

Abstract
Apelin has complex vasomotor actions inasmuch as the peptide may cause either vasodilation or vasoconstriction depending on the vascular bed and experimental conditions. In cerebral arteries, apelin inhibits endothelium-dependent relaxations mediated by nitric oxide (NO); however, its effects on relaxation to other endothelium-derived substances (e.g. prostacyclin, endothelium-derived hyperpolarizing factors(s) (EDHF)) are unknown. The present study was designed to determine effects of apelin on endothelium-dependent relaxations that are independent of NO in rat cerebral arteries. In arterial rings contracted with 5-HT, A23187 caused endothelium-dependent relaxation that was unaffected by inhibitors of eNOS, guanylyl cyclase or cyclooxygenase, but was attenuated by MS-PPOH, a selective inhibitor of cytochrome P450 catalyzed synthesis of epoxyeicosatrienoic acids (EETs) and by 14,15-EE(Z)E, an EET-receptor antagonist. Apelin inhibited A23187-induced relaxation, as well as relaxations evoked by exogenous 11,12- and 14,15-EET. These effects of apelin were mimicked by the selective BKCa channel blocker, iberiotoxin. The APJ receptor antagonist, F13A abolished the effects of apelin on A23187-induced relaxations. Both 11,12- and 14,15-EET also increased BKCa channel current density in isolated cerebral artery smooth muscle cells, effects that were inhibited in a similar manner by apelin and iberiotoxin. These findings provide evidence that apelin impairs endothelium-dependent relaxation of cerebral arteries by inhibiting an NO-independent pathway (i.e. “EDHF-like”) involving activation of smooth muscle cell BKCa channels by endothelium-derived EETs. Inhibition of such pathway may create an environment favoring vasoconstriction in cerebral arteries.
Keywords: apelin, BKCa channels, cerebral artery, EDHF, EETs, vasorelaxation
1. Introduction
Apelin is an endogenous vasoactive peptide that is gaining recognition as an important signaling molecule in the cardiovascular system [1]. Apelin binds to G-protein-coupled receptors known as APJ receptors [2], which are expressed in heart, blood vessels, and cardiovascular regulatory centers in the brain [3–6]. Principal sources of apelin include adipose tissue [7], atria [8], vascular endothelial cells [9], and central nervous system neurons [10]. Measurable levels of the peptide are detected in the circulation [8, 11], and elevated plasma levels of apelin have been reported in patients with diabetes, obesity, and ischemic heart disease [7, 12, 13]. Nonetheless, the precise role of apelin under normal physiologic conditions and/or in cardiovascular disease is not yet clear.
Apelin has complex effects on vascular function, inasmuch as the peptide may cause either vasoconstriction or vasodilation, depending on experimental conditions and the origin of the blood vessel or vascular bed under investigation [14]. In this regard, our present understanding of the vasomotor effects of apelin in the cerebral circulation, which is essential for delivering oxygen and nutrients to the brain, is very limited. In rat isolated cerebral arteries apelin inhibits endothelium-dependent relaxations that are mediated by nitric oxide (NO) [15]; however, the effects of apelin on relaxations to other endothelium-derived mediators (e.g. endothelium-derived hyperpolarizing factor(s) (EDHF), prostacyclin) in cerebral arteries are unknown. This is significant since it would predictably increase the risk of cerebral vascular dysfunction if multiple endothelium-dependent vasodilator pathways were impaired by apelin. For example, studies in diseased arteries indicate that NO-independent pathways can serve as a redundant mechanism for eliciting vasodilation under conditions when NO signaling is impaired (e.g. hypercholesterolemia, heart failure) [16–18], and disruptions in cerebral blood flow are linked to debilitating disorders such as stroke, cognitive impairment, and dementia [19]. Thus, in order to gain a more complete understanding of the cerebrovascular actions of apelin, the present study was designed to determine the effects of apelin, if any, on endothelium-dependent relaxations that are independent of NO release in cerebral arteries.
2. Materials and Methods
2.1. Animals and Tissue Preparation
Experiments were performed on tissues isolated from 12-week-old male Sprague-Dawley rats (Envigo RMS, Indianapolis, IN). Rats were housed at 22 ± 2°C on a 12 h-12 h light-dark cycle and provided with food and water ad libitum. All animal protocols were approved by the North Dakota State University Institutional Animal Care and Use Committee and were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The rats were euthanized with isoflurane anesthesia followed by decapitation. Brains were isolated immediately and placed into ice-cold physiological salt solution (PSS) with the following composition (in mM): 119 NaCl, 15 NaHCO3, 4.6 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 1.5 CaCl2 and 5.5 glucose). Middle cerebral arteries were isolated, cleaned of adherent tissue, and cut into ring segments (80–100 μm in diameter; 1.2 mm in length).
2.2. Vascular Function Studies
Cerebral arterial rings were suspended in myograph chambers for isometric tension recordings and were stretched to an initial tension of 2 mN [15]. Vascular reactivity was confirmed by evoking a contractile response to KCl (60 mM). In some rings, endothelial denudation was performed by gently rubbing the intimal surface of the vessels with a human hair. The absence or presence of functionally intact endothelium was confirmed by measuring relaxation in response to the endothelium-dependent vasodilator, bradykinin (10−7 M). Arterial rings that showed ≥90% relaxation to bradykinin were considered as endothelium-intact preparations. Responses to the vasodilators used in this study were obtained in arterial rings contracted with 5-HT (10−7 M). Inhibitors were added to the myograph chamber 20 min prior to contraction with 5-HT, with the exception of apelin (10−7 M), which was added 5 min after the contraction had stabilized. This concentration of apelin was selected because in previous studies it produced the greatest inhibitory effect on BKCa channel currents in rat cerebral smooth muscle cells [20] and on NO-induced relaxations in rat cerebral arteries [15]. After addition to the myograph chamber, the inhibitors remained in contact with the tissues for the remainder of the experiment. Experiments with inhibitors were conducted in parallel with untreated control rings taken from the same animal.
