

Abstract
Protein phosphatases regulate a wide array of proteins through post-translational modification and are required for a plethora of intracellular events in eukaryotes. While some core components of the protein phosphatase complexes are well characterized, many subunits of these large complexes remain unstudied. Here we characterize a loss-of-function allele of the protein phosphatase 1 regulatory subunit 35 (Ppp1r35) gene. Homozygous mouse embryos lacking Ppp1r35 are developmental delayed beginning at embryonic day (E) 7.5 and have obvious morphological defects at later stages. Mutants fail to initiate turning and do not progress beyond the size or staging of normal E8.5 embryos. Consistent with recent in vitro studies linking PPP1R35 with the microcephaly protein Rotatin and with a role in centrosome formation, we show that Ppp1r35 mutant embryos lack primary cilia. Histological and molecular analysis of Ppp1r35 mutants revealed that notochord development is irregular and discontinuous and consistent with a role in primary cilia, that the floor plate of the neural tube is not specified. Similar to other mutant embryos with defects in centriole function, Ppp1r35 mutants displayed increased cell death that is prevalent in the neural tube and an increased number of proliferative cells in prometaphase. We hypothesize that loss of Ppp1r35 function abrogates centriole homeostasis, resulting in a failure to produce functional primary cilia, cell death and cell cycle delay/stalling that leads to developmental failure. Taken together, these results highlight the essential function of Ppp1r35 during early mammalian development and implicate this gene as a candidate for human microcephaly.
Keywords: Ppp1r35, centriole, notochord, cilia, knockout, development
Introduction:
There are currently more than 21,000 protein coding genes identified in humans, many of which have unknown functions [1]. The mouse serves as the predominant model for assessing the role of mammalian genes during normal development. The goal of the Knockout Mouse Project (KOMP) [2], along with the International Mouse Phenotyping Consortium (IMPC) [3], is to gain insight into gene function by generating a null mutation in every protein coding gene in the mouse and characterizing the loss-of-function phenotype [4]. To begin to understand the role of protein phosphatase 1 regulatory subunit 35 (Ppp1r35), the IMPC determined that no homozygous null pups were recovered after birth nor were null embryos present at embryonic day 15.5 [3]. To further investigate the role of this crucial protein, we used the KOMP-generated null allele to characterize Ppp1r35 function during early embryonic development.
Many cellular processes must occur in a strictly regimented spatiotemporal manner for normal developmental progress. Following cleavage and implantation, gastrulation involves major embryonic morphogenetic movements that reorganize the single-layered epiblast into a multilayered gastrula containing the three primary germ cell layers: endoderm, ectoderm, and mesoderm. Gastrulation involves an epithelial-to-mesenchymal transition that is generated in the primitive streak, an embryonic structure that initiates at E6.5 and organizes the emerging germ layers. By E7.5, the primitive streak extends from the proximal/posterior portion of the embryo to the distal tip and forms the node, an important embryonic signaling center. The cells of the node, each containing a primary cilia, generate signals that result in appropriate left-right patterning [5, 6]. The notochord emanates from the node and consists of a rod of cells along the anterior-posterior (A/P) midline axis. The notochord acts as an organizer by secreting growth factors such as sonic hedgehog (Shh), which is important for patterning surrounding tissues. For example, secreted Shh patterns the ventral neural tube in a concentration dependent manner to specify the floor plate [7, 8]. Together the primitive streak, node, and notochord are essential for normal germ layer establishment and appropriate embryonic patterning which culminates in an organized E9.5 embryo that has initiated organogenesis.
Protein phosphatases provide functional post-translational regulation of various proteins by removing phosphate groups and are therefore essential for regulating many intracellular events in eukaryotic organisms [9]. Protein phosphatases are classified into two families that include protein tyrosine phosphatases (PTPs) and protein serine/threonine (Ser/Thr) phosphatases (PSPs). Approximately 107 PTPs have been classified in humans and only about 30 PSPs have been identified [10]. Within the PSP family, there are multiple sub-families including the phosphoprotein phosphatases (PPPs) [11]. The PPP sub-family contains multiple members including protein phosphatase 1 (PP1), PP2A, PP2B, PP4, PP5, PP6, and PP7 [10, 11]. PP1 is implicated in many essential cellular processes such as cell division and meiosis, cell cycle arrest and apoptosis, metabolism, protein synthesis, actin and actomyosin reorganization as well as the regulation of cell membrane receptors and signals [12]. The PP1 holoenzyme is composed of a large catalytic subunit (PP1C) and a regulatory subunit (R). The regulatory subunits direct the catalytic subunit to specific target substrates and/or act as substrates themselves [13]. There are currently 181 genes in the human genome that are classified as PPP1Rs [14]. The majority of regulatory subunits have not been studied in detail, and consequently their functional requirements and roles in development and disease remain poorly understood. Additionally, many of these subunits are named as such due to sequence homology/orthology but their actual role and/or interaction with PP complexes has not yet been determined.
A mass spectrometry study identified protein phosphatase 1 regulatory subunit 35 (PPP1R35) as a novel human centrosomal protein [15]. Centrioles are eukaryotic organelles composed of microtubules which are necessary for many cellular functions such as the formation of centrosomes, cilia, and flagella [16, 17]. Recent studies in human cells have demonstrated that PPP1R35 is necessary for centriole elongation during centriole duplication [18, 19]. PPP1R35 forms a complex with the centrosomal protein Rotatin (RTTN) [19], a protein that localizes to the luminal wall of the elongating centriole and stabilizes the machinery known to initiate cartwheel formation in the duplicating centrioles that includes STIL, SAS6, and CPAP (SAS4, CENPJ) [20].
Defects in centriolar proteins can result in human congenital ciliopathies [16, 21, 22] such as autosomal recessive primary microcephaly (MCPH), a developmental disorder characterized by decreased brain size [21, 23]. Murine loss-of-function studies of centrosome related genes including Rttn, Sas4, and STIL have demonstrated that each of these genes are required for embryonic development and display similar organogenesis stage null phenotypes, suggesting a common major defect [24–26]. Here we demonstrate that Ppp1r35 is required for organogenesis and show that mutant embryos have similar defects to several of the centrosome-related null animals including defects in primary cilia, notochord formation, neural tube specification/closure and increased neural tube cell death. Together our data supports the hypothesis that PPP1R35 interacts with RTTN and that Ppp1r35 is a candidate gene for MCPH.
Materials and methods:
Animals
The Ppp1r35 knockout was generated by the University of California, Davis (Ppp1r35tm1.1(KOMP)Vlcg, stock # MMRRC:049168-UCD). 1,112 base pairs of the Ppp1r35 genomic sequence was replaced with the lacZ reporter gene sequence, removing parts of exons 1–4, in the C57BL/6NJ background (Figure 1A). The p53-null allele was purchased from the Jackson Laboratory (Trp53tm1Tyj, stock # 002101).
Figure 1: Ppp1r35 expression during embryonic development.
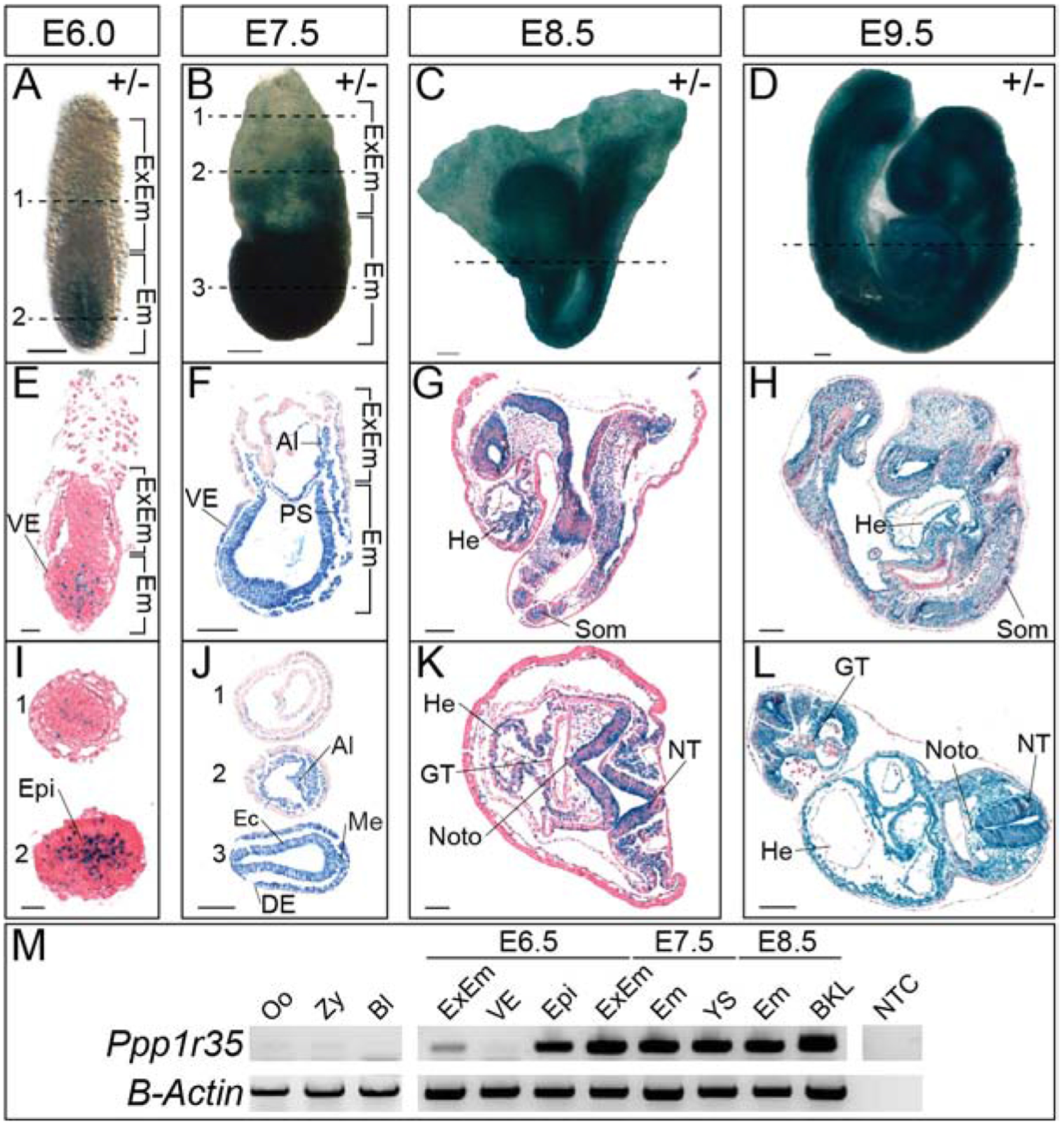
X-gal stained heterozygous embryos at E6.0 (A), E7.5 (B), E8.5 (C), and E9.5 (D). Sagittal (E-H) or transverse (I-L) of X-gal stained heterozygotes at E6.0–9.5. I) At E6.0, 1 and 2 denote sections indicated by the dashed line in (A). J) At E7.5, 1–3 denote sections indicated by the dashed lines in (B). M) RT-PCR of Ppp1r35 in various tissues and at the developmental stages denoted. β-actin is a loading control. +/− denotes Ppp1r35 heterozygous embryos. ExEm; extra-embryonic, Em; embryonic, VE; visceral endoderm, Epi; epiblast, Al; allantois, PS; primitive streak, Ec; ectoderm, DE; definitive endoderm, Me; mesoderm, He; heart, Som; somites, GT; gut tube, Noto; notochord, NT; neural tube, Oo; oocyte, Zy; zygote, Bl; blastocyst, YS; yolk sac, BKL; brain-kidney-liver, NTC; no template control. Scale bars in A-D, F-H, J, and L = 100μm, E&I = 20μm, and K= 50μm.
