

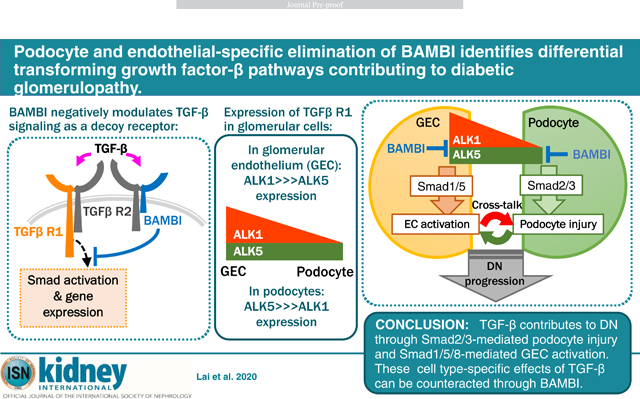
Abstract
Transforming growth factor-β (TGF-β) is a central mediator of diabetic nephropathy. The effect of TGF-β, mediated by the type I TGF-β receptor, ALK5, and subsequent Smad2/3 activation results in podocyte apoptosis and loss. Previously, we demonstrated that the genetic deletion of the BMP and Activin Membrane-Bound Inhibitor (BAMBI), a negative modulator TGF-β signaling, accelerates diabetic nephropathy in mice. This was associated with heightened ALK1-mediated activation of Smad1/5 in the glomerular endothelial cells (ECs). Therefore, to evaluate the glomerular cell-specific effects of TGF-β in diabetic nephropathy we examined the effects of the podocyte- or EC-specific loss of Bambi (Pod-Bambi−/− or EC-Bambi−/−) in streptozotocin-induced diabetic mice with endothelial nitric oxide synthase deficiency. Interestingly, although hyperglycemia and body weight loss were similar in all groups of diabetic mice, significant hypertension was present only in the diabetic EC-Bambi−/− mice. While the podocyte or EC-specific loss of BAMBI both accelerated the progression of diabetic nephropathy, the worsened podocyte injury and loss observed in the diabetic Pod-Bambi−/− mice were associated with enhanced Smad3 activation. Increased Smad1/5 activation and EC proliferation were apparent only in the glomeruli of diabetic EC-Bambi−/− mice. The enhanced Smad1/5 activation in diabetic EC-Bambi−/− mice was associated with increased glomerular expression of plasmalemma vesicle-associated protein, pointing to the involvement of immature or dedifferentiated glomerular ECs in diabetic nephropathy. Notably, diabetic EC-Bambi−/− mice displayed podocyte injury and loss that were comparable to diabetic Pod-Bambi−/− mice. Thus, our results highlight the glomerular cell-specific contribution of TGF-β signaling and the intricate cross-talk between injured glomerular cells in the progression of diabetic nephropathy.
Keywords: Diabetic nephropathy, TGF-β, podocyte, glomerular endothelial cells, PLVAP
Graphical Abstract
INTRODUCTION
Transforming growth factor-β (TGF-β) plays a major role in the development and progression of diabetic nephropathy (DN). Inhibition of TGF-β mitigates mesangial matrix overexpression, renal hypertrophy, and podocyte loss, and eventual glomerulosclerosis in rodent models of DN1–3. The signaling pathways involved have been attributed mostly to the canonical TGF-β signaling, which involves the binding of TGF-β to its cognate receptors (i.e., TGFBR1/ALK5, type I receptor, and TGFBR2, type II receptor) and the activation of Smad2/3 signaling pathways4. TGF-β activation of podocytes contributes to their loss, which is considered a reliable indicator for the progression of glomerulosclerosis5, 6. Surprisingly, while interfering with ALK5/Smad3 signaling in experimental diabetic animal studies markedly diminished the glomerular matrix changes, it had minimal effects on preventing the development of proteinuria in experimental models of DN7, 8.
Consistent with the role of TGF-β in DN, we recently showed that the genetic deletion of BMP and Activin Membrane-Bound Inhibitor (BAMBI), an endogenous pseudo-receptor inhibitor for pan TGF-β signaling9, worsened the diabetic glomerular injury in mice10. Interestingly, the worsened proteinuria and DN phenotype in Bambi−/− mice was associated with increased ACVRL1/ALK1 type I receptor-mediated Smad1/5 activation10, suggesting that the inhibition of both ALK1 and ALK5 pathways is necessary to curtail the TGF-β’s action on the diabetic glomerular injury. In this context, it is of interest that TGF-β’s effect on endothelial cells is mediated also by ALK1, a type I receptor whose expression is restricted to endothelial cells. ALK1 signaling results in the activation of Smad1/5-mediated signaling cascade, which works in an opposing manner to the ALK5-Smad2/3 pathway to promote endothelial cell activation and neo-angiogenesis11–14. Indeed, global endothelial dysfunction is closely associated with diabetes,15 and abnormal glomerular angiogenesis and decreased glomerular endothelial fenestration have been described in human and experimental models of DN14–19. The importance of endothelial TGF-β signaling in DN pathogenesis is further corroborated by our recent observation that the attenuation of TGF-β signaling by the elimination of leucine-rich α−2-glycoprotein 1 (LRG1) significantly mitigated the diabetic glomerulopathy and proteinuria in mice14. Thus, we posited that TGF-β might contribute to diabetic glomerulopathy in a dual manner: by activating the canonical ALK5-Smad2/3 pathway in podocytes to favor their loss and activating the ALK1-Smad1/5 pathway in GECs to favor their proinflammatory and proliferative signaling, resulting in glomerular leakiness and neo-angiogenesis. As both of these pathways would be enhanced by BAMBI loss, we examined the effects of podocyte-specific or endothelial cell-specific loss of BAMBI to ascertain the distinct contribution of ALK5 and ALK1 signaling in DN.
RESULTS
Glomerular expression of TGF-β signaling components
A prominent BAMBI mRNA transcript was observed in the glomeruli as compared to the tubulointerstitium in both mice and human kidney10, 20, 21, but its glomerular cell-specific expression was not known. To examine the glomerular cell-type expression of Bambi mRNA in comparison to other TGF-β receptor signaling components, we took advantage of the glomerular transcriptome datasets from eNOS-deficient mice, as used in this study. We utilized the bulk RNA sequencing (RNA-seq) data of isolated glomeruli, podocytes22, and GECs18, as well as the single-cell RNAseq (scRNA-seq) dataset of isolated glomerular cells from control and streptozotocin-induced diabetic eNOS−/− mice23. The bulk RNAseq analysis of isolated cells showed the expression of Bambi in both podocytes and GECs, with expression levels similar to Tgfbr1 (Alk5) mRNA (Fig. 1A). In comparison to these, the expression of both Acvrl1 (Alk1) and Tgfbr2 showed a much higher expression in the isolated glomeruli, likely as a result of their prominent expressions in GECs (Fig. 1A). The scRNA-seq from the same mouse model confirmed these observations in that similar expression of Bambi was observed in GECs, podocytes, and mesangial cells (Fig. 1B) The mean expression of Alk5 was higher in podocytes in comparison to the other glomerular cells, which further increased in diabetic kidneys (Fig. 1B). As expected, the expression of Alk1, typically expressed in the vascular endothelial cells, was markedly higher in GECs in comparison to the other glomerular cells in both control and diabetic kidneys, and interestingly, so was the expression of Tgfbr2 (Fig. 1B). The glomerular scRNA-seq dataset from Karaiskos et al24 of normal mouse kidneys corroborated these observations (Supp. Fig. 1A). Because BMP9 and BMP10 can also signal through the ALK1 receptor in addition to TGF-β, we also evaluated the expression of Tgfb1, Bmp9, and Bmp10 from the bulk RNA-seq analysis of isolated glomerular cells. Tgfb1 expression was present in isolated glomeruli, which appeared to be due to the predominant expression in GECs, whereas Bmp9 and Bmp10 mRNAs were barely detectable in isolated glomeruli, podocytes, or GECs (Supp. Fig. 1B). Although the activity of TGF-β is regulated largely by post-transcriptional modification25, the significant expression of Tgfb1 in glomeruli and the lack of Bmp9 and Bmp10 expression suggests that the glomerular ALK1 signaling occurs mainly through TGF-β1.
Figure 1. Expression of TGF-β signaling components in glomerular cells.

(A) Normalized expression of Bambi, Tgfbr2, Tgfbr1 (Alk5), and Acvrl1 (Alk1) mRNAs from bulk RNA-seq analyses of isolated glomeruli (n=4 samples), podocytes (n=4 samples), and GECs (n=3 samples) from eNOS−/− mice. Each sample consists of pooled cells from 3–4 mice (Fu et al., 2015 and Fu et al., 2018). RPKM, reads per kilobase million. (B) Expression of Tgfrb1 (Alk5), Acvrl1 (Alk1), Tgbf2, and Bambi mRNAs from scRNA-seq analysis of isolated glomerular cells from control and diabetic eNOS−/− mice (Fu et al. 2019). MCs, mesangial cells; Ctrl, control mice; DM, diabetic mice. (C) Western blot analysis of TGF-β signaling components in immortalized murine podocytes and GECs after stimulation with TGF-β (10ng/mL) for 30 minutes.
