

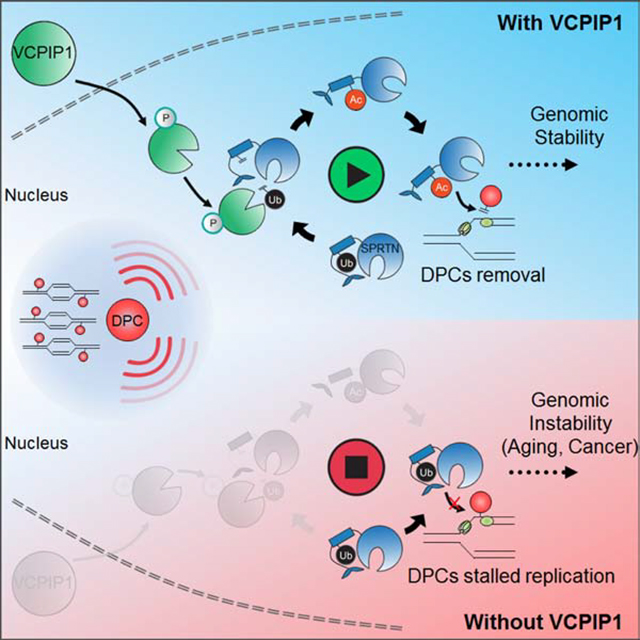
SUMMARY
DNA-protein crosslinks (DPCs) are highly toxic DNA lesions that threaten genomic integrity. Recent findings highlight that SPRTN, a specialized DNA-dependent metalloprotease, is a central player in proteolytic cleavage of DPCs. Previous studies suggest that SPRTN deubiquitination is important for its chromatin association and activation. However, the regulation and consequence of SPRTN deubiquitination remain unclear. Here, we report that in response to DPC induction, the deubiquitinase VCPIP1/VCIP135 is phosphorylated and activated by ATM/ATR. VCPIP1, in turn, deubiquitinates SPRTN and promotes its chromatin relocalization. The deubiquitination of SPRTN is required for its subsequent acetylation, which promotes SPRTN relocation to the site of chromatin damage. Furthermore, Vcpip1 knockout mice are prone to genomic instability and premature aging. We propose a model that two sequential post-translational modifications (PTMs) regulate SPRTN chromatin accessibility to repair DPCs and maintain genomic stability and healthy life span.
Keywords: DNA-protein crosslink, SPRTN, VCPIP1/VCIP135, ubiquitination, acetylation, aging
In Brief
Huang et al. discover the deubiquitinase VCPIP1 is required for SPRTN activation, and further demonstrate that SPRTN activation is regulated by two-step post-translational modification, deubiquitination followed by acetylation. This way, cells can keep SPRTN activity in check, thereby promoting DPC repair, genomic stability and healthy aging.
Graphiccal Abstract
INTRODUCTION
DNA-protein crosslinks (DPCs) are highly toxic DNA lesions, as they impede chromatin-based processes such as gene transcription and DNA replication, thereby generating genomic instability (Fu et al., 2011; Nakano et al., 2013). They are formed when cellular proteins are covalently trapped on DNA strands (Barker et al., 2005; Tretyakova et al., 2015). DPCs are generated by various crosslinking agents, including exogenous agents (e.g. UV-light, ionizing radiation, and platinum-based chemotherapeutics) and endogenous sources (e.g. formaldehyde and acetaldehyde) (Ide et al., 2011). In cells, there are three prominent repair pathways responsible for resolution of DPCs: direct crosslink hydrolysis, nuclease-dependent and protease-dependent repair (Stingele et al., 2017). Recently, protease-dependent DPC repair has emerged as a crucial mechanism for maintaining genomic integrity. The SPRTN/Wss1 DPC protease acts as DNA-dependent protease that protects proliferative cells from DPC toxicity (Duxin et al., 2014; Lopez-Mosqueda et al., 2016; Maskey et al., 2017; Morocz et al., 2017; Stingele et al., 2016; Stingele et al., 2014; Vaz et al., 2016). Importantly, SPRTN dependent DPC repair associates with the DNA replication machinery and removes DPCs during DNA synthesis (Vaz et al., 2016). Hypomorphic mutations in Sprtn cause genomic instability, early onset hepatocellular carcinoma and progeroid features in mice (Maskey et al., 2017; Maskey et al., 2014). In humans, SPRTN mutation is causally linked to Ruijs-Aalfs syndrome, which is an autosomal recessive genetic disorder with characteristics of premature aging, chromosome instability and development of hepatocellular carcinoma (Lessel et al., 2014).
Although progress has been made in detailing the function of SPRTN in DPC repair, the regulatory mechanism of this pathway remains largely unclear. The DPC-cleaving proteolytic activity of SPRTN is tightly controlled by several layers of regulatory switches: ubiquitin switch, DNA switch and auto-cleavage switch (Stingele et al., 2016; Stingele et al., 2017). The ubiquitin switch has been implicated with the most upstream regulation of SPRTN (Stingele et al., 2017). SPRTN is present in cells in two forms, unmodified and mono-ubiquitinated (Maskey et al., 2017; Stingele et al., 2016; Stingele et al., 2017; Vaz et al., 2016). As mono-ubiquitylated SPRTN is excluded from chromatin, induction of DPCs triggers SPRTN deubiquitination by an unknown deubiquitylating enzyme (DUB), which allows its relocalization to chromatin (Stingele et al., 2017). This switch is critical for SPRTN activation by the DNA switch. However, because the DUB that regulates SPRTN remains unidentified, how the ubiquitin switch of SPRTN is dynamically regulated to avoid excessive proteolysis on chromatin is unclear.
In this study, we report that SPRTN is jointly regulated by two post-translational modifications (PTMs): deubiquitination and acetylation. Upon DPC induction, the deubiquitinase VCPIP1/VCIP135 is activated by ATM/ATR, which then deubiquitinates SPRTN. Once SPRTN is deubiquitinated, a subsequent acetylation further promotes SPRTN relocation to the damaged chromatin sites. Strikingly, Vcpip1 knockout mice exhibit genomic instability and progeroid features, which are similar to the phenotypes observed in mice with Sprtn hypomorph. Our results reveal how SPRTN is tightly regulated by PTMs to facilitate its specific DPC repair activity to maintain genomic stability and healthy life span.
RESULTS
VCPIP1 is a SPRTN-interacting protein and is involved in the response to DPC
SPRTN is a specialized DNA-dependent metalloprotease that plays a central role in the repair of DPCs. SPRTN is activated by a ubiquitination switch mechanism, but the DUB that regulates this switch remains unidentified. We first utilized a panel of DUBs to screen for potential SPRTN-interacting DUBs (Figure S1A). Treatment with formaldehyde (FA), a common DPC-inducing agent that results in significant accumulation of general or specific DPCs, was used to induce DPCs (Conaway et al., 1996; Quievryn and Zhitkovich, 2000). The DUB screen was repeated three times, and, doing so, revealed VCPIP1 as the most consistent hit. We confirmed the interaction between VCPIP1 and SPRTN using co-immunoprecipitation experiments (Figures 1A–1B and S1B–S1C). This is also consistent with a previous report, VCPIP1 was found in a proteomic study of SPRTN-associated proteins (Ghosal et al., 2012). Moreover, after treating cells with three known DPC-inducing agents, FA, camptothecin (CPT) and cisplatin (Stingele et al., 2017), the interaction between VCPIP1 and SPRTN increased (Figures 1C and S1D–S1E). However, UV exposure did not result in significant SPRTN deubiquitination, as has been published previously (Stingele et al., 2016), and the interaction did not change (Figure S1F). Consistent with the interaction shown by co-immunoprecipitation, direct association between VCPIP1 and SPRTN in cells was confirmed by proximity ligation assay (PLA) and PLA foci signals were mostly in the nucleus and significantly increased after FA treatment (Figures 1D–1E). Thus, these data suggest that VCPIP1 is a SPRTN-interacting DUB and a candidate for SPRTN deubiquitination.
Figure 1. VCPIP1 is a SPRTN-interacting protein and is involved in DPC repair.

(A) HEK293T cell lysates were subjected to immunoprecipitation with control IgG or anti-VCPIP1 antibodies and immunoblotted with the indicated antibodies.
(B) HEK293T cells were transfected with empty vector (Vec) or Flag-SPRTN. Cell lysates were subjected to immunoprecipitation with Flag beads and immunoblotted with the indicated antibodies.
(C) HEK293T cells transfected with empty vector (Vec) or Flag-SPRTN were treated with or without formaldehyde (FA, 2 mM, 2 h). Cell lysates were subjected to immunoprecipitation with Flag beads and immunoblotted with the indicated antibodies.
(D-E) U2OS cells stably expressing Flag-SPRTN were left untreated or treated with FA (0.5 mM, 1 h). Proximity ligation assay (PLA) was performed using anti-Flag antibody vs. control IgG or anti-VCPIP1 antibodies. Nuclei were visualized with DAPI (blue). Representative images are shown in (D). Quantification of the foci signals is shown in (E). Error bars indicate mean ± SEM of three independent experiments (** p<0.01). Scale bars, 10 μm.
(F) Diagram of VCPIP1 wild-type and mutation constructs. WT: wild-type; N: N terminus; ΔOTU: deletion of OTU domain; C: C terminus.
(G) HEK293T cells transfected with deletion mutants of Flag-VCPIP1 outlined in (F) were subjected to co-immunoprecipitation as in (C).