2.3. Cerebral Artery Smooth Muscle Cell Isolation
Vascular smooth muscle cell isolation was carried out by placing freshly isolated arterial segments in ice-cold cell isolation solution containing (in mM): 60 NaCl, 80 Na-glutamate, 5 KCl, 2 MgCl2, 10 glucose, and 10 HEPES (pH 7.2). Arterial segments were first incubated in isolation solution containing 1.2 mg/ml papain (Worthington) and 2.0 mg/ml dithioerythritol (Sigma Aldrich) for 17 min at 37°C, followed by 0.8 mg/ml type II collagenase (Worthington) for 12 min at 37°C. After enzymatic digestion, arterial segments were washed and incubated in ice-cold cell isolation solution in order to stop further enzymatic reactions. Single cell suspensions were obtained by trituration using a fire-polished glass pipette. Smooth muscle cells were stored in ice-cold cell isolation solution and used within 6 h of isolation.
2.4. Electrophysiological Recording
Whole cell BKCa currents were recorded from freshly isolated cerebral arterial smooth muscle cells, using previously described procedures [15]. Briefly, smooth muscle cells were allowed to adhere to glass coverslips and superfused with bath solution of following composition (in mM) 145 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 5 HEPES, 10 glucose; pH 7.4 (NaOH). Glass patch pipettes (4–5 MΩ) were filled with pipette solution containing (in mM) 145 KCl, 5 NaCl, 0.37 CaCl2, 2 MgCl2, 10 HEPES, 1 EGTA, 7.5 glucose; pH 7.2 (KOH). All drugs were diluted in bath solution and perfused into the experimental chamber while recording. Standard recording conditions for BKCa currents were achieved by stepping in 10 mV increments from a holding potential of −60 to +80 mV. BKCa currents are expressed as current density (current divided by cell membrane capacitance).
2.5. Cyclic GMP Estimation
A competitive ELISA method was used to measure cyclic GMP levels in cerebral arteries. Arterial segments were initially equilibrated in PSS for 20 min at 37°C, followed by treatment with A23187 (10−6 M, 5 min) or bradykinin (10−7 M, 5 min). The tissues were then frozen in liquid nitrogen and homogenized in 0.1 M hydrochloric acid at 4°C. Tissue homogenates were centrifuged for 10 min at ≥600 g and cyclic GMP and total protein content were determined in the supernatant, as described in the direct cyclic GMP enzyme immunoassay kit (Assay Design, Ann Arbor, MI) and Pierce BCA protein assay kit (ThermoFisher Scientific), respectively. Cyclic GMP levels were expressed as picomoles per microgram of protein.
2.6. Data analysis
Relaxation responses are expressed as a percent of the initial tension induced by 5-HT (10−7 M). The drug concentration that produced 50% of its own maximal response (EC50 value) was determined, converted to the negative logarithm, and expressed as -log molar EC50 (pD2). Results are expressed as means ± SEM, and n refers to the number of animals unless otherwise stated. Values were compared by Student’s t-test or one-way ANOVA using Tukey’s test as post-hoc analysis for paired or unpaired observations, as appropriate, to determine significance between groups. Values were considered significantly different when p<0.05.
2.7. Drugs
The following drugs were used: bradykinin, diltiazem, 5-hydroxytryptamine (5-HT), indomethacin, and nitro-l-arginine (NLA) (Sigma Chemical, St. Louis, MO); iberiotoxin, and 1H-[1,2,4] oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) (Tocris, Ellisville, MO); apelin-13, and F13A (H-Gln-Arg-Pro-Arg-Leu-Ser-His-Lys-Gly-Pro-Met-Pro-Ala-OH trifluoroacetate salt) (Bachem, Torrance, CA), A23187 (Abcam, Cambridge, MA) and N-(methylsulfonyl)-2-(2-propynyloxy)-benzenehexanamide (MS-PPOH), 11,12-EET, 14,15-EET and 14,15-EE(Z)E (Cayman Chemicals, Ann Arbor, MI). All solutions were freshly prepared, stored on ice, and protected from light until used. All drugs were dissolved initially in double-distilled water with the exception of MS-PPOH, ODQ, and A23187, which were dissolved initially in DMSO, and indomethacin, which was dissolved initially in 1 mM sodium carbonate solution. Drugs were added to the myograph chambers in volumes not greater than 0.02 ml except 14,15-EE(Z)E, with a volume of 0.16 ml. Final molar concentrations in the myograph chamber are reported for each drug, unless otherwise stated.
3. Results
3.1. A23187 causes endothelium-dependent relaxations that are unrelated to NO and prostacyclin synthesis in isolated cerebral arteries:
The calcium ionophore, A23187 (10−9 – 3 × 10−6 M), caused concentration-dependent relaxation of isolated rat cerebral arteries with, but not without, intact endothelium (Figure 1). The NO synthase inhibitor, nitro-l-arginine (NLA, 3 × 10−5 M) had no effect on the response to A23187 (Figure 2A). A23187-induced relaxation was also unaffected by the cyclooxygenase inhibitor, indomethacin (10−5 M), either alone (Figure 2B) or in combination with NLA (Figure 2C). Relaxation responses to A23187 were insensitive to the soluble guanylyl cyclase inhibitor, ODQ (10−5 M) (Figure 3A), and exposure of freshly isolated cerebral arteries (with endothelium) to A23187 had no effect on intracellular cyclic GMP levels (Figure 3B). By comparison, the endothelium-dependent vasodilator, bradykinin, which acts via the release of endothelium-derived NO in cerebral arteries [15, 21, 22], caused a significant increase in intracellular cyclic GMP formation in these preparations (Figure 3B).
