

Abstract
Mitochondrial inner membrane protein (Mitofilin or Mic60) is a mitochondria-shaping protein that plays a key role in maintaining mitochondrial cristae structure and remodeling. We recently showed that Mitofilin knockdown in H9c2 myoblasts induces mitochondrial structural damage resulting in mitochondrial dysfunction that is responsible for cell death via apoptosis. Here, we investigated the role of Mitofilin regulation in ischemia/reperfusion (I/R) injury and studied the relationship between Mitofilin and Cyclophilin (CypD), a key regulator of mitochondrial permeability transition pore (mPTP) opening. C57Bl6 male mice hearts were subjected to different ischemia times (15, 30, or 45 min) followed by a 2h reperfusion period, or 45 min ischemia followed by 0, 15, 30, 60, or 120 min reperfusion to determine the impact of ischemia or reperfusion times on Mitofilin levels and its interaction with CypD. We found that the increase in myocardial infarct size and the reduction of mitochondrial calcium retention capacity were concomitant with Mitofilin reduction as a function of ischemic duration. We also found that 15 min reperfusion after 45 min ischemia was sufficient to cause a reduction of Mitofilin levels compared to sham, while 45 min ischemia alone was not enough to cause a significant decrease of Mitofilin. We revealed that the c-terminus coiled-coiled domain of Mitofilin is important for its interaction with CypD and the deletion of this identified sequence resulted in a loss of Mitofilin-CypD link, dissipation of mitochondrial membrane potential and increase in cell death. A decrease of the levels of Mitofilin was also associated with mitochondrial structural integrity damage, increased reactive oxygen species (ROS) production, and calpain activity. Our results indicate that Mitofilin physically binds to CypD in the inner mitochondrial membrane and the disruption of this interaction may play a critical role in the increase of mitochondrial dysfunction and initiation of myocytes’ death after I/R injury.
Keywords: Inner mitochondrial membrane (immt, mitofilin); Cyclophilin D; mitochondrial dysfunction; ischemia/reperfusion injury; mitochondrial permeability transition pore (mPTP); mitochondrial structural integrity and function
INTRODUCTION
Mitochondria are organelles found in large numbers in cells and are well-known as essential sites for biochemical processes of respiration and energy (ATP) production. However, in response to stress-induced by several disease conditions including metabolic syndrome, neurodegenerative diseases, aging, and ischemia/reperfusion (I/R) injury, mitochondrial dysfunction occurs. Mitochondria have a double membrane structure: the outer membrane (OMM) and the inner membrane (IMM) that is folded inward to form layers (cristae) and hosts the electron transport chain machinery, which generates membrane potential necessary for ATP generation. As with all biological organelles, the function of mitochondria is tightly linked with their structural integrity.
Recently, several proteins present in both the outer and inner membrane have been found to play a role in the molecular mechanisms regulating cristae biogenesis and architecture. Mitochondrial contact site (MICOS) complexes 12 and 27 (Mic12 and Mic27) have been identified as important factors that play an essential role in MICOS complex formation [1]. Coiled-Coil Helix Cristae Morphology 1 protein (CHCM1)/Coiled-coil-helix-coiled-coil-helix domain 6 (CHCM1/CHCHD6) has been reported to harbor a coiled-coil helix-domain at its c-terminal end and is predominantly localized to the mitochondrial inner membrane. Coiled-coil-helix-coiled-coil-helix domain 3 (CHCHD3) plays a key role in maintaining cristae integrity and mitochondrial function. Deletion of CHCHD3 led to a complete loss of both Mitofilin and Sam50 proteins [2]. Coiled-coil-helix-coiled-coil-helix domain 10 (CHCHD10) mutations led to MICOS complex disassembly, loss of mitochondrial cristae, decrease in nucleoid number, and disorganization [3].
Mitochondrial inner membrane protein (IMMT or Mic60), also referred to as Mitofilin, is a ubiquitously expressed mitochondrial membrane protein [4] that was initially characterized as a heart muscle protein based on the high abundance of its mRNA in rat embryonic heart [5]. Mitofilin has been proposed to be a critical organizer of mitochondrial cristae morphology and thus indispensable for normal mitochondrial function [6]. In humans, two cDNAs have been isolated that encode different isoforms of Mitofilin and both isoforms are derived by alternative splicing, and encode two proteins immt-1 and immt-2 of 88 and 90 kDa [7]. Malsburg et al., reported that Mitofilin is part of a large multi-subunit protein complex in the IMM, termed mitochondrial inner membrane organizing system (MINOS) that controls cristae morphology [8]. Further, apolipoprotein O (APOO) and apolipoprotein O-like protein (APOOL) were both identified as putative components of the Mitofilin/MINOS protein complex [9]. It has been proposed that MINOS integrity is required for the maintenance of the characteristic morphology of the IMM, with an inner boundary region closely faced to the OMM and cristae membranes [10].
More recently several studies using patients and animal models related mitochondrial cristae regulation with several diseases including obesity, neurodegenerative diseases, osteoporosis, diabetes, osteogenesis and cardiac dysfunctions [11];[12];[13]. Transgenic overexpression of Mitofilin has been found to preserve mitochondrial structure leading to a restoration of mitochondrial function [12] and promoting cardiac hypertrophy [14]. On the contrary, knockdown of Mitofilin in HeLa cells led to a fragmentation of the mitochondrial network and disorganization of the cristae, which caused cytochrome release and apoptosis [15]. Using cultured H9c2 cardiomyoblasts and HEK 293 cells treated with Mitofilin siRNA, we recently showed, that Mitofilin knockdown increases mitochondrial dysfunction and cell apoptosis via an AIF-PARP cleavage axis through activation of both calpain and PARP, leading to nuclear fragmentation and S phase cell cycle arrest [16]. The present study are focused to determine the impact of Mitofilin regulation in the impairment of mitochondrial structure morphology and function involved in the mechanism of the I/R-induced cardiomyocyte death.
In our previous study, we found that the mitochondrial dysfunction observed in response to Mitofilin knockdown was associated with enhanced reactive oxygen species (ROS) generation, depleted ATP production and dissipation of the mitochondrial membrane potential. These results indicated a possible involvement of the opening of the mitochondrial permeability transition pore (mPTP) [16] in Mitofilin knock down actions. The mPTP opening is well-recognized to play a crucial role in the mechanism of cell death after I/R injury [17–19]. Although several proteins have been reported to contribute to mPTP formation, its molecular identity and mechanism are highly debated and not completely understood [20]. Nevertheless, there is a consensus on the role of Cyclophilin D (a peptidyl-prolyl cis-trans isomerase, CypD) as a regulator of the mPTP opening [21]. This assertion is supported by reports indicating that knockdown of CypD in mice causes marked delay of the mPTP opening [22–24]. Mice lacking CypD have displayed reduced mitochondrial dysfunction and resistance to necrotic cell death induced by reactive oxygen species (ROS) and Ca2+ overload suggesting protection of mitochondrial structural integrity [24]. Furthermore, CypD-deficient mice exhibited a high level of resistance to I/R-induced cardiac injury. However, whether CypD’s role in the preservation of mitochondrial morphology influences the delay of mPTP opening still needs to be clarified. Although, CypD null mice (CypD null mitochondria) are more resistant to Ca2+ and ROS resulting in a marked desensitization of mPTP to Ca2+ and oxidative stress [24], nonetheless, these mice still display mPTP activation. Therefore, one should be careful in the interpretation of data obtained from experiments using genetic CypD inhibition, which only show desensitization but not complete blockage of mPTP [20].
Although the role of CypD in I/R injury has been studied using CypD null mice or heterozygote mice [22, 25], how I/R injury insult regulates CypD levels during the early moment of reperfusion is not clear. Wang et al., have shown that CypD expression was increased in response to I/R after (30, 60, or 90 min) [25]. It is not clearly explained whether these authors were referring to different times of ischemia or reperfusion. However, in both conditions, it is questionable whether an increase in CypD can happen after such a short time, as protein transcription and translation need a longer time to be implemented. Also, activation of mPTP opening that is associated with a reduction of mitochondrial calcium retention capacity can be observed after only 10 min reperfusion after ischemia [26, 27], it is, therefore, difficult to attribute this effect to upregulation of CypD protein expression. Hence, the impact of I/R injury insult in the levels of CypD during the reperfusion time course needs to be determined. Therefore, in this study, we measured the levels of both CypD and Mitofilin after, i) the same ischemia followed by variable duration of reperfusion, and variable duration of ischemia followed by a consistent duration of reperfusion.
Because both Mitofilin, a structural protein, and CypD are located in the IMM, in this study, we investigated whether there is a direct interaction between these two proteins. We discovered that Mitofilin levels reduces after I/R injury at the early moment of reperfusion. We report that reduction of Mitofilin levels due to I/R stress disrupts the direct association between Mitofilin and CypD in the IMM. Reduction in Mitofilin results in an increase in mitochondrial damage and sensitivity to calcium overload, increase in ROS production as well as dissipation of mitochondrial membrane potential (MMP) leading to mitochondrial dysfunction. The destruction of the Mitofilin-CypD interaction might play an important role in the initiation of mechanisms that subsequently promote cardiomyocyte death observed in I/R injury.
MATERIALS AND METHODS
Experimental protocols.
All protocols followed the Guide for the Care, and Use of Laboratory Animals (US Department of Health, NIH), and received UT Health Science Center at San Antonio Institutional Animal Care and Use Committee (IACUC) institutional approval. Animals were housed in the animal-specific pathogen-free facility at UTHSCSA’s main campus in cages with standard wood bedding and space for five mice. The animals had free access to food and drinking water and a 12-hour shift between light and darkness. The animals were selected randomly and the data analysis was performed by a blinded investigator.
Langendorff preparation and heart perfusion.
All materials used were purchased from Sigma-Aldrich (St. Louis, MO), unless otherwise stated. Mice were anesthetized by intraperitoneal injection of ketamine (80 mg-kg−1 i.p.) and xylazine (8 mg-kg−1 i.p.), and heparin (200 Ul/kg) was used to prevent blood coagulation. The surgical procedures and protocol were similar to those described in our previous study [28]. Hearts were surgically removed and immediately arrested in cold (4°C) Krebs Henseleit bicarbonate buffer (KH) solution (mM): glucose 11, NaCl 118, KCl 4.7, MgSO4 1.2, KH2PO4 1.2, NaHCO3 25 and CaCl2 3, pH 7.4. The aorta was rapidly cannulated and the heart was retrograde-perfused at a constant rate (3 ml/min) in the Langendorff mode using KH buffer bubbled with 95% O2/5% CO2 at 37 °C. After 30 minutes of equilibration, global normothermic ischemia was induced by stopping KH buffer supply to the aorta for 15, 30, or 45 minutes. Different reperfusion duration (15, 30, 60, also 120 min) were used. We preferentially considered good hearts the ones that reached a minimum left ventricular developed pressure (LVDP) of 80 mmHg at the end of the basal perfusion (before ischemia, data not shown). The mouse model of I/R we used consists of 30 min equilibration, 35 min normothermic ischemia by stopping buffer followed by120 min reperfusion. This I/R injury protocol typically results in ~50% of infarct size [28]. Sham hearts were not subjected to I/R injury but only were perfused for the same duration as the I/R injury protocol.
Ca2+-induced opening of the mitochondrial permeability transition pore (mPTP)
Mitochondria isolation.
After 10 min of reperfusion, hearts were excised while still beating and immediately placed in cold Krebs-Henseleit buffer. The right ventricle was discarded and the area at risk (AAR) myocardium was harvested for mitochondria isolation. All procedures were carried out at 4 °C as previously described in [29]. Briefly, the heart tissue was placed in isolation buffer A (mM): 70 sucrose, 210 mannitol, 1 EDTA and 50 Tris-HCl, pH 7.4. The tissue was finely minced with scissors and homogenized in the same buffer A (1 ml buffer/0.15 g of tissue) using Kontes and Potter-Elvehjem tissue grinders. The homogenate was centrifuged at 1,300 x g for 3 min, and the supernatant was filtered through cheesecloth and centrifuged at 10,000 x g for 10 min. The supernatant was discarded and the pellet was gently washed 3 times with 500 μl of buffer B (in mM): 150 sucrose, 50 KCl, 2 KH2PO4, 5 succinic acid, and 20 Tris/HCl, pH 7.4, and resuspended with 50 μl of the same buffer. Mitochondrial protein concentration was assayed using the Bradford method and adjusted to a final concentration of 25 mg/ml.
