

Abstract
Most terpene synthase reactions follow Markovnikov rules for formation of high energy carbenium ion intermediates. However, there are notable exceptions. For example, pentalenene synthase (PS) undergoes an initial anti-Markovnikov cyclization reaction followed by a 1,2-hydride shift to form an intermediate humulyl cation with positive charge on the secondary carbon C9 of the farnesyl diphosphate substrate. The mechanism by which these enzymes stabilize and guide regioselectivity of secondary carbocations has not heretofore been elucidated. In an effort to better understand these reactions, we grew crystals of apo-PS, soaked them with the non-reactive substrate analog 12,13-difluorofarnesyl diphosphate, and solved the x-ray structure of the resulting complex at 2.2 Å resolution. The most striking feature of the active site structure is that C9 is perfectly positioned to make a C-H…π interaction with the side chain benzene ring of residue F76; this would enhance hyperconjugation to stabilize a developing cation at C10 and thus support the anti-Markovnikov regioselectivity of the cyclization. The benzene ring is also positioned to catalyze H migration to C10 and stabilize a C9 carbocation. On the opposite face of C9, further cation stabilization is possible via interactions with the main chain carbonyl of I177 and neighboring intramolecular C6,C7-double bond. Mutagenesis experiments also support a role for residue 76 in these interactions, but most interesting is the F76W mutant, whose crystal structure clearly shows carbons C9 and C10 centered above the fused benzene and pyrrole rings of the indole side chain, respectively, such that a carbocation at either position could be stabilized in this complex, and two anti-Markovnikov products, pentalenene and humulene, are formed. Finally, we show that there is a rough correlation (although not absolute) of an aromatic side chain (F or Y) at position 76 in related terpene synthases from Streptomyces that catalyze similar anti-Markovnikov addition reactions.
Terpenes comprise the largest class of natural products with over 80,000 unique members identified to date.1–4 They are found largely in the plant world, but also serve important roles in animal and bacterial metabolism. They are important commercially and medicinally, and increasingly viewed as viable candidates for renewable biofuels.5
The committed step in the biosynthesis of all terpenes is catalyzed by a terpene synthase (or terpene cyclase).6 Typically, these remarkable enzymes take a linear, prenyl substrate and convert it into a cyclic hydrocarbon by generating and controlling the reactivity of high energy carbenium ion intermediates in reactions that often involve complicated carbon skeleton rearrangements, including methyl and hydride migrations. Class I terpene synthases employ prenyl diphosphate substrates and use metal ions to facilitate dissociation of the allylic diphosphate.6 Cyclization occurs via attack by π electrons from double bonds present in the prenyl chain.7, 8 The reactions are typically terminated by deprotonation to form an olefin or cation capture by a nucleophile in the active site such as a water molecule. Downstream enzymes, mostly P450s and acyl transferases, then modify the hydrocarbon scaffold to increase functionalization of the resulting end product in the pathway.9–11
Pentalenene synthase (PS) is a class I sesquiterpene synthase that catalyzes the committed step in biosynthesis of pentalenolactone antibiotics in several species of Streptomycetes.12, 13 The x-ray crystal structure of the enzyme from Streptomyces UC5319 was first solved by Christianson and coworkers in 1997.14 It is a 38 kDa monomer composed of a single domain with typical class I α-helical terpenoid cyclase fold and contains conserved Asp-rich (DDXXD) and NSE/DTE motifs for binding of the diphosphate moiety of the substrate along with three Mg2+ ions.6, 14 The Mg2+-dependent reaction mechanism has been studied extensively2, 15, 16 and begins with anti-Markovnikov attack of the 10,11 double bond on C1 of the farnesyl diphosphate substrate by either direct displacement of the diphosphate or through ionization and intermediary formation of a resonance-stabilized allylic carbenium ion on C1 (Fig. 1). This step in the reaction initially produces humulyl cation A, with positive charge located on the secondary carbon C10, and then humulyl cation B, following a 1,2-hydride shift to move the charge to C9. Concerted, asynchronous attack of the C2,C3 and C6,C7 alkenes leads to the protoilludyl cation intermediate,17–19 which is thought then to undergo rearrangement to the Z-secoilludyl cation, followed by a 1,2-hydride shift, transannular cyclization, and finally deprotonation to produce pentalenene (Fig. 1). This series of reaction steps produces a stereochemically dense, enantiomerically pure triquinane structure containing four chiral carbons.
Fig. 1. Comparison of reaction intermediates for two sesquiterpene synthases: germacrene A synthase and pentalenene synthase.

Both reactions begin with ionization or direct displacement of the allylic diphosphate (FPP substrate) with attack of the 10,11 double bond. Germacrene A synthase favors attack of the π electrons to form the expected Markovnikov product with positive charge on the more highly substituted carbon (C11 of the germacradienyl cation). In contrast, pentalenene synthase directs anti-Markovnikov attack such that the positive charge develops on the less substituted secondary carbon (C10) to produce first humulyl cation A, and then, following a 1,2-hydride shift, humulyl cation B with carbocation at position C9.
Our interest in this reaction is focused on how the enzyme directs anti-Markovnikov attack of C11 on C1 to produce first humulyl cation A, with the carbenium ion located on the secondary carbon C10, and then, following the 1,2-hydride shift, humulyl cation B, with the carbenium ion located on the secondary carbon at C9. How is it that the enzyme selects a pathway with the less stable secondary carbocations rather than allowing Markovnikov attack of C10 on C1 to produce the much more stable tertiary cation at C11, as for example happens in solution and with most of the other sesquiterpene synthase reactions (e.g., germacrene A synthase in Fig. 1)?20–23 How does the enzyme stabilize charge on these secondary carbon atoms? Despite the fact that pentalenene synthase was one of the first terpene cyclases to have its structure determined by x-ray crystallography,14 the enzymatic mechanism underlying regioselectivity in formation of these unstable carbenium ion intermediates on secondary carbon atoms is completely unknown.
RESULTS
To explore the mechanism underlying the initial anti-Markovnikov cyclization in the PS reaction, we first prepared a non-reactive substrate analog, 12,13-difluorofarnesyl diphosphate (DFFPP), by enzymatic elongation of 8,9-difluorogeranyl diphosphate (DFGPP)24, 25 with isopentenyl diphosphate (IPP; Fig. 2). The two electronegative fluorine atoms in DFFPP are positioned to inhibit reactivity of the π electrons in the C10,C11-double bond and thus block the initial cyclization reaction. Reaction of DFFPP with PS produced no hexane-extractable products, including those from elimination; apparently the diphosphate does not ionize in the absence of a competent nucleophilic C10,11 double bond, suggesting that the first step proceeds by direct displacement rather than generation of a farnesyl cation.
Fig. 2.

Enzymatic reaction scheme for production of 12,13-DFFPP from 8,9-DFGPP and IPP.
We then determined a structure for the enzyme with this ligand in the active site. While the structure of apo-PS was determined more than 20 years ago, no structure with ligand bound in the active site has been reported previously. We began by preparing crystals of the apo-enzyme and then soaking them with the DFFPP ligand. After 1 hr incubation, the crystals were immersed in liquid nitrogen, and the structure of the resulting complex was solved by x-ray diffraction in space group P63 to 2.2 Å resolution.
As has been described before for crystals of apo-PS (1PS1),14 there are two molecules in the asymmetric unit, with essentially identical structure (0.3 Å RMSD). Each monomer is composed of 11 α-helices (A-K). Electron density is not visible for residues 159–165, 225–234, and 240–249 in molecule A, and these amino acids were not modelled into the structure.
