

Abstract
Over the last 20 years, a number of studies have provided strong support for protein degradation mediated by the ubiquitin-proteasome system in synaptic plasticity and memory formation. In this system, target substrates become covalently modified by the small protein ubiquitin through a series of enzymatic reactions involving hundreds of different ligases. While some substrates will acquire only a single ubiquitin, most will be marked by multiple ubiquitin modifications, which link together at specific lysine sites or the N-terminal methionine on the previous ubiquitin to form a polyubiquitin chain. There are at least eight known linkage-specific polyubiquitin chains a target protein can acquire, many of which are independent of the proteasome, and these chains can be homogenous, mixed, or branched in nature, all of which result in different functional outcomes and fates for the target substrate. However, as the focus has remained on protein degradation, much remains unknown about the role of these diverse ubiquitin chains in the brain, particularly during activity- and learning-dependent synaptic plasticity. Here, we review the different types and functions of ubiquitin chains and summarize evidence suggesting a role for these diverse ubiquitin modifications in synaptic plasticity and memory formation. We conclude by discussing how technological limitations have limited our ability to identify and elucidate the role of different ubiquitin chains in the brain and speculate on the future directions and implications of understanding linkage-specific ubiquitin modifications in activity- and learning-dependent synaptic plasticity.
Keywords: Ubiquitin, memory, transcription, protein degradation
1. Introduction
The neurobiological basis of memory formation has been extensively studied, and we now have a good understanding of the molecular mechanisms involved in the processes of long-term memory formation, storage, and modification (Asok, Leroy, Rayman, and Kandel, 2019; Johansen, Cain, Ostroff, and LeDoux, 2011; Kandel, 2012; Park and Kaang, 2019). For example, short-term memories for learned associations are susceptible to disruption until completion of a time-dependent process known as consolidation (McGaugh, 2015), which requires de novo protein synthesis as broad-spectrum manipulation of this or upstream intracellular signaling cascades often impairs long-term memory (Bailey et al., 1999; Bekinschtein et al., 2007; Schafe et al., 1999; Shrestha et al., 2020). Furthermore, following retrieval, memories destabilize and are temporarily rendered labile, requiring new protein synthesis in order for the memory to be restabilized, a process referred to as reconsolidation (Nader and Hardt, 2009; Nader, Schafe, and Le Doux, 2000; Roesler, 2017). A number of transcriptional control pathways upstream of mRNA and protein synthesis have been implicated in the consolidation and reconsolidation processes, including PKA, PKC, ERK/MAP, CREB, CaMKII and mTOR (Cho et al., 2018; Giese and Mizuno, 2013; Hoeffer and Klann, 2010; Kandel, 2012; Medina and Viola, 2018; Park et al., 2020; Peng et al., 2010; Silva, Kogan, Frankland, and Kida, 1998; Zalcman, Federman, and Romano, 2018). As a result, it is clear that the memory storage process is complicated, requiring a large number of individual and interacting processes that ultimately converge on increased regulation of transcriptional and translational processes necessary for memory formation and modification.
While traditional models of activity- and learning-dependent synaptic plasticity had focused on the need for mRNA and protein synthesis (Johansen et al., 2011), current models now take into account the potential role for protein degradation in these processes (Hegde, 2017; Hegde, Haynes, Bach, and Beckelman, 2014; Jarome and Helmstetter, 2013; 2014; Park and Kaang, 2019). The ubiquitin-proteasome system (UPS) controls the majority of protein degradation in cells and is involved in a number of molecular processes through proteolytic and nonproteolytic mechanisms. However, the focus in eukaryotes has remained on the canonical protein degradation function that has been shown to be involved in cell cycle progression, transcription, and synaptic plasticity. For example, protein degradation has been shown to control homeostatic scaling, new dendritic spine growth in vitro, long-term depression (LTD) and long-term potentiation (LTP), the proposed cellular analog of memory (Bingol et al., 2010; Cai, Frey, Sanna, and Behnisch, 2010; Djakovic et al., 2012; Dong et al., 2008; Dorrbaum et al., 2020; Ehlers, 2003; Hamilton et al., 2012; Li, Korte, and Sajikumar, 2016). Furthermore, recent evidence suggests that protein degradation is a critical regulator of memory formation and stability. Within cells of multiple brain regions, learning increases the amount of degradation-specific polyubiquitination and proteasome catalytic activity and administration of catalytic inhibitors of the proteasome result in memory impairments for a variety of behavioral tasks (Artinian et al., 2008; Cullen, Ferrara, Pullins, and Helmstetter, 2017; Figueiredo et al., 2015; Jarome, Werner, Kwapis, and Helmstetter, 2011; Lopez-Salon et al., 2001; Reis, Jarome, and Helmstetter, 2013; Rodriguez-Ortiz et al., 2011) and disease models, such as drug addiction (Werner et al., 2019; Werner et al., 2018). Furthermore, protein degradation has been shown to control the destabilization of memory after retrieval, suggesting a critical role for it in the reconsolidation process (Ferrara et al., 2019; Jarome et al., 2011; Lee, 2008; Lee et al., 2008), and has been widely implicated in the reconsolidation of drug-associated behaviors (Massaly et al., 2013; Werner et al., 2015). Learning-induced NMDA receptor activation results in changes in the activity of several intracellular signaling pathways responsible for memory formation. Interestingly, activation of NMDA receptors also controls learning-dependent increases in ubiquitin-proteasome activity (Jarome et al., 2011; Rosenberg, Elkobi, Dieterich, and Rosenblum, 2016). Downstream of NMDA receptors, CaMKII has been shown to target the proteasome and increase its catalytic activity via phosphorylation of the regulatory subunit RPT6 at serine-120 (Jarome, Ferrara, Kwapis, and Helmstetter, 2016; Jarome, Kwapis, Ruenzel, and Helmstetter, 2013), indicating that the protein degradation process is intricately connected to intracellular signaling mechanisms previously reported to be critical for memory formation.
While strong evidence supports that ubiquitin-proteasome mediated protein degradation is a critical regulator of synaptic plasticity and memory formation, very little attention has been paid to the degradation-independent functions of this system in these processes. Importantly, while protein degradation is thought of as the canonical process, it is only one of many functions of protein ubiquitination as this modification can result in many other functions and cellular fates for the target substrate. In fact, strong evidence indicates that protein ubiquitination can regulate a wide variety of processes independent of the proteasome, including kinase activation, endocytosis, epigenetic processes, intracellular trafficking, and mRNA stability (Akutsu, Dikic, and Bremm, 2016; Komander and Rape, 2012; Swatek and Komander, 2016). Additionally, some ubiquitin modifications can even impede the protein degradation process. These diverse functions are a result of there being eight different linkage-specific polyubiquitin chains that a target protein can acquire, resulting in a complex “ubiquitin code.” In this review, we will identify the different types of ubiquitin modifications and discuss the known functions of them outside of the brain. Next, we review what is currently known about the role of these ubiquitin modifications in synaptic plasticity and memory formation. We conclude by discussing the technical issues that have limited the identification and manipulation of these diverse ubiquitin modifications in neurons and theorize about how these nonproteolytic functions of protein ubiquitination contribute to activity-and learning-dependent synaptic plasticity.
2. The Ubiquitin-Proteasome System
The UPS is a central component of eukaryotic cells responsible for controlling intracellular protein homeostasis via degradation (Cromm and Crews, 2017; Gaczynska and Osmulski, 2018; Hershko and Ciechanover, 1998) of misfolded proteins due to heat or oxidation stresses, worn-out long-lived proteins, or short-lived transcription factors are among the many UPS substrates (Eisele and Wolf, 2008). In this system, the multi-subunit 26S proteasome complex recognizes and degrades target proteins that are “tagged” by ubiquitin and this degradation process is one of the most versatile mechanisms in the cell as it can regulate a myriad of intracellular biological and pathological functions (Hegde, 2017; Mabb and Ehlers, 2010; Shaid, Brandts, Serve, and Dikic, 2013). As the proteasome complex has been extensively reviewed elsewhere (Bard et al., 2018; Bedford et al., 2010), this section will focus primarily on the protein ubiquitination process, though we will briefly discuss the proteasome structure.
2.1. The ubiquitination process
Ubiquitin protein tagging is catalyzed by an enzymatic cascade involving three families of enzymes, a ubiquitin activating enzyme (E1), ubiquitin conjugating enzymes (E2), and ubiquitin ligases (E3) (Gropper et al., 1991; Jentsch, 1992). The first step begins with the E1 enzyme forming a thioester bond between its active site cysteine residue and the C-terminus of the glycine residue of a free ubiquitin molecule using ATP. Prior to the binding of ATP, the E1 has a low affinity for the ubiquitin, however, the ubiquitin binding site may be more accessible after ATP-dependent conformational changes occur (Pickart, 2001). Each E1 carries two activated ubiquitin molecules, one as an adenylate and the other as a thiolester, the latter of which is then transferred to a cysteine residue of the E2 conjugating enzymes (Cromm and Crews, 2017; Eletr and Kuhlman, 2007; Kumari, Lee, and Jha, 2018). The E1 concentration is often less than the E2 concentration, but due to its high efficiency, the E1 produces an adequate amount of activated ubiquitin for the downstream conjugation reactions (Stewart, Ritterhoff, Klevit, and Brzovic, 2016). There are about 40 different E2 enzymes in humans, which share a conserved core domain and perform a wide range of functions in the cell. However, in general, E2s are mostly responsible for carrying the ubiquitin molecule to the next enzyme, the E3 ligase (Stewart et al., 2016).
The E3 ligase recognizes the target substrate and binds to it, transferring the E2-bound ubiquitin molecule to a lysine residue of the target substrate through an isopeptide bond (Stewart et al., 2016; Zheng and Shabek, 2017). E3 ligases can only interact with a limited number of E2 enzymes, while E2 enzymes can interact with several E3 ligases, which increases the specificity of the ubiquitination process. The E3 category of enzymes, with more than 600 species identified thus far, have been classified into three different families of proteins based on their mechanism to transfer the ubiquitin: Really Interesting New Gene (RING), Homologous to the E6AP Carboxyl Terminus (HECT), and RING-between-RING (RBR). E3 ligases containing the HECT domain and RBRs act as intermediates by accepting the ubiquitin from the E2 and forming a covalent E3-ubiquitin thioester bond before transferring it directly to the substrate. In contrast, the largest family among the three, RING domain ligases, function as a scaffold for the E2 by bringing it to the proximity of the substrate and catalyzing the ubiquitin transfer directly from the E2 enzyme to the substrate (Eletr and Kuhlman, 2007; Kumari et al., 2018; Metzger, Hristova, and Weissman, 2012). As a result, the ubiquitination is complex and requires the coordinated actions of hundreds of different protein ligases.
2.2. The proteasome
Once proteins are tagged by specific ubiquitin modifications, such as lysine 48 linkage (K48), the 26S proteasome complex can identify and degrade them. The complete 26S proteasome complex, which is named after its sedimentation coefficient (Tanaka, 2009), is made up of two subcomplexes: A 20S catalytic core particle (CP) with a 19S regulatory particle (RP) linked on either or both ends of it, which regulates its activity. The RP consists of about 20 different subunits that can be categorized into two groups, the Regulatory particle of non-ATPase (RPN) subunits and the Regulatory particle of triple-ATPase (RPT) subunits. At least nine RPN subunits form the lid subcomplex, and six RPT along with four RPN subunits form the base subcomplex of the 19S cap (Tanaka, 2009). The 20S complex is a highly conserved structure among eukaryotic species, consisting of two outer rings of α subunits and three inner catalytic rings formed by β subunits facing the lumen of the barrel (Kunjappu and Hochstrasser, 2014; Latham, Sekhar, and Kay, 2014). The center of the outer rings, called the α-annulus, is in an almost closed state, forming a gate to keep unwanted proteins from penetrating the catalytic barrel of the complex. Polyubiquitinated proteins of specific chain linkage are recognized by the 19S base and deubiquitinating enzymes on the lid remove the chain and recycles the ubiquitin molecules. Then, the base unfolds the protein via the mechanical pulling force its heterohexameric ATPase motor generates, opens the 20S channel via the α ring and shuttles the unfolded substrates into the core for degradation by the β subunits (Bard et al., 2018; Finley, Chen, and Walters, 2016; Liu and Jacobson, 2013; Tanaka, 2009). The target substrate is cut into oligopeptides ranging from 3–15 amino acid residues, which may be hydrolyzed to monomers by oligopeptidases later (Kloetzel and Ossendorp, 2004; Rabl et al., 2008; Tanaka, 2009).