Calcium Retention Capacity (CRC).
Adapted from previously described by [30], CRC was defined as the amount of Ca2+ required to trigger a massive Ca2+ release by mitochondria, which corresponds to mPTP opening. Mitochondrial CRC was measured spectrofluorometer using calcium green-5N (Invitrogen) and excitation and emission wavelengths set at 500 and 530 nm, respectively. Isolated mitochondria (250 μg of mitochondrial protein/ml) were added to a spectrofluorometer cuvette containing 2 ml of buffer B supplemented with 0.5 μΜ calcium green-5N under constant stirring. Upon the addition of mitochondria, there is a progressive reduction of the Ca2+ in the media due to mitochondrial Ca2+ uptake reaching a quasi-steady-state in ~90 s. At this time, Ca2+ pulses of 20 nmoles/mg of mitochondrial protein were added every 60 s to the cuvette. The Ca2+ pulses induced a peak of extra-mitochondrial Ca2+ concentration that returns to near-baseline levels as Ca2+ enters the mitochondrial matrix. With increase in mitochondria calcium loading, extra-mitochondrial Ca2+ starts accumulating until the addition of Ca2+ leads to a sustained Ca2+ increase indicating a massive release of mitochondrial Ca2+.
Rat H9c2 cardiomyoblast and HEK293 cell line culture
Rat H9c2 cardiomyoblasts and HEK293 cell line were purchased from the American Type Culture Collection (ATCC® Number CRL-1446 and CRL-11268 respectively) and cultured in Dulbecco’s modified Eagle’s medium (DMEM; Invitrogen Life Technologies) supplemented with 10% fetal bovine serum (FBS; Gibco-BRL, Grand Island, NY, USA), 100 U/ml penicillin/streptomycin and grown in an atmosphere of 5% CO2/95% humidified air at 37°C. The culture medium was changed every second day. Cells were used between passage 4 and 7, at 70-80% confluence. The cells were selected randomly, and the data analysis was performed by a blinded investigator.
Isolation of Ventricular Myocytes from Left Ventricle.
Animals were injected intraperitoneally with heparin (200 IU/kg) and 20 min later they were anesthetized with ketamine (80 mg-kg−1 i.p.) and xylazine (8 mg-kg−1 i.p.). Hearts were harvested and instantaneously arrested in ice-cold PBS (KCl 2 mM, KH2PO4 1.5 mM, NaCI 138 mM, Na2HPO4 8.1 mM) to remove excess blood. Hearts were transferred to ice-cold Tyrode’s solution [NaCl 130 mM, KCl 5.4 mM, MgCl2 1 mM, Na2HPO4 0.6 mM, Glucose 10 mM, Taurine 5 mM, 2,3-butanedione monoxime 10 mM, and HEPES 10 mM, pH 7.4, oxygenated with 95% (vol/vol) O2-5% (vol/vol) CO2], and mounted on a modified Langendorff apparatus at a constant pressure of 80 cm H2O. After 5 min of perfusion at 37 °C with Tyrode’s solution, the heart was perfused for 10 min with Tyrode’s solution containing 186 U/mL Collagenase Type-2 and 0.5 U/mL Protease Type-XIV, and then washed for 5 min with a high K+ buffer (KB) [KCl 25 mM, KH2PO4 10 mM, MgSO4 2 mM, Glucose 20 mM, Taurine 20 mM, Creatine 5 mM, K-Glutamate 100 mM, Aspartic acid 10 mM, EGTA 0.5 mM, Hepes 5 mM, and 1% (wt/vol) BSA, pH 7.2 oxygenated with 95% O2-5% (vol/vol) CO2]. After washing, the left ventricle was cut into pieces in KB solution to dissociate cells. Isolated ventriculocytes were filtered (100-μm strainer), and centrifuged 2 min at 1,000 × g for further use.
Generation of Mitofilin mutation plasmids
The mouse Mitofilin cDNA construct was cloned into the Myc-tagged pCDNA3.1 entry vector in the frame using the restriction enzyme XhoI/Kpnl (New England Biolabs, Ipswich, MA). Each deletion mutant (Mitofilin-mutation1 – 4) were constructed by PCR amplification using a Common Forward primer: 5’CCGCTCGAGGCCACCATGCTGCGGGCCTGTCAGTTA3’ and unique reverse primers specific to each deletion as follows: mutation 1 reverse: 5’CGGGGTAC CTGCACCCTCCACTGTTCGCCA3’, mutation 2 reverse: 5’CGGGGTACCAGCAATCTCTCTTTTCTTTGC3’, mutation 3 reverse: 5’CGGGGTACCAGTAGAGAGTTTGCCAGCTAG3’, mutation 4 reverse 5’CGGGGTACCATTCTCCATGGCATCTCTGAC3’. We amplified the cDNAs of full-length or each c-terminal deletion mutants of Mitofilin by PCR with the primers specified above and ligated them into the XhoI/KpnI site of the myc-PCMV6 vector. All constructs were verified using sequencing. Transfections were performed using the Lipofectamine 3000 transfection reagent according to the manufacturer’s protocol.
In situ detection of MPTP Opening
mPTP opening was also determined using a Transition Pore Assay Kit (MitoProbe; Life Technologies) based on the manufacturer’s instructions. mPTP opening in H9c2 cells was assessed by the Calcein-cobalt (Co2+) quenching technique that tracks the fluorescent dye Calcein trapped in the mitochondria [31],[32]. Co2+ is a metal channel selective fluorescence-quencher that does not cross the mitochondrial membrane. Calcein diffuses into cells passively and accumulates into the cytosol and mitochondria to liberate the highly polar fluorescent dye Calcein. Briefly, cells were stained with MPTP Staining Dye (Calcein) showing a cumulative fluorescence signal from both cytoplasm and mitochondria (Tube 2). Tube 1 contains a sample without treatment; it is used for instrument setup. In Tubes 3, samples were stained with 2 μM Calcein and 1 mM CoCl2 in Hank’s Balanced Salt Solution (HBSS)/Ca2+ at 37°C for 15 min while protected from light to determine mitochondrial fluorescence only. After two washes with HBSS/Ca2+, fluorescence Calcein of cells (~30,000) for each experiment was detected by high-content screening at 488/530 nm using an LSR II flow cytometer (BD Biosciences). The calcein fluorescence intensity in Calcein+Cobalt tubes was presented as the degree of MPTP activation and subsequent depolarization of the mitochondrial membrane.
Western Blot analysis
Equal amounts of protein were loaded in each well of 4-20% Tris-glycine gels (Bio-Rad) as recently described [33]. After electrophoresis for 90 min at 125 V of constant voltage, the gel was blotted onto a nitrocellulose membrane by electrophoretic transfer at 90 V of constant voltage for 1.5 h. The membrane was washed, blocked with 5% blocking solution, and probed with various primary antibodies at 4°C overnight. Antibodies: Mitofilin (Polyclonal, Proteintech 10179-1-AP, 1 μg/ml, and Monoclonal, Invitrogen (2E4AD5, 45-6400) at 2 μg/mL for WB), Cyclophilin D (Novus Biologicals, 455900, 1 μg/ml), Calpain 10 (Santa Cruz, (C-20), sc-48454 1 μg/ml), Calpain 1 Santa Cruz, (D-11), sc-271313 1 μg/ml), Myc-Tag Antibody (Cell Signaling, #2272 1 μg/ml), VDAC1 (Santa Cruz (B6) sc-390996, 5 μg/ml), GAPDH (Cell Signaling, D4C6R #9716, 5 μg/ml). After washing, membranes were incubated for 1h at room temperature with the corresponding fluorophore-conjugated secondary antibodies (goat anti-rabbit Alexa 680, 20 ng/ml; goat anti-mouse IR Dye 800CW, 10 ng/ml). The total protein loading was measured by immersing the membrane in the ponceau S solution (CAS No.: 6226-79-5). After washing, bands were visualized using an infrared fluorescence system (Odyssey Imaging System, Li-COR Biosciences).
Mitochondrial membrane potential
Mitochondrial membrane potential was qualitatively assessed in transfected rat myoblasts using JC-1 dye (Cayman, 15003) [34]. After, 48h transfection, media were carefully removed from twenty four-well plates and cells washed twice with PBS, 0.5 ml of DMEM containing FBS. Transfected cells were incubated with and without a cytotoxic agent hydrogen peroxide (H2O2) 0.5 mM) supplemented in the media for 4 hours. Cells were resuspended in DMEM containing FBS, stained by adding JC-1 (10 μg/ml) and cultured for 30 min at 37 °C with continuous gently shaking. At the end of the incubation, the media was removed, and cells were washed twice with normal PBS. To image the cells, 0.5 ml of DMEM containing FBS was added to each well and image taken with fluorescence microscopy.
Mitochondrial ROS production
Mitochondrial ROS production was assessed as described earlier [35]. Mitochondrial ROS generation was measured with a spectrofluorometer (560-nm excitation and 590-nm emission) from 0.125 mg/ml of mitochondrial protein incubated in a solution containing: 20 mM Tris, 250 mM sucrose, 1 mM EGTA, 1 mM EDTA, and 0.15% bovine serum albumin adjusted to pH 7.4 at 30 °C with continuous stirring. ROS was measured with the H2O2-sensitive dye Amplex red (10 μM) according to the manufacturer’s instructions (Invitrogen). Hydrogen peroxide (H2O2) levels were measured from a calibration curve obtained from the fluorescence emission intensity as a function of H2O2 concentration. The sodium salt of glutamate/malate (3 mM) was used to activate complex I of the mitochondrial electron transport chain.
Calpain activity assay
To measure calpain activity, we used the Calpain Activity Assay kit (Abcam, catalog no. ab65308) following the supplier’s instructions. Mitochondria were suspended in Extraction buffer, lysed and the concentration was determined. Equal concentrations of lysates were incubated with reaction buffer, calpain substrate, and inhibitor for 1 h at 37°C. Reading were then taken on a fluorescence spectrofluorometer at excitation/emission wavelengths of 400/505 nm.
Sucrose gradient analysis
Equal amounts of total lysed proteins from sham and after I/R hearts were carefully layered onto the top of a 10-45% (v/v) sucrose gradient. The sucrose gradient was prepared in PBS, pH 7.4, containing 1% Triton X-100 after ultracentrifugation at 50,000 rpm for 16 h at 4°C. Ten fractions (400 μl) were collected from the bottom of the tube. Mitofilin and CypD were detected by Western blot and protein loading assayed by silver staining.
Co-immunoprecipitation assay
Heart tissues obtained after I/R and rat myoblasts were lysed in lysis buffer (150 mM NaCI, 50 mM Tris, 5 mM EDTA, 10 mM HEPES, 0.1% octylphenyl-polyethylene glycol (IGEPAL CA-630), 0.25% sodium deoxycholate, pH 7.4, plus Complete Protease Inhibitor Cocktail Tablets). Lysates were centrifuged (10 min, 13,000 x g, 4 °C) and the supernatants were precleared with 10^l protein A/G resin/mg protein (1 h, 4 °C, shaking) (Pierce Biotechnology, Inc.) and centrifuged 2 min at 2,000 x g. The precleared lysates (1 mg protein) were incubated overnight at 4 °C with 10 μl antibody saturated protein A/G resin (2 μg Ab/10 μl resin, 2 h at 4 °C, shaking) in a final volume of 500 μl lysis buffer. Samples were centrifuged 2 min at 2,000 x g and washed five times with Buffer A. The immunoprecipitated proteins were eluted from the beads with 30 μl 3x Laemmli sample buffer (37 °C, 1 h). After centrifugation (3 min, 13,000 x g, 4 °C), immunoprecipitated proteins, as well as lysates (input), were analyzed by SDS/PAGE and immunoblotting (Western blot).
