

Abstract
Germline mutations in TP53 cause a rare high penetrance cancer syndrome, Li Fraumeni Syndrome (LFS). Here we identified a rare TP53 tetramerization domain missense mutation, c.1000G>C;p.G334R, in a family with multiple late-onset LFS-spectrum cancers. Twenty additional c.1000G>C probands and one c.1000G>A proband were identified, and available tumors showed biallelic somatic inactivation of TP53. The majority of families were of Ashkenazi Jewish descent, and the TP53 c.1000G>C allele was found on a commonly inherited Chromosome 17p13.1 haplotype. Transient transfection of the p.G334R allele conferred a mild defect in colony suppression assays. Lymphoblastoid cell lines from the index family in comparison to TP53 normal lines showed that while classical p53 target gene activation was maintained, a subset of p53 target genes (including PCLO, PLTP, PLXNB3 and LCN15) showed defective transactivation when treated with Nutlin-3a. Structural analysis demonstrated thermal instability of the G334R mutant tetramer, and the G334R mutant protein showed increased preponderance of mutant conformation. Clinical case review in comparison to classic LFS cohorts demonstrated similar rates of pediatric adrenocortical tumors and other LFS component cancers, but the latter at significantly later ages of onset. Our data show that TP53 c.1000G>C;p.G334R is found predominantly in Ashkenazi Jewish individuals, causes a mild defect in p53 function, and leads to low penetrance Li Fraumeni Syndrome.
Keywords: TP53, p53, inherited cancer syndrome, Li Fraumeni Syndrome, cancer genetics
Introduction
Li-Fraumeni Syndrome (LFS) (1) is caused by germline mutations in the TP53 gene that disrupt the tumor suppressive function of p53 (chromosome 17p13.1; OMIM 191170). p53 is a transcription factor responsible for genomic integrity and transactivation of downstream genes critical for cell cycle arrest, DNA repair, and/or apoptosis (2). Classic LFS is characterized by a diverse spectrum of malignancies that may begin in childhood and continues through life, including soft tissue and bone sarcomas, breast cancers, brain and adrenocortical tumors (ACT), and leukemia (1). Families with incomplete features of LFS are referred to as Li-Fraumeni-Like (LFL) for which several clinical definitions have been proposed (3). It is recommended that individuals with germline TP53 pathogenic mutations undergo high risk surveillance, with the option of tissue-specific prophylactic surgery (4). Recently, surveillance in TP53 carriers using whole body MRI has shown clinical utility (4).
The majority of cancer-associated TP53 mutations are missenses in the DNA binding domain (DBD) (5). However, pathogenic mutations can be found in the tetramerization domain (TD), resulting in alteration of p53 oligomerization, integrity of the tetramer, and DNA affinity (6,7). The most well documented germline TD missense mutation is the c.1010G>A, p.R337H (rs121912664) founder mutation clustered in the southern Brazilian population (8). A significant portion of families with c.1010G>A; p.R337H have a predominance of childhood ACT (9). Functionally, TD missense mutations have variable properties; some but not all variants reduce transactivation, inhibit oligomerization, or cause temperature or pH dependent tetramer instability (10,11).
Despite the robust functional characterization of the majority of possibleTP53 variants (12), significant challenges in clinically validated variant classification remain. Research beyond the DBD is limited and in vitro p53 studies may not be accurate at predicting clinical phenotype. Rare variants may not be seen at sufficient frequency within a single commercial laboratory to allow effective use of internal cross-family data in variant classification. Additionally, phenotypic and co-segregation data are not routinely pooled across laboratories nor made openly accessible to the clinical genetics community. These issues are particularly relevant with respect to rare hypomorphic mutations with potentially attenuated clinical phenotypes. Consequently, collaborations between academic centers and commercial laboratories are necessary to assemble enough cases for analysis of rare suspected pathogenic mutations. Presently, there are large scale initiatives to enhance data sharing and communications among gene-specific experts, as represented by National Institute of Healthy Initiative Clinical Genome Resource (ClinGen) (13); however, these initiatives are still in their infancy. We present aggregate data between multiple clinical sites and two laboratories supplemented with functional and structural studies for a rare TP53 tetramerization domain mutation, c.1000G>C; p.G334R, which has discordant classifications across clinical laboratories, and has been reviewed by ClinGen’s “TP53 variant curation expert panel” and determined to be a variant of uncertain significance based upon preexisting published data.
Materials and Methods
Case ascertainment
The index proband carrying TP53 c.1000G>C; p.G334R was identified in a research sequencing study (14), and all family members were studied under an Institutional Review Board approved protocol. Additional probands/families with TP53 c.1000G>C; p.G334R were ascertained from clinical practice sites, genetic testing laboratories and online resources. Clinical practice ascertainment was conducted via the National Society of Genetic Counselors (NSGC) ListServ and direct inquiry with academic medical centers. Genetic testing laboratory cases were ascertained from Ambry Genetics, Inc. (Aliso Viejo, CA, USA) and Memorial Sloan Kettering Cancer Center’s “Integrated Mutation Profiling of Actionable Cancer Targets” (MSKCC IMPACT) dataset. Online resources included review of the R20 release of the IARC TP53 database (5) and a literature search. From the total of twenty-two p.G334R cases, twenty-one have c.1000G>C and one proband from the IARC database has c.1000G>A. The study team and genetic testing laboratory reviewed all ascertained cases to minimize duplicative counting. Available data on proband and family clinical history and genetic testing was collected centrally. TP53 c.1000G>C;p.G334R classifications were ascertained via ClinVar (15), clinical genetic testing reports, and direct inquiry with Laboratory Directors. Published population datasets and functional studies were retrieved from the Catalogue for Somatic Mutations in Cancer (COSMIC; cancer.sanger.ac.uk), the IARC database (5), and gnomAD (16). Analysis of cancer spectrum in pathogenic and likely pathogenic TP53 mutation carriers in the Penn LFS cohort and IARC was as described (17), updated for the IARC R20 version. IARC probands with likely pathogenic or pathogenic mutations in TP53 were identified and divided into p.R337H carriers and non-R337H carriers, excluding p.G334R carriers. For Penn LFS, both p.R337H and p.G334R carriers were excluded as there were insufficient numbers of p.R337H carriers for independent analysis. Breast-cancer free survival curves were created using the earliest age of breast cancer diagnosis, or youngest age in IARC if unaffected with breast cancer, for all female probands. Cancer-free survival curves were created using the earliest age of cancer diagnosis, or youngest age in IARC if unaffected with cancer, for all probands. For analysis of cancer rates in probands, probands with multiple primary cancers were included in each applicable cancer group. For analysis of cancer rates in families, families were included if they carried a likely pathogenic or pathogenic mutation and had three or more generations in IARC (average number of individuals per family was six for non-R337H and five for R337H families). SEER data was downloaded from https://seer.cancer.gov/csr/1975_2014/, based on November 2016 SEER data submission, posted to the SEER web site, April 2017 (18).
TP53 Genotyping and cancer susceptibility gene analysis
TP53 genotypes were obtained by direct review of research sequencing (RS) results for the probands of families G334R-1 (14) and G334R-21 (19); by publication review for the probands of families G334R-8 (20) and G334R-20 (21); by review of clinical genetic testing (CGT) reports for the probands of families G334R-2 through 7; and by report from the laboratory director of the two genetic testing company cohorts (probands of families G334R-9 through 19). Germline whole exome sequencing (WES) was also performed for the G334R-2 proband, and the raw WES BAM file for G334R-21 germline was downloaded under approved dbGAP project #21931 and analyzed as described (22). Genotyping of additional G334R-1 family members was performed by Sanger sequencing of the genomic region containing the variant. Primers were developed using NCBI Primer Design software (Supplementary Table S1a), and PCR products were generated with GoTaq Hot Start Polymerase (Promega). Where available, the RS or CGT was reviewed for any mutations in other cancer susceptibility genes (Supplementary Table S2). TP53 genotype for the proband of family G334R-22 (IARC 996) was obtained from the IARC database (5).
Ancestry and haplotype analysis
Ancestry was ascertained by self-report in the clinical and laboratory cohorts. For haplotype analysis, a panel of fifteen microsatellite markers and single nucleotide polymorphisms (SNP) inside and outside the TP53 locus were used to assess chromosome 17p13 haplotype shared among available DNA samples from TP53 c.1000G>C carriers (Supplementary Table S1b). The presence of TP53 c.1000G>C allele was evaluated in Ashkenazi Jewish control population sequencing data (23), in the gnomAD database (v2.1.1 cancer and non-cancer cohorts) (16), in a germline genetic testing laboratory dataset (Ambry Genetics, Inc.) comprising 309,222 samples, and in a tumor genetic testing laboratory dataset (MSKCC IMPACT) comprising 21,729 samples.