(H) VCPIP1 was knocked down in HepG2 cells and immunoblot with the indicated antibodies was performed.
(I-K) Control (Ctrl) or VCPIP1-knockdown HepG2 cells were subjected to colony formation assay to assess the sensitivity to formaldehyde (FA), camptothecin (CPT) and cisplatin (Cis). Error bars indicate mean ± SD of three independent experiments (* p<0.05; ** p<0.01).
(L) U2OS cells were treated with FA (0.5 mM, 1 h) and subjected to immunofluorescence with anti-VCPIP1 antibody. Nuclei were visualized with DAPI (blue). The nuclear (nuc) and cytoplasmic (cyto) signals of VCPIP1 were quantified and ratio was calculated to assess VCPIP1 localization (right). Scale bars, 10 μm.
(M-N) Control or VCPIP1-knockdown U2OS cells were treated with CPT (1 μM, 30 min) and Top1cc signals were detected by immunofluorescence. Nuclei were visualized with DAPI (blue). Representative images are shown in (M). Quantification of the foci signals is shown in (N). Error bars indicate mean ± SEM of three independent experiments (** p<0.01). Scale bars, 10 μm.
(O) Control or VCPIP1-knockdown U2OS cells were treated with FA (250 μM, 1 h) and DPCs were measured as the ratio of crosslinked DNA compared to total DNA (* p<0.05; ** p<0.01).
See also Figure S1.
Next, to investigate which domain(s) of VCPIP1 interacts with SPRTN, we generated truncated VCPIP1 constructs and co-expressed them with SPRTN in HEK293T cells (Figure 1F). Only constructs containing the OTU domain, but not other regions of VCPIP1, retained the ability to interact with SPRTN, indicating that the OTU domain is essential for SPRTN binding (Figure 1G).
VCPIP1/VCIP135, an OTU family DUB, has been implicated in the reassembly of the Golgi and the endoplasmic reticulum following mitosis (Wang et al., 2004). VCPIP1 has also been reported to stabilize a catalytic light chain metalloprotease to accelerate botulinum neurotoxin intoxication in patients (Tsai et al., 2017). We sought to investigate the function of VCPIP1 in DPC repair. We knocked down VCPIP1 using two independent shRNAs and tested cellular sensitivity to FA, CPT and cisplatin. As shown in Figures 1H–1K and S1G–S1I, knockdown of VCPIP1 in two distinct cell lines resulted in hypersensitivity to these agents. In addition, VCPIP1 displayed cytoplasmic- to- nuclear translocation in response to FA (Figure 1L). The translocation of VCPIP1 upon FA treatment was induced in a time-dependent manner (Figure S1J). These results suggest that VCPIP1 is involved in FA-induced DPC repair.
To directly test whether VCPIP1 plays a role in DPC repair, we examined the repair of CPT-induced topoisomerase 1 cleavage complex (Top1cc), a covalent adduct of DPC, which can block advancing replication forks, ultimately resulting in toxic DNA damage such as DNA double-strand breaks and cell death (Pommier, 2006). We found that VCPIP1 deficiency resulted in enhanced Top1cc foci signals after CPT treatment (Figures 1M–1N), suggesting a role of VCPIP1 in Top1cc removal. We further measured the amount of DPCs accumulation in cells using the KCl/SDS precipitation assay (Stingele et al., 2016; Vaz et al., 2016). The loss of VCPIP1 led to an increase in DPC formation upon FA treatment (Figure 1O), suggesting that cells require VCPIP1 for the repair of DPCs. Collectively, these results indicate that VCPIP1 is a new regulator in the DPC repair pathway.
Deubiquitination of SPRTN by VCPIP1 is important for DPC repair
We hypothesized that VCPIP1 deubiquitinates SPRTN and promotes SPRTN-mediated DPC repair. As previously reported (Stingele et al., 2016), the level of mono-ubiquitinated SPRTN decreased when cells were exposed to FA (Figure 2A) or CPT (Figure S2A). Importantly, as shown in Figures 2B and S2B, knockdown of VCPIP1 abolished the deubiquitination induced by FA treatment, suggesting that DPC-induced deubiquitination of SPRTN is dependent on VCPIP1.
Figure 2. VCPIP1 deubiquitinates SPRTN and is important for SPRTN relocalization to chromatin upon DPCs.

(A) U2OS cells were treated for 2 h with the indicated dose of FA and Western blots were performed with the indicated antibodies.
(B) Control (Ctrl) or VCPIP1-knockdown U2OS cells were treated with or without FA (2 mM, 2 h) and Western blots were performed with the indicated antibodies.
(C) Control (Ctrl) or VCPIP1-knockdown HEK293T cells were transfected with vector (Vec), WT or C218A (CA) Flag-VCPIP1 and treated with FA (2 mM, 2 h). Western blots were performed with the indicated antibodies.
(D) Flag-SPRTN isolated from cells was incubated with GST-tagged purified wild-type (WT) VCPIP1 or catalytically inactive (CA) VCPIP1 in vitro and immunoblotted with the indicated antibodies.
(E) Control or VCPIP1-knockdown U2OS cells were treated with FA (2 mM, 2 h). Cells were either lysed directly in SDS-containing loading dye (total) or subjected to fractionation into soluble and chromatin components. Western blots were performed with the indicated antibodies.
(F) VCPIP1-knockdown HEK293T cells were transfected with vector (Vec), wild-type (WT) or C218A (CA) Flag-VCPIP1 and treated with FA (2 mM, 2 h). Cells were lysed as in (E) and immunoblotted with the indicated antibodies.
(G-H) VCPIP1-knockdown U2OS cells stably expressing vector (Vec), WT or C218A (CA) Flag-VCPIP1 were treated with CPT (1 μM, 30 min). Top1cc signals were detected by immunofluorescence. Nuclei were visualized with DAPI (blue). Representative images are shown in (G). Quantification of foci signals is shown in (H). Error bars indicate mean ± SEM of three independent experiments (** p<0.01; ns: not significant). Scale bars, 10 μm.
See also Figure S2.
To further confirm that the regulation of SPRTN by VCPIP1 is dependent on its catalytic activity, we reconstituted VCPIP1-deficient cells with VCPIP1 -wild type (WT) or VCPIP1 catalytically inactive (CA) mutant. We found that VCPIP1-WT, but not VCPIP1-CA, restored SPRTN deubiquitination upon DPC induction (Figure 2C). To determine whether VCPIP1 directly deubiquitinates SPRTN, we performed an in vitro deubiquitination assay. We found that recombinant VCPIP1-WT, but not VCPIP1-CA, deubiquitinated SPRTN in vitro (Figure 2D). These results suggest that VCPIP1 deubiquitinates SPRTN both in vitro and in cells.
Mono-ubiquitinated SPRTN is excluded from chromatin; DPC induces SPRTN deubiquitination, allowing its localization to chromatin (Stingele et al., 2016). Therefore, we further examined whether VCPIP1 affects chromatin recruitment of SPRTN. Indeed, upon DPC induction, knockdown of VCPIP1 reduced the relocalization of SPRTN pool to chromatin (Figure 2E), whereas VCPIP1 reconstitution reversed this phenotype (Figures 2F and S2C). This restoration of SPRTN chromatin localization required the catalytic activity of VCPIP1 (Figure 2F). In agreement with this data, VCPIP1-WT rather than VCPIP1-CA could promote Top1cc removal by SPRTN (Figures 2G–2H). Furthermore, knockdown of VCPIP1 did not further affect the cellular sensitivity to FA and CPT and Top1cc removal in SPRTN-depleted cells (Figures S2D–S2H), suggesting that VCPIP1 regulates DPC repair through SPRTN.
Considering that VCPIP1 is a VCP/p97 interacting protein (Figure S2I) (Uchiyama et al., 2002), and SPRTN has been previously shown to interact with VCP via its SHP-box during translesion synthesis (Davis et al., 2012; Mosbech et al., 2012), we asked whether VCP is involved in the regulation between SPRTN and VCPIP1. Knockdown of VCP did not change the SPRTN/VCPIP1 interaction (Figure S2J). In addition, knockdown or inhibition of VCP did not influence SPRTN deubiquitination upon DPC induction (Figures S2K–S2L). These results suggest that VCP is dispensable for VCPIP1 mediated SPRTN activation during DPC repair, although it might act as a downstream cofactor of SPRTN during proteolytic digestion (Fielden et al., 2020).
Taken together, our data indicate that VCPIP1 regulates SPRTN via its Ub protease activity and deubiquitination of SPRTN by VCPIP1 is important for its chromatin retention in response to DPC lesions.
VCPIP1 is phosphorylated by ATM/ATR upon DPC induction
Our results so far suggest that VCPIP1 deubiquitinates SPRTN, thereby triggering association of SPRTN with chromatin following DPC damage. This suggests that the VCPIP1 activity is induced towards SPRTN following DPC treatment. We further investigated whether and how VCPIP1 itself is regulated during DPC repair. DNA damage-responsive kinases ATM/ATR are able to be activated by DPCs (Ortega-Atienza et al., 2016; Wong et al., 2012). As shown in Figure 3A, following FA and CPT treatment, VCPIP1 was phosphorylated at its SQ/TQ motifs, which are consensus ATM/ATR phosphorylation sites. The FA induced phosphorylation was blocked by an ATM-specific inhibitor (KU55933), while CPT induced phosphorylation was blocked by the ATR-specific inhibitor (VX970). Nuclear VCPIP1 was phosphorylated at the SQ/TQ motif in wild-type but not ATM-deficient cells (Figure 3B). These results suggest that VCPIP1 is phosphorylated by ATM/ATR following DPC induction. To identify the candidate site that is phosphorylated upon DPC-induction, we further analyzed the VCPIP1 protein sequence and found only one Ser/Thr site fitting the SQ/TQ motif: S1207 (S1207). Mutation of Serine 1207 to Alanine (S1207A) resulted in complete abolishment the p-SQ/TQ signal, suggesting that S1207 is the major phosphorylation site of ATM/ATR in response to DPCs (Figure 3C).