Immunofluorescence staining
The cells passage 4-7, were plated on 8-well glass chamber slides and allowed to reach 70-80% confluence. The media was changed every other day. 24 hours prior to the Mitofilin siRNA transfection, the media was changed to OPTI-MEM serum and antibiotic-free media (Life Technologies). The next day, the cells were transfected with Mitofilin siRNA or scrambled siRNA according to the manufacturer’s protocol. After 24 h, the cells were fixed and stained with various antibodies: anti-Mitofilin antibody (Abcam, Cat: ab110329). For adult cardiomyocyte immunolabelling, freshly harvested cells were fixed and stained with anti-Mitofilin antibody (Abcam, Cat: ab110329). Secondary antibodies used were Alexa Fluor 488 Goat anti-rabbit (Abcam, Cat: ab150077), Alexa Fluor 488 Goat anti-mouse (Abcam, Cat: ab150113) and Alexa Fluor 647 Goat anti-mouse (Abcam, Cat: ab150119). Imaging was taken on a Zeiss Axiovert 200M inverted motorized fluorescence microscope (Carl Zeiss Microscope, Jena, Germany) and super-resolution microscopy.
Electron microscopy
The mouse heart tissues and isolated mitochondria from sham and after I/R were imaged by electron microscopy to observe mitochondrial quality and morphology. Heart tissues and mitochondria were immediately fixed in 2.5% (wt/vol) glutaraldehyde (Fluka) and stored in the same solution at 4 °C overnight. The dissected heart tissues and mitochondria were washed with PBS, post-fixed in 2% (wt/vol) osmium tetroxide for 2 h at room temperature. Fixed dissected tissues and mitochondria were dehydrated in a graded alcohol series and embedded in Eponate 12 medium. The blocks were cured at 60 °C for 48 h, and sections (70 nm) were cut with an RMC ultramicrotome, and mounted on Formvar-coated grids. The sections were double-stained with uranyl acetate and lead citrate, and finally examined and imaged with a 100CX JEOL transmission electron microscope.
Protein identification/relative quantification by mass spectrometry
To determine the proteins that link to Mitofilin, we performed immunoprecipitation with anti-Mitofilin on mitochondria fractions from non-ischemic wild type hearts. Proteins eluted from the Mitofilin pull-down were separated by SDS-PAGE prior to mass spectrometry analysis. Gels were run ~ 2 cm and then regions of interest in each lane were subdivided into slices that were individually reduced/alkylated and digested with trypsin. The digests were analyzed by HPLC-electrospray ionization-tandem mass spectrometry on an Orbitrap Velos Pro (Thermo Scientific). Mascot (Matrix Science) was used to search the UniProt_mouse database and a database of common contaminants. The Mascot results files for the gel slices in each lane were combined for subset searching of the identified proteins by X Tandem, cross-correlation with the Mascot results, and determination of protein and peptide identity probabilities by Scaffold (Proteome Software). Relative quantities were determined by spectral counting.
Bimolecular fluorescence complementation (BiFC)
To determine whether Mitofilin and Cyclophilin D binds in the inner mitochondrial membrane, we performed bimolecular fluorescence complementation (also known as BiFC) assay. BiFC is a technology typically used to validate protein interactions. It is based on the association of fluorescent protein fragments that are attached to components of the same macromolecular complex. pBiFC-VC155 and pBiFC-VN173 were gifts from Chang-Deng Hu (Addgene plasmid# 22010 and # 22011) [36]. We cloned flag-CyPD cDNA and Myc-Mitofilin cDNA into the VC155 and VN173 constructs, respectively. H9c2 rat myoblasts grown on coverslips were transfected with either the Myc-Mitofilin-pBiFC-VN173 or Flag-CyPD-pBiFC-VC155 or both constructs using Lipofectamine 3000 (Life Technologies) according to manufacturer’s instructions. 24 hour post-transfection, cells were fixed and observed with Olympus FV-1000 confocal/MPE (Olympus FV-1000 confocal/MPE) using the YFP channel.
Statistical analysis
Error bars are the standard errors of the mean (±SEM) for a minimum of five independent hearts for myocardial infarct size assessment, and a minimum of three independent experiments for biochemical analysis. For cardiac infarct size, mitochondrial CRC, MTT assay and JC-1 assay, means were compared between groups using one-way ANOVA. For in situ mPTP opening assay in cells, mitochondrial ROS production and calpain activity assay, the student’s t-test was performed. Under the ANOVA model, pair wise mean comparisons were judged significant using the Tukey studentized range criterion. SPSS, version 13.0 (SPSS Inc, Chicago, IL) was used to carry out the computations. Because all outcomes were continuous, results were summarized with means ± SEMs. P<0.05 was considered statistically significant.
Results
Mitofilin levels are reduced during early reperfusion and the degree of Mitofilin loss is dependent on the severity of the I/R stress.
To determine the impact of Mitofilin in the mechanism of I/R injury, we measured the level of Mitofilin in isolated mitochondria after 45 min ischemia (Isch) followed by reperfusion for 0, 15, 30, 60 and 120 min. Western blot performed in mitochondrial fractions showed that Mitofilin expression was reduced after I/R versus sham. In fact, Figure 1A shows that after 15 min reperfusion, Mitofilin expression is decreased to 73% compared to sham (100%). Because, the levels of Mitofilin were similar after ischemia without reperfusion versus sham, and was significantly reduced after only 15 min reperfusion, we concluded that Mitofilin loss occurs mostly during the early moment of reperfusion (before 15 min).
Figure 1. Reduction in Mitofilin expression is associated with an increase in myocardial infarct size.

A. Representative immunoblots and graph (mean of 4 separate experiments) showing reduction of Mitofilin levels in function of reperfusion durations of 0 (R0=I, ischemia only), 15, 30, 60, 120 min after the ischemic duration of 45 min compared to sham (non-ischemic). Values are expressed as mean±SEM; *P<0.05 versus. Control group, (n= 4 hearts/group). B. Representative immunoblots and graph (mean of 4 separate experiments) showing reduction of Mitofilin levels with an increase in ischemic duration (15, 30 and 45 min) followed by the same time of reperfusion (10 min) compared to sham (non-ischemic). *P<0.05 versus. control group, (n= 4 hearts/group). C. Confocal images showing localization of Mitofilin in isolated cardiomyocytes (top) and mitochondria (bottom) from non-ischemic and after 45 min ischemia followed by 30 reperfusion. Note that Mitofilin expression is reduced after ischemia compared to sham. D. Confocal and high-resolution microscopy (STED) images of isolated mitochondria labeled with Mitofilin. A2 and B2 are corresponding images of A1 and B1 taken with high-resolution microscopy. Note that Mitofilin spatial organization of mitochondrial cristae morphology is almost extinct after I/R as compared to sham, in which Mitofilin is still dense. E. Representative images of heart slices after TTC staining showing increased myocardial infarct size in sham (non-ischemic) and different durations of ischemic (15, 30 and 45 min) followed by the same reperfusion (2h). F. Graphs showing the percentage of myocardial infarct size in the left ventricle showing increased myocardial infarct size in sham (non-ischemic) and different durations of ischemia (15, 30 and 45 min) followed by the same reperfusion (2h). Values are expressed as mean±SEM; *P<0.05 versus. Control group, (n= 6 hearts/group). Note that I stands for ischemia, R0=I indicates ischemia alone, and R indicates reperfusion.
To confirm the role of I/R stress in the loss of Mitofilin, we compared Mitofilin levels after 15-, 30- and 45-min ischemia followed by the same duration of reperfusion (1 h) versus non-ischemic (sham). We found that Mitofilin was marginally reduced in 15 min ischemia group versus sham (95% versus 100%). However, after 30- and 45-min ischemia, Mitofilin levels were significantly reduced as compared to sham (77% and 65% versus 100%, respectively, (Fig. 1B). These results suggest that the degree of Mitofilin loss is dependent on the severity of I/R stress. Using immunocytochemistry and confocal microscopy approach, we observed the expression of Mitofilin in isolated cardiomyocytes and mitochondria in sham and after I/R groups. Figure 1C confirms that Mitofilin levels were reduced after I/R as compared to sham in both isolated cardiomyocytes and mitochondria. Furthermore, we determined the Mitofilin spatial distribution in mitochondria after I/R. To this end, we took images with confocal and the corresponding super-resolution microscopy (STED) for the same region of isolated mitochondria (Fig. 1D). We found that mitochondrial spatial organization of Mitofilin was disrupted after I/R with the reduction of Mitofilin expression compared to sham, in which Mitofilin was abundant and spatially well-organized along with the cristae morphology (Fig. 1D, A2 and B2). We measured myocardial infarct sizes in the function of ischemia duration, and we found that myocardial infarct size increases proportionally with ischemia duration similar to Mitofilin (Figs. 1E & 2C).
Figure 2. Reduction in CypD expression is associated with decreased Mitofilin levels and an increase in myocardial infarct size.
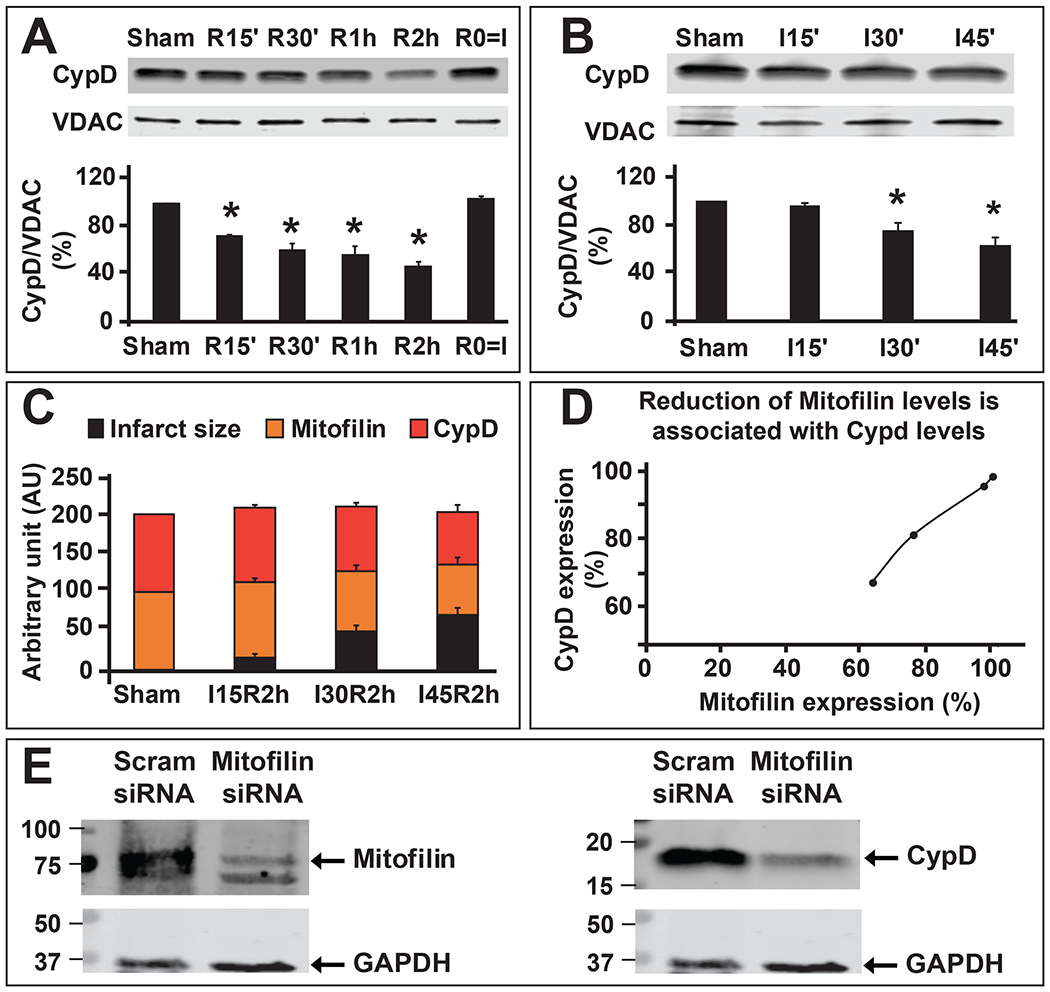
A. Representative immunoblots and graph (mean of 4 independent experiments) showing reduction of CypD levels after the durations of reperfusion for 0 (R0=I, ischemia only), 15, 30, 60, 120 min) 45 min of ischemia, compared to sham (non-ischemic). Values are expressed as mean±SEM; *P<0.05 versus. control group, (n= 4 hearts/group). B. Representative immunoblot and graph (mean of 4 independent experiments) showing reduction of CypD levels with an increase in ischemic duration (15, 30, and 45 min), compared to sham (non-ischemic) followed by the same time of reperfusion (10 min). Values are expressed as mean±SEM; *P<0.05 versus. control group, (n= 4 hearts/group). C. Graph showing the progression of myocardial infarct size (black) in the function of Mitofilin expression (orange) and CypD (red) in sham (non-ischemic) and after different durations of ischemia (15, 30, and 45 min) followed by two-hour reperfusion. Note that an increase in myocardial infarct size is inversely proportional to Mitofilin and CypD expressions after I/R. D. Graph showing the corresponding levels of Mitofilin and CypD levels. Note that a decrease in Mitofilin expression after I/R is proportional to the decrease in CypD levels suggesting a link in the regulation between both proteins during reperfusion. E. Immunoblots showing that reduction of Mitofilin levels (left) in H9c2 rats myoblasts transfected with Mitofilin siRNA is associated with a decrease in CypD levels (right).