3. The different types of protein ubiquitination chains
Ubiquitin is a highly conserved 76 amino acid protein that is ubiquitously expressed in cells. Four genes code for ubiquitin, Ubb, Ubc, Uba52, and Rps27a (Figure 1A). Ubb and Ubc are unique poly-cistronic genes in which the coding region is repeated 3 or 8 times, respectively, resulting in them coding for multiple ubiquitin that are linked together in a head-to-tail manner (Wiborg et al., 1985). Conversely, the N-terminal of Uba52 and Rps27a each code for a single ubiquitin that is fused at the C-terminal to the ribosomal proteins L40 and S27a, respectively (Redman and Rechsteiner, 1989; Webb, Baker, Coggan, and Board, 1994). These precursors are usually processed post-translationally into single ubiquitin molecules by deubiquitinating enzymes, though processing during translation has also been reported for Ubb and Ubc (Grou et al., 2015). While it is clear all the ubiquitin genes code for the same protein and are critical for cell viability (Kobayashi et al., 2016; Park and Ryu, 2014; Ryu et al., 2008), their functions are not necessarily redundant, and the importance of these four genes to ubiquitin homeostasis remains largely unknown. The ubiquitin protein contains eight different sites at which polyubiquitin chains can form: The first methionine on the N-terminal (M1) or lysine (K) 6, 11, 27, 29, 33, 48, and 63 (Figure 1B). While some proteins will acquire only a single ubiquitin modification, termed monoubiquitination, most substrates acquire large polyubiquitin chains (Figure 1C). The linkage for these polyubiquitin chains can be homogeneous or mixed, with each ubiquitin molecule being modified at only one site by the next ubiquitin. However, in some cases, a target substrate can acquire a branched polyubiquitin chain, which is when ubiquitin is modified at multiple sites by additional ubiquitin molecules (Akutsu et al., 2016; Komander and Rape, 2012; Swatek and Komander, 2016). As a result, the ubiquitin code is highly complex, and a given polyubiquitin linkage site may result in different cell fates or functions depending on whether the chain is homogeneous, mixed, or branched. In this section, we discuss the known biological functions of the seven homogeneous polyubiquitin chains. Additionally, we also discuss the cellular functions of mixed and branched polyubiquitin chains and monoubiquitination. A summary of the various functions regulated by different polyubiquitin chains is presented in Table 1.
Figure 1. The complexity of the ubiquitin code.
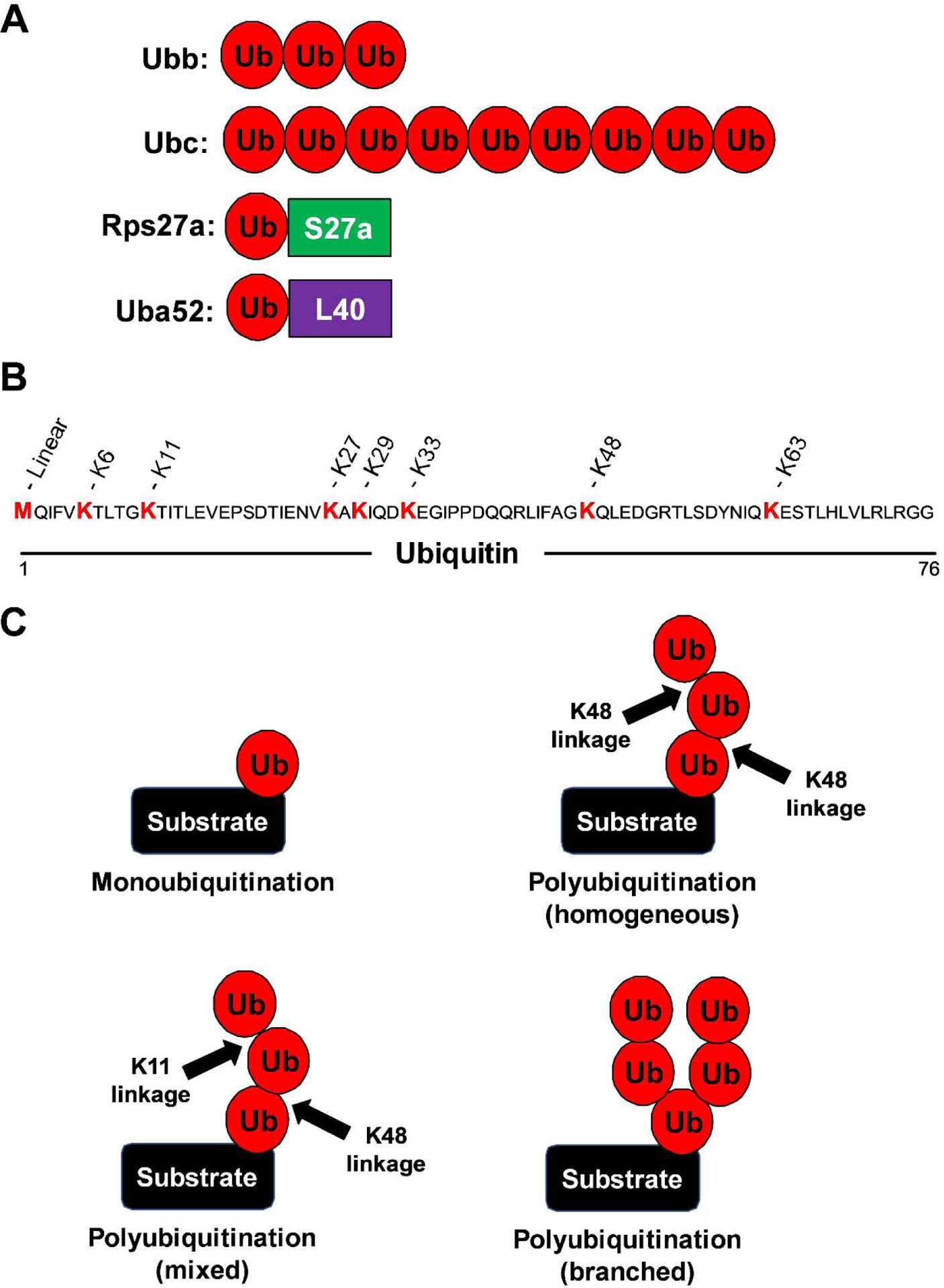
(A) Illustration of the four genes that code for ubiquitin and the number of ubiquitin molecules each code for. (B) The sequence of the 76 amino acid ubiquitin protein showing the 8 sites at which polyubiquitin chains can form. (C) The different structures of ubiquitin chains, including mono, homogeneous poly, mixed poly and branched poly forms.
Table 1:
The diverse functions of ubiquitin chains
| Ubiquitin linkage | Promote degradation | Impede degradation | DNA damage response | Kinase in/activation* | mRNA stability | Epigenetics | Trafficking | Endocytosis | Apoptosis | Autophagy |
|---|---|---|---|---|---|---|---|---|---|---|
| Mono | X | X | X | X | X | |||||
| M1/Linear | X | X | X | X | ||||||
| K6 | X | X | ||||||||
| K11 | X? | |||||||||
| K27 | X | X | X | |||||||
| K29 | X | X | X | |||||||
| K33 | X? | X | X | |||||||
| K48 | X | |||||||||
| K63 | X | X | X | X | X | X | ||||
| Mixed M1/K63 | X | |||||||||
| Branched K48/K63 | X | X | ||||||||
| Branched K48/K63/K11 | X | |||||||||
| Branched K11 | X |
This category includes protein binding activity
3.1. The canonical degradation chain: K48
The most abundant and well-studied polyubiquitin linkage site is K48, largely due to evidence that it is the canonical degradation chain (Chau et al., 1989). The proteasome can target proteins with at least four K48-linked ubiquitin molecules for degradation, and binding occurs at 19S subunits RPN10 and RPN13 (Grice and Nathan, 2016; Peth, Uchiki, and Goldberg, 2010). Importantly, the proteasome’s affinity for K48-linked ubiquitin chains is up to 100-fold greater than for mono- or di-ubiquitinated proteins (Clague and Urbe, 2010; Thrower, Hoffman, Rechsteiner, and Pickart, 2000). Additionally, the application of proteasome inhibitors results in a rapid accumulation of K48 polyubiquitinated proteins (Xu et al., 2009), furthering emphasizing the importance of this linkage site in the protein degradation process. To date, functions of K48-linked chains that are independent of the proteasome remain equivocal, and those that have been identified are usually associated with mixed or branched chains, as described in a later section.
3.2. The canonical degradation-independent chain: K63
K63 linkage sites have been the second most studied polyubiquitin chains and are considered to be the canonical protein degradation-independent chain. This designation resulted from studies showing that levels of K63 polyubiquitin conjugates remain largely unchanged following proteasome inhibition and, in general, the proteasome has low affinity for this ubiquitin chain (Jacobson et al., 2009; Nathan et al., 2013; Xu et al., 2009). Instead, this chain generally targets substrates for other cellular fates, and while the potential functions of K63 polyubiquitination in cells are diverse, the focus has generally been on control of protein kinase activation (Chen and Sun, 2009). For example, there is strong evidence for K63 chain involvement in the activation of the NF-κB transcriptional pathway (Wang et al., 2012), a well described regulator of synaptic plasticity and memory formation (Merlo, Freudenthal, Maldonado, and Romano, 2005; Yeh, Lin, Lee, and Gean, 2002). In the canonical pathway, protein K63 polyubiquitination can act as docking sites for other factors necessary for NF-κB activation, such as TAB2 (Kanayama et al., 2004; Kulathu et al., 2009), or can lead to the oligomerization of proteins such as RICK that also influences the activation of this transcriptional pathway (Hasegawa et al., 2008). Activation of NF-κB can also occur via K63 polyubiquitination of other proteins, such as TRAF2 and RIP1 (Bertrand et al., 2011; Skaug, Jiang, and Chen, 2009). Additionally, strong evidence suggests a role for K63 polyubiquitination in the activation of other protein kinases, such as AKT (Yang et al., 2009; Yang, Zhang, and Lin, 2010), further emphasizing the role of the K63 chain in kinase activation. K63 polyubiquitination has also been shown to be involved in the DNA damage response where K63 chains on histone proteins serve as recognition sites for downstream repair proteins (Lee et al., 2017). Furthermore, a large number of plasma membrane proteins have been identified as targets of K63 polyubiquitination, suggesting a role in intracellular trafficking (Erpapazoglou, Walker, and Haguenauer-Tsapis, 2014). This chain has also been shown to be involved in endocytosis, autophagy, and mitophagy (Cunningham et al., 2015; Shaid et al., 2013; Tanno and Komada, 2013). Thus, while the potential functions of the K48 chain are minimal, the K63 linkage site can target substrates for a variety of different cellular fates.
Despite the notion that K63 polyubiquitination is independent of the protein degradation process, some evidence suggests that the proteasome can target this ubiquitin chain. In vitro models have shown that K63 linked tetra-ubiquitin can be targeted by the proteasome for degradation (Hofmann and Pickart, 2001), an effect that has also been reported in vivo (Saeki et al., 2009). While the reason for this discrepancy is unclear, some recent evidence suggests that it may be due to branched ubiquitin chains. For example, K63 ubiquitination can act as a seed to recruit a K48/K63 branched ubiquitin chain, leading to degradation of the substrate by the proteasome (Ohtake, Tsuchiya, Saeki, and Tanaka, 2018). We discuss these branched chains in more detail in a later section; however, this does suggest that K63 polyubiquitination can lead to degradation of the substrate under certain conditions.
3.3. Noncanonical lysine chains: K6, K11, K27, K29, K33
K6 polyubiquitin chains do not accumulate following proteasome inhibition (Kim et al., 2011), thus are considered to be independent of the protein degradation process. Instead, this linkage site has been implicated in several cellular processes. Similar to K63, the K6 linkage site has been widely implicated in the DNA damage response (Morris and Solomon, 2004; Nishikawa et al., 2004; Wagner et al., 2011). Additionally, strong evidence suggests a role for K6 polyubiquitination in mitochondria homeostasis as this chain typically accumulates on depolarized mitochondria (Durcan et al., 2014). Consistent with this, mutations of K6 delayed mitophagy, similar to what was observed with K63 chains (Cunningham et al., 2015; Ordureau et al., 2015). Interestingly, some evidence indicates that K6 polyubiquitination can also inhibit the protein degradation process (Shang et al., 2005), suggesting a potentially unique function of this linkage site relative to many of the other ubiquitin chains. However, due to difficulties in its detection, much still remains unknown about the physiological roles of K6 polyubiquitin chains.
Unlike the K6 chain, K11 polyubiquitination has been well studied, and, in general, there is a strong consensus that this linkage site is involved in the protein degradation process (Matsumoto et al., 2010; Qin et al., 2014). However, this chain is unique relative to the K48 degradation signal in that proteasome affinity for K11 linkage is poor. Instead, K11 branched chains are generally a better signal for degradation (Grice et al., 2015; Meyer and Rape, 2014). This has led to questions regarding the actual function of homogeneous K11 chains as a lack of proteasome affinity suggests a role independent of protein degradation. Though indirect evidence points to a role in kinase activation and receptor trafficking (Hu et al., 2013; Swatek and Komander, 2016), to date, the functions of homogeneous K11 polyubiquitination remain unknown.
The K27 chain has been implicated in a variety of cellular processes. Similar to K11 and K48, K27 ubiquitin chains are associated with degradation of the substrate by the proteasome; however, this linkage site actually slows the degradation process (Birsa et al., 2014). Additionally, other studies suggest potential functions that are independent of the protein degradation process (Arimoto et al., 2010). For example, K27 polyubiquitination of NEMO serves as a docking site for Rhbdd3, a member of the rhomboid family of proteases, which also undergoes K27 polyubiquitination. This results in the recruitment of deubiquitinating enzymes that prevent K63 polyubiquitination of NEMO and the subsequent activation of NF-κB during the autoimmune response (Liu et al., 2014). Similar to K6 and K63 chains, K27 polyubiquitination has been implicated in the DNA damage response where it recruits DNA damage repair factors such as the p53 binding protein 1 (Gatti et al., 2015), suggesting a protein recruitment role of this polyubiquitin chain. Interestingly, some evidence indicates that K27 chains are not cleaved by most deubiquitinating enzymes, a unique characteristic relative to other linkage sites (Castaneda et al., 2016). This has made it difficult to identify the interacting proteins of the K27 linkage site, limiting our understanding of this noncanonical ubiquitin chain.