In silico analysis
In silico tools used to predict pathogenicity were SIFT, PolyPhen2_HDIV, PolyPhen2_HVAR, LRT, MutationTaster, MutationAssessor, BayesDel, AlignGVGD, FATHMM, GERP++, PhyloP, SiPhy. The interpretation of mutation effect and the molecular modelling of TP53 p.G334R was predicted using Pymol molecular graphics program (http://pymol.sourceforge.net) and SWISS-MODEL (https://swissmodel.expasy.org/). For the SWISS-MODEL analysis, the p53-G334R substitution was modeled onto the wild-type tetramer structure (p53-WT, PDB:1OLH) and the resultant structure prediction analyzed with MolProbity (24).
Tumor Profiling
DNA was extracted from tumors in family G334R-2 (breast cancer), G334-3 (adrenocortical carcinoma), G334R-4 (adrenocortical tumor), G334R-6 (adrenocortical tumor), G334R-17 (pancreatic cancer), G334R-18 (pancreatic cancer). G334R-2 breast tumor and normal blood leukocyte DNA was prepared as described (22). Tumor and normal DNA libraries underwent WES with downstream analysis for somatic mutational profiles using Mutect2, homologous recombination deficiency scores, and mutational signature analysis using deconstructSigs as described (22). DNA from G334R-3, and G334R-4 tumors underwent the Comprehensive Cancer Panel targeted massively parallel sequencing assay at the Division of Genomic Diagnostics at the Children’s Hospital of Philadelphia (25). DNA from the G334R-6 tumor and normal blood leukocyte DNA underwent amplification by PCR of the microsatellite marker VNTR(p53) for analysis of LOH. DNA from G334R-17 and G334R-18 tumors underwent clinical IMPACT sequencing at Memorial Sloan Kettering Cancer Center (26). The raw WES BAM file for G334R-21 tumor was downloaded under approved dbGAP project #21931 and analyzed as described (22).
Cell Lines and Culture Conditions
WT and G334R/+ lymphoblastoid cells (LCLs) were created from patients under an IRB approved protocol via immortalization using Epstein Barr Virus (EBV). G334R/+ LCLs were genotyped to confirm TP53 c.1000G>C;p.G334R mutation positivity using Sanger sequencing as above (Supplementary Table S1a), and all four cell lines underwent WES to confirm the absence of other TP53 mutations. LCLs were grown in RPMI 1640 (10-040-CV) (Corning Cellgro™, Corning, NY, USA) supplemented with 15% Fetal Bovine Serum (HyClone™, GE Healthcare Life Sciences), 4 mM L-Glutamine (Corning Cellgro™) and 1% Penicillin/Streptomycin (Corning Cellgro™). H1299 (lung adenocarcinoma, p53-null) cells were obtained from ATCC, multiple batches were frozen upon receipt and new aliquots were used for this study every six months. H1299 cells were grown in DMEM (10-013-CM) (Corning Cellgro™) with 10% FBS and 1% Pen/Strep. Saos-2 cells were a gift to GPZ from Dr. Varda Rotter, and identity confirmed by STR profiling (Supplementary Table S1c). Saos-2 cells were grown in Dulbecco’s modified Eagle’s medium with L-Glucose (ThermoFisher Scientific) supplemented with 10% fetal bovine serum, MEM Non-Essential Amino Acids solution (ThermoFisher Scientific), 2.5 mmol/L-Glutamine and penicillin-streptomycin. Cells were grown in a 5% CO2 incubator at 37°C. Cells were tested for mycoplasma every six months, as well as immediately prior to RNA-Seq, by the University of Pennsylvania Cell Center Services (Philadelphia, PA, USA) or prior to functional analysis by St. Jude Children’s Research Hospital.
Antibodies and Reagents
Antibodies used were purchased from: GAPDH (2118S), MDM2 (86934S), p21 (2947T), PUMA (12450T), and Cleaved Lamin A (2035S) (Cell Signaling, Danvers, MA, USA); p53 (DO-1) (OP-43), p53 (pAb240, mutant p53 conformation) (OP-29), PGC1-α (ST1202) (Millipore Sigma, Burlington, MA, USA); LCN15 (PA5-31997) (Thermo Fisher Scientific, Waltham, MA, USA); p53 (FL-393) (Bioss Antibodies, Woburn, MA, USA); and Plexin B3 (AF4958) (R&D Systems, Minneapolis, MN, USA). Nutlin-3a (SML-0580-5MG) was purchased from Millipore Sigma. Constructs containing the c.1000G>A and c.1000G>C alleles were created by site direct mutagenesis using QuikChange II kit (Stratagene, La Jolla, CA, USA) in the pCMV-Neo-Bam expression vector containing wild-type TP53 cDNA as template.
Immunoblotting and Immunofluorescence Analysis
For Western blot analysis, 50-100 μg of whole-cell lysate was resolved over SDS-PAGE gels using pre-cast 10% NuPAGE Bis-Tris gels (Thermo Fisher Scientific) and transferred onto PVDF membranes (IPVH00010, pore size: 0.45 μm) (Millipore Sigma) prior to analysis. Secondary antibodies conjugated to horseradish peroxidase were used at a dilution of 1:10,000 (Jackson Immunochemicals, West Grove, PA, USA). ECL (Amersham; RPN2232) was applied to blots for 2-5 minutes and protein levels were detected using autoradiography. Densitometry quantification of protein signals were performed using ImageJ software (National Institutes of Health). For indirect immunofluorescence, cells were fixed 48 hours post-transfection in 4% paraformaldehyde for 10 minutes at room temperature, followed by permeabilization in 0.25% Triton X-100 for 5 minutes at room temperature. Cells were blocked in 1% BSA/4% goat serum for 30 minutes at room temperature. Primary antibodies were used to label mutant or total p53 (Pab240 or p53FL-393, respectively; 1:200), incubated at 4°C overnight, followed by incubation in Alexa-488 and Alexa-594 conjugated secondary (1:400 or 1:200, respectively) for 45 minutes at room temperature. Slides were mounted with mounting media with DAPI and imaged on a Nikon TE2000 inverted microscope.
Transfections and p53 Promoter Transactivation Assay
Constructs containing the c.1000G>A and c.1000G>C alleles were created by site direct mutagenesis using QuikChange II kit (Stratagene, La Jolla, CA, USA) in the pCMV-Neo-Bam expression vector containing wild-type TP53 cDNA as template. Constructs encoding p53 wild-type (TP53-WT), p53-R175H (TP53-R175H), and p53-G334R (c.1000G>A and c.1000G>C) were transiently transfected into p53 deficient Saos-2 cells. Luciferase transactivation assay was performed as described (27).
Transfections and Colony Formation Assays
Transfections were performed on H1299 (p53-null) cells using the Lipofectamine LTX with Plus Reagent (Thermo Fisher Scientific) according to the manufacturer’s instructions. Cells were transfected with the respective p53 constructs. Forty-eight hours following transfection, 50,000 cells were plated in triplicate in 6-well plates in the presence of 400 μg/mL G418 (Cell Center Services). Crystal violet staining was performed as described (28).
RNA-Seq and Quantitative PCR
3′ mRNA-seq libraries were generated from DNase I-treated total RNA using the QuantSeq FWD library preparation kit (Lexogen) according to the manufacturer’s directions. Overall library size was determined using the Agilent Tapestation and the DNA 5000 Screentape (Agilent). Libraries were quantitated using real- time PCR (Kapa Biosystems). Libraries were pooled, and high-out-put single-read 75-base-pair next-generation sequencing was done on a NextSeq500 (Illumina). RNA-Seq data were aligned using Bowtie2 (29) against hg19 build of the human genome, and RSEM version 1.2.12 software (30) was used to estimate raw read counts for each gene using Ensemble v84 transcriptome information. DESeq2 (31) was used to estimate the significance of the differential expression between sample groups. Overall gene expression changes were considered significant if they passed a false discovery rate (FDR) threshold of <20%. Genes that significantly (FDR<5%) responded at 24 hours in WT p53, but had significantly (nominal p<0.05) less response in p.G334R p53 were considered and ranked by the fold difference in response. The RNA-Seq data were deposited to the Gene Expression Omnibus database (http://ncbi.nlm.nih.gov/geo) with accession number GSE143741. Quantitative RT-PCR was performed as described (32).