We further verified the expression of ALK5 and ALK1 receptors and the downstream regulatory Smad activation in immortalized murine podocytes26 (mPod) and glomerular endothelial cells (mGEC)18 (Fig. 1C). In these cultured cell lines, stimulation with TGF-β1 (10ng/mL) resulted in robust phosphorylation of Smad2 (Ser465/Ser467) in mPods, while substantially less Smad2 phosphorylation was observed in mGECs (Fig. 1C). However, the TGF-β1 induced Smad5 phosphorylation (Ser463/465) was only observed in mGECs (Fig. 1C). Therefore, we hypothesized that the increased TGF-β signaling in DN would predominantly elicit ALK5-Smad2/3 in podocytes and ALK1-Smad1/5 signaling in GECs. BAMBI, expressed in both cell types, would negatively modulate both pathways, and its loss would thereby amplify respective pathways in DN.
Generation of podocyte- and EC-specific loss of Bambi in mice
To test the above hypothesis, we generated a podocyte- and endothelial-specific Bambi-null mice. Podocyte-specific Bambi-null mice were generated by crossing Bambifl/fl mice with transgenic mice with podocin promoter-driven Cre expression (Nphs2-Cre). As there is no GEC-specific Cre transgenic model currently available, we generated pan-endothelial Bambi-null mice by crossing Bambifl/fl mice with transgenic mice with VE-Cadherin promoter-driven Cre expression (Cdh5-Cre). As Nphs2-Cre;Bambifl/fl and Cdh5-Cre;Bambifl/fl mice were in the relatively DN-resistant mixed C57BL/6J background, they were further crossed with eNOS−/− mice for the aggravation of the DN phenotype27, 28. Similar to the global Bambi−/− mice20, 21, 29, Nphs2-Cre;Bambifl/fl;eNOS−/− (hereafter referred to as Pod-Bambi−/−) mice and Cdh5-Cre;Bambifl/fl;eNOS−/− (EC-Bambi−/−) mice showed no major phenotype or abnormalities at baseline (data not shown). Lacking a reliable commercial antibody that detects the murine BAMBI protein by western blot or immunostaining analyses, we employed two different methods to confirm the cell-specific ablation of Bambi in Pod-Bambi−/− and EC-Bambi−/− mice. We first tested the cell type-specific Cre recombinase activity by crossing the Pod-Bambi−/− and EC-Bambi−/− mice with mTmG Cre reporter mice, resulting in Pod-Bambi+/−;mTmG and EC-Bambi+/−;mTmG mice. As Cre recombination results in the expression of enhanced green fluorescent protein (eGFP) from the mTmG reporter allele, in Pod-Bambi+/−;mTmG kidneys eGFP were evident in podocytes that co-expressed Wilms tumor-1 (WT-1), but not CD31+ (Supp. Fig. 2A, top panels). Conversely, eGFP expression in EC-Bambi+/−;mTmG kidneys co-localized with CD31 and not found in WT1+ cells (Supp. Fig. 2, bottom panels). Secondly, Bambi mRNA levels were assessed in primary podocytes that were cultivated from control and Pod-Bambi−/− mice, and in primary GECs from control and EC-Bambi−/− mice. Assessment of Bambi transcript by quantitative PCR (qPCR) confirmed the loss of Bambi in primary podocytes established from Pod-Bambi−/− mice in comparison to those from Bambi+/+ mice (Supp. Fig. 2B). A similar extent of reduction in Bambi mRNA was observed in primary GECs established from EC-Bambi−/− mice in comparison to those from Bambi+/+ mice (Supp. Fig. 2B). Together with the above data, these results confirmed an effective cell type-specific ablation of Bambi in Pod-Bambi−/− and EC-Bambi−/− mice.
Podocyte- and EC-specific loss of Bambi exacerbates diabetic glomerulopathy
Diabetes (DM) was induced in Bambi+/+, Pod-Bambi−/−, and EC-Bambi−/− mice with low-dose streptozotocin injections (+STZ). Citrate buffer vehicle-injected littermates (−STZ) were used as controls. All mice were euthanized at 16 weeks post-injection. The levels of hyperglycemia and body weight loss were similar between all three diabetic groups (Fig. 2A–B). Interestingly, at 16 weeks post DM, significant hypertension was observed only in the diabetic EC-Bambi−/− mice and not in the other diabetic groups (Supp. Table 1). This was somewhat surprising since we did not observe any significant changes in blood pressure in our previous study of diabetic mice with global BAMBI loss10, and since eNOS deficiency alone did not affect the mean blood pressure in the diabetic Bambi+/+ mice. Thus, the significant DM-induced hypertension only in EC-Bambi−/− mice is likely due to the increased pan-endothelial ALK1 signaling in the absence of BAMBI and the contribution of eNOS deficiency by mechanisms yet to be determined.
Figure 2. Albuminuria is worsened in the diabetic Pod- and EC-Bambi−/− mice.
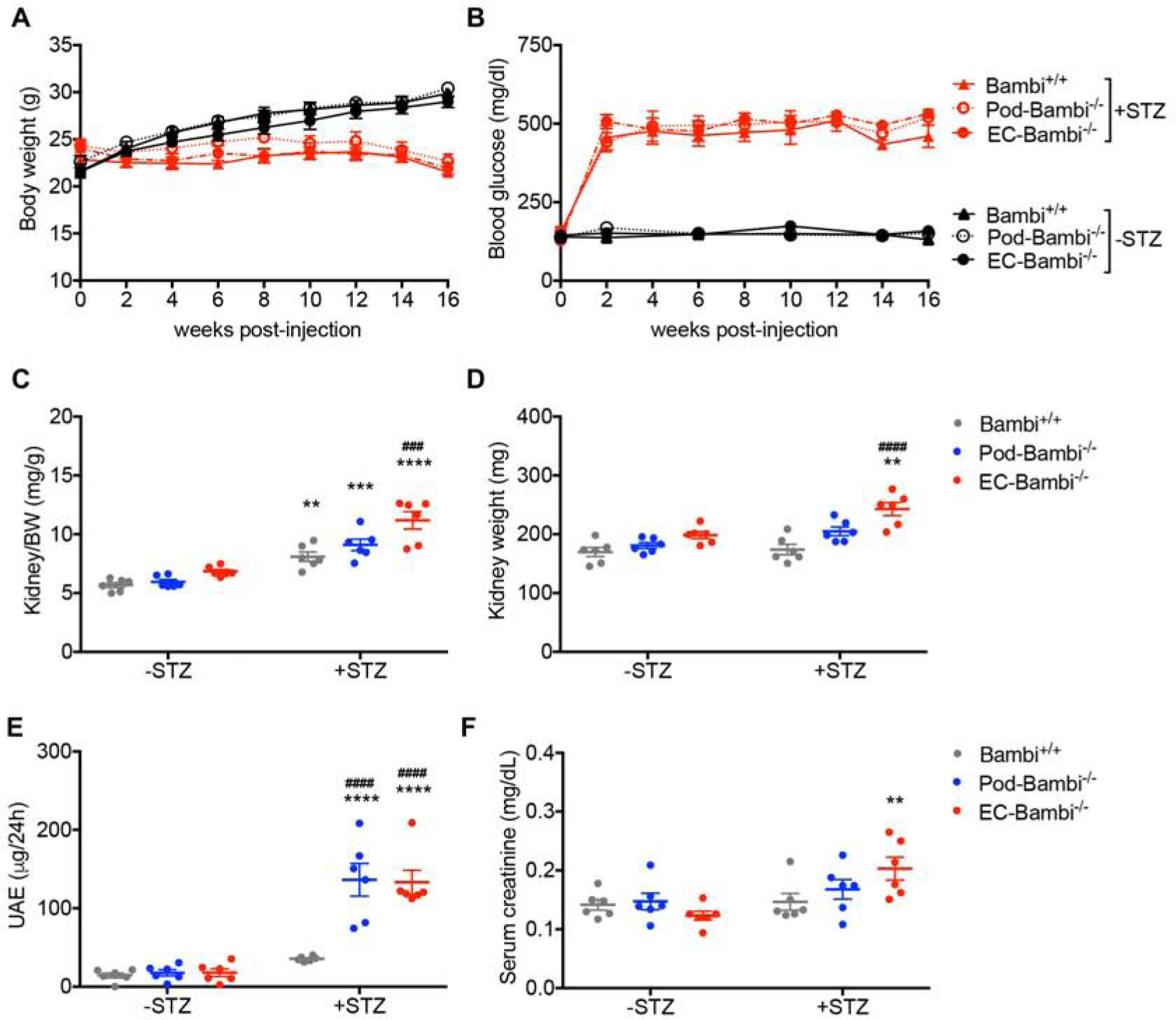
(A–B) Bodyweight (A) and blood glucose level (B) of control (−STZ) or diabetic (+STZ) Bambi+/+, Pod-Bambi−/−, and EC-Bambi−/− mice. (C–D) Kidney-to-body weight (BW) ratio (C) and kidney weight at 16 weeks post-DM. (E–F) 24-hour urinary albumin excretion (UAE) (E) and serum creatinine levels (F) at 16 weeks post-DM. n=6 mice per group. **p<0.01, ***p<0.001, and ****p<0.0001 vs. respective −STZ controls; ###p<0.001 and ####p<0.0001 vs. diabetic Bambi+/+ mice (2-way ANOVA with Tukey’s post hoc analysis).