Embryo retrieval and genotyping
Ppp1r35-null embryos were generated by heterozygote intercrosses. Males and females were housed together, and the presence of the vaginal plugs was defined as E0.5. All Ppp1r35 embryos were genotyped by PCR using primers (given 5′ to 3′): wild type (CCGAGGAGCAGGTACTGAAC and ATCCGATGCATGAGAAAGGT) and knockout (CGGTCGCTACCATTACCAGT and GGGCAGGAAAAGGGTAGAAG).
Double-mutant (Ppp1r35−/−, p53−/−) embryos were generated by crossing females heterozygous for Ppp1r35 with males heterozygous for p53. Embryos were genotyped by PCR using Ppp1r35 primers listed above, and p53 primers (given 5’ to 3’): wild type (AGGCTTAGAGGTGCAAGCTG and TGGATGGTGGTATACTCAGAGC) and knockout (CAGCCTCTGTTCCACATACACT and TGGATGGTGGTATACTCAGAGC).
All results presented were performed on at least 3 mutant embryos (often more). All experiments were approved by the University of Massachusetts Amherst Institutional Animal Care and Use Committee.
RNA extraction and RT-PCR analysis
RNA was extracted using Roche High Pure RNA Isolation Kit (Roche Diagnostics, 11828665001). cDNA synthesis was performed using Bio-Rad iScript cDNA Synthesis (Bio-Rad, 1708890). RT-PCR was performed for 35 cycles of 30 seconds at 60°C, 72°C and 95°C with the following primers with indicated gene names unless otherwise specified (given 5′ to 3′): Ppp1r35 (219 bp): (CAGCCTGGCTTTGAGTCTG and GCGTTCAGTACCTGCTCCTC); Yy1 (574 bp): (ACGACGACTACATAGAGCAGACG and ACGAACGCTTTGCCACACT); β-actin (460 bp): (GGCCCAGAGCAAGAGAGGTATCC and ACGCACGATTTCCCTCTCAGC).
X-gal (5-bromo-4-chloro-3-indolyl-β-D-galactopyranoside) staining
Freshly dissected embryos were fixed in X-gal buffer containing 0.2% glutaraldehyde and 1% formaldehyde on ice for 15 minutes and subjected to a modified protocol from Tremblay et al. (2000) [27]. Fixed embryos were washed with X-gal buffer (PBS, 5mM EGTA, 2mM MgCl:6H2O, 0.2% NP-40, 0.2mM deoxycholate) for 10 minutes, three times, and stained with X-gal stain (X-gal buffer, 5 mM potassium ferricyanide and 5 mM potassium ferrocyanide and 0.5 mg/ml X-gal) overnight at 37°C. Embryos were then dehydrated in ethanol, cleared in xylene, embedded with paraffin and sectioned at 7μm thick. The sectioned embryos were deparaffinized and rehydrated for subsequent processing. Eosin staining was performed by immersing rehydrated sectioned embryos in eosin for 15 seconds, followed by 95% ethanol washes for 2 minutes, 100% ethanol for 2 minutes, and lastly cleared in xylene. Slides were then sealed with Cytoseal 60. X-gal stained sections were imaged with a Panoramic MIDI II slide scanner (3DHISTECH).
Hematoxylin and eosin (H&E) staining
Freshly dissected embryos were fixed in 4% paraformaldehyde overnight, dehydrated in ethanol, cleared in xylene, embedded with paraffin and sectioned at 7μm, as previously described [28]. The sectioned embryos were deparaffinized and rehydrated for subsequent procedures. Slides were then stained with hematoxylin for 45 seconds, placed under gently running tap water for 1 minute, submerged in Scott’s Tap Water Substitute (20 h MgSO4, 3.5g NaHCO2/L dH2O) for 1 minute, and then washed in still tap water for another minute. The slides were quickly dipped into 95% ethanol, stained with eosin for 15 seconds, and then again washed in 95% ethanol with two 2-minute washes in 100% ethanol. Lastly, the slides were washed with xylenes three times for one minute each, and then sealed with Cytoseal 60. H&E stained sections were imaged with a Panoramic MIDI II slide scanner (3DHISTECH).
Immunofluorescence (IF)
Freshly dissected embryos were fixed in 4% paraformaldehyde overnight, dehydrated in ethanol, cleared in xylene, embedded with paraffin, and sectioned at 7μm thick. The sectioned embryos were deparaffinized and rehydrated for subsequent procedures. Antigen retrieval was performed by boiling the slides in citric acid buffer (pH 6) for 4 minutes and cooling to room temperature. The slides were then rinsed twice in phosphate buffered saline/0.01% Tween 20 (PBT) for 2 minutes and blocked with 0.5% milk in PBT for 2 hours at room temperature in a humid chamber. Primary antibodies were applied in 0.05% milk/PBT and incubated at 4°C overnight in a humid chamber. The slides were then rinsed three times with PBT for 15 minutes, followed by incubation with secondary antibodies at room temperature for 1 hour in a humid chamber. Next, the slides were rinsed with PBS for 15 minutes, three times. Nuclei were counterstained with DAPI (4′,6-diamidino-2-phenylindole) in PBS (1:10,000) for 3 minutes. Slides were rinsed with 1X PBS and sealed with ProLong Gold (Thermo Fisher Scientific, P36934). Fluorescent slides were imaged with Panoramic MIDI II slide scanner (3DHISTECH). The primary antibodies used and their dilution include: Brachyury (T) (Santa Cruz Biotechnology, SC-17743, 1:150); E-cadherin (BD Biosciences, 610181, 1:500); Phospho-p53 (ser15) (p53) (Cell Signaling Technology, 9284S, 1:100); ADP ribosylation factor like GTPase 13B (ARL13B) (Proteintech, 17711–1-AP, 1:250); Forkhead box protein A2 (FOXA2) (Santa Cruz Biotechnology, SC-6554, 1:250); Laminin (Sigma Aldrich, L9393, 1:250); Histone H3 (phospho S10) (PH3) (Abcam, ab5176, 1:500).
The secondary antibodies used include: Alexa Fluor 488 donkey anti-goat (Thermo Fisher Scientific, A21206, 1:500); Alexa Fluor 488 donkey anti-rabbit (Thermo Fisher Scientific, A11034, 1:500); Alexa Fluor 546 donkey anti-mouse (Thermo Fisher Scientific, A10036, 1:500); Alexa Fluor 546 donkey anti-goat (Thermo Fisher Scientific, A11056, 1:500); Alexa Fluor 647 donkey anti-rabbit (Thermo Fisher Scientific, A31573, 1:500); Alexa Fluor 647 donkey anti-goat (Thermo Fisher Scientific, A21447, 1:500); DAPI (Molecular Probes, 1:10,000).
PH3 positive cells were counted using ImageJ [29].