Together, these results indicate that I/R stress-induced Mitofilin degradation and loss during the early moment of reperfusion and the degree of loss is dependent on the severity of the ischemic stress.
Mitofilin interacts with CypD in the inner mitochondrial membrane.
Mitofilin and CypD are localized to the IMM. We found that the levels of Mitofilin correlatively reduces as those of CypD after ischemia-reperfusion (Figs. 1A & B and 2A–D). Downregulation of Mitofilin using siRNA also caused a markedly decrease in CypD levels (Fig. 2E), suggesting that regulation of Mitofilin expression correspondingly impacts CypD levels. To determine whether these two proteins are associated in the IMM, we first performed mass spectrometry analysis on mitochondria from sham mice hearts as shown in Fig. 3. We identified a set of IMM proteins that bind to Mitofilin. Among the proteins identified were CypD, and several well-known proteins located in the IMM including but not limited to ATP synthase F1 subunit alpha and beta, MICOS complex subunits 19, 25, 26, 27, Voltage-dependent anion-selective channel 1, 2, and 3 as well as Adenine nucleotide translocator 1 (Fig. 3). To confirm our Mass spectrometry result, we performed immunoprecipitation (IP) with anti-Mitofilin antibody and we detected CypD with Western blot (IB) analysis, the reverse co-IP performed with anti-CypD antibody pulled down Mitofilin that was detected with Western blot analysis (Fig. 4A). However, we found that when mitochondria from I/R hearts were used, the pulled-down fractions of Mitofilin or CypD were reduced as compared to sham (Fig. 4A). Next, we checked the close association between these proteins using immunocytochemistry followed by confocal microscopy. Figure 4B shows the immunostaining of isolated mitochondria labeled with anti-Mitofilin (red), anti-CypD (green), and the overlay of both signals (yellow). We found that in sham (non-ischemic) mitochondria, Mitofilin predominantly co-localizes with CypD. However, when mitochondria isolated from I/R heart were used we observed a decrease in the number of Mitofilin that were associated with CypD. To confirm whether the colocalization signal was not coming from the secondary antibodies, we imaged mitochondria that were not co-labeled with Mitofilin antibody and observed the absence of the Mitofilin specific signal and the presence of CypD signal (Fig. 4C). To examine whether these two proteins interact intricately in the IMM, we performed a sucrose gradient analysis on mitochondrial fractions. In sham, we found that CypD (18 KDa) was typically detected in lower MW fractions (10 to 6) and in the highest MW fractions (1 and 2) it is association with Mitofilin (86 KDa) (Fig. 4D). This result, in agreement with our IP and immuno-labeling results, indicates a tight association between Mitofilin and CypD in the IMM. Importantly, with the same amount of protein ((Fig. 4E), arrow), immunoblot showed a dramatic reduction of Mitofilin-CypD interaction after I/R (pink arrows). To confirm an eventual direct interaction between these two IMM proteins, we used a bimolecular fluorescence complementation technique (BiFC) [37]. We determined the protein-protein interactions between Mitofilin and CypD using the VC155 and VN173, which are YFP protein fragments that can complement each other to form a functional fluorophore [38]. When fused to two different proteins, if both proteins interact or close spatial proximity, the two protein fragments (VC155 and VN173) of YFP assemble and emit fluorescence [37, 38]. Figure 5 shows that either the VC155-CypD or the VN 173-Mitofilin constructs failed to emit YFP fluorescence. However, co-expression of both constructs successfully emitted YFP fluorescence as compared to both constructs transfected separately (Fig. 5, bottom). We observed enhanced YFP fluorescence surrounding the nuclei of the cells, the region where mitochondria are predominantly localized in cells (Fig. 5, bottom), suggesting that both Mitofilin and CypD interact and are in close proximity.
Figure 3. Identification of mitochondrial proteins interacting with Mitofilin.

Left: Immunoblot showing different regions of a 4-20% gel stained with coomassie blue dye obtained for mass spectrometry after immunoprecipitation of non-ischemic mitochondria with an anti-Mitofilin antibody. Right: List of selected important mitochondria proteins that interact with Mitofilin identified by Mass spectrometry. Note the identification of CypD as a protein associated with Mitofilin.
Figure 4. Mitofilin co-localizes with CypD in the inner mitochondrial membrane.
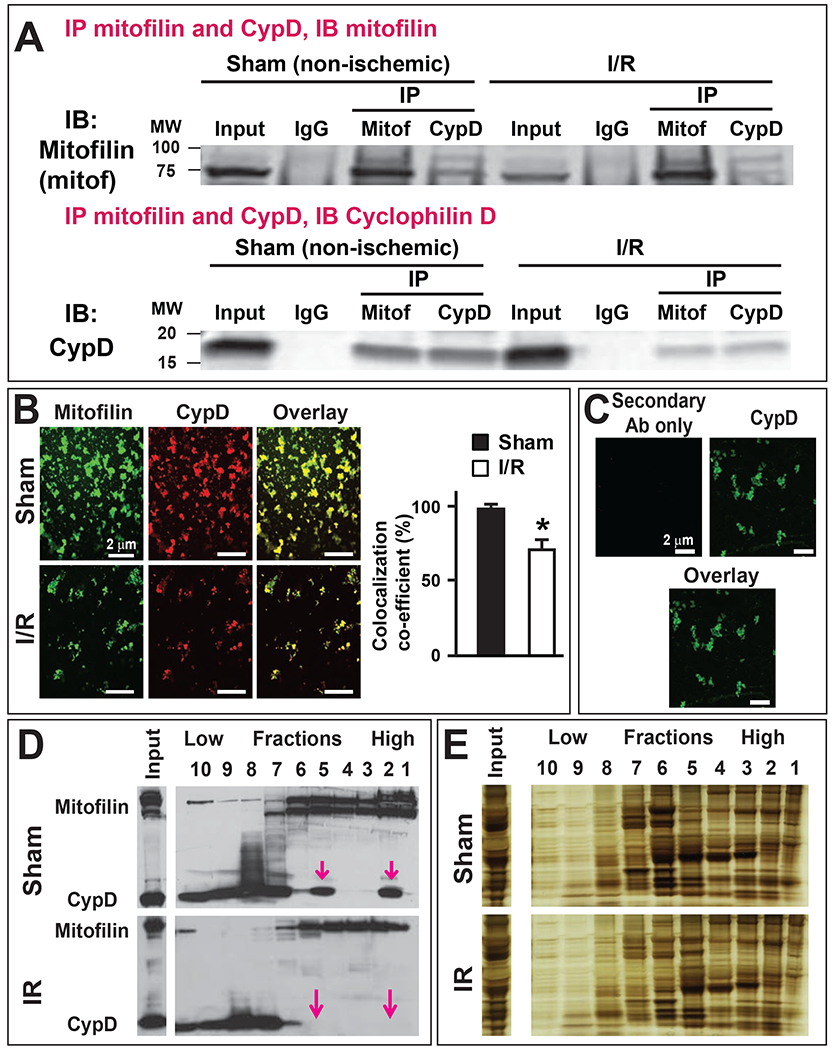
A. Immunoblots showing the link between Mitofilin and CypD in the inner mitochondrial membrane (IMM) in sham and after I/R (35 min ischemia followed by 30 min reperfusion. Top: Immunoprecipitation (IP) in mitochondrial lysate fractions with anti-Mitofilin antibody immunoprecipitated CypD that was revealed by Western analysis, and left: reverse Co-IP with anti-CypD confirm their interaction as Mitofilin was found in the immunoprecipitate. n= 3 independent experiments. Note that the immunoprecipitate fractions after I/R were reduced compared to sham (non-ischemic) suggesting reduction of the interaction between the two proteins. B. Confocal microscopy images of freshly isolated mitochondria from sham and after I/R labeled with Mitofilin (green) and CypD (red) and the overlay of both proteins (yellow). The graph shows the colocalization co-efficiency of the overlay indicating that the co-localization between Mitofilin and CypD is reduced after I/R (35 min ischemia followed by 30 min reperfusion), n= 3 experiments C. Confocal microscopy images of freshly isolated mitochondria from sham labeled with CypD (green) and the overlay. Note that the absence of Mitofilin (red) was used as a negative control to determine the specificity of the antibodies, n= 3 experiments D. Immunoblots showing the distribution of Mitofilin and CypD after sucrose gradient fractioning in normal (sham, top) and after I/R (bottom) mitochondria. Ten fractions were collected from the bottom of the tube (high molecular weight (MW) proteins) to the top (low molecular weight proteins). Note that after I/R, co-localized Mitofilin-CypD seen in some high MW fractions (2 and 5) is almost absent as compared to sham (pink arrows), n= 3 experiments E. Silver staining showing the protein loading control in D, n= 3 experiments.
Figure 5. Mitofilin directly interacts with CypD in the inner mitochondrial membrane.
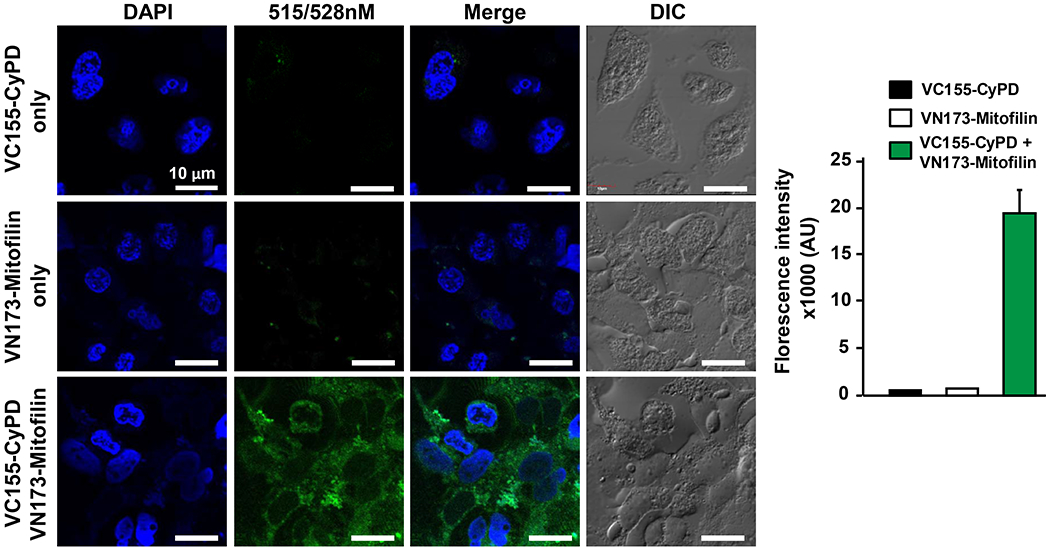
Bimolecular fluorescence complementation (BiFC) assay showing Mitofilin directly binding to CypD in the inner mitochondrial membrane. H9c2 rat myoblasts were transfected with either the Myc-Mitofilin-pBiFC-VN173 (middle) or Flag-CyPD-pBiFC-VC155 (top) or both constructs (bottom) using Lipofectamine 3000 according to manufacturer instructions. Image of cells was taken using Olympus FV-1000 confocal/MPE (Olympus FV-1000 confocal/MPE) using the YFP channel. DAPI was used to stain cell nuclei, while Differential Interference Contrast (DIC) imaging was used to obtain brightfield images. Note that the green fluorescence was only significantly increased being visible when both constructs were transfected indicating a direct contact between both proteins, n= 4 minimum images/group were taken from different wells.
Together, these results indicate that Mitofilin and CypD are tightly associated with the IMM and loss of Mitofilin seems to lead to a reduction of CypD level direct protein-protein interaction.