The kidney-to-body weight ratios increased in all diabetic groups at 16 weeks post-DM (Fig. 2C), consistent with the loss of body weight in diabetic mice in comparison to control mice (Fig. 2A), but also due to the increase in kidney weights especially in the diabetic EC-Bambi−/− mice (Fig. 2D). The level of albuminuria, as determined by 24-hour albumin excretion, was only modestly increased in the diabetic Bambi+/+ mice, but a robust increase was observed in the diabetic Pod-Bambi−/− and EC-Bambi−/− mice (Fig. 2E). This is consistent with the enhanced proteinuria in mice with a global BAMBI loss as reported previously10. Both serum creatinine and BUN levels were significantly increased only in the diabetic EC-Bambi−/− mice, whereas only a mild, non-significant increase was observed in the other diabetic groups (Fig. 2F and Supp. Table 2), indicating that the endothelial loss of Bambi markedly worsened renal function in DN. Histopathological analysis of periodic acid-Schiff (PAS)-stained kidney sections showed enlarged glomeruli and increased mesangial matrix in all diabetic mice in comparison to the non-diabetic controls (Fig. 3A–B). However, both glomerular hypertrophy and mesangial matrix expansion were accentuated in the diabetic Pod-Bambi−/− and even more so in the diabetic EC-Bambi−/− mice, consistent with the worsening of DN in absence of either podocyte of endothelial expression of BAMBI (Fig. 3A–B).
Figure 3. Glomerular injury is worsened in the diabetic Pod- and EC-Bambi−/− mice.
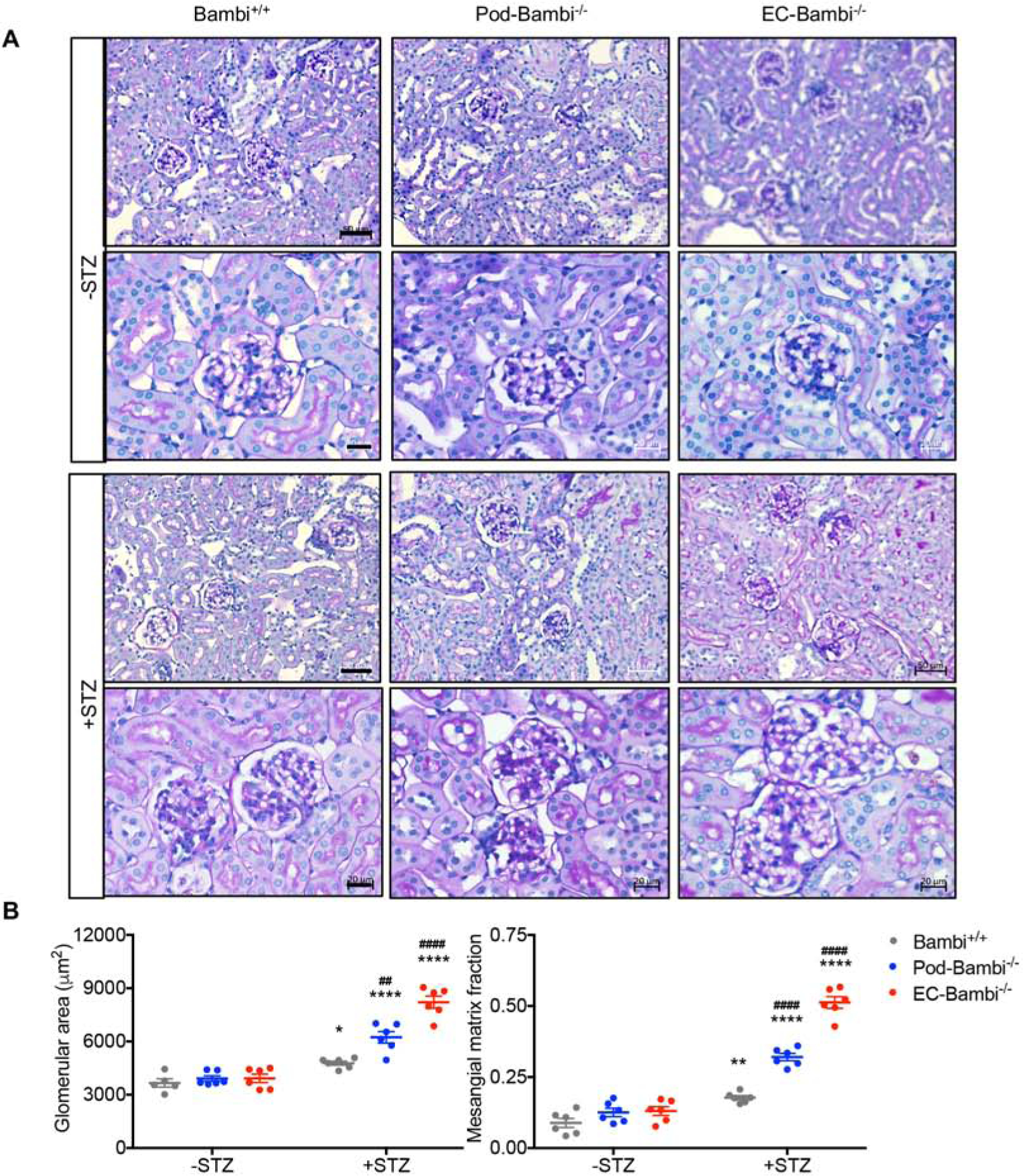
(A) Representative images of periodic acid-Schiff (PAS)-stained kidneys at low magnification (top panels; scale bar, 50μm) and high magnification (bottom panels; scale bar, 20μm). (B) Quantification of glomerular area (left) and the mesangial matrix fraction (right). n=6 mice per group with at least 30 glomeruli evaluated per mouse. *p<0.05 and ****p<0.0001 vs. respective −STZ controls; ##p<0.01 and ####p<0.0001 vs. diabetic Bambi+/+ mice (2-way ANOVA with Tukey’s post hoc analysis).
Podocyte- and EC-specific loss of Bambi exacerbates podocyte injury and loss in diabetic mice
Consistent with the above observations, ultrastructural analysis by transmission electron microscopy (TEM) showed significant glomerular basement membrane (GBM) thickening in all diabetic mice in comparison to the nondiabetic controls (Fig. 4A–B), but this too was further exacerbated in diabetic pod-Bambi−/− mice and to greater extent in diabetic EC-Bambi−/− mice. Significant foot process effacement was noted in the glomeruli of all diabetic mice (Fig. 4A–B), and as anticipated, diabetic pod-Bambi−/− mice showed worsened podocyte foot process effacement in comparison to the diabetic Bambi+/+ mice. Surprisingly, this was also true in the diabetic EC-Bambi−/− mice as compared to the control diabetic Bambi+/+ mice (Fig. 4A–B). To assess the extent of podocyte loss in DN, we performed semi-quantitative measurements of podocytes per glomerular cross-section using p57Kip2 (p57)30, 31 and Wilm’s tumor-1 (WT-1) as podocyte markers (Fig. 5A–B and Supp. Fig. 3). The analysis consistently showed a decline in the number of podocytes in all diabetic mice in comparison to non-diabetic mice, but significantly more so in the diabetic Pod-Bambi−/− and EC-Bambi−/− mice (Fig. 5A–B and Supp. Fig. 3A–B). Interestingly, terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL)-positive podocytes, though few cells in number and not in all glomeruli, was observed more readily in diabetic pod-Bambi−/− mouse kidneys that were rarely observed in the other diabetic or control groups (Supp. Fig. 3B–C), suggesting that the mechanisms of podocyte loss vary between podocyte or endothelial loss of BAMBI in diabetic mice.
Figure 4. GBM thickening and foot process effacement in the diabetic Pod- and EC-Bambi−/− mice.