Whole-mount in situ hybridization (WISH)
Freshly dissected embryos were fixed in 4% paraformaldehyde for 2–4 hours on ice and subjected to WISH [30]. Embryos were dehydrated in methanol and stored up to 1 month. Embryos were rehydrated in a series of methanol washes in PBT. Subsequently, the embryos were rinsed twice in PBT for 5 minutes and bleached in 6% hydrogen peroxide for 1 hour. Embryos were then rinsed with PBT, treated with Proteinase K for 6–8 minutes and incubated with glycine in PBT for 5 minutes. After rinsing twice with PBT for 5 minutes, embryos were fixed with 4% paraformaldehyde with 0.02% glutaraldehyde for 20 minutes. Embryos were rinsed twice in PBT for 5 minutes and incubated in 1:1 hybridization buffer/PBT for 10 minutes and subsequently in hybridization buffer for 10 minutes. Embryos were then incubated with 150ng RNA probes in hybridization buffer at 70°C overnight. The next day, embryos were washed with solutions containing formamide, 20X SSC, 1M citric acid, 20% SDS and Tween 20 for 30 minutes at 70°C for the first three washes, and at 65°C for the last three washes. Following this step, the embryos were rinsed in maleic acid buffer (MAB) for 5 minutes, three times. The embryos were then blocked in MAB with 2% Boehringer block for 1 hour at room temperature and then incubated with the antibody solution containing Anti-Digoxigenin-AP (1:2,000, Roche Diagnostics, 11093274910), heat inactivated sheep serum (Sigma-Aldrich, S2263) and levamisole (Sigma-Aldrich, L9756) at 4°C overnight. The following day, the embryos were washed six times with MAB for at least 1 hour. The embryos were then rinsed with alkaline phosphatase buffer containing 1M Tris-HCl pH 9.5, 5M NaCl, 1M MgCl2 and Tween 20 for 10 minutes, three times, and developed in BM purple solution (Roche Diagnostics, 11442074001) until the color has developed. The embryos were then washed with 2mM EDTA/PBT, fixed in 4% paraformaldehyde overnight, dehydrated in a series of ethanol, cleared in xylene, embedded with paraffin, and sectioned at 7μm thick.
Probes used: Shh [31].
Results:
Generation of Ppp1r35-null embryos.
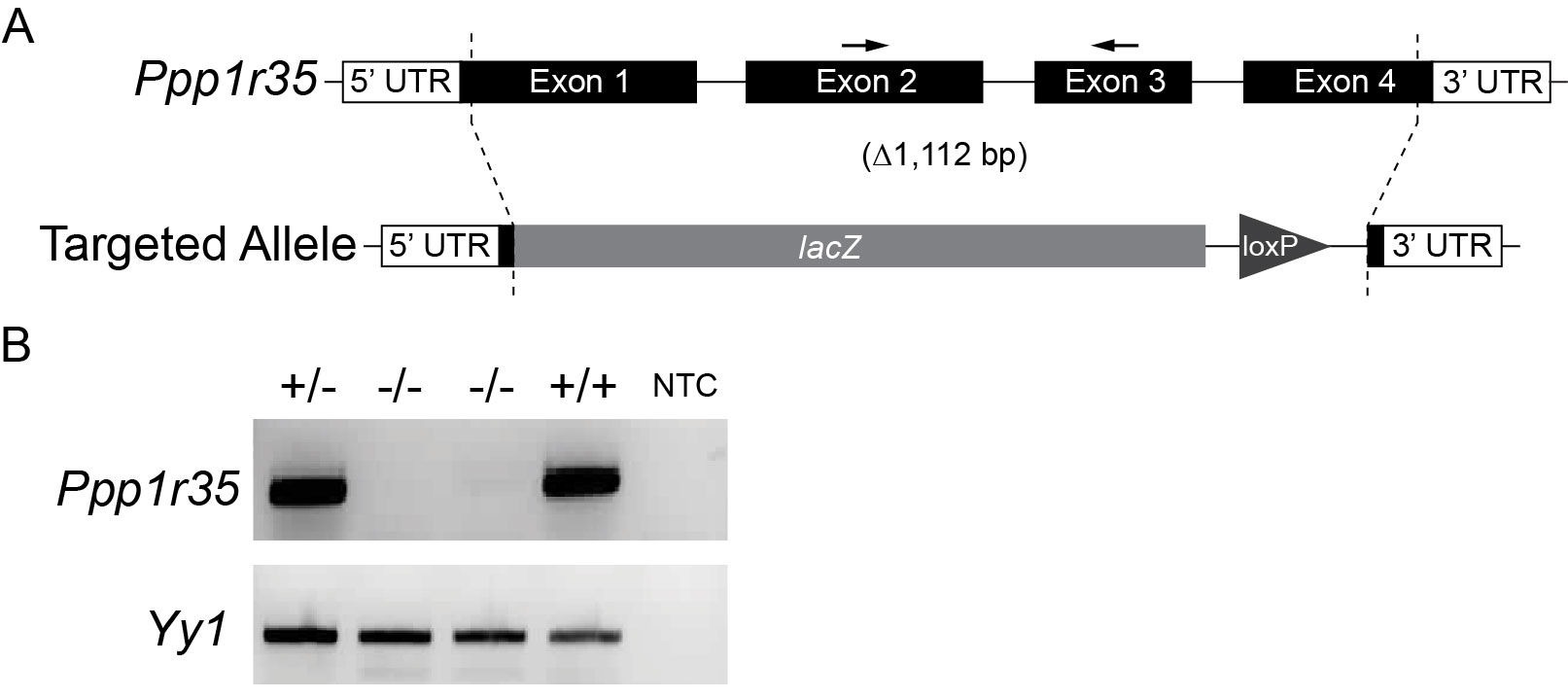
A knockout allele generated on the C57Bl6/N background (Ppp1r35tm1.1(KOMP)Vlcg) was used to analyze the role of Ppp1r35 in murine embryonic development [3]. The knockout is an in-frame deletion of 1,112 base pairs that includes part of exon 1, intron 1, exon 2, intron 2, exon 3, intron 3, and part of exon 4 and inserts a beta-galactosidase (lacZ) to create a reporter allele (Supplementary Figure S1A). The International Mouse Phenotyping Consortium (IMPC) reported that mice homozygous for the Ppp1r35 allele are not recovered at birth (0/110 pups) or at E15.5 indicating that homozygous embryos (hereby referred to as mutants, “−/−”, or “null”) are lethal during early embryogenesis [3]. Heterozygous Ppp1r35 animals were crossed to produce embryos at various stages of development. At all stages, up to and including E9.5, mutant embryos were recovered at expected Mendelian ratios (25% homozygous mutant, 49% heterozygous, 26% homozygous wild type). RT-PCR was used to confirm that the Ppp1r35 transcript is absent in homozygous mutant embryos (Supplementary Figure S1B).
Expression of Ppp1r35 during development.
The lacZ reporter gene was used to investigate the expression of Ppp1r35 in heterozygous embryos. LacZ positive cells are present in the epiblast and some extra-embryonic ectoderm cells but is absent in the visceral endoderm (VE) in E6.0 pre-streak stage embryos (Figure 1A,E,I). At E7.5, the reporter is expressed in all nascent embryonic derivatives (Figure 1B), however, the VE conspicuously lacks reporter expression (Figure 1B,F,J), consistent with the E6.0 pattern. The lack of expression in the VE is supported by RT-PCR with intron spanning primers designed to detect endogenous Ppp1r35 (Figure 1M). At E7.5, the reporter is robustly expressed in the embryonic ectoderm and mesoderm derivatives, including the primitive streak and allantois (Figure 1F,J). A similar expression pattern is observed at E8.5 and E9.5 with lacZ expression in mesoderm-derived structures including the heart, notochord, and somites as well as the ectoderm-derived neural tube (Figure 1G–L). Interestingly, expression in the definitive endoderm-derived gut tube is relatively absent at E8.5 but returns in the foregut, but not the hindgut, by E9.5 (Figure 1K,L). To summarize, Ppp1r35 is expressed throughout much of the embryo at E7.5, in mesoderm and ectoderm-derived structures at E8.5, and throughout much of the embryo at E9.5, but is absent in the extra-embryonic VE at all stages examined. Two caveats to the lacZ reporter-based expression should be noted. The first is the nature of the constructed allele; expression may potentially be altered by the deletion at the locus. The second is possible perdurance of the beta-galactosidase protein from the lacZ reporter; the reported expression may be temporally extended beyond the endogenous Ppp1r35.
Ppp1r35-null embryos fail to turn and progress beyond ~E8.5.
To investigate the function of Ppp1r35 during embryonic development, timed heterozygous by heterozygous crosses of adult mice were used to generate embryos between E7.5 and E10.5 (Figure 2A). At E7.5, mutant embryos are phenotypically normal but are slightly smaller and delayed compared to littermates. By E8.5, Ppp1r35−/− embryos appear to gastrulate, and now possess a defined anterior-posterior axis and features typical of ~E8.0 stage (headfolds, heart, amnion) but are considerably smaller than their wild type (WT) E8.5 littermates (Figure 2A). By E9.5, all mutants have obvious morphological abnormalities (Figure 2A). E9.5 Ppp1r35-null embryos fail to turn and resemble E8.5 wild type embryos in size and in embryonic orientation (Figure 2A). Other obvious defects include enlargement of the pericardial sac, pooled blood in the posterior, and failed neural tube closure. Mutant embryos are found at E10.5 but have not progressed beyond that observed at E9.5 (Figure 2A). These data indicate that Ppp1r35−/− embryos are capable of initiating and completing gastrulation as well as specifying the anterior/posterior and the dorsal/ventral axis, but that in the absence of Ppp1r35, developmental delays result in the failure to progress past E8.5–9.0.
Figure 2: Ppp1r35 knockout mouse embryos show severe morphological defects compared to their littermates.