Mitofilin binds to CypD at the C-terminal site.
We discovered that Mitofilin and CypD interact in the IMM. To investigate the mechanism of the Mitofilin-CypD interaction, we determined the potential site in Mitofilin that binds to CypD. We mutated four residues in Mitofilin as shown in Fig. 6A. Mitofilin mutants were transfected in HEK293 cells (Fig. 6B) and the molecular weight of all the constructs confirmed with Western blot analysis using Myc-tagged antibodies (Fig. 6C). Using IP with anti Myc antibody followed by probing with anti CypD antibody, we exclusively detected CypD when IP was performed in lysates obtained from cells transfected with the full-length-Mitofilin-Myc (FL) and Mitofilin-mutant 4 (C-terminus deletion) (Fig. 7A–E). In fact, while in all transfections with different Mitofilin-mutants the corresponding Mitofilin-mutated protein was detected, however, CypD was only detected in the FL and Mitofilin-mutant 4 pull-downs. Conversely, when transfected cells were treated with a cytotoxic agent hydrogen peroxide (H2O2, 0.5 mM) for 4 h, MTT assay that reflects the number of viable cells showed a similar number of live cells in both full-length and Mitofilin-mutant 4 transfected cells. While the number of viable cells was significantly reduced in the other Mitofilin-mutant transfected cells (Fig. 7F). These results were corroborated by measuring the mitochondrial membrane potential using JC-1 dye. The red fluorescence indicates healthy mitochondria, while the green fluorescence indicates depolarized mitochondria. Figure 8 shows that mitochondrial membrane potential (MMP) measured in live cells that were transfected with full-length (FL) and the Mitofilin-mutant 4 plasmids exhibited higher MMP than the three other Mitofilin-mutants. In fact, cells transfected with FL and the Mitofilin-mutant 4 plasmids displayed high ratio of aggregate (red)/monomer (green) florescent intensity compared to the other three mutants 1, 2, and 3 (Fig. 8D). We found that after treatment H2O2, Cells transfected with mutation 1, 2, and 3 constructs display a reduced red fluorescence intensity than cell transfected with Mutant 4 plasmid, which was much similar to transfection with the full-length plasmid. However, the red fluorescence was slightly (but not significantly) reduced in untreated cells transfected with mutation 1, 2, and 3 compared to those transfected with mutant 4 and FL (Fig. 8B). In the other hands, H2O2-treated cells transfected with mutation 1, 2, and 3 constructs display a much higher green fluorescence intensity, which indicates a higher mitochondrial depolarization, than cell transfected with Mutant 4 plasmid, which was much similar to transfection with the full-length plasmid (Fig. 8C). However, the green fluorescence was not changed in untreated cells transfected with mutation 1, 2, and 3 compared to those transfected with mutant 4 and FL. Together, these observations indicate that a coiled-coil domain in the c-terminus of Mitofilin plays an essential role in the Mitofilin-CypD binding.
Figure 6. Mitofilin mutation plasmids used to determine Mitofilin region that bind to CypD.

A. Schematic representations of the structure of Myc-tagged Mitofilin mutants; 5: Full length (FL) Mitofilin, 4: Mitofilin-mutant 4, c-terminal (aa 476 – 757) deleted, 3: Mitofilin-mutant 3, c-terminal and C-C deleted (397 – 757) deleted, 2: Mitofilin-mutation2, (317 – 757) deleted, 1: Mitofilin-mutant 1 (279 – 757) deleted. B. Mutation efficiency of deletion in PCR mediated by high-fidelity DNA polymerase. C. Immunoblots showing the transfection efficiency of all the mutation plasmids in HEK293 cells.
Figure 7. Mitofilin binds to CypD via c-terminal.
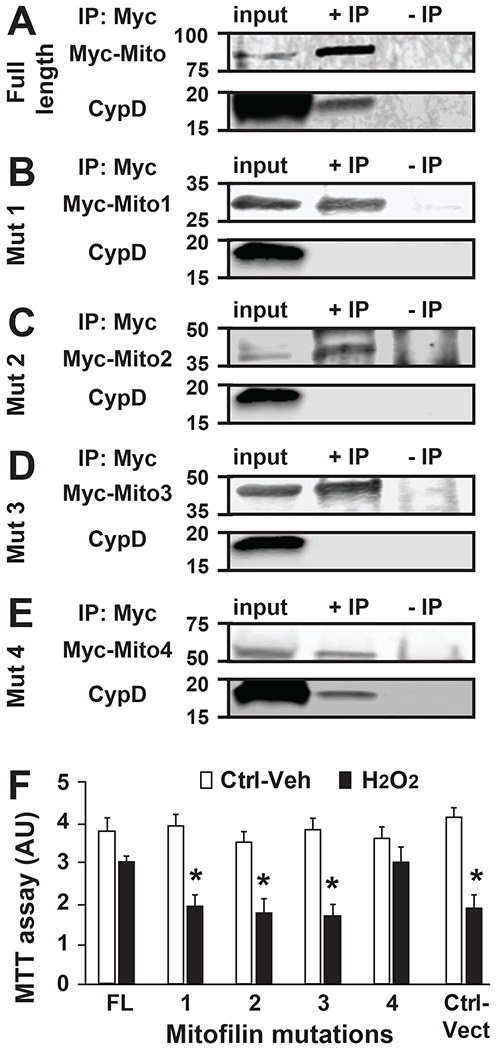
A-E. Immunoblots showing the C-terminal site in Mitofilin that bind to CypD. Mitofilin-mutated plasmids (1 to 5) were transfected in HEK293 cells and immunoprecipitation performed with Mitofilin-inserted-Myc in whole-cell lysate was followed by Western blot with anti-CypD antibody that allowed detection of CypD only when IP were performed in cells transfected with full-length-Mitofilin-Myc and Mitofilin-mutation4-Myc plasmid (A and E). This result indicates that the Mitofilin site that binds to CypD is located on the C-terminal that faces mitochondrial matrix IP: immunoprecipitation; IB: Western blot (immunoblot). F. Graph showing the level of cell death measure by MTT assay in HEK293 cells transfected with Mitofilin mutants as well as the full-length and treated with vehicle and the cytotoxic agent H2O2. Note that after treatment with H2O2, the cell death was reduced when the Mitofilin-CypD link was not disrupted (FL and 4) compared to plasmids (1, 2, and 3) and control vector. This observation suggests that the Mitofilin-CypD interaction in the IMM have a protective role against cell death. Values are expressed as mean±SEM; *P<0.05 full length (FL)-transfected plasmid (control) (n= 3/group triplicated in each). IP: immunoprecipitation; IB: Western blot (immunoblot).
Figure 8. Disruption of Mitofilin-Cyclophilin interaction dissipates mitochondrial membrane potential.
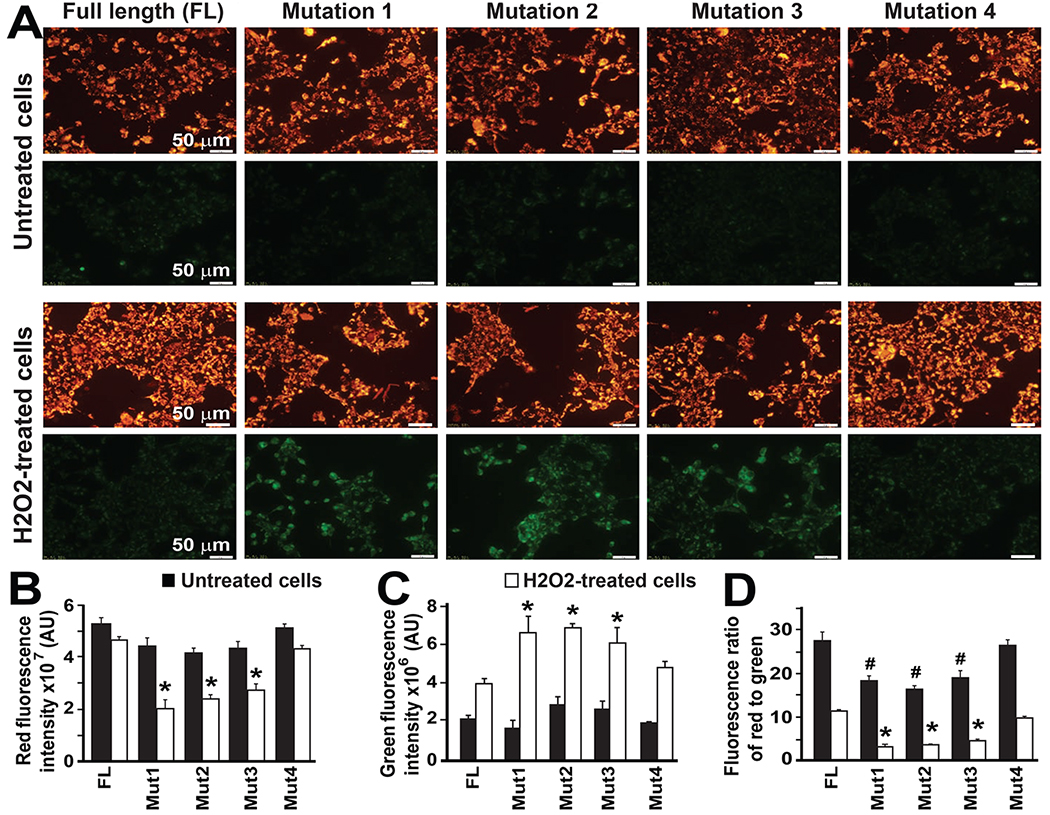
A. H9c2 rat myoblasts were transfected with Mitofilin-mutated plasmids (as in Figs. 6 & 7) and treated with or without a cytotoxic agent hydrogen peroxide (H2O2, 0.5 mM) for 4h. Cells were thereafter stained with JC-1 for 30 min and imaged with fluorescence microscopy. Polarized mitochondria are marked by punctate orange-red fluorescent staining. After depolarization, the orange-red punctate staining is replaced by diffuse green monomer fluorescence. The green fluorescence is present in cells transfected with mutations 1, 2, and 3 plasmids, indicating an increase in the number of depolarized mitochondria. B. Graph showing the red fluorescence intensity in normal H9c2 cells and after H2O2 treatment. Note the reduction in the red fluorescence in H2O2-treated cells (white bars) transfected with the mutants 1, 2, and 3 compared to the mutant 4 and the FL. In untreated cells (black bars) transfected with the mutants 1, 2, and 3, the red fluorescence is only slightly reduced versus mutant 4 and the FL. Values are expressed as mean ±SEM; *P<0.05 full-length (FL)-transfected plasmid (control), n= 3 experiments. C. Graph showing the green fluorescence intensity in normal H9c2 cells and after H2O2 treatment. Note the increase in the green fluorescence in H2O2-treated cells (white bars) transfected with the mutants 1, 2, and 3 compared to the mutant 4 and the FL indicating depolarization of mitochondria. In untreated cells (black bars) the green fluorescence was not different between the group versus the FL. Values are expressed as mean±SEM; *P<0.05 full-length (FL)-transfected plasmid (control), n= 3 experiments. D. Graph showing the fluorescence ratio of aggregate JC-1 dye (red)/monomer JC-1 dye (green) intensity in normal H9c2 cells and after H2O2 treatment. Note the reduction in the ratio of aggregate (red)/monomer (green) in both normal (black bars) and H2O2-treated cells (white bars) transfected with the mutants 1, 2, and 3 compared to the mutant 4 and the FL. Values are expressed as mean±SEM; *P<0.05 full-length (FL)-transfected plasmid (H2O2-treated cells), #P<0.05 full-length (FL)-transfected plasmid (untreated cells), n= 3 experiments.
Mitofilin regulation correlates with mitochondrial Ca2+ retention capacity after I/R.
The mPTP opening is well known to be a crucial effector that mediates cell death by apoptosis and necrosis [39, 40]. To determine whether Mitofilin reduction after I/R impacts the opening of mPTP, we first measured the mitochondrial calcium retention capacity (CRC) in sham and after different durations (15, 30 and 45 min) of ischemia followed by the same duration of reperfusion (10 min). We found that compared to sham, all the I/R groups displayed reduced mitochondrial CRC as a function of ischemia duration (Fig. 9A & B). Also, the reduction of mitochondrial CRC was proportional to the reduction of Mitofilin levels (Fig. 9C). To confirm whether Mitofilin regulation influences the opening of mPTP, we measured mPTP opening in H9c2 rat myoblasts transfected with Mitofilin siRNA and Mitofilin overexpression vector. We found that the mPTP opening was more active in cells transfected with Mitofilin siRNA as compared to scrambled siRNA (Fig. 9D). In contrast, cells transfected with Mitofilin-overexpression plasmid exhibited reduced in mPTP opening as compared to the PCMV6 vector (control) (Fig. 9E). Together, these results indicate that the reduction of Mitofilin levels during I/R is associated with a decrease in mitochondrial CRC (Fig. 9C). Therefore, Mitofilin regulation might influence the opening of mPTP.