As shown in Fig. 3, there is well-defined electron density in the active site of the enzyme at 1 σ contour corresponding to the inactive substrate analog DFFPP. The ligand binds only to molecule A of the asymmetric unit. As is the case with all of the structures reported here, there is an unidentified electron density in the active-site of the apo-enzyme that is displaced upon soaking with the DFFPP ligand. The diphosphate moiety of DFFPP is anchored at the conserved Asp-rich (DDXXD) and NSE/DTE motifs that are required for binding of the trinuclear metal cluster, but only two of the three Mg2+ ions are visible in the active site (metal ions are shown in Fig. 4). The prenyl chain extends deep into the binding pocket, makes a U-turn at the C6,C7-double bond, and then folds back on itself to position C11 4.0 Å directly across from C1 where attack of the C10,C11 π electrons on the developing allylic carbenium ion would proceed with the expected inversion of configuration at C1.2 The active site architecture is organized such that C1 is located closer to C11 (4.0 Å) than to C10 (4.3 Å), consistent with the anti-Markonikov sense of the cyclization (Fig. 5). Most strikingly, C9 of the prenyl chain, the eventual site of the reactive secondary carbocation, is positioned 3.5 Å above the center of the benzene ring of residue F76 with a C–H bond pointed directly toward the ring (Fig. 5). This 2.5 Å C-H…π interaction26 with the benzene ring would initially stabilize a developing cation at C10 during anti-Markovnikov cyclization by enhanced hyperconjugation from the C9–H bond, i.e., the electrons of the C9–H bond are rendered more available for sharing with the empty C10 p orbital because the benzene π system is donating its electrons to H9 (Fig. 6). Computational studies27–29 have previously predicted that such an interaction could stabilize the C10 (humulyl A) cation, greatly stabilize the transition state for H migration to C10, and stabilize the eventual cation at C9, but to our knowledge this is the first experimental evidence supporting the computational results. The carbocation–π interactions described in Fig. 6 are best considered as hybrid C-H…π26 and cation–π interactions.27–37 The arrangement of the ligand is conducive to further interactions that would stabilize a cation at C9. The C6,7 double bond is oriented toward the face of C9 opposite F76 and could confer a homoallylic cation–π interaction, similar to that observed in gas phase calculations.17 Finally, the mainchain carbonyl oxygen of I177, which is exposed at the break in helix G, is oriented toward the same face of C9 as the C6,C7-double bond at a distance of 3.4 Å, well positioned to stabilize the C9 carbocation through a charge-dipole interaction (Fig. S1). The helix G break is a common feature of class I terpene synthases. The possible role of the helix G break in providing dipoles for stabilization of cation intermediates was first proposed by Noel and coworkers for the reaction of epi-aristolochene synthase38 and then generalized by Pandit et al.39 and Dickschat and coworkers.40, 41
Fig. 3. Stereo view of DFFPP ligand in the active site of PS.

Fourier electron density in blue mesh is contoured at 1 σ. Protein is shown in green, diphosphate moiety of DFFPP in orange, prenyl chain in yellow, and the two fluorine atoms in white. The side chain of N219 has been included to aide with orientation. Carbon atoms 1, 9, 10, and 11 of the DFFPP ligand are labeled.
Fig. 4. Stereo view of DFFPP ligand and metal ions in the active site of PS.

Protein is shown in green, diphosphate moiety of DFFPP in orange, prenyl chain in yellow, and the two fluorine atoms in white. Carbon atoms of the DFFPP ligand are labeled.
Fig. 5.

Stereo view of the DFFPP ligand in the active site of PS showing the distance separating carbon C1 from carbons C10 and C11, and the C9-H…π interaction with the benzene ring of F76.
Fig. 6.

Scheme showing C-H…π and cation-π stabilization in the transfer of charge from C10 to C9 in the PS reaction.
F77 also contributes an aromatic side chain to the active site architecture (Fig. 4), but the closest distance of C9 to any of the F77 sidechain carbons is 5.1 Å, and the fact that the hydrogens of the C9-H bonds are pointing away from F77 make it unlikely that F77 contributes to carbocation stabilization at C9. By contrast, the C10-H bond projects toward the F77 benzene ring, suggestive of a vinylic C+-H···π interaction that could stabilize a cation at C10. A similar C+-H···π interaction has been predicted from quantum chemical computations to stabilize a norbornyl cation, but the distance observed here (3.3 Å) is significantly greater than that with the norbornyl cation model system27 or a neutral sp2 C-H···π interaction42 (both about 2.6 Å). In addition, the observed 20-fold loss of activity with a PS F77Y mutant15 does not support a role for F77 in C+-H···π or cation-π interactions.
To further probe the role of F76 in guiding reactivity of the C10,C11-π electrons to favor an anti-Markovnikov product, we constructed several PS mutants with amino acid substitutions at position 76 (Table 1). X-ray crystal structures were solved for each mutant after soaking with DFFPP (Table 2). The overall structures were identical to the WT enzyme, and any differences were limited to the site of mutation (Fig. 7). The activities and product distributions for mutants in which various aromatic amino acids replace F76 are shown in Table 1 (overnight incubations performed to examine product distributions), Figs. S2–S4 (GC-MS identification of the products), and Fig. S5 (activities under initial rate conditions). The F76A mutant, as reported by others, expresses well but is poorly soluble and not active.15 For these reasons, the Ala mutant was not pursued here. In contrast to F76A, all mutants with aromatic amino acids at position 76 display significant activity. F76Y displays near WT activity, as is expected from the calculated electrostatic potentials and gas phase Na+-ion binding energies for phenol versus benzene.36, 37 In addition, the Y76 side chain is found in an almost identical position in the structure of the DFFPP-bound mutant compared with the WT protein (Fig. 7A and B).
Table 1. Activity and product distribution of WT and mutant PS.
All values were determined by GC/MS, expressed relative to the amount of pentalenene produced by the WT enzyme (i.e., 100). Assays (overnight) were performed as described in Methods. Values are averages of two independent experiments with indicated range.
| Product |  |
 |
 |
|---|---|---|---|
| WT | 100 | 0 | 0 |
| F76Y | 100 ± 8 | 0 | 0 |
| F76W | 32 ± 8 | 9 ± 2 | 0 |
| F76H | 18 ± 1 | 3 ± 1 | 11 ± 1 |
Table 2.
Crystallographic Data Collection and Refinement Statistics
| WT-MGapo | WT-DFFPP | F76Y-apo | F76Y-DFFPP | F76W-apo | F76W-DFFPP | F76H-apo | F67H-DFFPP | |
|---|---|---|---|---|---|---|---|---|
| PDB ID | 6WKC | 6WKD | 6WKE | 6WKF | 6WKG | 6WKH | 6WKI | 6WKJ |
| Data collection statistics | ||||||||
| Space group | P21 | P63 | P63 | P63 | P63 | P63 | P63 | P63 |
| Wavelength (Å) | 0.9774 | 1.0 | 0.9774 | 0.9774 | 0.9774 | 0.9774 | 0.9774 | 0.9774 |
| Resolution range (Å) | 20 – 1.65 | 20 – 2.20 | 20 – 2.40 | 20 – 2.50 | 20.0 – 2.30 | 20.0 – 2.55 | 20 – 2.35 | 20.0 – 2.30 |
| Highest resolution shell (Å) | 1.74 – 1.65 | 2.32 – 2.20 | 2.53 – 2.40 | 2.64 – 2.50 | 2.42 – 2.30 | 2.69 – 2.55 | 2.48 – 2.35 | 2.42 – 2.30 |
| Unit cell parameters (Å) | a = 60.7, | a = 182.5, | a = 181.8, | a = 183.3, | a = 183.2, | a = 185.0, | a = 181.5, | a = 182.0, |
| b = 73.8, | b = 182.5, | b = 181.8, | b = 183.3, | b = 183.2, | b = 185.0, | b = 181.5, | b = 182.0, | |
| c = 81.2; | c = 56.2; | c = 56.4; | c = 56.7; | c = 56.5; | c = 56.9; | c = 56.5; | c = 56.4; | |
| α = γ = 90 | α = β = 90 | α = β = 90 | α = β = 90 | α = β = 90 | α = β = 90 | α = β = 90 | α = β = 90 | |
| β = 100.2 | γ = 120 | γ = 120 | γ = 120 | γ = 120 | γ = 120 | γ = 120 | γ = 120 | |
| Total reflections | 566358 | 1114810 | 853949 | 755975 | 951335 | 726238 | 911638 | 952163 |
| Unique reflections | 83734 | 54634 | 41985 | 38010 | 48511 | 36655 | 44617 | 47794 |
| Completeness %a | 98.9 (98.0) | 99.9 (100) | 99.8 (100) | 99.8 (100) | 99.8 (100) | 99.8 (100) | 99.8 (100) | 99.8 (100) |
| Rmerge %a | 9.4 (91) | 11.4 (172) | 12.8 (152) | 16.5 (112) | 13.6 (146) | 13.7 (167) | 11.1 (179) | 10.9 (150) |
| CC(1/2)a | 1.0 (0.7) | 1.0 (0.8) | 1.0 (0.8) | 1.0 (0.9) | 1.0 (0.8) | 1.0 (0.7) | 1.0 (0.7) | 1.0 (0.8) |
| I/σ (I)a | 14.9 (2.0) | 22.2 (2.2) | 21.6 (2.4) | 9.4 (2.3) | 16.9 (2.5) | 15.9 (2.1) | 23.5 (2.0) | 20.0 (2.3) |
| Redundancya | 6.8 (6.7) | 20.4 (21.0) | 20.3 (21.1) | 19.9 (20.5) | 19.6 (20.2) | 19.8 (20.2) | 20.4 (20.5) | 19.9 (20.3) |
| Refinement statistics | ||||||||
| Resolution range (Å) | 20 – 1.65 | 20 – 2.20 | 20 – 2.4 | 20.0 – 2.5 | 20 – 2.30 | 20 – 2.55 | 20 – 2.35 | 20 – 2.3 |
| No. of reflections used | 83593 | 54616 | 41971 | 37825 | 48499 | 36638 | 44601 | 47769 |
| Rcryst % | 16.7 | 20.1 | 19.8 | 20.8 | 21.6 | 22.5 | 22.0 | 21.1 |
| Rfree % | 19.3 | 21.9 | 23.2 | 23.5 | 23.7 | 25.6 | 24.1 | 24.6 |
| Protein atoms | 4842 | 4611 | 4494 | 4430 | 4455 | 4377 | 4493 | 4355 |
| Ligand atoms | 0 | 26 | 0 | 26 | 0 | 26 | 0 | 26 |
| Metal atoms | 4 | 2 | 0 | 0 | 0 | 0 | 0 | 0 |
| Water molecules | 1171 | 363 | 234 | 97 | 304 | 101 | 172 | 212 |
| r.m.s.d. in bond lengths (Å) | 0.005 | 0.003 | 0.003 | 0.003 | 0.002 | 0.002 | 0.002 | 0.004 |
| r.m.s.d. in bond angles (°) | 0.8 | 0.5 | 0.5 | 0.5 | 0.4 | 0.4 | 0.4 | 0.6 |
| CCoverall | 0.9 | 0.9 | 0.9 | 0.8 | 0.9 | 0.8 | 0.8 | 0.9 |
| RSCCligand | - | 0.9 | - | 0.7 | - | 0.7 | - | 0.6 |
Highest resolution shell values are given in parentheses.
