

Abstract
Acellular nerve allografts (ANAs) are increasingly used to repair nerve gaps following injuries. However, these nerve scaffolds have yet to surpass the regenerative capabilities of cellular nerve autografts; improved understanding of their regenerative mechanisms could improve design. Due to their acellular nature, both angiogenesis and diverse cell recruitment is necessary to repopulate these scaffolds to promote functional regeneration. We determined the contribution of angiogenesis to initial cellular repopulation of ANAs used to repair nerve gaps, as well as the signaling that drives a significant portion of this angiogenesis. Wild-type (WT) mice with nerve gaps repaired using ANAs that were treated with an inhibitor of VEGF receptor signaling severely impaired angiogenesis within ANAs, as well as hampered cell repopulation and axon extension into ANAs. Similarly, systemic depletion of hematogenous-derived macrophages, but not neutrophils, in these mice models severely impeded angiogenesis and subsequent nerve regeneration across ANAs suggesting hematogenous-derived macrophages were major contributors to angiogenesis within ANAs. This finding was reinforced using CCR2 knockout (KO) models. As macrophages represented the majority of CCR2 expressing cells, a CCR2 deficiency impaired angiogenesis and subsequent nerve regeneration across ANAs. Furthermore, an essential role for CCL2 during nerve regeneration across ANAs was identified, as nerves repaired using ANAs had reduced angiogenesis and subsequent nerve regeneration in CCL2 KO vs WT mice. Our data demonstrate the CCL2/CCR2 axis is important for macrophage recruitment, which promotes angiogenesis, cell repopulation, and subsequent nerve regeneration and recovery across ANAs used to repair nerve gaps.
Keywords: acellular nerve allograft, angiogenesis, macrophage, peripheral nerve, regeneration
Introduction
Severe nerve damage frequently results in a gap between the nerve ends, resulting in loss of function (Robinson, 2000). While small nerve gaps can be repaired directly, longer gaps require the use of nerve grafts or other devices to bridge the gap (Ijkema-Paassen et al., 2004; Rbia and Shin, 2017). More recently, “off-the-shelf” substitutes for nerve grafts are increasingly used in the clinic, as autografts have inherent disadvantages, such as need for a donor site to harvest patients’ own nerve tissue. Yet, a prevailing concern when using these substitutes clinically is that as their length increases, axon regeneration across the substitute diminishes. This phenomenon of reduced nerve regeneration across longer substitutes bridging a nerve gap has been observed across artificial nerve conduits and acellular nerve scaffolds in animal models (Mokarram et al., 2017; Saheb-Al-Zamani et al., 2013). And, this relationship regarding longer substitute length, exhibited as fewer instances of meaningful recovery, is also observed in the clinic (Leckenby et al., 2020; Pan et al., 2019b). Therefore, further knowledge regarding the biology of regeneration across substitutes is critical toward understanding these shortcomings and improving designs.
A majority of clinically used substitutes are initially acellular biomaterials and scaffolds. Therefore, they require repopulation by host cells, including Schwann cells (SCs) and endothelial cells (ECs), to promote axon regeneration across (Pan et al., 2019a; Poppler et al., 2016). Acellular nerve allografts (ANAs) are one such substitute, where nerves from allogenic donors are decellularized to generate a scaffold for cellular repopulation and regeneration while minimizing antigenicity. ANAs also represent a useful substitute to understand the processes mediating nerve regeneration across long nerve gap, as they are a flexible material that accommodates critically-sized distances (i.e. ≥1 cm) in small animal models. In previous studies, we used ANAs of different lengths to identify why ANAs become functionally limited by their length. We found decreased angiogenesis and effective vascularization within long (4 cm) compared to short (2 cm) ANAs bridging rat nerve gaps. This decreased vascularization of long ANAs was followed by decreased SC and T cell accumulation within ANAs, and ultimately failed axon regeneration across the ANA (Pan et al., 2019a). Insufficient angiogenesis and lack of proper vascularization are widely known to inhibit tissue regeneration (Hankenson et al., 2011; Madeddu, 2005). Therefore, while our observations did not demonstrate reduced angiogenesis as a cause of failure, these associations suggest such a link.
Greater understanding of the contributions of angiogenesis across biomaterials and scaffolds bridging a nerve gap, as well as what promotes or limits angiogenesis, could be key to understanding how to promote robust nerve regeneration across critically sized nerve gaps. The process of angiogenesis is largely promoted by sprouting of existing vessels to a hypoxic region via ECs, which then align and connect to the existing vasculature (Shweiki et al., 1992). In the case of a small nerve gap (~3 mm), the innate immune system is responsible for promoting this process within the gap. Specifically, macrophages infiltrate the nerve injury site, and produce pro-angiogenic factors, such as vascular endothelial cell growth factor (VEGF), due to the hypoxic environment, which drives EC functions to promote angiogenesis within the gap (Cattin et al., 2015; Cattin and Lloyd, 2016). However, the extent to which the innate immune system, and macrophages, promote angiogenesis within initially acellular biomaterials used to repair a critically sized nerve gap is unknown. Furthermore, because of the larger volume requiring angiogenesis, there may be roles for other cells of innate immune response to facilitate angiogenesis within ANAs. For example, neutrophils are the earliest leukocytes to arrive at an injury site, and can promote macrophage recruitment or otherwise alter gene expression in macrophages (Beck-Schimmer et al., 2005; Warnatsch et al., 2015).