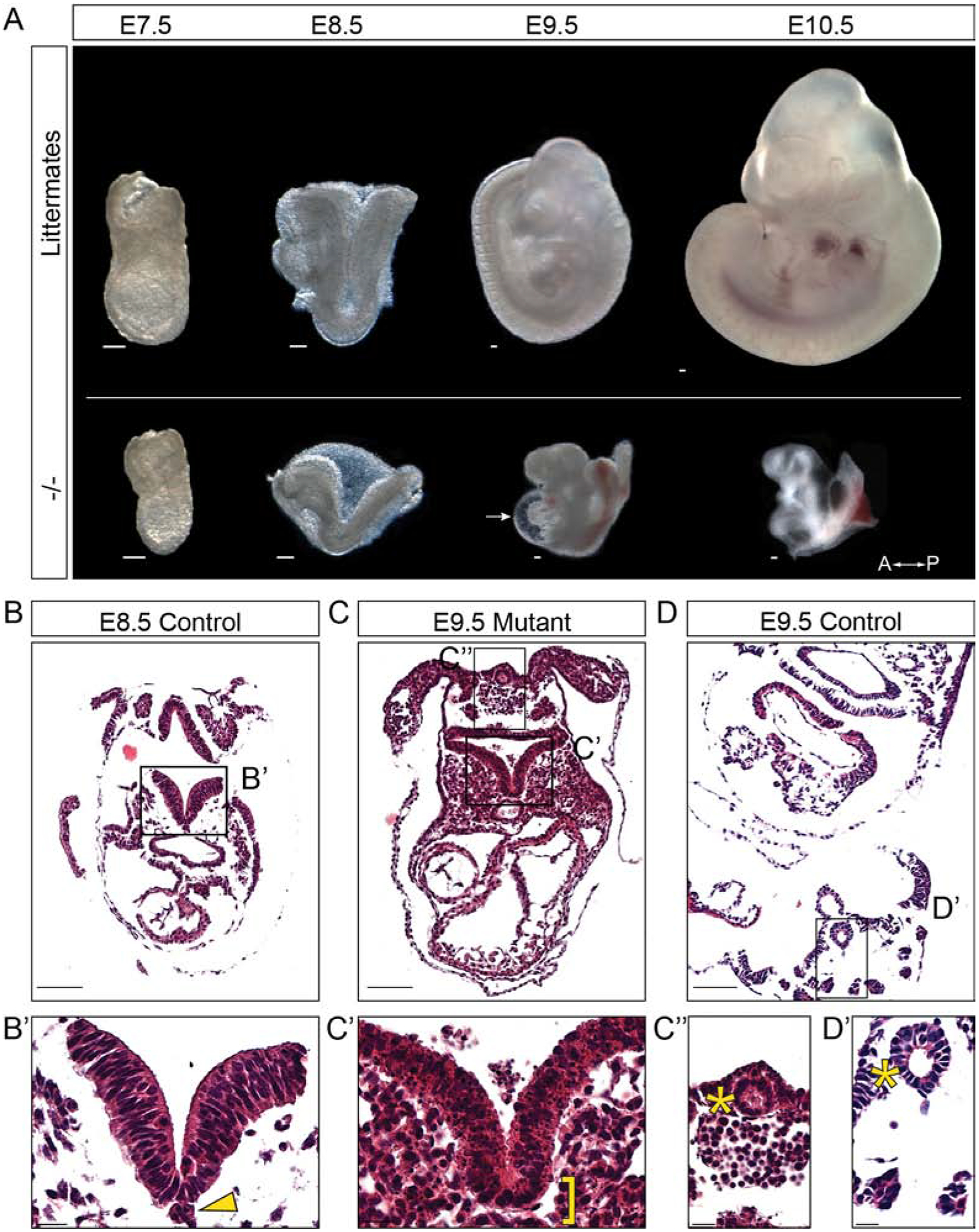
A) Ppp1r35 mutant embryos compared to littermates collected at E7.5–10.5. Hematoxylin & eosin staining performed on transverse sections of E8.5 (B,B’) and E9.5 (D,D’) control embryos compared to an E9.5 mutant (C,C’,C”) embryo. Arrow denotes the pericardial sac. A-P indicates anterior and posterior orientation of the embryos. The arrowhead points to notochord and bracket indicates expected location of the notochord. The asterisk indicates the closed hindgut of the E9.5 mutant and E9.5 control. −/− denotes Ppp1r35 homozygous embryos. Littermates or ctrl denotes embryos wild type or heterozygous for Ppp1r35. Scale bars in A,B,C,D = 100μm and B’,C’,C”,D’ = 20μm.
The morphological defects in Ppp1r35 mutants were next examined using hematoxylin and eosin (H&E) staining of sectioned embryos. Such analysis of mutants at E9.5 reveals numerous embryonic abnormalities (Figure 2C). The E9.5 mutants are compared to developmentally matched E8.5 controls as well as E9.5 littermates. During normal E9.5 development, the neural folds hinge inward and close to complete the neural tube. In E9.5 mutants, the neural folds fail to hinge inward and do not make contact, presumably underlying the complete failure of neural tube closure. Interestingly, while the cells of the neural tube in wild type E8.5 embryos display large intact nuclei (Figure 2B’), the nuclei of cells within the E9.5 mutant neural tube are fragmented and pyknotic, suggesting apoptosis (Figure 2C’). Furthermore, while the E8.5 stage matched controls and E9.5 littermates have an easily distinguishable notochord/notochordal plate along the midline of the A/P axis, those present in mutants are inconsistent and are identifiable in some sections but not in adjacent sections from the same embryo (compare asterisk in Figure 2B’ to C’). Finally, although the gut tube of the E8.5 controls is mainly open, the hindgut of E9.5 mutants has closed and is similar to that of normal E9.5 embryos (asterisks in Figure 2C”, 2D’). Together, these data document the dramatic morphological abnormalities that may contribute to the embryonic lethality observed in Ppp1r35 mutants.
Ppp1r35 is necessary for proper primary cilia formation.
Cell culture studies have suggested that PPP1R35 is a centrosomal protein involved in centrosome assembly [18, 19]. Common features of embryos that are defective in centrosome components are midgestation lethality and embryonic defects caused by the loss or compromised primary cilia [25]. Primary cilia are found in the node, notochord, and neural tube/plate of the E8.0 embryo, where they play important roles in tissue or embryonic patterning. To detect primary cilia, immunofluorescence (IF) was performed with an anti-ADP-ribosylation factor-like protein 13B (ARL13B) antibody on E8.0–8.5 embryos. ARL13B positive primary cilia extend ventrally from the cells of the nod and cells within the neural tube display positive ARL13B puncta in control littermates (Figure 3A,A’,C,C’). Although Ppp1r35 mutant embryos have an identifiable node, the cells of the node lack ARL13B positive primary cilia and cells in the neural tube lack ARL13B puncta (Figure 3B,B’,D,D’). These results suggest that Ppp1r35 is required to form primary cilia and that ciliogenesis is defective in mutant embryos, supporting the hypothesis that Ppp1r35 a component of the centrosome.
Figure 3: Ppp1r35 is necessary for primary cilia formation in the developing embryo.
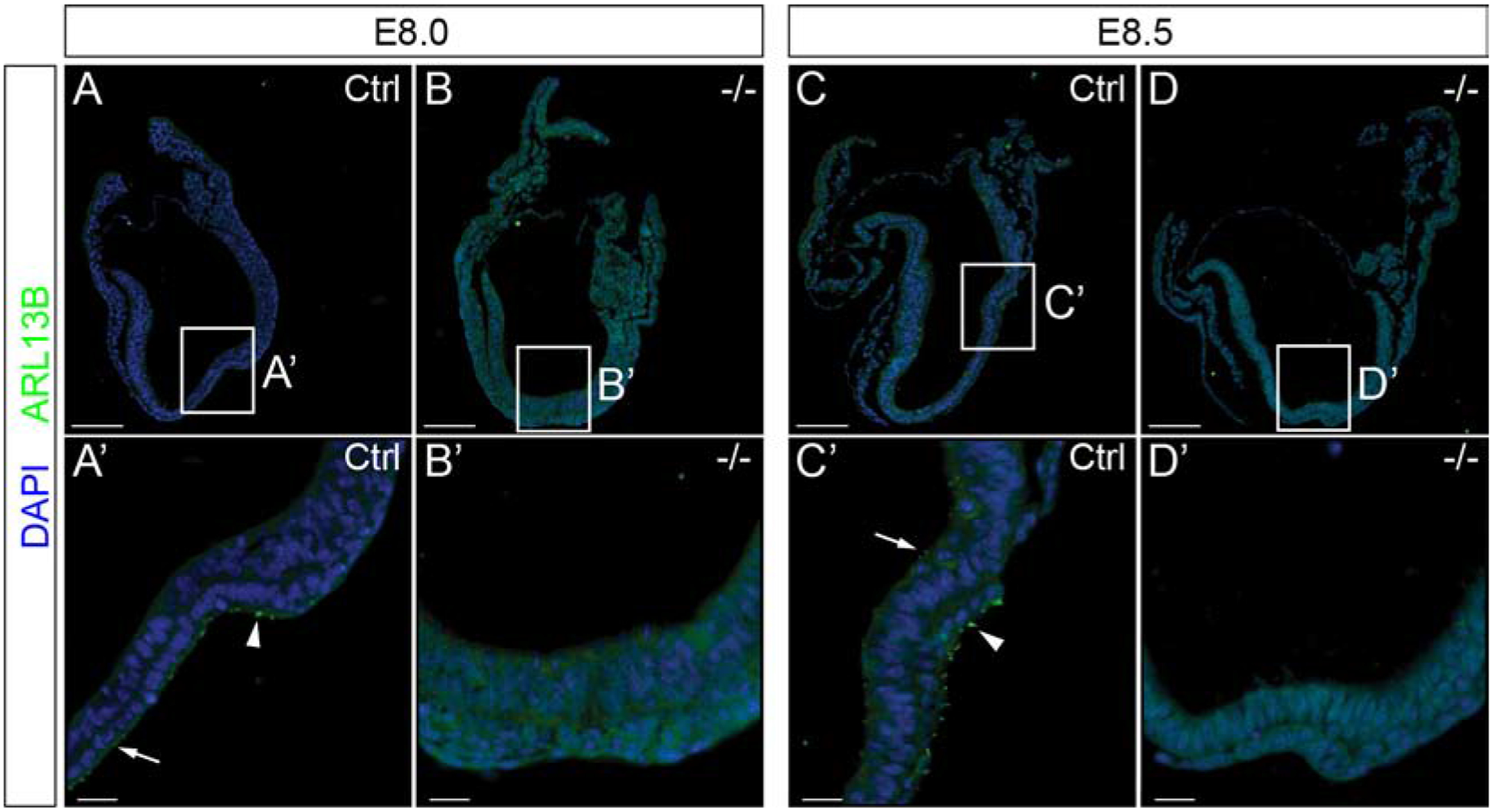
Embryos collected at E8.0 and E8.5 were labeled using ARL13B (green), and nuclei counterstained with DAPI (blue). Ppp1r35 E8.0 and 8.5 control embryos have ARL13B-positive cilia as expected (A,A’) and (C,C’). However, mutant embryos have a complete loss of ARL13B-positive primary cilia (B,B’) (D,D’). Arrowhead denotes nodal cilia and an arrow points to positive puncta. White box indicates the location of the node and corresponds to A’,B’,C’, and D’ respectively. −/− denotes Ppp1r35 homozygous embryos. Ctrl denotes embryos wild type or heterozygous for Ppp1r35. ARL13B mutant embryos: n=5. Scale bars for A-D = 100 μm and A’-D’ = 20 μm.