Structural Analysis
The G334R mutation was introduced into a previously described p53 tetramerization domain construct (33), modified to include a TEV cleavage site using Quikchange mutagenesis (Agilent). Proteins were expressed in E. coli BL21(DE3). Cultures were grown at 37°C until the OD600 reached ~0.8. The cultures were then moved to 16°C and protein expression was induced via the addition of 0.5mM IPTG. Cultures were harvested after overnight expression via centrifugation. Cell pellets were resuspended in a buffer containing 50mM Tris, 500mM NaCl, and 0.5mM TCEP at pH 7.5. Cells were lysed via sonication. Lysate was cleared via centrifugation at 30,000 g for 30 minutes and then purified via Ni2+ chromatography. Proteins were eluted from the resin with an elution buffer containing 25mM Tris, 100mM NaCl, 300mM imidazole, and 0.5mM TCEP at pH 7.5. The 6x-his tag was then removed by overnight incubation at 4°C with TEV while dialyzing against a buffer containing 20mM sodium phosphate and 250mM NaCl at pH 7.2. The tag was removed by orthogonal Ni2+ chromatography. Thermal denaturation curves were collected by measuring the molar ellipticity at 222nm on a Jasco 101 spectropolarimeter as a function of temperature. CD melting curves were then fit to a two-state model to determine the melting temperature and stability of the proteins according to the method of Greenfield using Prism (34). All CD experiments were performed with a Jasco J-1500 Spectrometer at a protein concentration of 10 μM.
Statistical Analysis of Data
Unless otherwise stated, all experiments were carried out in triplicate (n=3) on all cell lines. For in vitro studies, the two-tailed unpaired Student t-test was performed. All in vitro data are reported as the mean ± standard deviation unless stated otherwise. Clinical data were compared using Fisher’s exact tests. Statistical analysis were performed using GraphPad Prism. p values are as indicated: * = p<0.05, ** = p<0.01, and *** = p<0.001(Fig. 2), and *p<0.01; **p<0.003 (Bonferroni corrected, Fig. 4).
Fig. 2: Analysis of p53 function in TP53 G334R mutant cell lines.
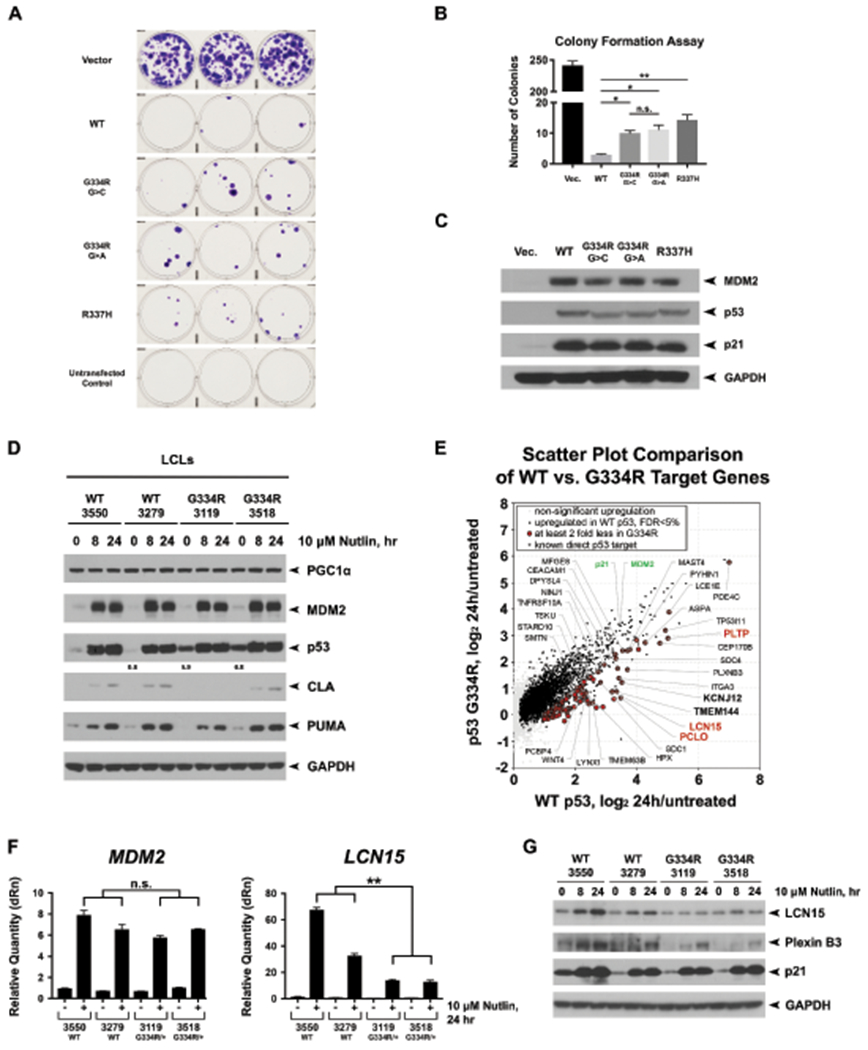
(a) Colony formation assays in H1299 cells transfected with wild-type TP53 (WT), CMV vector alone, and expression constructs containing the c.1000G>C;p.G334R allele (G334R G>C), the c.1000G>A;p.G334R allele (G334R G>A), the p.R337H allele, as well as mock transfected cells. Colonies stained with crystal violet and counted as per inset. Shown are representative images of triplicate wells from the same experiment. All experiments were performed in triplicate and the average numbers of colonies from two biological and three technical replicates are depicted in (b), including standard deviation. (c) Confirmation that transfected H1299 cells expressed equivalent levels of each p53 protein; the slight shift in mobility of p53 is due to Pro72Arg, which does not alter colony suppression. (d) Two TP53 wild-type lymphoblastoid cell lines (LCLs) and LCLs made from the cousin pair of Family G334R-1 were treated for the indicated times with 10μM Nutlin-3a (Nutlin). Western blot of PGC1α, MDM2, p53, cleaved Lamin A (CLA), PUMA, and GAPDH. (e) Scatter plot highlighting the top differentially regulated genes between WT and G334R/+ cells treated with Nutlin. The data depict average changes of two replicates from two WT and p.G334R/+ LCLs treated with 10 μM Nutlin-3a (Nutlin) for 24 hours versus untreated condition. Known direct p53 targets with significantly less response of at least 2 fold in p.G334R/+ cells are highlighted. (f) Quantitative PCR analysis of MDM2 and LCN15 expression levels in two WT and two p.G334R LCLs after 24 hours of 10 μM Nutlin. (g) Western blots of LCN15 and PLXNB3. Data shown are representative of three technical replicates and two biological replicates. *p<0.05. **p<0.01. ***p<0.001
Fig. 4: Age of cancer onset and cancer spectrum in probands and families with TP53 c.1000G>C;p.G334R.
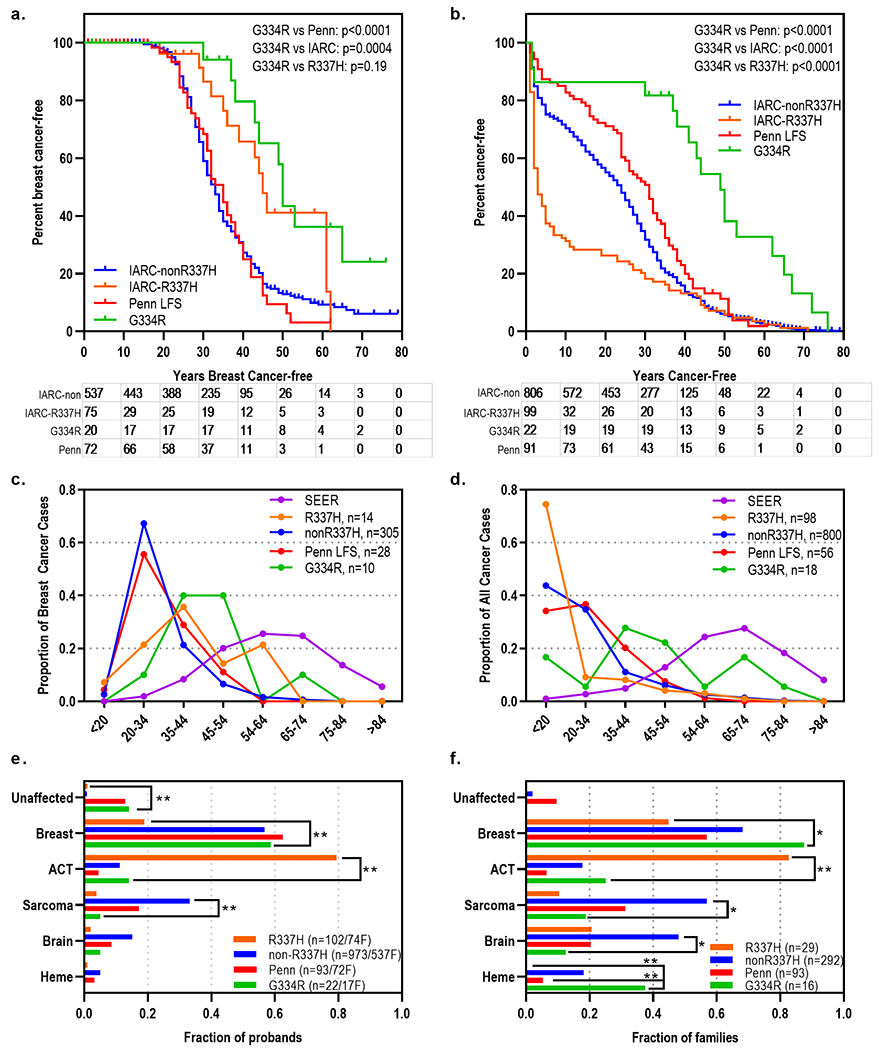
(a) Kaplan-Meier curves showing fraction of probands without a breast cancer diagnosis from families with TP53 c.1000G>C;p.G334R compared to an institutional LFS cohort and the IARC dataset, split into carriers and non-carriers of p.R337H. (b) Kaplan-Meier curves showing fraction of probands without any cancer diagnosis from families with TP53 c.1000C>G;p.G334R compared to an institutional LFS cohort and the IARC dataset, split into carriers and non-carriers of p.R337H. (c) Proportion of probands with age of onset of breast cancer in the indicated deciles from TP53 c.1000G>C;p.G334R families compared to an institutional LFS cohort, the IARC dataset split into carriers and non-carriers of p.R337H, and national SEER data. (d) Proportion of probands with age of onset of any cancer in the indicated deciles from TP53 c.1000G>C;p.G334R families compared to an institutional LFS cohort, the IARC dataset split into carriers and non-carriers of p.R337H, and national SEER data. (e) Fraction of probands with the indicated LFS-component cancers from TP53 c.1000G>C;p.G334R families compared to an institutional LFS cohort and the IARC dataset, split into carriers and non-carriers of p.R337H. (f) Fraction of families with at least one family member with the indicated LFS-component cancers from TP53 c.1000G>C;p.G334R families compared to an institutional LFS cohort and the IARC dataset, split into carriers and non-carriers of p.R337H. *p<0.01; **p<0.003 (Bonferroni corrected).
