

Abstract
While glia are essential for regulating the homeostasis in the normal brain, their dysfunction contributes to neurodegeneration in many brain diseases, including Parkinson’s disease (PD). Recent studies have identified that PD-associated genes are expressed in glial cells as well as neurons and have crucial roles in microglia and astrocytes. Here, we discuss the role of microglia and astrocytes dysfunction in relation to PD-linked mutations and their implications in PD pathogenesis. A better understanding of microglia and astrocyte functions in PD may provide insights into neurodegeneration and novel therapeutic approaches for PD.
Keywords: Microglia, Astrocyte, Glia, Neuroinflammation, Neurodegeneration, Parkinson’s disease
Introduction
Parkinson’s disease (PD) is the second most common neurodegenerative disorder. The degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc) is a hallmark and is responsible for the motor impairments of the disease (Moore et al., 2005). Aging is a dominant risk factor for PD, with a sharp increase in incidence after age 60 years (de Lau and Breteler, 2006). After identifying the mutations responsible for the disease in the SNCA gene that encodes the α-synuclein protein in 1997 (Polymeropoulos et al., 1997), many other pathogenic mutations associated with PD have been identified. (For review see Bandres-Ciga et al., 2020; Klein and Westenberger, 2012; Trinh and Farrer, 2013). A subset of PD cases are monogenic forms caused by mutations in specific genes, including SNCA, parkin, PTEN-induced kinase 1 (PINK1), leucine rich repeat kinase 2 (LRRK2), and DJ-1 among others (Martin et al., 2011). In addition, unique variants of LRRK2 and GBA with incomplete penetrance are strong risk factors for PD in certain populations (Hernandez et al., 2016). Genome-wide association studies (GWAS) also suggest that both adaptive and innate immunity may play a role in PD pathogenesis (Chang et al., 2017a; Hamza et al., 2010; Holmans et al., 2013; Pierce and Coetzee, 2017).
In the brain, neurons communicate with glial cells to maintain parenchymal structure and function. Included among these cell types are microglia, the resident macrophages in the brain, representing 5–10% of total central nervous system (CNS) cells, and astrocytes, the most abundant cell type in the brain (Frost and Schafer, 2016; Sofroniew and Vinters, 2010). Although glia are essential to CNS homeostasis, (Schwartz et al., 2013; Wyss-Coray and Mucke, 2002), there is emerging evidence that microglial and astrocytic dysfunctions contribute to PD pathogenesis and progression. Recently, it has been shown that many of the PD-related genes are also expressed in glial cells as well as neurons (Booth et al., 2017; Zhang et al., 2016), suggesting that mutated gene products in microglia and/or astrocytes could contribute to PD etiology. Here, we focus on the evidence that implicating α-synuclein, PINK1, parkin, LRRK2, DJ-1 and GBA in microglial and astrocytic dysfunction during PD pathogenesis.
Microglia in Parkinson’s disease
Microglia are the resident macrophages and primary immune cells of the CNS. Averaging across brain regions, the density of microglia in a non-diseased human brain is about 10%, making them a significant cellular population (Mittelbronn et al., 2001). Microglia undergo regulated cycles of renewal to maintain appropriate overall densities, which may also regulate relative proportions of different microglial phenotypes (Askew et al., 2017). Furthermore, individual microglia are dynamic, mobile, and vigilant surveillants of tissue damage or infection, thus serving crucial roles for maintaining parenchymal homeostasis (Nimmerjahn et al., 2005). An increased focus on microglia in PD and other neurodegenerative diseases owes largely to their roles as mediators of immunity, especially their capacity to initiate neuroinflammation in response to pro-inflammatory molecules.
Whereas neuroinflammation has traditionally been assumed to be an epiphenomenon of neurodegenerative diseases, there are many studies identifying causal links between these states. Neuroinflammation and associated ‘reactive’ microglia have long been recognized as elements of PD (McGeer et al., 1988), but any causal relationship has been problematic to decipher. The more recent discovery that microglia and the innate immune system are essential for synaptic pruning was a major demonstration of their ability to impart changes to the neural world around them and suggested that mechanistically analogous processes could contribute to both neurological and psychiatric illnesses (Hong et al., 2016; Sekar et al., 2016; Stevens et al., 2007; Vainchtein et al., 2018). Similarly, microglia-derived inflammation may induce astrocytes to adopt neurotoxic functions or to lose neurotrophic or synaptoptrophic functionality (Liddelow et al., 2017).
Importantly, microglia exhibit significant spatiotemporal variation with respect to their transcriptional signatures, although the relevance of this diversity with respect to health and disease is only beginning to be considered (Hammond et al., 2019; Olah et al., 2018). Microglial phenotypes in the midbrain are not identical to those elsewhere in the CNS, which probably has important implications for the selective vulnerability of dopamine neurons in PD (Surmeier et al., 2017). One study has demonstrated this on a granular scale by showing that the transcriptional, morphological, and functional phenotypes of neurons in the SNpc differ from those of the ventral tegmental area (VTA), a related dopaminergic region that exhibits less degeneration in PD (De Biase et al., 2017). This report also showed enrichment of disease-associated transcripts specifically in the SNpc microglia of healthy mice, suggesting that these microglia are predisposed to adopt maladaptive functions in pathologic contexts. However, regional microglial subtype distribution is itself dependent on signals from other cells in the vicinity, meaning microglial ontogeny is negotiated in a complex and non-cell autonomous manner (Gosselin et al., 2017). Therefore, changes in these subtypes throughout aging or disease should be understood in relation to corresponding alterations in neighboring glia and neurons.
Genetically guided investigations of PD have also long pointed toward the immune compartment as a likely substrate of disease-relevant variation. Meta-analyses of GWAS have implicated a growing number of loci associated with sporadic PD that are important for core microglial behaviors. For example, both protein-protein and gene expression enrichment approaches implicate immune signal transduction pathway elements and the interferon-gamma response as being overrepresented among PD risk loci (Nalls et al., 2018). This points to canonical immune signaling and inflammation as being relevant to sporadic PD. However, most other cellular pathways associated with these risk loci are also important to microglial functions, including proteasomal protein catabolism, stress responses, lysosomal function, and autophagy (Chang et al., 2017b; Nalls et al., 2018). Because monogenic forms of PD are also thought to impact these cellular behaviors, they may help provide insight into how microglia sustain pathobiological processes that lead to neurodegeneration. In the remainder of this section, we explore the relation between microglial function and several key genes that cause PD when mutated.
SNCA (α-Synuclein)
The intraneuronal inclusions of α-synuclein protein, commonly referred to as Lewy bodies (LB) or Lewy neurites (LN), are pathological hallmark of PD. α-Synuclein is encoded by the SNCA gene, which is a genetic risk factor for both sporadic and familial forms of PD (International Parkinson Disease Genomics et al., 2011). Missense mutations and multiplications (duplications or triplications) of the gene cause PD (Ibanez et al., 2004; Ibanez et al., 2009; Polymeropoulos et al., 1997; Singleton et al., 2003). Under physiological conditions, α-synuclein regulates the trafficking of synaptic vesicles and the formation of the SNARE complex in the presynaptic terminals, but in its pathologically state, α-synuclein undergoes aggregation and fibrillization that leads to neurotoxicity in PD (Burre et al., 2014; Cookson, 2009; Jo et al., 2004). The microglial response to excess or mutant α-synuclein species is the subject of ongoing investigations. Extracellular α-synuclein oligomers function as damage-associated molecular patterns (DAMPs), activating innate immune receptors on the surface of microglia, including toll-like receptor 2 (TLR2) (Kim et al., 2013a). α-Synuclein is also reported to bind Fc gamma receptor IIB (FcγRIIB) on microglial surfaces and reduce microglial phagocytosis, which could impair the clearance of aggregated species or other parenchymal debris (Choi et al., 2015b). Fibrillar α-synuclein activates the NF-κB pathway in microglia (Yun et al., 2018), which is central to the inflammatory microglial response. A recent study showed that the uptake of α-synuclein fibrils by microglia is regulated by Fyn kinase—which was genetically linked to PD in a recent GWAS analysis (Nalls et al., 2018)—and class B scavenger receptor CD36 (Panicker et al., 2019). The same investigation showed that this signaling mechanism led to NLRP3 inflammasome priming and activation, thereby driving IL-1β release by microglia. Finally, a TLR9 agonist was shown to cause dopamine cell loss in mice in a glucocorticoid receptor (GR)-dependent manner, whereas TLR9 deletion confers resistance in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) neurotoxin mouse model of PD (Maatouk et al., 2018). Thus, aggregated α-synuclein induces pro-inflammatory microglial behaviors via both classic innate immune receptors and interactions with intracellular signaling cascades.
Several other immune pathways and cellular functions are reported to interact meaningfully with α-synuclein in microglia, both in vitro and in vivo. For example, α-synuclein carrying the A30P, E46K, and A53T mutations stimulate microglial cytokine secretion and activate multiple mitogen-activated protein (MAP) kinase pathways in vitro, including p38, ERK(1/2), and JNK (Hoenen et al., 2016; Klegeris et al., 2008). Multiple studies have also shown dramatic ROS production induced by mutant α-synuclein (Jiang et al., 2015; Zhang et al., 2007). Animal models have also implicated microglial reactivity in PD; for example, human A53T α-synuclein overexpression increased nigral neurodegeneration and neuroinflammation in mice lacking the fractalkine receptor (CX3CR1) (Castro-Sanchez et al., 2018). In the rodent intrastriatal α-synuclein preformed fibril (PFF)-injection model, microgliosis is observed in the substantia nigra prior to the onset of dopamine cell loss in rats, suggesting early microglial reactivity may be an important event and translational target in this model (Duffy et al., 2018). Other studies have observed sustained microglial reactivity in α-synuclein PFF-injected mice (Yun et al., 2018). In marmosets, overexpression of wild-type or A53T α-synuclein caused long-lasting (> 1 year) microglial morphology changes, suggesting again a prolonged microglial engagement in α-synucleinopathy (Barkholt et al., 2012). Given that brainstem LPS- and α- synuclein-induced inflammation are probably not identical (Couch et al., 2011), there may be microglial immune programs of particular relevance to α-synucleinopathy, which remain an active area of investigation.