Altered Shh expression and notochord loss in Ppp1r35 mutant embryos.
It is well established that functional primary cilia are required to transmit sonic hedgehog signals in Shh-responsive cells [32, 33]. For example, Shh, which is secreted from the notochord, is required to induce the floorplate from the ventral neural tube [34]. Disruption in ciliogenesis results in a complete failure of floorplate emergence, despite normal Shh expression in the notochord [35]. Whole-mount in situ hybridization (WISH) was used to examine Shh, which is normally expressed in the notochord/notochordal plate, dorsal endoderm and floorplate [31]. E7.5 WT embryos express Shh in the notochordal plate (Figure 4A), however, E7.5 mutants are completely lacking Shh expression (Figure 4B). Consistent with a developmental delay, E8.0–8.5 mutants express Shh in a pattern similar to that of E7.5-E8.0 controls, respectively (compare Figure 4A,C to D,F). At E8.5, control embryos have a defined Shh-positive notochord (Figure 4E), and by E9.5 display Shh ventrally in the endoderm and dorsally in the notochord and floorplate (Figure 4G). In contrast to normal patterning, the E9.5 mutant embryo displays a single discontinuous stripe of Shh expression in the presumptive notochord (Figure 4H). To further investigate the midline defects, we examined expression of forkhead box protein A2 (FOXA2), a transcription factor expressed in and required for the proper formation of the node, notochord, gut tube, and floor plate [36, 37]. At E9.5, some FOXA2-expressing notochord can be identified in mutants (Figure 4J). However, the FOXA2-positive notochord intermittently disappears, even from one section to the adjacent section. When FOXA2-positive notochord is present it is usually mispositioned and shifted lateral to the neural tube (Figure 4J’–J”‘). It is clear that FoxA2 is required downstream of Shh signaling in the newly specified floorplate [34], however, we do not see FOXA2 expression in the mutant neural tube (Figure 4J’–J”‘).
Figure 4: Shh expression and notochord are disrupted in Ppp1r35 mutant embryos at E9.5.
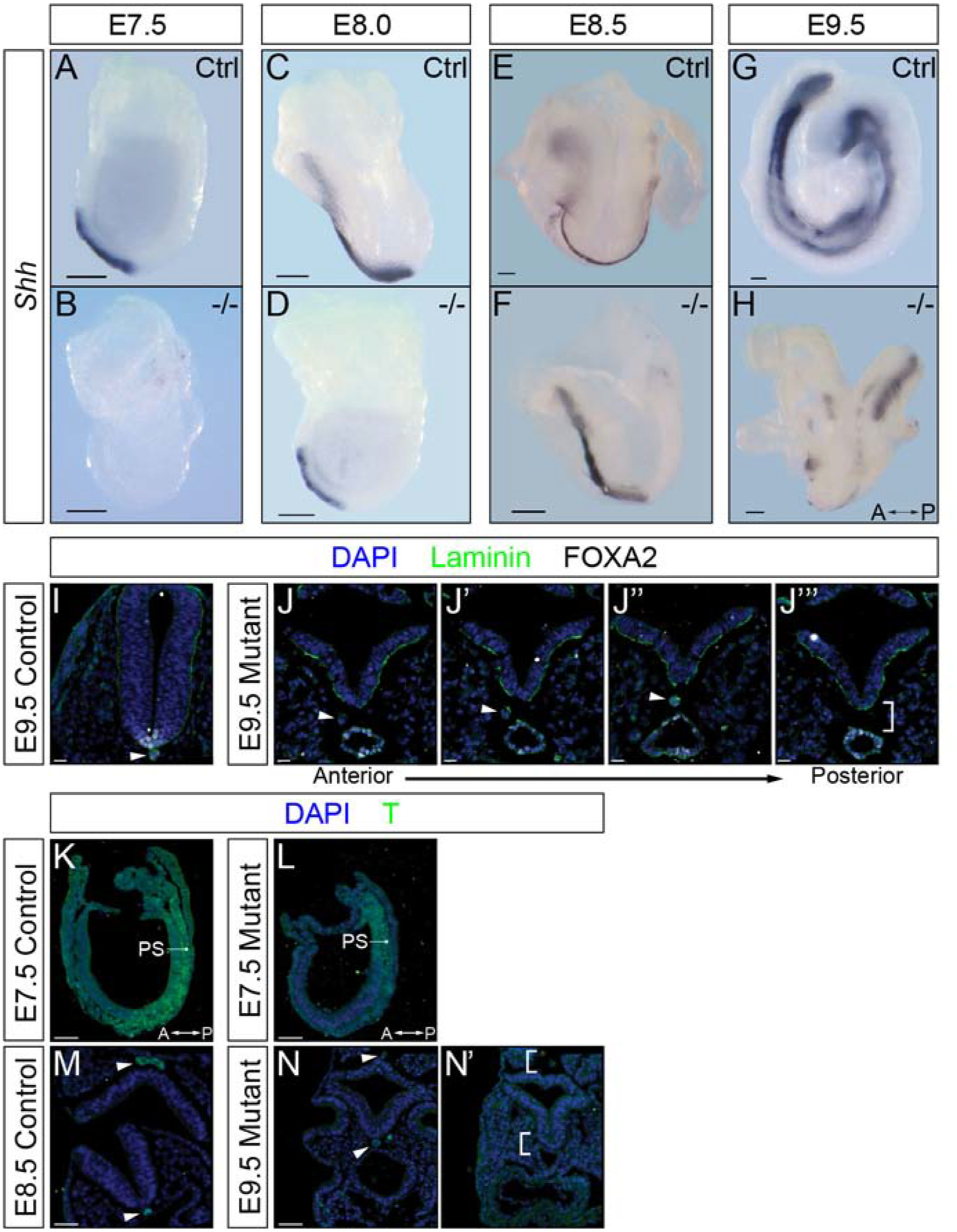
Whole-mount in situ hybridization to detect sonic hedgehog (Shh) expression in littermates and Ppp1r35 mutant embryos collected at E7.5 (A,B), E8.0 (C,D), E8.5 (E,F) and E9.5 (G,H). Shh mutant embryos: E7.5 n=3, E8.0 n=2, E8.5 n=3, E9.5 n=3. Immunofluorescence of transversely sectioned E9.5 embryos using laminin (green), FOXA2 (white) and nuclei counterstained with DAPI (blue) (I,J-J”‘). FOXA2 mutant embryos: n=3. T (green) and DAPI (blue) staining of sectioned E7.5 sagittal control (K), E7.5 sagittal mutant (L), E8.5 transverse control (M) and E9.5 transverse mutant (N,N’). T mutant embryos: E7.5 n=3, E9.5 n=3. A-P indicates the anterior-posterior axis of the E9.5 mutant embryo. Arrowhead indicates FOXA2 and T positive notochord. Brackets indicate expected location of the notochord. −/− denotes Ppp1r35 homozygous embryos. Ctrl denotes wild type or heterozygous Ppp1r35 embryos. Scale bars for A-H = 100 μm, I-J” = 20 μm, K-N’ = 50 μm. The arrows indicate the position of the anterior and posterior (A-P) axis.
We next examined expression of a second critical transcription factor, brachyury (T). Beginning at E6.5, T is expressed in primitive streak and in the nascent mesoderm. Similar to WT littermates, the E7.5 mutant embryos have T-positive cells in the primitive streak and nascent mesoderm (Figure 4K,L). By E8.5, T is normally restricted to the notochord (Figure 4M) [38]. Because the E9.5 mutants are delayed and more similar to E8.5 embryos, T expression is relegated to the inconsistent notochord along the anterior/posterior axis (Figure 4N,N’). These results suggest that T is appropriately expressed and maintained in the primitive streak, nascent mesoderm and consistent with the results presented above, demonstrates that the notochord is indeed fragmented. It is interesting to note that in transverse sections of mutant embryos that lack notochord, the spacing between the gut and neural tube is markedly reduced and in some cases the germ layers appear to touch (brackets in Figure 2C’). These results support the hypothesis posited by others that a role of the notochord is to secrete repulsive cues that function to maintain appropriate spacing between adjacent tissue layers [39].