Figure 3. Phosphorylation of VCPIP1 promotes its activation.

(A) HEK293T cells stably expressing Flag-VCPIP1 were pretreated with DMSO or 25 μM Ku55933 (ATMi) or 80 nM VX970 (ATRi) for 2 h. Cells were then left untreated or treated with CPT (10 μM) or FA (2 mM). After an additional 1 h, cell lysates were subjected to immunoprecipitation with Flag beads, left untreated or treated with phosphatase, and immunoblot with phospho-SQTQ (p-SQTQ) antibody. ATM and ATR kinase activities were determined by assessing phospho-Chk2 (p-Chk2) and phospho-Chk1 (p-Chk1) levels, respectively.
(B) Control or ATM-knockout HEK293T cells were transfected with Flag-VCPIP1 and left untreated or treated with FA (2 mM, 2 h). Flag-VCPIP1 was immunoprecipitated from nuclear lysates, and immunoblot with phospho-SQTQ (p-SQTQ) antibody was performed.
(C) HEK293T cells stably expressing WT or S1207A Flag-VCPIP1 were left untreated or treated with CPT (10 μM, 1 h) or FA (2 mM, 2 h). Flag-VCPIP1 was immunoprecipitated and immunoblot with phospho-SQTQ (p-SQTQ) antibody was performed.
(D) VCPIP1-knockdown HepG2 cells stably expressing WT or S1207A Flag-VCPIP1 were treated with or without FA (2 mM, 2 h). Western blots were performed with the indicated antibodies.
(E-F) Cells from (D) were subjected to colony formation assay to assess the sensitivity of cells to FA or CPT. Error bars indicate mean ± SD of three independent experiments (* p<0.05; ** p<0.01).
(G) WT or the S1207A mutant VCPIP1 was purified from HEK293T cells treated with or without FA (2 mM, 2 h). The deubiquitinase activity of VCPIP1 was assessed using Ub-AMC assay.
Next, we further investigated the importance of S1207 phosphorylation of VCPIP1 during DPC repair. VCPIP1-WT or VCPIP1-SA mutant was reconstituted into cells in which endogenous VCPIP1 had been knocked down. As shown in Figure 3D, the VCPIP1-SA, but not VCPIP1-WT, significantly compromised the deubiquitination of SPRTN upon DPC induction. In addition, the VCPIP1-SA, but not VCPIP1-WT, failed to rescue VCPIP1-deficient cells’ hypersensitivity to FA or CPT treatment (Figures 3E–3F). This observation led us to hypothesize that VCPIP1 phosphorylation affects its activity. To test this, we performed an in vitro deubiquitination assay. VCPIP1-WT showed much higher activity towards Ub-AMC after FA treatment, while the S1207A mutant had minimal change in activity after FA treatment (Figure 3G). Taken together, these data suggest that VCPIP1 phosphorylation by ATM/ATR is important for VCPIP1 activation following DPC induction.
SPRTN is acetylated at residue K230 by DPC-inducing agents
The induction of DPCs triggers deubiquitination of SPRTN, which allows SPRTN access to chromatin (Stingele et al., 2016). However, the deubiquitinated SPRTN under undamaged conditions is not sufficient for its localization to chromatin. Thus, we set out to investigate whether there are other post-translational modifications of SPRTN that contribute to its chromatin association. Using anti-Ac antibodies, we found that DPC induction by FA increased SPRTN acetylation (Figure 4A). We identified one acetylated lysine residue (K230) by mass spectrometry, the signal for which was strongly increased upon FA treatment (Figure S3A). The K230 residue is conserved across many species and is located in the region between the protease domain (SprT) and the SHP domain, which has been shown to have DNA-binding activity (aa 200–250, Figure 4B) (Li et al., 2019; Stingele et al., 2016; Vaz et al., 2016). To confirm that it is the major acetylated residue of SPRTN, lysine 230 was replaced by arginine (KR) or glutamine (KQ) (mimicking hyperacetylated SPRTN) by mutagenesis. As shown in Figures 4C and S3B, the acetylation level of the SPRTN-KR mutant was significantly decreased in comparison to the SPRTN-WT upon DPC damage, indicating that SPRTN is acetylated at residue K230 in response to DPC induction. We also noticed that the acetylated SPRTN always showed one band that corresponded to the deubiquitinated SPRTN (Figures 4A, 4C and S3B).
Figure 4. Acetylation of SPRTN is critical for SPRTN-mediated DPCs repair and is dependent on SPRTN deubiquitination.

(A) HEK293T cells transfected with Flag-SPRTN were treated with FA (2 mM, 2 h). Cell lysates were subjected to immunoprecipitation with Flag beads and immunoblotted with the indicated antibodies.
(B) The alignment of the amino-acid sequence of SPRTN among different species. BOVIN: Bos Taurus; XENTR: Xenopus tropicalis; CAEEL: Caenorhabditis elegans.
(C-D) HEK293T cells transfected with WT, K230R or K230Q Flag-SPRTN were treated with or without FA (2 mM, 2 h). Cells were lysed as in (A) and immunoblotted with the indicated antibodies.
(E-F) SprtnF/F; Cre-ERT2 MEFs stably expressing control vector (Vec), human WT, K230R or K230Q SPRTN were treated with MeOH or 4-OHT for 48 h, followed by CPT (1 μM, 30 min). Top1cc signals were detected by immunofluorescence. Nuclei were visualized with DAPI (blue). Representative images are shown in (E). Quantification of foci signals is shown in (F) (** p<0.01). Scale bars, 10 μm.
(G) Cells from (E) (treated with MeOH or 4-OHT for 48 h) were treated with FA (250 μM, 1 h) and DPCs were isolated. DPCs were measured as the ratio of crosslinked DNA compared to total DNA.
(H) Survival assays for cells from (E) (treated with MeOH or 4-OHT for 48 h) exposed to FA or CPT. Error bars indicate mean ± SD of three independent experiments (** p<0.01).
(I) Control or VCPIP1-knockdown HEK293T cells transfected with Flag-SPRTN were treated with or without FA (2 mM, 2 h). Cells were lysed as in (A) and immunoblotted with the indicated antibodies.
See also Figures S3–S4.
K230 acetylation of SPRTN is critical for SPRTN-mediated DPC repair
To investigate whether K230 acetylation of SPRTN affects its activity upon DPC induction, we evaluated the ability of the KR and KQ mutant SPRTN to localize to chromatin. Notably, as shown in Figures 4D and S3C, SPRTN-WT and SPRTN-KQ were recruited to chromatin following FA treatment, consistent with the observations with deubiquitinated SPRTN. The KQ mutant also showed increased chromatin association in unstressed cells. In contrast, the SPRTN-KR mutant hardly accumulated on the chromatin after FA treatment, suggesting that acetylation controls SPRTN’s access to chromatin upon DPC induction.
To establish the importance of K230 acetylation of SPRTN for DPC repair, human SPRTN-WT, SPRTN-KR or SPRTN-KQ constructs were expressed in conditional Sprtn knockout mouse embryonic fibroblasts (MEFs). The loss of SPRTN resulted in the accumulation of Top1cc foci, as has been published previously (Maskey et al., 2017). We found that SPRTN-WT or SPRTN-KQ, but not the SPRTN-KR mutant, suppressed the accumulation of Top1cc foci (Figures 4E–4F and S3D–S3G). Moreover, Sprtn knockout or ectopic expression of SPRTN-KR in Sprtn knockout MEFs resulted in an almost complete failure to repair DPCs, whereas DPCs were repaired in a time-dependent manner in Sprtn knockout MEFs expressing SPRTN-WT or SPRTN-KQ (Figure 4G). We also found that WT and the KQ mutant, but not the KR mutant rescued SPRTN-deficient cells’ hypersensitivity towards FA as well as CPT (Figures 4H and S3H). Altogether, these results suggest a critical function of K230 acetylation of SPRTN in the resolution of DPCs.
SPRTN deubiquitination is a prerequisite for SPRTN acetylation
Since we noticed that the acetylated SPRTN always corresponded to the deubiquitinated SPRTN band on Western blot (Figures 4A and 4C), we next explored whether there is a crosstalk between deubiquitination and acetylation of SPRTN during DPC repair. We found that knockdown of VCPIP1 decreased acetylation of SPRTN upon DPC induction (Figure 4I), suggesting that SPRTN acetylation requires SPRTN deubiquitination. Consistent with this, VCPIP1 depleted cells reconstituted with VCPIP1-CA showed decreased acetylation of SPRTN upon DPC damage when compared to VCPIP1 depleted cells reconstituted with VCPIP1 WT (Figure S4A), suggesting that VCPIP1 catalytic activity is important for SPRTN acetylation. On the other hand, mutation of K230 did not affect SPRTN deubiquitination (Figures 4C–4D). These results suggest that SPRTN deubiquitination is a prerequisite for SPRTN acetylation, which then promotes SPRTN chromatin association.