Figure 9. Mitofilin expression modulates mitochondrial permeability transition pore opening.
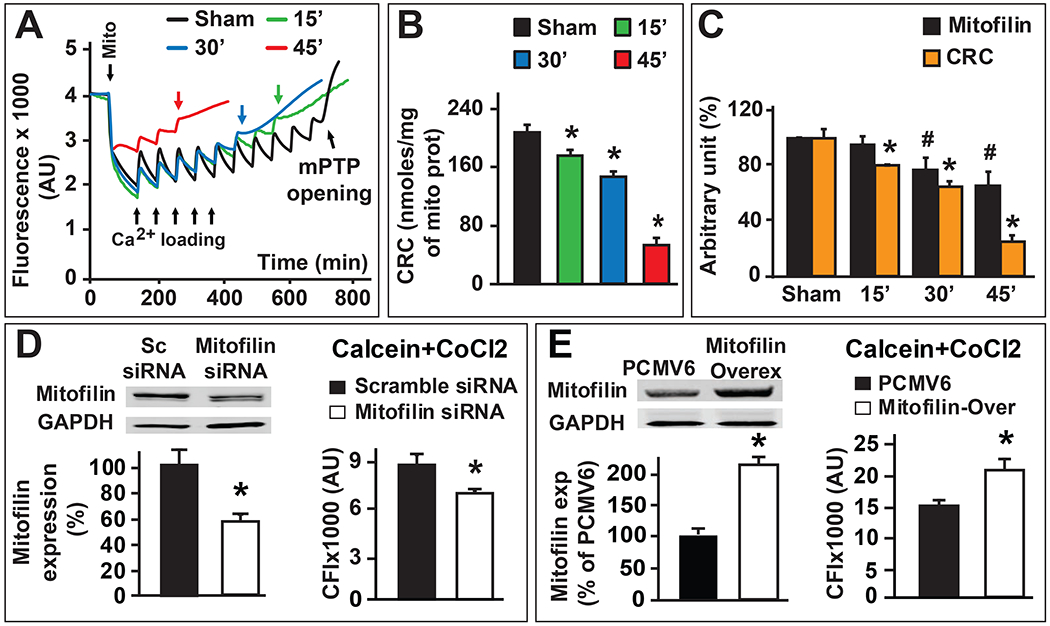
A. Typical spectrofluorometric recordings of Ca2+ overload in mitochondria isolated from hearts after 35 min ischemia followed by different reperfusion duration 15 min (green) 30 min (blue) and 45 min (red) and sham group (black). Arrowheads mark mitochondria addition and the initial mitochondrial Ca2+ uptake. Subsequent 20 nmol-mg−1 of protein Ca2+ pulses were delivered until a spontaneous massive release was observed, presumably to the opening of mPTP (arrows). B. Graphs of mean Ca2+ retention capacity values (amount of Ca2+ load needed to induce mPTP opening) in mitochondria isolated from hearts after 35 min ischemia followed by different reperfusion duration 15 min (green) 30 min (blue) and 45 min (red) and sham group (black). Values are expressed as mean ± SEM; *P < 0.05 I/R versus sham group, n= 4/group. C. Graph showing the progression of mitochondrial calcium retention capacity (CRC, orange) in the function of Mitofilin expression (black) in sham (non-ischemic) and after different durations of ischemia (15, 30 and 45 min) followed by two hour reperfusion. Note that a decrease in mitochondrial calcium retention capacity is proportionally associated with the reduction of Mitofilin after I/R. Values are expressed as mean ± SEM; *P < 0.05 I/R versus sham group (CRC), #P < 0.05 I/R versus sham group (Mitofilin), n= 4/group. D. Graph showing calcein fluorescence intensity in mitochondria after H9c2 cells were treated with Calcein/Cobalt indicating the degree of MPTP activation and subsequent depolarization of the mitochondrial membrane. The graph shows a decrease in fluorescence in cells transfected with Mitofilin siRNA compared to scrambled siRNA. This result suggests that the mPTP is more active and mitochondria more depolarized in cells transfected with Mitofilin siRNA compared to scramble siRNA. Values are expressed as mean ± SEM; *P < 0.05 I/R versus sham scrambled siRNA, n= 3 experiments. E. Graph showing calcein fluorescence intensity in mitochondria after H9c2 cells were treated with Calcein/Cobalt indicating the degree of MPTP activation and subsequent depolarization of the mitochondrial membrane. The graph shows an increase in fluorescence in cells transfected with Mitofilin overexpressed cells compared to cells transfected with PCMV6 suggesting that the mPTP is less active and mitochondria less depolarized in cells transfected Mitofilin overexpressed cells compared to cells transfected with PCMV6. Values are expressed as mean ± SEM; *P < 0.05 I/R versus sham scrambled siRNA, n= 3 experiments.
Ischemia/reperfusion increases ROS production and calpain activity.
Mitofilin plays a key role in maintaining mitochondrial cristae structure and remodeling and deletion of Mitofilin induces mitochondrial dysfunction in a cell line. We probed whether cell insult, which reduces Mitofilin levels, also exhibits mitochondrial structural changes. EM images were taken in cells transfected with different mutants (Fig. 10). We found that cells that were transfected with mutated constructs 1, 2, and 3 showed rounded mitochondria, in which cristae morphology were disrupted compared to non-transfected cells and cells transfected with mutant 4 and FL (Fig. 10A). This observation suggests that the destruction of the Mitofilin-CypD link promotes mitochondria cristae damage. These observations are similar to those others and we have observed in mitochondria after I/R, where cristae morphology were damaged when compared to sham [28]. We report here that I/R insult is associated with an increase in calpain10, but not calpain 1 (cytosolic isoform) levels (Fig. 10B), the calcium-activated enzyme (calpain) activity (Fig. 10C), and mitochondrial reactive oxygen species (ROS) production when complex I was activated (Fig. 10D). However, when complex II was activated, I/R induced a decrease in ROS production (Fig. 10E).
Figure 10. I/R insult increases calpain activity and ROS production when complex I, but not complex II, is activated.
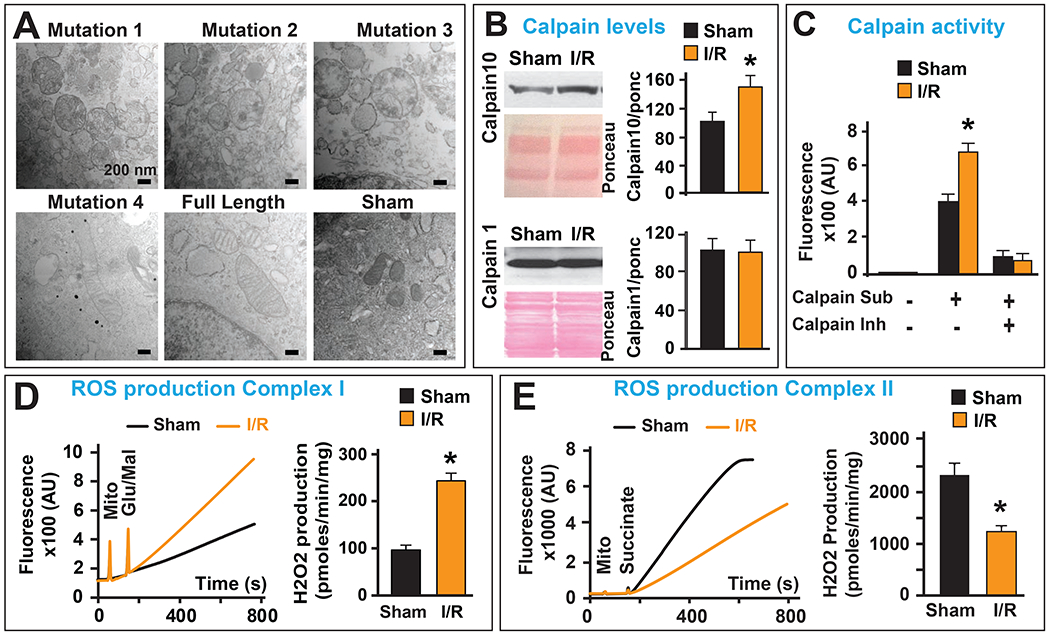
A. Electron microscopy images of mitochondria in cells transfected with the five mutations and normal (untransfected) showing an increase in cristae disruption after transfection with mutants (1, 2, and 3) but not in mutated construct 4, full length and untransfected cells. B. Representative immunoblots and graphs (mean of 3 separate experiments) showing an increase in mitochondrial calpain 10, but not calpain 1 levels after I/R compared to sham (non-ischemic). Pink staining represents Ponceau (Ponc) as a loading control. Values are expressed as mean±SEM; *P<0.05 I/R versus sham (n= 3/group). C. graph showing an increase in mitochondrial calpain activity after I/R compared to sham (non-ischemic). Values are expressed as mean±SEM; *P<0.05 I/R versus sham (n= 3/group). D. Recording and graph of H2O2 release from mitochondria isolated from sham versus I/R hearts and activated with glutamate/malate the substrate of complex I. Note that the ROS production was increased after I/R compared to sham (non-ischemic). E. Typical recording and graph of H2O2 release from mitochondria isolated from sham versus I/R hearts and activated with succinate the substrate of complex II. Note that the ROS production was decreased after I/R compared to sham (non-ischemic). Important: D-E results from our previous report in Am J Cardiovasc Dis. 2016 Sep 15;6(3):93-108.
Discussion
In this study, we discovered that the inner mitochondrial membrane protein (Mitofilin) directly binds to CypD, a well-known regulator of the mPTP opening, through its C-terminal, in the inner mitochondrial membrane (IMM) and this interaction is reduced during the early moments of reperfusion. We report that Mitofilin expression decreases concomitantly with CypD levels after I/R, probably due to the disruption of the direct link between these two proteins. We found that the extent of myocardial infarct size is inversely proportional to the reduction of Mitofilin levels after I/R.
Mitofilin deletion in HEK293T and HeLa cells has been found to disorganize cristae structures and loosen cristae junctions in the mitochondria [15]. In Caenorhabditis elegans, Han’s group reported the existence of two Mitofilin isoforms that have non-overlapping functions in controlling mitochondrial cristae formation. These observations were in agreement with reports by Griefers et al., on isolated mitochondria and mitoplasts from human osteosarcoma cells that indicated that Mitofilin is a transmembrane protein of the inner mitochondrial membrane expressed as two isoforms [7]. In fact, a sequence analysis by Frey’s group after 35S-labeled Mitofilin was transported into isolated yeast mitochondria suggested that Mitofilin is anchored in the IMM with an amino-terminal transmembrane domain, while the majority of the protein is extruding into the intermembrane space [7]. These results point to the crucial role of Mitofilin in controlling mitochondrial cristae morphology [6]. It is not surprising that the stable cells with Mitofilin deficiency were found to be more susceptible to apoptosis upon several apoptotic stimuli. This state was accompanied with redistribution of cytochrome c and accelerated release, but irrelevant to the activation of pro-apoptotic member Bax in the mitochondrial outer membrane [15]. In line with these observations, we recently reported that Mitofilin knockdown in H9c2 rat myoblasts by siRNA increases calpain activity that presumably leads to mitochondrial structural degradation and critical reduction of mitochondrial function, which is responsible for the increase in cell death by apoptosis. This apoptosis was not caspase-dependent, but via an AIF-PARP mechanism and associated with nuclear fragmentation, and S phase arrest of the cell cycle [16]. These two studies suggest that Mitofilin might have a preponderant role in the cell death.