Notochord morphogenesis is altered in Ppp1r35 mutant embryos.
During late headfold stages (~E8.0) the embryo is unturned and the cells that will contribute to the notochord and notochordal plate, are contiguous with the endoderm on the ventral surface of the embryo. During the 1–4 somite stages (~E8.25) in an anterior to posterior wave, the cells of the notochordal plate detach from this epithelial layer using a process termed resolution. Resolution is accompanied by the dorsal migration of the newly detached cells and the subsequent formation of the rod that defines the notochord [40, 41]. To examine this process in Ppp1r35 mutants, we examined FOXA2, normally expressed in the endoderm, notochord and floorplate at these stages, and laminin, a component of the basement membrane. To compensate for the embryonic delay, E8.75 mutant embryos were compared to similarly staged E8.5 controls. Normally, when the notochord resolves from the dorsal midline of the endoderm, laminin surrounds its dorsal and lateral sides (Figure 5A,A’,A”) [40, 42]. When a notochord is apparent in mutant embryos, laminin is atypically distributed. The mutants displayed in Figure 5B and C represent adjacent sections from 2 embryos. The sections in B and B’ have notochord that lacks laminin on the left lateral side, while the notochord that is apparent in the mutant in C and C’ lacks laminin. These data suggest that the loss of Ppp1r35 results in abnormal notochord resolution and provide support for the hypothesis that the basal lamina is integral in this process [42].
Figure 5: Notochord morphogenesis is disrupted in Ppp1r35 mutant embryos.
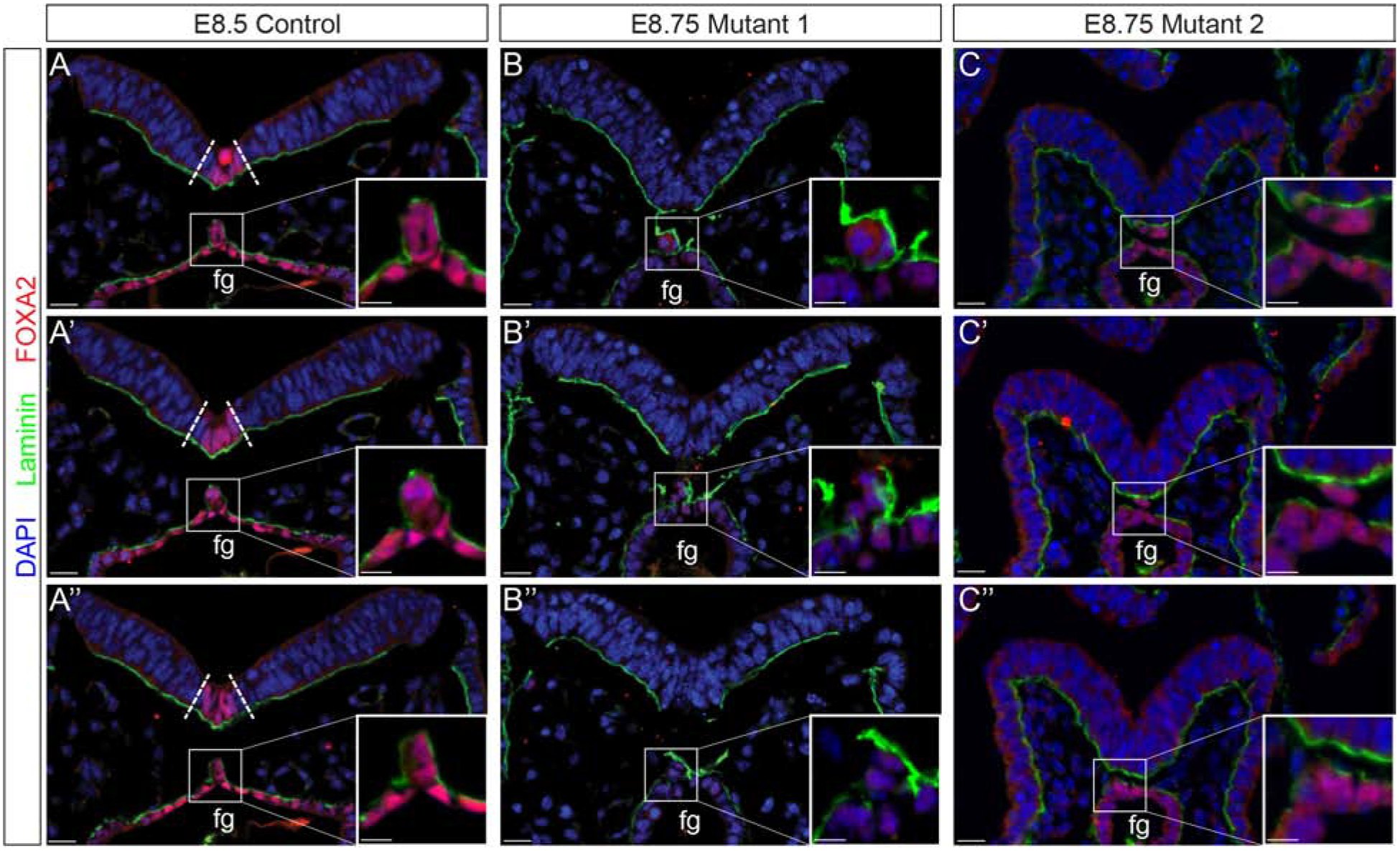
Immunofluorescent analysis of laminin (green), FOXA2 (red), and nuclei counterstained with DAPI (blue). Consecutive sections through a stage-matched E8.5 control reveals a FOXA2-positive floor plate (boundary defined by dotted lines) and a contiguous notochord that has laminin distributed laterally and dorsally (A-A”). Although the two E8.75 two mutants vary slightly they both lack a FOXA2-positive floor plate and display altered notochord resolution (B-B”, C-C”). Fg; foregut. Ctrl denotes embryos wild type or heterozygous for Ppp1r35. −/− denotes Ppp1r35 homozygous embryos. Boxes indicate a higher magnification of the notochord. FOXA2-labeled mutant embryos: n=3. Scale bars = 20 μm.
Cell death and proliferation in the developing embryo.
To understand the causes underlying the developmental delay observed in Ppp1r35 mutant embryos, we examined apoptosis and proliferation in mutant embryos and stage-matched controls. Phospho-S10 Histone H3 (PH3) was used to evaluate proliferation in mutant and control embryos (Figure 6A,B). Both contain many PH3-positive nuclei, however high magnification imaging reveals that a significantly higher number of nuclei in the mutant display a distinctive nuclear “halo” pattern of PH3 than in control (Figure 6D). PH3 localization can be used to determine specific cell cycle stages (Figure 6C) [25, 43], and this halo pattern is indicative of prometaphase (Figure 6B’,C). We therefore hypothesize that the loss of Ppp1r35 causes cells to stall or arrest in prometaphase.
Figure 6: Ppp1r35 mutants display increased apoptosis in the neural tube and a significant, embryo-wide, increase in prometaphase cells.