K29 chains have been largely implicated in functions independent of the protein degradation process, though some exceptions have been noted. Proteasome inhibition results in an accumulation of K29 polyubiquitinated proteins (Kim et al., 2011), suggesting that this mark could be targeted for substrate degradation by the proteasome. However, this relationship is complicated by evidence indicating that proteasome subunit RPN13 is ubiquitinated with a K29 chain in response to proteasomal stress (Besche et al., 2014), which prevents further substrate engagement with the proteasome, indicating that accumulation of this polyubiquitin chain following proteasome inhibition may occur as a result of mechanisms independent of K29-targeted substrate degradation. Independent of the protein degradation process, K29 chains have been implicated in the regulation of Wnt/β-catenin signaling (Fei et al., 2013), a critical regulator of memory formation (Maguschak and Ressler, 2011; Tan et al., 2013). This polyubiquitin chain has also been implicated in mRNA stability and epigenetic regulation via its targeting of the histone lysine demethylase KDM4D (Jin et al., 2016; Zhou, Geng, Luo, and Lou, 2013), the latter of which targets histone H3 lysine 9, a well described regulator of synaptic plasticity and memory formation (Gupta-Agarwal et al., 2012; Gupta-Agarwal, Jarome, Fernandez, and Lubin, 2014; Schaefer et al., 2009).
K33-linked polyubiquitin chains are considered to be independent of the protein degradation process, instead being primarily involved in intracellular trafficking. For example, this chain stabilizes Actin for post-Golgi transport and Coronin 7 for recruitment to the trans-Golgi network (Yuan et al., 2014). Additionally, K33 chains have also been shown to regulate T-cell receptor-ζ function by controlling its phosphorylation and protein binding abilities (Huang et al., 2010). It is important to note that some evidence does suggest that the K33 polyubiquitin mark can lead to protein degradation (Kim et al., 2013; Michel et al., 2015; Xu et al., 2009); however, this has never been directly proven.
3.4. Atypical, lysine-independent chain: M1
Unlike the polyubiquitin chains described above, linear (M1) chain linkage does not occur at a lysine site. Instead, in linear chains the linkage site is the first methionine on the N-terminal. Another unique characteristic of linear polyubiquitination is that only a single E3 ligase can conjugate this chain, which is known as LUBAC (Spit, Rieser, and Walczak, 2019). The physiological roles of linear polyubiquitination have been a hot topic in recent years, with most evidence suggesting functions independent of the protein degradation process. For example, LUBAC has been heavily studied in the activation of the NF-κB transcriptional pathway. LUBAC was shown to target NEMO, conjugating a linear polyubiquitin mark that was necessary for interaction with the TNF-R1 signaling complex. This resulted in the stabilization of the complex and activation of the NF-κB pathway (Haas et al., 2009; Tokunaga et al., 2009). The linear chain then acts as a scaffold to recruit other essential factors for NF-κB activation, including IKK, to the NEMO complex (Rahighi et al., 2009; Tokunaga, 2013; Tokunaga et al., 2011). Additionally, linear ubiquitination has been shown to activate NF-κB via targeting of other proteins, including Optineurin (Nakazawa et al., 2016). Genetic loss of linear polyubiquitination partially impairs NF-κB activation (Gerlach et al., 2011; Seymour et al., 2007), though other studies have reported no change or increases in NF-κB activity following manipulations of LUBAC (Zak et al., 2011). While the reasons for these conflicting results are unclear, it is possible that this could be due to the redundant role of K63 polyubiquitination in NF-κB activation or evidence that linear polyubiquitination can be involved in integrin activation (Rantala et al., 2011). Regardless, it is clear that the linear linkage site at least partially contributes to the regulation of NF-κB activation.
Considering its role in TNF signaling, the linear chain has also been shown to regulate apoptosis via targeting of cFLIP, a master anti-apoptotic regulator and resistance factor, protecting it from proteasome-mediated degradation (Tang et al., 2018). Interestingly, unlike K6, K27, and K63 polyubiquitin chains, evidence suggests linear linkage is not involved in the DNA damage response (Zhao and Ulrich, 2010). It should also be noted that while generally considered to be independent of the protein degradation process, some evidence suggests that linear polyubiquitination can target substrates for degradation by the proteasome (Zhao and Ulrich, 2010), though this has not yet been confirmed by other studies.
3.5. Branched/mixed ubiquitin chains
Complicating our understanding of the diverse functions of specific polyubiquitin chains is evidence that some substrates can carry multiple ubiquitin of mixed linkage. Adding another layer of complexity, these mixed (or homeogenous) chains can be also be branched, whereby two separate polyubiquitin chains are formed on the same substrate. In general, these mixed and branched chains have been challenging to study as the current methodology cannot distinguish a homogeneous chain from one that is mixed or branched, essentially making them “invisible” to detection (Swatek and Komander, 2016). Despite this, we have been able to learn about the physiological role of at least a few mixed and branched polyubiquitin chains (Figure 2). For example, strong evidence suggests that K63 chains can be modified by M1 chains in a mixed or branched structure (Emmerich et al., 2013). The basis for this is that LUBAC has been shown to bind to polyubiquitin chains of different compositions, and one of the main components of this complex, HOIP (RNF31), can bind ubiquitin that is part of an existing chain (Stieglitz et al., 2013). This mixed K63/M1 chain could explain the apparently redundant role of each of these linkage sites in NF-κB activation via NEMO. A branched chain of K48/K63/K11 has been shown to impede while a branched K11 chain facilitates degradation of the substrate by the proteasome (Kim et al., 2007; Meyer and Rape, 2014). Conversely, despite the well-known role of K48 homogeneous chains in the protein degradation process (Ohtake et al., 2016), branched K48/K63 chains have been shown to facilitate NF-κB activation or lead to substrate degradation, as described in Section 3.2 (Ohtake et al., 2018). Mixed chains of K11/K48 are primarily a degradation signal (Grice et al., 2015; Meyer and Rape, 2014). The K11/K63 mixed polyubiquitin chains can lead to endocytosis (Boname et al., 2010). Some other identified mixed polyubiquitin chains include K11/K29, K6/K48, K27/K48, K29/K48, and K11/K33, though the functions of these chains are unknown.
Figure 2. Known functions of mixed and branched polyubiquitin chains.
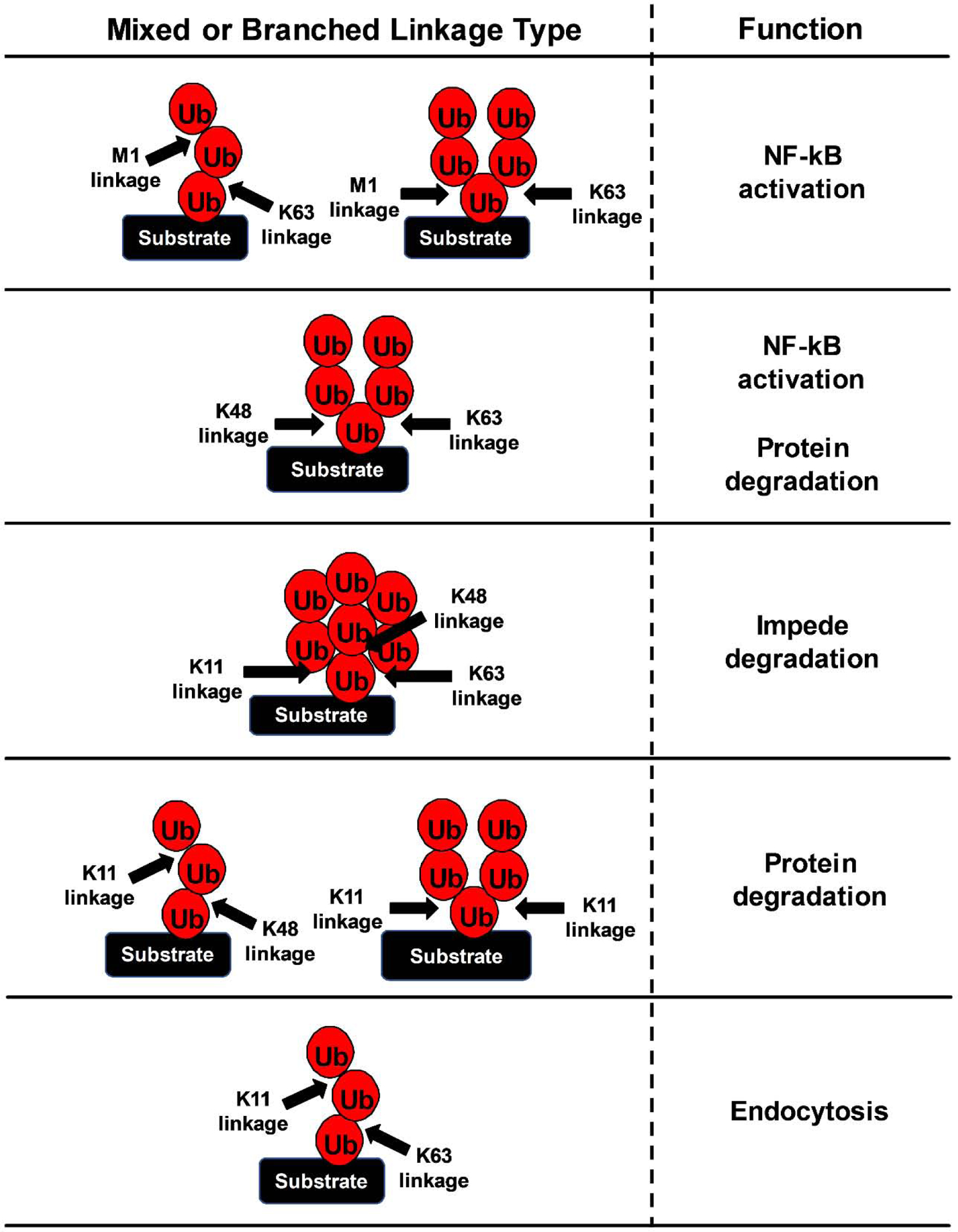
Illustration of mixed and branched polyubiquitin chains (left) and their known functions (right). Mixed chains are indicated by a single ubiquitin (Ub) column attached to the base ubiquitin. Branched chains are indicated by multiple (2–3) ubiquitin columns attached to the base ubiquitin. Arrows denote the linkage type for that specific site.
3.6. Monoubiquitination
In general, there is a strong consensus that monoubiquitination is involved in substrate localization, endocytosis, transcriptional control, and epigenetic regulation (Hicke, 2001). For example, the transcription factor FOXO4 can be monoubiquitinated, which results in re-localization to the nucleus and an increase in its activity (van der Horst et al., 2006). Some of the more well-described monoubiquitin targets are histones H2A and H2B, the latter which regulates the chromatin structure via recruitment of other epigenetic modifiers such as histone H3 methylation (Chandrasekharan, Huang, and Sun, 2009; Sun and Allis, 2002). While monoubiquitin modifications typically do not target proteins for degradation, some recent evidence suggests that it can under certain circumstances. In particular, some substrates that acquire multiple monoubiquitin marks can be degraded by the proteasome (Dimova et al., 2012; Lu et al., 2015), though this is complicated by evidence that not all multi-monoubiquitinated proteins undergo protein degradation (Lu et al., 2015). Why the reason for this sparing of some but not other multi-monoubiquitinated proteins remains equivocal, some evidence suggests that it may be related to the position of unfolded regions within the target substrate (Prakash et al., 2004; Shabek et al., 2012).
4. The role of protein ubiquitination in synaptic plasticity and memory formation
Despite the diversity of ubiquitin chains and the various functions they serve in cellular signaling, few studies have directly examined the role of polyubiquitin modifications in synaptic plasticity and memory formation (Table 2). As the primary focus has been on protein degradation, K48-linked chains have received the most attention, though some recent evidence implicates K63-linked and linear polyubiquitin chains in synaptic plasticity and/or memory storage processes. However, to date, no study has examined if K6, K11, K27, K29, and K33 polyubiquitin chains are altered as a result of or involved in activity- and learning-dependent synaptic plasticity. Furthermore, the potential involvement of mixed or branched polyubiquitin chains in any cellular process in the brain remains unknown. In this section, we will review the known roles of K48, K63, and linear polyubiquitination in synaptic plasticity and memory formation. Additionally, we will briefly discuss evidence implicating protein monoubiquitination in these processes.