Results
Whole exome sequencing (WES) identified TP53 c.1000G>C;p.G334R in a pair of third-degree relatives (14) from a family (G334R-1) with multiple LFS-component cancers mostly occurring in the fourth to ninth decades, as well as five family members with multiple primary malignancies (Fig. 1a, Supplementary Fig. S1, Table 1). In silico modelling programs based on amino acid substitution characteristics and phylogeny predicted c.1000G>C;p.G334R was damaging (Supplementary Table S3). No additional predicted pathogenic mutations were shared by the cousin pair by WES analysis (Supplementary Table S4). Seven other independent families with c.1000G>C were ascertained from clinical genetics practices (Table 1, Supplementary Fig. S1). One family showed segregation of LFS-component cancers to third-degree relatives (G334R-6, Fig. 1b, Supplementary Fig S1). While most affected individuals had adult-onset cancers (Table 1), six individuals from four families had pediatric adrenocortical tumors (ACTs), including one family with pediatric ACTs in a sibling pair (G334R-4, Fig. 1c, Supplementary Fig. S1). In total, eight individuals from four families had multiple primary tumors (Supplementary Fig. S1). All probands were negative for other cancer predisposition gene mutations when tested (Table 1, Supplementary Table S2), except one family also segregated the AJ founder mutation BRCA2 c.5974delT;p.Ser1982fs (G334R-4) (Supplementary Fig. S1, Supplementary Table S2).
Fig. 1: Families with TP53 c.1000G>C;p.G334R.
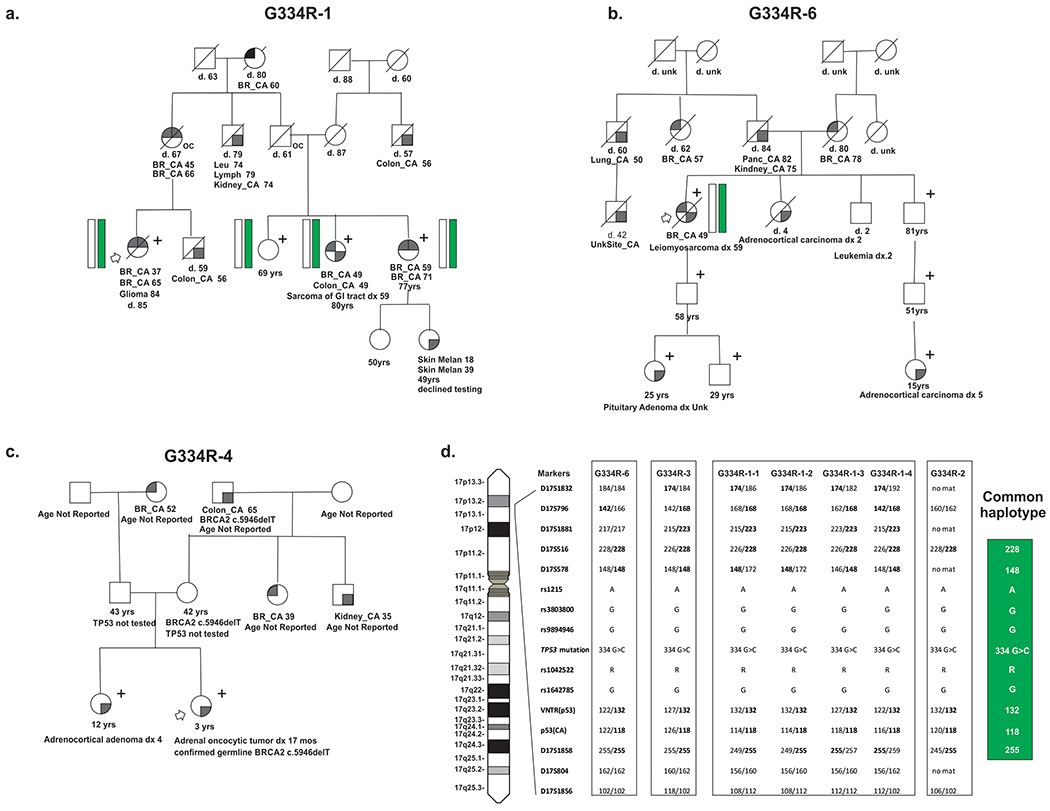
(a) Abbreviated pedigree of the index family, G334R-1 showing three mutation positive individuals (denoted with “+”) and two obligate carriers (mother and maternal uncle of proband, denoted with “oc”). Both proband and mother have multiple primary LFS-component cancers (breast, sarcoma, colon, and brain cancers). Mutation positive individuals (“+”) underwent either targeted panel testing clinically or site-specific TP53 testing. (b) Abbreviated pedigree of family G334R-6 showing tracking of the mutation (denoted with “+”) from a proband with multiple primary LFS-component cancers (breast, sarcoma) to her great-nephew with an ACT. Mutation positive individuals (“+”) underwent either targeted panel testing clinically or site-specific TP53 testing. (c) Abbreviated pedigree of family G334R-4 showing a mutation positive sib ship (denoted with “+”) both with ACTs. Mutation positive individuals (“+”) underwent either targeted panel testing clinically or site-specific TP53 testing. (d) Identification of a Chromosome 17 c.1000G>C haplotype from position 6,653,587 to 8,868,384 (GRCh37/hg19) in four independent families. Schematic diagram of chromosome 17 showing the identification and location (based on GRCh37) of genotyped SNPs and microsatellites analyzed in haplotype determination. The shared common haplotype is represented by a green bar in the pedigrees. Families used for identification of the haplotype are G334R-1, -2, -3, -6.