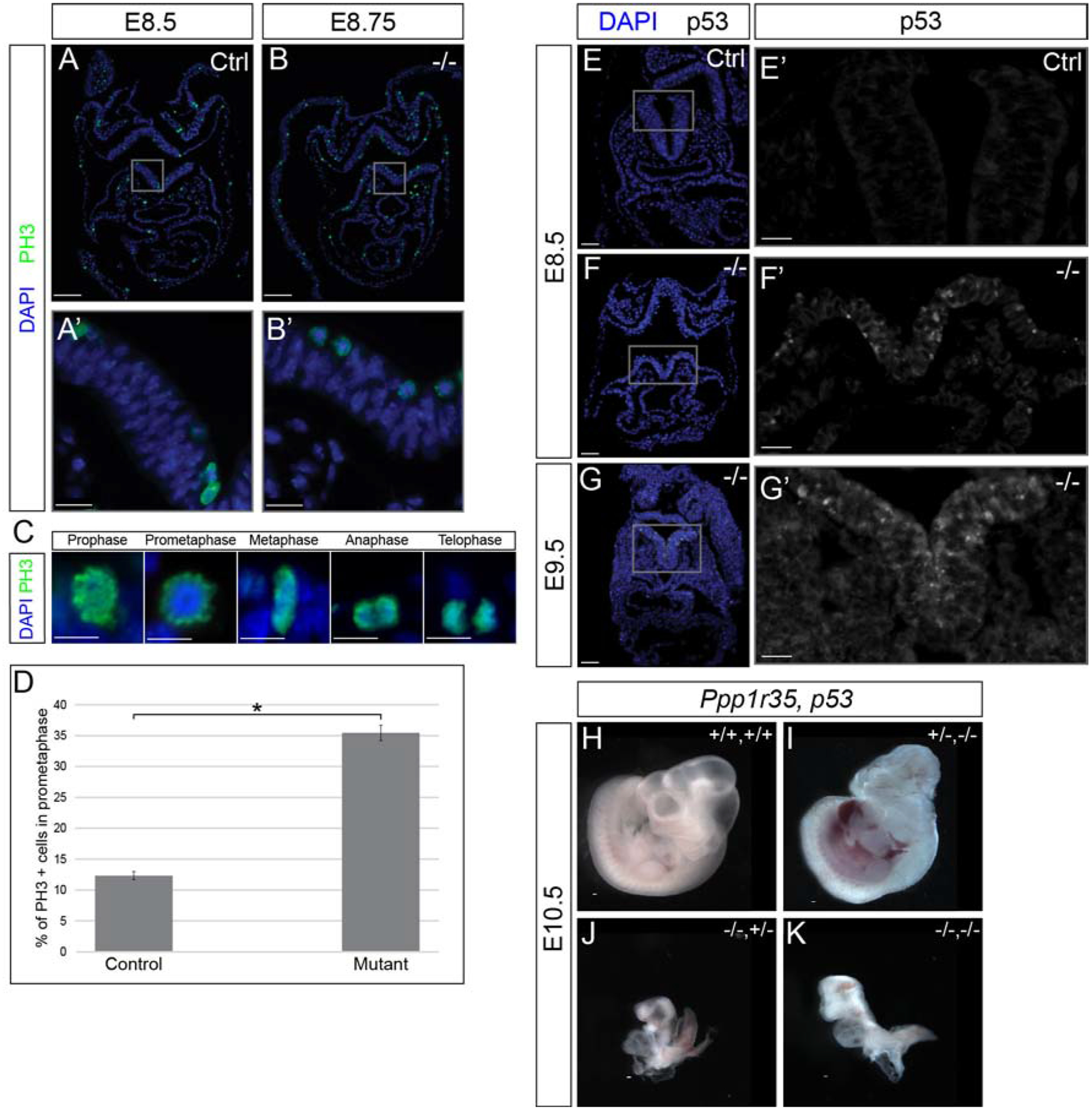
Immunofluorescent analysis of cell proliferation using PH3 (green), counterstained with DAPI (blue). E8.5 stage-matched control used for the E8.75 mutant embryo (A,B). PH3 mutant embryos: n=3. PH3 positive cells undergoing the stages of mitosis (C). Significantly more PH3-positive cells in prometaphase are observed in mutant embryos (compare A’ to B’, quantified in D). Analysis of active p53 (white; E-G’) and p53 with nuclei (DAPI; blue) (E,F,G). No/rare p53 positive cells are found in control E8.5 embryos (E,E’). Many p53-positive cells are observed in the neural tube of the E8.5 (F,F’) and E9.5 (G,G’) Ppp1r35 mutants. p53 mutant embryos: n=3. No obvious phenotypic rescue was observed in Ppp1r35;p53 double-knockout embryos collected at E10.5 (H-K). −/− denotes Ppp1r35 homozygous embryos. Littermates or ctrl denotes embryos wild type or heterozygous for Ppp1r35. +/+,+/+ (WT Ppp1r35,WT p53); +/−,−/− (Het Ppp1r35, Mut p53); −/−,+/− (Mut Ppp1r35, Het p53); −/−,−/− (Mut Ppp1r35, Mut p53). Scale bars for A and B = 100 μm, A’ and B’ = 20 μm, C = 10 μm, E,F,G = 50 μm, E’,F’,G’ = 20 μm, H-K = 100μm. Error bars indicate SEM. Statistically significant comparisons are indicated with a bracket and star where *P<0.001. (P= 9.31E-16).
Anti-Phospho-p53 (ser15) (p53) was next used to examine apoptosis in mutants. While control embryos contain only rare p53-positive cells (Figure 6E,E’), E8.5–9.5 mutant embryos have extensive apoptosis in the neural tube (Figure 6F,F’,G,G’), confirming that the nuclear fragmentation observed in (Figure 2C’) represent cells undergoing apoptosis. Together these data demonstrate that Ppp1r35 deficiency results in increased apoptosis that is specific to the neural tube.
p53 knockout fails to rescue Ppp1r35 knockout phenotype.
Defects in centrosome-related proteins have been shown to initiate p53-dependent cell cycle arrest in cultured human cells [44]. Due to the drastically increased number of p53-positive cells in the neural tube of E8.5–9.5 mutant embryos, we hypothesized that the increased cell death could underlie the developmental delay. In an attempt to rescue the phenotype of the Ppp1r35-null embryos, we generated Ppp1r35−/−; p53−/− double-mutant embryos. This strategy has been used to partially rescue the embryonic phenotypes produced by loss of other centrosome-related genes including Kif20b and Sas4 [25, 45]. While double-mutant embryos collected at E10.5 are slightly larger than Ppp1r35-null embryos, no significant change in the phenotype is observed (Figure 6J,K). Therefore, we conclude that while there is greatly increased cell death in Ppp1r35 mutant embryos, p53-mediated apoptosis is not a major driving factor of the overall phenotype.
Discussion:
Here we show that Ppp1r35 is required for early development and that null embryos fail at the onset of organogenesis. Previous studies have linked PPP1R35 to centrosome formation in cultured human cells [18, 19]. In support of this proposed role, we provide the first evidence that murine Ppp1r35 is necessary for the formation of primary cilia in vivo. Primary cilia formed on the cell surface from the mother centriole of the centrosome are integral to the ability of a cell to respond to Hedgehog signaling [46]. Consistent with a role for Ppp1r35 in centrosome assembly, our data show that Ppp1r35 mutant embryos are deficient in primary cilia assembly. The notochord, although fragmented in mutants, expresses Shh and should be capable of floorplate induction in the adjacent neural tube. The failure of the mutant neural tube to express FoxA2 in response to Shh signals provides further evidence that proper cilia formation and function are required for neural tube patterning. Indeed, neural tube patterning defects are found in other mutants that lack primary cilia [35, 47, 48].
By E9.5, the Ppp1r35 mutant embryos have a discontinuous notochord. Despite being an evolutionarily conserved feature of chordates, morphogenesis of the notochord is not well studied. At approximately E8.0–8.5 of murine development, the cells that will become notochord, termed the notochordal plate, are continuous with the endoderm and share a basal lamina [5, 40]. Between E8.5–9.5, the notochordal plate uses an unknown mechanism to ingress from the endoderm layer. During ingression, the notochord remains connected to the endoderm by the basal lamina [5, 40]. By E10.5, the notochord has resolved from the endoderm and adheres to the neural tube [5, 40]. In Ppp1r35 mutants, the notochord is inconsistently present and alterations to the basal lamina are also observed. These data suggest that the loss of Ppp1r35 alters notochord evagination and may offer a window of inquiry towards further understanding normal notochord morphogenesis.
Defects in centrosomal proteins have been linked to the neurodevelopmental disorder autosomal recessive primary microcephaly (MCPH) [21–23]. A recent paper investigating PPP1R35 identified Rotatin, a gene involved in centriole elongation, as a critical partner to PPP1R35 [19]. Mice lacking Rttn as well as other centrosome-related genes linked to MCPH, including Sas4 and STIL, each have null phenotypes that are similar Ppp1r35-null embryos [24–26, 49]. For example, like Ppp1r35-null embryos, embryos that are null for each of these genes fail to properly pattern or close their neural tube and display a fragmented notochord. Importantly, and unlike embryos with mutations in genes that are solely involved in primary cilia formation [25, 35], Sas4, STIL and Rttn arrest by E8.5 and never turn. Finally, although not examined in Rttn-deficient embryos, Sas4 and STIL mutants display widespread increased apoptosis that in the case of Sas4, is accompanied by a prolonged prometaphase [24–26]. Although Sas4 mutants can be partially rescued by loss of p53 [25] resulting in embryos that turn and develop to approximately E9.5, Ppp1r35 −/− embryos do not display a similar rescue in the context of p53 loss. This result suggests that the defects observed in Ppp1r35 −/− embryos are not due to p53-mediated apoptosis. This difference could be due to differences in the genetic background of each of the mutant lines or could be a phenotype the distinguishes the role of Ppp1r35 from that of Sas4.
Due to the phenotypic similarities described herein, as well as the cited biochemical and cell biological evidence, we suggest that the phenotype observed in Ppp1r35-null embryos is due to defects in centrosome function or biogenesis and/or interaction with other centrosomal proteins such as Rttn. Together, these data support a role for Ppp1r35 in MCPH. Understanding the precise role for microcephaly-related proteins in normal development may aid in therapeutic approaches for human patients.
Supplementary Material
Supplementary Figure S1 Legend: Ppp1r35 knockout A) Schematic representation of the Ppp1r35 wild type allele (upper) and targeted Ppp1r35tm1.1(KOMP)Vlcg allele (lower). The knockout allele was generated by replacing 1,112 bp of genomic sequence and inserting a lacZ reporter cassette. Arrows indicate primers used for RT-PCR in (B) which verifies the expression of Ppp1r35 mRNA in wild type (+/+) and heterozygous (+/−) embryos, and the absence of detectable Ppp1r35 mRNA in homozygous (−/−) mutant embryos. Yy1 was used as a positive control to show successful cDNA production from mutant embryos. NTC denotes “no template control”.
{kind=link}
{kind=link}
Highlights:
Homozygous Ppp1r35 mutant embryos fail to turn by E9.5 with defective neural tube, notochord, floorplate morphology.
Mutant embryos lack primary cilia.
Cell cycle defects are consistent with a role for Ppp1r35 in centriole function.
These results implicate Ppp1r35 as a candidate involved in human microcephaly.
Acknowledgements:
We thank members of the Mager and Tremblay labs for providing useful input throughout the project. This work was supported by R01HD083311 to JM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1.Pertea M, et al. , Thousands of large-scale RNA sequencing experiments yield a comprehensive new human gene list and reveal extensive transcriptional noise. bioRxiv, 2018: p. 332825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.UCDavis. KOMP Repository Knockout Mouse Project. 2019; Available from: http://www.KOMP.org.
- 3.Consortium IMP Gene: Ppp1r35. 2019; Available from: https://www.mousephenotype.org/data/genes/MGI:1922853.