Figure 1. A23187 causes endothelium-dependent relaxation of isolated cerebral arteries.

(A) Representative original tracings of isometric tension recordings from rat isolated cerebral arteries (with and without endothelium) in response to cumulative addition of increasing concentrations of A23187, followed by diltiazem (10−5 M). (B) Mean data demonstrating A23187-induced relaxation in cerebral arteries with endothelium (E+), which was abolished in the endothelium denuded (E-) segments. Data are expressed as a percentage of the initial increase in tension induced by 5-HT (10−7 M). Each point represents the mean ± S.E.M. (n=6). *p <0.05 vs. with endothelium (E+).
Figure 2. Relaxation response to A23187 is independent of nitric oxide and prostacyclin in isolated cerebral arteries.

Log concentration-response curves to A23187 in the absence and presence of (A) nitro-l-arginine (NLA, 3 × 10−5 M); (B) indomethacin (Indo, 10−5 M); and (C) combination of NLA and indomethacin. Data are expressed as a percentage of the initial increase in tension induced by 5-HT (10−7 M). Values are represented as means ± S.E.M. (n = 6).
Figure 3. Role of guanylyl cyclase/cGMP in A23187-induced relaxation.

(A) Log concentration-response curves to A23187 in the absence and presence of ODQ (10−5 M); data are expressed as a percentage of the initial increase in tension induced by 5-HT (10−7 M). (B) Bar graph representing the effects of A23187 (10−6 M) and bradykinin (10−7 M) on intracellular cGMP formation in cerebral arteries. All data are represented as means ± S.E.M. (n=5). *p <0.05 vs. basal.
Experiments were then performed in order to identify possible mediators involved in the relaxation response to A23187. A23187-induced relaxation was unaffected by the presence of the hydrogen peroxide (H2O2) scavenger, PEG-catalase (500 U/ml) [23]. The pD2 values for A23187 were 6.96 ± 0.2 vs 7.04 ± 0.3 and Emax values = 69 ± 7 vs 59 ± 10% in the absence and presence of PEG-catalase, respectively (n=5; P>0.05). By contrast, the relaxation response to A23187 was markedly impaired by MS-PPOH (10−5 M), an inhibitor of the production of EETs by cytochrome P450 epoxygenase (Figure 4A), and by the EET receptor antagonist, 14,15-EE(Z)E (10−5 M) (Figure 4B).
Figure 4. Effect of MS-PPOH and 14,15-EE(Z)E on A23187-induced relaxation of isolated cerebral arteries.

Log concentration-response curves to A23187 in the absence and presence of (A) MS-PPOH (10−5 M, n=6) and (B) 14,15 EE(Z)E (10−5 M, n=7). Data are expressed as a percentage of the initial increase in tension induced by 5-HT (10−7 M). Values are represented as means ± S.E.M. *p<0.05 vs. A23187 alone (i.e. in the absence of inhibitor).
3.2. Apelin inhibits responses to A23187 and EETs in cerebral arteries:
A23187-induced relaxations were inhibited in the presence of apelin (10−7 M) (pD2: 7.08 ± 0.21 vs. 6.44 ± 0.11 without and with apelin, respectively; n=7, p<0.05) (Figure 5A). However, in the presence of the EET synthesis inhibitor, MS-PPOH (10−5 M), apelin had no effect on A23187-induced relaxation (pD2: 5.97 ± 0.21 vs. 6.19 ± 0.23 without and with apelin, respectively; n=6, p>0.05). The selective BKCa channel blocker, iberiotoxin (IBTx), had an inhibitory effect similar to that of apelin (pD2: 7.25 ± 0.12 vs. 6.81 ± 0.16 without and with IBTx, respectively; n=6, p<0.05) (Figure 5B). Combined treatment with apelin (10−7 M) plus IBTx (10−7 M) had no further inhibitory effect on A23187-induced relaxation than was observed with either inhibitor alone (Figure 5C). The APJ receptor antagonist, F13A (10−7 M) (Lee et al., 2005), which by itself had no effect on the response to A23187, abolished the inhibitory effect of apelin on A23187-induced relaxation (Figure 5D).
Figure 5. Apelin inhibits A23187-induced relaxation of isolated cerebral arteries.

Log concentration-response curves to A23187 in the absence and presence of (A) apelin (10−7 M; n=7); (B) iberiotoxin (IBTx, 10−7 M; n=6); (C) combination of apelin and IBTx; (n=6); and (D) apelin with and without F13A (10−7 M); n=6). Data are expressed as a percentage of the initial increase in tension induced by 5-HT (10−7 M). Values are represented as means ± S.E.M. *p<0.05 vs. A23187 alone.
Two EET regioisomers, 11,12-EET and 14,15-EET, were used to evaluate the effects of apelin on responses to EETs. 11,12-EET (10−6 M) and 14,15-EET (10−6 M) each caused relaxation of cerebral arteries contracted with 5-HT (10−7 M) (Figure 6). The relaxation responses evoked by both EETs were inhibited to a similar extent in the presence of apelin (10−7 M) (Figure 6A,B) or iberiotoxin (10−7 M) (Figure 6C,D). Both 11,12-EET and 14,15-EET also caused a significant increase in whole cell BKCa channel currents measured in freshly isolated cerebral arterial smooth muscle cells (Figures 7 and 8). Similar to the results obtained in the vascular functional studies, EET-induced increases in BKCa channel currents were inhibited in the presence of either apelin or IBTx (Figures 7 and 8).