Table 2:
Ubiquitin modifications in synaptic plasticity and memory formation
| Ubiquitin linkage | Neuronal stimulation | Learning | Critical for memory |
|---|---|---|---|
| Mono | Increase | Increase | Yes - positive regulator |
| Multimono | ? | Decrease | Yes - negative regulator |
| M1/Linear | ? | Increase | ? |
| K6 | ? | ? | ? |
| K11 | ? | ? | ? |
| K27 | ? | ? | ? |
| K29 | ? | ? | ? |
| K33 | ? | ? | ? |
| K48 | Decrease | Increase | Yes - positive regulator |
| K63 | Decrease | Increase | ? |
| Mixed | ? | ? | ? |
| Branched | ? | ? | ? |
4.1. K48 polyubiquitination
Considering the focus on protein degradation in activity- and learning-dependent synaptic plasticity, several studies have examined changes in K48 polyubiquitination in the brain. Stimulation of cultured hippocampal neurons with NMDA decreased K48 polyubiquitination in dendritic spines, consistent with increased proteasome activity and protein degradation (Bingol et al., 2010). Furthermore, auditory and contextual fear learning transiently increased K48 polyubiquitination in the amygdala in a learning-dependent manner (Jarome et al., 2013; Jarome et al., 2011), which is largely localized to the nucleus and cytoplasm (Orsi et al., 2019). A similar increase in K48-linked polyubiquitination has been observed in the medial prefrontal cortex following trace fear conditioning (Reis et al., 2013). Additionally, retrieval of contextual and auditory fear memories increases K48 polyubiquitination in the amygdala (Ferrara et al., 2019; Jarome et al., 2011), which is largely localized to synapses (Orsi et al., 2019). While no study has directly manipulated K48 ubiquitin chains in the brain, which is due to the diversity of ubiquitin ligases involved in conjugation of this linkage site, indirect evidence from proteasome inhibition strongly implicates K48 polyubiquitination in synaptic plasticity and memory formation and modification (Dong, Bach, Haynes, and Hegde, 2014; Dong et al., 2008; Ehlers, 2003; Ferrara et al., 2019; Jarome, Ferrara, Kwapis, and Helmstetter, 2015; Jarome et al., 2011; Lee, 2008; Lee et al., 2008). Furthermore, while the protein targets of K48 polyubiquitination during memory formation and storage have yet to be identified, a number of strong candidates exist based off of proteasome targeting including the synaptic scaffolds SHANK and GKAP, the RNAi-induced silencing complex (RISC) factor MOV10 and the epigenetic modifier HDAC7 (Jarome et al., 2011; Jing et al., 2017; Lee et al., 2008).
4.2. K63 polyubiquitination
Unlike K48-linked chains, K63 polyubiquitination has seldom been examined in the brain. One study found that K63 polyubiquitination is abundant in the mouse forebrain and hippocampus and that in cultured hippocampal neurons, PSD95 can undergo K63 polyubiquitination (Ma et al., 2017). This modification of PSD95 occurred via the coordinated actions of the E2 complex UBC13/UEV1a and E3 ligase TRAF6 and could be reversed by the deubiquitinating enzyme CYLD. Neuronal stimulation resulted in a rapid decrease in K63 conjugates globally and locally at synapses and silencing CYLD abolished chemically-induced long-term depression (LTD), suggesting that loss of K63 polyubiquitination may be related to K48-linked chain degradation during synaptic plasticity. Consistent with this relationship but with an inverse pattern, recently, our group found that learning in a contextual fear conditioning paradigm increased K63 polyubiquitination in the amygdala, which correlated with increased K48-linked polyubiquitination (Orsi et al., 2019). Interestingly, these changes in K63 polyubiquitin were present in the nucleus, but not in the cytoplasm or at synapses, suggesting that it could be involved in transcriptional control or the DNA damage response, as the latter has been shown to occur as a result of learning (Madabhushi et al., 2015). Additionally, we also found that in the amygdala, K63 polyubiquitination increased at synapses and in the nucleus, but not in the cytoplasm, following retrieval (Orsi et al., 2019). Collectively, these results suggest potentially unique and contradicting functions for K63 polyubiquitination in synaptic plasticity and memory formation. However, these remain the only investigations of how K63-linked ubiquitin chains may be involved in activity-and learning-dependent synaptic plasticity. Furthermore, the protein targets and functional significance of K63 polyubiquitination to memory formation or other forms of synaptic plasticity has yet to be elucidated.
4.3. Linear polyubiquitination
Similar to K63 polyubiquitination, linear ubiquitination has been largely unexplored in the brain, and, to date, only one study has examined the role of this polyubiquitin chain in activity- or learning-dependent synaptic plasticity. Our group found that following contextual fear conditioning linear polyubiquitination increased in the nucleus, but not cytoplasm or at synapses, in the amygdala (Orsi et al., 2019). These results support a potential role of linear polyubiquitination in transcriptional control during memory formation, though this has never been directly tested. We also found that memory retrieval again increased linear polyubiquitination, though this was selective to synapses, suggesting a potential role for this ubiquitin modification in the reconsolidation process. However, similar to K63-linked chains, the protein targets and functional significance of linear polyubiquitination to synaptic plasticity and memory formation have yet to be elucidated.
4.4. Monoubiquitination
While less studied than polyubiquitination, some evidence suggests that protein monoubiquitination is also critical for synaptic plasticity and memory formation. The first study to investigate this modification in the brain found that neuronal stimulation increased nonproteolytic monoubiquitination of CPEB3, which occurred via the E3 ligase Neuralized1, and this was critical for the consolidation of hippocampus-dependent spatial memories (Pavlopoulos et al., 2011). Additionally, one recent study found that TrkB, the receptor for BDNF, carried a multimonoubiquitin modification, which was subsequently deubiquitinated (removed) in stimulated hippocampal neurons (Guo et al., 2017). Furthermore, inhibiting removal of the multimonoubiquitin modification from TrkB impaired hippocampus-dependent spatial memory, suggesting that multimonoubiquitin modifications may act to limit memory formation. However, beyond this, little is known about the role of protein single or multi monoubiquitination in activity-and learning-dependent synaptic plasticity.
5. Technical limitations and future directions
Despite recent evidence that some degradation-independent polyubiquitin chains are likely involved in synaptic plasticity and memory formation, much still remains unknown about the ubiquitin code during these processes. This largely results from several technical limitations, which have hindered identifying and manipulating diverse polyubiquitin chains in the brain. However, as technology has started to advance in this field, it is becoming increasingly more likely that many different polyubiquitin linkage sites will be revealed to play a significant role in various cellular processes during activity- and learning-dependent synaptic plasticity. In this section, we review the technical limitations that have slowed the study of the ubiquitin code in brain tissue and what the next steps the field should take to remedy the resulting lack in knowledge. Additionally, we speculate on the potential functions of diverse polyubiquitin chains in synaptic plasticity and memory formation.
5.1. Technological limitations in identifying and manipulating diverse polyubiquitin chains in the brain
While recent technological advancements have increased our understanding of the ubiquitination process in various cell and tissue types (Choi et al., 2019; Michel, Swatek, Hospenthal, and Komander, 2017), a number of limitations have hindered our understanding of diverse polyubiquitin chains in the brain. One of the main technological limitations is in identifying different polyubiquitin linkage sites. For example, one of the most widely published polyubiquitin antibodies, the FK2 clone, cannot distinguish between the many different linkage sites, resulting in the quantification of overall polyubiquitination irrespective of the type of chain a target protein has acquired (Fujimuro, Sawada, and Yokosawa, 1994). However, recent developments of linkage-specific polyubiquitin antibodies have helped to bridge this technology gap (Newton et al., 2008) and, to date, commercially available antibodies are now available that can detect K11, K27, K48, K63 and M1/linear in a broad range of species and tissue types. Additionally, the development of Tandem Ubiquitin Binding Entities (TUBEs), when combined with mass spectrometry, now allow for the identification of K48, K63, and M1-targeted protein substrates (Hjerpe et al., 2009). Despite this, the spectrum of commercially available antibodies and TUBE reagents remains limited, and some of the less studied linkage sites, such as K6, K29, and K33, remain difficult to quantify. Different polyubiquitin chains can also be identified via electrophoretic mobility as each linkage site migrates differently during SDS-PAGE (Emmerich and Cohen, 2015; Komander et al., 2009). Furthermore, linkage-specific ubiquitin chains on a protein of interest can be determined by using immobilized ubiquitin-binding domains (UBD) of defined ubiquitin chain specificity, followed by immunoblotting. This method is capable of detecting M1, K29, K33, and K63 chains on a target protein (Scott et al., 2015). Finally, polyubiquitin chains can also be identified by treatment of the tissue with specific deubiquitinating enzymes, as some have a high affinity for only one type of linkage site (Mevissen et al., 2013). Despite this, a majority of these methods, except for the simultaneous use of multiple UBDs and TUBES, are incapable of deciphering between homogeneous and mixed polyubiquitin chains and no method is able to identify whether the linkage type is branched. Consequently, while these technological developments have aided in the recent study of linkage-specific polyubiquitin chains in synaptic plasticity and memory formation (Jarome et al., 2011; Orsi et al., 2019), a number of methodological barriers still exist.
Even when identified, a major hurdle in understanding the role of a specific polyubiquitin chain in synaptic plasticity comes from the inability to specifically manipulate that linkage site in a given brain region. A common strategy is to target the E3 ligase responsible for conjugating that specific polyubiquitin chain to the target substrate (Tokunaga et al., 2011), which could be achieved via controlled knockout or RNAi, and has been effective at elucidating the role of specific ubiquitin conjugating enzymes to various behavioral processes and disease states (e.g., Pick, Malumbres, and Klann, 2013; Pick, Wang, Mayfield, and Klann, 2013; Werner et al., 2018; Yao et al., 2011). However, this is complicated by the diversity and promiscuous nature of E3 ligases as, with the exception of linear polyubiquitination, multiple E3 ligases are capable of conjugating specific ubiquitin chains to a target substrate. Thus, this approach often provides very little information about the role of a specific polyubiquitin linkage site in the biological process examined. This issue potentially could be partially overcome by combining ubiquitin E3 ligase manipulation with mass spectrometry, which could identify the specific type of polyubiquitin chains altered by loss of the protein. However, again it still remains likely that the E3 ligase could be regulating multiple polyubiquitin linkage sites. A better approach would be to make point mutations in specific linkage sites on ubiquitin, a method that has been widely used in yeast and various human cell lines but has yet to be applied to the brain (e.g., Meza Gutierrez et al., 2018; Petroski and Deshaies, 2005). A potential concern with these point mutations could be cell viability following mutations that lack temporal and spatial control as various polyubiquitin chains regulate cellular processes with critical roles in early brain development and loss of even a single ubiquitin coding gene can be lethal (Bowerman and Kurz, 2006; Chen et al., 2011; Hallengren, Chen, and Wilson, 2013; Kobayashi et al., 2016; Park and Ryu, 2014; Ryu et al., 2008). Thus, even if the levels of a specific polyubiquitin chain are shown to change in brain tissue during activity- or learning-dependent synaptic plasticity, specific manipulation of this linkage site can be difficult. However, it is possible that this issue could be circumvented by rapidly expanding technology, such as viral-mediated overexpression of ubiquitin dominant negative point mutations or via use of recently developed single base-pair DNA and RNA editing tools, including CRISPR-dCas9nickase or CRISPR-Cas13 (Cox et al., 2017; Navabpour, Kwapis, and Jarome, 2020; Shen et al., 2014), which could control endogenous ubiquitin mutation expression in the adult brain. Consistent with this, recent evidence confirms that various CRISPR platforms can successfully control gene expression selectively in the adult brain via stereotaxic delivery (Butler, Johnston, Kaur, and Lubin, 2019; Kwapis et al., 2018; Savell et al., 2019), which would allow these point mutations to be expressed after the critical developmental period. Thus, while the current tools cannot manipulate specific polyubiquitin linkage sites in the brain, the technology is there for such approaches to be developed.
5.2. Furthering our understanding of linkage-specific polyubiquitin chains in synaptic plasticity and memory formation
Despite the difficulties in identifying and manipulating various polyubiquitin chains in the brain, increasing evidence suggests that they likely play a significant role in activity- and learning-dependent synaptic plasticity. The initial focus of future studies should be on quantifying changes in these different polyubiquitin chains following neuronal stimulation or behavioral training. As stated above, while K48, K63, and M1 chains have been reported to increase as a function of neuronal stimulation and/or learning (Bingol et al., 2010; Ma et al., 2017; Orsi et al., 2019), K11 and K27 have yet to be studied despite the available of antibodies to detect them. Additionally, for those that have been reported to have altered expression, a majority of the target proteins have yet to be identified (Jarome et al., 2011; Lee et al., 2008; Ma et al., 2017). This information is essential, though, to fully understand the functional role of linkage-specific polyubiquitin modifications in synaptic plasticity and memory formation, especially for the chains that are known to have many functions (K63, M1). Furthermore, a better understanding of how these chains interact during these processes is particularly important since some linkage sites have been shown to regulate specific cellular processes via a mixed confirmation, such as K63/M1 (Emmerich et al., 2013). Finally, manipulation of specific polyubiquitin linkage sites will be critical to define the functional significance of any ubiquitin chain to synaptic plasticity and memory formation, which may require the development of new technology, as discussed above.