- 4.Dickinson ME, et al. , High-throughput discovery of novel developmental phenotypes. Nature, 2016. 537: p. 508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jurand A, Some aspects of the development of the notochord in mouse embryos. J Embryol Exp Morphol, 1974. 32(1): p. 1–33. [PubMed] [Google Scholar]
- 6.Lee JD and Anderson KV, Morphogenesis of the node and notochord: the cellular basis for the establishment and maintenance of left-right asymmetry in the mouse. Developmental dynamics : an official publication of the American Association of Anatomists, 2008. 237(12): p. 3464–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chamberlain CE, et al. , Notochord-derived Shh concentrates in close association with the apically positioned basal body in neural target cells and forms a dynamic gradient during neural patterning. Development, 2008. 135(6): p. 1097. [DOI] [PubMed] [Google Scholar]
- 8.Corallo D, Trapani V, and Bonaldo P, The notochord: structure and functions. Cellular and Molecular Life Sciences, 2015. 72(16): p. 2989–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Verbinnen I, Ferreira M, and Bollen M, Biogenesis and activity regulation of protein phosphatase 1. Biochemical Society Transactions, 2017. 45(1): p. 89. [DOI] [PubMed] [Google Scholar]
- 10.Shi Y, Serine/Threonine Phosphatases: Mechanism through Structure. Cell, 2009. 139(3): p. 468–484. [DOI] [PubMed] [Google Scholar]
- 11.Peti W, Nairn AC, and Page R, Structural basis for protein phosphatase 1 regulation and specificity. The FEBS journal, 2013. 280(2): p. 596–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ceulemans H and Bollen M, Functional Diversity of Protein Phosphatase-1, a Cellular Economizer and Reset Button. Physiological Reviews, 2004. 84(1): p. 1–39. [DOI] [PubMed] [Google Scholar]
- 13.Egloff MP, et al. , Structural basis for the recognition of regulatory subunits by the catalytic subunit of protein phosphatase 1. Embo j, 1997. 16(8): p. 1876–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Committee HGN Gene group: Protein phosphatase 1 regulatory subunits (PPP1R) 2019, August 14; Available from: https://www.genenames.org/data/genegroup/#!/group/694.
- 15.Jakobsen L, et al. , Novel asymmetrically localizing components of human centrosomes identified by complementary proteomics methods. The EMBO Journal, 2011. 30(8): p. 1520–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nigg EA and Raff JW, Centrioles, Centrosomes, and Cilia in Health and Disease. Cell, 2009. 139(4): p. 663–678. [DOI] [PubMed] [Google Scholar]
- 17.Azimzadeh J and Marshall WF, Building the centriole. Current biology : CB, 2010. 20(18): p. R816–R825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fong CS, Ozaki K, and Tsou M-FB, PPP1R35 ensures centriole homeostasis by promoting centriole-to-centrosome conversion. Molecular biology of the cell, 2018. 29(23): p. 2801–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sydor AM, et al. , PPP1R35 is a novel centrosomal protein that regulates centriole length in concert with the microcephaly protein RTTN. eLife, 2018. 7: p. e37846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chen H-Y, et al. , Human microcephaly protein RTTN interacts with STIL and is required to build full-length centrioles. Nature Communications, 2017. 8(1): p. 247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barbelanne M and Tsang WY, Molecular and cellular basis of autosomal recessive primary microcephaly. BioMed research international, 2014. 2014: p. 547986–547986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Faheem M, et al. , Molecular genetics of human primary microcephaly: an overview. BMC medical genomics, 2015. 8 Suppl 1(Suppl 1): p. S4–S4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Woods CG, Bond J, and Enard W, Autosomal recessive primary microcephaly (MCPH): a review of clinical, molecular, and evolutionary findings. American journal of human genetics, 2005. 76(5): p. 717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faisst AM, et al. , Rotatin is a novel gene required for axial rotation and left–right specification in mouse embryos. Mechanisms of Development, 2002. 113(1): p. 15–28. [DOI] [PubMed] [Google Scholar]
- 25.Bazzi H and Anderson KV, Acentriolar mitosis activates a p53-dependent apoptosis pathway in the mouse embryo. Proceedings of the National Academy of Sciences of the United States of America, 2014. 111(15): p. E1491–E1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Izraeli S, et al. , The SIL gene is required for mouse embryonic axial development and left–right specification. Nature, 1999. 399(6737): p. 691–694. [DOI] [PubMed] [Google Scholar]
- 27.Tremblay KD, et al. , Formation of the definitive endoderm in mouse is a Smad2-dependent process. Development, 2000. 127(14): p. 3079. [DOI] [PubMed] [Google Scholar]
- 28.Wallingford MC, Angelo JR, and Mager J, Morphogenetic analysis of peri-implantation development. Developmental Dynamics, 2013. 242(9): p. 1110–1120. [DOI] [PubMed] [Google Scholar]
- 29.Schindelin J, et al. , Fiji: an open-source platform for biological-image analysis. Nature Methods, 2012. 9(7): p. 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tremblay KD, Dunn NR, and Robertson EJ, Mouse embryos lacking Smad1 signals display defects in extra-embryonic tissues and germ cell formation. Development, 2001. 128(18): p. 3609. [DOI] [PubMed] [Google Scholar]
- 31.Echelard Y, et al. , Sonic hedgehog, a member of a family of putative signaling molecules, is implicated in the regulation of CNS polarity. Cell, 1993. 75(7): p. 1417–1430. [DOI] [PubMed] [Google Scholar]
- 32.Bangs F and Anderson KV, Primary Cilia and Mammalian Hedgehog Signaling. Cold Spring Harbor perspectives in biology, 2017. 9(5): p. a028175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wheway G, Nazlamova L, and Hancock JT, Signaling through the Primary Cilium. Frontiers in cell and developmental biology, 2018. 6: p. 8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiang C, et al. , Cyclopia and defective axial patterning in mice lacking sonic hedgehog gene function. Nature, 1996. 383(6599): p. 407–13. [DOI] [PubMed] [Google Scholar]
- 35.Huangfu D, et al. , Hedgehog signalling in the mouse requires intraflagellar transport proteins. Nature, 2003. 426(6962): p. 83–87. [DOI] [PubMed] [Google Scholar]
- 36.Ang S-L and Rossant J, HNF-3β is essential for node and notochord formation in mouse development. Cell, 1994. 78(4): p. 561–574. [DOI] [PubMed] [Google Scholar]
- 37.Weinstein DC, et al. , The winged-helix transcription factor HNF-3β is required for notochord development in the mouse embryo. Cell, 1994. 78(4): p. 575–588. [DOI] [PubMed] [Google Scholar]
- 38.Wilkinson DG, Bhatt S, and Herrmann BG, Expression pattern of the mouse T gene and its role in mesoderm formation. Nature, 1990. 343(6259): p. 657–659. [DOI] [PubMed] [Google Scholar]
- 39.Domyan Eric T., et al. , Roundabout Receptors Are Critical for Foregut Separation from the Body Wall. Developmental Cell, 2013. 24(1): p. 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balmer S, Nowotschin S, and Hadjantonakis A-K, Notochord morphogenesis in mice: Current understanding & open questions. Developmental dynamics : an official publication of the American Association of Anatomists, 2016. 245(5): p. 547–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fausett SR, Brunet LJ, and Klingensmith J, BMP antagonism by Noggin is required in presumptive notochord cells for mammalian foregut morphogenesis. Developmental Biology, 2014. 391(1): p. 111–124. [DOI] [PubMed] [Google Scholar]
- 42.Pulina M, Liang D, and Astrof S, Shape and position of the node and notochord along the bilateral plane of symmetry are regulated by cell-extracellular matrix interactions. Biology open, 2014. 3(7): p. 583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Goto H, et al. , Aurora-B phosphorylates Histone H3 at serine28 with regard to the mitotic chromosome condensation. Genes Cells, 2002. 7(1): p. 11–7. [DOI] [PubMed] [Google Scholar]
- 44.Mikule K, et al. , Loss of centrosome integrity induces p38—p53—p21-dependent G1—S arrest. Nature Cell Biology, 2007. 9(2): p. 160–170. [DOI] [PubMed] [Google Scholar]
- 45.Little JN and Dwyer ND, p53 deletion rescues apoptosis and microcephaly in a Kif20b mouse mutant. bioRxiv, 2018: p. 272393. [Google Scholar]
- 46.Wheway G, Nazlamova L, and Hancock JT, Signaling through the Primary Cilium. Frontiers in Cell and Developmental Biology, 2018. 6(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huangfu D and Anderson KV, Cilia and Hedgehog responsiveness in the mouse. Proceedings of the National Academy of Sciences of the United States of America, 2005. 102(32): p. 11325–11330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bay SN and Caspary T, What are those cilia doing in the neural tube? Cilia, 2012. 1(1): p. 19-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Melloy PG, et al. , No turning, a mouse mutation causing left-right and axial patterning defects. Dev Biol, 1998. 193(1): p. 77–89. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1 Legend: Ppp1r35 knockout A) Schematic representation of the Ppp1r35 wild type allele (upper) and targeted Ppp1r35tm1.1(KOMP)Vlcg allele (lower). The knockout allele was generated by replacing 1,112 bp of genomic sequence and inserting a lacZ reporter cassette. Arrows indicate primers used for RT-PCR in (B) which verifies the expression of Ppp1r35 mRNA in wild type (+/+) and heterozygous (+/−) embryos, and the absence of detectable Ppp1r35 mRNA in homozygous (−/−) mutant embryos. Yy1 was used as a positive control to show successful cDNA production from mutant embryos. NTC denotes “no template control”.