To gain further insight into how the acetylation of SPRTN is achieved, we screened a panel of acetyltransferases and identified PCAF and GCN5 as major acetyltransferases for SPRTN (Figure S4B). Depletion of PCAF and GCN5 also inhibited SPRTN acetylation, chromatin association and Top1cc repair upon DPC induction (Figures S4C–S4F). Interestingly, VCPIP1 interacted with PCAF and GCN5 in a DPC damage-inducible manner (Figure S4G). Knockdown of VCPIP1 greatly reduced the interaction between SPRTN and PCAF/GCN5 in the presence of DPCs (Figure S4H), suggesting that VCPIP1 is required to bring acetyltransferases to SPRTN in a DPC-dependent manner.
Taken together, our findings reveal that post-translational modification of SPRTN is critical for its recruitment to chromatin in the presence of DPCs, a process that is tightly controlled by its deubiquitination followed by acetylation. The tandem deubiquitination and acetylation ensures that SPRTN can precisely localize to chromatin to repair DPCs.
VCPIP1 is critical for DPC repair, genomic stability and healthy aging in mice
To explore the physiologic relevance of VCPIP1, we generated a mouse model in which a “knockout” allele was inserted in the Vcpip1 gene (Figures S5A–S5B). Vcpip1−/− mice were born at near Mendelian ratios, although the birth rate was slightly decreased (Figure S5C). MEFs derived from Vcpip1−/− embryo expressed no detectable VCPIP1 protein and showed significant hypersensitivity to DPC-inducing agents, FA, CPT and Cisplatin (Figure 5A). Knockout of Vcpip1 also enhanced the accumulation of Top1cc foci signals (Figures 5B–5C), and impeded spontaneous DPC repair ability (Figure 5D), indicating that VCPIP1 deficiency results in failure to repair DPCs. Vcpip1−/− cells displayed more chromosomal abnormalities than those of Vcpip1+/+ cells (Figures 5E–5F). We also examined micronuclei (an indication of genomic instability) in mouse normochromic erythrocytes (NCEs) in vivo. As shown in Figures 5G and S5D, Vcpip1−/− NCEs showed significantly increased micronuclei compared to wild-type controls. In addition, we isolated the livers of 4-month-old mice and assessed Top1cc foci signals. Significant accumulation of Top1cc foci signals was observed in the livers of Vcpip1−/− mice (Figures 5H–5I), indicating that Vcpip1−/− livers exhibit impaired DPC repair at a young age. Increased γ-H2AX foci signals, a pan marker for DNA damage, was also observed in the livers of 4-month-old Vcpip1−/− mice (Figures S5E–S5F). These data suggest that Vcpip1−/− mice have genomic instability.
Figure 5. VCPIP1 is critical for DPC repair, genomic stability and healthy aging.

(A) Survival assays for Vcpip1+/+ and Vcpip1−/− MEFs exposed to formaldehyde (FA), camptothecin (CPT) and cisplatin (Cis). Error bars indicate mean ± SD of three independent experiments (* p<0.05; ** p<0.01). The expression level of VCPIP1 was assessed by Western blot (top left).
(B-C) Vcpip1+/+ or Vcpip1−/− MEFs were treated with CPT (1 μM, 30 min) and Top1cc signals were detected by immunofluorescence. Nuclei were visualized with DAPI (blue). Representative images are shown in (B). Quantification of foci signals is shown in (C) (** p<0.01). Scale bars, 10 μm.
(D) Vcpip1+/+ or Vcpip1−/− MEFs were treated with FA (250 μM, 1 h) and DPCs were measured as the ratio of crosslinked DNA compared to total DNA (* p<0.05).
(E-F) Vcpip1−/− cells showed genomic instability. (E) Representative images of metaphases prepared from Vcpip1+/+ and Vcpip1−/− MEFs. Arrows indicate chromosomal abnormalities. (F) Quantification of the percentage of chromosomal abnormalities. Metaphase spreads (50) were evaluated for each genotype. Scale bars, 10 μm.
(G) Quantification of micronucleated normochromic erythrocytes from Vcpip1+/+ and Vcpip1−/− mice blood (** p<0.01).
(H-I) Immunohistochemistry of Top1cc signals from the livers of 4-month-old Vcpip1+/+ and Vcpip1−/− mice. Nuclei were visualized with Hoechst (blue). Representative images are shown in (H). Quantification of foci signals is shown in (I) (** p<0.01). Scale bars, 10 μm.
(J) Representative images of the eyes of female Vcpip1+/+ and Vcpip1−/− mice. Note cataract in Vcpip1−/− mice.
(K) Incidence of cataract formation over time in Vcpip1+/+ (n=29) and Vcpip1−/− (n=22) mice (** p<0.01).
(L) Representative images of 6-month-old female Vcpip1+/+ and Vcpip1−/− mice. Note lordokyphosis in the Vcpip1−/− mouse as indicted by the dotted red line.
(M) Incidence of lordokyphosis in Vcpip1+/+ (n=29) and Vcpip1−/− (n=22) mice (* p<0.05).
(N) Inguinal adipose tissue (IAT) from 6-month-old Vcpip1+/+ or Vcpip1−/− mice (n=3) were analyzed by SA-β-Gal staining.
(O) Body weight analysis of female Vcpip1+/+ and Vcpip1−/− mice (n=4) (** p<0.01).
(P) Overall survival curves for Vcpip1+/+ (n=29) and Vcpip1−/− (n=22) mice (* p<0.05).
See also Figure S5.
Premature aging phenotypes have been observed in the Sprtn hypomorphic mice (Maskey et al., 2014) and patients with SPRTN mutations (Lessel et al., 2014). Hence, we asked whether Vcpip1 knockout renders mice prone to premature aging. We first examined the senescence of MEFs during serial passages by SA-β-Gal staining. As shown in Figures S5G–S5H, β-Gal positive cells appeared earlier in passages of Vcpip1−/− cells compare to Vcpip1+/+ cells. Strikingly, by 10 month of age, Vcpip1−/− mice exhibited significantly higher incidence of cataract formation (Figures 5J–5K) and developed lordokyphosis (Figures 5L–5M), while none were observed in age matched cohort of wild-type mice. Furthermore, inguinal adipose tissue (IAT) from Vcpip1−/− mice displayed significant senescence as assessed by SA-β-Gal (Figure 5N). Another aging-related phenotype observed in Vcpip1−/− mice (both male and female) is decreased body weight compared to wild-type mice (Figures 5O and S5I). The median overall survival of Vcpip1−/− mice was reduced in comparison to wild-type mice as well (Figure 5P). These results indicate that loss of VCPIP1 promotes premature aging in mice.
Overall, Vcpip1 knockout mice showed genomic instability and premature aging phenotypes that phenocopied Sprtn hypomorphic mice. These data support an important function of VCPIP1 in DPC repair, maintenance of genomic stability and protection against aging.
DISCUSSION
It has been shown that SPRTN protease activity is tightly regulated by several mechanisms to prevent uncontrolled proteolysis of proteins not at DPC lesions (Kojima and Machida, 2020; Reinking et al., 2020; Stingele et al., 2017). First, SPRTN undergoes the ubiquitin switch (Stingele et al., 2016). Mono-ubiquitinated SPRTN is excluded from chromatin; DPC induction triggers SPRTN deubiquitination, allowing its localization to chromatin. Once SPRTN is recruited to chromatin, the DNA switch can be activated and controls SPRTN enzyme activity in a replication dependent manner (Stingele et al., 2016; Vaz et al., 2016). DPCs bypass by the helicase CMG results in polymerase stalling and ssDNA exposure (Sparks et al., 2019). The polymerase approach targets and recruits SPRTN behind the replication fork (Larsen et al., 2019). Then exposed ssDNA at DPCs fully activates proteolytic activity. SPRTN also switches itself off using its autocleavage activity when DPC repair is complete (Stingele et al., 2016; Vaz et al., 2016). The ubiquitin switch appears to be an early upstream mechanism of SPRTN activation, which can adjust the level of chromatin-accessible SPRTN in response to DPCs. However, the DUB for SPRTN remains unknown and how this modification is regulated is largely unknown. Here we have characterized that VCPIP1, an OTU family DUB, is a SPRTN-interacting protein. Loss of VCPIP1 resulted in cellular hypersensitivity to DPC-inducing agents, as well as Top1cc and DPC accumulation, suggesting that VCPIP1 plays a critical role in DPC repair. We therefore propose that VCPIP1 is the ‘missing’ DUB in the SPRTN-mediated DPC repair pathway. Indeed, mechanistically, DPC induction triggers VCPIP1 activation by DNA damage-responsive kinases ATM/ATR, which then deubiquitinates SPRTN to repair DPCs. Thus, the identification of VCPIP1 as a deubiquitinase for SPRTN elucidates the dynamic regulation of deubiquitination of SPRTN for DPC repair.
