia and Chronic Pain. Front Pharmacol 2019;10:543 doi 10.3389/fphar.2019.00543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang J, Pendergast AM. The Emerging Role of ABL Kinases in Solid Tumors. Trends Cancer 2015;1(2):110–23 doi 10.1016/j.trecan.2015.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Greuber EK, Smith-Pearson P, Wang J, Pendergast AM. Role of ABL family kinases in cancer: from leukaemia to solid tumours. Nat Rev Cancer 2013;13(8):559–71 doi 10.1038/nrc3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Malone A, Langabeer S, O’Marcaigh A, Storey L, Bacon CL, Smith OP. A Doctor(s) dilemma: ETV6-ABL1 positive acute lymphoblastic leukaemia. Brit J Haematol 2010;151(1):101–2 doi 10.1111/j.1365-2141.2010.08323.x. [DOI] [PubMed] [Google Scholar]
- 9.Quentmeier H, Cools J, MacLeod RAF, Marynen P, Uphoff CC, Drexler HG. e6-a2 BCR-ABL1 fusion in T-cell acute lymphoblastic leukemia. Leukemia 2005;19(2):295–6 doi 10.1038/sj.leu.2403595. [DOI] [PubMed] [Google Scholar]
- 10.Ritchie DS, McBean M, Westerman DA, Kovalenko S, Seymour JF, Dobrovic A. Complete molecular response of e6a2 BCR-ABL-positive acute myeloid leukemia to imatinib then dasatinib. Blood 2008;111(5):2896–8 doi 10.1182/blood-2007-08-107508. [DOI] [PubMed] [Google Scholar]
- 11.Hochhaus A, Larson RA, Guilhot F, Radich JP, Branford S, Hughes TP, et al. Long-Term Outcomes of Imatinib Treatment for Chronic Myeloid Leukemia. N Engl J Med 2017;376(10):917–27 doi 10.1056/NEJMoa1609324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milojkovic D, Apperley J. Mechanisms of Resistance to Imatinib and Second-Generation Tyrosine Inhibitors in Chronic Myeloid Leukemia. Clin Cancer Res 2009;15(24):7519–27 doi 10.1158/1078-0432.CCR-09-1068. [DOI] [PubMed] [Google Scholar]
- 13.Heinrich MC, Corless CL, Blanke CD, Demetri GD, Joensuu H, Roberts PJ, et al. Molecular correlates of imatinib resistance in gastrointestinal stromal tumors. J Clin Oncol 2006;24(29):4764–74 doi 10.1200/JCO.2006.06.2265. [DOI] [PubMed] [Google Scholar]
- 14.Eilers G, Czaplinski JT, Mayeda M, Bahri N, Tao D, Zhu M, et al. CDKN2A/p16 Loss Implicates CDK4 as a Therapeutic Target in Imatinib-Resistant Dermatofibrosarcoma Protuberans. Mol Cancer Ther 2015;14(6):1346–53 doi 10.1158/1535-7163.MCT-14-0793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Metzgeroth G, Walz C, Erben P, Popp H, Schmitt-Graeff A, Haferlach C, et al. Safety and efficacy of imatinib in chronic eosinophilic leukaemia and hypereosinophilic syndrome: a phase-II study. Br J Haematol 2008;143(5):707–15 doi 10.1111/j.1365-2141.2008.07294.x. [DOI] [PubMed] [Google Scholar]
- 16.Metzgeroth G, Erben P, Martin H, Mousset S, Teichmann M, Walz C, et al. Limited clinical activity of nilotinib and sorafenib in FIP1L1-PDGFRA positive chronic eosinophilic leukemia with imatinib-resistant T674I mutation. Leukemia 2012;26(1):162–4 doi 10.1038/leu.2011.181. [DOI] [PubMed] [Google Scholar]
- 17.Kantarjian HM, Giles F, Gattermann N, Bhalla K, Alimena G, Palandri F, et al. Nilotinib (formerly AMN107), a highly selective BCR-ABL tyrosine kinase inhibitor, is effective in patients with Philadelphia chromosome-positive chronic myelogenous leukemia in chronic phase following imatinib resistance and intolerance. Blood 2007;110(10):3540–6 doi 10.1182/blood-2007-03-080689. [DOI] [PubMed] [Google Scholar]
- 18.Mughal TI, Schrieber A. Principal long-term adverse effects of imatinib in patients with chronic myeloid leukemia in chronic phase. Biologics 2010;4:315–23 doi 10.2147/BTT.S5775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mauro MJ. Defining and managing imatinib resistance. Hematology Am Soc Hematol Educ Program 2006:219–25 doi 10.1182/asheducation-2006.1.219. [DOI] [PubMed] [Google Scholar]