Fig. 7. Structure of the active site surrounding residue 76 in WT and three mutants of PS.
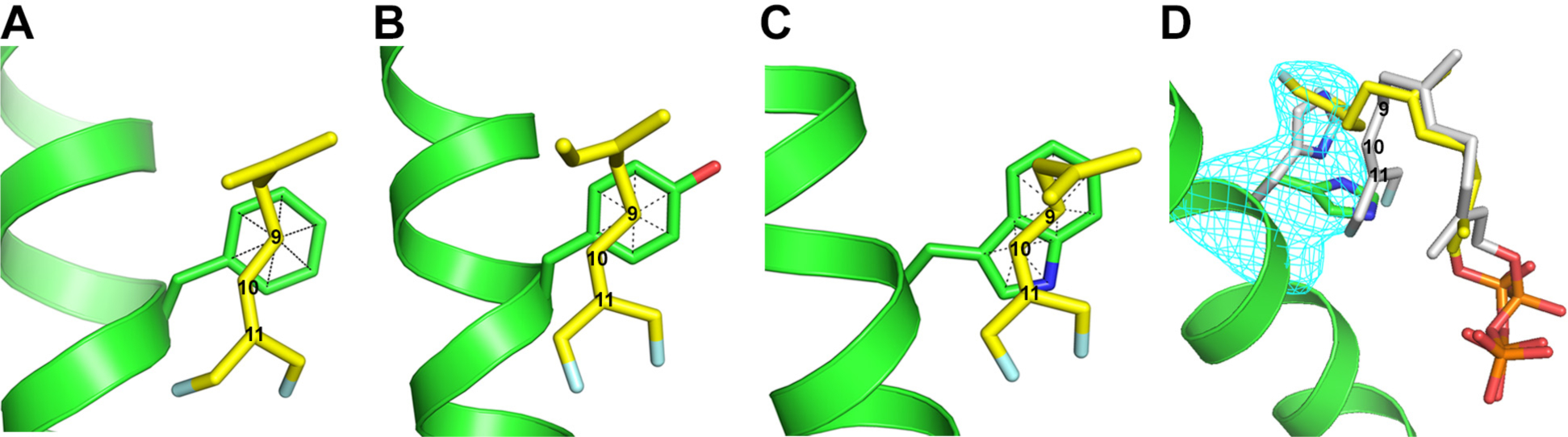
A, WT; B, F76Y; C, F76W; and D, F76H. The electron density (blue mesh) for F76H shown in panel D is a Polder map contoured at 3 σ and indicates the presence of at least two rotamer conformations for the 76 side chain, with one (30%) corresponding to the orientation of Phe in the WT protein (grey). Electron density was observed for DFFPP only in an extended conformation (yellow) corresponding to the new rotamer conformation of the H76 side chain (green). The grey conformation, corresponding to the ligand in WT, was modelled in to avoid steric conflict with the grey His76 rotamer. Carbon atoms 9, 10, and 11 of the DFFPP ligand are labeled.
The F76W mutant intriguingly generates a significant amount of α-humulene in addition to pentalenene (Table 1). α-humulene can be generated from humulyl cation A by abstraction of a proton from C9 (see Fig. 1 and Table 1), suggesting that F76W may stabilize a carbenium ion at C10 as well as at C9. Consistent with this suggestion, the structure of the DFFPP-bound F76W mutant clearly shows carbons C9 and C10 centered above the fused benzene and pyrrole rings of the indole side chain (Fig, 7C), respectively, such that carbocations at both positions should be stabilized in this complex. The lesser preference for stabilizing the cation at C9 over C10 may be responsible for the loss of selectivity; because the enzyme is not set up to promote alkene attack on a cation at C10, this intermediate, if long-lived, may be more prone to elimination side reactions. Alternatively, the tryptophan indole43 is roughly 20 pKa units more basic than benzene44 and 8 pKa units more basic than an alkene (e.g., isobutylene)44 and may serve as a general base to facilitate deprotonation of humulyl cation A or B.
The F76H mutant is interesting. As can be seen in Table 1, F76H produces not only the two anti-Markovnikov products, pentalenene and α-humulene, but also a significant amount of germacrene A, a product resulting from Markovnikov attack of C10 on C1 of the FPP substrate (Fig. 1 and Table 1). Significantly, the crystal structure of the F76H/DFFPP complex displays at least two conformations of the imidazole side chain of H76 (Fig. 7D), one of which is in a similar location to that of the benzene ring of F76 in the WT protein. While density for the prenyl chain is not well defined, the two conformations of the imidazole side chain of H76 (30% and 70% by occupancy) must be associated with two dramatically different conformations of the bound DFFPP ligand because the new conformation (70%) is sterically incompatible with the ligand conformation observed in the WT protein. Thus, the mutation causes re-sculpting of the active site such that a different conformation of the ligand is stabilized in the binding pocket. These data indicate that Phe76 plays a structural role in the ligand binding pocket in addition to its role in stabilizing development of positive charge on C9, and the multiple conformations of the H76 side chain suggests a rationale, at least in general terms, for the generation of multiple products differing in regioselectivity of the initial cyclization reaction.
Discussion
The active sites of terpene synthases are often lined with aromatic amino acid side chains, and it has long been suspected that carbocation intermediates along the reaction coordinate are stabilized through cation-π interactions involving these amino acid residues.6 Polar residues are not well-suited for this purpose as they would be at risk of covalent modification with the highly reactive carbenium ion intermediates leading to suicide inactivation of the enzymes, as has been observed for a number of different terpene synthases by Noel and coworkers.45 The presumed involvement of cation-π interactions in terpene synthase reactions has been supported by numerous studies showing that mutation of aromatic amino acid residues in terpene synthase active sites severely disrupts function of the enzymes, but the enzyme active site contour is often defined in large part by the steric bulk of aromatic amino acids, and it can be difficult to sort the effects of mutation on electrostatics from the role played by side chains in stabilizing the reactive conformation of the flexible isoprenoid substrate. In addition, with notable exceptions,46, 47 the mutagenesis data are not always supported by structural studies linking a particular aromatic residue to a carbocation intermediate in the reaction coordinate, which is crucial to understanding the mechanism by which terpene synthases guide regioselectivity in the generation of specific products from substrates which have the potential to produce a large number of different products.