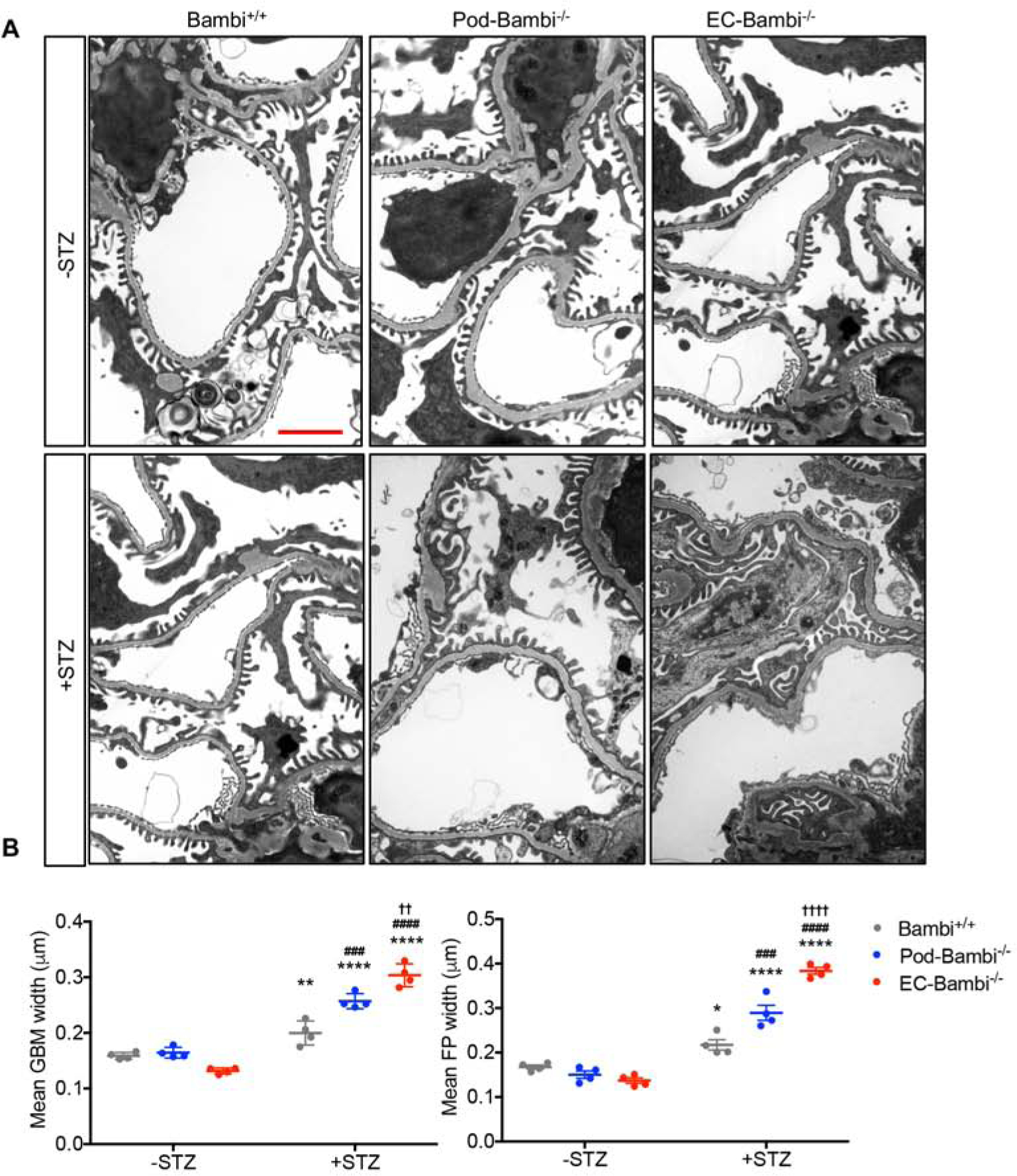
(A) Representative transmission EM images of control and diabetic mouse kidneys at 5k magnification (scale bar, 2μm). (B) Quantification of mean glomerular basement membrane (GBM) and podocyte foot process (FP) widths. n=4 mice per group with 15 fields evaluated per mouse. *p<0.05 and ****p<0.0001 vs. respective −STZ controls; ###p<0.001 and ####p<0.0001 vs. diabetic Bambi+/+ mice (2-way ANOVA with Tukey’s post hoc analysis).
Figure 5. Podocyte loss is worsened in the diabetic Pod- and EC-Bambi−/− mice.

(A) Representative images of p57 immunofluorescence of control and diabetic mouse kidneys. Scale bar, 20μm. (B) Quantification of total number p57+ cells per glomerular cross-section per experimental group (left, n=240–256 glomeruli counts per group) and the average number of p57+ cells per glomerular cross-section per mouse (right, n=6 mice per group, at least 40 glomeruli counted per mouse). *p<0.05 and ****p<0.0001 vs. respective −STZ controls; #p<0.05 and ####p<0.0001 vs. diabetic Bambi+/+ mice (2-way ANOVA with Tukey’s post hoc analysis).
EC-specific loss of Bambi increases GEC proliferation in diabetic mice
We previously showed that global loss of Bambi−/− in mice worsened DN progression, which was associated with increased glomerular ALK1-Smad1/5 signaling10, 20, 29. We also showed that the increased TGF-β/ALK1 signaling in early diabetic kidney disease increases GEC proliferation and angiogenesis to promote disease progression14. We thus postulated that the endothelial loss of BAMBI would heighten ALK1-mediated GEC activation and proliferation in diabetic kidneys. Indeed, immunostaining of endothelial cells with CD31 showed a markedly increased CD31+ area in the glomeruli of diabetic EC-Bambi−/− mice in comparison to the other diabetic groups (Fig. 6A–B). Moreover, a significant number of glomerular cells expressing the proliferation marker Ki-67 was detected in diabetic EC-Bambi−/− mice (Fig. 6A–B), although Ki-67+ cells in the tubulointerstitial cells were readily observed in all diabetic mouse kidneys.
Figure 6. GEC proliferation is markedly increased only in the diabetic EC-Bambi−/− mice.
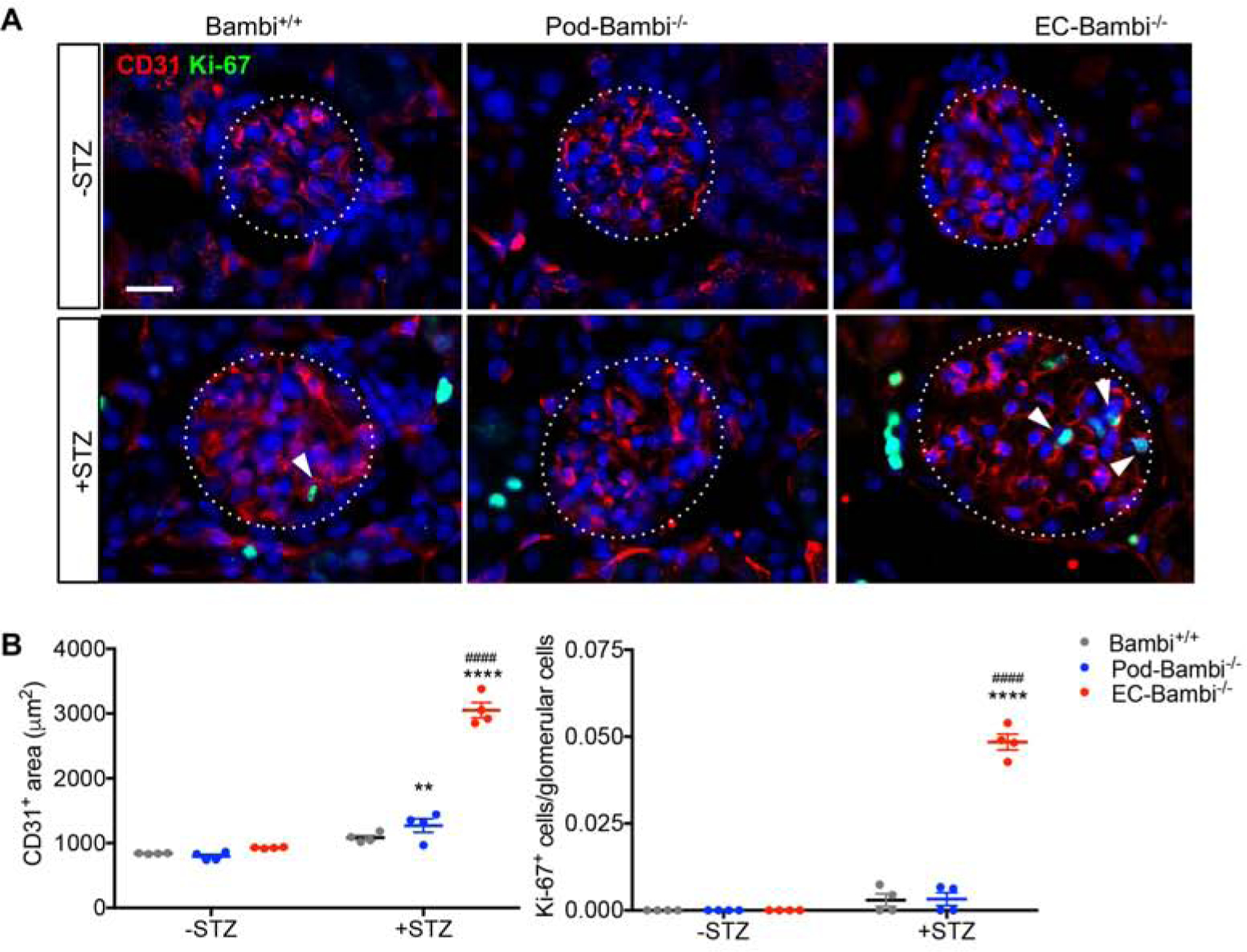
(A) Representative images of Ki-67 (green) and CD31 (red) immunofluorescence of control and diabetic mouse kidneys. Glomeruli are outlined with dotted lines. Arrowheads show examples of Ki-67+ cells in the glomeruli. Scale bar, 20μm. (B–C) Quantification of CD31+ area (B) and the fraction of Ki-67+ cells (C) per glomerular cross-section per mouse. n=4 mice per group with at least 40 glomeruli evaluated per mouse. **p<0.01 and ****p<0.0001 vs. respective −STZ controls; ####p<0.0001 vs. diabetic Bambi+/+ mice (2-way ANOVA with Tukey’s post hoc analysis).