From previous work (Lessel et al., 2014; Maskey et al., 2017; Stingele et al., 2016; Vaz et al., 2016) and our findings in this manuscript, there is an emerging question: There is a basal level of deubiquitinated SPRTN under undamaged conditions. Yet, why are these deubiquitinated SPRTN excluded from chromatin? We propose that deubiquitination of SPRTN, although required, is not sufficient for its chromatin localization. Besides deubiquitination, we sought to identify other potential post-translational modifications that are important for SPRTN activation. We found that SPRTN is acetylated in response to DPC induction and showed that K230 acetylation of SPRTN is critical for the recruitment of this protease to chromatin. Intriguingly, the residue K230 of SPRTN is located in the DNA binding region. How acetylation of SPRTN helps its chromatin accumulation is unclear, as DNA is negatively charged and acetylation of SPRTN would theoretically weaken its DNA binding. It is possible that additional factors would be able to bind acetylated SPRTN and help its chromatin accumulation. Of note, deletion of VCPIP1 decreases the level of acetylated SPRTN, suggesting that acetylation occurs after its deubiquitination upon DPCs. Intriguingly, upon DPC induction, the recruitment of acetyltransferases to SPRTN is regulated by VCPIP1, perhaps explaining the coupling of deubiquitination and acetylation and why the basal deubiquitinated SPRTN under undamaged conditions does not become acetylated. Thus, our findings demonstrate a tightly regulated mechanism: the activation of SPRTN not only requires deubiquitination but also subsequent acetylation.
Previous studies showed that hypomorphic mutations in Sprtn cause premature aging and tumorigenesis in mice, while complete Sprtn knockout is embryonically lethal (Maskey et al., 2017; Maskey et al., 2014). Although Vcpip1−/− mice were born normal, they were prone to genomic instability and premature aging, which are similar to phenotypes observed in Sprtn hypomorphic mice. Vcpip1 knockout also caused hypersensitivity to DPC-inducing agents, Top1cc accumulation, chromosomal abnormality and micronuclei formation, suggesting that VCPIP1 is critical in DPC repair and maintenance of genomic stability. The lack of embryonic lethality in Vcpip1 knockout mice might be due to the presence of SPRTN, which can still be partially activated. Overall, our results in mice support our model that VCPIP1 is a DUB that regulates SPRTN for DPC repair.
Our data strongly suggest that VCPIP1-SPRTN signaling axis is the key regulation in DPC repair. As VCPIP1-SPRTN signaling protects cells from DPC induction, it seems likely that inhibition of VCPIP1-SPRTN could be employed to sensitize cells to different kinds of DPC-inducing agents. Taken into consideration that DPCs are widely induced by various chemotherapeutics such as cisplatin, CPT and PARP inhibitors, our results provide a promising strategy for inhibition of this signaling pathway for combination therapies.
In conclusion, we present evidence that VCPIP1 acts as a deubiquitinase for SPRTN, and demonstrate how VCPIP1 is activated and regulated in SPRTN-mediated DPC repair. Moreover, we report that in the presence of DPCs, SPRTN undergoes successive deubiquitination followed by acetylation. We propose a model that sequential deubiquitination and subsequent acetylation of SPRTN ensures its access to chromatin to promote DPC repair process, genomic stability and healthy aging.
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Zhenkun Lou (Lou.Zhenkun@mayo.edu).
Materials Availability
All reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and Code Availability
Original imaging data (including microscopy, gels and western blots) have been deposited to Mendeley Data (http://dx.doi.org/10.17632/5n33btjncz.1).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Vcpip1 knockout mice
Vcpip1 knockout (KO) mice were generated by co-injections of Cas9 2NLS Nuclease, a crRNA/tracrRNA duplex (Synthego), and a single-strand DNA (ssDNA) donor into the cytoplasm of C57BL/6NHsd zygotes. 3–4-week-old C57BL/6NHsd females were used as zygote donors. The ssDNA donors contained stop codons in all three frames to ensure a premature stop when inserted (Gagnon et al., 2014). The cr/tracrRNA targets and ssDNA donor sequences used are as follows: guide targets Vcpip1, TTCGGTCCCTTCGTTTCGAA; ssDNA donor for Vcpip1, CCGGAAAGGATTCTTCGGTCCCTTCGTTTCCTACAACAGCTTAATTAAGGTTTAAACGCCATGACGAAAGGCCCCCCGGAGAAGCCGCCGCCGCCAGAGA. The crRNA/tracrRNA duplexes were prepared following the manufacture’s instruction. The injection mixes contained 0.33 μM Cas9 2NLS Nuclease, 0.33 μM cr/tracrRNA duplex and 50 ng/μL ssDNA donor in 10 mM Tris, 0.1 mM EDTA, pH 7.5. Surviving embryos were transferred to oviducts of pseudo-pregnant ICR mice. Pseudo-pregnant ICR mice were 30–40 g weight females (over 6-week-old). Genotyping was performed by PCR analyses of tail DNA using two primers: forward (5’-AAAAGGAAAGCCATTCGCCCTG-3’), reverse (5’-GAGGCCCATCACCTTTACCAGT-3’). The PCR products are 472 base pair (bp) for the wild-type allele and 507 bp for the KO allele. The PCR products of KO allele can be screened with Pac I/Pme I digestion. All the pups were screened by genotyping with primers flanking the targeting sites followed by sequencing of the PCR products. The reverse primer was using for sequencing. All of the animal procedures were approved by Mayo Clinic Institutional Animal Care and Use Committee.
Cell lines
Cell lines used in this study are listed in the Key Resources Table.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-VCPIP1 | Cell Signaling Technology | Cat# 88153; RRID:AB_2800115 |
| Rabbit polyclonal anti-VCPIP1 (C2C3) | Genetex | Cat# GTX107169; RRID:AB_1952546 |
| Mouse monoclonal anti-human SPRTN | (Maskey et al., 2017; Maskey et al., 2014) | N/A |
| Mouse monoclonal anti-Top1cc | (Patel et al., 2016) | N/A |
| Rabbit polyclonal anti-phospho-SQTQ | Cell Signaling Technology | Cat# 2851; RRID:AB_330318 |
| Rabbit polyclonal anti-Lysine Acetylated (ACK) | ROCKLAND | Cat# 600–401-939; RRID:AB_2205731 |
| Mouse monoclonal anti-Flag | Sigma-Aldrich | Cat# F1804; RRID:AB_262044 |
| Rabbit polyclonal anti-Flag | Sigma-Aldrich | Cat# F7425; RRID:AB_439687 |
| Rabbit polyclonal anti-Calnexin | Sigma-Aldrich | Cat# C4731; RRID:AB_476845 |
| Rabbit monoclonal anti-SP1 (D4C3) | Cell Signaling Technology | Cat# 9389; RRID:AB_11220235 |
| Mouse monoclonal anti-α-Tubulin | Sigma-Aldrich | Cat# T5168; RRID:AB_477579 |
| Rabbit monoclonal anti-Histone H3 (D1H2) | Cell Signaling Technology | Cat# 4499; RRID:AB_10544537 |
| Mouse monoclonal anti-Myc | Cell Signaling Technology | Cat# 2276; RRID:AB_331783 |
| Rabbit monoclonal anti-PCAF (C14G9) | Cell Signaling Technology | Cat# 3378; RRID:AB_2128409 |
| Rabbit monoclonal anti-GCN5 (C26A10) | Cell Signaling Technology | Cat# 3305; RRID:AB_2128281 |
| Rabbit monoclonal anti-phospho-Chk1 (Ser345) (133D3) | Cell Signaling Technology | Cat# 2348; RRID:AB_331212 |
| Rabbit monoclonal anti-phospho--Chk2 (Thr68) (C13C1) | Cell Signaling Technology | Cat# 2197; RRID:AB_2080501 |
| Mouse monoclonal anti-phospho-Histone H2A.X (Ser139) | Sigma-Aldrich | Cat# 05–636; RRID:AB_309864 |
| Mouse monoclonal anti-β-Actin | Sigma-Aldrich | Cat# A1978; RRID:AB_476692 |
| Rabbit polyclonal anti-VCP | Santa Cruz | Cat# sc-20799; RRID:AB_793930 |
| Goat Anti-Mouse IgG (H+L) HRP | Jackson ImmunoResearch | Cat# 115–035-146; RRID:AB_2307392 |
| Goat anti-Rabbit IgG (H+L) HRP | Jackson ImmunoResearch | Cat# 111–035-144; RRID:AB_2307391 |
| Goat Anti-Mouse IgG (H+L) HRP,light chain specific | Jackson ImmunoResearch | Cat# 115–035-174; RRID:AB_2338512 |