In addition, while it has been shown that macrophages may be important for angiogenesis, the sources of macrophage, as well as the chemokines for recruiting these macrophages, are not clear in the context of ANA repaired nerve injury. Two major sources of macrophages have been described. Tissue resident macrophages are niche-specific macrophages that reside in various tissues, including nerve. Following injury, these macrophages increase their numbers within the tissue through local proliferation (Sager et al., 2016). Hematogenous-derived macrophages are generated from monocytes that became mature following recruitment from blood to the tissue site. These monocytes and/or macrophages are recruited through a variety of chemokines. In particular, signaling through C-C chemokine receptor type 2 (CCR2) has been shown to be critical to a substantial portion of their recruitment (Siebert et al., 2000). And, CCR2 signaling is important for recruitment of macrophages to nerve following injury (Lindborg et al., 2017; Siebert et al., 2000). Multiple ligands can engage the CCR2 receptor to signal monocyte/macrophage influx including CCL2, CCL7, and CCL8 (Crown et al., 2006). In particular, chemokine (C-C motif) ligand 2 (CCL2), also referred to as monocyte chemoattractant protein 1 (MCP1), is produced in nerve following injury and has been shown to recruit macrophages to nerve (Martini et al., 2008; Van Steenwinckel et al., 2015). Thus, CCL2 represents a possible candidate for the recruitment of macrophages into biomaterials, such as ANAs, repairing a nerve gap. Defining what macrophage populations, and how these macrophages are recruited, can thus inform our understanding of processes driving angiogenesis across biomaterials and scaffolds repairing nerve gaps.
Here, we explored the contribution of angiogenesis to cell repopulation and subsequent nerve regeneration following repair across an initially acellular nerve scaffold (ANA) bridging a critically sized nerve gap. We then dissected the role of hematogenous-derived macrophages and neutrophils in promoting angiogenesis and subsequent nerve regeneration. In addition, we asked how hematogenous-derived macrophages accumulate within the scaffold, and if CCL2/CCR2 signaling contributes to these processes.
Materials and methods
Animals care and use
Commercially-available male mice 7 to 10 weeks old (20–25g, Jackson Laboratories, Bar Harbor, ME) were utilized for all experiments. Wild-type (WT; C57BL/6J), CCR2 KO (JAX stock #004999 (Boring et al., 1997)), and CCL2 KO (JAX stock #004434 (Lu et al., 1998)) mice were randomized to groups for experimental treatments. Mice were randomized to groups for experimental treatments. Randomized C57BL/6J mice were used as donor mice to derive ANAs. Surgical procedures and peri-operative care measures were conducted in compliance with the AAALAC accredited Washington University Institutional Animal Care and Use Committee (IACUC) and the National Institutes of Health guidelines. All animals were housed in a central animal care facility and provided with free access to food (PicoLab rodent diet 20, Purina Mills Nutrition International, St. Louis, MO) and water.
Surgical procedures
For surgical procedures, mice were anesthesized using a cocktail of ketamine (100 mg/kg; Fort Dodge Animal Health, Fort Dodge, IA) and dexmedetomidine (0.5 mg/kg; Pfizer Animal Health, Exton, PA). For mice serving as ANA donors, following euthanization, their sciatic nerves were transected proximally at the level of exiting nerve roots, and distally just beyond the sciatic trifurcation. In experimental mice receiving the ANAs, the right sciatic nerve was exposed and transected 5 mm proximal to the distal trifurcation. The indicated grafts (10 mm) were reverse oriented (distal end of donor graft facing proximal nerve stump of recipient) sutured into the nerve gap with 11–0 nylon microsuture (Sharpoint, Reading, PA). A two-layer closure of muscle and skin was performed using 6–0 vicryl and 4–0 nylon suture, respectively. Atipamezole solution (0.1mg/kg; Zoetis, Florham Park, NJ) was administered for anesthesia reversal. The animals were recovered on a warming pad and monitored for postoperative complications before returning them to a central animal care facility. Postoperative pain was managed using Buprenorphine SR™ (0.05 mg/kg; ZooPharm, Windsor, CO). Animals were monitored daily post-operatively for signs of infection and/or distress. At the appropriate endpoints, animals were euthanized via cervical dislocation under anesthesia, and their tissues collected for respective studies (see below).
ANA processing
ANAs were decellularized using a modified series of detergents in the method described previously (Poppler et al., 2016). Briefly, nerves isolated from donor animals were repeatedly washed in deionized water and detergents in a sodium phosphate buffer: Triton X-100, sulfobetaine-16 (SB-16), and sulfobetaine-10 (SB-10). All grafts were washed and stored in 10 mM phosphate-buffered 50 mM sodium solution at 4°C and used within 1 week.
VEGF Receptor (VEGFR) inhibition with cabozantinib
In select mice as indicated in the results, angiogenesis was inhibited using cabozantinib, a selective VEGFR2 inhibitor (Selleckchem, Houston, TX). Six days after surgery, mice were given cabozantinib (100mg/kg, in water) via oral gavage twice a day until fourteen days after surgery, when they were sacrificed. Control mice received water without the inhibitor.
Clodronate liposome depletion of macrophages
In select mice as indicated in the results, macrophages were depleted using clodronate liposome (Liposoma B.V., Amsterdam, Netherland), which selectively targets and kills macrophages. Starting either 2 days prior to surgery or 4 days after surgery, 200uL of clodronate liposome were administered intraperitoneally (i.p.) once every three days. Control mice were administered phosphate buffered saline (PBS).
Macrophage labeling with Dil-liposome (in supplementary material)
In select mice as indicated in the results, macrophages were labeled using Dil liposome (Liposoma B.V., Amsterdam, Netherland). Thirteen days after surgery, 200uL of Dil-liposome were administered i.p. once in WT or CCR2KO mice. The next day, mice were sacrificed to collect ANAs for analysis.
Neutrophil depletion using Ly6G antibody
In select mice as indicated in the results, neutrophils were depleted through the administration of Ly6G neutralizing antibodies (Bio X Cell, Lebanon, NH). Two days prior to surgery rat anti-mouse Ly6G antibody (clone 1A8) or its corresponding control isotype (clone LTF-2), were given intraperitoneally. Each mouse was given 250 μg of antibody diluted to 100 μL using PBS. These were continued once every other day until the fourteen-day endpoint. Depletion of neutrophils was verified by flow cytometry analysis of spleen.