- 20.Demetri GD, Lo Russo P, MacPherson IR, Wang D, Morgan JA, Brunton VG, et al. Phase I dose-escalation and pharmacokinetic study of dasatinib in patients with advanced solid tumors. Clin Cancer Res 2009;15(19):6232–40 doi 10.1158/1078-0432.CCR-09-0224. [DOI] [PubMed] [Google Scholar]
- 21.Daud AI, Krishnamurthi SS, Saleh MN, Gitlitz BJ, Borad MJ, Gold PJ, et al. Phase I study of bosutinib, a src/abl tyrosine kinase inhibitor, administered to patients with advanced solid tumors. Clin Cancer Res 2012;18(4):1092–100 doi 10.1158/1078-0432.CCR-11-2378. [DOI] [PubMed] [Google Scholar]
- 22.Demetri GD, Casali PG, Blay JY, von Mehren M, Morgan JA, Bertulli R, et al. A phase I study of single-agent nilotinib or in combination with imatinib in patients with imatinib-resistant gastrointestinal stromal tumors. Clin Cancer Res 2009;15(18):5910–6 doi 10.1158/1078-0432.CCR-09-0542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reddy EP, Aggarwal AK. The ins and outs of bcr-abl inhibition. Genes Cancer 2012;3(5–6):447–54 doi 10.1177/1947601912462126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hantschel O Allosteric BCR-ABL inhibitors in Philadelphia chromosome-positive acute lymphoblastic leukemia: novel opportunities for drug combinations to overcome resistance. Haematologica 2012;97(2):157–9 doi 10.3324/haematol.2012.061812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Eide CA, Zabriskie MS, Savage Stevens SL, Antelope O, Vellore NA, Than H, et al. Combining the Allosteric Inhibitor Asciminib with Ponatinib Suppresses Emergence of and Restores Efficacy against Highly Resistant BCR-ABL1 Mutants. Cancer Cell 2019;36(4):431–43 e5 doi 10.1016/j.ccell.2019.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hantschel O, Nagar B, Guettler S, Kretzschmar J, Dorey K, Kuriyan J, et al. A myristoyl/phosphotyrosine switch regulates c-Abl. Cell 2003;112(6):845–57 doi 10.1016/s0092-8674(03)00191-0. [DOI] [PubMed] [Google Scholar]
- 27.2019. Search results for: abl-001; Also searched for Asciminib. <https://clinicaltrials.gov/ct2/results?cond=&term=abl-001&cntry=&state=&city=&dist=>.
- 28.Zhang J, Adrian FJ, Jahnke W, Cowan-Jacob SW, Li AG, Iacob RE, et al. Targeting Bcr-Abl by combining allosteric with ATP-binding-site inhibitors. Nature 2010;463(7280):501–6 doi 10.1038/nature08675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Mauro MJ, Lang F, Kim D-W, Cortes JE, Hughes TP, Hochhaus A, et al. Clinical development of asciminib (ABL001) in chronic myeloid leukemia (CML): A randomized phase 3 study vs. bosutinib. Journal of Clinical Oncology 2018;36(15_suppl):TPS7081–TPS doi 10.1200/JCO.2018.36.15_suppl.TPS7081. [DOI] [Google Scholar]
- 30.Bildik G, Acilan C, Sahin GN, Karahuseyinoglu S, Oktem O. C-Abl is not activated in DNA damage-induced and Tap63-mediated oocyte apoptosis in human ovary. Cell Death Dis 2018;9(10):943 doi 10.1038/s41419-018-1026-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Choi Y, Seeliger MA, Panjarian SB, Kim H, Deng X, Sim T, et al. N-myristoylated c-Abl tyrosine kinase localizes to the endoplasmic reticulum upon binding to an allosteric inhibitor. J Biol Chem 2009;284(42):29005–14 doi 10.1074/jbc.M109.026633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wylie AA, Schoepfer J, Jahnke W, Cowan-Jacob SW, Loo A, Furet P, et al. The allosteric inhibitor ABL001 enables dual targeting of BCR-ABL1. Nature 2017;543(7647):733–7 doi 10.1038/nature21702. [DOI] [PubMed] [Google Scholar]
- 33.Kim SH, Kim MO, Kim KR. Anti-growth Effects of Imatinib and GNF5 via Regulation of Skp2 in Human Hepatocellular Carcinoma Cells. J Cancer Prev 2018;23(4):170–5 doi 10.15430/JCP.2018.23.4.170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Malki WH, Gouda AM, Ali HEA, Al-Rousan R, Samaha D, Abdalla AN, et al. Structural-based design, synthesis, and antitumor activity of novel alloxazine analogues with potential selective kinase inhibition. Eur J Med Chem 2018;152:31–52 doi 10.1016/j.ejmech.2018.04.029. [DOI] [PubMed] [Google Scholar]