Parkin and PINK1
Most PD-related parkin (encoded by PARK2 gene) and PINK1 (encoded by PINK1 gene) mutations are loss-of-function mutations that are associated with early onset familial PD (Dawson and Dawson, 2010; Kawajiri et al., 2011). Parkin functions as a ubiquitin E3 ligase and PINK1 functions as a serine/threonine protein kinase and play critical roles in mitochondrial quality control. Mutations in parkin and PINK1 dysregulate mitochondrial quality control leading to the neurodegeneration in PD (Panicker et al., 2017; Pickles et al., 2018; Pickrell and Youle, 2015; Scarffe et al., 2014). How this might alter microglial biology is only starting to be understood and represents a largely untapped target of research. Mitochondria are at the nexus of many key immunometabolic and innate immune pathways, such that their proper function is essential to the regulation of immunity. For example, mitochondria serve as physical scaffolding apparatuses for the mitochondrial antiviral signaling (MAVS) protein pathway, which mounts a type I interferon (IFN-I) response to viral RNA detection (Seth et al., 2005). Damaged or dysfunctional mitochondria can also perpetuate inflammatory responses by generating ROS or releasing mitochondrial damage-associated molecular patterns (DAMPs). Thus, in the absence of healthy mitochondrial function, microglia may be biased toward a more inflammatory phenotype. For example, the NLRP3 inflammasome, a multi-protein ensemble that mounts massive inflammation via IL-1β and IL-18, is activated by mitochondrial ROS in the context of impaired mitophagy (Nakahira et al., 2011; Zhou et al., 2011). This process is parkin-dependent in macrophages, suggesting that its presence in microglia may be important in parkin-linked PD (Zhong et al., 2016). NLRP3 activation exacerbates tau and Aβ pathology in mouse models (Heneka et al., 2013; Ising et al., 2019; Venegas et al., 2017) and may be involved in α-synucleinopathy induced neurodegeneration as well (Gordon et al., 2018; Panicker et al., 2019), suggesting a potential inflammatory link for aggregates in neurodegenerative diseases. However, roles for parkin, PINK1, or mitochondrial quality control in these specific mechanisms remain speculative. Parkin deficiency may enhance NF-κB-dependent NLRP3 activation in microglia (Mouton-Liger et al., 2018), but the upstream mediators of this mechanism were not conclusively shown. One study found that caspase-1, a downstream NLRP3 effector, cleaves parkin to inactivate it, though this was not shown in microglia (Kahns et al., 2003). Interestingly, another group found that PINK1 could bind to multiple IL-1R signal transduction proteins, suggesting that PINK1 could act downstream of the inflammasome to modulate cellular responses to IL-1β, but microglia were not examined specifically in these studies either (Lee and Chung, 2012; Lee et al., 2012).
Parkin and PINK1 are also both involved the repression of mitochondrial antigen presentation (MitAP), another critical intersection of mitochondria and immunity (Matheoud et al., 2016). Antigenic molecules are typically displayed on cell-surface major histocompatibility complex (MHC) proteins and detected by T cells, enabling the latter to sample the internal chemical milieu of a cell and sense self or non-self particles (Guermonprez et al., 2002). MHC pathways are the principal molecular mechanism for antigen presentation; intuitively, MitAP appears to be an analogous system for the presentation of mitochondrial antigens, where externalization is mediated by mitochondria-derived vesicles (MDV) intermediaries (Matheoud et al., 2016). Loss of parkin or PINK1 increased the production of MDV for MitAP, demonstrating a role for these PD-linked genes in regulating a potential source of autoimmune dysfunction. Given that genetic variants in the human leukocyte antigen (HLA) system—which encode MHC proteins—have been linked to PD, aberrant antigen recognition is a potential source of autoimmune inflammation with causal relevance (Nalls et al., 2018). This in turn may initiate or sustain microglial reactivity and neuroinflammation. Thus, these findings represent an exciting avenue of research into how MitAP may help elucidate a role for the adaptive immune system in PD.
Mouse models of PD or dopaminergic function have also suggested links between microglial dysfunction and parkin or PINK1. Germline parkin−/− mice exhibit increased motor deficits and SNc neurodegeneration in response to systemic LPS injections over time (Frank-Cannon et al., 2008) and parkin expression is reportedly downregulated by LPS in an NF-κB-dependent manner in both microglia and neurons (Tran et al., 2011). Transcriptomic analysis of PINK1−/− mice brains is consistent with inflammatory module induction, particularly in older mice, which may drive microglial reactivity (Torres-Odio et al., 2017). Other research has focused on how loss of parkin or PINK1 alter the behavior of microglia in vitro. Parkin knockout in BV2 microglia reduced necroptosis in one study, possibly through ubiquitination of necroptosis machinery (Dionisio et al., 2019). Failure to undergo necroptotic cell death may prolong inflammation and theoretically preclude the replacement of pro-inflammatory microglia with immunoregulatory microglia, thereby exacerbating neurological disease (Lloyd et al., 2019). Another study found that PARKIN−/− murine glial cultures had relatively more microglia and fewer astrocytes than wild type cultures (Solano et al., 2008). Similarly, PINK1−/− glial cultures exhibited increased nitric oxide production and reduced anti-inflammatory IL-10 production, though whether this can be specifically attributed to microglia was unclear (Sun et al., 2018). Collectively, however, these studies hint at likely roles for parkin and PINK1 in microglia in the pathogenesis of PD.
DJ-1
The PARK7 gene exhibits a diverse range of intracellular functionalities (Dolgacheva et al., 2019), making it difficult to link to specific microglial behaviors. Furthermore, only a few studies have examined potential microglia-relevant effects of PARK7 mutation. Intranigral injection of LPS elicits greater dopaminergic neurodegeneration in Park7 KO mice than wild-type mice, suggesting a broad role for Park7 in the regulation of nigral inflammatory reactivity (Chien et al., 2016). However, a related study did not see any parkinsonism or dopamine cell loss induced by systemic LPS exposure (Nguyen et al., 2013). Park7 KO microglia exhibit increased IL-1β and IL-6 secretion in response to dopamine exposure, suggesting an interaction between local dopaminergic levels and DJ-1 function in microglia (Trudler et al., 2014). This study also showed increased ROS production and monoamine oxidase (MAO) levels in the Park7 KO microglia and that a MAO inhibitor could normalize the microglial phenotype, implicating monoamine metabolism and ROS more specifically in the process. One group has shown that Park7 KO reduces IFNγ-induced activity of suppressor of cytokine signaling 1 (SOCS1) in astrocytes and microglia, possibly implicating the interferon system in DJ-1-mediated inflammation (Kim et al., 2013b; Kim et al., 2014). We are aware of only one study that has analyzed the impact of Park7 KO on microglial autophagy and α-synuclein phagocytosis and degradation, which did observe an impairment in these processes (Nash et al., 2017).
LRRK2
Mutations in LRRK2/PARK8 are associated with both monogenic and sporadic forms of PD, while their penetrance is incomplete and age-dependent (Kluss et al., 2019). For example, the most common mutation, G2019S has an age range of penetrance increasing from 17 % at 50 years old to 85 % at 70 years old. Moreover, some carriers never develop PD (Lee et al., 2017; San Luciano et al., 2010). Autosomal-dominant mutations in LRRK2 may represent the most compelling inflammation-based model of genetic PD, as LRRK2 has been widely implicated in immune cell function. LRRK2 is a serine/threonine kinase with diverse functions and targets (Islam and Moore, 2017). PD causing mutations in LRRK2 enhance its kinase activity and contribute to neurodegeneration (Berwick et al., 2019; Cookson, 2015). Among PD patients with LRRK2 mutations, serum levels of inflammatory cytokines differentiates patients with different disease presentations, suggesting close connections among LRRK2, disease progression, and the immune system (Brockmann et al., 2016; Brockmann et al., 2017; Dzamko et al., 2012). Even in sporadic or idiopathic PD, LRRK2 levels are increased in PD patient monocytes, lymphocytes (Cook et al., 2017), and neutrophils (Atashrazm et al., 2019). Both idiopathic and LRRK2-associated PD (G2019S and R1441G) individuals had increased cyclooxygenase-2 activity in patient-derived fibroblasts in another study (Lopez de Maturana et al., 2014). Finally, LRRK2 genetic variants may explain part of the noted co-morbidities between PD and autoimmune conditions such as Crohn’s disease, further implicating immune-based mechanisms in LRRK2 pathobiology (Hui et al., 2018; Witoelar et al., 2017). Animal studies largely corroborate these findings. For example, LRRK2 appears to modulate the overall inflammatory and oxidative stress responses in microglia treated with α-synuclein fibrillar aggregates (Russo et al., 2019). Similarly, LRRK2 deficient rats are resistant to α-synuclein mediated dopaminergic neurodegeneration with reduction in proinflammatory responses (Daher et al., 2014).
Despite the apparent immunological import of LRRK2, the relevance of microglial LRRK2 to in vivo microglial function in PD is controversial. A study on this topic found that mice overexpressing the R1441G LRRK2 mutation showed dopamine cell loss and exacerbated peripheral inflammation in response to systemic LPS injection, demonstrating synergy between inflammation and LRRK2 dysfunction in PD (Kozina et al., 2018). However, the authors found no evidence for LRRK2 expression in microglia that were acutely isolated from the experimental mice; rather, flow-sorting showed enrichment only in neurons (Kozina et al., 2018). Independent neuropathological studies have failed to detect LRRK2 mRNA (Sharma et al., 2011) and LRRK2 protein (Dzamko et al., 2017) in the microglia of PD patients with LRRK2 mutations. These results complicate the interpretation of microglia-specific LRRK2 investigations that rely upon primary or immortalized microglia studied in vitro, such as those discussed below. It is now understood that microglial phenotypes are profoundly altered upon removal from a normal CNS environment, which could explain these discrepancies (Butovsky et al., 2014; Butovsky and Weiner, 2018).