Figure 6. Apelin-inhibits EET-induced relaxation of isolated cerebral arteries.

Bar graphs representing relaxation responses to 11,12 and 14,15 EET in rat isolated cerebral arteries (without endothelium) in the absence and presence of apelin (10−7 M) (A and B) or iberiotoxin (10−7 M) (C and D). Data are expressed as a percentage of the initial increase in tension induced by 5-HT (10−7 M). Values are represented as means ± S.E.M (6A-C: n=6; 6D: n=4). *p<0.05 vs. 11,12- or 14,15-EET alone (i.e. in the absence of inhibitor).
Figure 7. 11,12-EET causes an increase in BKCa channel current density in isolated cerebral smooth muscle cells.

Whole cell BKCa currents were recorded in freshly isolated vascular smooth muscle cells in response to successive voltage pulses of 800 ms duration, increasing in 10 mV increments from −60 mV to +80 mV before and after treatment with 11,12-EET (10−6 M) with and without apelin (10−7 M) or iberiotoxin (IBTx, 10−7 M). (A) Representative tracings depicting current recordings from a single smooth muscle cell before and after treatment with 11,12-EET (10−6 M, 2 min) alone, or with apelin (10−7 M, 5 min) or IBTx (10−7 M, 5 min); (B) Summary I-V curve plots of BKCa currents at baseline and after treatment with 11,12-EET (10−6 M) without and with apelin (10−7 M) or IBTx (10−7 M); and (C) Bar graph summarizing the effect of apelin (10−7 M) or IBTx (10−7 M) on 11,12-EET (10−6 M)-induced average current density (pA/pF) at +80 mV. Values are presented as mean ± SEM (n=6). *p<0.05 vs. control current density; #p<0.05 vs. 11,12-EET-induced current density.
Figure 8. 14,15-EET causes an increase in BKCa channel current density in isolated cerebral smooth muscle cells.

Whole cell BKCa currents were recorded in freshly isolated vascular smooth muscle cells in response to successive voltage pulses of 800 ms duration, increasing in 10 mV increments from −60 mV to +80 mV before and after treatment with 14,15-EET (10−6 M) with and without apelin (10−7 M) or Iberiotoxin (IBTx, 10−7 M). (A) Representative tracings depicting current recordings from a single smooth muscle cell before and after treatment with 14,15-EET (10−6 M, 2 min) alone, or with apelin (10−7 M, 5 min) or IBTx (10−7 M, 5 min); (B) Summary I-V curve plots of BKCa currents at baseline and after treatment with 14,15-EET (10−6 M) without and with apelin (10−7 M) or IBTx (10−7 M, 5 min); and (C) Bar graph summarizing the effect of apelin (10−7 M) or IBTx (10−7 M) on 14,15-EET (10−6 M)-induced average current density (pA/pF) at +80 mV. Values are presented as mean ± SEM (n=6). *p<0.05 vs. control current density; #p<0.05 vs. 14,15-EET-induced current density.
4. Discussion
The novel finding of the present study is that apelin inhibits endothelium-dependent relaxations that are resistant to inhibitors of eNOS, guanylyl cyclase, and cyclooxygenase in rat cerebral arteries. These NO-independent relaxations appear to be mediated, at least in part, by the synthesis and release of an EET(s) from endothelial cells, which in turn increase BKCa currents in cerebral arterial smooth muscle cells. This view is further supported by the observations that apelin attenuated exogenously added EET-induced smooth muscle relaxation and activation of BKCa currents in a manner similar to iberiotoxin. Taken together, the results provide evidence that apelin impairs endothelium-dependent relaxation of cerebral arteries by inhibiting an NO-independent pathway likely involving activation of smooth muscle cell BKCa channels by endothelium-derived EETs (Figure 9).
Figure 9. Inhibition of an endothelium-dependent, NO-independent vasodilator pathway by apelin in cerebral arteries.
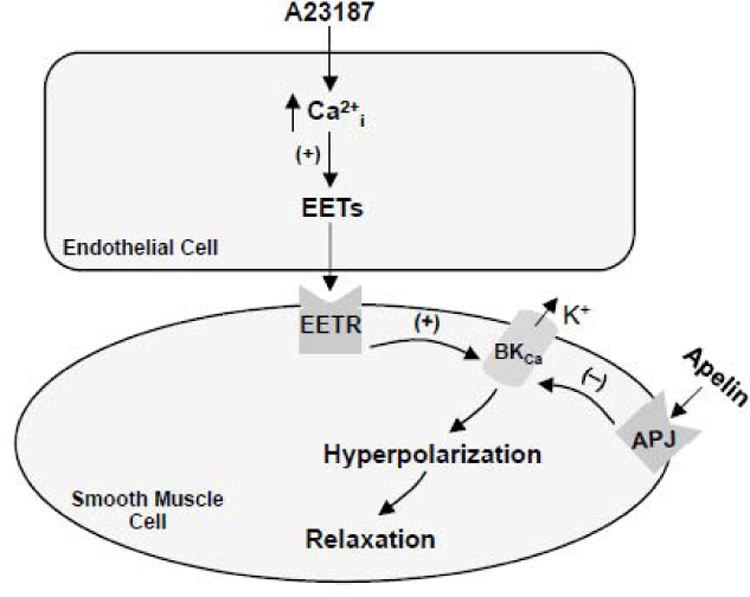
Increased levels of intracellular free calcium in response to the calcium ionophore, A23187, stimulates the production of epoxyeicosatrienoic acids (EETs) in cerebral artery endothelial cells. EETs diffuse to the underlying smooth muscle cells where binding to their cell membrane receptors (EETR) leads to activation of large conductance, calcium-activated K (BKCa) channels, efflux of potassium ions, membrane hyperpolarization and smooth muscle relaxation. Binding of apelin to APJ receptors on cerebral artery smooth muscle cells inhibits BKCa channel activation, thereby preventing smooth muscle hyperpolarization and relaxation in response to EETs.