An important example demonstrating the feasibility of cation-π interactions in terpene synthases was provided in a study by Christianson and coworkers in which a crystal structure of the enzyme epi-isozizaene synthase in complex with 3 Mg2+ ions, inorganic pyrophosphate, and the benzyltriethylammonium cation showed the quaternary ammonium ion surrounded by three phenylalanine side chains in the active site, with the positively charged nitrogen located about 5 Å from the center of each aromatic side chain.48 Aza-analogs of substrates have also been used,49 but they often bind in non-productive conformations for the reaction, and it can be difficult to know if the observed cation-π interaction is truly mimicking that of an intermediate along the reaction coordinate or if the observed interaction is forced as a consequence of introducing a positively charged ligand into the active site of the enzyme. In contrast, the results presented here provide compelling evidence for a carbocation-π interaction (especially highlighting the C–H…π component of this interaction) controlling regioselectivity for a developing carbenium ion in the anti-Markovnikov cyclization step of the PS reaction. This stabilization is brought about by interaction with the benzene ring of the F76 side chain. Following a soak of the apo-PS crystal, the DFFPP substrate analog was found in the active site with C9–H positioned 2.5 Å above the center of the benzene ring of the side chain, well positioned to participate in a C–H…π interaction that should stabilize a developing C10 carbocation.26–29 After catalyzing the hydrogen shift to C10, the F76 aromatic ring can stabilize the C9 carbocation from one face, with mainchain carbonyl dipole of I177 and neighboring intramolecular C6,C7-double bond stabilizing the opposite face. Importantly, this active site geometry is on display with a ligand in which C9 is a neutral carbon atom, and DFFPP is bound in a productive conformation for initial C1/C11 bond formation to proceed with correct stereo- and regioselectivity.
The arrangement of atoms may have relevance for another aspect of the initial cyclization reaction. The fact that C10 is found slightly outside of the perimeter of the benzene ring suggests that a full positive charge may not develop at C10. In this case, C11 attack on C1 might occur concurrently with 1,2-hydrogen shift from C9 to C10 (Fig. 8). In this case, positive charge may only slightly develop on C10, but the F76 benzene ring would still stabilize positive charge at C9 and the migrating hydrogen.28, 29 This proposal is supported by the crystal structure and product profile of the F76W mutant in which the indole ring of the side chain is in a better position to support development of positive charge on C10, and the product profile shifts to include ahumulene, which likely results from deprotonation of a humulyl cation A intermediate (Fig. 1).
Fig. 8.

Scheme showing C11/C1 cyclization with concerted 1,2-hydride shift from C9 to C10.
Finally, we address how conserved the F76 residue is in terpene synthases. The search is restricted to Streptomyces, which contain the largest group of bacterial sesquiterpene synthases. There is a rough correlation with enzymes catalyzing reactions that begin with anti-Markovnikov attack and development of positive charge on C9 (Figs. 9 and 10). Pentalenene synthase, cucumene synthase,50 isohirsut-4-ene synthase,51 and (+)-isoafricanol synthase52 all begin with attack of C11 on C1, followed by a 1,2 hydride shift resulting in a carbenium ion intermediate with positive charge on C9, and all have either a Phe or Tyr residue at position 76 (pentalenene synthase numbering). In contrast, α-eudesmol synthase,53 a-amorphene synthase,53 selina-4(15),7(11)-diene synthase,41 daucadiene synthase,54 (+)-T-muurolol synthase,55 germacradiene-11-ol synthase,53 (−)-d-cadinene synthase,55 and (+)-epicubenol synthase56 all have a non-aromatic amino acid residue at position 76 and none catalyze reactions that are initiated with anti-Markovnikov addition reactions. However, the correlation is not absolute. Epi-isozizaene synthase,57 (−)-epi-α-bisabolol synthase,58 avermitilol synthase,59 and (−)-germacradien-4-ol synthase58 all have an aromatic amino acid side chain at position 76, but none catalyze a reaction beginning with an anti-Markovnikov attack, and africanene synthase54 does not have an aromatic residue at position 76, but does catalyze a reaction beginning with an anti-Markovnikov attack and development of positive charge on C9.
Fig. 9. Multiple sequence alignment of sesquiterpene synthases from Streptomyces sp.

Full length sequences for pentalenene synthase (AAA19131.1), cucumene synthase (B5GLM7), isohirsut-4-ene synthase (slt18_1880), (+)-isoafricanol synthase (from S. malaysiensis), epi-isozizaene synthase (Q9K499), epi-α-bisabolol synthase (AB621339), avermitilol synthase (Q82RR7), (−)-germacradien-4-ol synthase (AB621338), α-eudesmol synthase (SCNRRL3882_07544), α-amorphene (SCNRRL3882_07041), selina-4(15),7(11)-diene synthase (B5HDJ6), dauca-8,11-diene synthase (SVEN_0552), (+)-T-muurolol synthase (B5GW45), germacrenediene-11-ol-synthase (SCNRRL3882_01776), (−)-d-cadinene synthase (B5GS26), (+)-epicubenol synthase (B1W477), africanene synthase (SCLAV_p0985), and (+)-caryolan-1-ol synthase (B1W019.1) were aligned. The sequence for pentalenene synthase was used as reference for preparing the figure, with the column containing F76 in pentalenene synthase enclosed in a red box. Sequences were aligned using MUSCLE74,75 and visualized with Jalview 2.76
Fig. 10. Chemical structures of compounds produced by the enzymes in the multiple sequence alignment.

Structures enclosed within the dotted lines are produced by terpene synthases that have a Phe or Tyr residue at the position corresponding to F76 in PS.
(+)-Caryolan-1-ol synthase (CS)60, from Streptomyces griseus, is an interesting case (Fig. 11). CS catalyzes formation of the tertiary alcohol caryolanol from FPP. As with PS, the reaction scheme begins with anti-Markovnikov attack of C11 on C1 of the farnesyl substrate, but in this case the secondary carbocation remains localized to C10 such that subsequent attack of C2 onto C10 forms a fused four-membered cyclobutane ring instead of the five-membered ring of the PS reaction.
Fig. 11.

Reaction scheme for (+)-caryolan-1-ol synthase.
Our interest in this reaction stems from its initial similarity to the PS reaction coupled with subsequent differences to generate a distinct product. Notably, the S. griseus enzyme, which shares 29% amino acid identity with PS, has an Ala in the same position as F76 in PS (Fig. 9).60 Given that the two proteins share only 29% sequence identity, it seems unlikely that substitution of Ala with Phe at this position would create a pentalenene synthase (and we have confirmed that suspicion, the mutant enzyme behaves the same as WT), but we also guess that exploration of the CS reaction will deepen significantly our understanding of how terpene synthases control regioselectivity in the cyclization reactions.
Methods
Synthesis of DMAPP, IPP, and E,E-FPP.
The synthesis and purification of DMAPP, IPP, and E,E-FPP from the corresponding allylic alcohols (Sigma-Aldrich, MO) was performed as previously described for substrates and substrate analogs of the enzyme (+)-limonene synthase.24, 61, 62 The purity of the diphosphate products was assessed by proton, carbon, and phosphorus NMR. NMR spectra were recorded on a Varian 400-MR spectrometer (9.4 T, 400 MHz) in D2O adjusted to pD ~8.4 with ND4OD. 1H and 13C chemical shifts are reported in parts per million (ppm) downfield from TMSP (trimethylsilyl propionic acid). 31P chemical shifts are reported in ppm relative to 85% ο-phosphoric acid. J coupling constants are reported in units of frequency (hertz) with multiplicities listed as s (singlet), d (doublet), dd (doublet of doublets), t (triplet), q (quartet), m (multiplet), br (broad), and app (apparent).
Dimethylallyl diphosphate (DMAPP): 1H NMR (400 MHz, D2O/ND4OD) δH 1.73 (3 H, s, CH3), 1.77 (3 H, s, CH3), 4.46 (2 H, app t, J = 6.8 Hz at C1), 5.46 (1 H, t, J = 7.4 Hz at C2); 13C NMR (100 MHz, D2O/ND4OD) δC 20.13, 27.79, 65.37 (d, JCP = 5.3 Hz), 122.72 (d, JCP = 8.5 Hz), 142.71; 31P NMR (162 MHz, D2O/ND4OD) δP −6.76 (d, JPP = 22.1 Hz, P1), −10.46 (dt, JPP = 22.1 Hz, JPH = 6.8 Hz P2).