These concerns acknowledged, it is still important to understand the effects of LRRK2 mutations on microglial function, which have not been definitively excluded as etiological events in human PD. Additionally, there remains a clear relation to neuroinflammation and its attendant effects on microglia. Bacterial LPS increases LRRK2 expression in primary rodent microglia and LRRK2 inhibition reduces LPS-induced cytokine induction (Gillardon et al., 2012; Kim et al., 2012; Moehle et al., 2012). Interestingly, LRRK2 appears to regulate microglial motility via focal adhesion kinase such that gain-of-function mutations (e.g. G2019S) reduce motility, thus impaired microglial relocalization to local insults could facilitate LRRK2-associated neurodegeneration (Choi et al., 2015a). This idea was recapitulated in another study showing that Lrrk2−/− microglia increase migratory behaviors and that altered responses to fractalkine (CX3CL1) may mediate this phenotype (Ma et al., 2016). The kinase activity of mutant LRRK2 also increased mitochondrial fission in microglia, leading to impaired mitochondrial dynamics and higher TNFα production that could be rescued with a LRRK2 kinase inhibitor (Ho et al., 2018). Collectively, these studies are consistent with roles for LRRK2 in cytoskeleton-associated and inflammatory cellular behaviors in microglia.
Similarly, given the extensive literature describing the roles of LRRK2 in neuronal vesicle trafficking (Cookson, 2016), it has been suggested that LRRK2 mutations could alter microglial vesicle trafficking to increase inflammatory cytokine release or externalization of surface receptors with pro-inflammatory signaling capabilities (Russo et al., 2014). Microglia derived from LRRK2-G2019S PD patients also increase phagocytic function mediated by cytoskeleton remodeling factors, consistent with an immobile and inflammatory “ameboid” microglia phenotype (Kim et al., 2018). Consistent with this, another study found that LRRK2 knockdown reduced autophagic flux in murine immortalized microglia (Schapansky et al., 2014). Therefore, normal LRRK2 function is likely important for endolysosomal function in microglia. Interestingly, LRRK2 knockout increased phagocytosis of α-synuclein by murine primary microglia in another study, but the authors did not characterize other impacts on autophagic function, making it difficult to compare these results to the others here (Maekawa et al., 2016). Future work may clarify more comprehensively how autophagy or vesicle trafficking are specifically altered in microglia by PD-specific mutations.
GBA (Glucocerebrosidase)
Glucocerebrosidase (encoded by GBA gene) is a lysosomal enzyme, whose homozygous mutations are implicated in a lysosomal storage disorder such as Gaucher’s disease (GD) and heterozygous mutations are common risk factors for PD (Sidransky et al., 2009). Its mutations reduce the GBA enzymatic (GCase) activity that may lead to impaired lysosomal protein degradation and increased exosomal release of α-synuclein (Magalhaes et al., 2016; Tremblay et al., 2019). Lysosome function is important to microglial biology given the critical role microglia play in clearing parenchymal debris and pro-inflammatory particles, such as pathologic α-synuclein. Moreover, genetic variants that ontologically sort into lysosomal and autophagy function are consistently identified in GWAS for PD (Chang et al., 2017b). GBA-associated parkinsonism is further genetically modified by other lysosomal genes, such as CTSB (cathepsin B) (Blauwendraat et al., 2019), a gene that is dramatically upregulated by disease-limiting microglial subtypes (Keren-Shaul et al., 2017). Relative to sporadic PD cases, PD patients with GBA mutations exhibit increased plasma levels of inflammatory markers and cytokines, including IL-8 and macrophage inflammatory protein 1α (Chahine et al., 2013). Glucocerebrosidase accumulation may also activate complement, which could exacerbate microglia-mediated neuronal dysfunction (Pandey et al., 2017). Indeed, systemic GCase inhibition was observed in one study to increase α-synuclein aggregation in the substantia nigra and to upregulate complement C1q (Rocha et al., 2015). Although GCase function in microglia has not been very closely investigated on a mechanistic level, macrophages from Gaucher patients homozygous for the PD-linked N370S mutation exhibit impaired autophagy, lysosome dysfunction, and consequent NLRP3 inflammasome hypersensitivity (Aflaki et al., 2016). A demonstration of this mechanism in microglia could help provide a mechanistic link between Gaucher variants and neuroinflammation mediated by NLRP3 in PD. Thus, microglial GCase function in health and disease remains an under-investigated area with high potential for insightful discoveries.
Astrocytes in Parkinson’s disease
Astrocytes are the most abundant glial cells in the central nervous system (CNS) and play a variety of physiological roles, including secretion of neurotrophic molecules, regulation of synaptic transmission, maintaining homeostasis of water and ions, and regulation of the permeability of the blood-brain barrier (BBB) (Allaman et al., 2011; Sofroniew and Vinters, 2010; Wilton et al., 2019). Since glial-derived neurotrophic factor (GDNF) promotes the survival and differentiation of dopaminergic neurons (Lin et al., 1993) and BBB is disrupted in PD patients (Cabezas et al., 2014; Gray and Woulfe, 2015), it is not surprising that the loss of normally supporting astrocyte roles (loss-of-function) is implicated in the onset and progression of PD. In contrast, reactive astrocytes formed by response to stimulus or injuries in the CNS promotes PD pathogenesis by toxic gain-of-function (Phatnani and Maniatis, 2015; Sofroniew, 2009). These reactive astrocytes release a variety of chemokines and cytokines such as tumor necrosis factor alpha (TNF-α) and interleukin-1 beta (IL-1β), which are neurotoxic (Lau and Yu, 2001; Leal et al., 2013). A recent study shows that activated microglia induce neurotoxic A1 astrocyte by secreting interleukin-1α (IL-1α), TNF-α and C1q and that A1 astrocytes are found in postmortem brains of human neurodegenerative diseases including PD (Liddelow et al., 2017). Moreover, pathological α-synuclein contributes to formation of A1 astrocytes and preventing α- synuclein-induced microglial activation and A1 astrocyte conversion protected against dopaminergic neurodegeneration and behavioral deficits in a mouse model of sporadic PD (Yun et al., 2018). Taken together, these findings demonstrate that both dysfunctional astrocyte and reactive astrogliosis contribute to PD pathogenesis and progression.
Recent studies have revealed the regional heterogeneity of astrocytes (For review see Ben Haim and Rowitch, 2017; Khakh and Deneen, 2019; Khakh and Sofroniew, 2015) by RNA profiling (Lanjakornsiripan et al., 2018; Morel et al., 2017), proteomic analysis (Chai et al., 2017) or single-cell analysis (Batiuk et al., 2020). These observations have compared astrocyte populations across diverse brain region, indicating the cellular, molecular, and functional heterogeneity of astrocytes in the adult brain. For example, hippocampal and striatal astrocytes have unique functional and morphological properties. Astrocytes from striatum exhibit less gap junction coupling, K+ currents and interaction with neurons that those from the hippocampus (Chai et al., 2017). These regional differences of astrocytes may contribute to selective vulnerability in PD, while the relation of those changes with aging or disease progression should be addressed. In this section, we review the implication of PD-associated genes in astrocyte dysfunction in PD.
α-Synuclein
Although α-synuclein is expressed at low levels in astrocytes when compared to neurons (Zhang et al., 2014; Zhang et al., 2016), α-synuclein deficiency disrupted astrocyte fatty acid uptake and trafficking (Castagnet et al., 2005), suggesting that α-synuclein has a physiological role in astrocytes. As noted above, pathologic α-synuclein accumulates in postmortem PD brains (Braak et al., 2007; Song et al., 2009; Wakabayashi et al., 2000). Because of cell-to-cell transmission of α-synuclein and its predominant expression in neurons (Hansen et al., 2011; Zhang et al., 2014; Zhang et al., 2016), it is thought that astrocytes accumulate α-synuclein released from the neurons. Studies from co-culture of primary astrocytes with human neuroblastoma cells secreting α-synuclein showed uptake of α-synuclein and formation of inclusion bodies in astrocytes (Lee et al., 2010b). Thus, α-synuclein acts as an exogenous stimulator of astrocytes. Astrocytes with accumulated α-synuclein have been shown to produce proinflammatory cytokines such as IL-1, IL-6 and TNF-α, as well as chemokines such as C-X-C motif ligand 1 (CXCL1) in cultures (Lee et al., 2010a). This α-synuclein-induced proinflammatory response is dependent on Toll-like receptor 4 (TLR4), while astrocytes do not seem to require TLR4 for uptake of extracellular α-synuclein (Fellner et al., 2013; Rannikko et al., 2015). In addition, a mouse that selectively expresses A53T α-synuclein in astrocytes showed non-cell autonomous killing of neurons and they developed a neurodegenerative movement disorder (Gu et al., 2010). Presymptomatic and symptomatic accumulation of α-synuclein aggregates in astrocytes disrupted astrocytic functions, such as glutamate uptake and blood-brain barrier regulation as well as induced microglial activation in the midbrain, brainstem and spinal cord, where a significant loss of dopaminergic and motor neurons was observed, suggesting that accumulation of α-synuclein in astrocytes leads to neuroinflammation and reactive astrogliosis that contribute to PD neurodegeneration (Gu et al., 2010).
More recently, another mechanism for the formation of neurotoxic reactive astrocytes that are induced by activated microglia was identified (Liddelow et al., 2017). Both in primary culture and mouse models, pathologic α-synuclein activated microglia to secrete Il-1α, TNF- α and C1q, followed by the induction of reactive A1 astrocytes that caused neuronal cell death in cultures and neurodegeneration in vivo (Yun et al., 2018). Moreover, prevention of microglial-mediated conversion of astrocytes by a glucagon-like peptide-1 receptor (GLP1R) agonist was neuroprotective against the loss of dopaminergic neurons and behavioral deficits in the α-synuclein preformed fibril (α-syn PFF) mouse model of sporadic PD (Yun et al., 2018), providing evidence for an indirect role of α-synuclein in astrocyte-mediated neurodegeneration via microglial activation in PD.