Endothelial cells play a central role in regulating vasomotor tone by releasing vasoactive substances that cause relaxation of the underlying vascular smooth muscle [24]. These include NO and prostacyclin, which act primarily via increased intracellular levels of cyclic GMP and cyclic AMP, respectively, in vascular smooth muscle cells. A third pathway is mediated by endothelium-derived hyperpolarizing factors (EDHFs) that act, in part, by increasing outward K currents. In the present study, A23187 caused endothelium-dependent relaxations that were unaffected by inhibitors of the production of NO (i.e. NLA) or prostacyclin (i.e. indomethacin), either alone or in combination. Inhibition of guanylyl cyclase with ODQ also had no effect on A23187-induced relaxation, nor did A23187 cause an increase in cyclic GMP levels in cerebral arteries, as would be expected in the presence of elevated NO. The lack of effect of A23187 on cyclic GMP levels was not due to inability of the cerebral endothelial cells to produce NO, nor was it due to a lack of responsiveness of cerebral smooth muscle to endothelium-derived NO, since bradykinin was capable of increasing cyclic GMP levels in these preparations. In total, these findings make it unlikely that the response to A23187 is mediated by either NO or prostacyclin and are consistent with an EDHF-like pathway being an important contributor to endothelium-dependent relaxations evoked by the calcium ionophore in cerebral arteries.
The mechanism by which apelin inhibits this EDHF-like pathway is not completely clear, but it is likely due to the ability of apelin to inhibit BKCa currents in cerebral arterial smooth muscle cells by increasing PI3 kinase-dependent phosphorylation of Akt to inhibit BKCa channel function [15, 20]. Indeed, the potent and selective BKCa channel blocker, iberiotoxin, mimicked the effect of apelin on A23187-induced relaxation, and combined treatment with apelin plus iberiotoxin had no greater effect on the response to A23187 than either inhibitor alone, consistent with a shared mechanism of action for iberiotoxin and apelin. In many blood vessels endothelial cells metabolize arachidonic acid via CYP450 epoxygenase pathways to form EETs [25, 26], which relax vascular smooth muscle by activating BKCa channels [26, 27]. Evidence for an endothelium-derived EET in A23187-induced relaxation of cerebral arteries is provided by the findings that the response to A23187 was markedly impaired by the selective EET receptor antagonist, 14,15-EE(Z)E, and by inhibition of CYP450 epoxygenase with MS-PPOH. Moreover, authentic 11,12- and 14,15-EET each caused relaxation of isolated cerebral arteries and increased BKCa currents in isolated cerebral arterial smooth muscle cells. Notably, the effects of apelin on both the functional and electrophysiological effects of the EETs were mimicked by iberiotoxin. By contrast, a role for H2O2, another putative EDHF known to act via BKCa channel activation [28, 29], is unlikely since the H2O2 scavenger, PEG-catalase, had no effect on A23187-induced relaxation.
An alternative possibility is that apelin could act on endothelial cells to inhibit the release of EETs in rat cerebral arteries; however, the observation that apelin inhibits relaxation induced by exogenously added EETs suggests a site of action for apelin other than, or in addition to, EET release. Since the primary mechanism by which EETs relax vascular smooth muscle is by activation of BKCa channels followed by hyperpolarization, our observation that apelin attenuates the EET-induced increase in BKCa channel activity in cerebral smooth muscle cells provides a plausible mechanism that could account for the inhibitory action of apelin on relaxation to either endogenous or exogenously added EETs.
It is generally held that apelin has favorable effects on the heart and peripheral circulation (e.g. vasodilation, decreased blood pressure, positive inotropic effect) [30–34]. These beneficial effects notwithstanding, the results of the present study indicate that apelin, by inhibiting an EDHF-like pathway, may have detrimental effects in the cerebral circulation. In many peripheral vascular beds, the role of EDHF(s) in controlling arterial diameter increases as the size of the vessel decreases [24, 35]. In cerebral arteries, however, both NO- and EDHF-dependent pathways are important contributors to regulating vasomotor tone under normal physiological conditions [36–38]. Moreover, both pathways are approximately equieffective in relaxing cerebral arterial smooth muscle [36–39]. Thus, the inhibitory effect of apelin is concerning since NO-independent (i.e. EDHF-like) pathways not only play a central role in regulating vasomotor tone under normal physiologic conditions but may also serve as a compensatory mechanism to maintain endothelium-dependent relaxation when the NO-dependent component is reduced or abolished in pathologic conditions (e.g. heart failure, hypercholesterolemia [16, 17]). In accordance with this concept and with regard to the role of endothelium-derived EETs specifically, there is strong evidence in the literature that the EET-vasodilator pathway is indeed enhanced and plays a compensatory role in animal models of cerebrovascular disorders [39, 40] and in hypertensive human patients [41, 42]. Moreover, apelin also inhibits endothelium-dependent relaxations mediated by NO in cerebral arteries [15]. Thus, the ability of apelin to inhibit both NO and non-NO endothelium-dependent pathways would likely create a net vasoconstrictor environment in cerebral arteries. This is significant as it would predictably increase the risk of cerebral vascular dysfunction if multiple endothelium-dependent vasodilator pathways were impaired by apelin. Adverse cerebrovascular effects could also be of concern in the development of apelin and apelin-mimetics as novel therapeutic agents that, based on their putative beneficial cardiovascular effects, are in preclinical development and clinical trials for disorders such as myocardial infarction, ischemia-reperfusion injury, and heart failure [31, 32, 43, 44]. The effects of elevated apelin levels and/or such drugs on cerebral arteries may be particularly relevant in patients with a single nucleotide polymorphism in the gene encoding the human APJ receptor that results in increased apelin/APJ receptor signaling and is associated with an increased risk of brain infarction [45].