Immunohistochemistry (IHC)
To assess cell populations and protein expression within ANAs, nerve samples were explanted at indicated endpoints and immediately placed in 4% paraformaldehyde in phosphate-buffer overnight followed by immersion in 30% sucrose in PBS solution for 24–48 hours. Samples were then frozen in OCT Compound (VWR, Radnor, PA) and sectioned at 15 μm onto pretreated charged glass slides. Sections were rehydrated with PBS and blocked using 5% normal goat serum diluted in PBS before primary antibody staining. Primary antibodies were used to stain for endothelial cells (CD31), T cells (CD3), axons (NF200), Schwann cells (S100), myelin basic protein (MBP), and macrophages (CD68, CD206). Primary antibody in 5% serum buffer was applied and incubated at 4°C overnight with specific antibodies and concentrations outlined in Supplemental Table 1. Sections were then washed in PBS and stained for the appropriate fluorochrome-conjugated secondary antibodies for 1 h at room temperature. All sections were mounted with Fluoroshield mounting medium with DAPI (Abcam, Boston, MA) and then imaged using the Fluoview FV1000 confocal microscope and acquisition system (Olympus, Waltham, MA) at overall 200x (20x water immersion objective) or 600x magnification (60x oil immersion objective). A minimum of three sections were analyzed and averaged for each tissue area using ImageJ (NIH) to obtain a value for each single animal (n=1). For percent area, an ImageJ macro was used to quantify the percentage of the area in a standardized field that was positive for the marker measured. For cell counts, field size was kept standard at a 60x objective (600x magnification overall), and colocalization of the primary marker(s) with DAPI was considered a positive cell. Data reported for cell counts was reflected as % cells, where cells colocalized with the marker(s) of interest was expressed as proportion of all DAPI+ cells in the standardized field.
Flow cytometry
To identify and quantify immune cell populations, at the indicated endpoints after surgery, mice were sacrificed and their ANAs, blood (collected via cardiac puncture), or spleen harvested. Nerve tissues and spleen were digested using a cocktail of 1 mL digestion buffer (0.1% collagenase, 0.05% DNase in 2% fetal bovine serum (FBS)/ Dulbeccòs Modified Eagle Media (DMEM)). Samples were incubated for 20 minutes at 37°C with constant agitation, and following digestion, the samples were re-suspended in FACS buffer (2% FBS, 0.1% EDTA in phosphate buffered saline). RBCs from blood or spleen were removed using RBC lysis buffer. After filtering through 75 um membrane, the cells were incubated with Fc block for 10 minutes followed by an antibody cocktail specific to the cell of interest (Supplemental Table 2) for 1 hour before analysis with flow cytometry using BD Fortessa. At least 50,000 events were acquired for flow cytometry. Data were analyzed using FlowJo. Macrophages were gated by CD45+, CD11b+, CD64+, F4/80+. Monocytes were gated on CD45+, CD11b+, CD64-, Ly6C+. Neutrophils were gated by CD45+, CD11b+, Ly6G+. Single color stained compensation beads were used as control.
Gene analysis (qRT-PCR) (for supplemental material)
To quantify gene expression from sorted cells within ANAs, total RNA was prepared from ANA explants. RNA was extracted using Trizol (Life Technologies), chloroform and a RNeasy Kit (Qiagen, Valencia, CA) according to manufacturer’s instructions. RNA concentration was determined on a NanoDrop 1000 Spectrophotometer (Thermo Scientific, Wilmington, DE). The cDNA was generated with SuperScript II reverse transcriptase (Invitrogen, Carlsbad, CA). Real-time PCR was performed using a Step One Plus thermocycler (Applied Biosystems, Foster City, CA) using Taqman Master Mix (Applied Biosystems) reagents with CCL2 primer (Assay ID: Mm00441242_m1, Thermofisher Scientific, Waltham, MA). The expression levels of genes were normalized to Actb. Data were analyzed using Step One Software v2.2.2 (Applied Biosystems, Foster City, CA).
Relative Muscle Mass
To further quantify the extent of nerve regeneration, relative gastrocnemius muscle mass was measured as it is indicative of reinnervation of denervated muscles. After nerve harvest at 6 weeks, the gastrocnemius muscles were harvested from the experimental and contralateral sides. Wet muscle weight was recorded on each side, and the ratio of the ipsilateral to contralateral muscle weight was calculated.
Grid walk assessment
At week 6 post-operatively, mice underwent a grid walk assessment to determine their functional recovery. Mice were placed on an elevated mesh or grid with a grid size measuring 3.5cm by 3.5cm. After mice have acclimated to the grid for at least 5 mins, they were recorded with a video camera for at least 4 minutes moving upon the grid. From the video considering the injured limb, the total number of steps with that foot, and total steps that resulted in a foot placement missing the mesh and going through the grid (slipped steps) was measured. Foot fault was calculated as the proportion of slipped steps to total steps.
Statistical analysis
Statistical analyses were performed using GraphPad Prism. Each animal was considered an ‘n’ value. All data were compiled as mean ± standard deviation. Data were tested for normality using the Kolmogorov–Smirnov test. Student’s t test was performed for analysis between 2 groups. A significance level of p < 0.05 was used in all statistical tests performed.