| Goat anti-Rabbit IgG (H+L) HRP,light chain specific | Jackson ImmunoResearch | Cat# 211–032-171; RRID:AB_2339149 |
| Alexa Fluor 488-labeled Goat anti-Mouse IgG (H+L) | Jackson ImmunoResearch | Cat# 115–545-062; RRID:AB_2338845 |
| Rhodamine Red-X-labeled Goat anti-Mouse IgG (H+L) | Jackson ImmunoResearch | Cat# 115–295-146; RRID:AB_2338766 |
| Normal Rabbit IgG | Sigma-Aldrich | Cat# 12–370; RRID:AB_145841 |
| FITC Rat anti-Mouse CD71 | Thermo Fisher Scientific | Cat# 11–0711-82; RRID:AB_465124 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Formaldehyde | Sigma-Aldrich | Cat# 8.18708 |
| Camptothecin | Sigma-Aldrich | Cat# C9911 |
| Cisplatin | Sigma-Aldrich | Cat# P4394 |
| ATM kinase inhibitor KU55933 | Abcam | Cat# ab120637 |
| ATR kinase inhibitor VX970 | National Cancer Institute (Rockville, MD) | N/A |
| ATPase VCP/p97 Inhibitor NMS-873 | TOCRIS | Cat# 6180 |
| (Z)-4-Hydroxytamoxifen (4-OHT) | Sigma-Aldrich | Cat# H7904 |
| Protein A/G PLUS-Agarose | Santa Cruz | Cat# sc-2003 |
| Anti-Flag M2 Affinity Gel | Sigma-Aldrich | Cat# A2220 |
| Colcemid | Gibco | Cat# 15210040 |
| Heparin | Calbiochem | Cat# 375095 |
| Isopropyl β-D-1-thiogalactopyranoside (IPTG) | Thermo Fisher Scientific | Cat# 15529019 |
| DAPI | Thermo Fisher Scientific | Cat# D1306 |
| Heochst 33258 | Thermo Fisher Scientific | Cat# H3569 |
| PI/RNase Staining Solution | Thermo Fisher Scientific | Cat# F10797 |
| Ub-AMC | Boston biochem | Cat# U-550 |
| Critical Commercial Assays | ||
| QuikChange Site-Directed Mutagenesis Kit | Agilent Technologies | Cat# 200518 |
| Duo-link in situ PLA Kit | Sigma-Aldrich | Cat# DUO92101 |
| Subcellular Protein Fractionation Kit | Thermo Fisher Scientific | Cat# 78840 |
| Senescence β-Galactosidase Staining Kit | Cell Signaling Technology | Cat# 9860 |
| Qubit dsDNA HS Assay Kit | Thermo Fisher Scientific | Cat# Q32851 |
| Deposited Data | ||
| Mendeley dataset | This paper | http://dx.doi.org/10.17632/5n33btjncz.1 |
| Experimental Models: Cell Lines | ||
| Human: HEK293T | ATCC | Cat# CRL-11268; RRID:CVCL_1926 |
| Human: U2OS | ATCC | Cat# HTB-96; RRID:CVCL_0042 |
| Human: HepG2 | ATCC | Cat# HB-8065; RRID:CVCL_0027 |
| Human: HCT-116 | ATCC | Cat# CCL-247; RRID:CVCL_0291 |
| MEF: SprtnF/F;Cre-ERT2 | (Maskey et al., 2017; Maskey et al., 2014) | N/A |
| MEF: Vcpip1+/+ | This paper | N/A |
| MEF: Vcpip1−/− | This paper | N/A |
| Experimental Models: Organisms/Strains | ||
| Mouse: Vcpip1+/+. C57BL/6NHsd | This paper | N/A |
| Mouse: Vcpip1−/−. C57BL/6NHsd | This paper | N/A |
| Oligonucleotides | ||
| Primers for genotyping Vcpip1 allele: Forward, 5’-AAAAGGAAAGCCATTCGCCCTG-3’; Reverse, 5’-GAGGCCCATCACCTTTACCAGT-3’ | This paper | N/A |
| shVCPIP1–1: 5’-CCGGGTGCTACATCGTCCTATTATTCTCGAGAATAATAGGACGATGTAGCACTTTTTG-3’ | Sigma-Aldrich | TRCN0000236141 |
| shVCPIP1–2: 5’-CCGGGAAAGTTGTCCACACTATATTCTCGAGAATATAGTGTGGACAACTTTCTTTTTG-3’ | Sigma-Aldrich | TRCN0000236140 |
| shSPRTN-1: 5’-CCGGCTATGTCAAACGAGCTACTAACTCGAGTTAGTAGCTCGTTTGACATAGTTTTTG-3’ | Sigma-Aldrich | TRCN0000073310 |
| shSPRTN-2: 5’-CCGGGTACAACCACAGCTCAGAATTCTCGAGAATTCTGAGCTGTGGTTGTACTTTTTG-3’ | Sigma-Aldrich | TRCN0000073311 |
| shVCP-1: 5’-CCGGGATGGATGAATTGCAGTTGTTCTCGAGAACAACTGCAATTCATCCATCTTTTT-3’ | Sigma-Aldrich | TRCN0000004252 |
| shVCP-2: 5’-CCGGAGGGAGGTAGATATTGGAATTCTCGAGAATTCCAATATCTACCTCCCTTTTTT-3’ | Sigma-Aldrich | TRCN0000004253 |
| shPCAF-1: 5’- CCGGGCAGACTTACAGCGAGTCTTTCTCGAGAAAGACTCGCTGTAAGTCTGCTTTTT-3’ | Sigma-Aldrich | TRCN0000018528 |
| shPCAF-2: 5’- CCGGGCAGATACCAAACAAGTTTATCTCGAGATAAACTTGTTTGGTATCTGCTTTTT-3’ | Sigma-Aldrich | TRCN0000018529 |
| ShGCN5–1: 5’- CCGGGCGCATGCCTAAGGAGTATATCTCGAGATATACTCCTTAGGCATGCGCTTTTTG-3’ | Sigma-Aldrich | TRCN0000307319 |
| ShGCN5: 5’- CCGGGCTGAACTTTGTGCAGTACAACTCGAGTTGTACTGCACAAAGTTCAGCTTTTTG-3’ | Sigma-Aldrich | TRCN0000286981 |
| Recombinant DNA | ||
| PLVX3-FLAG-VCPIP1/ VCPIP1(C218A)/ VCPIP1(S1207A) | This paper | N/A |
| PLVX3-FLAG-VCPIP1-N(1–361aa)/ VCPIP1-C(362–1222aa)/ VCPIP1-ΔOTU(Δ208–361aa) | This paper | N/A |
| PLVX3-FLAG-SPRTN/ SPRTN(K230R)/ SPRTN(K230Q) | This paper | N/A |
| PEFF-MYC-SPRTN | This paper | N/A |
| PGEX-4T-2-VCPIP1/VCPIP1(C218A) | This paper | N/A |
| MYC-PCAF/GCN5/Tip60/MOF/P300 | Gift from Dr. Jun Huang (Zhejiang University) | N/A |
| Software and Algorithms | ||
| ImageJ | NIH | https://imagej.nih.gov/ij/ |
| Prism 8 | GraphPad | https://www.graphpad.com/ |
| FlowJo (10.1) | FlowJo LLC | https://www.flowjo.com/ |
Primary Vcpip1+/+ and Vcpip1−/− mouse embryonic fibroblasts (MEFs) were isolated from E13.5 embryos produced by crossing heterozygous Vcpip1 mice (2-month-old) and immortalized by serial passaging. HEK293T and MEFs were cultured in DMEM medium with 10% fetal bovine serum (FBS). HepG2 was cultured in EMEM medium with 10% FBS. U2OS and HCT-116 cells were cultured in McCoy’s 5A and RPMI1640 with 10% FBS, respectively. To induce the knockout allele in SprtnF/F;Cre-ERT2 MEFs, cells were treated with 2 μM (Z)-4-Hydroxytamoxifen (4-OHT) dissolved in methanol (MeOH) for 2 days.
METHOD DETAILS
Plasmids, transfection and lentiviral infection
HA-FLAG-VCPIP1 was purchased from Addgene (Plasmid #22592). VCPIP1 C218A was a gift from Dr. Allan M. Weissman (National Cancer Institute). These plasmids were subcloned into PLVX3 lentiviral or pGEX-4T-2 vectors (Clontech). Flag-SPRTN was described previously (Maskey et al., 2014) and subcloned into PLVX3 lentiviral plasmid. VCPIP1 S1207A, SPRTN K230R, and SPRTN K230Q mutants were generated using a QuikChange Site-Directed Mutagenesis Kit (Agilent Technologies). VCPIP1 truncations were generated by PCR and subcloned into PLVX3 lentiviral plasmid. VCPIP1 shRNA (NM_025054), SPRTN shRNA (NM_032018), VCP shRNA (NM_007126), PCAF shRNA (NM_003884) and GCN5 shRNA (NM_021078) were purchased from Sigma-Aldrich. Cells were transfected with TransIT-X2 (Mirus) according to manufacturer’s instructions. Lentiviral infection of cells was performed as described previously (Huang et al., 2017).
Immunofluorescence
For standard immunofluorescence staining, cells were cultured on coverslips 24 h before experiments. Cells were fixed with 4% paraformaldehyde, permeabilized with 0.5% Triton X-100, incubated with primary antibody and subsequently incubated with the corresponding Alexa Fluor 488 or 594-conjugated secondary IgG antibodies. Cellular nuclei were stained with DAPI. The cover slips were mounted onto glass slides with anti-fade solution and visualized using a Nikon eclipse 80i fluorescence microscope.
For Top1cc immunofluorescence, experiments were performed as described previously (Maskey et al., 2017; Patel et al., 2016). Briefly, U2OS or MEF cells were cultured on coverslips and treated with 1 μM camptothecin (CPT) for 30 min. Cells were washed in precooled PBS, fixed with 4% paraformaldehyde for 15 min on ice, permeabilized with 0.25% Triton X-100 for 15 min on ice and treated with 1% sodium dodecyl sulfate (SDS) for 5 min at RT. Samples were then washed five times in wash buffer (0.1% Triton X-100, 0.1% BSA in PBS), blocked with blocking buffer (10% milk in 150 mM NaCl and 10 mM Tris-HCl, pH 7.4) and then incubated with anti-Top1cc antibody (1:100) for 1 h. Samples were washed six times with wash buffer and incubated with secondary IgG antibody for 30 minutes. Cellular nuclei were stained with DAPI. The cover slips were mounted onto glass slides with anti-fade solution and visualized using a Nikon eclipse 80i fluorescence microscope. For immunostaining of tissue samples, fresh tissues were embedded in optimal cutting temperature compound (Sakura Finetek), frozen in liquid nitrogen, sectioned onto slides and fixed with 4% paraformaldehyde for 15 min at 4 °C. The immunostaining followed the procedures described above for the cell lines. Cellular nuclei were stained with Hoechst 33258.