As with any study, there are some limitations to be considered. Firstly, though the pharmacological evidence strongly supports the notion that A23187-induced relaxation is mediated by an endothelium-derived EET(s), detection of these substances by sensitive and specific chemical methods will be needed to confirm this. Nonetheless, the data clearly show that regardless of the mediator(s) involved, apelin inhibits endothelium-dependent relaxations that are independent of NO and prostacyclin in cerebral arteries. Secondly, since membrane potential was not recorded we cannot state with certainty that smooth muscle cell hyperpolarization occurred in isolated cerebral arteries under our experimental conditions, thus we use the term “EDHF-like” to describe the response. Lastly, cerebral arteries from male rats were used in this study and we cannot definitively state that apelin has similar effects on arteries from females. However, it will be important to confirm these findings in cerebral arteries from females since sex hormones play a pivotal role in regulating mechanisms that control vasomotor tone.
In conclusion, the results of the present study provide evidence that apelin impairs endothelium-dependent relaxation of cerebral arteries by inhibiting an NO-independent (i.e. EDHF-like) pathway involving activation of smooth muscle cell BKCa channels by endothelium-derived EETs. Inhibition of such pathway may create an environment favoring vasoconstriction in cerebral arteries.
Highlights.
Apelin causes vasodilation in most arteries
In cerebral arteries, apelin inhibits a specific endothelium-dependent
vasodilator pathway
Effect of apelin is due to impaired activation of smooth muscle BKCa
channels by EETs
Inhibition of this pathway may create an environment favoring
vasoconstriction in cerebral arteries
Acknowledgements:
This work was supported by the National Institutes of Health National Heart, Lung, and Blood Institute [Grant HL124338].
Abbreviations:
- APJR
apelin receptor
- BCA
bicinchoninic acid
- BKCa
channel large conductance calcium-activated potassium channel
- CYP450
Cytochrome P450
- eNOS
endothelial nitric oxide synthase
- EDHF
endothelium-derived hyperpolarizing factors(s)
- EETs
epoxyeicosatrienoic acids
- F13A
H-Gln-Arg-Pro-Arg-Leu-Ser-His-Lys-Gly-Pro-Met-Pro-Ala-OH
- cGMP
cyclic guanosine monophosphate
- 5-HT
5-hydroxytryptamine
- IBTx
iberiotoxin
- MS-PPOH
N-(methylsulfonyl)-2-(2-propynyloxy)-benzenehexanamide
- NO
nitric oxide
- NLA
nitro-l-arginine
- ODQ
oxadiazolo[4,3-a]quinoxalin-1-one
- PSS
physiological salt solution
- TBS
Tris buffered saline
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declarations of interest: none
The authors have no competing interests to declare.
References
- [1].O’Carroll AM, Lolait SJ, Harris LE, Pope GR, The apelin receptor APJ: journey from an orphan to a multifaceted regulator of homeostasis, J Endocrinol 219(1) (2013) R13–35. [DOI] [PubMed] [Google Scholar]
- [2].Tatemoto K, Hosoya M, Habata Y, Fujii R, Kakegawa T, Zou MX, Kawamata Y, Fukusumi S, Hinuma S, Kitada C, Kurokawa T, Onda H, Fujino M, Isolation and characterization of a novel endogenous peptide ligand for the human APJ receptor, Biochem Biophys Res Commun 251(2) (1998) 471–6. [DOI] [PubMed] [Google Scholar]
- [3].Medhurst AD, Jennings CA, Robbins MJ, Davis RP, Ellis C, Winborn KY, Lawrie KW, Hervieu G, Riley G, Bolaky JE, Herrity NC, Murdock P, Darker JG, Pharmacological and immunohistochemical characterization of the APJ receptor and its endogenous ligand apelin, J Neurochem 84(5) (2003) 1162–72. [DOI] [PubMed] [Google Scholar]
- [4].Katugampola SD, Maguire JJ, Matthewson SR, Davenport AP, [(125)I]-(Pyr(1))Apelin-13 is a novel radioligand for localizing the APJ orphan receptor in human and rat tissues with evidence for a vasoconstrictor role in man, Br J Pharmacol 132(6) (2001) 1255–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kleinz MJ, Skepper JN, Davenport AP, Immunocytochemical localisation of the apelin receptor, APJ, to human cardiomyocytes, vascular smooth muscle and endothelial cells, Regul Pept 126(3) (2005) 233–40. [DOI] [PubMed] [Google Scholar]
- [6].Hosoya M, Kawamata Y, Fukusumi S, Fujii R, Habata Y, Hinuma S, Kitada C, Honda S, Kurokawa T, Onda H, Nishimura O, Fujino M, Molecular and functional characteristics of APJ. Tissue distribution of mRNA and interaction with the endogenous ligand apelin, J Biol Chem 275(28) (2000) 21061–7. [DOI] [PubMed] [Google Scholar]
- [7].Boucher J, Masri B, Daviaud D, Gesta S, Guigne C, Mazzucotelli A, Castan-Laurell I, Tack I, Knibiehler B, Carpene C, Audigier Y, Saulnier-Blache JS, Valet P, Apelin, a newly identified adipokine up-regulated by insulin and obesity, Endocrinology 146(4) (2005) 1764–71. [DOI] [PubMed] [Google Scholar]