Results
Endothelial cell accumulation and angiogenesis occurs prior to appreciable Schwann cell repopulation of ANAs
We first established the time-course of EC migration and vessel formation within ANAs repairing a relatively-long nerve gap (10 mm) for a mouse model. This relationship was described with respect to SC repopulation of ANAs, based on previous work that described EC migration preceding SC repopulation of a small, unrepaired nerve gap (~2–3 mm) (Cattin et al., 2015). Based on the extent of CD31 area (ECs), ECs and moderate vessel formation were present but limited within ANAs 10 days following repair. Subsequently, the extent of CD31 increased significantly over time. CD31 area increased to nearly 5% of total area within ANAs by day 14, and further increased to 8% by day 18 (Fig. 1). Concurrently, the formation of elongated vessels became prominently clear over this same time period. Similarly, the extent of SCs (S100 area) within ANAs closely followed EC accumulation and increased in proportion to the extent of vessel formation. While no S100+ cells were detectable on day 10 within ANAs, SCs increased by day 14 onward (Fig. 1). Overall, these data demonstrated appreciable angiogenesis closely preceded SC repopulation of ANAs, and angiogenesis progressed during ongoing nerve regeneration across ANAs.
Fig. 1.
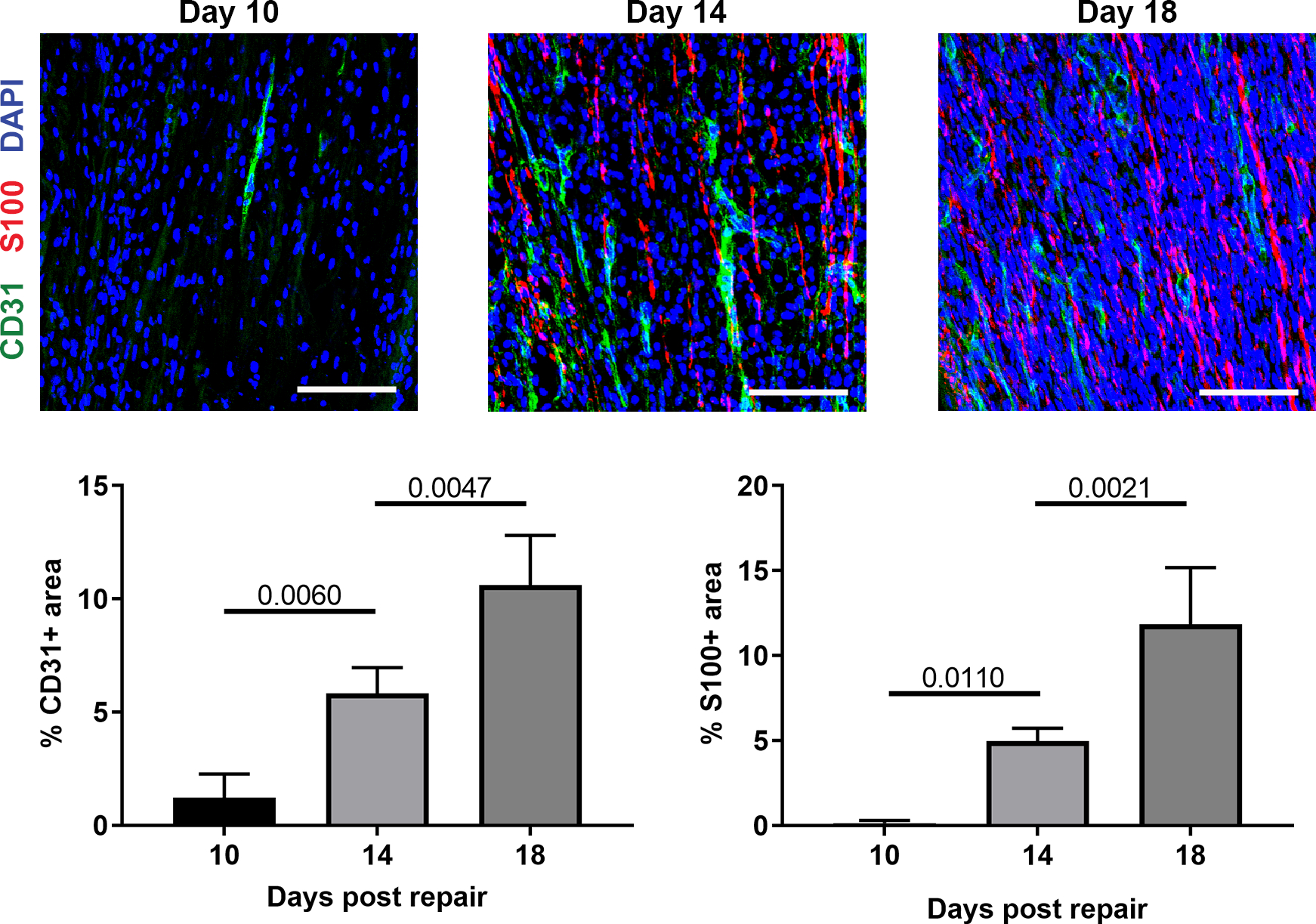
Angiogenesis within ANAs slightly precedes Schwann cell accumulation. Representative immunofluorescence images and quantification of endothelial cells (CD31) and Schwann cells (S100) within ANAs post-repair. Scale bars represent 50 μm. Mean ± SD, n=4/group; p values shown.
Blockade of VEGF signaling inhibits Schwann cell and T cell repopulation of ANAs
Given the association between angiogenesis and SC accumulation within ANAs, the contribution of angiogenesis to general cell migration and repopulation of ANAs was assessed. Two weeks following nerve gap repair using ANAs, WT mice given cabozantinib to inhibit VEGF signaling showed drastically reduced EC quantities (CD31 area) within ANAs compared to untreated mice (Fig. 2A). Furthermore, no blood vessels could be discerned within the treated-group ANAs. As a result, these ANAs also had severely reduced SC accumulation (S100 area; Fig. 2A), and reduced axonal extension (NF200 area) into ANAs (Fig. 2B). As we previously found T cells accumulate within ANAs and contribute to regeneration (Pan et al., 2019a), we also examined the accumulation of T cells (CD3) within ANAs at this 2 week endpoint. Following cabozantinib treatment, there was essentially no T cell accumulation (CD3+ cells) within ANAs (Fig. 2C). Conversely, macrophages were only modestly impacted by cabozantinib. While the total number of macrophages (CD68+ cells) within ANAs was reduced in cabozantinib treated compared to untreated mice, there was an increase in the proportion of macrophages (% CD68 cells) relative to all cells (DAPI+; Fig. 2D). These data demonstrated that VEGF signaling was critical to EC accumulation and angiogenesis within ANAs, which in turn promoted SC and T cell repopulation, but had only moderate influence on macrophage accumulation.