Table 1:
Characteristics of families and probands found to carry TP53 c.1000C>G; p.G334R substitution
| Satisfies LFS Criteria7 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Fam ID1 | Site2 | Race / AJ3 | Sex | Allele | TP53 Sequencing; BRCA status4 | Clinical Testing / Variant Class5 | Proband cancer history (age at diagnosis)6 | Cancer Family History (age of diagnosis)6 | Classic | Chompret | LFL-Birch | LFL-Eeles | Tumor Studies8 |
| Clinical Cohort | |||||||||||||
| G334R-1 (ref.14) | Penn | EA/AJ | F | G>C | RS/CGT Negative | Ambry / VUS | MP (BC 37yr, BC 65yr, GBM 84yr) | MP (BC 45yr,BC 66yr, Obl); MP (BC 49yr, CRC 49yr, Sarc 52yr (Mut+); MP (BC 59yr,BC 71yr, Mut+); CRC 56yr; MP (Leukemia 74yr, Kidney 74yr, Lymphoma 79yr); BC 60yr; MP (Mel 18yr, Mel 39) | - | X | - | X | NA |
| G334R-2 | Penn | EA/AJ | F | G>C | CGT Negative | Myriad / VUS | BC (Her2+) 30yr | BC 50yr; BC 55yr; Leukemia 55yr | - | X | - | - | Somatic TP53 p.N247I; HRD score=75, MutSigs 1,2,10 |
| G334R-3 | COH / SJ | EA/AJ | F | G>C | CGT Not tested | Ambry / VUS | ACC 18mo | BC 42yr; BC 40yr; Thyroid 32yr | - | X | - | ? | TP53 LOH |
| G334R-4 | CHOP | EA/AJ | F | G>C | TT; CGT BRCA2+ | CHOP/ LP | ACT 17 mo | ACT 4y (Mut+) | - | X | ? | X | TP53 LOH & BRCA2 c.5946delT heterozygous |
| G334R-5 | Cooper | EA/AJ | F | G>C | CGT Negative | GeneDx / VUS | BC 53yr, meningioma | MP (BC 61yr, Lung 84); BC 42yr; BC unk, CRC unk, BC unk, BC 42yr, BC unk | - | - | - | ? | NA |
| G334R-6 | DFCI/ Stanford/ SJ | EA/AJ | F | G>C | CGT Not tested | COH / LP | MP (BC 49yr, Sarc 59yr) | ACC 5yr (Mut+); PA Unk (Mut+); ACC 2yr; Leukemia 2yr; MP (Kidney 75yr, Panc 82yr); BC 57yr; Lung 50yr; Unk site 42yr | - | X | X | X | NA; ACT of TDR - TP53 LOH |
| G334R-7 (ref.10) | CHOP / COH / SK | EA/AJ | F | G>C | CGT Not tested | Sick Kids / LP | ACC 21 mo | Sarc 41yr; Lung 40yr; Lung 58yr | ? | X | ? | ? | NA |
| G334R-8 (ref.20) | DFCI | EA/AJ | F | G>C | RS/CGT Negative | Penn / LP | BC (Her2+) 44yr | BC Unk; Panc 49yr; Brain 12yr; BC 50yr; CRC 60yr; MP (BBC >50yr) | - | ? | ? | ? | NA |
| Genetic Testing Diagnostic Laboratory Cohort (Ambry Genetics Laboratory, Inc.) | |||||||||||||
| G334R-9 | Ambry | EA/AJ | M | G>C | CGT Negative | Ambry / VUS | MP (Kidney 41yr, Panc 69yr) | BC 68yr; BC 60s; BC 50s; Heme 60s | - | - | - | ? | NA |
| G334R-10 | Ambry | Mixed Race/ AJ | F | G>C | CGT Negative | Ambry / VUS | Unaffected 31yr | BC 41yr; Panc 70s | - | - | - | - | NA |
| G334R-11 | Ambry | EA/AJ | F | G>C | CGT Negative | Ambry / VUS | MP (BC 50yr, BC 64yr) | BC 35yr; BC 68yr; MP (BC 41yr, BC 45yr); BC 40s; BC <50yr; Prostate 67yr; BC 60yr | - | - | - | ? | NA |
| G334R-12 | Ambry | EA/AJ | F | G>C | CGT Negative | Ambry / VUS | BC 43yr | Heme <10yr; Lung 62yr; Prostate Unk; BC Unk; BC Unk; BC Unk | - | - | ? | ? | NA |
| G334R-13 | Ambry | EA/AJ | F | G>C | CGT Negative | Ambry / VUS | Unaffected 53yr | BC 53yr; BC Unk; CRC Unk; BC 55yr; Prostate 55yr; BC 60s | - | - | - | ? | NA |
| G334R-14 | Ambry | EA/ Unk | F | G>C | CGT Negative | Ambry / VUS | Skin 62yr | Eso 70yr; Panc 84yr; Lung Unk; Mel 62yr; Heme 62yr; BC Unk; Heme 62yr; BC unk; BC Unk; Brain 62; Gastric 48yr; BC 42yr | - | - | - | ? | NA |
| G334R-15 | Ambry | EA/ Unk | F | G>C | CGT Negative | Ambry / VUS | BC 50yr | Pros 60yr; BC 70yr; MP (BC 50yr, BC 68yr, BC 77yr); CRC 80yr | - | - | - | ? | NA |
| G334R-16 | Ambry | EA/ Unk | F | G>C | CGT Negative | Ambry / VUS | BC 65yr | MP (BC 45yr,BC 47yr); BC 65yr; BC 78yr; CRC 80yr | - | - | - | ? | NA |
| MSKCC IMPACT Cohort | |||||||||||||
| G334R-17 | MSKCC | EA/AJ | M | G>C | CGT Negative | MSKDMP / VUS | Panc, 67yr | NA | ? (All criteria) | Somatic TP53 p.K319X (met) | |||
| G334R-18 | MSKCC | EA/AJ | F | G>C | CGT Negative | MSKDMP / VUS | Panc, 72yr | NA | ? (All criteria) | Somatic TP53 E171H* & H168_T170dup | |||
| G334R-19 | MSKCC | AA/ non-AJ | F | G>C | CGT Negative | MSKDMP / VUS | Pituitary adenoma, 36yr | NA | ? (All criteria) | NA | |||
| IARC/Literature Review | |||||||||||||
| G334R-20 (ref.21) | Mayo | Unk /Unk | F | G>C | RS Negative | NA | BC 38yr | NA | ? (All criteria) | NA | |||
| G334R-21 (ref.19) | TCGA | EA/ Unk | F | G>C | RS Negative | Unk / LP | OC 76yr | NA | ? (All criteria) | TP53 LOH (83% VAF) | |||
| G334R-22 (ref.11) | IARC | Unk/ Unk | Unk | G>A | IARC Unknown | Unk/Unk | ACC 2yr | NA | ? | X | ? | ? | NA |
Families which have been previously reported: Proband of family 1(14); Proband of family 7(10); Proband (IARC ID: RAT13-3-I-1) of family 8/IARC-854(20); Proband of family 20(21); Proband (IARC ID: KAN14-1) of family 21/IARC-946(19), Proband (IARC ID: WAS15-12-1) of family 22/IARC-996(11).
Penn: Penn Medicine; CHOP: Children’s Hospital of Philadelphia; COH: City of Hope; DFCI: Dana Farber Cancer Institute; MSKCC: Memorial Sloan Kettering Cancer Center; MSKDMP: Memorial Sloan Kettering Diagnostic Molecular Pathology; SJ: St.Jude; SK: Sick Kids. Family G334R-9 was also reported in IARC. Probands or family members of families 1, 3, and 7 also had testing at Ambry Genetics but are not included in the Clinical Diagnostic Laboratory Cohort.
EA: European American; AJ: Ashkenazi Jewish; Unk: Unknown; Cau: Caucasian
CGT: Clinical Genetic Testing; RS: Research Sequencing; TT: Tumor testing; IARC: International Agency for Research on Cancer
VUS: variant of uncertain significance; LP: likely pathogenic; P: pathogenic
Individuals reported for lineage suspected to carry the mutation, relationships to proband and other cancer family history found in Supplemental Figure 2 and Supplemental Table 2; yr: years; mo: months; MP: multiple primary cancers; BC: breast cancer; ACC: adrenocortical carcinoma, ACT: adrenocortical tumor; Sarc: sarcoma; GBM: glioblastoma; CRC: colorectal; Eso: esophageal; Heme: hematological malignancy NOS; Mel: melanoma; OC: Ovarian Cancer; Panc: pancreatic; PA: Pituitary Adenoma; Obl: obligate carrier of mutation; Mut+: positive for TP53 c.1000G>C
LFS Criteria(3);”X”: Criteria satisfied; “-“: Deduction can be made that diagnostic criteria not met based upon absent reported family history; “?”: Unknown, cannot assess diagnostic criteria (met/not met) due to lack of established lineage or available family history data
LOH: Loss of heterozygosity; cnLOH: Copy neutral Loss of Heterozygosity; ACT: adrenocortical tumor; VAF: Variant allele frequency
NA: Data Not Available
Thirteen additional TP53 c.1000G>C;p.G334R carriers were identified from two genetic testing laboratory cohorts and literature review (Table 1). Eight probands were identified among 309,922 (0.003%) individuals undergoing germline testing at Ambry Genetics Laboratories, and three probands were identified among 21,729 (0.014%) individuals undergoing tumor genomic profiling at MSKCC. Three probands were unaffected but from multi-cancer families. Eleven probands had a variety of tumor types in the fifth to eighth decades, including two with multiple primary tumors. No other pathogenic mutations nor variants of uncertain significance were identified in other cancer predisposition genes, including BRCA1/2 in these probands (Table 1, Supplementary Table S2).