Parkin and PINK1
The expression and distribution of astrocytic parkin is selectively increased by intracellular stress. The expression of PINK1 has an important role in the astrogliogenesis during brain development (Choi et al., 2016; Ledesma et al., 2002), suggesting that mutations in parkin and PINK1 in astrocyte may play a specific role on PD pathogenesis. In mouse astrocytes, the pro-inflammatory cytokine IL-1β decreased the level of parkin, whereas TNF-α induced its upregulation (Khasnavis and Pahan, 2014). Parkin regulation in the astrocytic inflammatory response is dependent on nitric oxide (NO) generated by inducible NO synthase (iNOS) (Khasnavis and Pahan, 2014). Astrocytes deficient in parkin display stress-induced elevation of nucleotide-oligomerization domain receptor 2 (NOD2), a cytosolic receptor integrating ER stress and inflammation (Singh et al., 2018). Parkin−/− astrocytes showed increased ER stress and cytokine release, as well as reduced astrocytic secretion of neurotrophic factors, which make neurons more susceptible to neurotoxins (Singh et al., 2018; Solano et al., 2008). Furthermore, Parkin−/− astrocytes exhibited decreased proliferation and increased proapoptotic protein expression (Solano et al., 2008). Parkin−/− astrocyte also may cause neurotoxicity through increased levels of damaged mitochondria (Schmidt et al., 2011).
PINK1 deficiency also may contribute to astrocyte dysfunction, at least in part by mitochondrial defects. PINK1−/− reduced astrocytic differentiation and glial fibrillary acidic protein (GFAP)-positive astrocytes in mouse brain (Choi et al., 2016). In PINK1−/− astrocyte cultures, increased p38 activation and decreased epidermal growth factor receptor (EGFR) expression and AKT activation appeared to be linked to the proliferation defects of astrocyte (Choi et al., 2013). In addition, mitochondrial defects, as demonstrated by decreased mitochondrial mass and membrane potential, increased intracellular reactive oxygen species (ROS) level, and decreased ATP production also contribute to decreased proliferation in Pink1 KO astrocytes (Choi et al., 2013). PINK1 also regulates the inflammatory response in astrocytes. Loss of PINK1 enhanced pro-inflammatory astrocyte functions, such as increased iNOS, NO, TNF-α and IL-1β expression via NF-kB signaling, which cause neuronal death in co-cultured system (Sun et al., 2018).
DJ-1
Since DJ-1 is predominantly expressed in astrocytes compared to neurons in human brain (Bandopadhyay et al., 2004), a role of DJ-1 in astrocyte biology has been widely studied. In PD brains, DJ-1 is upregulated in reactive astrocytes (Rizzu et al., 2004). Overexpression of DJ-1 in astrocytes protected neurons against rotenone-induced death, whereas knockdown or knockout of DJ-1 impaired the neuroprotective capacity of astrocytes and decreased neuronal survival (Lev et al., 2013; Mullett and Hinkle, 2009). It has been suggested that DJ-1 may have multiple neuroprotective roles in astrocytes.
Astrocytic DJ-1 is important for mitochondrial function and regulating oxidative stress. Knockdown of DJ-1 reduced mitochondrial motility in astrocytes that are observed in rotenone treated cells (Larsen et al., 2011). Moreover, rotenone-induced decrease of mitochondrial fission and membrane potential were exacerbated by astrocytic DJ-1 knockdown (Larsen et al., 2011). The role of DJ-1 in astrocyte-mediated neuroprotection is dependent on mitochondrial complex I (Mullett and Hinkle, 2011). Since DJ-1 serves as a redox sensor that recognize oxidative stress (Cao et al., 2014), DJ-1 mutations may contribute to PD pathogenesis by dysregulating nitrosative stress in astrocytes. DJ-1−/− astrocytes generated greater than 10 times more NO than littermate controls through p38 and iNOS-dependent mechanism, which are neurotoxic (Waak et al., 2009). In contrast, overexpression of DJ-1 protect astrocytic function and prevent oxidative stress. Astrocytic DJ-1 overexpression in zebrafish model upregulated proteins associated with redox regulation and mitochondrial respiration and prevented NO generation (Froyset et al., 2018). Moreover, rats overexpressing human DJ-1 in astrocytes were protected from rotenone-induced neurodegeneration and displayed a marked reduction in oxidative stress (Pelletier et al., 1988). Taken together, DJ-1 prevent the production of oxidative stress in astrocytes.
Another role of DJ-1 in astrocyte is the inflammatory response. DJ-1−/− astrocytes induced the proinflammatory mediators such as cyclooxygenase-2 (COX-2) and IL-6 and subsequent neuronal death in response to LPS treatment (Waak et al., 2009). DJ-1 selectively influences the TLR4 but not the TLR3 pathway to regulate astrocyte inflammation (Waak et al., 2009). It has been found that DJ-1 protein associated with lipid rafts, a highly organized membrane microdomains enriched in cholesterol and sphingolipids that play a role in synaptic transmission, endocytosis, exocytosis and signal transduction (Kim et al., 2013c; Munro, 2003). DJ-1−/− astrocytes disrupted lipid raft assembly and impaired TLR4 endocytosis that may affect astrocyte inflammation (Kim et al., 2013c). DJ-1 also negatively regulates the inflammatory response of astrocytes in response to interferon-gamma (IFN-γ) as a scaffold protein that facilitates p-STAT1 interaction with its phosphatase SHP-1 that preventing prolonged STAT1 activation (Kim et al., 2013b). IFN-γ treatment induced more neuronal damage in DJ-1−/− KO brain slices than in WT (Kim et al., 2013b), supporting that the loss of DJ-1 function may increase the risk of PD by enhancing brain inflammation.
LRRK2
LRRK2 is constitutively expressed in astrocyte and microglia as well as neurons in the human brain (Miklossy et al., 2006; Sharma et al., 2011). Although there are few studies about the role LRRK2 in astrocytes, astrocytic LRRK2 is involved in the autophagy-lysosome pathway like in neurons (di Domenico et al., 2019; Henry et al., 2015; Manzoni et al., 2013). The initiation of macroautophagy is sustained by activation of ULK1 and ULK2 complexes, which are inhibited by mTOR, and the nucleation and maturation of autophagic vesicle is mediated by Beclin-1-Vps34-Vps15 core complexes and other proteins (Kang et al., 2011). Inhibition of LRRK2 kinase activity induces autophagy that is independent on mTOR in primary astrocytes (Manzoni et al., 2013). Chemical inhibition of LRRK2 kinase activity also resulted in the stimulation of macroautophagy independent of mTOR and ULK1, but dependent on the activation of Beclin-1 (Manzoni et al., 2016), suggesting that LRRK2 is implicated in either autophagosome formation or maturation. Moreover, in a study using induced pluripotent stem cell (iPSC)-derived astrocytes containing familial LRRK2 G2019S mutation, dysfunctional chaperone-mediated autophagy (CMA), impaired macroautophagy, and progressive α-synuclein accumulation has been observed in PD astrocytes (di Domenico et al., 2019). LRRK2 itself also regulates lysosomal size, number and function in astrocytes (Henry et al., 2015). Expression of PD-related pathogenic LRRK2 mutations (G2019S, R1441C or Y1699C) produced enlarged lysosomes and diminished lysosomal capacity in a kinase-dependent manner in primary astrocytes (Henry et al., 2015). Since both autophagic and lysosomal dysfunction appear in neurodegenerative diseases including PD (Menzies et al., 2017), astrocytic LRRK2 mutations might be implicated in PD via dysregulating autophagy-lysosome pathway. A recent study using a KI mouse with deficient phosphorylation of LRKK2 S910 and S935 observed in both cell culture models and postmortem brain tissues of PD patients (Dzamko et al., 2017; Nichols et al., 2010) showed that striatal PD pathology was accompanied by a decreased number of astrocytes, whereas the number of microglia remained unchanged (Zhao et al., 2018). This study further suggest that the LRRK2 phosphorylation may have direct implications for astrocyte function.
GBA (Glucocerebrosidase, GBA)
Since in both GD and PD patients brain GBA mutations caused astrogliosis as well as abnormal α-synuclein inclusion (Wong et al., 2004), GBA may also have a role in astrocyte on pathogenesis. Consistent with this, Gba KO mice showed astroglial activation within nigrostriatal pathways, accompanied by abnormal α-synuclein accumulation (Ginns et al., 2014). The iPSC-derived astrocytes containing GBA mutations also showed astroglial activation and impaired lysosomal cathepsin activity, leading to aggregated α-synuclein accumulation (Aflaki et al., 2020). Moreover, specific GBA deficiency in neural and glial progenitor cells in mouse brain resulted in the increase of cathepsin lysosomal protease expression in both astrocytes and neurons and the redistribution of cathepsin in areas of astrogliosis and neuronal loss (Vitner et al., 2010). In a study using GBA D409V knockin mouse astrocytes, TLR4-dependent inflammatory responses and lysosomal dysfunction has been observed, which are normalized by inhibition of LRRK2 kinase activity, suggesting functional intracellular crosstalk between GBA and LRRK2 in astrocytes (Sanyal et al., 2020). Another study showed that autophagic and proteasomal machinery are defective in primary astrocytes lacking GBA (Osellame et al., 2013). In addition, Gba KO neurons and astrocytes showed mitochondrial dysfunction such as impaired respiration and decreased membrane potential and mitochondrial fragmentation that is more severe in astrocytes than in neurons (Osellame and Duchen, 2013; Osellame et al., 2013).