Fig. 2.
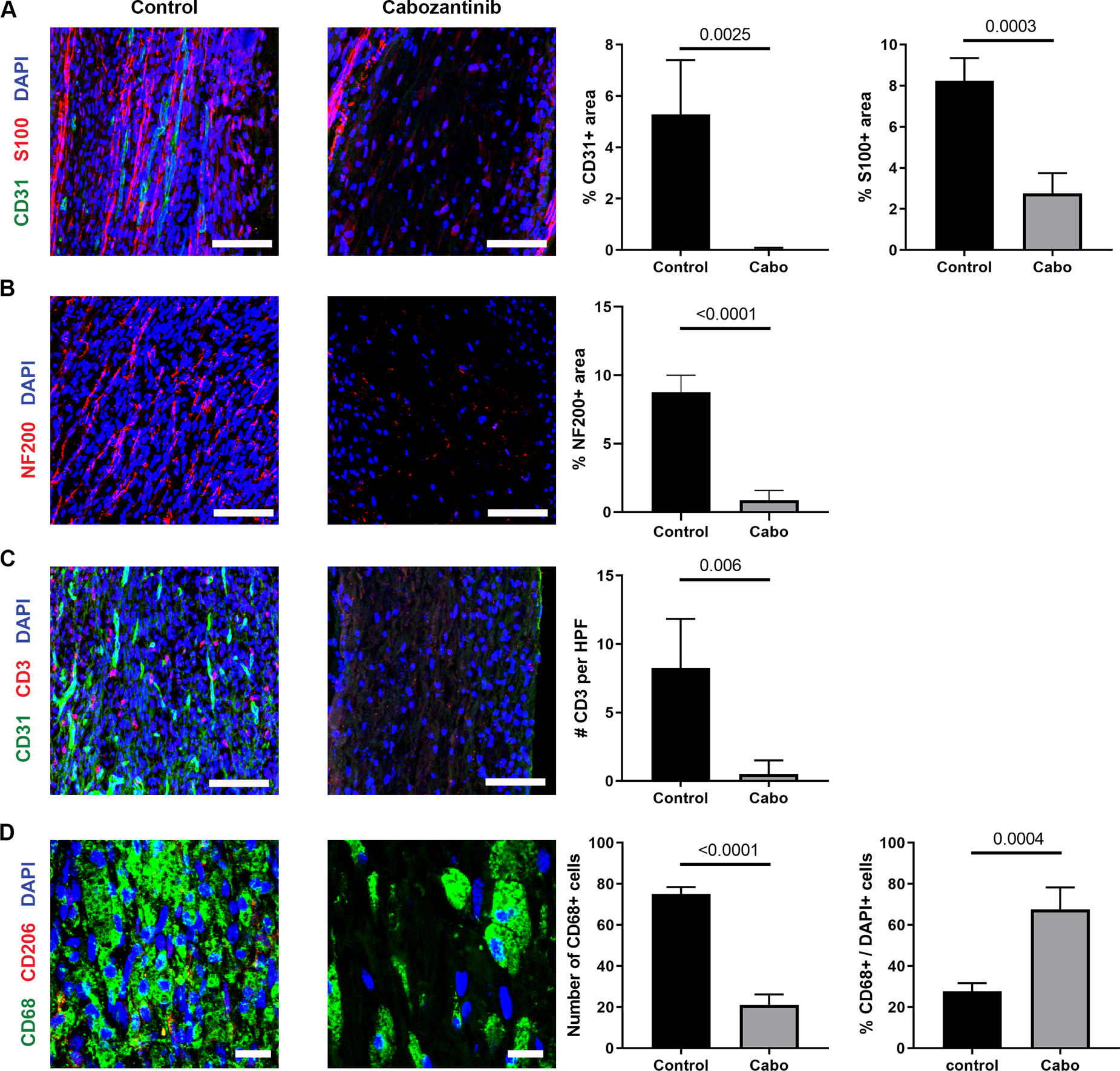
Angiogenesis within ANAs is required for cell repopulation and axon regeneration. Representative immunofluorescence staining and quantification of endothelial cells (CD31; A & C), Schwann cells (S100; A), axons (NF200; B), T cells (CD3; C), and macrophages (CD68 and CD206; D) within ANAs 14 days post-repair from WT mice receiving cabozantinib or water (control). Scale bars represent 50 μm (A-C) or 10 μm (D). Mean ± SD, n=4/group; p values shown.
Cells of the innate immune system accumulate prior to ECs and their numbers remain elevated during angiogenesis
Given the presence of macrophages in ANAs despite blocking VEGF signaling, we reasoned that macrophages, and other cells of the innate immune system, could accumulate within ANAs before EC migration and contribute to angiogenesis. At 10, 14, and 18 days post repair following nerve repair using ANAs, we used flow cytometry to assess these cell populations relative to all leukocytes (CD45+; Fig. 3A) finding that myeloid cells (CD11b+) were the predominate leukocytes within ANAs. Of myeloid cells, macrophages (CD45+, CD11b+, CD64+, F4/80+) consisted of the majority (Fig. 3B). Also, the proportion of macrophages and monocytes (CD45+, CD11b+, CD64-, Ly6C+) remained relatively stable within ANAs over time, while the proportion of neutrophils (CD45+, CD11b+, Ly6G+) were highest day 10 post injury and ANA repair, before declining throughout the regeneration process (Fig. 3B). To visualize the changes in macrophages over time with respect to all cells, we assessed macrophages (CD68+) using IHC within the ANAs 10, 14, and 18 days post repair, and found that their proportion among all cells (DAPI+) were greatest at day 14 before declining by day 18 (Fig. 3D; middle graph). And, considering macrophage polarization, principally the M2-like phenotype (CD206+), the proportion of macrophages that expressed CD206 of macrophages (CD68+) were highest at day 10 (Fig. 3C–D).