Immunoprecipitation and Western blotting
Cells were lysed with NETN buffer (20 mM Tris-HCl, pH 8.0, 100 mM NaCl, 1 mM EDTA, 0.5% Nonidet P-40) containing 50 mM β-glycerophosphate, 10 mM NaF and 1 mg mL−1 each of pepstatin A and aprotinin. Whole cell lysates were centrifuged at 12000 rpm for 10 min. Cell lysates were incubated with antibody and protein A/G PLUS agarose beads (Santa Cruz) or Anti-Flag M2 Affinity Gel (Sigma) for 2 h or overnight at 4 °C. After incubation, the immunocomplexes were washed with NETN buffer and separated by SDS–PAGE. Western blotting was performed following standard procedures.
Proximity ligation assay (PLA)
Cells were washed in precooled PBS, fixed with 4% paraformaldehyde on ice for 15 min, permeabilized with 0.1% Triton X-100 on ice for 10 min. Then PLA was performed by a Duo-link in situ PLA kit (Sigma) according to the manufacturer’s protocol. Briefly, samples were blocked in blocking solution at 37 °C for 1 h and then incubated with the mixture of primary antibodies (1:1000) at 4 °C overnight. Then the probes were incubated at 37 °C for 1 h, followed by hybridization, ligation, amplification, and detection. Cellular nuclei were stained with DAPI. The cover slips were mounted onto glass slides with anti-fade solution and visualized using a Nikon eclipse 80i fluorescence microscope.
Chromatin fractionation
Chromatin fractionation was performed as described previously (Lou et al., 2006). In brief, cells were collected and resuspended in low salt buffer (10mM Tris-HCl, pH 7.4, 0.2 mM MgCl2, 50 mM β-glycerophosphate, 10 mM NaF and 1 mg mL−1 each of pepstatin A and aprotinin) containing 1% Triton X-100 on ice for 15 min. After centrifugation (14000 rpm, 10 min), the supernatant contained the soluble proteins, and the pellet contained the chromatin-bound proteins. The pellets were then resuspended in 0.2 N HCl on ice for 15 min, sonicated and finally neutralized with 1 M Tris-HCl, pH 8.0. The amounts of each fraction were analyzed by SDS-PAGE and Western blotting.
Subcellular fractionation
Subcellular fractionation was performed by a subcellular protein fractionation kit (Thermo Fisher) according to the manufacturer’s instruction. The amounts of each fraction were analyzed by SDS-PAGE and Western blotting. Fractionation efficiency was determined by assessing α-Tubulin (cytosolic), Calnexin (ER membrane), SP1 (nuclear soluble) and H3 (chromatin) levels, respectively.
Senescence assay
Senescent cells were identified by a senescence-associated β-galactosidase kit (SA-β-Gal) (Cell Signaling) according to the manufacturer’s instruction. For senescence induced by spontaneous immortalization, MEFs at passages 3, 6, and 9 were harvested and fixed for SA-β-Gal staining. For determining the SA-β-Gal activity in mice, inguinal adipose tissue (IAT) was isolated from mice and fixed for SA-β-Gal staining.
DNA-protein crosslink detection
The detection of DNA-protein crosslinks was performed as described previously (Stingele et al., 2016; Vaz et al., 2016). Briefly, MEFs were pretreated with 250 μM formaldehyde (FA) for 1 h. Cells were lysed in denaturing lysis buffer (2% SDS, 20 mM Tris/HCl pH 7.5) and stored at −80 °C until further processing. After collecting all samples, lysates were thawed at 55 °C and sonicated. Cellular proteins were then precipitated in assay buffer (200 mM KCl, 20 mM Tris, pH 7.5) and incubated on ice for 5 min. The precipitate was pelleted by centrifugation and the supernatant was used for quantifying soluble DNA. The pellet was resuspended in assay buffer and resolved by shaking at 55 °C for 5 min, cooled on ice for 5 min, and precipitated by centrifugation. The wash procedure outlined above was repeated three times and finally the pellet was resuspended in 400 μL of assay buffer. Proteins were digested by adding 0.2 mg/mL Proteinase K at 55 °C for 2 h. 10 μL BSA (50 mg/mL) was added to samples, cooled on ice for 5 min and then centrifuged. The final supernatant was used for quantifying crosslinked DNA. Soluble and crosslinked DNA were quantified by the Qubit dsDNA HS assay. The amount of DPCs was calculated as the ratio between DNA precipitated by SDS/KCl to total DNA (SDS/KCl precipitated plus soluble DNA).
Colony formation
800–1000 cells were plated in triplicate in each well of 6 well plates. 16 hours later, cells were treated with either formaldehyde (FA), camptothecin (CPT) or cisplatin (Cis) and left for 10–14 days at 37 °C to allow colony formation. Colonies were stained with Giemsa solution (Sigma) and counted. Results were normalized to plating efficiencies.
Metaphase spreads
Vcpip1+/+ and Vcpip1−/− MEFs were incubated with 20 ng/mL colcemid (KaryoMAX, GibcoBRL) at 37 °C for 2 h. Cells were harvested and suspended in prewarmed hypotonic buffer (75 mM KCl ) at 37 °C for 25 min which induced cellular distention. After centrifugation, cells were fixed with Carnoy’s buffer (methanol: acetic acid in 3:1 ratio) at RT for 10 min. Fixed cells were centrifuged and then resuspended in Carnoy’s buffer twice. The final supernatant, containing the fixed mitotic cells, were dropped on to slides and dried for 10 min. Slides were stained with Giemsa solution (Sigma).
Micronucleus assay
The micronucleus assay was performed as described previously (Garaycoechea et al., 2018). Briefly, mice blood (8–12 weeks of age) was mixed with 100 μL PBS containing 1,000 U mL−1 of heparin (Calbiochem). Blood suspension was then added to 1 mL of methanol and stored at −80 °C overnight until further processing. 1 mL of fixed blood cells was washed with 6 mL of bicarbonate buffer (0.9% NaCl, 5.3 mM NaHCO3). Cells were suspended in 100 μL of bicarbonate buffer with 1 μL of FITC-conjugated CD71 antibody (Thermo Fisher) at 4 °C for 45 min. After centrifugation, pellets were washed with bicarbonate buffer and resuspended in 5 μg mL−1 PI/RNase Staining Solution (Thermo Fisher). Samples were analyzed immediately on an Nxt Attune FACS analyzer (Thermo Fisher) and data analyzed with FlowJo.
Recombinant protein expression and in vitro deubiquitination assay
The recombinant GST-VCPIP1 and VCPIP1 C218A were induced in BL21 cells with 0.2 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) at 18.5 °C for 20 h. The cells were then lysed using appropriate lysis buffer supplemented with Protease Inhibitor Cocktail (Roche). For the preparation of the substrate protein, HEK293T cells were transfected with Flag-SPRTN expression vector. The mono- ubiquitinated SPRTN proteins were purified from the cell extracts with anti-Flag beads. For in vitro deubiquitination assay, mono-ubiquitinated SPRTN proteins were incubated with recombinant VCPIP1 proteins in a deubiquitination buffer (50 mM Tris-HCl pH 8.0, 50 mM NaCl, 1 mM EDTA, 10 mM DTT, 5% glycerol) at 30 °C for 4 h.
Ub-AMC assay
In vitro deubiquitinase enzymatic assays using Ub-AMC (Boston Biochem) were performed in 50 μL reaction buffer (20 mM HEPES-KOH pH 7.8, 20 mM NaCl, 0.1 mg/mL ovalbumin, 0.5 mM EDTA, and 10 mM DTT) at 25 °C. Fluorescence was monitored in an Infinite® M1000 PRO Fluorometer (TECAN).
QUANTIFICATION AND STATISTICAL ANALYSIS
All experiments were repeated at least 3 independent times otherwise stated in the figure legend. Two-tailed unpaired Student’s t tests or Mann Whitney U tests were applied for comparisons between two groups. Statistical analyses were performed using GraphPad Prism software. The data are presented as the means ± SEM except where stated otherwise. The differences with *p < 0.05 or **p < 0.01 were considered statistically significant.
KEY RESOURCES TABLE
Supplementary Material
Highlights.