Conclusion
Since PD is characterized by the degeneration of dopaminergic neurons in the substantia nigra pars compacta (SNpc), the majority of PD research has been focused on selective loss of neurons and trying to understand this vulnerability to identify targets for neuroprotection. Less effort, however, has been spent in understanding the role of glial cells in PD pathogenesis. There are growing evidence that glial cells, including microglia and astrocytes, likely contribute to PD pathogenesis by both the loss of their normal homeostatic functions and/or the gain of neurotoxic functions. There are many PD-associated genes and the list is growing. Since many of the PD-related genes are expressed in glial cells as well as neurons, non-cell-autonomous mechanisms of those genes in microglia and astrocytes likely contribute to degenerative process and support the importance to further understand the role of glial cells in PD. It is also important to identify reciprocal pathways between PD-associated gene products rather than a single pathogenic pathway in glial cells, which might affect neuroinflammation and neuronal death either directly or indirectly. Overall, a better picture of the non-cell-autonomous interactions between glial cells and neurons will provide important insights into neurodegeneration. Moreover, since PD pathologies are prevented by inhibition of microglial activation and the conversion of resting astrocytes to the neurotoxic reactive A1 astrocytes (Dawson and Dawson, 2019; Yun et al., 2018), strategic approaches targeting these non-cell autonomous mechanisms in PD are promising avenues of therapeutic development and intervention.
Figure 1. Neuroinflammation in Parkinson’s Disease.
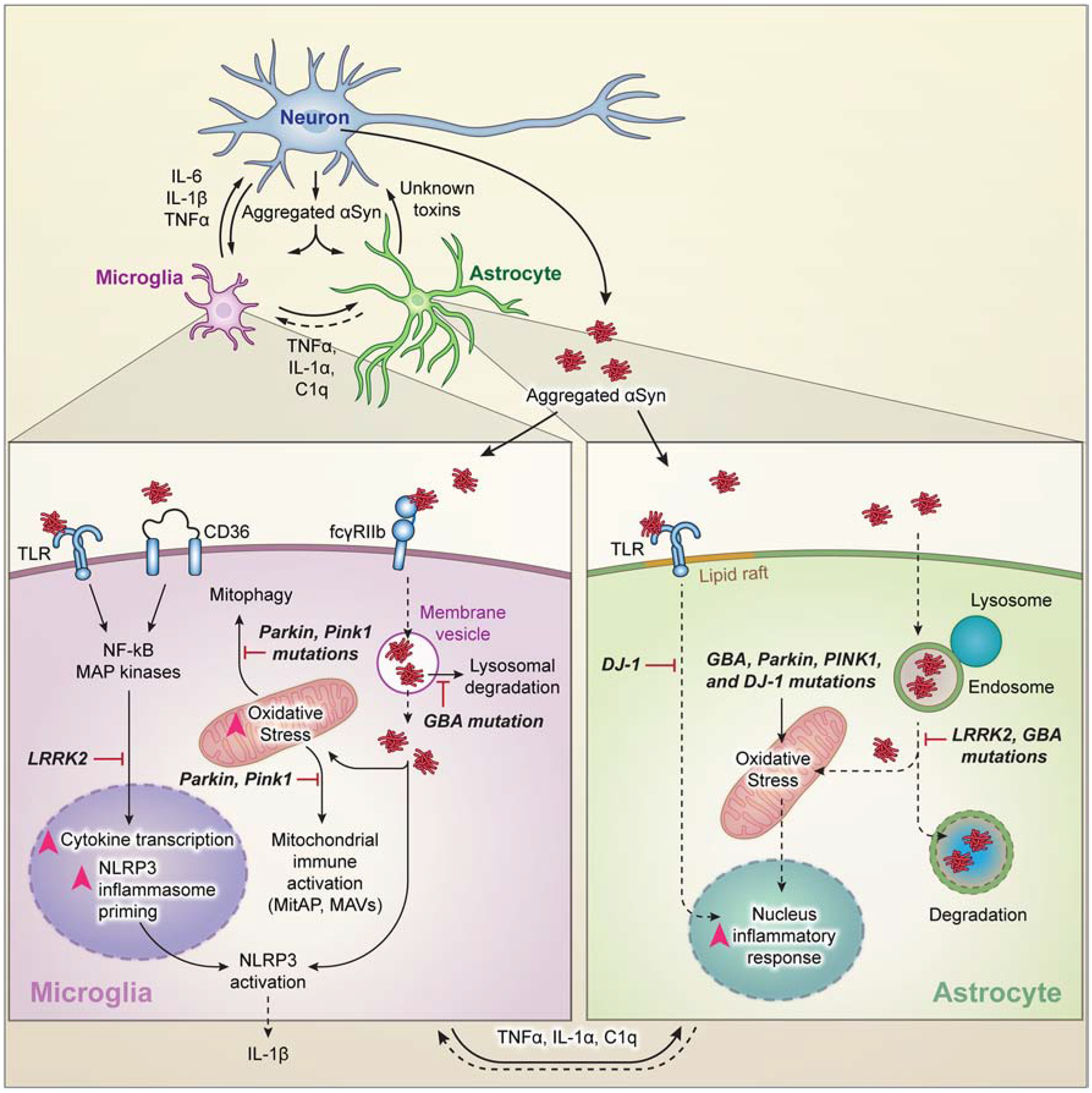
Pathologic α-Synuclein (α-Syn) is released from neurons and transmitted to and activates microglia and astrocytes. PD-associated GBA or LRRK2 mutations impair lysosomal protein degradation, which causes accumulation of α-syn. This leads to oxidative stress and proinflammatory responses. Mutations in Parkin, PINK1 and DJ-1 contribute to enhanced oxidative stress and proinflammatory responses in microglia and astrocytes. This culminates in the release of proinflammatory mediators derived from activated microglia, astrocytes or both, or unknown toxins released from reactive astrocytes that promote dopaminergic neuronal degeneration in PD.
Acknowledgments
This work was supported by NIH/NINDS grant NS38377 and the JPB Foundation. T.M.D. is the Leonard and Madlyn Abramson Professor in Neurodegenerative Diseases. The authors thank I-Hsun Wu for assistance with the illustrations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interests
T.M.D. and V.L.D. are founders of and hold shares of stock options and equity in Neuraly, Inc., and they are inventors of some of the technology discussed in this article, which Neuraly, Inc. has licensed from Johns Hopkins University. T.M.D. and V.L.D. are founders of, and are interim Chief Scientific Officer and Chief Executive Officer of, respectively, hold equity in, serve on the Board of Directors of, and are compensated for their roles as consultants to Valted Seq Inc. T.M.D. and V.L.D. are founders of Valted, LLC and hold an ownership equity interest in the company and the value of patents owned by Valted, LLC could be affected by this article. T.M.D. and V.L.D. are consultants to Inhibikase Therapeutics and own stock options in the company. T.M.D. is a paid consultant to Sun Pharmaceutical Industries Ltd.
References
- Aflaki E, et al. , 2016. Lysosomal storage and impaired autophagy lead to inflammasome activation in Gaucher macrophages. Aging Cell. 15, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aflaki E, et al. , 2020. A characterization of Gaucher iPS-derived astrocytes: Potential implications for Parkinson’s disease. Neurobiol Dis. 134, 104647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaman I, et al. , 2011. Astrocyte-neuron metabolic relationships: for better and for worse. Trends Neurosci. 34, 76–87. [DOI] [PubMed] [Google Scholar]
- Askew K, et al. , 2017. Coupled Proliferation and Apoptosis Maintain the Rapid Turnover of Microglia in the Adult Brain. Cell Reports. 18, 391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atashrazm F, et al. , 2019. LRRK2-mediated Rab10 phosphorylation in immune cells from Parkinson’s disease patients. Mov Disord. 34, 406–415. [DOI] [PubMed] [Google Scholar]
- Bandopadhyay R, et al. , 2004. The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson’s disease. Brain. 127, 420–30. [DOI] [PubMed] [Google Scholar]
- Bandres-Ciga S, et al. , 2020. Genetics of Parkinson’s disease: An introspection of its journey towards precision medicine. Neurobiol Dis. 137, 104782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkholt P, et al. , 2012. Long-term polarization of microglia upon alpha-synuclein overexpression in nonhuman primates. Neuroscience. 208, 85–96. [DOI] [PubMed] [Google Scholar]
- Batiuk MY, et al. , 2020. Identification of region-specific astrocyte subtypes at single cell resolution. Nat Commun. 11, 1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Haim L, Rowitch DH, 2017. Functional diversity of astrocytes in neural circuit regulation. Nat Rev Neurosci. 18, 31–41. [DOI] [PubMed] [Google Scholar]
- Berwick DC, et al. , 2019. LRRK2 Biology from structure to dysfunction: research progresses, but the themes remain the same. Mol Neurodegener. 14, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blauwendraat C, et al. , 2019. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth HDE, et al. , 2017. The Role of Astrocyte Dysfunction in Parkinson’s Disease Pathogenesis. Trends Neurosci. 40, 358–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, et al. , 2007. Development of alpha-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathol. 114, 231–41. [DOI] [PubMed] [Google Scholar]
- Brockmann K, et al. , 2016. Inflammatory profile in LRRK2-associated prodromal and clinical PD. J Neuroinflammation. 13, 122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brockmann K, et al. , 2017. Inflammatory profile discriminates clinical subtypes in LRRK2-associated Parkinson’s disease. Eur J Neurol. 24, 427–e6. [DOI] [PubMed] [Google Scholar]
- Burre J, et al. , 2014. alpha-Synuclein assembles into higher-order multimers upon membrane binding to promote SNARE complex formation. Proc Natl Acad Sci U S A. 111, E4274–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, et al. , 2014. Identification of a unique TGF-beta-dependent molecular and functional signature in microglia. Nat Neurosci. 17, 131–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butovsky O, Weiner HL, 2018. Microglial signatures and their role in health and disease. Nat Rev Neurosci. 19, 622–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas R, et al. , 2014. Astrocytic modulation of blood brain barrier: perspectives on Parkinson’s disease. Front Cell Neurosci. 8, 211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, et al. , 2014. The oxidation states of DJ-1 dictate the cell fate in response to oxidative stress triggered by 4-hpr: autophagy or apoptosis? Antioxid Redox Signal. 21, 1443–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castagnet PI, et al. , 2005. Fatty acid incorporation is decreased in astrocytes cultured from alpha-synuclein gene-ablated mice. J Neurochem. 94, 839–49. [DOI] [PubMed] [Google Scholar]
- Castro-Sanchez S, et al. , 2018. Cx3cr1-deficiency exacerbates alpha-synuclein-A53T induced neuroinflammation and neurodegeneration in a mouse model of Parkinson’s disease. Glia. 66, 1752–1762. [DOI] [PubMed] [Google Scholar]
- Chahine LM, et al. , 2013. Clinical and biochemical differences in patients having Parkinson disease with vs without GBA mutations. JAMA Neurol. 70, 852–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chai H, et al. , 2017. Neural Circuit-Specialized Astrocytes: Transcriptomic, Proteomic, Morphological, and Functional Evidence. Neuron. 95, 531–549 e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, et al. , 2017a. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nat Genet. 49, 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang D, et al. , 2017b. A meta-analysis of genome-wide association studies identifies 17 new Parkinson’s disease risk loci. Nature Genetics. 49, 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien CH, et al. , 2016. Microglia-Derived Cytokines/Chemokines Are Involved in the Enhancement of LPS-Induced Loss of Nigrostriatal Dopaminergic Neurons in DJ-1 Knockout Mice. PLoS One. 11, e0151569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I, et al. , 2016. PINK1 expression increases during brain development and stem cell differentiation, and affects the development of GFAP-positive astrocytes. Mol Brain. 9, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I, et al. , 2015a. LRRK2 G2019S mutation attenuates microglial motility by inhibiting focal adhesion kinase. Nat Commun. 6, 8255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I, et al. , 2013. PINK1 deficiency attenuates astrocyte proliferation through mitochondrial dysfunction, reduced AKT and increased p38 MAPK activation, and downregulation of EGFR. Glia. 61, 800–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi YR, et al. , 2015b. FcgammaRIIB mediates the inhibitory effect of aggregated alpha-synuclein on microglial phagocytosis. Neurobiol Dis. 83, 90–9. [DOI] [PubMed] [Google Scholar]