Fig. 3.
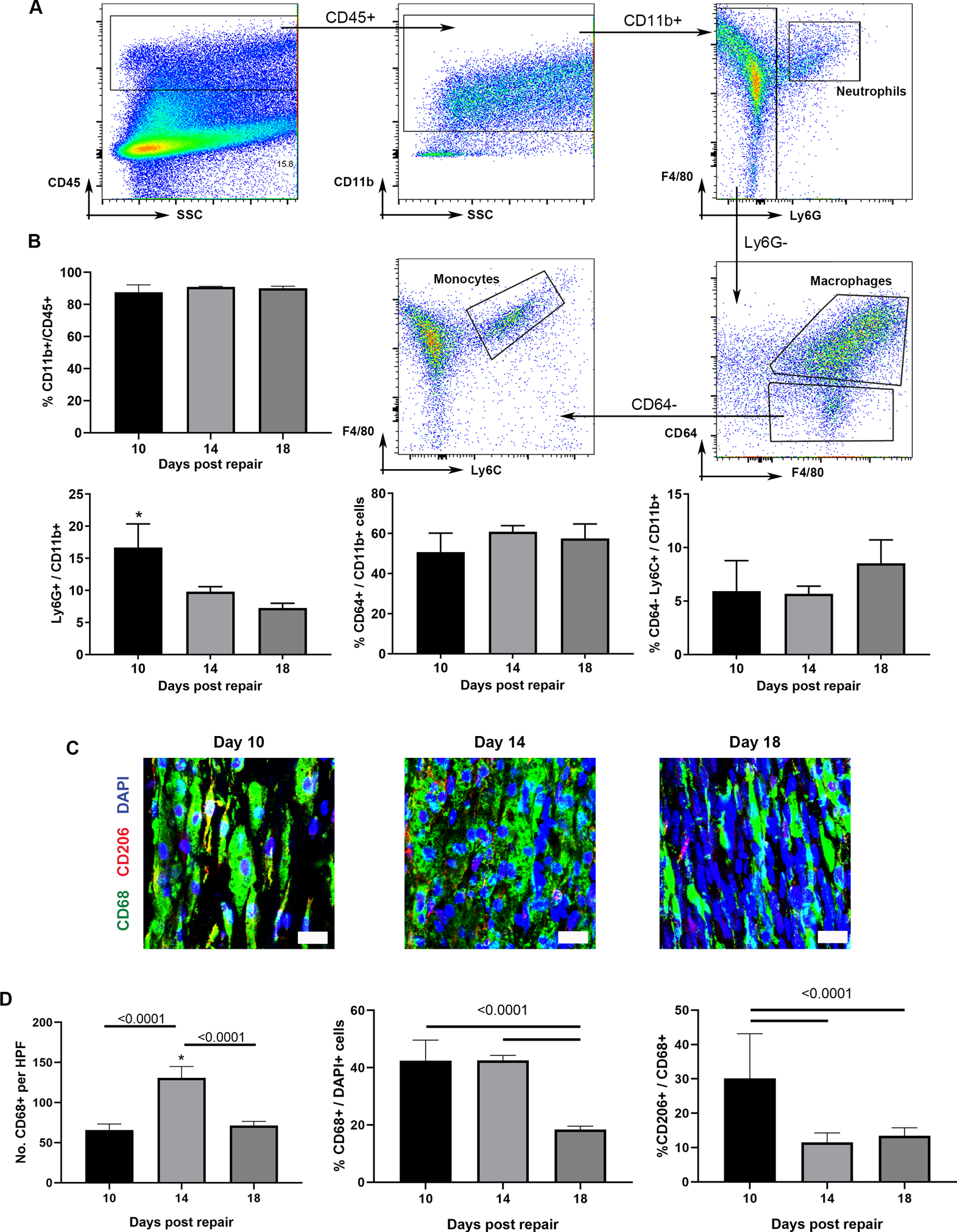
Myeloid cells robustly accumulate early within ANAs. A) Flow cytometry gating strategy to quantify leukocytes (CD45+) populations from within ANAs: myeloid cells (CD11b+), neutrophils (CD45+, CD11b+, Ly6G+), macrophages (CD45+, CD11b+, CD64+, F4/80+), and monocytes (CD45+, CD11b+, CD64-, Ly6C+). For each flow plot, arrows between flow cytometry plots represent further sorting of outlined cell populations for subsequent plots. B) Analysis from ANAs post-repair. Representative immunofluorescence staining (C) and quantification (D) of macrophages (CD68 and CD206) within ANAs post-repair. Scale bars represent 10 μm. Mean ± SD, n=4/group; p values shown.
Macrophages, but not neutrophils, have essential contributions to angiogenesis within ANAs
As macrophages are recruited within ANAs despite blockade of VEGF signaling and prior to EC migration within ANAs, the roles of both neutrophils, which precede macrophages, as well as macrophages, in angiogenesis was determined. The role of neutrophils was assessed by their depletion in WT mice administered Ly6G antibody, which depleted systemic neutrophil levels by 8-fold measured at a 2-week endpoint (Supplemental Fig. 1). Despite this substantial neutrophil depletion by 2 weeks, no differences in EC quantities (CD31 area) or vessel morphology were observed within ANAs from treated vs isotype control mice (Fig. 4A). Likewise, SC accumulation (S100 area) within ANAs of treated mice was not different vs control (Fig. 4B), nor was macrophage accumulation (CD68 area) within ANAs (Fig. 4C).