DPC-induced deubiquitination of SPRTN is dependent on VCPIP1
VCPIP1 is activated by ATM/ATR in response to DPCs
SPRTN activation is regulated by sequential deubiquitination and acetylation
Loss of VCPIP1 results in genomic instability and premature aging
ACKNOWLEDGEMENTS
We thank the Mayo Clinic Transgenic and Knockout Core for generating Vcpip1 knockout mice. We thank Allan M. Weissman (National Cancer Institute) for providing VCPIP1 constructs. We thank Scott H. Kaufmann (Mayo Clinic) for providing anti-Top1cc antibody. We thank members of the Lou lab for comments and discussion throughout the project. This work was supported in part by grant from the National Institutes of Health (CA203561, Z.L.) and Mayo Edward C. Kendall Fellowship in Biochemistry Award (J.H.).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Barker S, Weinfeld M, and Murray D (2005). DNA-protein crosslinks: their induction, repair, and biological consequences. Mutat Res 589, 111–135. [DOI] [PubMed] [Google Scholar]
- Conaway CC, Whysner J, Verna LK, and Williams GM (1996). Formaldehyde mechanistic data and risk assessment: endogenous protection from DNA adduct formation. Pharmacol Ther 71, 29–55. [DOI] [PubMed] [Google Scholar]
- Davis EJ, Lachaud C, Appleton P, Macartney TJ, Nathke I, and Rouse J (2012). DVC1 (C1orf124) recruits the p97 protein segregase to sites of DNA damage. Nat Struct Mol Biol 19, 1093–1100. [DOI] [PubMed] [Google Scholar]
- Duxin JP, Dewar JM, Yardimci H, and Walter JC (2014). Repair of a DNA-protein crosslink by replication-coupled proteolysis. Cell 159, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fielden J, Wiseman K, Torrecilla I, Li S, Hume S, Chiang SC, Ruggiano A, Narayan Singh A, Freire R, Hassanieh S, et al. (2020). TEX264 coordinates p97- and SPRTN-mediated resolution of topoisomerase 1-DNA adducts. Nat Commun 11, 1274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu YV, Yardimci H, Long DT, Ho TV, Guainazzi A, Bermudez VP, Hurwitz J, van Oijen A, Scharer OD, and Walter JC (2011). Selective bypass of a lagging strand roadblock by the eukaryotic replicative DNA helicase. Cell 146, 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon JA, Valen E, Thyme SB, Huang P, Akhmetova L, Pauli A, Montague TG, Zimmerman S, Richter C, and Schier AF (2014). Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One 9, e98186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garaycoechea JI, Crossan GP, Langevin F, Mulderrig L, Louzada S, Yang F, Guilbaud G, Park N, Roerink S, Nik-Zainal S, et al. (2018). Alcohol and endogenous aldehydes damage chromosomes and mutate stem cells. Nature 553, 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghosal G, Leung JW, Nair BC, Fong KW, and Chen J (2012). Proliferating cell nuclear antigen (PCNA)-binding protein C1orf124 is a regulator of translesion synthesis. J Biol Chem 287, 34225–34233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang JZ, Chen M, Chen, Gao XC, Zhu S, Huang H, Hu M, Zhu H, and Yan GR (2017). A Peptide Encoded by a Putative lncRNA HOXB-AS3 Suppresses Colon Cancer Growth. Mol Cell 68, 171–184 e176. [DOI] [PubMed] [Google Scholar]
- Ide H, Shoulkamy MI, Nakano T, Miyamoto-Matsubara M, and Salem AM (2011). Repair and biochemical effects of DNA-protein crosslinks. Mutat Res 711, 113–122. [DOI] [PubMed] [Google Scholar]
- Kojima Y, and Machida YJ (2020). DNA-Protein Crosslinks from Environmental Exposure: Mechanisms of Formation and Repair. Environ Mol Mutagen. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen NB, Gao AO, Sparks JL, Gallina I, Wu RA, Mann M, Raschle M, Walter JC, and Duxin JP (2019). Replication-Coupled DNA-Protein Crosslink Repair by SPRTN and the Proteasome in Xenopus Egg Extracts. Mol Cell 73, 574–588 e577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessel D, Vaz B, Halder S, Lockhart PJ, Marinovic-Terzic I, Lopez-Mosqueda J, Philipp M, Sim JC, Smith KR, Oehler J, et al. (2014). Mutations in SPRTN cause early onset hepatocellular carcinoma, genomic instability and progeroid features. Nat Genet 46, 1239–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li FX, Raczynska JE, Chen Z, and Yu HT (2019). Structural Insight into DNA-Dependent Activation of Human Metalloprotease Spartan. Cell Rep 26, 3336–+. [DOI] [PubMed] [Google Scholar]
- Lopez-Mosqueda J, Maddi K, Prgomet S, Kalayil S, Marinovic-Terzic I, Terzic J, and Dikic I (2016). SPRTN is a mammalian DNA-binding metalloprotease that resolves DNA-protein crosslinks. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou Z, Minter-Dykhouse K, Franco S, Gostissa M, Rivera MA, Celeste A, Manis JP, van Deursen J, Nussenzweig A, Paull TT, et al. (2006). MDC1 maintains genomic stability by participating in the amplification of ATM-dependent DNA damage signals. Mol Cell 21, 187–200. [DOI] [PubMed] [Google Scholar]
- Maskey RS, Flatten KS, Sieben CJ, Peterson KL, Baker DJ, Nam HJ, Kim MS, Smyrk TC, Kojima Y, Machida Y, et al. (2017). Spartan deficiency causes accumulation of Topoisomerase 1 cleavage complexes and tumorigenesis. Nucleic Acids Res 45, 4564–4576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maskey RS, Kim MS, Baker DJ, Childs B, Malureanu LA, Jeganathan KB, Machida Y, van Deursen JM, and Machida YJ (2014). Spartan deficiency causes genomic instability and progeroid phenotypes. Nat Commun 5, 5744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morocz M, Zsigmond E, Toth R, Enyedi MZ, Pinter L, and Haracska L (2017). DNA-dependent protease activity of human Spartan facilitates replication of DNA-protein crosslink-containing DNA. Nucleic Acids Res 45, 3172–3188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosbech A, Gibbs-Seymour I, Kagias K, Thorslund T, Beli P, Povlsen L, Nielsen SV, Smedegaard S, Sedgwick G, Lukas C, et al. (2012). DVC1 (C1orf124) is a DNA damage-targeting p97 adaptor that promotes ubiquitin-dependent responses to replication blocks. Nature Structural & Molecular Biology 19, 1084–+. [DOI] [PubMed] [Google Scholar]
- Nakano T, Miyamoto-Matsubara M, Shoulkamy MI, Salem AM, Pack SP, Ishimi Y, and Ide H (2013). Translocation and stability of replicative DNA helicases upon encountering DNA-protein cross-links. J Biol Chem 288, 4649–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortega-Atienza S, Wong VC, DeLoughery Z, Luczak MW, and Zhitkovich A (2016). ATM and KAT5 safeguard replicating chromatin against formaldehyde damage. Nucleic Acids Res 44, 198–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AG, Flatten KS, Peterson KL, Beito TG, Schneider PA, Perkins AL, Harki DA, and Kaufmann SH (2016). Immunodetection of human topoisomerase I-DNA covalent complexes. Nucleic Acids Res 44, 2816–2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pommier Y (2006). Topoisomerase I inhibitors: camptothecins and beyond. Nat Rev Cancer 6, 789–802. [DOI] [PubMed] [Google Scholar]
- Quievryn G, and Zhitkovich A (2000). Loss of DNA-protein crosslinks from formaldehyde-exposed cells occurs through spontaneous hydrolysis and an active repair process linked to proteosome function. Carcinogenesis 21, 1573–1580. [PubMed] [Google Scholar]
- Reinking HK, Hofmann K, and Stingele J (2020). Function and evolution of the DNA-protein crosslink proteases Wss1 and SPRTN. DNA Repair (Amst) 88, 102822. [DOI] [PubMed] [Google Scholar]
- Sparks JL, Chistol G, Gao AO, Raschle M, Larsen NB, Mann M, Duxin JP, and Walter JC (2019). The CMG Helicase Bypasses DNA-Protein Cross-Links to Facilitate Their Repair. Cell 176, 167–181 e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, Bellelli R, Alte F, Hewitt G, Sarek G, Maslen SL, Tsutakawa SE, Borg A, Kjaer S, Tainer JA, et al. (2016). Mechanism and Regulation of DNA-Protein Crosslink Repair by the DNA-Dependent Metalloprotease SPRTN. Mol Cell 64, 688–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stingele J, Bellelli R, and Boulton SJ (2017). Mechanisms of DNA-protein crosslink repair. Nat Rev Mol Cell Biol 18, 563–573. [DOI] [PubMed] [Google Scholar]
- Stingele J, Schwarz MS, Bloemeke N, Wolf PG, and Jentsch S (2014). A DNA-dependent protease involved in DNA-protein crosslink repair. Cell 158, 327–338. [DOI] [PubMed] [Google Scholar]
- Tretyakova NY, Groehler A.t., and Ji S (2015). DNA-Protein Cross-Links: Formation, Structural Identities, and Biological Outcomes. Acc Chem Res 48, 1631–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai YC, Kotiya A, Kiris E, Yang M, Bavari S, Tessarollo L, Oyler GA, and Weissman AM (2017). Deubiquitinating enzyme VCIP135 dictates the duration of botulinum neurotoxin type A intoxication. Proc Natl Acad Sci U S A 114, E5158–E5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchiyama K, Jokitalo E, Kano F, Murata M, Zhang X, Canas B, Newman R, Rabouille C, Pappin D, Freemont P, et al. (2002). VCIP135, a novel essential factor for p97/p47-mediated membrane fusion, is required for Golgi and ER assembly in vivo. J Cell Biol 159, 855–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaz B, Popovic M, Newman JA, Fielden J, Aitkenhead H, Halder S, Singh AN, Vendrell I, Fischer R, Torrecilla I, et al. (2016). Metalloprotease SPRTN/DVC1 Orchestrates Replication-Coupled DNA-Protein Crosslink Repair. Mol Cell 64, 704–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Satoh A, Warren G, and Meyer HH (2004). VCIP135 acts as a deubiquitinating enzyme during p97-p47-mediated reassembly of mitotic Golgi fragments. J Cell Biol 164, 973–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong VC, Cash HL, Morse JL, Lu S, and Zhitkovich A (2012). S-phase sensing of DNA-protein crosslinks triggers TopBP1-independent ATR activation and p53-mediated cell death by formaldehyde. Cell Cycle 11, 2526–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Original imaging data (including microscopy, gels and western blots) have been deposited to Mendeley Data (http://dx.doi.org/10.17632/5n33btjncz.1).