Consistent with proliferating and immature GECs in DN, we observed the appearance of plasmalemma vesicle-associated protein (PLVAP, also known as MECA-32) in the glomeruli of diabetic mice, with most prominent expression in the diabetic EC-Bambi−/− mice (Fig. 7A–B, Supp. Fig. 4A). PLVAP, an integral transmembrane glycoprotein protein typically expressed in the diaphragms of endothelial fenestrae, transcellular channels, and caveolae32, 33, is normally present in the peritubular capillary endothelial cells32, but absent in the mature GECs of adult kidneys. However, its expression is detected during renal development in the immature GECs34. We found that Plvap mRNA expression was significantly upregulated only in the isolated glomeruli from the diabetic Pod-Bambi−/− and EC-Bambi−/− mice, but barely detectable in the glomeruli of control mice of all 3 groups (Supp. Fig. 4A), suggesting that PLVAP expression might be correlated with increased severity of diabetic glomerulopathy. These findings were corroborated by immunohistochemistry for PLVAP. As expected, PLVAP expression was found in the peritubular capillaries of all mice but was negative in glomeruli from non-diabetic control mice (Fig. 7A–B). In control diabetic Bambi+/+ mice, which displayed only a mild diabetic glomerulopathy, fewer PLVAP+ cells in the glomeruli were visible (Fig. 7A–B). In contrast, in the diabetic Pod-Bambi−/− and particularly in EC-Bambi−/− mice greater number of glomerular cells were positive for PLVAP expression, although not all glomeruli (Fig. 7A–B). Thus, the appearance of PLVAP in some of the glomeruli in diabetic mice appeared to be associated with advanced DN. Indeed, we also observed that PLVAP is re-expressed in the glomeruli of human diabetic kidneys by immunostaining (Fig. 7C, Supp. Fig. 4B–C). While the staining for PLVAP in control kidney tissue (unaffected areas of kidney tumor nephrectomies from nondiabetic patients) was only seen in peritubular capillaries, kidney samples of all diabetic patients showed areas of segmental staining for PLVAP in the glomeruli (Supp. Fig. 4B), irrespective of whether they had established DN (serum creatinine ≥ 1.5 mg/dl and albuminuria ≥300 mg/dL; Supp. Table 3). Interestingly, in some glomeruli of these patients, the PLVAP+ cells were particularly visible at the vascular pole (Fig. 7C, Supp. Fig. 4B). Similar to what we observed in mouse kidneys, PLVAP expression in glomeruli was focal and segmental in the human tissues with histologically confirmed DN.
Figure 7. Plasmalemma vesicle-associated protein (PLVAP) expression is increased in the glomeruli of diabetic Pod- and EC-Bambi−/− mice.

(A) Representative immunohistochemistry images of PLVAP in control and diabetic mouse kidneys. Glomeruli are outlined with dotted lines. Scale bar, 50μm. (B) Quantification of glomerular PLVAP+ cells per experimental group. n=120–159 glomeruli per experimental group from 4 mice per group (left) and the average number of glomerular PLVAP+ cells per mouse (right). n=4 mice per group with 24–40 glomeruli evaluated per mouse. (**p<0.01 and ***p<0.0001 vs. respective −STZ controls; ##p<0.01, ###p<0.001, and ####p<0.0001 vs. diabetic Bambi+/+ mice (2-way ANOVA with Tukey’s post hoc analysis). (C) Immunofluorescence image of PLVAP (green) co-labeled with endothelial marker isolectin B4 (red) in human DN kidney. Scale bar, 100μm. A magnified view of the dotted area in the left panel is shown on the right.
Podocyte- and EC-specific loss of Bambi exacerbates oxidative stress in the glomeruli of diabetic mice
TGF-β signaling has been shown to increase mitochondrial reactive oxygen species (ROS) production in different cell types, including renal cells in the diabetic kidneys35–37. Moreover, increased oxidative stress in GECs was shown to be associated with worsened DN and with increased ALK1 pathway14, 18, 19. We therefore examined the extent of oxidative stress in the diabetic kidneys in absence of BAMBI by 8-oxo-2’-deoxyguanosine (8-oxo-dG) detection. A significant amount of 8-oxo-dG was detected only in the glomeruli of diabetic mice (Fig. 8A–B), but consistent with the above data of increased severity of DN in Pod-Bambi−/− and EC-Bambi−/− mice, its expression was further augmented in their kidneys. Co-staining of CD31 showed enhanced overlapping areas of 8-oxo-dG and CD31 in the glomeruli of all diabetic mice, but this too was most evident in the diabetic EC-Bambi−/− mouse kidneys amongst the diabetic groups (Fig. 8A).
Figure 8. Oxidative stress damage is increased in the glomeruli of diabetic Pod- and EC-Bambi−/− mice.
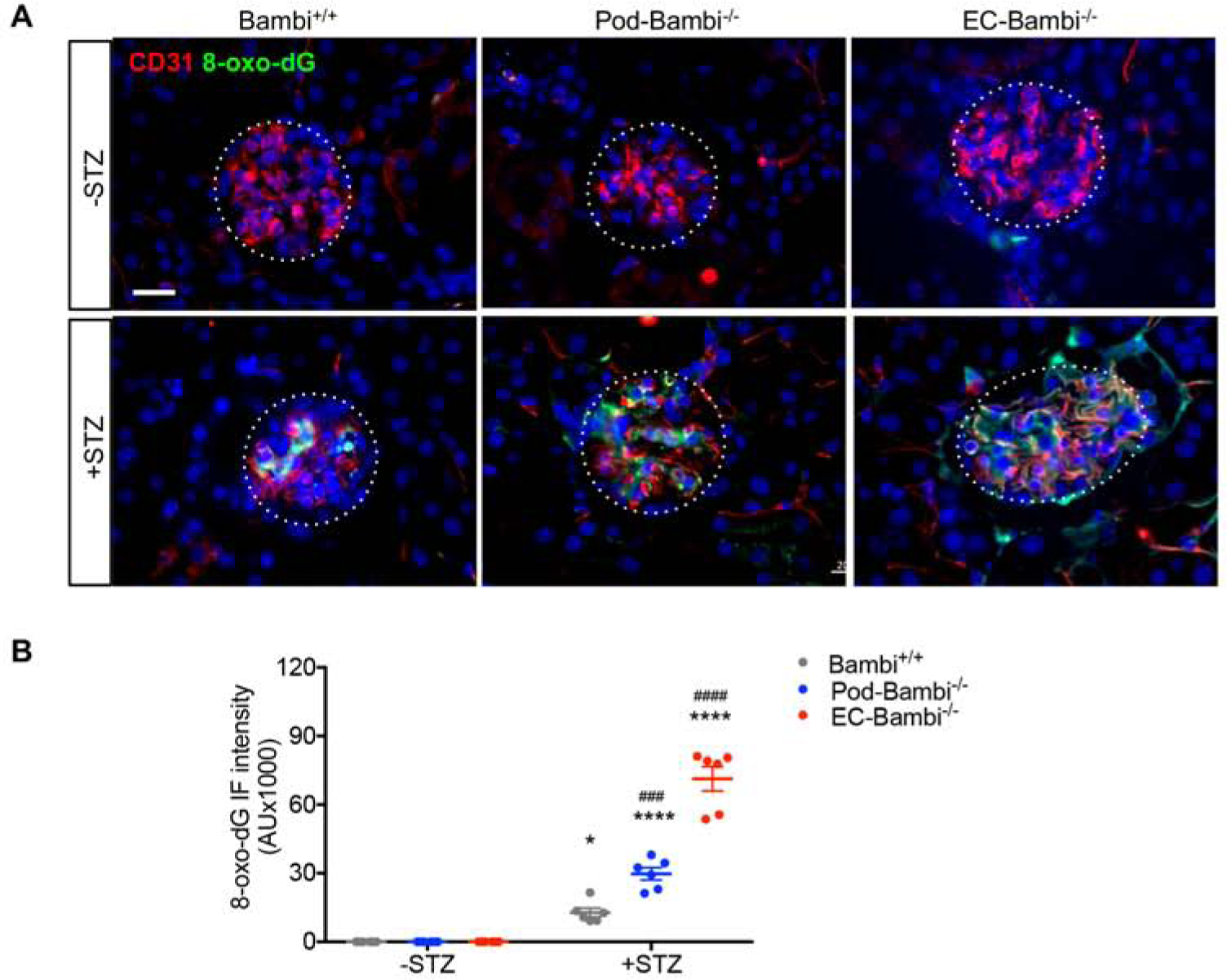
(A) Representative images of 8-oxo-dG (green) and CD31 (red) immunofluorescence. Glomeruli are outlined with dotted lines. Scale bar, 20μm. (B) Quantification of 8-oxo-dG intensity per mouse. AU, arbitrary units. n=6 mice per group with at least 40 glomeruli evaluated per mouse. *p<0.05 and ****p<0.0001 vs. respective −STZ controls; ###p<0.001 and ####p<0.0001 vs. diabetic Bambi+/+ mice (2-way ANOVA with Tukey’s post hoc analysis).