- [8].Földes G, Horkay F, Szokodi I, Vuolteenaho O, Ilves M, Lindstedt KA, Mäyränpää M, Sármán B, Seres L, Skoumal R, Lakó-Futó Z, deChâtel R, Ruskoaho H, Tóth M, Circulating and cardiac levels of apelin, the novel ligand of the orphan receptor APJ, in patients with heart failure, Biochem Biophys Res Commun 308(3) (2003) 480–5. [DOI] [PubMed] [Google Scholar]
- [9].Kleinz MJ, Davenport AP, Immunocytochemical localization of the endogenous vasoactive peptide apelin to human vascular and endocardial endothelial cells, Regul Pept 118(3) (2004) 119–25. [DOI] [PubMed] [Google Scholar]
- [10].Reaux A, Gallatz K, Palkovits M, Llorens-Cortes C, Distribution of apelin-synthesizing neurons in the adult rat brain, Neuroscience 113(3) (2002) 653–62. [DOI] [PubMed] [Google Scholar]
- [11].Chen MM, Ashley EA, Deng DX, Tsalenko A, Deng A, Tabibiazar R, Ben-Dor A, Fenster B, Yang E, King JY, Fowler M, Robbins R, Johnson FL, Bruhn L, McDonagh T, Dargie H, Yakhini Z, Tsao PS, Quertermous T, Novel role for the potent endogenous inotrope apelin in human cardiac dysfunction, Circulation 108(12) (2003) 1432–9. [DOI] [PubMed] [Google Scholar]
- [12].Castan-Laurell I, Dray C, Attané C, Duparc T, Knauf C, Valet P, Apelin, diabetes, and obesity, Endocrine 40(1) (2011) 1–9. [DOI] [PubMed] [Google Scholar]
- [13].Habchi M, Duvillard L, Cottet V, Brindisi MC, Bouillet B, Beacco M, Crevisy E, Buffier P, Baillot-Rudoni S, Verges B, Petit JM, Circulating apelin is increased in patients with type 1 or type 2 diabetes and is associated with better glycaemic control, Clin Endocrinol (Oxf) 81(5) (2014) 696–701. [DOI] [PubMed] [Google Scholar]
- [14].Mughal A, O’Rourke ST, Vascular effects of apelin: Mechanisms and therapeutic potential, Pharmacol Ther 190 (2018) 139–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mughal A, Sun C, O’Rourke ST, Apelin reduces nitric oxide-induced relaxation of cerebral arteries by inhibiting activation of large-conductance, calcium-activated K Channels, J Cardiovasc Pharmacol 71(4) (2018) 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Najibi S, Cohen RA, Enhanced role of K+ channels in relaxations of hypercholesterolemic rabbit carotid artery to NO, Am J Physiol 269(3 Pt 2) (1995) H805–11. [DOI] [PubMed] [Google Scholar]
- [17].Malmsjo M, Bergdahl A, Zhao XH, Sun XY, Hedner T, Edvinsson L, Erlinge D, Enhanced acetylcholine and P2Y-receptor stimulated vascular EDHF-dilatation in congestive heart failure, Cardiovasc Res 43(1) (1999) 200–9. [DOI] [PubMed] [Google Scholar]
- [18].Clark SG, Fuchs LC, BK(Ca) channels compensate for loss of NOS-dependent coronary artery relaxation in cardiomyopathy, Am J Physiol Heart Circ Physiol 279(6) (2000) H2598–603. [DOI] [PubMed] [Google Scholar]
- [19].Iadecola C, Duering M, Hachinski V, Joutel A, Pendlebury ST, Schneider JA, Dichgans M, Vascular Cognitive Impairment and Dementia: JACC Scientific Expert Panel, J Am Coll Cardiol 73(25) (2019) 3326–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Modgil A, Guo L, O’Rourke ST, Sun C, Apelin-13 inhibits large-conductance Ca2+-activated K+ channels in cerebral artery smooth muscle cells via a PI3-kinase dependent mechanism, PLoS One 8(12) (2013) e83051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Erdös B, Miller AW, Busija DW, Impaired endothelium-mediated relaxation in isolated cerebral arteries from insulin-resistant rats, Am J Physiol Heart Circ Physiol 282(6) (2002) H2060–5. [DOI] [PubMed] [Google Scholar]
- [22].Bai N, Moien-Afshari F, Washio H, Min A, Laher I, Pharmacology of the mouse-isolated cerebral artery, Vascul Pharmacol 41(3) (2004) 97–106. [DOI] [PubMed] [Google Scholar]
- [23].Sato A, Sakuma I, Gutterman DD, Mechanism of dilation to reactive oxygen species in human coronary arterioles, Am J Physiol Heart Circ Physiol 285(6) (2003) H2345–54. [DOI] [PubMed] [Google Scholar]
- [24].Vanhoutte PM, Shimokawa H, Feletou M, Tang EH, Endothelial dysfunction and vascular disease - a 30th anniversary update, Acta Physiol (Oxf) 219(1) (2017) 22–96. [DOI] [PubMed] [Google Scholar]
- [25].Roman RJ, P-450 metabolites of arachidonic acid in the control of cardiovascular function, Physiol Rev 82(1) (2002) 131–85. [DOI] [PubMed] [Google Scholar]
- [26].Campbell WB, Gebremedhin D, Pratt PF, Harder DR, Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors, Circ Res 78(3) (1996) 415–23. [DOI] [PubMed] [Google Scholar]
- [27].Earley S, Heppner TJ, Nelson MT, Brayden JE, TRPV4 forms a novel Ca2+ signaling complex with ryanodine receptors and BKCa channels, Circ Res 97(12) (2005) 1270–9. [DOI] [PubMed] [Google Scholar]
- [28].Barlow RS, White RE, Hydrogen peroxide relaxes porcine coronary arteries by stimulating BKCa channel activity, Am J Physiol 275(4) (1998) H1283–9. [DOI] [PubMed] [Google Scholar]