Isopentenyl diphosphate (IPP): 1H NMR (400 MHz, D2O/ND4OD) δH 1.78 (3 H, s, CH3 at C4), 2.40 (2 H, t, J = 6.4 Hz at C2), 4.07 (2 H, app q, J = 6.7 Hz at C1), 4.85 (1H, s, H at C5), 4.87 (1H, s, H at C5); 13C NMR (100 MHz, D2O/ND4OD) δC 24.58, 40.77 (d, JCP = 7.6 Hz), 66.90 (d, JCP = 5.4 Hz), 114.32, 146.85; 31P NMR (162 MHz, D2O/ND4OD) δP −6.60 (d, JPP = 22.0 Hz, P1), −10.60 (d, JPP = 22.0 Hz, JPH = 6.6 Hz, P2).
E,E-Farnesyl Diphosphate (FPP): 1H NMR (400 MHz, D2O/ND4OD) δH 1.63 (6H, two s, CH3), 1.70 (3 H, s, CH3), 1.73 (3 H, s, CH3), 2.00–2.23 (8 H, m, H at C4, C5, C8, C9), 4.48 (2 H, app t, J = 6.5 Hz, H at C1), 5.16–5.26 (2 H, m, H at C6, and C10), 5.48 (1 H, t, J = 7.2 Hz at C2); 13C NMR (100 MHz, D2O/ND4OD) δC 18.11, 18.51, 19.83, 27.71, 28.51, 28.63, 41.65, 41.71, 65.33 (d, JCP = 5.2 Hz), 122.82 (d, JCP = 8.5 Hz), 127.17, 127.33, 136.40, 139.57, 145.61; 31P NMR (162 MHz, D2O/ND4OD) δP −5.76 (d, JPP = 22.2 Hz, P1), −9.59 (d, JPP = 22.2 Hz, JPH = 6.5 Hz, P2).
Preparation of FPPS and PS.
Genes for farnesyl pyrophosphate synthase (fpps) from T. cruzi (GenBank AF312690) and pentalenene synthase (ps) from Streptomyces sp. UC5319 (GenBank UO5213) were codon optimized for expression in E. coli and purchased from Synbio Technologies (New Jersey). The genes were in a pET-28a (+) vector as a NcoI (5’-end) and EcoRI (3’-end) cartridge with a glycine codon immediately after the start methionine and followed by a His6 tag and tobacco etch virus (TEV) protease cut site.
The plasmids were used to transform BL21 (DE3) competent cells, and colonies selected for growth on LB/agar plates containing 50 μg/mL kanamycin. A single colony was used to inoculate 10–15 mL of LB containing 50 μg/mL kanamycin, and then incubated overnight at 37 °C with shaking at 220 rpm. The overnight culture was used to inoculate 1 L of LB containing 50 μg/mL kanamycin. General procedures for purification of the proteins were performed as previously described for (+)-limonene synthase.62 Procedures specific for each protein were essentially as previously described.13, 63
FPPS. Inoculated cultures were grown with shaking at 37 °C to OD600 ≈ 0.6 and induced with 1 mM IPTG for 6 hr, as described previously.63 Harvested cells were re-suspended in 20 mM Tris buffer, pH 7.9, containing 500 mM NaCl and 5 mM DTT (Buffer A), sonicated, and clarified by centrifugation. The resulting lysate was loaded at a rate of 2 mL/min onto a prepacked 5 mL HiTrapFF Ni-Sepharose column (GE Healthcare Life Sciences, MA) that had been equilibrated with Buffer A. The column was washed with 30 mL of 20 mM imidazole in Buffer A and then eluted with a linear gradient of 20–250 mM imidazole in Buffer A. Fractions containing FPPS were pooled, the imidazole removed by repeated concentration using Amicon Ultra - 15 filtration units with a 10 kDa cutoff followed by dilution with Buffer A, and the sample aliquoted (100 μM), then flash frozen with liquid nitrogen, and stored at −80 °C until further use.
PS.13 1 L cultures were grown at 37 °C with shaking at 220 rpm to an OD600 ~0.4–0.6, then moved to a pre-cooled shaking incubator at 16 °C, and induced with 0.5 mM IPTG after 1 hr, and incubated overnight. Cells were harvested by centrifugation and resuspended in 50 mM bis-tris propane buffer, pH 7.5, containing 150 mM KCl, and 10 mM MgCl2 (Buffer B), sonicated, and the supernatant fraction clarified by centrifugation. The resulting lysate was loaded at a rate of 2 mL/min onto a prepacked 5 mL HiTrapFF Ni-Sepharose column (GE Healthcare Life Sciences, MA) that had been equilibrated in Buffer B. The column was then washed with ~30 mL of Buffer B containing 20 mM imidazole, and the protein eluted with a linear gradient of 0.02–1.0 M imidazole in Buffer B. For PS mutants, which tended to be less stable in the presence of high imidazole concentrations, fractions were collected in 50-mL Falcon tubes containing 10 mL of Buffer B to dilute the imidazole. Eluted protein was buffer exchanged into low salt buffer (50 mM Tris base, 5 mM DTT, 10% glycerol at pH 8.2) and loaded onto a 5 mL prepacked Q-sepharose ion exchange column (GE Healthcare Life Sciences, MA) at 1 mL/min. Protein was eluted with a linear NaCl gradient ranging from 0 to 1 M. Fractions containing PS were pooled, the NaCl removed by repeated concentration using Amicon Ultra - 15 filtration units with a 10 kDa cutoff followed by dilution with Buffer B, and the sample aliquoted (100 μM), then flash frozen with liquid nitrogen, and stored at −80 °C until further use.
Preparation and Characterization of 12,13-Difluorofarnesyl Diphosphate (DFFPP).
DFFPP was prepared enzymatically from DFGPP (Fig. 2). 10 mM 8,9-DFGPP,24, 25 10 mM IPP, and 10 mM MgCl2 were incubated with 5 μM FPPS in 450 μL of 20 mM Tris buffer, pH 7.9, containing 500 mM NaCl overnight at RT, and the crude reaction mixture then stored at −20 °C. The thawed sample was filtered by centrifugation through an Amicon Ultra filter with 10 kDa molecular mass cutoff, and the flow-through fraction collected and lyophilized to dryness. After re-suspension of the lyophilized powder in 500 μL of D2O, and recording of a 1H NMR spectrum, the presence of 3 vinyl protons matched a previously reported spectrum25 of 12,13-DFFPP and indicated that the enzymatic elongation of 8,9-DFGPP to 12,13-DFFPP had been successful. The sample was then lyophilized, re-suspended in 50 μL H2O, and stored at −20 °C until use. To determine if PS was unreactive with DFFPP, 1 μM FPPS, 1 μM PS, 200 μM DFGPP, and 200 μM IPP was prepared in a final volume of 1 mL of buffer B and overlaid with 1 mL of hexanes and incubated overnight at RT (Fig. S6d). A control reaction containing 200 μM DMAPP in lieu of 8,9 DFGPP and 400 μM IPP was prepared in the same fashion (Fig. S6a). To ensure FPPS converts 8,9 DFGPP and IPP into DFFPP, 1 μM FPPS was mixed with 200 μM 8,9 DFGPP, 200 μM IPP in a total volume of 1 mL of buffer B and incubated overnight at RT. 100 μL of 1N HCl was then added to the reaction and incubated at RT for 2 hr to convert the diphosphates into the corresponding alcohols. The reaction mixtures were then neutralized with 100 μL of 1 N NaOH before analyzing the sample by GC-MS (Fig. S6e & f). A control reaction containing 1 μM FPPS, 200 μM DMAPP, and 400 μM IPP was also prepared and treated with acid, extracted with 1 mL hexanes, and the isoprenoid alcohols analyzed by GC-MS (Fig. S6b & c).
Enzyme Activity.