In this study, we determined the role of Mitofilin in the initiation of myocardial injury after I/R. We found that Mitofilin expression is reduced after I/R, and the reduction is directly correlated with the duration of reperfusion. The expression of Mitofilin during reperfusion was dependent on the ischemia duration (Fig. 1). In fact, in hearts where duration of ischemia was longer, which caused a much larger myocardial infarct size, the level of Mitofilin reduces in tandem. We postulate an inversely proportional relationship between the loss of Mitofilin during the early moment of reperfusion and extent of myocardial infarction due to I/R injury (Fig. 2). Our results are in agreement with earlier reports by Hollander’s group on diabetes mellitus-associated cardiac and mitochondria dysfunction [12]. Thapa et al. reported that transgenic overexpression of Mitofilin preserves the mitochondrial structure, leading to a restoration of mitochondrial function and attenuation of cardiac contractile dysfunction in the diabetic heart [12]. Along with those lines, a potential role for Mitofilin in the left ventricular hypertrophy of hemodialysis patients [41] as well as in neurodegenerative disease (Parkinson’s disease) [42] has been reported.
Although, there is now a consensus on the role of Mitofilin in maintaining the cristae morphology, how Mitofilin acts to fulfill its role in the IMM is not completely elucidated. Recently, Mitofilin isoforms were shown to be part of hetero-oligomeric protein complexes that have been termed the mitochondrial inner membrane organizing system (MINOS) [10, 43]. Song’s team revealed that Mic60/Mitofilin homeostasis regulated by Yme1L was central to the mitochondrial contact site and cristae organizing system (MICOS) assembly, which is required for maintenance of mitochondrial morphology and organization of mtDNA nucleoids [44]. The general acquiescence of the literature on Mitofilin point to Mitofilin acting via its association with other proteins in the IMM. In line with this idea, we investigated whether Mitofilin interacts with CypD, a regulator of the mPTP opening. The mPTP opening is a crucial event in the mechanism of cell death after I/R [45]. We discovered in this study that Mitofilin and CypD, both located in the IMM, physically interact with each other (Figs. 3–5). We revealed that the levels of CypD alongside Mitofilin reduces after I/R and are inversely proportional to the degree of myocardial infarct size (Figs. 1 & 2). This result is in agreement with the idea that Mitofilin acts in a big protein complex of the IMM including SAM50, metaxins 1 and 2, DnaJC11, and ApoO [46].
Using a systemic mutation approach, we determined the potential site where CypD interact with Mitofilin. We performed four deletion mutations in Mitofilin (Fig. 6). Using IP with Myc antibodies to pull down Mitofilin or its mutated variants, followed by Western blot with anti CypD antibody, we show that Mitofilin binds CypD at the C-terminus that faces mitochondrial matrix (Fig. 7). Figures 7 and 8 show that transfection with Mitofilin mutated-constructs 3 and 4 exhibit opposite results in cell viability. Mutation 4 transfected cells displayed similar phenotype (increase in live cells) as the full-length construct, while Mutation 3 similarly to Mutations 1 and 2 exhibits opposite result (decrease in live cells, and more damaged mitochondria (Fig. 10A)) to the full-length after H2O2 treatment. These results suggest that a coiled-coil domain (between 397 and 476 AA) of Mitofilin act as a putative site of Mitofilin and CypD interaction. Further characterization within this coiled-coil domain is required to determine the residues in Mitofilin that interacts with CypD. To confirm the hypothesis that a direct interaction between both proteins occurs in the IMM, we performed a bimolecular fluorescence complementation assay (Fig. 5). We found that transfection with VC155-CypD or the VN173-Mitofilin constructs alone failed to emit YFP fluorescence. However, co-transfection of both plasmids increased YFP fluorescence in the periphery of the nuclei of the cells, the region where mitochondria are predominantly localized in cells (Fig. 5, bottom), suggesting that both Mitofilin and CypD are in a close proximity.
Using biochemical approaches including immunoprecipitation, immuno-organelle-chemistry, and sucrose gradient, we revealed that the Mitofilin-CypD link is abridged after I/R (Fig. 4). As CypD plays the role of regulator of the mPTP opening, one might hypothesize that Mitofilin, could exhibit an anchor-like role by stabilizing CypD structural integrity. The fact that the C-terminus of Mitofilin is important for its interaction with CypD could also point to an adaptor role for Mitofilin in the IMM. Therefore, disruption of the Mitofilin-CypD link during reperfusion might favor the induction of mitochondrial permeability transition, which is associated with mPTP opening. This idea is supported by the fact that the reduction of Mitofilin with an increase in ischemia duration correlates with a decrease in mitochondrial Ca2+ retention capacity required to induce mPTP opening (Fig. 9). In H9c2 cells, we found that Mitofilin knockdown and overexpression oppositely influenced the activation of mPTP opening (Fig. 9D & E) suggesting that Mitofilin regulation might act upstream of CypD. Our findings contrast with those obtained using CypD-deficient mice that exhibited a high level of resistance to I/R-induced cardiac injury [24]. The reason for this discrepancy might come from the use of a genetically modified animal model that may possess several structural and phenotypical modifications versus a reduction of the protein levels during an acute insult.
Since Mitofilin expression at the end of ischemia was similar to sham (non-ischemic), we, therefore, postulate that factors activated during reperfusion might promote a catalytic activity of mitochondrial proteins. However, it is also feasible that ischemia insult might weaken the Mitofilin-CypD link, which is perturbed and washed out during reperfusion. We could not decipher the precise mechanism responsible for the disruption of the Mitofilin-CypD link during reperfusion. Nevertheless, we predict an increase in enzymatic activity as we observed an increase in mitochondrial calpain activity during the early reperfusion compared to non-ischemic hearts (Fig. 10C). In fact, we revealed that the levels of calpain 10, the mitochondrial subtype, but not calpain 1, the cytosolic subtype, were higher after I/R when compared to sham (Fig. 10D). This observation suggests a decrease in the autolysis of calpain 10, which can explain the increase in calpain cleavage activity after I/R. However, the tentative trial with two calpain inhibitors (Calpeptin and MDL-28170) did not yield any conclusive results. Further studies are required to elucidate the reasons for the disruption of Mitofilin-CypD interaction during reperfusion, which might provide new mechanisms associated with cell death after I/R that could be related to regulation of ROS production as seen Fig. 10D–E.
In conclusion, we report here that ischemia/reperfusion insult promotes Mitofilin loss in the IMM resulting in increased mitochondrial dysfunction and myocardial infarct size. Mitofilin physically binds to CypD in the IMM and disruption of this link during reperfusion play a crucial role in the opening of mPTP, a key event in the initiation of cardiomyocyte death responsible for myocardial infarction after I/R.
Supplementary Material
Figure 11. Graphic abstract summarizing the proposed mechanism implicating Mitofilin degradation during stress induced cell death.
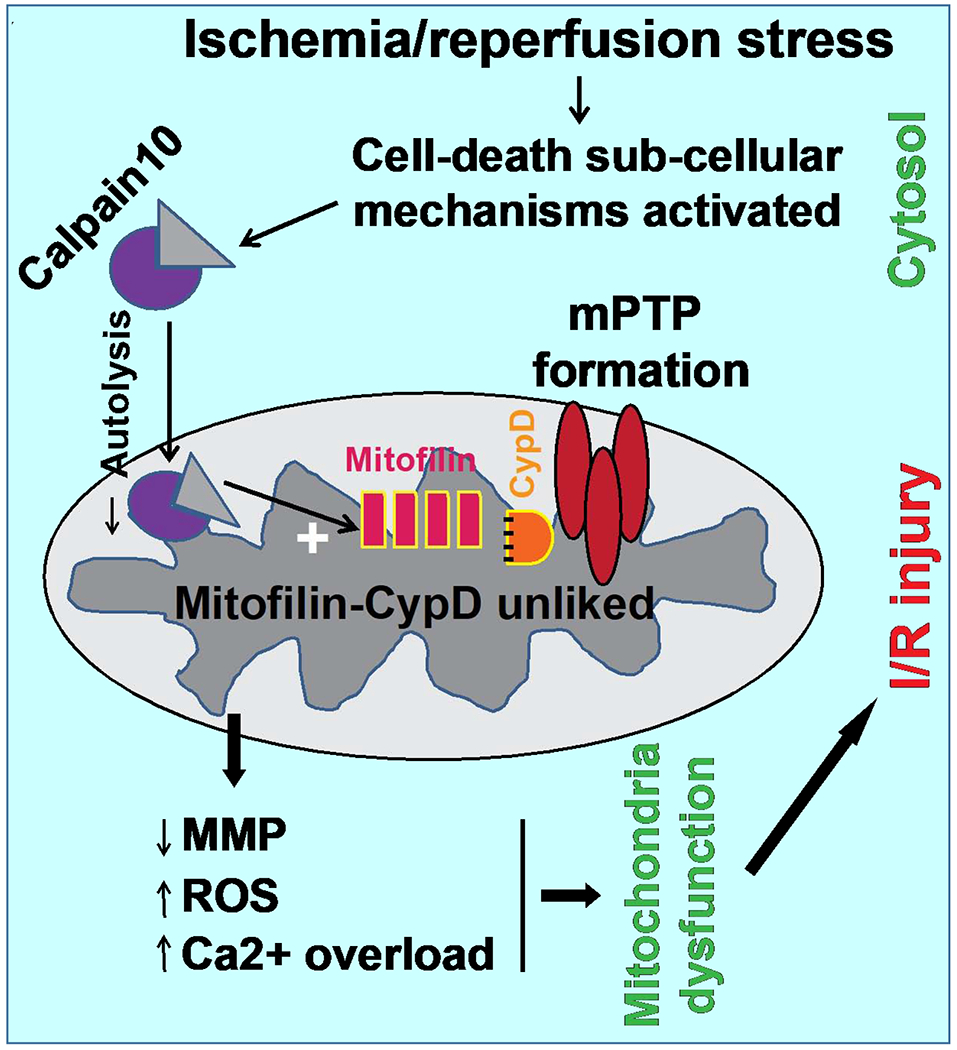
A. During physiological conditions, inactivation of subcellular signaling leads to reduce mitochondrial calpain10 activity resulting in the protection of Mitofilin catalysis. Mitofilin interacting with CypD is postulated to reduce mitochondrial ROS production and calcium overload that maintain higher mitochondrial membrane potential and the mPTP is inactivated, thereby sub-serving no cell death. B. Activation of subcellular signaling during I/R increases calpain10 cleavage of Mitofilin that disrupts Mitofilin-CypD interaction augmenting mitochondrial ROS production and calcium overload that dissipate mitochondrial membrane potential and mPTP formation, which subsequently causes cell death and I/R injury.
Highlights.
Ischemia/reperfusion stress reduces Mitofilin levels
Mitofilin reduction increases myocardial infarct size
Mitofilin binds to Cyclophilin D at the c- terminus
Regulation of mitofilin expression impacts Ca2+-induced mPTP opening
Acknowledgements
The authors thank Drs. Mohammad E Kabir and Madungwe B. Ngonizashe for their help in the study.
Funding Sources
This work was supported by the American Heart Association (AHA) grant 17SDG33100000 (JCB); NIH grant HL138093 and Voeckler fund (JCB).
ABBREVIATIONS
- immt, Mic60, mitofilin
Inner mitochondrial membrane
- MICOS
mitochondrial contact site and cristae organizing system
- IMM
inner mitochondrial membrane
- ROS
reactive oxygen species
- I/R
ischemia/reperfusion
- mPTP
mitochondrial permeability transition pore
- CRC
calcium retention capacity
- MMP
mitochondrial membrane potential
- CypD
Cyclophilin D
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no conflicts of interest.