Ancestry was available for 16 probands with TP53 c.1000G>C, p.G334R. All eight families in the clinical cohort, and seven of eight from the two genetic testing cohorts, reported AJ ancestry (Table 1). The mutation was found at an approximately ten-fold enrichment in AJ versus non-AJ individuals in both genetic testing cohorts (0.023% AJ vs 0.001% of Caucasian non-AJ and 0.07% AJ vs 0.005% of non-AJ, Supplementary Table S5). The TP53 c.1000G>C mutation was not reported in the non-cancer cohort of gnomAD (v2.1.1), including 10,036 AJ alleles (16) (Supplementary Table S3), nor 1154 chromosomes from an AJ cancer-free cohort (23). Microsatellite and SNP analysis demonstrated that nine individuals from four families shared a common chromosome 17p13.1 haplotype (Fig. 1d).
In three available ACTs and the ovarian cancer, TP53 loss of heterozygosity was identified (Table 1). Somatic biallelic inactivation of BRCA2 was not found in the ACT also carrying a BRCA2 mutation (Table 1). Two pancreatic cancers and one breast cancer had a second somatic TP53 mutation (Table 1), although mutational phase was unavailable. The breast cancer sample from G334R-2 underwent WES, demonstrating typical breast tumor mutational signatures, high levels of genomic instability, and no other breast cancer driver mutations, characteristics consistent with p53-mutant breast tumors (35) (Table 1).
Previous functional studies of another p.G334R allele, c.1000G>A, have shown normal to impaired transactivation activity (42-145% of wild-type activity) dependent on cell type (6,7,11,12). In the interest of rigor, we created plasmids encoding both c.1000G>A and c.1000G>C alleles. In transient transfection assays, these showed comparable transactivation activity toward a canonical p53 promoter, similar to wild-type p53 (Supplementary Fig. S2). However, in colony suppression assays, these two p.G334R alleles showed mildly impaired suppression ability compared to wild-type TP53 (Fig. 2a–c) and functioned at a level similar to another tetramerization domain mutant p.R337H. We then studied immortalized lymphoblastoid cell lines (LCLs) created from the two cousins of the index family, compared to two TP53 wild-type LCLs from unrelated individuals of AJ descent. WES of these four cell lines did not identify any additional TP53 or other cancer-related mutations (Supplementary Table S4). p53 steady state levels were noted to be higher in G334R mutant lines compared to wild-type LCLs when treated with both the MDM2 inhibitor Nutlin-3a and cisplatin (Supplementary Fig. S3). The G334R mutant cell lines retained the ability to induce MDM2 and p21 in response to eight and 24-hours of treatment with Nutlin-3a, as expected (11,12) (Fig. 2d,f,g). RNA-Seq data from the four lines confirmed that transactivation of canonical p53 target genes in response to Nutlin at 24 hours was comparable in G334R mutant compared to wild-type LCLs (Supplementary Fig. S4). However, a subset of p53 target genes induced by Nutlin treatment in TP53 wild-type LCLs showed significantly impaired induction in G334R LCLs (Supplementary Table S6); these genes include PCLO, PLTP, PLXNB3 and LCN15 (Fig. 2e–g).
To investigate the biochemical impact of the p.G334R substitution, three-dimensional modelling of the p53-G334R mutant TD was performed, confirming that Gly-334 is the hinge residue located in the loop connecting the α-helix and β-sheet in each protomer (36,37) (Fig. 3a). MolProbity analysis predicted a potentially incompatible clash of Arg-333 of the A-protomer with Glu-349 of the C-protomer (Clash score 1.75). We therefore assessed thermal stability of the G334R tetramer, and found that G334R exhibits mildly decreased thermal stability compared to wild-type (Fig. 3b). We next tested the possibility that this amino acid substitution altered the folding of p53. Using the pAb240 conformation-specific antibody, we found that a significant percentage of cells transfected with either p.G334R allele showed evidence for p53 protein in the mutant conformation (Fig. 3c–d). This suggests an explanation for the increased steady-state level of G334R in untreated LCLs (Fig. 2d). These combined data suggest that the G334R mutant protein may be defective in transcription of a subset of genes due to increased propensity to adopt a mutant conformation.
Fig. 3: Structural analysis of p53-G334R tetramer.
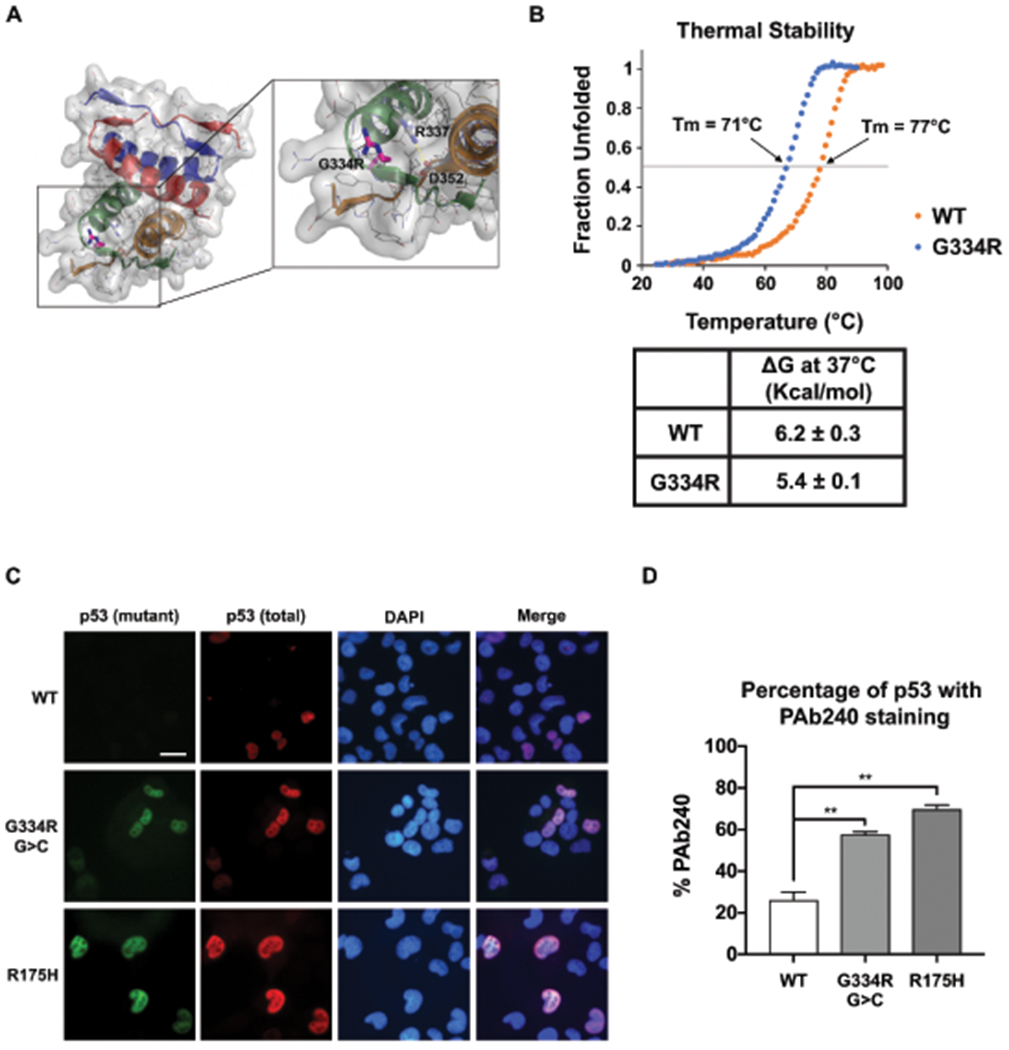
(a) Structural modelling analysis of p53-G334R mutant tetramer. Pymol analysis (PDB, Protein data Bank code 1PES) showing the tetramerization domain comprising residues 310-360. G334 is in a surface exposed loop. Carbon, Nitrogen, and Oxygen atoms are depicted in gray, blue, and red, respectively. The glycine to arginine mutation at position 334 is shown in magenta sticks. The side-chains of R337 and D352 are shown as sticks to highlight the close proximity of this salt bridge to residue 334. The salt bridge is denoted with a dashed yellow line. The surface depicts the space occupied by the non-mutated protein. The cartoon representation of each monomer subunit are colored differently to highlight the symmetry of the tetramer. (b) Thermal stability analysis of wild-type (WT) versus p53-G334R tetramer demonstrating fraction of the tetramer that is denatured with increasing temperature. Inset demonstrates the deltaG of WT versus p53-G334 mutant tetramer. (c) H1299 cells were transfected with TP53 vector (WT) and vectors containing the c.1000G>C;p.G334R allele (G334R G>C), the c.1000G>A;p.G334R allele (G334R G>A), and the p.R337H allele. Cells were subjected to immuno-fluorescence (IF) analysis of total (FL-393) and mutant conformation (pAb240) p53 staining. All experiments were done in triplicate. (d) Quantification of the fraction of cells containing any staining for pAb240 and mutant conformation (pAb240) p53 staining.