Enzyme assays were performed using the discontinuous single-vial method described by O’Maille et al.,64 with the exception that hexanes were used for the organic layer rather than ethyl acetate. Hexane extractable terpene products were identified and quantified using GC-MS (Agilent Technologies 7890A GC System coupled with a 5975C VL MSD with a triple-axis detector) as previously described by us and others.62, 65 Pulsed-splitless injection was used to inject 5 μL samples onto an HP-5ms (5%-phenyl)-methylpolysiloxane capillary GC column (Agilent Technologies, CA; 30 m × 250 μm × 0.25 μm) at an inlet temperature of 220 °C, transfer temperature of 240 °C, and run at constant pressure using helium as the carrier gas. Samples were initially held at an oven temperature of 50 °C for 1 min, followed by a linear temperature gradient of 10 °C/min to 220 °C, which was then held for 10 min.
The assays were run under two different conditions: 1. Overnight reactions to determine product distribution; and 2. Initial-rate reactions to determine kcat for the major product. The concentrations of the different products were estimated on the basis of a standard curve for α-humulene.
Overnight reactions.
1 μM of PS (either WT, F76Y, F76W, or F76H) was mixed with 200 μM of E,E-FPP in a total reaction volume of 1 mL buffer B. The solution was overlaid with 1 mL hexanes (Sigma-Aldrich, MO) and incubated overnight at RT. The solutions were vortexed to stop the reaction and extract products into the organic layer which was analyzed by GC-MS. Purchased standards of β-elemene (Cayman Chemical, MI) and α-humulene (Sigma-Aldrich, MO) were used to determine identity of major, non-pentalenene peaks from reactions of the PS mutants, where product formation was followed in scan mode.
Initial rates.
Initial rates were determined under Vmax conditions, which was confirmed in each case by running the reactions at two different substrate concentrations. Enzyme (10 nM for PS WT and F76Y; 100 nM for F76W, and F76H) was mixed with 50 μM E,E-FPP (75 μM for F76W) in 1 mL buffer B, overlaid with 1 mL hexanes, and incubated at RT for various times (WT 3, 6, and 9 min; F76Y 1, 2, and 3 min; F76H 1.5, 3, 5, and 7 min; and F76W 5, 10, and 15 min) before vortexing to stop the reaction and extract products into the organic layer for analysis by GC-MS as described above, where product formation was followed in select ion monitoring (SIM) mode, specifically set to monitor a mass at 204.1.
Crystallization.
Crystallization trials were performed by sitting drop vapor diffusion at room temperature using Hampton (Hampton Research, CA) and Jena Bioscience (Jena Bioscience, Jena, Germany) sparse matrix crystallization screens. Drops were set with a Phoenix robot (Art Robbins Instruments, CA) by mixing protein (7.5 mg/mL) in 50 mM Bis-Tris propane, pH 7.5, 150 mM KCl and 10 mM MgCl2 with crystallization mother liquor in a 1:1 ratio. Crystals of WT-Mg appeared after about 3 weeks in 8–14% w/v PEG4000, 100 mM HEPES, pH 6.5–7.5, 200 mM MgCl2, 15% ethylene glycol and 10% 2-propanol at 20 °C while WT and mutant crystals formed in 0.8–1.2 M sodium tartrate, 100 mM Tris, pH 7.5–9, and 5 mM DTT.
Data Collection, Processing, and Refinement.
WT-Mg crystals were soaked in reservoir solution containing 15% glycerol as cryoprotectant before flash freezing in liquid nitrogen. To generate the DFFPP complexes, WT and mutant crystals were soaked with reservoir solution containing 1 mM DFFPP, 10 mM MgCl2, and 15% glycerol for 1 hr at room temperature before flash-freezing in liquid nitrogen. Diffraction data were collected at 100 K with beam line 5.0.2 at the Advanced Light Source (Lawrence Berkeley National Laboratory, Berkeley, CA) using a Pilatus3 X 6M detector (DECTRIS Ltd., Switzerland). Data sets were integrated using XDS66 and scaled using SCALA version 3.367 from the CCP4 software suite version 7.0.68, 69 All diffraction data sets, except WT-Mg, were processed in space group P63. WT-Mg data were processed in P21. Complete data collection statistics are listed in Table 2.
The WT structure was solved by molecular replacement using PHASER version 2.870 with molecule A of the published structure for PS (PDB entry 1PS1) as a search model. All other structures were solved using the final refined model of WT as a search model. The coordinates of WT have not been deposited, as the structure is essentially identical to that of PDB entry 1PS1. The molecular replacement solutions contain two molecules in the asymmetric unit. Rigid body refinement followed by positional and B-factor refinement was carried out using phenix.refine71 from the PHENIX software suite version 1.16.72 Simulated annealing was included in earlier refinements to minimize the initial model bias. Manual model building was done using COOT version 0.8.73 After initial refinement, difference Fourier electron density was observed for the analog in the active site of the protein complexes. The ligand was modeled using Jligand version 1.0 from CCP4 software suite version 7.0 and the generated coordinates and restraints were used for further refinements. Water molecules were included in the final refinement after satisfying the criteria of 3 σ Fo –Fc and 1 σ 2Fo – Fc. Several iterative cycles of refinement were carried out before final submission of data. Data collection and final refinement statistics are given in Table 2. Data sets for the WT-Mg (PDB entry 6WKC), WT-DFFPP (PDB entry 6WKD), F76Y (PDB entry 6WKE), F76Y-DFFPP (PDB entry 6WKF), F76W (PDB entry 6WKG) and F76W-DFFPP (PDB entry 6WKH), F76H (PDB entry 6WKI), and F76H-DFFPP (PDB entry 6WKJ) have been submitted to the Protein Data Bank. All crystal structure figures in this paper were prepared using PyMol version 2.3 (Schrödinger LLC, Portland, OR).
Supplementary Material
Acknowledgements
We are grateful to the staff at the Advanced Light Source-Berkeley Center for Structural Biology for their assistance in X-ray data collection. The Advanced Light Source is funded by the Director, Office of Science, Office of Basic Energy Sciences, of the United States Department of Energy under contract DE-AC02-05CH11231. The Berkeley Center for Structural Biology is supported in part by grants from the NIGMS, National Institutes of Health. We would like to thank Prof. Barry Snider for helpful discussions.
Funding
This work was supported by National Institutes of Health Grants T32GM007596 (J.O.M.) and the National Science Foundation CAREER program CHE-1253363 (I.J.K.).
ABBREVIATIONS
- FPP
farnesyl diphosphate
- DFFPP
12,13-difluorofarnesyl diphosphate
- PS
pentalenene synthase
- CS
caryolan-1-ol synthase
Footnotes
Competing interests
The authors declare no competing financial interest.
Accession Codes
Accession codes are as follows: pentalenene synthase (PS) from Streptomyces exfoliatus UC5319, UniProt Q55012; and farnesyl pyrophosphate synthase from Trypanosoma cruzi (FPPS), UniProt Q95WL3. The atomic coordinates and structure factors for the DFFPP-PS complex have been deposited in the Protein Data Bank. RCSB PDB entry 6WKC (WT-Mg), 6WKD (WT-DFFPP), 6WKE (F76Y), 6WKF (F76Y-DFFPP), 6WKG (F76W), 6WKH (F76W-DFFPP), 6WKI (F76H), 6WKJ (F76H-DFFPP) for the respective models.
Supporting Information
Figure showing the interaction between I177 and DFFPP, product distribution profile for PS WT and PS F76Y, product distribution profile for PS F76W, product distribution profile for PS F76H, kcat values for PS WT and F76 mutants and characterization of enzymatically prepared DFFPP.