5.3. Potential functions of linkage-specific polyubiquitin chains in synaptic plasticity and memory formation
Even as evidence has started to emerge supporting a role for diverse polyubiquitin chains in synaptic plasticity and memory formation, questions still remain regarding what the functional significance of these modifications are to these processes. However, based on work in cultures or the periphery and what is currently known about the molecular mechanisms of activity- and learning-dependent synaptic plasticity, a number of intriguing potential candidate functions exist. For example, although only studied outside the brain, to date, various polyubiquitin chains (K63, M1) can activate the NF-κB transcriptional pathway, which plays a significant role in the long-term memory and synaptic plasticity (Kaltschmidt and Kaltschmidt, 2015). Also, considering the recently identified important role of double-stranded DNA breaks in chromatin remodeling during synaptic plasticity and memory formation (Madabhushi et al., 2015) and the well described role of various polyubiquitin chains in this process outside the brain (K6, K27, K63), there is a need for further studies to address the question of how specific polyubiquitin modifications regulate the DNA damage response during these processes (Merlo, Cuchillo-Ibañez, Parlato, and Rammes, 2016). Furthermore, mitochondria are a cellular organelle implicated in many psychological processes, especially ones that increase neuronal firing rates, such as learning and memory (Hebert-Chatelain et al., 2016) and synaptic plasticity (Todorova and Blokland, 2017), since it regulates cellular respiration and energy production. As we discussed earlier, various polyubiquitin chains have a role in mitochondria homeostasis (K6, K63). Based on this, there is a critical need to investigate the role of mitochondria polyubiquitination in synaptic plasticity and memory formation. Some ubiquitination marks also play a substantial role in receptor trafficking (mono, K33, K63), such as the deubiquitination of RTKs, membrane-bound receptors that facilitate the communication between cells and their environment, which triggers their endocytosis and recycling (Critchley et al., 2018). Interestingly, strong evidence indicates that RTKs have multiple functions in memory formation and synaptic plasticity (Giese and Mizuno, 2013); however, whether ubiquitination/deubiquitination of RTKs critical to these processes remains unknown. Thus, there are numerous ways in which diverse polyubiquitin modifications could contribute to activity- and learning-dependent synaptic plasticity, which should help guide future studies attempting to elucidate the role of the complex ubiquitin code in memory formation.
6. Conclusions
Strong support exists suggesting a role for ubiquitin-proteasome mediated protein degradation in synaptic plasticity and memory formation. While most protein substrates will be marked for degradation by a K48-linked polyubiquitin modification, there are at least seven other known linkage-specific polyubiquitin chains (M1, K6, K11, K27, K29, K33, K63) that a target protein can acquire, which can be independent of the proteasome and instead impede protein degradation or regulate kinase activation, endocytosis, the DNA damage response, mRNA stability, or apoptosis. Furthermore, in addition to homogenous chains, mixed or branched polyubiquitin modifications can target substrates for cellular fates that are dependent or independent of the protein degradation process. However, as the focus has remained on protein degradation, much remains unknown about the role of these diverse polyubiquitin chains in the brain during activity and learning-dependent synaptic plasticity. Here, we reviewed the known functions of these diverse polyubiquitin chains and evidence implicating some of them in synaptic plasticity and memory formation. Additionally, we discussed the technical limitations and recent advancements in this field, which may help with the identification and study of linkage-specific polyubiquitination chains in the brain. Collectively, the studies outlined in this review suggest diverse polyubiquitin modifications are likely involved in various cellular processes that are independent of protein degradation and underlie memory formation.
Highlights.
Polyubiquitin chains occur at 8 different linkage sites and can be mixed or branched
Many of these chains are involved in processes independent of the proteasome
The K48, K63 and M1 chains have been implicated in memory formation
K6, K11, K27, K29, K33 and mixed/branched chains have yet to be studied in the brain
Acknowledgements
This work was supported by National Institutes of Health grants MH120569 and MH120498 and startup funds from the College of Agricultural and Life Sciences and the School of Neuroscience at Virginia Tech (TJJ).
Abbreviations
- AKT
Protein kinase B
- BDNF
Brain-derived neurotrophic factor
- CaMKII
Calcium/calmodulin-dependent protein kinase II
- cFLIP
Cellular flice-like inhibitor protein
- CPEB3
Cytoplasmic polyadenylation element binding protein 3
- CREB
Cyclic AMP response element binding protein
- CYLD
Cylindromatosis tumor-suppressor protein
- ERK/MAPK
Extracellular signal regulated kinase
- FOX04
Forkhead box protein 04 peptide
- GKAP
Disks large-associated protein 1
- HDAC7
Histone deacetylase 7
- HOIP (RNF31)
Ring Finger 31
- IKK
The inhibitor of nuclear factor kappa B kinase
- KDM4D
Histone lysine demethylase 4D
- LUBAC
Linear ubiquitin chain assembly complex
- MOV10
Mov10 RISC complex RNA helicase
- mTOR
Mammalian target of rapamycin
- NEMO
NF-kappa B essential modulator
- NF-kB
Nuclear factor kappa B
- PKA
Protein kinase A
- PKC
Protein Kinase C
- PSD95
Postsynaptic density protein 95
- Rhbdd3
Rhomboid domain containing 3
- RICK
RIP like interacting CLARP kinase
- RIP1
Receptor interacting protein
- RPN10
26S proteasome non-ATPase regulatory subunit 4
- RPN13
Proteasomal ubiquitin receptor ADRM1
- Rps27a
Ribosomal protein S27a
- RPT6
Regulatory particle triple-A ATPase 6
- RTK
Receptor Tyrosine Kinase
- SHANK
SH3 and multiple ankyrin repeat domains
- TAB2
TGF-beta activated kinase 1 MAP3K7 binding protein 2
- TNF-R1
Tumor necrosis factor receptor 1
- TRAF2
TNF receptor associated factor 2
- TRAF6
TNF receptor associated factor 6
- TrkB
Tropomyosin receptor kinase B
- Uba52
Ubiquitin A-52 Residue Ribosomal Protein Fusion Product 1
- Ubb
Ubiquitin B
- Ubc
Ubiquitin C
- Wnt
Wingless and Int-1
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Akutsu M, Dikic I, & Bremm A (2016). Ubiquitin chain diversity at a glance. J Cell Sci, 129, 875–880. [DOI] [PubMed] [Google Scholar]
- Arimoto K, Funami K, Saeki Y, Tanaka K, Okawa K, Takeuchi O, Akira S, Murakami Y, & Shimotohno K (2010). Polyubiquitin conjugation to NEMO by triparite motif protein 23 (TRIM23) is critical in antiviral defense. Proc Natl Acad Sci U S A, 107, 15856–15861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artinian J, McGauran AM, De Jaeger X, Mouledous L, Frances B, & Roullet P (2008). Protein degradation, as with protein synthesis, is required during not only long-term spatial memory consolidation but also reconsolidation. Eur J Neurosci, 27, 3009–3019. [DOI] [PubMed] [Google Scholar]
- Asok A, Leroy F, Rayman JB, & Kandel ER (2019). Molecular Mechanisms of the Memory Trace. Trends Neurosci, 42, 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey DJ, Kim JJ, Sun W, Thompson RF, & Helmstetter FJ (1999). Acquisition of fear conditioning in rats requires the synthesis of mRNA in the amygdala. Behav Neurosci, 113, 276–282. [DOI] [PubMed] [Google Scholar]
- Bard JAM, Goodall EA, Greene ER, Jonsson E, Dong KC, & Martin A (2018). Structure and Function of the 26S Proteasome. Annual Review of Biochemistry, 87, 697–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford L, Paine S, Sheppard PW, Mayer RJ, & Roelofs J (2010). Assembly, structure, and function of the 26S proteasome. Trends Cell Biol, 20, 391–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekinschtein P, Cammarota M, Igaz LM, Bevilaqua LR, Izquierdo I, & Medina JH (2007). Persistence of long-term memory storage requires a late protein synthesis- and BDNF- dependent phase in the hippocampus. Neuron, 53, 261–277. [DOI] [PubMed] [Google Scholar]
- Bertrand MJ, Lippens S, Staes A, Gilbert B, Roelandt R, De Medts J, Gevaert K, Declercq W, & Vandenabeele P (2011). cIAP1/2 are direct E3 ligases conjugating diverse types of ubiquitin chains to receptor interacting proteins kinases 1 to 4 (RIP1–4). PLoS One, 6, e22356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besche HC, Sha Z, Kukushkin NV, Peth A, Hock EM, Kim W, Gygi S, Gutierrez JA, Liao H, Dick L, & Goldberg AL (2014). Autoubiquitination of the 26S proteasome on Rpn13 regulates breakdown of ubiquitin conjugates. EMBO J, 33, 1159–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bingol B, Wang CF, Arnott D, Cheng D, Peng J, & Sheng M (2010). Autophosphorylated CaMKIIalpha acts as a scaffold to recruit proteasomes to dendritic spines. Cell, 140, 567–578. [DOI] [PubMed] [Google Scholar]
- Birsa N, Norkett R, Wauer T, Mevissen TE, Wu HC, Foltynie T, Bhatia K, Hirst WD, Komander D, Plun-Favreau H, & Kittler JT (2014). Lysine 27 ubiquitination of the mitochondrial transport protein Miro is dependent on serine 65 of the Parkin ubiquitin ligase. J Biol Chem, 289, 14569–14582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boname JM, Thomas M, Stagg HR, Xu P, Peng J, & Lehner PJ (2010). Efficient internalization of MHC I requires lysine-11 and lysine-63 mixed linkage polyubiquitin chains. Traffic, 11, 210–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowerman B, & Kurz T (2006). Degrade to create: developmental requirements for ubiquitin-mediated proteolysis during early C. elegans embryogenesis. Development, 133, 773–784. [DOI] [PubMed] [Google Scholar]
- Butler AA, Johnston DR, Kaur S, & Lubin FD (2019). Long noncoding RNA NEAT1 mediates neuronal histone methylation and age-related memory impairment. Sci Signal, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai F, Frey JU, Sanna PP, & Behnisch T (2010). Protein degradation by the proteasome is required for synaptic tagging and the heterosynaptic stabilization of hippocampal late-phase long-term potentiation. Neuroscience, 169, 1520–1526. [DOI] [PubMed] [Google Scholar]
- Castaneda CA, Dixon EK, Walker O, Chaturvedi A, Nakasone MA, Curtis JE, Reed MR, Krueger S, Cropp TA, & Fushman D (2016). Linkage via K27 Bestows Ubiquitin Chains with Unique Properties among Polyubiquitins. Structure, 24, 423–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekharan MB, Huang F, & Sun ZW (2009). Ubiquitination of histone H2B regulates chromatin dynamics by enhancing nucleosome stability. Proc Natl Acad Sci U S A, 106, 16686–16691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chau V, Tobias JW, Bachmair A, Marriott D, Ecker DJ, Gonda DK, & Varshavsky A (1989). A multiubiquitin chain is confined to specific lysine in a targeted short-lived protein. Science, 243, 1576–1583. [DOI] [PubMed] [Google Scholar]
- Chen PC, Bhattacharyya BJ, Hanna J, Minkel H, Wilson JA, Finley D, Miller RJ, & Wilson SM (2011). Ubiquitin homeostasis is critical for synaptic development and function. J Neurosci, 31, 17505–17513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZJ, & Sun LJ (2009). Nonproteolytic functions of ubiquitin in cell signaling. Mol Cell, 33, 275–286. [DOI] [PubMed] [Google Scholar]
- Cho J, Sypniewski KA, Arai S, Yamada K, Ogawa S, & Pavlides C (2018). Fear memory consolidation in sleep requires protein kinase A. Learn Mem, 25, 241–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YS, Bollinger SA, Prada LF, Scavone F, Yao T, & Cohen RE (2019). High-affinity free ubiquitin sensors for quantifying ubiquitin homeostasis and deubiquitination. Nat Methods, 16, 771–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clague MJ, & Urbe S (2010). Ubiquitin: same molecule, different degradation pathways. Cell, 143, 682–685. [DOI] [PubMed] [Google Scholar]
- Cox DBT, Gootenberg JS, Abudayyeh OO, Franklin B, Kellner MJ, Joung J, & Zhang F (2017). RNA editing with CRISPR-Cas13. Science, 358, 1019–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Critchley WR, Pellet-Many C, Ringham-Terry B, Harrison MA, Zachary IC, & Ponnambalam S (2018). Receptor Tyrosine Kinase Ubiquitination and De-Ubiquitination in Signal Transduction and Receptor Trafficking. Cells, 7, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cromm PM, & Crews CM (2017). Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cell chemical biology, 24, 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cullen PK, Ferrara NC, Pullins SE, & Helmstetter FJ (2017). Context memory formation requires activity-dependent protein degradation in the hippocampus. Learn Mem, 24, 589–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham CN, Baughman JM, Phu L, Tea JS, Yu C, Coons M, Kirkpatrick DS, Bingol B, & Corn JE (2015). USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol, 17, 160–169. [DOI] [PubMed] [Google Scholar]