BAMBI suppresses TGF-β-mediated ALK5 signaling in podocytes and ALK1 signaling in GECs in diabetic kidneys
Given the preponderance of Alk1 expression versus Alk5 expression in GECs (Fig. 1A), we postulated that the loss of BAMBI would mostly affect the ALK1-Smad1/5 pathway in GECs, whereas in podocytes that do not express ALK1, the canonical ALK5-Smad2/3 pathway would be affected. We therefore examined the activation of Smad2/3 (ALK5-mediated) and Smad1/5 (ALK5-mediated) in the glomeruli of control and diabetic mice by immunostaining for phosphorylated Smad3 and Smad1/5 proteins (p-Smad3 and p-Smad1/5). Consistent with increased TGF-β signaling in the diabetic kidneys, increased p-Smad3 or p-Smad1/5 immunofluorescence was detected in the glomeruli of all diabetic mice, but not in the non-diabetic control mice (Fig. 9A–C). This was not due to the change in the level of total Smad proteins, as the expression of Smad3 and Smad1 proteins were not significantly different between the non-diabetic versus diabetic mouse kidneys (Supp. Fig. 5). As anticipated, the loss of podocyte BAMBI resulted in prominent p-Smad3 expression, but not that of p-Smad1/5, while the loss of endothelial BAMBI resulted in significantly enhanced p-Smad1/5 expression without change in the p-Smad3 expression (Fig. 9A–C). Co-staining with podocyte and EC marker indeed confirmed the cell-specific increase of phospho-Smad proteins in the glomeruli of diabetic mice (Supp. Fig. 6). We also noted that there were increased p-Smad3 and p-Smad1/5 staining in the tubulointerstitial space, consistent with TGF-β activation in the tubulointerstitial compartment in DN38, 39. Overall, these findings are consistent with the notion that in TGF-β signaling occurs predominantly through ALK5 and Smad3 in podocytes and ALK1 and Smad1/5 in GECs in DN.
Figure 9: Increased Smad3 activation in diabetic Pod-Bambi−/− mice and increased Smad1/5 activation in diabetic EC-Bambi−/− mice.

(A) immunofluorescence images of p-Smad3 in the glomeruli of control and diabetic mouse kidneys. (B) Immunofluorescence images of p-Smad1/5 in the glomeruli of control and diabetic mouse kidneys. DNA is counterstained in blue. Glomeruli are outlined with dotted lines. Scale bar, 20μm. (C) Quantification of the fraction of p-Smad3+ cells (left) or p-Smad1/5+ cells (right) in the glomerular cross-section of control and diabetic mice. n=6 mice per group with at least 40 glomeruli evaluated per mouse. *p<0.05 and ****p<0.0001 vs. −STZ controls; ####p<0.0001 vs. diabetic Bambi+/+ mice (2-way ANOVA with Tukey’s post hoc analysis).
DISCUSSION
Despite the abundant evidence of TGF-β as a major contributor to diabetic glomerulopathy36, 39, interfering with the downstream canonical Smad3 signaling by genetic or pharmacological manipulations were not effective at reducing urinary albumin excretion in diabetic mouse models7, 8, suggesting that other aspects of TGF-β signaling remained unaffected. Our earlier work showed that the loss of the negative regulator of TGF-β signal transduction, BAMBI, exacerbated the albuminuria and DN in mice, which was associated with enhanced ALK1-Smad1/5 signaling10. As the pleiotropic actions of TGF-β can be detrimental as well as protective in kidney cell homeostasis40, 41, by using the podocyte- and endothelial-specific Bambi−/− mice, in this study we sought to better define the contribution of differential TGF-β signaling pathways in podocytes and glomerular endothelial cells in DN. Our results now show that the podocyte-specific loss of BAMBI resulted in a heightened activation of ALK5-Smad2/3 signaling in podocytes and worsened podocyte loss, proteinuria, and DN severity in diabetic mice. EC-specific loss of BAMBI instead resulted in a heightened activation of ALK1-Smad1/5 signaling in endothelial cells and was associated with increased GEC proliferation, worsened proteinuria and renal function. Notably, it was also associated with increased podocyte loss in diabetic mice, underscoring the intricate cross-talk between glomerular cells38, 42–45 and strengthening the concept of the glomerulus as an integrated functional unit. A well-established cross-talk mediator between podocyte and GECs is the vascular endothelial growth factor (VEGF). Both the overexpression as well as decreased generation of VEGF-A by podocytes can result in glomerular pathology with de-differentiation, proliferation or loss of endothelial cells and glomerulosclerosis, including in DN44, 46–49. Recent studies have also highlighted the importance of endothelin (Edn) in the podocyte-to-GEC cross-talk19, 50, 51. In a mouse model of FSGS in which the podocyte-specific TGFBR1 (ALK5) activation induces progressive glomerular disease and renal failure, the release of Edn1 by podocytes and engagement of Edn receptor A (Ednra) in GECs was shown to induce mitochondrial oxidative stress and dysfunction, which in turn leads to release of as yet unidentified factor(s) that mediate the damage and depletion of adjacent podocytes50, 51. Qi et al showed that a similar endothelial-to-podocyte cross-talk could also underlie the segmental lesions in DN19. It is also unclear at present which GEC-derived mediators are responsible for the podocyte loss in the diabetic EC-Bambi−/− mice. However, while the present study was not designed to identify potential candidates for the cross-talk, our results nevertheless underscore the importance of GEC and podocytes cross-talk in the spreading of the injury between the cell types, irrespective of which cell had been injured initially.
Changes in the glomerular endothelium appear to precede those in the podocytes during DM in both human biopsies52 and in experimental mice susceptible to diabetic glomerulopathy19. However, identifying changes in the glomerular endothelium of diabetic kidneys requires both time-consuming and technically demanding studies of determining the density and percentage of endothelial fenestrae, which are also susceptible to variations of sample preparations (e.g. variations in fixation, embedding, and sectioning techniques). We posited that either the endothelial de-differentiation or the neo-angiogenesis in the glomeruli in DN might be associated with the earlier developmental pattern, characterized by a fenestrated endothelium with a PLVAP+ diaphragm. Indeed, we found that while all the glomeruli of non-diabetic mice were negative for PLVAP, some of the glomeruli from Pod-Bambi−/− and especially EC-Bambi−/− diabetic mice showed robust PLVAP staining in a capillary pattern. While the mechanism and consequence of the increased glomerular endothelial PLVAP expression will need further studies, PLVAP is shown to be a marker of immature or leaky retinal vessels and is transcriptionally regulated by canonical Wnt/β-catenin signaling during development53. In addition, overexpression of VEGF in podocytes results in impaired endothelial fenestrations and neo-angiogenesis, which also has been noted to be associated with de novo expression of PLVAP in glomerular endothelial cells54. Thus, PLVAP expression in glomeruli with diabetic glomerulopathy may reflect neo-angiogenesis with immature endothelial cells, which in turn may contribute to diabetic glomerulopathy. Interestingly, the increased glomerular PLVAP expression found in kidneys of diabetic patients was particularly concentrated near the vascular pole in some of the glomeruli of DM patients. These observations are in line with previous reports of neo-vascularization occurring at the vascular pole in diabetic patients55, 56, and suggests that changes in the glomerular PLVAP expression may indicate the early endothelial changes in in the diabetic kidney. Thus, while the potential of PLVAP as a marker for diabetic glomerulopathy needs to be further examined, it appears to be a novel finding for glomerular endothelial dysfunction in DN.