References
- [1].Buckingham J, Cooper C, and Purchase R (2016) Natural products desk reference, CRC Press, Taylor & Francis Group, Boca Raton. [Google Scholar]
- [2].Cane DE (1990) Enzymatic Formation of Sesquiterpenes, Chemical Reviews 90, 1089–1103. [Google Scholar]
- [3].Dewick PM (2002) The biosynthesis of C5-C25 terpenoid compounds, Nat Prod Rep 19, 181–222. [DOI] [PubMed] [Google Scholar]
- [4].Fraga BM (2005) Natural sesquiterpenoids, Nat Prod Rep 22, 465–486. [DOI] [PubMed] [Google Scholar]
- [5].Chuck CJ, and Donnelly J (2014) The compatibility of potential bioderived fuels with Jet A-1 aviation kerosene, Applied Energy 118, 83–91. [Google Scholar]
- [6].Christianson DW (2017) Structural and Chemical Biology of Terpenoid Cyclases, Chem Rev 117, 11570–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Cane DE (1985) Isoprenoid Biosynthesis - Stereochemistry of the Cyclization of Allylic Pyrophosphates, Accounts Chem Res 18, 220–226. [Google Scholar]
- [8].Davis EM, and Croteau R (2000) Cyclization enzymes in the biosynthesis of monoterpenes, sesquiterpenes, and diterpenes, Top Curr Chem 209, 53–95. [Google Scholar]
- [9].Kaspera R, and Croteau R (2006) Cytochrome P450 oxygenases of Taxol biosynthesis, Phytochem Rev 5, 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Pateraki I, Heskes AM, and Hamberger B (2015) Cytochromes P450 for terpene functionalisation and metabolic engineering, Adv Biochem Eng Biotechnol 148, 107–139. [DOI] [PubMed] [Google Scholar]
- [11].Wong NR, Liu X, Lloyd H, Colthart AM, Ferrazzoli AE, Cooper DL, Zhuang Y, Esquea P, Futcher J, Pochapsky TM, Matthews JM, and Pochapsky TC (2018) A new approach to understanding structure-function relationships in cytochromes P450 by targeting terpene metabolism in the wild, J Inorg Biochem 188, 96–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cane DE, and Pargellis C (1987) Partial purification and characterization of pentalenene synthase, Archives of biochemistry and biophysics 254, 421–429. [DOI] [PubMed] [Google Scholar]
- [13].Cane DE, Sohng JK, Lamberson CR, Rudnicki SM, Wu Z, Lloyd MD, Oliver JS, and Hubbard BR (1994) Pentalenene synthase. Purification, molecular cloning, sequencing, and high-level expression in Escherichia coli of a terpenoid cyclase from Streptomyces UC5319, Biochemistry 33, 5846–5857. [DOI] [PubMed] [Google Scholar]
- [14].Lesburg CA, Zhai GZ, Cane DE, and Christianson DW (1997) Crystal structure of pentalenene synthase: Mechanistic insights on terpenoid cyclization reactions in biology, Science 277, 1820–1824. [DOI] [PubMed] [Google Scholar]
- [15].Seemann M, Zhai G, de Kraker JW, Paschall CM, Christianson DW, and Cane DE (2002) Pentalenene synthase. Analysis of active site residues by site-directed mutagenesis, J Am Chem Soc 124, 7681–7689. [DOI] [PubMed] [Google Scholar]
- [16].Seemann M, Zhai GZ, Umezawa K, and Cane D (1999) Pentalenene synthase. Histidine-309 is not required for catalytic activity, J Am Chem Soc 121, 591–592. [Google Scholar]
- [17].Gutta P, and Tantillo DJ (2006) Theoretical studies on farnesyl cation cyclization: Pathways to pentalenene, J Am Chem Soc 128, 6172–6179. [DOI] [PubMed] [Google Scholar]
- [18].Lodewyk MW, Willenbring D, and Tantillo DJ (2014) Pentalenene formation mechanisms redux, Org Biomol Chem 12, 887–894. [DOI] [PubMed] [Google Scholar]
- [19].Zu LS, Xu MM, Lodewyk MW, Cane DE, Peters RJ, and Tantillo DJ (2012) Effect of Isotopically Sensitive Branching on Product Distribution for Pentalenene Synthase: Support for a Mechanism Predicted by Quantum Chemistry, J Am Chem Soc 134, 11369–11371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Hong YJ, and Tantillo DJ (2010) Formation of beyerene, kaurene, trachylobane, and atiserene diterpenes by rearrangements that avoid secondary carbocations, J Am Chem Soc 132, 5375–5386. [DOI] [PubMed] [Google Scholar]
- [21].Hong YJ, and Tantillo DJ (2011) How many secondary carbocations are involved in the biosynthesis of avermitilol?, Org Lett 13, 1294–1297. [DOI] [PubMed] [Google Scholar]
- [22].Tantillo DJ (2010) The carbocation continuum in terpene biosynthesis--where are the secondary cations?, Chem Soc Rev 39, 2847–2854. [DOI] [PubMed] [Google Scholar]
- [23].Tantillo DJ (2017) Importance of Inherent Substrate Reactivity in Enzyme-Promoted Carbocation Cyclization/Rearrangements, Angew Chem Int Ed Engl 56, 10040–10045. [DOI] [PubMed] [Google Scholar]
- [24].Morehouse BR, Kumar RP, Matos JO, Yu Q, Bannister A, Malik K, Temme JS, Krauss IJ, and Oprian DD (2019) Direct Evidence of an Enzyme-Generated LPP Intermediate in (+)-Limonene Synthase Using a Fluorinated GPP Substrate Analog, ACS Chem Biol 14, 2035–2043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Yu F, Miller DJ, and Allemann RK (2007) Probing the reaction mechanism of aristolochene synthase with 12,13-difluorofarnesyl diphosphate, Chem Commun (Camb), 4155–4157. [DOI] [PubMed] [Google Scholar]
- [26].Nishio M, Umezawa Y, Fantini J, Weiss MS, and Chakrabarti P (2014) CH-pi hydrogen bonds in biological macromolecules, Phys Chem Chem Phys 16, 12648–12683. [DOI] [PubMed] [Google Scholar]
- [27].Hong YJ, and Tantillo DJ (2007) Perturbing the structure of the 2-norbornyl cation through C-H…N and C-H…pi interactions, J Org Chem 72, 8877–8881. [DOI] [PubMed] [Google Scholar]
- [28].Hong YJ, and Tantillo DJ (2013) C-H…π interactions as modulators of carbocation structure - implications for terpene biosynthesis, Chem Sci 4, 2512–2518. [Google Scholar]
- [29].Hong YJ, and Tantillo DJ (2015) Tension between Internal and External Modes of Stabilization in Carbocations Relevant to Terpene Biosynthesis: Modulating Minima Depth via C-H…pi Interactions, Org Lett 17, 5388–5391. [DOI] [PubMed] [Google Scholar]
- [30].Dougherty DA (1996) Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp, Science 271, 163–168. [DOI] [PubMed] [Google Scholar]
- [31].Dougherty DA (2007) Cation-pi interactions involving aromatic amino acids, J Nutr 137, 1504S–1508S; discussion 1516S-1517S. [DOI] [PubMed] [Google Scholar]
- [32].Heidrich D (2002) Do isopropyl and tert-butyl cations form pi complexes with benzene?, Angew Chem Int Ed Engl 41, 3208–3210. [DOI] [PubMed] [Google Scholar]
- [33].Jenson C, and Jorgensen WL (1997) Computational investigations of carbenium ion reactions relevant to sterol biosynthesis, J Am Chem Soc 119, 10846–10854. [Google Scholar]
- [34].Kolboe S (2012) Computational study of isopropylbenzenium ions, J Phys Chem A 116, 3710–3716. [DOI] [PubMed] [Google Scholar]
- [35].Kumpf RA, and Dougherty DA (1993) A mechanism for ion selectivity in potassium channels: computational studies of cation-pi interactions, Science 261, 1708–1710. [DOI] [PubMed] [Google Scholar]
- [36].Mecozzi S, West AP Jr., and Dougherty DA (1996) Cation-pi interactions in aromatics of biological and medicinal interest: electrostatic potential surfaces as a useful qualitative guide, Proc Natl Acad Sci U S A 93, 10566–10571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wheeler SE, and Houk KN (2009) Substituent effects in cation/pi interactions and electrostatic potentials above the centers of substituted benzenes are due primarily to through-space effects of the substituents, J Am Chem Soc 131, 3126–3127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Starks CM, Back K, Chappell J, and Noel JP (1997) Structural basis for cyclic terpene biosynthesis by tobacco 5-epi-aristolochene synthase, Science 277, 1815–1820. [DOI] [PubMed] [Google Scholar]
- [39].Pandit J, Danley DE, Schulte GK, Mazzalupo S, Pauly TA, Hayward CM, Hamanaka ES, Thompson JF, and Harwood HJ Jr. (2000) Crystal structure of human squalene synthase. A key enzyme in cholesterol biosynthesis, The Journal of biological chemistry 275, 30610–30617. [DOI] [PubMed] [Google Scholar]
- [40].Baer P, Rabe P, Citron CA, de Oliveira Mann CC, Kaufmann N, Groll M, and Dickschat JS (2014) Hedycaryol synthase in complex with nerolidol reveals terpene cyclase mechanism, Chembiochem 15, 213–216. [DOI] [PubMed] [Google Scholar]
- [41].Baer P, Rabe P, Fischer K, Citron CA, Klapschinski TA, Groll M, and Dickschat JS (2014) Induced-fit mechanism in class I terpene cyclases, Angew Chem Int Ed Engl 53, 7652–7656. [DOI] [PubMed] [Google Scholar]