- Dimova NV, Hathaway NA, Lee BH, Kirkpatrick DS, Berkowitz ML, Gygi SP, Finley D, & King RW (2012). APC/C-mediated multiple monoubiquitylation provides an alternative degradation signal for cyclin B1. Nat Cell Biol, 14, 168–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djakovic SN, Marquez-Lona EM, Jakawich SK, Wright R, Chu C, Sutton MA, & Patrick GN (2012). Phosphorylation of Rpt6 regulates synaptic strength in hippocampal neurons. J Neurosci, 32, 5126–5131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Bach SV, Haynes KA, & Hegde AN (2014). Proteasome modulates positive and negative translational regulators in long-term synaptic plasticity. J Neurosci, 34, 3171–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong C, Upadhya SC, Ding L, Smith TK, & Hegde AN (2008). Proteasome inhibition enhances the induction and impairs the maintenance of late-phase long-term potentiation. Learn Mem, 15, 335–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrbaum AR, Alvarez-Castelao B, Nassim-Assir B, Langer JD, & Schuman EM (2020). Proteome dynamics during homeostatic scaling in cultured neurons. Elife, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Durcan TM, Tang MY, Perusse JR, Dashti EA, Aguileta MA, McLelland GL, Gros P, Shaler TA, Faubert D, Coulombe B, & Fon EA (2014). USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J, 33, 2473–2491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD (2003). Activity level controls postsynaptic composition and signaling via the ubiquitin-proteasome system. Nat Neurosci, 6, 231–242. [DOI] [PubMed] [Google Scholar]
- Eisele F, & Wolf DH (2008). Degradation of misfolded protein in the cytoplasm is mediated by the ubiquitin ligase Ubr1. FEBS Lett, 582, 4143–4146. [DOI] [PubMed] [Google Scholar]
- Eletr ZM, & Kuhlman B (2007). Sequence determinants of E2-E6AP binding affinity and specificity. J Mol Biol, 369, 419–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerich CH, & Cohen P (2015). Optimising methods for the preservation, capture and identification of ubiquitin chains and ubiquitylated proteins by immunoblotting. Biochem Biophys Res Commun, 466, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emmerich CH, Ordureau A, Strickson S, Arthur JS, Pedrioli PG, Komander D, & Cohen P (2013). Activation of the canonical IKK complex by K63/M1-linked hybrid ubiquitin chains. Proc Natl Acad Sci U S A, 110, 15247–15252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erpapazoglou Z, Walker O, & Haguenauer-Tsapis R (2014). Versatile roles of k63-linked ubiquitin chains in trafficking. Cells, 3, 1027–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fei C, Li Z, Li C, Chen Y, Chen Z, He X, Mao L, Wang X, Zeng R, & Li L (2013). Smurf1-mediated Lys29-linked nonproteolytic polyubiquitination of axin negatively regulates Wnt/beta-catenin signaling. Mol Cell Biol, 33, 4095–4105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara NC, Jarome TJ, Cullen PK, Orsi SA, Kwapis JL, Trask S, Pullins SE, & Helmstetter FJ (2019). GluR2 endocytosis-dependent protein degradation in the amygdala mediates memory updating. Sci Rep, 9, 5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueiredo LS, Dornelles AS, Petry FS, Falavigna L, Dargel VA, Kobe LM, Aguzzoli C, Roesler R, & Schroder N (2015). Two waves of proteasome-dependent protein degradation in the hippocampus are required for recognition memory consolidation. Neurobiol Learn Mem, 120, 1–6. [DOI] [PubMed] [Google Scholar]
- Finley D, Chen X, & Walters KJ (2016). Gates, Channels, and Switches: Elements of the Proteasome Machine. Trends in biochemical sciences, 41, 77–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimuro M, Sawada H, & Yokosawa H (1994). Production and characterization of monoclonal antibodies specific to multi-ubiquitin chains of polyubiquitinated proteins. FEBS Lett, 349, 173–180. [DOI] [PubMed] [Google Scholar]
- Gaczynska M, & Osmulski PA (2018). Targeting Protein-Protein Interactions in the Ubiquitin-Proteasome Pathway. Adv Protein Chem Struct Biol, 110, 123–165. [DOI] [PubMed] [Google Scholar]
- Gatti M, Pinato S, Maiolica A, Rocchio F, Prato MG, Aebersold R, & Penengo L (2015). RNF168 promotes noncanonical K27 ubiquitination to signal DNA damage. Cell Rep, 10, 226–238. [DOI] [PubMed] [Google Scholar]
- Gerlach B, Cordier SM, Schmukle AC, Emmerich CH, Rieser E, Haas TL, Webb AI, Rickard JA, Anderton H, Wong WW, Nachbur U, Gangoda L, Warnken U, Purcell AW, Silke J, & Walczak H (2011). Linear ubiquitination prevents inflammation and regulates immune signalling. Nature, 471, 591–596. [DOI] [PubMed] [Google Scholar]
- Giese KP, & Mizuno K (2013). The roles of protein kinases in learning and memory. Learn Mem, 20, 540–552. [DOI] [PubMed] [Google Scholar]
- Grice GL, Lobb IT, Weekes MP, Gygi SP, Antrobus R, & Nathan JA (2015). The Proteasome Distinguishes between Heterotypic and Homotypic Lysine-11-Linked Polyubiquitin Chains. Cell Rep, 12, 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grice GL, & Nathan JA (2016). The recognition of ubiquitinated proteins by the proteasome. Cell Mol Life Sci, 73, 3497–3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gropper R, Brandt RA, Elias S, Bearer CF, Mayer A, Schwartz AL, & Ciechanover A (1991). The ubiquitin-activating enzyme, E1, is required for stress-induced lysosomal degradation of cellular proteins. J Biol Chem, 266, 3602–3610. [PubMed] [Google Scholar]
- Grou CP, Pinto MP, Mendes AV, Domingues P, & Azevedo JE (2015). The de novo synthesis of ubiquitin: identification of deubiquitinases acting on ubiquitin precursors. Sci Rep, 5, 12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo YY, Lu Y, Zheng Y, Chen XR, Dong JL, Yuan RR, Huang SH, Yu H, Wang Y, Chen ZY, & Su B (2017). Ubiquitin C-Terminal Hydrolase L1 (UCH-L1) Promotes Hippocampus-Dependent Memory via Its Deubiquitinating Effect on TrkB. J Neurosci, 37, 5978–5995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta-Agarwal S, Franklin AV, Deramus T, Wheelock M, Davis RL, McMahon LL, & Lubin FD (2012). G9a/GLP histone lysine dimethyltransferase complex activity in the hippocampus and the entorhinal cortex is required for gene activation and silencing during memory consolidation. J Neurosci, 32, 5440–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta-Agarwal S, Jarome TJ, Fernandez J, & Lubin FD (2014). NMDA receptor- and ERK-dependent histone methylation changes in the lateral amygdala bidirectionally regulate fear memory formation. Learn Mem, 21, 351–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas TL, Emmerich CH, Gerlach B, Schmukle AC, Cordier SM, Rieser E, Feltham R, Vince J, Warnken U, Wenger T, Koschny R, Komander D, Silke J, & Walczak H (2009). Recruitment of the linear ubiquitin chain assembly complex stabilizes the TNF-R1 signaling complex and is required for TNF-mediated gene induction. Mol Cell, 36, 831–844. [DOI] [PubMed] [Google Scholar]
- Hallengren J, Chen PC, & Wilson SM (2013). Neuronal ubiquitin homeostasis. Cell Biochem Biophys, 67, 67–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamilton AM, Oh WC, Vega-Ramirez H, Stein IS, Hell JW, Patrick GN, & Zito K (2012). Activity-dependent growth of new dendritic spines is regulated by the proteasome. Neuron, 74, 1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa M, Fujimoto Y, Lucas PC, Nakano H, Fukase K, Nunez G, & Inohara N (2008). A critical role of RICK/RIP2 polyubiquitination in Nod-induced NF-kappaB activation. EMBO J, 27, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hebert-Chatelain E, Desprez T, Serrat R, Bellocchio L, Soria-Gomez E, Busquets-Garcia A, Zottola ACP, Delamarre A, Cannich A, & Vincent PJN (2016). A cannabinoid link between mitochondria and memory. 539, 555–559. [DOI] [PubMed] [Google Scholar]
- Hegde AN (2017). Proteolysis, synaptic plasticity and memory. Neurobiol Learn Mem, 138, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegde AN, Haynes KA, Bach SV, & Beckelman BC (2014). Local ubiquitin-proteasome-mediated proteolysis and long-term synaptic plasticity. Front Mol Neurosci, 7, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, & Ciechanover A (1998). The ubiquitin system. Annu Rev Biochem, 67, 425–479. [DOI] [PubMed] [Google Scholar]
- Hicke L (2001). Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol, 2, 195–201. [DOI] [PubMed] [Google Scholar]
- Hjerpe R, Aillet F, Lopitz-Otsoa F, Lang V, England P, & Rodriguez MS (2009). Efficient protection and isolation of ubiquitylated proteins using tandem ubiquitin-binding entities. EMBO Rep, 10, 1250–1258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeffer CA, & Klann E (2010). mTOR signaling: at the crossroads of plasticity, memory and disease. Trends Neurosci, 33, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann RM, & Pickart CM (2001). In vitro assembly and recognition of Lys-63 polyubiquitin chains. J Biol Chem, 276, 27936–27943. [DOI] [PubMed] [Google Scholar]
- Hu H, Brittain GC, Chang JH, Puebla-Osorio N, Jin J, Zal A, Xiao Y, Cheng X, Chang M, Fu YX, Zal T, Zhu C, & Sun SC (2013). OTUD7B controls noncanonical NF-kappaB activation through deubiquitination of TRAF3. Nature, 494, 371–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Jeon MS, Liao L, Yang C, Elly C, Yates JR 3rd, & Liu YC (2010). K33-linked polyubiquitination of T cell receptor-zeta regulates proteolysis-independent T cell signaling. Immunity, 33, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson AD, Zhang NY, Xu P, Han KJ, Noone S, Peng J, & Liu CW (2009). The lysine 48 and lysine 63 ubiquitin conjugates are processed differently by the 26 s proteasome. J Biol Chem, 284, 35485–35494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Ferrara NC, Kwapis JL, & Helmstetter FJ (2015). Contextual Information Drives the Reconsolidation-Dependent Updating of Retrieved Fear Memories. Neuropsychopharmacology, 40, 3044–3052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Ferrara NC, Kwapis JL, & Helmstetter FJ (2016). CaMKII regulates proteasome phosphorylation and activity and promotes memory destabilization following retrieval. Neurobiol Learn Mem, 128, 103–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, & Helmstetter FJ (2013). The ubiquitin-proteasome system as a critical regulator of synaptic plasticity and long-term memory formation. Neurobiol Learn Mem, 105, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, & Helmstetter FJ (2014). Protein degradation and protein synthesis in long-term memory formation. Front Mol Neurosci, 7, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Kwapis JL, Ruenzel WL, & Helmstetter FJ (2013). CaMKII, but not protein kinase A, regulates Rpt6 phosphorylation and proteasome activity during the formation of long-term memories. Front Behav Neurosci, 7, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarome TJ, Werner CT, Kwapis JL, & Helmstetter FJ (2011). Activity dependent protein degradation is critical for the formation and stability of fear memory in the amygdala. PLoS One, 6, e24349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jentsch S (1992). The ubiquitin-conjugation system. Annu Rev Genet, 26, 179–207. [DOI] [PubMed] [Google Scholar]
- Jin J, Xie X, Xiao Y, Hu H, Zou Q, Cheng X, & Sun SC (2016). Epigenetic regulation of the expression of Il12 and Il23 and autoimmune inflammation by the deubiquitinase Trabid. Nat Immunol, 17, 259–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jing X, Sui WH, Wang S, Xu XF, Yuan RR, Chen XR, Ma HX, Zhu YX, Sun JK, Yi F, Chen ZY, & Wang Y (2017). HDAC7 Ubiquitination by the E3 Ligase CBX4 Is Involved in Contextual Fear Conditioning Memory Formation. J Neurosci, 37, 3848–3863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen JP, Cain CK, Ostroff LE, & LeDoux JE (2011). Molecular mechanisms of fear learning and memory. Cell, 147, 509–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaltschmidt B, & Kaltschmidt C (2015). NF-KappaB in Long-Term Memory and Structural Plasticity in the Adult Mammalian Brain. Frontiers in molecular neuroscience, 8, 69–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanayama A, Seth RB, Sun L, Ea CK, Hong M, Shaito A, Chiu YH, Deng L, & Chen ZJ (2004). TAB2 and TAB3 activate the NF-kappaB pathway through binding to polyubiquitin chains. Mol Cell, 15, 535–548. [DOI] [PubMed] [Google Scholar]
- Kandel ER (2012). The molecular biology of memory: cAMP, PKA, CRE, CREB-1, CREB-2, and CPEB. Mol Brain, 5, 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim HT, Kim KP, Lledias F, Kisselev AF, Scaglione KM, Skowyra D, Gygi SP, & Goldberg AL (2007). Certain pairs of ubiquitin-conjugating enzymes (E2s) and ubiquitin-protein ligases (E3s) synthesize nondegradable forked ubiquitin chains containing all possible isopeptide linkages. J Biol Chem, 282, 17375–17386. [DOI] [PubMed] [Google Scholar]