Another interesting observation, as well as a limitation of the study, is that significant diabetes-induced hypertension was observed only in the EC-Bambi−/− mice. Hypertension was not noted in the diabetic mice with global knockout of Bambi in our previous studies10. The reason for hypertension in the diabetic EC-Bambi−/− mice in this study may be related to the enhanced TGF-β/ALK1 signaling in the endothelium of the entire vasculature in absence of endothelial expression of BAMBI and eNOS. Conceivably, this could include the enhanced ALK1-mediated endothelial proliferation and signaling, increasing peripheral resistance, and increased TGF-β-induced endothelial cell generation of endothelin57, resulting in elevated blood pressure. The change in the vascular endothelium induced by the pan-endothelial loss of BAMBI and ensuing hypertension is likely to have contributed to a significant worsening of the DN in the EC-Bambi−/− mice. Another limitation is that since there are no GEC-specific Cre transgenic mice yet available, the contribution of TGF-β/ALK1 only in the glomerular endothelium in DN cannot be dissected at present. Despite these shortcomings, the unexpected finding of hypertension and worsening of diabetic glomerulopathy in the diabetic EC-Bambi−/− mice will be of considerable interest in future studies designed to examine the potential relationship and contribution of hypertension and endothelial dysfunction in DN progression.
In conclusion, our data show that TGF-β promotes DN by promoting ALK5-Smad2/3-mediated podocyte injury and ALK1-Smad1/5-mediated endothelial activation and proliferation. These cell type-specific effects of TGF-β are attenuated through the modulatory actions of BAMBI, which therefore may also become a target for potential intervention against DN progression.
METHODS
Mouse models
All mouse experiments were performed under the guidelines of and approved by the Institutional Animal Care and Use Committee at the Icahn School of Medicine at Mount Sinai. All mouse strains used in this study were purchased through the Jackson Laboratory (Bar Harbor, ME). Podocyte- and EC-specific Bambi knockout mice were generated by crossing Bambifl/fl mice (B6;129S1-Bambitm1Jian/J, #009389) with eNOS-deficient mice (B6.129P2-Nos3tm1Unc/J; stock number 002684) and further crossing the progenies with either podocin-Cre (B6.Cg-Tg(NPHS2-cre)295Lbh/J; stock number 008205) or VE-Cadherin-Cre (B6;129-Tg(Cdh5-cre)1Spe/J; #017968). To check the efficiency of Cre recombinase, NPHS2-cre;Bambifl/fl or Cdh5-cre;Bambifl/fl were crossed with mTmG Cre reporter mice (B6.129(Cg)-Gt(ROSA)26Sortm4(ACTB-tdTomato,-EGFP)Luo/J mT/mG mice, #007676). All mice in the study are in the mixed C57BL/6J and 129 strain. To establish diabetes, 8–10 week old mice were intraperitoneally injected with streptozotocin (STZ, 50μg/g body weight) (Sigma-Aldrich, St Louis, MO) for 5 consecutive days or with citrate buffer as vehicle control. Mice were euthanized at 16 weeks after STZ injection.
Kidney histology
Kidneys were removed and fixed with 4% paraformaldehyde 16 hours at 4°C. The 4μm sections were cut from paraffin-embedded kidney tissues. Sections were stained with periodic acid-Schiff (PAS) for histology analysis. Assessment of the mesangial and glomerular cross-sectional areas was performed by pixel counts per section under 200x and 400x magnifications (Zeiss AX10 microscope, Carl Zeiss, Thornwood, NY).
Kidney function measurements
24-hour urine collections in the metabolic cages were used for the determination of urinary albumin excretion. Urine albumin was quantified by using the ELISA kit from Bethyl Laboratories, Inc. (Houston, TX). Blood urea content was measured using a commercially available kit (BioAssay Systems). Serum creatinine was measured by the capillary electrophoresis method, performed by O’Brien Research Core at the University of Texas South Western.
Statistical Analysis
Data are expressed as mean±SD. For comparison of means between groups, 2-way ANOVA with Tukey post-test was applied. For comparisons of means between two groups, two-tailed, unpaired t-tests were performed. Prism 7 software (GraphPad, La Jolla, CA) was used for statistical analyses.
Supplementary Material
Translational statement:
The role of TGF-β in the development of diabetic kidney disease has been mostly assigned to the development of fibrosis. By analyzing different TGF-β signaling pathways in the glomeruli with the elimination of BAMBI, a modifier of TGF-β signaling in either podocytes of endothelial cells, we now demonstrate that signaling of TGF-β in podocytes contributes to podocyte loss through the Smad3-mediated signaling, while in glomerular endothelial cells TGF-β controls their proliferation and thereby neo-angiogenesis through Smad1/5 pathways. Thus, future therapeutic interventions directed at TGF-β signaling in diabetic glomerular disease will have to take into account the different signaling mechanisms for podocytes and endothelial cells. Furthermore, the cross-talk between podocytes and endothelial cells and play an important role in diabetic glomerular disease.
Funding:
AQC is supported by the National Natural Science Foundation of China(#81800637) and Natural Science Foundation of Fujian Province (2019J01560); JCH is supported by NIH/NIDDK R01DK078897, NIH R01DK088541, and VA Merit Award; KL is supported by NIH/NIDDK R01DK117913.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DISCLOSURE: The authors declare that they have no conflict of interest.
Additional detailed methods are included in the Supplementary Material.
SUPPLEMENTARY MATERIAL:
Supplementary information is available on Kidney International’s website.
REFERENCES
- 1.Sharma K, Jin Y, Guo J, et al. Neutralization of TGF-beta by anti-TGF-beta antibody attenuates kidney hypertrophy and the enhanced extracellular matrix gene expression in STZ-induced diabetic mice. Diabetes 1996; 45: 522–530. [DOI] [PubMed] [Google Scholar]
- 2.Ziyadeh FN, Hoffman BB, Han DC, et al. Long-term prevention of renal insufficiency, excess matrix gene expression, and glomerular mesangial matrix expansion by treatment with monoclonal antitransforming growth factor-beta antibody in db/db diabetic mice. Proceedings of the National Academy of Sciences of the United States of America 2000; 97: 8015–8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen S, Iglesias-de la Cruz MC, Jim B, et al. Reversibility of established diabetic glomerulopathy by anti-TGF-beta antibodies in db/db mice. Biochemical and biophysical research communications 2003; 300: 16–22. [DOI] [PubMed] [Google Scholar]
- 4.Massague J TGF-beta signal transduction. Annu Rev Biochem 1998; 67: 753–791. [DOI] [PubMed] [Google Scholar]
- 5.Schiffer M, Bitzer M, Roberts IS, et al. Apoptosis in podocytes induced by TGF-beta and Smad7. J Clin Invest 2001; 108: 807–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Susztak K, Raff AC, Schiffer M, et al. Glucose-induced reactive oxygen species cause apoptosis of podocytes and podocyte depletion at the onset of diabetic nephropathy. Diabetes 2006; 55: 225–233. [PubMed] [Google Scholar]
- 7.Li J, Qu X, Yao J, et al. Blockade of endothelial-mesenchymal transition by a Smad3 inhibitor delays the early development of streptozotocin-induced diabetic nephropathy. Diabetes 2010; 59: 2612–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang A, Ziyadeh FN, Lee EY, et al. Interference with TGF-beta signaling by Smad3-knockout in mice limits diabetic glomerulosclerosis without affecting albuminuria. American journal of physiology Renal physiology 2007; 293: F1657–1665. [DOI] [PubMed] [Google Scholar]
- 9.Onichtchouk D, Chen YG, Dosch R, et al. Silencing of TGF-beta signalling by the pseudoreceptor BAMBI. Nature 1999; 401: 480–485. [DOI] [PubMed] [Google Scholar]
- 10.Fan Y, Li X, Xiao W, et al. BAMBI elimination enhances alternative TGF-beta signaling and glomerular dysfunction in diabetic mice. Diabetes 2015; 64: 2220–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goumans MJ, Mummery C. Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice. The International journal of developmental biology 2000; 44: 253–265. [PubMed] [Google Scholar]
- 12.Pardali E, ten Dijke P. Transforming growth factor-beta signaling and tumor angiogenesis. Frontiers in bioscience 2009; 14: 4848–4861. [DOI] [PubMed] [Google Scholar]
- 13.Oh SP, Seki T, Goss KA, et al. Activin receptor-like kinase 1 modulates transforming growth factor-beta 1 signaling in the regulation of angiogenesis. Proceedings of the National Academy of Sciences of the United States of America 2000; 97: 2626–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hong Q, Zhang L, Fu J, et al. LRG1 Promotes Diabetic Kidney Disease Progression by Enhancing TGF-beta-Induced Angiogenesis. J Am Soc Nephrol 2019; 30: 546–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sena CM, Pereira AM, Seica R. Endothelial dysfunction - a major mediator of diabetic vascular disease. Biochimica et biophysica acta 2013; 1832: 2216–2231. [DOI] [PubMed] [Google Scholar]