References
- [1].Zerbes RM, Hoss P, Pfanner N, van der Laan M, Bohnert M, Distinct Roles of Mic12 and Mic27 in the Mitochondrial Contact Site and Cristae Organizing System, J Mol Biol 428(8) (2016) 1485–92. [DOI] [PubMed] [Google Scholar]
- [2].Darshi M, Mendiola VL, Mackey MR, Murphy AN, Koller A, Perkins GA, Ellisman MH, Taylor SS, ChChd3, an inner mitochondrial membrane protein, is essential for maintaining crista integrity and mitochondrial function, J Biol Chem 286(4) (2011) 2918–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Genin EC, Plutino M, Bannwarth S, Villa E, Cisneros-Barroso E, Roy M, Ortega-Vila B, Fragaki K, Lespinasse F, Pinero-Martos E, Auge G, Moore D, Burte F, Lacas-Gervais S, Kageyama Y, Itoh K, Yu-Wai-Man P, Sesaki H, Ricci JE, Vives-Bauza C, Paquis-Flucklinger V, CHCHD10 mutations promote loss of mitochondrial cristae junctions with impaired mitochondrial genome maintenance and inhibition of apoptosis, EMBO Mol Med 8(1) (2016) 58–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Odgren PR, Toukatly G, Bangs PL, Gilmore R, Fey EG, Molecular characterization of mitofilin (HMP), a mitochondria-associated protein with predicted coiled coil and intermembrane space targeting domains, J. Cell Sci 109 (Pt 9) (1996) 2253–2264. [DOI] [PubMed] [Google Scholar]
- [5].Icho T, Ikeda T, Matsumoto Y, Hanaoka F, Kaji K, Tsuchida N, A novel human gene that is preferentially transcribed in heart muscle, Gene 144(2) (1994) 301–6. [DOI] [PubMed] [Google Scholar]
- [6].John GB, Shang Y, Li L, Renken C, Mannella CA, Selker JM, Rangell L, Bennett MJ, Zha J, The mitochondrial inner membrane protein mitofilin controls cristae morphology, Mol Biol Cell 16(3) (2005) 1543–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gieffers C, Korioth F, Heimann P, Ungermann C, Frey J, Mitofilin is a transmembrane protein of the inner mitochondrial membrane expressed as two isoforms, Exp. Cell Res 232(2) (1997) 395–399. [DOI] [PubMed] [Google Scholar]
- [8].von der MK, Muller JM, Bohnert M, Oeljeklaus S, Kwiatkowska P, Becker T, Loniewska-Lwowska A, Wiese S, Rao S, Milenkovic D, Hutu DP, Zerbes RM, Schulze-Specking A, Meyer HE, Martinou JC, Rospert S, Rehling P, Meisinger C, Veenhuis M, B. Warscheid d.K., van I, Pfanner N, Chacinska A, van der LM Dual role of mitofilin in mitochondrial membrane organization and protein biogenesis, Dev. Cell 21(4) (2011) 694–707. [DOI] [PubMed] [Google Scholar]
- [9].Weber TA, Koob S, Heide H, Wittig I, Head B, van der Bliek A, Brandt U, Mittelbronn M, Reichert AS, APOOL is a cardiolipin-binding constituent of the Mitofilin/MINOS protein complex determining cristae morphology in mammalian mitochondria, PLoS One 8(5) (2013) e63683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Zerbes RM, d.K. van I, Veenhuis M, Pfanner N, van der LM, Bohnert M, Mitofilin complexes: conserved organizers of mitochondrial membrane architecture, Biol. Chem 393(11) (2012) 1247–1261. [DOI] [PubMed] [Google Scholar]
- [11].Baseler WA, Dabkowski ER, Williamson CL, Croston TL, Thapa D, Powell MJ, Razunguzwa TT, Hollander JM, Proteomic alterations of distinct mitochondrial subpopulations in the type 1 diabetic heart: contribution of protein import dysfunction, Am J Physiol Regul Integr Comp Physiol 300(2) (2011) R186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Thapa D, Nichols CE, Lewis SE, Shepherd DL, Jagannathan R, Croston TL, Tveter KJ, Holden AA, Baseler WA, Hollander JM, Transgenic overexpression of mitofilin attenuates diabetes mellitus-associated cardiac and mitochondria dysfunction, J Mol Cell Cardiol 79 (2015) 212–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Gorr MW, Wold LE, Mitofilin: Key factor in diabetic cardiomyopathy?, J Mol Cell Cardiol 85 (2015) 292–3. [DOI] [PubMed] [Google Scholar]
- [14].Xu Y, Ma H, Shao J, Wu J, Zhou L, Zhang Z, Wang Y, Huang Z, Ren J, Liu S, Chen X, Han J, A Role for Tubular Necroptosis in Cisplatin-Induced AKI, J Am Soc Nephrol 26(11) (2015) 2647–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Yang RF, Zhao GW, Liang ST, Zhang Y, Sun LH, Chen HZ, Liu DP, Mitofilin regulates cytochrome c release during apoptosis by controlling mitochondrial cristae remodeling, Biochem. Biophys. Res. Commun 428(1) (2012) 93–98. [DOI] [PubMed] [Google Scholar]
- [16].Madungwe NB, Feng Y, Lie M, Tombo N, Liu L, Kaya F, Bopassa JC, Mitochondrial Inner Membrane Protein (Mitofilin) Knockdown Induces Cell Death by Apoptosis Via an AIF-PARP-Dependent Mechanism and Cell Cycle Arrest, Am J Physiol Cell Physiol (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Argaud L, Gateau-Roesch O, Chalabreysse L, Gomez L, Loufouat J, Thivolet-Bejui F, Robert D, Ovize M, Preconditioning delays Ca2+-induced mitochondrial permeability transition, Cardiovasc. Res 61(1) (2004) 115–122. [DOI] [PubMed] [Google Scholar]
- [18].Argaud L, Gateau-Roesch O, Raisky O, Loufouat J, Robert D, Ovize M, Postconditioning inhibits mitochondrial permeability transition, Circulation 111(2) (2005) 194–197. [DOI] [PubMed] [Google Scholar]
- [19].Li J, Yan Z, Fang Q, A Mechanism Study Underlying the Protective Effects of Cyclosporine-A on Lung Ischemia-Reperfusion Injury, Pharmacology 100(1-2) (2017) 83–90. [DOI] [PubMed] [Google Scholar]
- [20].Bernardi P, The mitochondrial permeability transition pore: a mystery solved?, Front Physiol 4 (2013) 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Baines CP, Molkentin JD, STRESS signaling pathways that modulate cardiac myocyte apoptosis, J. Mol. Cell Cardiol 38(1) (2005) 47–62. [DOI] [PubMed] [Google Scholar]
- [22].Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, Hetz C, Danial NN, Moskowitz MA, Korsmeyer SJ, Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia, Proc Natl Acad Sci U S A 102(34) (2005) 12005–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, Inohara H, Kubo T, Tsujimoto Y, Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death, Nature 434(7033) (2005) 652–8. [DOI] [PubMed] [Google Scholar]
- [24].Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, Brunskill EW, Sayen MR, Gottlieb RA, Dorn GW, Robbins J, Molkentin JD, Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death, Nature 434(7033) (2005) 658–62. [DOI] [PubMed] [Google Scholar]
- [25].Wang X, Carlsson Y, Basso E, Zhu C, Rousset CI, Rasola A, Johansson BR, Blomgren K, Mallard C, Bernardi P, Forte MA, Hagberg H, Developmental shift of cyclophilin D contribution to hypoxic-ischemic brain injury, J Neurosci 29(8) (2009) 2588–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Griffiths EJ, Halestrap AP, Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion, Biochem J 307 ( Pt 1) (1995) 93–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Di Lisa F, Menabo R, Canton M, Barile M, Bernardi P, Opening of the mitochondrial permeability transition pore causes depletion of mitochondrial and cytosolic NAD+ and is a causative event in the death of myocytes in postischemic reperfusion of the heart, J Biol Chem 276(4) (2001) 2571–5. [DOI] [PubMed] [Google Scholar]
- [28].Feng Y, Madungwe NB, da Cruz Junho CV, Bopassa JC, Activation of G protein-coupled oestrogen receptor 1 at the onset of reperfusion protects the myocardium against ischemia/reperfusion injury by reducing mitochondrial dysfunction and mitophagy, Br J Pharmacol 174(23) (2017) 4329–4344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Feng Y, Madungwe NB, Imam Aliagan AD, Tombo N, Bopassa JC, Liproxstatin-1 protects the mouse myocardium against ischemia/reperfusion injury by decreasing VDAC1 levels and restoring GPX4 levels, Biochem Biophys Res Commun 520(3) (2019) 606–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ichas F, Jouaville LS, Sidash SS, Mazat JP, Holmuhamedov EL, Mitochondrial calcium spiking: a transduction mechanism based on calcium-induced permeability transition involved in cell calcium signalling, FEBS Lett 348(2) (1994) 211–5. [DOI] [PubMed] [Google Scholar]
- [31].Petronilli V, Miotto G, Canton M, Brini M, Colonna R, Bernardi P, Di LF, Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence, Biophys. J 76(2) (1999) 725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kroemer G, Galluzzi L, Brenner C, Mitochondrial membrane permeabilization in cell death, Physiol Rev 87(1) (2007) 99–163. [DOI] [PubMed] [Google Scholar]
- [33].Kabir ME, Singh H, Lu R, Olde B, Leeb-Lundberg LM, Bopassa JC, G Protein-Coupled Estrogen Receptor 1 Mediates Acute Estrogen-Induced Cardioprotection via MEK/ERK/GSK-3beta Pathway after Ischemia/Reperfusion, PLoS One 10(9) (2015) e0135988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Elefantova K, Lakatos B, Kubickova J, Sulova Z, Breier A, Detection of the Mitochondrial Membrane Potential by the Cationic Dye JC-1 in L1210 Cells with Massive Overexpression of the Plasma Membrane ABCB1 Drug Transporter, Int J Mol Sci 19(7) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Madungwe NB, Zilberstein NF, Feng Y, Bopassa JC, Critical role of mitochondrial ROS is dependent on their site of production on the electron transport chain in ischemic heart, Am J Cardiovasc Dis 6(3) (2016) 93–108. [PMC free article] [PubMed] [Google Scholar]
- [36].Shyu YJ, Liu H, Deng X, Hu CD, Identification of new fluorescent protein fragments for bimolecular fluorescence complementation analysis under physiological conditions, Biotechniques 40(1) (2006) 61–6. [DOI] [PubMed] [Google Scholar]
- [37].Miller KE, Kim Y, Huh WK, Park HO, Bimolecular Fluorescence Complementation (BiFC) Analysis: Advances and Recent Applications for Genome-Wide Interaction Studies, J Mol Biol 427(11) (2015) 2039–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Lonn P, Landegren U, Close Encounters - Probing Proximal Proteins in Live or Fixed Cells, Trends Biochem Sci 42(7) (2017) 504–515. [DOI] [PubMed] [Google Scholar]
- [39].Bopassa JC, Ferrera R, Gateau-Roesch O, Couture-Lepetit E, Ovize M, PI 3-kinase regulates the mitochondrial transition pore in controlled reperfusion and postconditioning, Cardiovasc Res 69(1) (2006) 178–85. [DOI] [PubMed] [Google Scholar]
- [40].Bopassa JC, Michel P, Gateau-Roesch O, Ovize M, Ferrera R, Low-pressure reperfusion alters mitochondrial permeability transition, Am. J. Physiol Heart Circ. Physiol 288(6) (2005) H2750–H2755. [DOI] [PubMed] [Google Scholar]
- [41].Wu QS, He Q, He JQ, Chao J, Wang WY, Zhou Y, Lou JZ, Kong W, Chen JF, The role of mitofilin in left ventricular hypertrophy in hemodialysis patients, Ren Fail 40(1) (2018) 252–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Van Laar VS, Otero PA, Hastings TG, Berman SB, Potential Role of Mic60/Mitofilin in Parkinson’s Disease, Front Neurosci 12 (2018) 898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Zerbes RM, Bohnert M, Stroud DA, von der MK, Kram A, Oeljeklaus S, Warscheid B, Becker T, Wiedemann N, Veenhuis M, d.K. van I, Pfanner N, van der LM, Role of MINOS in mitochondrial membrane architecture: cristae morphology and outer membrane interactions differentially depend on mitofilin domains, J. Mol. Biol 422(2) (2012) 183–191. [DOI] [PubMed] [Google Scholar]
- [44].Li H, Ruan Y, Zhang K, Jian F, Hu C, Miao L, Gong L, Sun L, Zhang X, Chen S, Chen H, Liu D, Song Z, Mic60/Mitofilin determines MICOS assembly essential for mitochondrial dynamics and mtDNA nucleoid organization, Cell Death Differ 23(3) (2016) 380–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Gateau-Roesch O, Argaud L, Ovize M, Mitochondrial permeability transition pore and postconditioning, Cardiovasc. Res 70(2) (2006) 264–273. [DOI] [PubMed] [Google Scholar]
- [46].Koob S, Reichert AS, Novel intracellular functions of apolipoproteins: the ApoO protein family as constituents of the Mitofilin/MINOS complex determines cristae morphology in mitochondria, Biol Chem 395(3) (2014) 285–96. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.