Fig. 4.
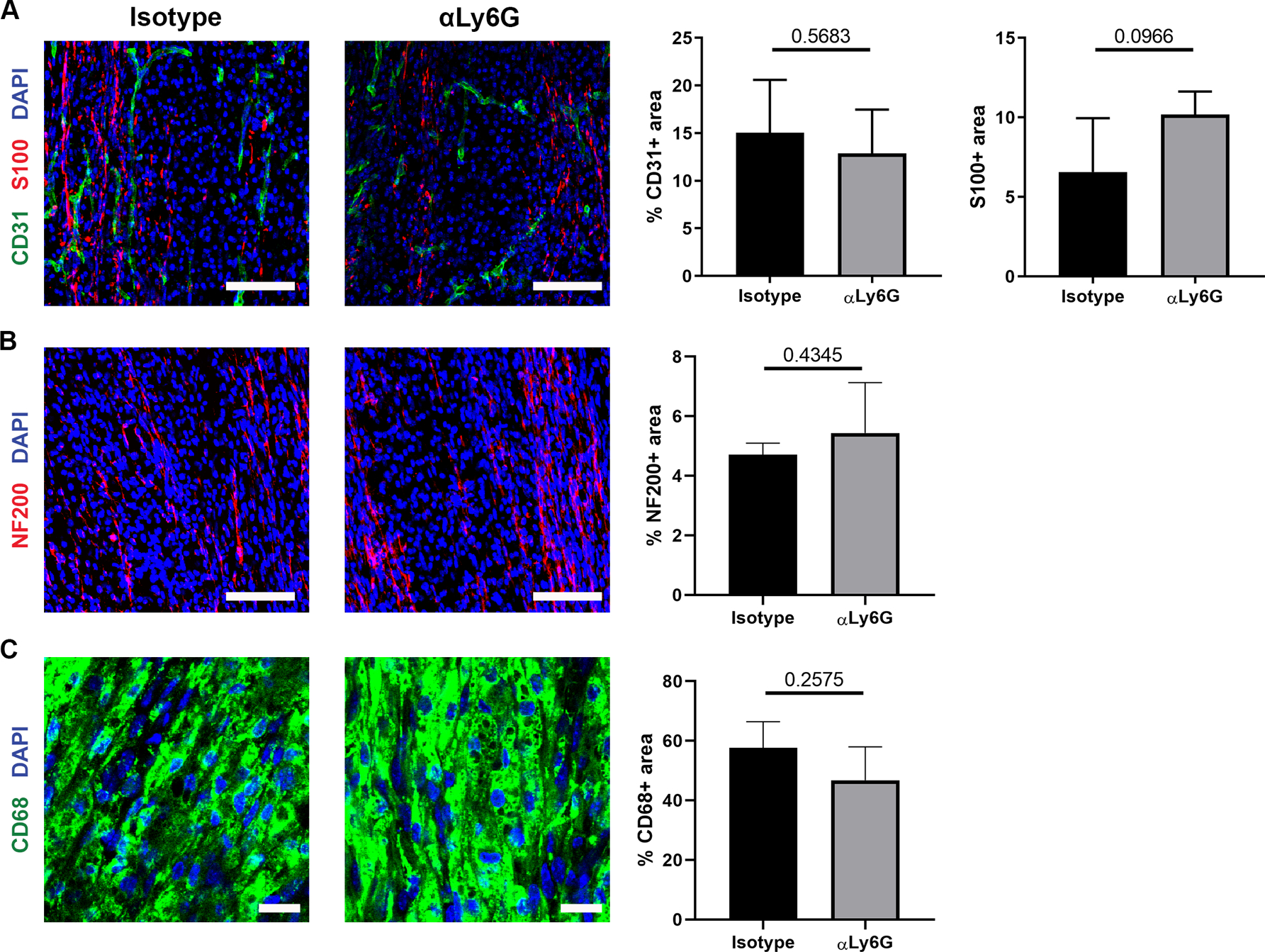
Neutrophils have limited roles in promoting angiogenesis and early regeneration within ANAs. Representative immunofluorescence staining and quantification of endothelial cells (CD31; A), Schwann cells (S100; A), axons (NF200; B), and macrophages (CD68; C) within ANAs post-repair from mice receiving isotype antibody (control) or monoclonal antibody against Ly6G to deplete neutrophils. Scale bars represent 50 μm (A-B) or 10 μm (C). Mean ± SD, n=4/group; p values shown.
Given the lack of impact due to neutrophil depletion, we next examined the role of macrophages. Macrophage were depleted through administering clodronate liposomes to WT mice up to the 2-week endpoint, which severely reduced macrophage accumulation within the ANAs in a duration-dependent manner (Fig. 5A). Longer duration of clodronate liposome administration led to greater depletion of macrophages (based on CD68 area). Regardless of duration of clodronate liposome administration, however, EC quantities (CD31 area) and vessel formation was severely reduced within ANAs from treated compared to untreated mice (Fig. 5B). In turn, SC accumulation (S100 area) within ANAs from treated mice were also reduced compared to untreated mice (Fig. 5B), as was axon extension (NF200 area) within ANAs (Fig. 5C). Altogether, these data demonstrated that macrophages, and not neutrophils, have essential contributions driving angiogenesis within ANAs, where this angiogenesis is associated with SC repopulation and axon extension within ANAs.
Fig. 5.