Our experimental evidence indicates that TP53 c.1000G>C;p.G334R has a partial defect in p53 function, similar to another hypomorphic TD mutation, p.R337H. Concordant with this mild defect, the median age of onset of all cancers, including breast cancers, in p.G334R probands was significantly later than probands in the Penn LFS and IARC non-p.R337H cohorts (Fig. 4a–b), though earlier compared to general population data from SEER (Fig 4c–d). While the median age of all cancers was significantly later in p.G334R compared to p.R337H probands, the median age of breast cancer was similar (Fig. 4a–b). Overall, the lifetime cumulative incidence of cancer is similar across all four cohorts. The rate of ACTs was similar in the p.G334R probands compared to Penn LFS and IARC non-p.R337H probands, although significantly lower than the rate in p.R337H probands (Fig. 4e–f). Conversely while breast cancer rates were similar in p.G334R, Penn LFS and IARC non-p.R337H probands, they were significantly higher than in p.R337H probands. Sarcomas were statistically less likely in p.G334R probands compared to the IARC non-p.R337H probands, although observed at a similar rate to p.R337H probands (Fig. 4e). Analysis of family data confirms these observations seen in probands (Fig. 4f). In addition, the family data shows a statistically significant lower incidence of brain cancer compared to IARC non-p.R337H families and a statistically significant higher rate of hematological malignancies in p.G334R families compared to Penn LFS and p.R337H families (Fig. 4e–f).
Discussion
Here-in we report that TP53 c.1000G>C;p.G334R is an AJ-predominant mutation causing a mild defect in p53 function likely due to decreased thermo-stability, which leads to a lower penetrance Li Fraumeni Syndrome phenotype. The TP53 p.G334R mutation demonstrates variable penetrance, with enrichment of the canonical LFS cancer, pediatric ACTs, in some families; whereas, other families predominantly show later onset classic LFS cancers, such as breast cancer and hematological malignancies, or multiple unaffected carriers.
Pediatric ACTs are extremely rare, diagnosed at 0.3 cases per million per year (38) and are highly predictive of an underlying germline TP53 mutation (11). Therefore, the high rate of pediatric ACT in our p.G334R cohort, particularly the sibling pair both with pediatric ACTs, is strong support for this mutation being causative of the observed phenotype. Overall, the later onset presentation of adult-onset cancers coupled with pediatric ACT enrichment seen with p.G334R probands and families is similar to that observed with another tetramerization domain mutation p.R337H (39), although the ACT enrichment is not as striking in the p.G334R cohort compared to p.R337H. The low incidence of sarcoma in our p.G334R cohort, has also been observed in p.R337H lineages as compared to classic LFS families (39).
Further studies of p.G334R and other tetramerization domain mutations under different physiological conditions are necessary to understand the etiology of the variable penetrance of mutations in this domain. It is possible that lineage specific factors, cell-type specific changes or other physiological conditions, such as aging, lead to decreased thermo-stability of the p53-G334R mutant tetramer only in specific target tissues. Pathogenesis in p53-G334R mutant cells may relate to the defective transactivation of the subset of p53 target genes that are not responsive to Nutlin in our analyses, and suggest that phenotyping of TP53 tetramerization domain mutations should differ from that used for DNA binding domain mutations (39). Finally, the cancer predisposition associated with the TP53 c.1000G>C; p.G334R allele may be due to genetic modifiers that affect cancer risk either in concert with or independently from the p53-G334R mutation. Modifiers both within TP53 (PIN3 and PEX4 polymorphisms) and outside of TP53 (MDM2 SNP309-G) are known to influence tumor risk and age of onset in TP53 germline carriers (40–42). Given the apparent progressively younger of age of onset of cancer in many of our p.G334R families, this or other mechanisms of genetic anticipation may also play an important role in p.G334R families (43). Similarly, modifier effects have been demonstrated with the TD Brazilian founder, p.R337H (44,45). Whether a variant(s) in linkage disequilibrium with c.1000G>C is acting cooperatively to alter p53 function remains unanswered.
Our data also show that the TP53 c.1000G>C mutation is present on a commonly inherited haplotype in nine individuals from four independent families and seen predominantly in individuals of Ashkenazi Jewish descent, suggesting that it is a founder mutation. While a number of founder mutations have been described in the Ashkenazi Jewish population (46), this is the first TP53 founder mutation to be described in this population. Future studies as more carriers are identified are necessary to investigate the age of this founder haplotype.
At present, discordant classification of TP53 c.1000G>C and other presumed hypomorphic alleles in TP53 and other cancer susceptibility genes are profoundly challenging in clinical genetics. Implementation of cancer screening modalities have demonstrated morbidity and mortality benefits for many patients with high penetrance inherited cancer susceptibility gene mutations (47–49). However, the best medical management strategies for patients with hypomorphic alleles are currently unclear. Reduced intensity or a bimodal screening approach based on age is likely preferable to either full screening or none at all. It is critical that translational approaches, as in this study, are applied more broadly to hypomorphic alleles in cancer susceptibility genes with the goal of precision implementation of appropriate life-saving cancer screening and treatment modalities.
Supplementary Material
Significance:
TP53 c.1000C>G;p.G334R is a pathogenic, Ashkenazi Jewish-predominant mutation associated with a familial multiple cancer syndrome in which carriers should undergo screening and preventive measures to reduce cancer risk.
Acknowledgements
We thank Jinling Wang (St. Jude Children’s Research Hospital) for technical assistance. Research support for this study was generously provided by National Institutes of Health Grants K08CA215312 (KNM), P30CA016520 (Abramson Cancer Center), R01CA102184 (MEM), K99CA241367 (TB), K08CA234394 (TPS), KL2TR00187903 (LCP), R01CA242218 (JNW), RC4CA153828 (JNW), R01CA225662 (FJC); Basser Center for BRCA at the University of Pennsylvania (KNM, KLN, SMD); Breast Cancer Research Foundation (FJC, SLD, KLN, JNW, JEG, KO); Burroughs Wellcome Foundation (KNM); International Pediatric Adrenocortical Tumor Registry (St. Jude IPACTR); American Lebanese Syrian Associated Charities (EMP, AHP, RCR, RWK, GZ).
Footnotes
Conflict of Interest Declaration: The authors declare no competing interests.