- Cook DA, et al. , 2017. LRRK2 levels in immune cells are increased in Parkinson’s disease. NPJ Parkinsons Dis. 3, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR, 2009. alpha-Synuclein and neuronal cell death. Mol Neurodegener. 4, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR, 2015. LRRK2 Pathways Leading to Neurodegeneration. Curr Neurol Neurosci Rep. 15, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cookson MR, 2016. Cellular functions of LRRK2 implicate vesicular trafficking pathways in Parkinson’s disease. Biochem Soc Trans. 44, 1603–1610. [DOI] [PubMed] [Google Scholar]
- Couch Y, et al. , 2011. The acute inflammatory response to intranigral alpha-synuclein differs significantly from intranigral lipopolysaccharide and is exacerbated by peripheral inflammation. J Neuroinflammation. 8, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daher JP, et al. , 2014. Abrogation of alpha-synuclein-mediated dopaminergic neurodegeneration in LRRK2-deficient rats. Proc Natl Acad Sci U S A. 111, 9289–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson TM, Dawson VL, 2010. The role of parkin in familial and sporadic Parkinson’s disease. Mov Disord. 25 Suppl 1, S32–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson VL, Dawson TM, 2019. Promising disease-modifying therapies for Parkinson’s disease. Sci Transl Med. 11. [DOI] [PubMed] [Google Scholar]
- De Biase LM, et al. , 2017. Local Cues Establish and Maintain Region-Specific Phenotypes of Basal Ganglia Microglia. Neuron. 95, 341–356.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lau LM, Breteler MM, 2006. Epidemiology of Parkinson’s disease. Lancet Neurol. 5, 525–35. [DOI] [PubMed] [Google Scholar]
- di Domenico A, et al. , 2019. Patient-Specific iPSC-Derived Astrocytes Contribute to Non-Cell-Autonomous Neurodegeneration in Parkinson’s Disease. Stem Cell Reports. 12, 213–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dionisio PEA, et al. , 2019. Loss of Microglial Parkin Inhibits Necroptosis and Contributes to Neuroinflammation. Mol Neurobiol. 56, 2990–3004. [DOI] [PubMed] [Google Scholar]
- Dolgacheva LP, et al. , 2019. Role of DJ-1 in the mechanism of pathogenesis of Parkinson’s disease. J Bioenerg Biomembr. 51, 175–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy MF, et al. , 2018. Lewy body-like alpha-synuclein inclusions trigger reactive microgliosis prior to nigral degeneration. J Neuroinflammation. 15, 129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzamko N, et al. , 2017. LRRK2 levels and phosphorylation in Parkinson’s disease brain and cases with restricted Lewy bodies. Mov Disord. 32, 423–432. [DOI] [PubMed] [Google Scholar]
- Dzamko N, et al. , 2012. The IkappaB kinase family phosphorylates the Parkinson’s disease kinase LRRK2 at Ser935 and Ser910 during Toll-like receptor signaling. PLoS One. 7, e39132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fellner L, et al. , 2013. Toll-like receptor 4 is required for alpha-synuclein dependent activation of microglia and astroglia. Glia. 61, 349–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frank-Cannon TC, et al. , 2008. Parkin deficiency increases vulnerability to inflammation-related nigral degeneration. J Neurosci. 28, 10825–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost JL, Schafer DP, 2016. Microglia: Architects of the Developing Nervous System. Trends Cell Biol. 26, 587–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Froyset AK, et al. , 2018. Astroglial DJ-1 over-expression up-regulates proteins involved in redox regulation and is neuroprotective in vivo. Redox Biol. 16, 237–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillardon F, et al. , 2012. Parkinson’s disease-linked leucine-rich repeat kinase 2(R1441G) mutation increases proinflammatory cytokine release from activated primary microglial cells and resultant neurotoxicity. Neuroscience. 208, 41–8. [DOI] [PubMed] [Google Scholar]
- Ginns EI, et al. , 2014. Neuroinflammation and alpha-synuclein accumulation in response to glucocerebrosidase deficiency are accompanied by synaptic dysfunction. Mol Genet Metab. 111, 152–62. [DOI] [PubMed] [Google Scholar]
- Gordon R, et al. , 2018. Inflammasome inhibition prevents α-synuclein pathology and dopaminergic neurodegeneration in mice. Science Translational Medicine. 10, eaah4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosselin D, et al. , 2017. An environment-dependent transcriptional network specifies human microglia identity. Science. 356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray MT, Woulfe JM, 2015. Striatal blood-brain barrier permeability in Parkinson’s disease. J Cereb Blood Flow Metab. 35, 747–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu XL, et al. , 2010. Astrocytic expression of Parkinson’s disease-related A53T alpha-synuclein causes neurodegeneration in mice. Mol Brain. 3, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guermonprez P, et al. , 2002. Antigen presentation and T cell stimulation by dendritic cells. Annu Rev Immunol. 20, 621–67. [DOI] [PubMed] [Google Scholar]
- Hammond TR, et al. , 2019. Single-Cell RNA Sequencing of Microglia throughout the Mouse Lifespan and in the Injured Brain Reveals Complex Cell-State Changes. Immunity. 50, 253–271 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamza TH, et al. , 2010. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nat Genet. 42, 781–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen C, et al. , 2011. alpha-Synuclein propagates from mouse brain to grafted dopaminergic neurons and seeds aggregation in cultured human cells. J Clin Invest. 121, 715–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, et al. , 2013. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 493, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henry AG, et al. , 2015. Pathogenic LRRK2 mutations, through increased kinase activity, produce enlarged lysosomes with reduced degradative capacity and increase ATP13A2 expression. Hum Mol Genet. 24, 6013–28. [DOI] [PubMed] [Google Scholar]
- Hernandez DG, et al. , 2016. Genetics in Parkinson disease: Mendelian versus non-Mendelian inheritance. J Neurochem. 139 Suppl 1, 59–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho DH, et al. , 2018. LRRK2 Kinase Activity Induces Mitochondrial Fission in Microglia via Drp1 and Modulates Neuroinflammation. Exp Neurobiol. 27, 171–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoenen C, et al. , 2016. Alpha-Synuclein Proteins Promote Pro-Inflammatory Cascades in Microglia: Stronger Effects of the A53T Mutant. PLoS One. 11, e0162717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmans P, et al. , 2013. A pathway-based analysis provides additional support for an immune-related genetic susceptibility to Parkinson’s disease. Hum Mol Genet. 22, 1039–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S, et al. , 2016. Complement and microglia mediate early synapse loss in Alzheimer mouse models. Science. 352, 712–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui KY, et al. , 2018. Functional variants in the LRRK2 gene confer shared effects on risk for Crohn’s disease and Parkinson’s disease. Sci Transl Med. 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibanez P, et al. , 2004. Causal relation between alpha-synuclein gene duplication and familial Parkinson’s disease. Lancet. 364, 1169–71. [DOI] [PubMed] [Google Scholar]
- Ibanez P, et al. , 2009. Alpha-synuclein gene rearrangements in dominantly inherited parkinsonism: frequency, phenotype, and mechanisms. Arch Neurol. 66, 102–8. [DOI] [PubMed] [Google Scholar]
- International Parkinson Disease Genomics, C., et al. , 2011. Imputation of sequence variants for identification of genetic risks for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet. 377, 641–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ising C, et al. , 2019. NLRP3 inflammasome activation drives tau pathology. Nature. 575, 669–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islam MS, Moore DJ, 2017. Mechanisms of LRRK2-dependent neurodegeneration: role of enzymatic activity and protein aggregation. Biochem Soc Trans. 45, 163–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang T, et al. , 2015. P2X7 receptor is critical in alpha-synuclein--mediated microglial NADPH oxidase activation. Neurobiol Aging. 36, 2304–2318. [DOI] [PubMed] [Google Scholar]
- Jo E, et al. , 2004. alpha-Synuclein-synaptosomal membrane interactions: implications for fibrillogenesis. Eur J Biochem. 271, 3180–9. [DOI] [PubMed] [Google Scholar]
- Kahns S, et al. , 2003. Caspase-1 and caspase-8 cleave and inactivate cellular parkin. J Biol Chem. 278, 23376–80. [DOI] [PubMed] [Google Scholar]
- Kang R, et al. , 2011. The Beclin 1 network regulates autophagy and apoptosis. Cell Death Differ. 18, 571–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawajiri S, et al. , 2011. Genetic mutations and functions of PINK1. Trends Pharmacol Sci. 32, 573–80. [DOI] [PubMed] [Google Scholar]
- Keren-Shaul H, et al. , 2017. A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell. 169, 1276–1290.e17. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Deneen B, 2019. The Emerging Nature of Astrocyte Diversity. Annu Rev Neurosci. 42, 187–207. [DOI] [PubMed] [Google Scholar]