- 16.Nakagawa T, Kosugi T, Haneda M, et al. Abnormal angiogenesis in diabetic nephropathy. Diabetes 2009; 58: 1471–1478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Satchell SC, Braet F. Glomerular endothelial cell fenestrations: an integral component of the glomerular filtration barrier. American journal of physiology Renal physiology 2009; 296: F947–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fu J, Wei C, Zhang W, et al. Gene expression profiles of glomerular endothelial cells support their role in the glomerulopathy of diabetic mice. Kidney Int 2018; 94: 326–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qi H, Casalena G, Shi S, et al. Glomerular Endothelial Mitochondrial Dysfunction Is Essential and Characteristic of Diabetic Kidney Disease Susceptibility. Diabetes 2017; 66: 763–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guillot N, Kollins D, Gilbert V, et al. BAMBI regulates angiogenesis and endothelial homeostasis through modulation of alternative TGFbeta signaling. PLoS One 2012; 7: e39406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xavier S, Gilbert V, Rastaldi MP, et al. BAMBI is expressed in endothelial cells and is regulated by lysosomal/autolysosomal degradation. PLoS One 2010; 5: e12995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fu J, Wei C, Lee K, et al. Comparison of Glomerular and Podocyte mRNA Profiles in Streptozotocin-Induced Diabetes. J Am Soc Nephrol 2016; 27: 1006–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fu J, Akat KM, Sun Z, et al. Single-Cell RNA Profiling of Glomerular Cells Shows Dynamic Changes in Experimental Diabetic Kidney Disease. J Am Soc Nephrol 2019; 30: 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karaiskos N, Rahmatollahi M, Boltengagen A, et al. A Single-Cell Transcriptome Atlas of the Mouse Glomerulus. J Am Soc Nephrol 2018; 29: 2060–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moustakas A, Heldin CH. The regulation of TGFbeta signal transduction. Development 2009; 136: 3699–3714. [DOI] [PubMed] [Google Scholar]
- 26.Mundel P, Reiser J, Zuniga Mejia Borja A, et al. Rearrangements of the cytoskeleton and cell contacts induce process formation during differentiation of conditionally immortalized mouse podocyte cell lines. Experimental cell research 1997; 236: 248–258. [DOI] [PubMed] [Google Scholar]
- 27.Mohan S, Reddick RL, Musi N, et al. Diabetic eNOS knockout mice develop distinct macro- and microvascular complications. Lab Invest 2008; 88: 515–528. [DOI] [PubMed] [Google Scholar]
- 28.Nakagawa T, Sato W, Glushakova O, et al. Diabetic endothelial nitric oxide synthase knockout mice develop advanced diabetic nephropathy. J Am Soc Nephrol 2007; 18: 539–550. [DOI] [PubMed] [Google Scholar]
- 29.Guillot N, Kollins D, Badimon JJ, et al. Accelerated reendothelialization, increased neovascularization and erythrocyte extravasation after arterial injury in BAMBI−/− mice. PLoS One 2013; 8: e58550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Andeen NK, Nguyen TQ, Steegh F, et al. The phenotypes of podocytes and parietal epithelial cells may overlap in diabetic nephropathy. Kidney Int 2015; 88: 1099–1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pichaiwong W, Hudkins KL, Wietecha T, et al. Reversibility of structural and functional damage in a model of advanced diabetic nephropathy. J Am Soc Nephrol 2013; 24: 1088–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stan RV, Kubitza M, Palade GE. PV-1 is a component of the fenestral and stomatal diaphragms in fenestrated endothelia. Proceedings of the National Academy of Sciences of the United States of America 1999; 96: 13203–13207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Herrnberger L, Seitz R, Kuespert S, et al. Lack of endothelial diaphragms in fenestrae and caveolae of mutant Plvap-deficient mice. Histochem Cell Biol 2012; 138: 709–724. [DOI] [PubMed] [Google Scholar]
- 34.Ichimura K, Stan RV, Kurihara H, et al. Glomerular endothelial cells form diaphragms during development and pathologic conditions. J Am Soc Nephrol 2008; 19: 1463–1471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jha JC, Banal C, Chow BS, et al. Diabetes and Kidney Disease: Role of Oxidative Stress. Antioxid Redox Signal 2016; 25: 657–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang AS, Hathaway CK, Smithies O, et al. Transforming growth factor-beta1 and diabetic nephropathy. American journal of physiology Renal physiology 2016; 310: F689–F696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Badal SS, Danesh FR. New insights into molecular mechanisms of diabetic kidney disease. American journal of kidney diseases : the official journal of the National Kidney Foundation 2014; 63: S63–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sharma K, McGowan TA. TGF-beta in diabetic kidney disease: role of novel signaling pathways. Cytokine Growth Factor Rev 2000; 11: 115–123. [DOI] [PubMed] [Google Scholar]
- 39.Ziyadeh FN. Mediators of diabetic renal disease: the case for tgf-Beta as the major mediator. J Am Soc Nephrol 2004; 15 Suppl 1: S55–57. [DOI] [PubMed] [Google Scholar]
- 40.Gewin L The many talents of transforming growth factor-beta in the kidney. Current opinion in nephrology and hypertension 2019; 28: 203–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sureshbabu A, Muhsin SA, Choi ME. TGF-beta signaling in the kidney: profibrotic and protective effects. American journal of physiology Renal physiology 2016; 310: F596–F606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu J, Lee K, Chuang PY, et al. Glomerular endothelial cell injury and cross talk in diabetic kidney disease. American journal of physiology Renal physiology 2015; 308: F287–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lennon R, Hosawi S. Glomerular cell crosstalk. Current opinion in nephrology and hypertension 2016; 25: 187–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Bartlett CS, Jeansson M, Quaggin SE. Vascular Growth Factors and Glomerular Disease. Annu Rev Physiol 2016; 78: 437–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Daehn IS. Glomerular Endothelial Cells Stress and Cross-Talk With Podocytes in the Development of Diabetic Kidney Disease. Front Med (Lausanne) 2018; 5: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eremina V, Baelde HJ, Quaggin SE. Role of the VEGF--a signaling pathway in the glomerulus: evidence for crosstalk between components of the glomerular filtration barrier. Nephron Physiol 2007; 106: p32–37. [DOI] [PubMed] [Google Scholar]
- 47.Eremina V, Sood M, Haigh J, et al. Glomerular-specific alterations of VEGF-A expression lead to distinct congenital and acquired renal diseases. J Clin Invest 2003; 111: 707–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sison K, Eremina V, Baelde H, et al. Glomerular structure and function require paracrine, not autocrine, VEGF-VEGFR-2 signaling. J Am Soc Nephrol 2010; 21: 1691–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sivaskandarajah GA, Jeansson M, Maezawa Y, et al. Vegfa protects the glomerular microvasculature in diabetes. Diabetes 2012; 61: 2958–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Daehn I, Casalena G, Zhang T, et al. Endothelial mitochondrial oxidative stress determines podocyte depletion in segmental glomerulosclerosis. J Clin Invest 2014; 124: 1608–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ebefors K, Wiener RJ, Yu L, et al. Endothelin receptor-A mediates degradation of the glomerular endothelial surface layer via pathologic crosstalk between activated podocytes and glomerular endothelial cells. Kidney Int 2019; 96: 957–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Weil EJ, Lemley KV, Mason CC, et al. Podocyte detachment and reduced glomerular capillary endothelial fenestration promote kidney disease in type 2 diabetic nephropathy. Kidney Int 2012; 82: 1010–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liebner S, Corada M, Bangsow T, et al. Wnt/beta-catenin signaling controls development of the blood-brain barrier. J Cell Biol 2008; 183: 409–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Suyama M, Miyazaki Y, Matsusaka T, et al. Forced expression of vascular endothelial growth factor-A in podocytes decreases mesangial cell numbers and attenuates endothelial cell differentiation in the mouse glomerulus. Clin Exp Nephrol 2018; 22: 266–274. [DOI] [PubMed] [Google Scholar]
- 55.Kanesaki Y, Suzuki D, Uehara G, et al. Vascular endothelial growth factor gene expression is correlated with glomerular neovascularization in human diabetic nephropathy. American journal of kidney diseases : the official journal of the National Kidney Foundation 2005; 45: 288–294. [DOI] [PubMed] [Google Scholar]
- 56.Osterby R, Asplund J, Bangstad HJ, et al. Neovascularization at the vascular pole region in diabetic glomerulopathy. Nephrol Dial Transplant 1999; 14: 348–352. [DOI] [PubMed] [Google Scholar]
- 57.Kurihara H, Yoshizumi M, Sugiyama T, et al. Transforming growth factor-beta stimulates the expression of endothelin mRNA by vascular endothelial cells. Biochemical and biophysical research communications 1989; 159: 1435–1440. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.