- Kim JB, Kim SY, Kim BM, Lee H, Kim I, Yun J, Jo Y, Oh T, Jo Y, Chae HD, & Shin DY (2013). Identification of a novel anti-apoptotic E3 ubiquitin ligase that ubiquitinates antagonists of inhibitor of apoptosis proteins SMAC, HtrA2, and ARTS. J Biol Chem, 288, 12014–12021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, Harper JW, & Gygi SP (2011). Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol Cell, 44, 325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kloetzel P-M, & Ossendorp F. J. C. o. i. i. (2004). Proteasome and peptidase function in MHC-class-I-mediated antigen presentation. 16, 76–81. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Oshima S, Maeyashiki C, Nibe Y, Otsubo K, Matsuzawa Y, Nemoto Y, Nagaishi T, Okamoto R, Tsuchiya K, Nakamura T, & Watanabe M (2016). The ubiquitin hybrid gene UBA52 regulates ubiquitination of ribosome and sustains embryonic development. Sci Rep, 6, 36780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komander D, & Rape M (2012). The ubiquitin code. Annu Rev Biochem, 81, 203–229. [DOI] [PubMed] [Google Scholar]
- Komander D, Reyes-Turcu F, Licchesi JD, Odenwaelder P, Wilkinson KD, & Barford D (2009). Molecular discrimination of structurally equivalent Lys 63-linked and linear polyubiquitin chains. EMBO Rep, 10, 466–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulathu Y, Akutsu M, Bremm A, Hofmann K, & Komander D (2009). Two-sided ubiquitin binding explains specificity of the TAB2 NZF domain. Nat Struct Mol Biol, 16, 1328–1330. [DOI] [PubMed] [Google Scholar]
- Kumari N, Lee KK, & Jha SJN (2018). Targeting the Ubiquitin Proteasome System in Cancer. 203. [Google Scholar]
- Kunjappu MJ, & Hochstrasser M (2014). Assembly of the 20S proteasome. Biochimica et Biophysica Acta (BBA) - Molecular Cell Research, 1843, 2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwapis JL, Alaghband Y, Kramar EA, Lopez AJ, Vogel Ciernia A, White AO, Shu G, Rhee D, Michael CM, Montellier E, Liu Y, Magnan CN, Chen S, Sassone-Corsi P, Baldi P, Matheos DP, & Wood MA (2018). Epigenetic regulation of the circadian gene Per1 contributes to age-related changes in hippocampal memory. Nat Commun, 9, 3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Latham MP, Sekhar A, & Kay LE (2014). Understanding the mechanism of proteasome 20S core particle gating. Proceedings of the National Academy of Sciences, 111, 5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BL, Singh A, Mark Glover JN, Hendzel MJ, & Spyracopoulos L (2017). Molecular Basis for K63-Linked Ubiquitination Processes in Double-Strand DNA Break Repair: A Focus on Kinetics and Dynamics. J Mol Biol, 429, 3409–3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JL (2008). Memory reconsolidation mediates the strengthening of memories by additional learning. Nat Neurosci, 11, 1264–1266. [DOI] [PubMed] [Google Scholar]
- Lee SH, Choi JH, Lee N, Lee HR, Kim JI, Yu NK, Choi SL, Kim H, & Kaang BK (2008). Synaptic protein degradation underlies destabilization of retrieved fear memory. Science, 319, 1253–1256. [DOI] [PubMed] [Google Scholar]
- Li Q, Korte M, & Sajikumar S (2016). Ubiquitin-Proteasome System Inhibition Promotes Long-Term Depression and Synaptic Tagging/Capture. Cereb Cortex, 26, 2541–2548. [DOI] [PubMed] [Google Scholar]
- Liu C-W, & Jacobson AD (2013). Functions of the 19S complex in proteasomal degradation. Trends in biochemical sciences, 38, 103–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Han C, Xie B, Wu Y, Liu S, Chen K, Xia M, Zhang Y, Song L, Li Z, Zhang T, Ma F, Wang Q, Wang J, Deng K, Zhuang Y, Wu X, Yu Y, Xu T, & Cao X (2014). Rhbdd3 controls autoimmunity by suppressing the production of IL-6 by dendritic cells via K27-linked ubiquitination of the regulator NEMO. Nat Immunol, 15, 612–622. [DOI] [PubMed] [Google Scholar]
- Lopez-Salon M, Alonso M, Vianna MR, Viola H, Mello e Souza T, Izquierdo I, Pasquini JM, & Medina JH (2001). The ubiquitin-proteasome cascade is required for mammalian long-term memory formation. Eur J Neurosci, 14, 1820–1826. [DOI] [PubMed] [Google Scholar]
- Lu Y, Lee BH, King RW, Finley D, & Kirschner MW (2015). Substrate degradation by the proteasome: a single-molecule kinetic analysis. Science, 348, 1250834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Ruan H, Peng L, Zhang M, Gack MU, & Yao WD (2017). Proteasome-independent polyubiquitin linkage regulates synapse scaffolding, efficacy, and plasticity. Proc Natl Acad Sci U S A, 114, E8760–E8769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mabb AM, & Ehlers MD (2010). Ubiquitination in postsynaptic function and plasticity. Annu Rev Cell Dev Biol, 26, 179–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madabhushi R, Gao F, Pfenning AR, Pan L, Yamakawa S, Seo J, Rueda R, Phan TX, Yamakawa H, Pao PC, Stott RT, Gjoneska E, Nott A, Cho S, Kellis M, & Tsai LH (2015). Activity-Induced DNA Breaks Govern the Expression of Neuronal Early-Response Genes. Cell, 161, 1592–1605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maguschak KA, & Ressler KJ (2011). Wnt signaling in amygdala-dependent learning and memory. J Neurosci, 31, 13057–13067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massaly N, Dahan L, Baudonnat M, Hovnanian C, Rekik K, Solinas M, David V, Pech S, Zajac JM, Roullet P, Mouledous L, & Frances B (2013). Involvement of protein degradation by the ubiquitin proteasome system in opiate addictive behaviors. Neuropsychopharmacology, 38, 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto ML, Wickliffe KE, Dong KC, Yu C, Bosanac I, Bustos D, Phu L, Kirkpatrick DS, Hymowitz SG, Rape M, Kelley RF, & Dixit VM (2010). K11-linked polyubiquitination in cell cycle control revealed by a K11 linkage-specific antibody. Mol Cell, 39, 477–484. [DOI] [PubMed] [Google Scholar]
- McGaugh JL (2015). Consolidating memories. Annu Rev Psychol, 66, 1–24. [DOI] [PubMed] [Google Scholar]
- Medina JH, & Viola H (2018). ERK1/2: A Key Cellular Component for the Formation, Retrieval, Reconsolidation and Persistence of Memory. Front Mol Neurosci, 11, 361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo D, Cuchillo-Ibañez I, Parlato R, & Rammes G (2016). DNA Damage, Neurodegeneration, and Synaptic Plasticity. Neural plasticity, 2016, 1206840–1206840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merlo E, Freudenthal R, Maldonado H, & Romano A (2005). Activation of the transcription factor NF-kappaB by retrieval is required for long-term memory reconsolidation. Learn Mem, 12, 23–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metzger MB, Hristova VA, & Weissman AM (2012). HECT and RING finger families of E3 ubiquitin ligases at a glance. Journal of Cell Science, 125, 531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mevissen TE, Hospenthal MK, Geurink PP, Elliott PR, Akutsu M, Arnaudo N, Ekkebus R, Kulathu Y, Wauer T, El Oualid F, Freund SM, Ovaa H, & Komander D (2013). OTU deubiquitinases reveal mechanisms of linkage specificity and enable ubiquitin chain restriction analysis. Cell, 154, 169–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer HJ, & Rape M (2014). Enhanced protein degradation by branched ubiquitin chains. Cell, 157, 910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meza Gutierrez F, Simsek D, Mizrak A, Deutschbauer A, Braberg H, Johnson J, Xu J, Shales M, Nguyen M, Tamse-Kuehn R, Palm C, Steinmetz LM, Krogan NJ, & Toczyski DP (2018). Genetic analysis reveals functions of atypical polyubiquitin chains. Elife, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MA, Elliott PR, Swatek KN, Simicek M, Pruneda JN, Wagstaff JL, Freund SM, & Komander D (2015). Assembly and specific recognition of k29- and k33-linked polyubiquitin. Mol Cell, 58, 95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michel MA, Swatek KN, Hospenthal MK, & Komander D (2017). Ubiquitin Linkage-Specific Affimers Reveal Insights into K6-Linked Ubiquitin Signaling. Mol Cell, 68, 233–246 e235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris JR, & Solomon E (2004). BRCA1 : BARD1 induces the formation of conjugated ubiquitin structures, dependent on K6 of ubiquitin, in cells during DNA replication and repair. Hum Mol Genet, 13, 807–817. [DOI] [PubMed] [Google Scholar]
- Nader K, & Hardt O (2009). A single standard for memory: the case for reconsolidation. Nat Rev Neurosci, 10, 224–234. [DOI] [PubMed] [Google Scholar]
- Nader K, Schafe GE, & Le Doux JE (2000). Fear memories require protein synthesis in the amygdala for reconsolidation after retrieval. Nature, 406, 722–726. [DOI] [PubMed] [Google Scholar]
- Nakazawa S, Oikawa D, Ishii R, Ayaki T, Takahashi H, Takeda H, Ishitani R, Kamei K, Takeyoshi I, Kawakami H, Iwai K, Hatada I, Sawasaki T, Ito H, Nureki O, & Tokunaga F (2016). Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat Commun, 7, 12547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nathan JA, Kim HT, Ting L, Gygi SP, & Goldberg AL (2013). Why do cellular proteins linked to K63-polyubiquitin chains not associate with proteasomes? EMBO J, 32, 552–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navabpour S, Kwapis JL, & Jarome TJ (2020). A neuroscientist’s guide to transgenic mice and other genetic tools. Neurosci Biobehav Rev, 108, 732–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, Dugger D, Gordon N, Sidhu SS, Fellouse FA, Komuves L, French DM, Ferrando RE, Lam C, Compaan D, Yu C, Bosanac I, Hymowitz SG, Kelley RF, & Dixit VM (2008). Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell, 134, 668–678. [DOI] [PubMed] [Google Scholar]
- Nishikawa H, Ooka S, Sato K, Arima K, Okamoto J, Klevit RE, Fukuda M, & Ohta T (2004). Mass spectrometric and mutational analyses reveal Lys-6-linked polyubiquitin chains catalyzed by BRCA1-BARD1 ubiquitin ligase. J Biol Chem, 279, 3916–3924. [DOI] [PubMed] [Google Scholar]
- Ohtake F, Saeki Y, Ishido S, Kanno J, & Tanaka K (2016). The K48-K63 Branched Ubiquitin Chain Regulates NF-kappaB Signaling. Mol Cell, 64, 251–266. [DOI] [PubMed] [Google Scholar]
- Ohtake F, Tsuchiya H, Saeki Y, & Tanaka K (2018). K63 ubiquitylation triggers proteasomal degradation by seeding branched ubiquitin chains. Proc Natl Acad Sci U S A, 115, E1401–E1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordureau A, Heo JM, Duda DM, Paulo JA, Olszewski JL, Yanishevski D, Rinehart J, Schulman BA, & Harper JW (2015). Defining roles of PARKIN and ubiquitin phosphorylation by PINK1 in mitochondrial quality control using a ubiquitin replacement strategy. Proc Natl Acad Sci U S A, 112, 6637–6642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orsi SA, Devulapalli RK, Nelsen JL, McFadden T, Surineni R, & Jarome TJ (2019). Distinct subcellular changes in proteasome activity and linkage-specific protein polyubiquitination in the amygdala during the consolidation and reconsolidation of a fear memory. Neurobiol Learn Mem, 157, 1–11. [DOI] [PubMed] [Google Scholar]
- Park A, Jacob AD, Walters BJ, Park S, Rashid AJ, Jung JH, Lau J, Woolley GA, Frankland PW, & Josselyn SA (2020). A time-dependent role for the transcription factor CREB in neuronal allocation to an engram underlying a fear memory revealed using a novel in vivo optogenetic tool to modulate CREB function. Neuropsychopharmacology, 45, 916–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park CW, & Ryu KY (2014). Cellular ubiquitin pool dynamics and homeostasis. BMB Rep, 47, 475–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, & Kaang BK (2019). Balanced actions of protein synthesis and degradation in memory formation. Learn Mem, 26, 299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlopoulos E, Trifilieff P, Chevaleyre V, Fioriti L, Zairis S, Pagano A, Malleret G, & Kandel ER (2011). Neuralized1 activates CPEB3: a function for nonproteolytic ubiquitin in synaptic plasticity and memory storage. Cell, 147, 1369–1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng S, Zhang Y, Zhang J, Wang H, & Ren B (2010). ERK in learning and memory: a review of recent research. Int J Mol Sci, 11, 222–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peth A, Uchiki T, & Goldberg AL (2010). ATP-dependent steps in the binding of ubiquitin conjugates to the 26S proteasome that commit to degradation. Mol Cell, 40, 671–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroski MD, & Deshaies RJ (2005). Mechanism of lysine 48-linked ubiquitin-chain synthesis by the cullin-RING ubiquitin-ligase complex SCF-Cdc34. Cell, 123, 1107–1120. [DOI] [PubMed] [Google Scholar]
- Pick JE, Malumbres M, & Klann E (2013). The E3 ligase APC/C-Cdh1 is required for associative fear memory and long-term potentiation in the amygdala of adult mice. Learn Mem, 20, 11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pick JE, Wang L, Mayfield JE, & Klann E (2013). Neuronal expression of the ubiquitin E3 ligase APC/C-Cdh1 during development is required for long-term potentiation, behavioral flexibility, and extinction. Neurobiol Learn Mem, 100, 25–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickart CM (2001). Mechanisms underlying ubiquitination. Annu Rev Biochem, 70, 503–533. [DOI] [PubMed] [Google Scholar]