- [29].Zhang DX, Borbouse L, Gebremedhin D, Mendoza SA, Zinkevich NS, Li R, Gutterman DD, H2O2-induced dilation in human coronary arterioles: role of protein kinase G dimerization and large-conductance Ca2+-activated K+ channel activation, Circ Res 110(3) (2012) 471–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Maguire JJ, Kleinz MJ, Pitkin SL, Davenport AP, [Pyr1]apelin-13 identified as the predominant apelin isoform in the human heart: vasoactive mechanisms and inotropic action in disease, Hypertension 54(3) (2009) 598–604. [DOI] [PubMed] [Google Scholar]
- [31].Barnes GD, Alam S, Carter G, Pedersen CM, Lee KM, Hubbard TJ, Veitch S, Jeong H, White A, Cruden NL, Huson L, Japp AG, Newby DE, Sustained cardiovascular actions of APJ agonism during renin-angiotensin system activation and in patients with heart failure, Circulation Heart Failure 6(3) (2013) 482–91. [DOI] [PubMed] [Google Scholar]
- [32].Japp AG, Cruden NL, Barnes G, van Gemeren N, Mathews J, Adamson J, Johnston NR, Denvir MA, Megson IL, Flapan AD, Newby DE, Acute cardiovascular effects of apelin in humans: potential role in patients with chronic heart failure, Circulation 121(16) (2010) 1818–27. [DOI] [PubMed] [Google Scholar]
- [33].Salcedo A, Garijo J, Monge L, Fernández N, Luis García-Villalón A, Sánchez Turrión V, Cuervas-Mons V, Diéguez G, Apelin effects in human splanchnic arteries. Role of nitric oxide and prostanoids, Regul Pept 144(1–3) (2007) 50–5. [DOI] [PubMed] [Google Scholar]
- [34].Mughal A, Sun C, O’Rourke ST, Activation of large conductance, calcium-activated potassium channels by nitric oxide mediates apelin-induced relaxation of isolated rat coronary arteries, J Pharmacol Exp Ther 366(2) (2018) 265–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Feletou M, The Endothelium: Part 2: EDHF-Mediated Responses “The Classical Pathway”, Morgan & Claypool Life Sciences Publisher, San Rafael (CA), 2011. [PubMed] [Google Scholar]
- [36].You J, Johnson TD, Marrelli SP, Mombouli JV, Bryan RM Jr., P2u receptor-mediated release of endothelium-derived relaxing factor/nitric oxide and endothelium-derived hyperpolarizing factor from cerebrovascular endothelium in rats, Stroke 30(5) (1999) 1125–33. [DOI] [PubMed] [Google Scholar]
- [37].Marrelli SP, Mechanisms of endothelial P2Y(1)- and P2Y(2)-mediated vasodilatation involve differential [Ca2+]i responses, Am J Physiol Heart Circ Physiol 281(4) (2001) H1759–66. [DOI] [PubMed] [Google Scholar]
- [38].Schildmeyer LA, Bryan RM Jr., Effect of NO on EDHF response in rat middle cerebral arteries, Am J Physiol Heart Circ Physiol 282(2) (2002) H734–8. [DOI] [PubMed] [Google Scholar]
- [39].Marrelli SP, Khorovets A, Johnson TD, Childres WF, Bryan RM Jr., P2 purinoceptor-mediated dilations in the rat middle cerebral artery after ischemia-reperfusion, Am J Physiol 276(1) (1999) H33–41. [DOI] [PubMed] [Google Scholar]
- [40].Abderrahmane A, Salvail D, Dumoulin M, Garon J, Cadieux A, Rousseau E, Direct activation of K(Ca) channel in airway smooth muscle by nitric oxide: involvement of a nitrothiosylation mechanism?, Am J Respir Cell Mol Biol 19(3) (1998) 485–97. [DOI] [PubMed] [Google Scholar]
- [41].Taddei S, Ghiadoni L, Virdis A, Buralli S, Salvetti A, Vasodilation to bradykinin is mediated by an ouabain-sensitive pathway as a compensatory mechanism for impaired nitric oxide availability in essential hypertensive patients, Circulation 100(13) (1999) 1400–5. [DOI] [PubMed] [Google Scholar]
- [42].Taddei S, Versari D, Cipriano A, Ghiadoni L, Galetta F, Franzoni F, Magagna A, Virdis A, Salvetti A, Identification of a cytochrome P450 2C9-derived endothelium-derived hyperpolarizing factor in essential hypertensive patients, J Am Coll Cardiol 48(3) (2006) 508–15. [DOI] [PubMed] [Google Scholar]
- [43].Wang W, McKinnie SM, Patel VB, Haddad G, Wang Z, Zhabyeyev P, Das SK, Basu R, McLean B, Kandalam V, Penninger JM, Kassiri Z, Vederas JC, Murray AG, Oudit GY, Loss of Apelin exacerbates myocardial infarction adverse remodeling and ischemia-reperfusion injury: therapeutic potential of synthetic Apelin analogues, Journal of American Heart Association 2(4) (2013) e000249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Narayanan S, Harris DL, Maitra R, Runyon SP, Regulation of the apelinergic system and its potential in cardiovascular disease: peptides and small molecules as tools for discovery, J Med Chem 58(20) (2015) 7913–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hata J, Matsuda K, Ninomiya T, Yonemoto K, Matsushita T, Ohnishi Y, Saito S, Kitazono T, Ibayashi S, Iida M, Kiyohara Y, Nakamura Y, Kubo M, Functional SNP in an Sp1-binding site of AGTRL1 gene is associated with susceptibility to brain infarction, Hum Mol Genet 16(6) (2007) 630–9. [DOI] [PubMed] [Google Scholar]