- [42].Nishio M (2004) CH/π hydrogen bonds in crystals, CrystEngComm 6, 130–158. [Google Scholar]
- [43].Hinman RL, and Lang J (1964) The Protonation of Indoles. Basicity Studies. The Dependence of Acidity Functions on Indicator Structure, J Am Chem Soc 86, 3796–3806. [Google Scholar]
- [44].Lawlor DA, More O’Ferrall RA, and Rao SN (2008) Stabilities and partitioning of arenonium ions in aqueous media, J Am Chem Soc 130, 17997–18007. [DOI] [PubMed] [Google Scholar]
- [45].Kersten RD, Diedrich JK, Yates JR 3rd, and Noel JP (2015) Mechanism-Based Post-Translational Modification and Inactivation in Terpene Synthases, ACS Chem Biol 10, 2501–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Blank PN, Barrow GH, Chou WKW, Duan L, Cane DE, and Christianson DW (2017) Substitution of Aromatic Residues with Polar Residues in the Active Site Pocket of epi-Isozizaene Synthase Leads to the Generation of New Cyclic Sesquiterpenes, Biochemistry 56, 5798–5811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Chen M, Al-lami N, Janvier M, D’Antonio EL, Faraldos JA, Cane DE, Allemann RK, and Christianson DW (2013) Mechanistic insights from the binding of substrate and carbocation intermediate analogues to aristolochene synthase, Biochemistry 52, 5441–5453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Aaron JA, Lin X, Cane DE, and Christianson DW (2010) Structure of epi-isozizaene synthase from Streptomyces coelicolor A3(2), a platform for new terpenoid cyclization templates, Biochemistry 49, 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Koksal M, Hu H, Coates RM, Peters RJ, and Christianson DW (2011) Structure and mechanism of the diterpene cyclase ent-copalyl diphosphate synthase, Nat Chem Biol 7, 431–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Blank PN, Pemberton TA, Chow JY, Poulter CD, and Christianson DW (2018) Crystal Structure of Cucumene Synthase, a Terpenoid Cyclase That Generates a Linear Triquinane Sesquiterpene, Biochemistry 57, 6326–6335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Flynn CM, and Schmidt-Dannert C (2018) Sesquiterpene Synthase-3-Hydroxy-3-Methylglutaryl Coenzyme A Synthase Fusion Protein Responsible for Hirsutene Biosynthesis in Stereum hirsutum, Appl Environ Microbiol 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Rabe P, Samborskyy M, Leadlay PF, and Dickschat JS (2017) Isoafricanol synthase from Streptomyces malaysiensis, Org Biomol Chem 15, 2353–2358. [DOI] [PubMed] [Google Scholar]
- [53].Kracht ON, Correia Cordeiro RS, Hakansson M, Stockmann J, Sander D, Bandow J, Senges CHR, Logan DT, and Kourist R (2019) Discovery of three novel sesquiterpene synthases from Streptomyces chartreusis NRRL 3882 and crystal structure of an alphα-eudesmol synthase, J Biotechnol 297, 71–77. [DOI] [PubMed] [Google Scholar]
- [54].Yamada Y, Kuzuyama T, Komatsu M, Shin-Ya K, Omura S, Cane DE, and Ikeda H (2015) Terpene synthases are widely distributed in bacteria, Proc Natl Acad Sci U S A 112, 857–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Hu Y, Chou WK, Hopson R, and Cane DE (2011) Genome mining in Streptomyces clavuligerus: expression and biochemical characterization of two new cryptic sesquiterpene synthases, Chem Biol 18, 32–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Nakano C, Tezuka T, Horinouchi S, and Ohnishi Y (2012) Identification of the SGR6065 gene product as a sesquiterpene cyclase involved in (+)-epicubenol biosynthesis in Streptomyces griseus, J Antibiot (Tokyo) 65, 551–558. [DOI] [PubMed] [Google Scholar]
- [57].Lin X, and Cane DE (2009) Biosynthesis of the sesquiterpene antibiotic albaflavenone in Streptomyces coelicolor. Mechanism and stereochemistry of the enzymatic formation of epi-isozizaene, J Am Chem Soc 131, 6332–6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Nakano C, Kudo F, Eguchi T, and Ohnishi Y (2011) Genome mining reveals two novel bacterial sesquiterpene cyclases: (−)-germacradien-4-ol and (−)-epi-alpha-bisabolol synthases from Streptomyces citricolor, Chembiochem 12, 2271–2275. [DOI] [PubMed] [Google Scholar]
- [59].Chou WK, Fanizza I, Uchiyama T, Komatsu M, Ikeda H, and Cane DE (2010) Genome mining in Streptomyces avermitilis: cloning and characterization of SAV_76, the synthase for a new sesquiterpene, avermitilol, J Am Chem Soc 132, 8850–8851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Nakano C, Horinouchi S, and Ohnishi Y (2011) Characterization of a novel sesquiterpene cyclase involved in (+)-caryolan-1-ol biosynthesis in Streptomyces griseus, The Journal of biological chemistry 286, 27980–27987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kumar RP, Morehouse BR, Matos JO, Malik K, Lin H, Krauss IJ, and Oprian DD (2017) Structural Characterization of Early Michaelis Complexes in the Reaction Catalyzed by (+)-Limonene Synthase from Citrus sinensis Using Fluorinated Substrate Analogues, Biochemistry 56, 1716–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Morehouse BR, Kumar RP, Matos JO, Olsen SN, Entova S, and Oprian DD (2017) Functional and Structural Characterization of a (+)-Limonene Synthase from Citrus sinensis, Biochemistry 56, 1706–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Montalvetti A, Bailey BN, Martin MB, Severin GW, Oldfield E, and Docampo R (2001) Bisphosphonates are potent inhibitors of Trypanosoma cruzi farnesyl pyrophosphate synthase, The Journal of biological chemistry 276, 33930–33937. [DOI] [PubMed] [Google Scholar]
- [64].O’Maille PE, Chappell J, and Noel JP (2004) A single-vial analytical and quantitative gas chromatography-mass spectrometry assay for terpene synthases, Analytical Biochemistry 335, 210–217. [DOI] [PubMed] [Google Scholar]
- [65].Ajikumar PK, Xiao WH, Tyo KE, Wang Y, Simeon F, Leonard E, Mucha O, Phon TH, Pfeifer B, and Stephanopoulos G (2010) Isoprenoid pathway optimization for Taxol precursor overproduction in Escherichia coli, Science 330, 70–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Kabsch W (2010) Xds, Acta Crystallogr D Biol Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Evans P (2006) Scaling and assessment of data quality, Acta crystallographica. Section D, Biological crystallography, 72–82. [DOI] [PubMed] [Google Scholar]
- [68].Potterton E, Briggs P, Turkenburg M, and Dodson E (2003) A graphical user interface to the CCP4 program suite, Acta crystallographica. Section D, Biological crystallography 59, 1131–1137. [DOI] [PubMed] [Google Scholar]
- [69].Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A, McNicholas SJ, Murshudov GN, Pannu NS, Potterton EA, Powell HR, Read RJ, Vagin A, and Wilson KS (2011) Overview of the CCP4 suite and current developments, Acta crystallographica. Section D, Biological crystallography 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, and Read RJ (2007) Phaser crystallographic software, J Appl Crystallogr 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, and Adams PD (2012) Towards automated crystallographic structure refinement with phenix.refine, Acta crystallographica. Section D, Biological crystallography 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, and Zwart PH (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta crystallographica. Section D, Biological crystallography 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Emsley P, Lohkamp B, Scott WG, and Cowtan K (2010) Features and development of Coot, Acta crystallographica. Section D, Biological crystallography 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Edgar RC (2004) MUSCLE: a multiple sequence alignment method with reduced time and space complexity, BMC Bioinformatics 5, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Edgar RC (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput, Nucleic Acids Res 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Waterhouse AM, Procter JB, Martin DM, Clamp M, and Barton GJ (2009) Jalview Version 2--a multiple sequence alignment editor and analysis workbench, Bioinformatics 25, 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.