- 35.Hughes TP, Mauro MJ, Cortes JE, Minami H, Rea D, DeAngelo DJ, et al. Asciminib in Chronic Myeloid Leukemia after ABL Kinase Inhibitor Failure. New Engl J Med 2019;381(24):2315–26 doi 10.1056/NEJMoa1902328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.2017. A Study of NOV1501 (ABL001) in Subject With Advanced Solid Tumors. <https://clinicaltrials.gov/ct2/show/study/NCT03292783?term=abl-001&draw=2&rank=5>. [Google Scholar]
- 37.Lee J, Kim S, Lee SJ, Park SH, Park JO, Ha E, et al. Phase 1a study results investigating the safety and preliminary efficacy of ABL001 (NOV1501), a bispecific antibody targeting VEGF and DLL4 in metastatic gastrointestinal (GI) cancer. Journal of Clinical Oncology 2019;37(15) doi DOI 10.1200/JCO.2019.37.15_suppl.3023. [DOI] [Google Scholar]
- 38.Gu JJ, Rouse C, Xu X, Wang J, Onaitis MW, Pendergast AM. Inactivation of ABL kinases suppresses non-small cell lung cancer metastasis. JCI Insight 2016;1(21):e89647 doi 10.1172/jci.insight.89647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meirson T, Genna A, Lukic N, Makhnii T, Alter J, Sharma VP, et al. Targeting invadopodia-mediated breast cancer metastasis by using ABL kinase inhibitors. Oncotarget 2018;9(31):22158–83 doi 10.18632/oncotarget.25243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tripathi R, Fiore LS, Richards DL, Yang Y, Liu J, Wang C, et al. Abl and Arg mediate cysteine cathepsin secretion to facilitate melanoma invasion and metastasis. Sci Signal 2018;11(518) doi 10.1126/scisignal.aao0422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hoj JP, Mayro B, Pendergast AM. A TAZ-AXL-ABL2 Feed-Forward Signaling Axis Promotes Lung Adenocarcinoma Brain Metastasis. Cell Rep 2019;29(11):3421–34 e8 doi 10.1016/j.celrep.2019.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Adrián FJ, Ding Q, Sim T, Velentza A, Sloan C, Liu Y, et al. Allosteric inhibitors of Bcr-abl-dependent cell proliferation. Nat Chem Biol 2006;2(2):95–102 doi 10.1038/nchembio760. [DOI] [PubMed] [Google Scholar]
- 43.Eadie LN, Saunders VA, Branford S, White DL, Hughes TP. The new allosteric inhibitor asciminib is susceptible to resistance mediated by ABCB1 and ABCG2 overexpression in vitro. Oncotarget 2018;9(17):13423–37 doi 10.18632/oncotarget.24393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Khatri A, Gu JJ, McKernan CM, Xu X, Pendergast AM. ABL kinase inhibition sensitizes primary lung adenocarcinomas to chemotherapy by promoting tumor cell differentiation. Oncotarget 2019;10(20):1874–86 doi 10.18632/oncotarget.26740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Elrashedy AA, Ramharack P, Soliman MES. The Perplexity of Synergistic Duality: Inter-Molecular Mechanisms of Communication in BCR-ABL1. Anticancer Agents Med Chem 2019. doi 10.2174/1871520619666190620120144. [DOI] [PubMed] [Google Scholar]
- 46.2014. A Phase I Study of Oral ABL001 in Patients With CML or Ph+ ALL. <https://clinicaltrials.gov/ct2/show/NCT02081378>.
- 47.2018. Study of Efficacy and Safety of Asciminib in Combination With Imatinib in Patients With Chronic Myeloid Leukemia in Chronic Phase (CML-CP). <https://clinicaltrials.gov/ct2/show/NCT03578367>.
- 48.DeAngelo DJ, Mauro MJ, Kim DW, Cortes J, Rea D, Hughes TP, et al. Combination of Asciminib plus Nilotinib or Asciminib plus Dasatinib in Previously Treated Chronic Myeloid Leukemia (CML) Patients: Phase 1 Study Results. Cl Lymph Myelom Leuk 2019;19:S290–S1 doi DOI 10.1016/j.clml.2019.07.236. [DOI] [Google Scholar]
- 49.Talpaz M, Cortes J, Lang F, Kim DW, Rea D, Mauro MJ, et al. Combination of Asciminib, a Novel and Specific BCR-ABL1 Inhibitor, Plus Imatinib in Previously Treated Chronic Myeloid Leukemia (CML) Patients: Phase 1 Study Results. Cl Lymph Myelom Leuk 2019;19:S287–S8 doi DOI 10.1016/j.clml.2019.07.231. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.