- Prakash S, Tian L, Ratliff KS, Lehotzky RE, & Matouschek A (2004). An unstructured initiation site is required for efficient proteasome-mediated degradation. Nat Struct Mol Biol, 11, 830–837. [DOI] [PubMed] [Google Scholar]
- Qin Y, Zhou MT, Hu MM, Hu YH, Zhang J, Guo L, Zhong B, & Shu HB (2014). RNF26 temporally regulates virus-triggered type I interferon induction by two distinct mechanisms. PLoS Pathog, 10, e1004358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabl J, Smith DM, Yu Y, Chang S-C, Goldberg AL, & Cheng Y (2008). Mechanism of gate opening in the 20S proteasome by the proteasomal ATPases. Molecular cell, 30, 360–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahighi S, Ikeda F, Kawasaki M, Akutsu M, Suzuki N, Kato R, Kensche T, Uejima T, Bloor S, Komander D, Randow F, Wakatsuki S, & Dikic I (2009). Specific recognition of linear ubiquitin chains by NEMO is important for NF-kappaB activation. Cell, 136, 1098–1109. [DOI] [PubMed] [Google Scholar]
- Rantala JK, Pouwels J, Pellinen T, Veltel S, Laasola P, Mattila E, Potter CS, Duffy T, Sundberg JP, Kallioniemi O, Askari JA, Humphries MJ, Parsons M, Salmi M, & Ivaska J (2011). SHARPIN is an endogenous inhibitor of beta1-integrin activation. Nat Cell Biol, 13, 1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redman KL, & Rechsteiner M (1989). Identification of the long ubiquitin extension as ribosomal protein S27a. Nature, 338, 438–440. [DOI] [PubMed] [Google Scholar]
- Reis DS, Jarome TJ, & Helmstetter FJ (2013). Memory formation for trace fear conditioning requires ubiquitin-proteasome mediated protein degradation in the prefrontal cortex. Front Behav Neurosci, 7, 150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Ortiz CJ, Balderas I, Saucedo-Alquicira F, Cruz-Castaneda P, & Bermudez-Rattoni F (2011). Long-term aversive taste memory requires insular and amygdala protein degradation. Neurobiol Learn Mem, 95, 311–315. [DOI] [PubMed] [Google Scholar]
- Roesler R (2017). Molecular mechanisms controlling protein synthesis in memory reconsolidation. Neurobiol Learn Mem, 142, 30–40. [DOI] [PubMed] [Google Scholar]
- Rosenberg T, Elkobi A, Dieterich DC, & Rosenblum K (2016). NMDAR-dependent proteasome activity in the gustatory cortex is necessary for conditioned taste aversion. Neurobiol Learn Mem, 130, 7–16. [DOI] [PubMed] [Google Scholar]
- Ryu KY, Sinnar SA, Reinholdt LG, Vaccari S, Hall S, Garcia MA, Zaitseva TS, Bouley DM, Boekelheide K, Handel MA, Conti M, & Kopito RR (2008). The mouse polyubiquitin gene Ubb is essential for meiotic progression. Mol Cell Biol, 28, 1136–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeki Y, Kudo T, Sone T, Kikuchi Y, Yokosawa H, Toh-e A, & Tanaka K (2009). Lysine 63-linked polyubiquitin chain may serve as a targeting signal for the 26S proteasome. EMBO J, 28, 359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savell KE, Bach SV, Zipperly ME, Revanna JS, Goska NA, Tuscher JJ, Duke CG, Sultan FA, Burke JN, Williams D, Ianov L, & Day JJ (2019). A Neuron-Optimized CRISPR/dCas9 Activation System for Robust and Specific Gene Regulation. eNeuro, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer A, Sampath SC, Intrator A, Min A, Gertler TS, Surmeier DJ, Tarakhovsky A, & Greengard P (2009). Control of cognition and adaptive behavior by the GLP/G9a epigenetic suppressor complex. Neuron, 64, 678–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafe GE, Nadel NV, Sullivan GM, Harris A, & LeDoux JE (1999). Memory consolidation for contextual and auditory fear conditioning is dependent on protein synthesis, PKA, and MAP kinase. Learn Mem, 6, 97–110. [PMC free article] [PubMed] [Google Scholar]
- Scott D, Oldham NJ, Strachan J, Searle MS, & Layfield R (2015). Ubiquitin-binding domains: mechanisms of ubiquitin recognition and use as tools to investigate ubiquitin-modified proteomes. Proteomics, 15, 844–861. [DOI] [PubMed] [Google Scholar]
- Seymour RE, Hasham MG, Cox GA, Shultz LD, Hogenesch H, Roopenian DC, & Sundberg JP (2007). Spontaneous mutations in the mouse Sharpin gene result in multiorgan inflammation, immune system dysregulation and dermatitis. Genes Immun, 8, 416–421. [DOI] [PubMed] [Google Scholar]
- Shabek N, Herman-Bachinsky Y, Buchsbaum S, Lewinson O, Haj-Yahya M, Hejjaoui M, Lashuel HA, Sommer T, Brik A, & Ciechanover A (2012). The size of the proteasomal substrate determines whether its degradation will be mediated by mono- or polyubiquitylation. Mol Cell, 48, 87–97. [DOI] [PubMed] [Google Scholar]
- Shaid S, Brandts CH, Serve H, & Dikic I (2013). Ubiquitination and selective autophagy. Cell Death Differ, 20, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shang F, Deng G, Liu Q, Guo W, Haas AL, Crosas B, Finley D, & Taylor A (2005). Lys6-modified ubiquitin inhibits ubiquitin-dependent protein degradation. J Biol Chem, 280, 20365–20374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen B, Zhang W, Zhang J, Zhou J, Wang J, Chen L, Wang L, Hodgkins A, Iyer V, Huang X, & Skarnes WC (2014). Efficient genome modification by CRISPR-Cas9 nickase with minimal off-target effects. Nat Methods, 11, 399–402. [DOI] [PubMed] [Google Scholar]
- Shrestha P, Ayata P, Herrero-Vidal P, Longo F, Gastone A, LeDoux JE, Heintz N, & Klann E (2020). Cell-type-specific drug-inducible protein synthesis inhibition demonstrates that memory consolidation requires rapid neuronal translation. Nat Neurosci, 23, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva AJ, Kogan JH, Frankland PW, & Kida S (1998). CREB and memory. Annu Rev Neurosci, 21, 127–148. [DOI] [PubMed] [Google Scholar]
- Skaug B, Jiang X, & Chen ZJ (2009). The role of ubiquitin in NF-kappaB regulatory pathways. Annu Rev Biochem, 78, 769–796. [DOI] [PubMed] [Google Scholar]
- Spit M, Rieser E, & Walczak H (2019). Linear ubiquitination at a glance. J Cell Sci, 132. [DOI] [PubMed] [Google Scholar]
- Stewart MD, Ritterhoff T, Klevit RE, & Brzovic PS (2016). E2 enzymes: more than just middle men. Cell research, 26, 423–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieglitz B, Rana RR, Koliopoulos MG, Morris-Davies AC, Schaeffer V, Christodoulou E, Howell S, Brown NR, Dikic I, & Rittinger K (2013). Structural basis for ligase-specific conjugation of linear ubiquitin chains by HOIP. Nature, 503, 422–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun ZW, & Allis CD (2002). Ubiquitination of histone H2B regulates H3 methylation and gene silencing in yeast. Nature, 418, 104–108. [DOI] [PubMed] [Google Scholar]
- Swatek KN, & Komander D (2016). Ubiquitin modifications. Cell Res, 26, 399–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Y, Yu D, Busto GU, Wilson C, & Davis RL (2013). Wnt signaling is required for long-term memory formation. Cell Rep, 4, 1082–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K (2009). The proteasome: overview of structure and functions. Proceedings of the Japan Academy. Series B, Physical and biological sciences, 85, 12–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Joo D, Liu G, Tu H, You J, Jin J, Zhao X, Hung MC, & Lin X (2018). Linear ubiquitination of cFLIP induced by LUBAC contributes to TNFalpha-induced apoptosis. J Biol Chem, 293, 20062–20072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanno H, & Komada M (2013). The ubiquitin code and its decoding machinery in the endocytic pathway. J Biochem, 153, 497–504. [DOI] [PubMed] [Google Scholar]
- Thrower JS, Hoffman L, Rechsteiner M, & Pickart CM (2000). Recognition of the polyubiquitin proteolytic signal. EMBO J, 19, 94–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todorova V, & Blokland A (2017). Mitochondria and Synaptic Plasticity in the Mature and Aging Nervous System. Current neuropharmacology, 15, 166–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tokunaga F (2013). Linear ubiquitination-mediated NF-kappaB regulation and its related disorders. J Biochem, 154, 313–323. [DOI] [PubMed] [Google Scholar]
- Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata S, Tanaka K, Nakano H, & Iwai K (2011). SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature, 471, 633–636. [DOI] [PubMed] [Google Scholar]
- Tokunaga F, Sakata S, Saeki Y, Satomi Y, Kirisako T, Kamei K, Nakagawa T, Kato M, Murata S, Yamaoka S, Yamamoto M, Akira S, Takao T, Tanaka K, & Iwai K (2009). Involvement of linear polyubiquitylation of NEMO in NF-kappaB activation. Nat Cell Biol, 11, 123–132. [DOI] [PubMed] [Google Scholar]
- van der Horst A, de Vries-Smits AM, Brenkman AB, van Triest MH, van den Broek N, Colland F, Maurice MM, & Burgering BM (2006). FOXO4 transcriptional activity is regulated by monoubiquitination and USP7/HAUSP. Nat Cell Biol, 8, 1064–1073. [DOI] [PubMed] [Google Scholar]
- Wagner SA, Beli P, Weinert BT, Nielsen ML, Cox J, Mann M, & Choudhary C (2011). A proteome-wide, quantitative survey of in vivo ubiquitylation sites reveals widespread regulatory roles. Mol Cell Proteomics, 10, M111 013284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Gao Y, Li L, Jin G, Cai Z, Chao JI, & Lin HK (2012). K63-linked ubiquitination in kinase activation and cancer. Front Oncol, 2, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webb GC, Baker RT, Coggan M, & Board PG (1994). Localization of the human UBA52 ubiquitin fusion gene to chromosome band 19p13.1-p12. Genomics, 19, 567–569. [DOI] [PubMed] [Google Scholar]
- Werner CT, Milovanovic M, Christian DT, Loweth JA, & Wolf ME (2015). Response of the Ubiquitin-Proteasome System to Memory Retrieval After Extended-Access Cocaine or Saline Self-Administration. Neuropsychopharmacology, 40, 3006–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner CT, Mitra S, Martin JA, Stewart AF, Lepack AE, Ramakrishnan A, Gobira PH, Wang ZJ, Neve RL, Gancarz AM, Shen L, Maze I, & Dietz DM (2019). Ubiquitin-proteasomal regulation of chromatin remodeler INO80 in the nucleus accumbens mediates persistent cocaine craving. Sci Adv, 5, eaay0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner CT, Viswanathan R, Martin JA, Gobira PH, Mitra S, Thomas SA, Wang ZJ, Liu JF, Stewart AF, Neve RL, Li JX, Gancarz AM, & Dietz DM (2018). E3 Ubiquitin-Protein Ligase SMURF1 in the Nucleus Accumbens Mediates Cocaine Seeking. Biol Psychiatry, 84, 881–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiborg O, Pedersen MS, Wind A, Berglund LE, Marcker KA, & Vuust J (1985). The human ubiquitin multigene family: some genes contain multiple directly repeated ubiquitin coding sequences. EMBO J, 4, 755–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu P, Duong DM, Seyfried NT, Cheng D, Xie Y, Robert J, Rush J, Hochstrasser M, Finley D, & Peng J (2009). Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell, 137, 133–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WL, Wang J, Chan CH, Lee SW, Campos AD, Lamothe B, Hur L, Grabiner BC, Lin X, Darnay BG, & Lin HK (2009). The E3 ligase TRAF6 regulates Akt ubiquitination and activation. Science, 325, 1134–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang WL, Zhang X, & Lin HK (2010). Emerging role of Lys-63 ubiquitination in protein kinase and phosphatase activation and cancer development. Oncogene, 29, 4493–4503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao I, Takao K, Miyakawa T, Ito S, & Setou M (2011). Synaptic E3 ligase SCRAPPER in contextual fear conditioning: extensive behavioral phenotyping of Scrapper heterozygote and overexpressing mutant mice. PLoS One, 6, e17317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh SH, Lin CH, Lee CF, & Gean PW (2002). A requirement of nuclear factor-kappaB activation in fear-potentiated startle. J Biol Chem, 277, 46720–46729. [DOI] [PubMed] [Google Scholar]
- Yuan WC, Lee YR, Lin SY, Chang LY, Tan YP, Hung CC, Kuo JC, Liu CH, Lin MY, Xu M, Chen ZJ, & Chen RH (2014). K33-Linked Polyubiquitination of Coronin 7 by Cul3-KLHL20 Ubiquitin E3 Ligase Regulates Protein Trafficking. Mol Cell, 54, 586–600. [DOI] [PubMed] [Google Scholar]
- Zak DE, Schmitz F, Gold ES, Diercks AH, Peschon JJ, Valvo JS, Niemisto A, Podolsky I, Fallen SG, Suen R, Stolyar T, Johnson CD, Kennedy KA, Hamilton MK, Siggs OM, Beutler B, & Aderem A (2011). Systems analysis identifies an essential role for SHANK-associated RH domain-interacting protein (SHARPIN) in macrophage Toll-like receptor 2 (TLR2) responses. Proc Natl Acad Sci U S A, 108, 11536–11541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zalcman G, Federman N, & Romano A (2018). CaMKII Isoforms in Learning and Memory: Localization and Function. Front Mol Neurosci, 11, 445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, & Ulrich HD (2010). Distinct consequences of posttranslational modification by linear versus K63-linked polyubiquitin chains. Proc Natl Acad Sci U S A, 107, 7704–7709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng N, & Shabek N. J. A. r. o. b. (2017). Ubiquitin ligases: structure, function, and regulation. 86, 129–157. [DOI] [PubMed] [Google Scholar]
- Zhou HL, Geng C, Luo G, & Lou H (2013). The p97-UBXD8 complex destabilizes mRNA by promoting release of ubiquitinated HuR from mRNP. Genes Dev, 27, 1046–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]