References
- 1.Li FP, Fraumeni JF Jr. Soft-tissue sarcomas, breast cancer, and other neoplasms. A familial syndrome? Ann Intern Med 1969;71:747–52 [DOI] [PubMed] [Google Scholar]
- 2.Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009;9:749–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.McBride KA, Ballinger ML, Killick E, Kirk J, Tattersall MH, Eeles RA, et al. Li-Fraumeni syndrome: cancer risk assessment and clinical management. Nat Rev Clin Oncol 2014;11:260–71 [DOI] [PubMed] [Google Scholar]
- 4.Kratz CP, Achatz MI, Brugieres L, Frebourg T, Garber JE, Greer MC, et al. Cancer Screening Recommendations for Individuals with Li-Fraumeni Syndrome. Clin Cancer Res 2017;23:e38–e45 [DOI] [PubMed] [Google Scholar]
- 5.Bouaoun L, Sonkin D, Ardin M, Hollstein M, Byrnes G, Zavadil J, et al. TP53 Variations in Human Cancers: New Lessons from the IARC TP53 Database and Genomics Data. Hum Mutat 2016;37:865–76 [DOI] [PubMed] [Google Scholar]
- 6.Kawaguchi T, Kato S, Otsuka K, Watanabe G, Kumabe T, Tominaga T, et al. The relationship among p53 oligomer formation, structure and transcriptional activity using a comprehensive missense mutation library. Oncogene 2005;24:6976–81 [DOI] [PubMed] [Google Scholar]
- 7.Kato S, Han SY, Liu W, Otsuka K, Shibata H, Kanamaru R, et al. Understanding the function-structure and function-mutation relationships of p53 tumor suppressor protein by high-resolution missense mutation analysis. Proc Natl Acad Sci U S A 2003;100:8424–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pinto EM, Billerbeck AE, Villares MC, Domenice S, Mendonca BB, Latronico AC. Founder effect for the highly prevalent R337H mutation of tumor suppressor p53 in Brazilian patients with adrenocortical tumors. Arq Bras Endocrinol Metabol 2004;48:647–50 [DOI] [PubMed] [Google Scholar]
- 9.Ribeiro RC, Sandrini F, Figueiredo B, Zambetti GP, Michalkiewicz E, Lafferty AR, et al. An inherited p53 mutation that contributes in a tissue-specific manner to pediatric adrenal cortical carcinoma. Proc Natl Acad Sci U S A 2001;98:9330–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fischer NW, Prodeus A, Tran J, Malkin D, Gariepy J. Association Between the Oligomeric Status of p53 and Clinical Outcomes in Li-Fraumeni Syndrome. J Natl Cancer Inst 2018;110:1418–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wasserman JD, Novokmet A, Eichler-Jonsson C, Ribeiro RC, Rodriguez-Galindo C, Zambetti GP, et al. Prevalence and functional consequence of TP53 mutations in pediatric adrenocortical carcinoma: a children’s oncology group study. J Clin Oncol 2015;33:602–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet 2018;50:1381–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, et al. ClinGen--the Clinical Genome Resource. N Engl J Med 2015;372:2235–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Maxwell KN, Wubbenhorst B, D’Andrea K, Garman B, Long JM, Powers J, et al. Prevalence of mutations in a panel of breast cancer susceptibility genes in BRCA1/2-negative patients with early-onset breast cancer. Genet Med 2015;17:630–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Landrum MJ, Lee JM, Benson M, Brown G, Chao C, Chitipiralla S, et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res 2016;44:D862–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Karczewski KJ, Francioli LC, Tiao G, Cummings BB, Alföldi J, Wang Q, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. bioRxiv 2019:531210 [Google Scholar]
- 17.MacFarland SP, Zelley K, Long JM, McKenna D, Mamula P, Domchek SM, et al. Earlier Colorectal Cancer Screening May Be Necessary In Patients With Li-Fraumeni Syndrome. Gastroenterology 2019;156:273–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howlader NNA, Krapcho M, Miller D, Bishop K, Kosary CL, Yu M, Ruhl J, Tatalovich Z, Mariotto A, Lewis DR, Chen HS, Feuer EJ, Cronin KA (eds). 2017. SEER Cancer Statistics Review, 1975-2014. [Google Scholar]
- 19.Kanchi KL, Johnson KJ, Lu C, McLellan MD, Leiserson MD, Wendl MC, et al. Integrated analysis of germline and somatic variants in ovarian cancer. Nature communications 2014;5:3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rath MG, Masciari S, Gelman R, Miron A, Miron P, Foley K, et al. Prevalence of germline TP53 mutations in HER2+ breast cancer patients. Breast Cancer Res Treat 2013;139:193–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Couch FJ, Hart SN, Sharma P, Toland AE, Wang X, Miron P, et al. Inherited mutations in 17 breast cancer susceptibility genes among a large triple-negative breast cancer cohort unselected for family history of breast cancer. J Clin Oncol 2015;33:304–11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maxwell KN, Wubbenhorst B, Wenz BM, De Sloover D, Pluta J, Emery L, et al. BRCA locus-specific loss of heterozygosity in germline BRCA1 and BRCA2 carriers. Nature communications 2017;8:319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vijai J, Topka S, Villano D, Ravichandran V, Maxwell KN, Maria A, et al. A Recurrent ERCC3 Truncating Mutation Confers Moderate Risk for Breast Cancer. Cancer discovery 2016;6:1267–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr 2010;66:12–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Surrey LF, MacFarland SP, Chang F, Cao K, Rathi KS, Akgumus GT, et al. Clinical utility of custom-designed NGS panel testing in pediatric tumors. Genome medicine 2019;11:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 2015;17:251–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Quinn EA, Maciaszek JL, Pinto EM, Phillips AH, Berdy D, Khandwala M, et al. From uncertainty to pathogenicity: clinical and functional interrogation of a rare TP53 in-frame deletion. Cold Spring Harb Mol Case Stud 2019;5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Barnoud T, Budina-Kolomets A, Basu S, Leu JI, Good M, Kung CP, et al. Tailoring Chemotherapy for the African-Centric S47 Variant of TP53. Cancer Res 2018;78:5694–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Methods 2012;9:357–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li B, Dewey CN. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 2011;12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Jennis M, Kung CP, Basu S, Budina-Kolomets A, Leu JI, Khaku S, et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes Dev 2016;30:918–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.DiGiammarino EL, Lee AS, Cadwell C, Zhang W, Bothner B, Ribeiro RC, et al. A novel mechanism of tumorigenesis involving pH-dependent destabilization of a mutant p53 tetramer. Nat Struct Biol 2002;9:12–6 [DOI] [PubMed] [Google Scholar]
- 34.Greenfield NJ. Using circular dichroism collected as a function of temperature to determine the thermodynamics of protein unfolding and binding interactions. Nat Protoc 2006;1:2527–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Donehower LA, Soussi T, Korkut A, Liu Y, Schultz A, Cardenas M, et al. Integrated Analysis of TP53 Gene and Pathway Alterations in The Cancer Genome Atlas. Cell reports 2019;28:1370–84 e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Higashimoto Y, Asanomi Y, Takakusagi S, Lewis MS, Uosaki K, Durell SR, et al. Unfolding, aggregation, and amyloid formation by the tetramerization domain from mutant p53 associated with lung cancer. Biochemistry 2006;45:1608–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kamada R, Nomura T, Anderson CW, Sakaguchi K. Cancer-associated p53 tetramerization domain mutants: quantitative analysis reveals a low threshold for tumor suppressor inactivation. J Biol Chem 2011;286:252–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lalli E, Figueiredo BC. Pediatric adrenocortical tumors: what they can tell us on adrenal development and comparison with adult adrenal tumors. Front Endocrinol (Lausanne) 2015;6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mastellaro MJ, Seidinger AL, Kang G, Abrahao R, Miranda ECM, Pounds SB, et al. Contribution of the TP53 R337H mutation to the cancer burden in southern Brazil: Insights from the study of 55 families of children with adrenocortical tumors. Cancer 2017;123:3150–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bougeard G, Baert-Desurmont S, Tournier I, Vasseur S, Martin C, Brugieres L, et al. Impact of the MDM2 SNP309 and p53 Arg72Pro polymorphism on age of tumour onset in Li-Fraumeni syndrome. J Med Genet 2006;43:531–3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fang S, Krahe R, Lozano G, Han Y, Chen W, Post SM, et al. Effects of MDM2, MDM4 and TP53 codon 72 polymorphisms on cancer risk in a cohort study of carriers of TP53 germline mutations. PLoS One 2010;5:e10813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bond GL, Hu W, Bond EE, Robins H, Lutzker SG, Arva NC, et al. A single nucleotide polymorphism in the MDM2 promoter attenuates the p53 tumor suppressor pathway and accelerates tumor formation in humans. Cell 2004;119:591–602 [DOI] [PubMed] [Google Scholar]
- 43.Tabori U, Nanda S, Druker H, Lees J, Malkin D. Younger age of cancer initiation is associated with shorter telomere length in Li-Fraumeni syndrome. Cancer Res 2007;67:1415–8 [DOI] [PubMed] [Google Scholar]
- 44.Marcel V, Palmero EI, Falagan-Lotsch P, Martel-Planche G, Ashton-Prolla P, Olivier M, et al. TP53 PIN3 and MDM2 SNP309 polymorphisms as genetic modifiers in the Li-Fraumeni syndrome: impact on age at first diagnosis. J Med Genet 2009;46:766–72 [DOI] [PubMed] [Google Scholar]
- 45.Assumpcao JG, Seidinger AL, Mastellaro MJ, Ribeiro RC, Zambetti GP, Ganti R, et al. Association of the germline TP53 R337H mutation with breast cancer in southern Brazil. BMC Cancer 2008;8:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cox DM, Nelson KL, Clytone M, Collins DL. Hereditary cancer screening: Case reports and review of literature on ten Ashkenazi Jewish founder mutations. Mol Genet Genomic Med 2018;6:1236–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ballinger ML, Best A, Mai PL, Khincha PP, Loud JT, Peters JA, et al. Baseline Surveillance in Li-Fraumeni Syndrome Using Whole-Body Magnetic Resonance Imaging: A Meta-analysis. JAMA oncology 2017;3:1634–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Villani A, Shore A, Wasserman JD, Stephens D, Kim RH, Druker H, et al. Biochemical and imaging surveillance in germline TP53 mutation carriers with Li-Fraumeni syndrome: 11 year follow-up of a prospective observational study. Lancet Oncol 2016;17:1295–305 [DOI] [PubMed] [Google Scholar]
- 49.Maxwell KN, Domchek SM. Cancer treatment according to BRCA1 and BRCA2 mutations. Nat Rev Clin Oncol 2012;9:520–8 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.