- Khakh BS, Sofroniew MV, 2015. Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci. 18, 942–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khasnavis S, Pahan K, 2014. Cinnamon treatment upregulates neuroprotective proteins Parkin and DJ-1 and protects dopaminergic neurons in a mouse model of Parkinson’s disease. J Neuroimmune Pharmacol. 9, 569–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim B, et al. , 2012. Impaired inflammatory responses in murine Lrrk2-knockdown brain microglia. PLoS One. 7, e34693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim C, et al. , 2013a. Neuron-released oligomeric α-synuclein is an endogenous agonist of TLR2 for paracrine activation of microglia. Nature Communications. 4, 1562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JH, et al. , 2013b. DJ-1 facilitates the interaction between STAT1 and its phosphatase, SHP-1, in brain microglia and astrocytes: A novel anti-inflammatory function of DJ-1. Neurobiol Dis. 60, 1–10. [DOI] [PubMed] [Google Scholar]
- Kim JH, et al. , 2014. Suppression of miR-155 Expression in IFN-gamma-Treated Astrocytes and Microglia by DJ-1: A Possible Mechanism for Maintaining SOCS1 Expression. Exp Neurobiol. 23, 148–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KS, et al. , 2013c. DJ-1 associates with lipid rafts by palmitoylation and regulates lipid rafts-dependent endocytosis in astrocytes. Hum Mol Genet. 22, 4805–17. [DOI] [PubMed] [Google Scholar]
- Kim KS, et al. , 2018. Regulation of myeloid cell phagocytosis by LRRK2 via WAVE2 complex stabilization is altered in Parkinson’s disease. Proc Natl Acad Sci U S A. 115, E5164–E5173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klegeris A, et al. , 2008. Alpha-synuclein activates stress signaling protein kinases in THP-1 cells and microglia. Neurobiol Aging. 29, 739–52. [DOI] [PubMed] [Google Scholar]
- Klein C, Westenberger A, 2012. Genetics of Parkinson’s disease. Cold Spring Harb Perspect Med. 2, a008888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kluss JH, et al. , 2019. LRRK2 links genetic and sporadic Parkinson’s disease. Biochem Soc Trans. 47, 651–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozina E, et al. , 2018. Mutant LRRK2 mediates peripheral and central immune responses leading to neurodegeneration in vivo. Brain. 141, 1753–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanjakornsiripan D, et al. , 2018. Layer-specific morphological and molecular differences in neocortical astrocytes and their dependence on neuronal layers. Nat Commun. 9, 1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen NJ, et al. , 2011. DJ-1 knock-down impairs astrocyte mitochondrial function. Neuroscience. 196, 251–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau LT, Yu AC, 2001. Astrocytes produce and release interleukin-1, interleukin-6, tumor necrosis factor alpha and interferon-gamma following traumatic and metabolic injury. J Neurotrauma. 18, 351–9. [DOI] [PubMed] [Google Scholar]
- Leal MC, et al. , 2013. Interleukin-1beta and tumor necrosis factor-alpha: reliable targets for protective therapies in Parkinson’s Disease? Front Cell Neurosci. 7, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ledesma MD, et al. , 2002. Astrocytic but not neuronal increased expression and redistribution of parkin during unfolded protein stress. J Neurochem. 83, 1431–40. [DOI] [PubMed] [Google Scholar]
- Lee AJ, et al. , 2017. Penetrance estimate of LRRK2 p.G2019S mutation in individuals of non-Ashkenazi Jewish ancestry. Mov Disord. 32, 1432–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, Chung KC, 2012. PINK1 positively regulates IL-1beta-mediated signaling through Tollip and IRAK1 modulation. J Neuroinflammation. 9, 271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, et al. , 2012. PINK1 stimulates interleukin-1beta-mediated inflammatory signaling via the positive regulation of TRAF6 and TAK1. Cell Mol Life Sci. 69, 3301–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, et al. , 2010a. Alpha-synuclein stimulation of astrocytes: Potential role for neuroinflammation and neuroprotection. Oxid Med Cell Longev. 3, 283–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HJ, et al. , 2010b. Direct transfer of alpha-synuclein from neuron to astroglia causes inflammatory responses in synucleinopathies. J Biol Chem. 285, 9262–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev N, et al. , 2013. Knocking out DJ-1 attenuates astrocytes neuroprotection against 6-hydroxydopamine toxicity. J Mol Neurosci. 50, 542–50. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, et al. , 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin LF, et al. , 1993. GDNF: a glial cell line-derived neurotrophic factor for midbrain dopaminergic neurons. Science. 260, 1130–2. [DOI] [PubMed] [Google Scholar]
- Lloyd AF, et al. , 2019. Central nervous system regeneration is driven by microglia necroptosis and repopulation. Nat Neurosci. 22, 1046–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez de Maturana R, et al. , 2014. Leucine-rich repeat kinase 2 modulates cyclooxygenase 2 and the inflammatory response in idiopathic and genetic Parkinson’s disease. Neurobiol Aging. 35, 1116–24. [DOI] [PubMed] [Google Scholar]
- Ma B, et al. , 2016. LRRK2 modulates microglial activity through regulation of chemokine (C-X3-C) receptor 1 -mediated signalling pathways. Hum Mol Genet. 25, 3515–3523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maatouk L, et al. , 2018. TLR9 activation via microglial glucocorticoid receptors contributes to degeneration of midbrain dopamine neurons. Nat Commun. 9, 2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maekawa T, et al. , 2016. Leucine-rich repeat kinase 2 (LRRK2) regulates alpha-synuclein clearance in microglia. BMC Neurosci. 17, 77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magalhaes J, et al. , 2016. Autophagic lysosome reformation dysfunction in glucocerebrosidase deficient cells: relevance to Parkinson disease. Hum Mol Genet. 25, 3432–3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni C, et al. , 2013. Inhibition of LRRK2 kinase activity stimulates macroautophagy. Biochim Biophys Acta. 1833, 2900–2910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni C, et al. , 2016. mTOR independent regulation of macroautophagy by Leucine Rich Repeat Kinase 2 via Beclin-1. Sci Rep. 6, 35106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin I, et al. , 2011. Recent advances in the genetics of Parkinson’s disease. Annu Rev Genomics Hum Genet. 12, 301–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matheoud D, et al. , 2016. Parkinson’s Disease-Related Proteins PINK1 and Parkin Repress Mitochondrial Antigen Presentation. Cell. 166, 314–327. [DOI] [PubMed] [Google Scholar]
- McGeer PL, et al. , 1988. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 38, 1285–91. [DOI] [PubMed] [Google Scholar]
- Menzies FM, et al. , 2017. Autophagy and Neurodegeneration: Pathogenic Mechanisms and Therapeutic Opportunities. Neuron. 93, 1015–1034. [DOI] [PubMed] [Google Scholar]
- Miklossy J, et al. , 2006. LRRK2 expression in normal and pathologic human brain and in human cell lines. J Neuropathol Exp Neurol. 65, 953–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mittelbronn M, et al. , 2001. Local distribution of microglia in the normal adult human central nervous system differs by up to one order of magnitude. Acta Neuropathol. 101, 249–55. [DOI] [PubMed] [Google Scholar]
- Moehle MS, et al. , 2012. LRRK2 inhibition attenuates microglial inflammatory responses. J Neurosci. 32, 1602–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore DJ, et al. , 2005. Molecular pathophysiology of Parkinson’s disease. Annu Rev Neurosci. 28, 57–87. [DOI] [PubMed] [Google Scholar]
- Morel L, et al. , 2017. Molecular and Functional Properties of Regional Astrocytes in the Adult Brain. J Neurosci. 37, 8706–8717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mouton-Liger F, et al. , 2018. Parkin deficiency modulates NLRP3 inflammasome activation by attenuating an A20-dependent negative feedback loop. Glia. 66, 1736–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullett SJ, Hinkle DA, 2009. DJ-1 knock-down in astrocytes impairs astrocyte-mediated neuroprotection against rotenone. Neurobiol Dis. 33, 28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullett SJ, Hinkle DA, 2011. DJ-1 deficiency in astrocytes selectively enhances mitochondrial Complex I inhibitor-induced neurotoxicity. J Neurochem. 117, 375–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munro S, 2003. Lipid rafts: elusive or illusive? Cell. 115, 377–88. [DOI] [PubMed] [Google Scholar]
- Nakahira K, et al. , 2011. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nature Immunology. 12, 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nalls MA, et al. , 2018. Parkinson’s disease genetics: identifying novel risk loci, providing causal insights and improving estimates of heritable risk. bioRxiv. [Google Scholar]
- Nash Y, et al. , 2017. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J Neurochem. 143, 584–594. [DOI] [PubMed] [Google Scholar]
- Nguyen TA, et al. , 2013. Analysis of inflammation-related nigral degeneration and locomotor function in DJ-1(−/−) mice. J Neuroinflammation. 10, 50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols RJ, et al. , 2010. 14-3-3 binding to LRRK2 is disrupted by multiple Parkinson’s disease-associated mutations and regulates cytoplasmic localization. Biochem J. 430, 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nimmerjahn A, et al. , 2005. Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo — Resting Microglial Cells Are Highly Dynamic Surveillants of Brain Parenchyma in Vivo — Supporting Online Material. 308, 1314–1319. [DOI] [PubMed] [Google Scholar]
- Olah M, et al. , 2018. A transcriptomic atlas of aged human microglia. Nature Communications. 9, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osellame LD, Duchen MR, 2013. Defective quality control mechanisms and accumulation of damaged mitochondria link Gaucher and Parkinson diseases. Autophagy. 9, 1633–5. [DOI] [PubMed] [Google Scholar]
- Osellame LD, et al. , 2013. Mitochondria and quality control defects in a mouse model of Gaucher disease--links to Parkinson’s disease. Cell Metab. 17, 941–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandey MK, et al. , 2017. Complement drives glucosylceramide accumulation and tissue inflammation in Gaucher disease. Nature. 543, 108–112. [DOI] [PubMed] [Google Scholar]
- Panicker N, et al. , 2017. Activation mechanisms of the E3 ubiquitin ligase parkin. Biochem J. 474, 3075–3086. [DOI] [PubMed] [Google Scholar]
- Panicker N, et al. , 2019. Fyn kinase regulates misfolded alpha-synuclein uptake and NLRP3 inflammasome activation in microglia. J Exp Med. 216, 1411–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier L, et al. , 1988. HgC12 induces T and B cells to proliferate and differentiate in BN rats. Clin Exp Immunol. 71, 336–42. [PMC free article] [PubMed] [Google Scholar]
- Phatnani H, Maniatis T, 2015. Astrocytes in neurodegenerative disease. Cold Spring Harb Perspect Biol. 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickles S, et al. , 2018. Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol. 28, R170–R185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickrell AM, Youle RJ, 2015. The roles of PINK1, Parkin, and mitochondrial fidelity in parkinson’s disease. Neuron. 85, 257–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce S, Coetzee GA, 2017. Parkinson’s disease-associated genetic variation is linked to quantitative expression of inflammatory genes. PLoS One. 12, e0175882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymeropoulos MH, et al. , 1997. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science. 276, 2045–7. [DOI] [PubMed] [Google Scholar]
- Rannikko EH, et al. , 2015. Exogenous alpha-synuclein induces toll-like receptor 4 dependent inflammatory responses in astrocytes. BMC Neurosci. 16, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzu P, et al. , 2004. DJ-1 colocalizes with tau inclusions: a link between parkinsonism and dementia. Ann Neurol. 55, 113–8.14705119 [Google Scholar]
- Rocha EM, et al. , 2015. Sustained Systemic Glucocerebrosidase Inhibition Induces Brain alpha-Synuclein Aggregation, Microglia and Complement C1q Activation in Mice. Antioxid Redox Signal. 23, 550–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo I, et al. , 2014. LRRK2 and neuroinflammation: partners in crime in Parkinson’s disease? J euroinflammation. 11, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo I, et al. , 2019. Transcriptome analysis of LRRK2 knock-out microglia cells reveals alterations of inflammatory- and oxidative stress-related pathways upon treatment with alpha-synuclein fibrils. Neurobiol Dis. 129, 67–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San Luciano M, et al. , 2010. Clinical expression of LRRK2 G2019S mutations in the elderly. Mov Disord. 25, 2571–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanyal A, et al. , 2020. Lysosome and Inflammatory Defects in GBA1-Mutant Astrocytes Are Normalized by LRRK2 Inhibition. Mov Disord. 35, 760–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarffe LA, et al. , 2014. Parkin and PINK1: much more than mitophagy. Trends Neurosci. 37, 315–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schapansky J, et al. , 2014. Membrane recruitment of endogenous LRRK2 precedes its potent regulation of autophagy. Hum Mol Genet. 23, 4201–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt S, et al. , 2011. Genetic mouse models for Parkinson’s disease display severe pathology in glial cell mitochondria. Hum Mol Genet. 20, 1197–211. [DOI] [PubMed] [Google Scholar]
- Schwartz M, et al. , 2013. How do immune cells support and shape the brain in health, disease, and aging? J Neurosci. 33, 17587–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sekar A, et al. , 2016. Schizophrenia risk from complex variation of complement component 4. Nature. 530, 177–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth RB, et al. , 2005. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 122, 669–82. [DOI] [PubMed] [Google Scholar]
- Sharma S, et al. , 2011. LRRK2 expression in idiopathic and G2019S positive Parkinson’s disease subjects: a morphological and quantitative study. Neuropathol Appl Neurobiol. 37, 777–90. [DOI] [PubMed] [Google Scholar]
- Sidransky E, et al. , 2009. Multicenter analysis of glucocerebrosidase mutations in Parkinson’s disease. N Engl J Med. 361, 1651–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh K, et al. , 2018. Parkin targets NOD2 to regulate astrocyte endoplasmic reticulum stress and inflammation. Glia. 66, 2427–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singleton AB, et al. , 2003. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 302, 841. [DOI] [PubMed] [Google Scholar]
- Sofroniew MV, 2009. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 32, 638–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sofroniew MV, Vinters HV, 2010. Astrocytes: biology and pathology. Acta Neuropathol. 119, 7–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solano RM, et al. , 2008. Glial dysfunction in parkin null mice: effects of aging. J Neurosci. 28, 598–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song YJ, et al. , 2009. Degeneration in different parkinsonian syndromes relates to astrocyte type and astrocyte protein expression. J Neuropathol Exp Neurol. 68, 1073–83. [DOI] [PubMed] [Google Scholar]
- Stevens B, et al. , 2007. The Classical Complement Cascade Mediates CNS Synapse Elimination. Cell. 131, 1164–1178. [DOI] [PubMed] [Google Scholar]
- Sun L, et al. , 2018. Lack of PINK1 alters glia innate immune responses and enhances inflammation-induced, nitric oxide-mediated neuron death. Sci Rep. 8, 383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, et al. , 2017. Selective neuronal vulnerability in Parkinson disease. Nat Rev Neurosci. 18, 101–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres-Odio S, et al. , 2017. Progression of pathology in PINK1-deficient mouse brain from splicing via ubiquitination, ER stress, and mitophagy changes to neuroinflammation. J Neuroinflammation. 14, 154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran TA, et al. , 2011. Lipopolysaccharide and tumor necrosis factor regulate Parkin expression via nuclear factor-kappa B. PLoS One. 6, e23660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tremblay ME, et al. , 2019. Glial phagocytic clearance in Parkinson’s disease. Mol Neurodegener. 14, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trinh J, Farrer M, 2013. Advances in the genetics of Parkinson disease. Nat Rev Neurol. 9, 445–54. [DOI] [PubMed] [Google Scholar]
- Trudler D, et al. , 2014. DJ-1 deficiency triggers microglia sensitivity to dopamine toward a pro-inflammatory phenotype that is attenuated by rasagiline. J Neurochem. 129, 434–47. [DOI] [PubMed] [Google Scholar]
- Vainchtein ID, et al. , 2018. Astrocyte-derived interleukin-33 promotes microglial synapse engulfment and neural circuit development. Science. 1273, 1269–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venegas C, et al. , 2017. Microglia-derived ASC specks crossseed amyloid-β in Alzheimer’s disease. Nature. 552, 355–361. [DOI] [PubMed] [Google Scholar]
- Vitner EB, et al. , 2010. Altered expression and distribution of cathepsins in neuronopathic forms of Gaucher disease and in other sphingolipidoses. Hum Mol Genet. 19, 3583–90. [DOI] [PubMed] [Google Scholar]
- Waak J, et al. , 2009. Regulation of astrocyte inflammatory responses by the Parkinson’s disease-associated gene DJ-1. FASEB J. 23, 2478–89. [DOI] [PubMed] [Google Scholar]
- Wakabayashi K, et al. , 2000. NACP/alpha-synuclein-positive filamentous inclusions in astrocytes and oligodendrocytes of Parkinson’s disease brains. Acta Neuropathol. 99, 14–20. [DOI] [PubMed] [Google Scholar]
- Wilton DK, et al. , 2019. Neuron-Glia Signaling in Synapse Elimination. Annu Rev Neurosci. 42, 107–127. [DOI] [PubMed] [Google Scholar]
- Witoelar A, et al. , 2017. Genome-wide Pleiotropy Between Parkinson Disease and Autoimmune Diseases. JAMA Neurol. 74, 780–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong K, et al. , 2004. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab. 82, 192–207. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T, Mucke L, 2002. Inflammation in neurodegenerative disease--a double-edged sword. Neuron. 35, 419–32. [DOI] [PubMed] [Google Scholar]
- Yun SP, et al. , 2018. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med. 24, 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, et al. , 2007. Microglial PHOX and Mac-1 are essential to the enhanced dopaminergic neurodegeneration elicited by A30P and A53T mutant alpha-synuclein. Glia. 55, 1178–88. [DOI] [PubMed] [Google Scholar]
- Zhang Y, et al. , 2014. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J Neurosci. 34, 11929–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, et al. , 2016. Purification and Characterization of Progenitor and Mature Human Astrocytes Reveals Transcriptional and Functional Differences with Mouse. Neuron. 89, 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, et al. , 2018. Nigrostriatal pathology with reduced astrocytes in LRRK2 S910/S935 phosphorylation deficient knockin mice. Neurobiol Dis. 120, 76–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong Z, et al. , 2016. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell. 164, 896–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, et al. , 2011. A role for mitochondria in NLRP3 inflammasome activation. Nature. 469, 221–226. [DOI] [PubMed] [Google Scholar]