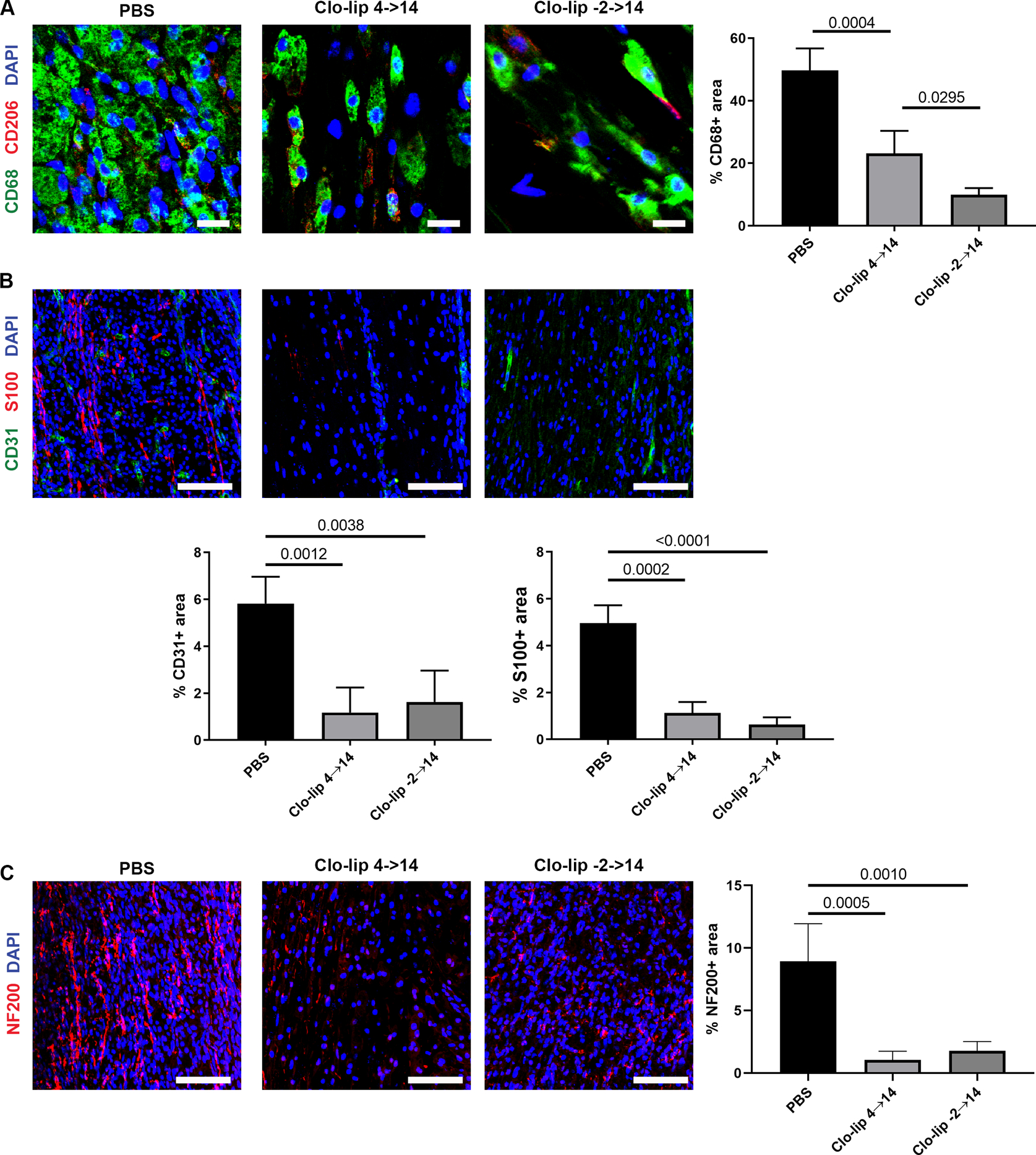
Macrophages promote angiogenesis and regeneration within ANAs. Representative immunofluorescence staining and quantification of macrophages (CD68 and CD206; A), endothelial cells (CD31; B), Schwann cells (S100; B), and axons (NF200; C), within ANAs 14 days post-repair from WT mice administered clodronate liposomes or PBS (control). Scale bars represent 10 μm (A) or 50 μm (B-C). Mean ± SD, n=4/group; p values shown.
Given the impact of clodronate liposomes on macrophages accumulating within ANAs, Dil encapsulated liposomes were used to identify the macrophage source primarily targeted by liposomes that accumulate within ANAs. In WT mice, a single dose of Dil encapsulated liposome labeled a plurality of cells within ANAs, but minimal of cells in the distal nerve to the attached ANA (Supplemental Fig. 2A). Through flow cytometry it was revealed that less than 4% of macrophages were Dil+ in a mice model that does not recruit hematogenous macrophages, compared to nearly 15% of macrophages in WT mice (CCR2 KO; Supplemental Fig. 2B). This suggested that liposomal clodronate has a greater effect on hematogenous-derived macrophages. Furthermore, uninjured mice that received clodronate liposomes had no differences in macrophages quantities within nerve compared to mice that received PBS (Supplemental Fig. 2C). Overall, these results suggested that clodronate liposomes had a greater effect on hematogenous-derived macrophages than tissue resident macrophages, thus motivating studies to identify the contributions of hematogenous-derived macrophages to regeneration across ANAs.
Hematogenous-derived macrophages impact angiogenesis and nerve regeneration across ANAs
CCR2 KO mice were then used to determine if hematogenous-derived macrophages contribute to angiogenesis and regeneration within ANAs repairing nerves. First, flow cytometry of cells expressing CCR2 confirmed macrophages represented cells with the greatest expression level of CCR2 (Supplemental Fig. 3). Furthermore, it was confirmed that nerve repaired using ANA in CCR2 KO mice accumulated fewer macrophages (based on CD68 area) within the ANAs by 2 weeks (Fig 6A). Consistent with the reduction of macrophages on CCR2 KO, at 2 weeks CCR2 KO mice ANAs had reduced angiogenesis (CD31 area) compared to WT mice ANAs (Fig. 6B). Similarly, CCR2 KO mice ANAs had reduced SC accumulation (S100 area), as well as reduced axon extension (NF200 area) within the ANA (Fig. 6B–C).
Fig. 6.
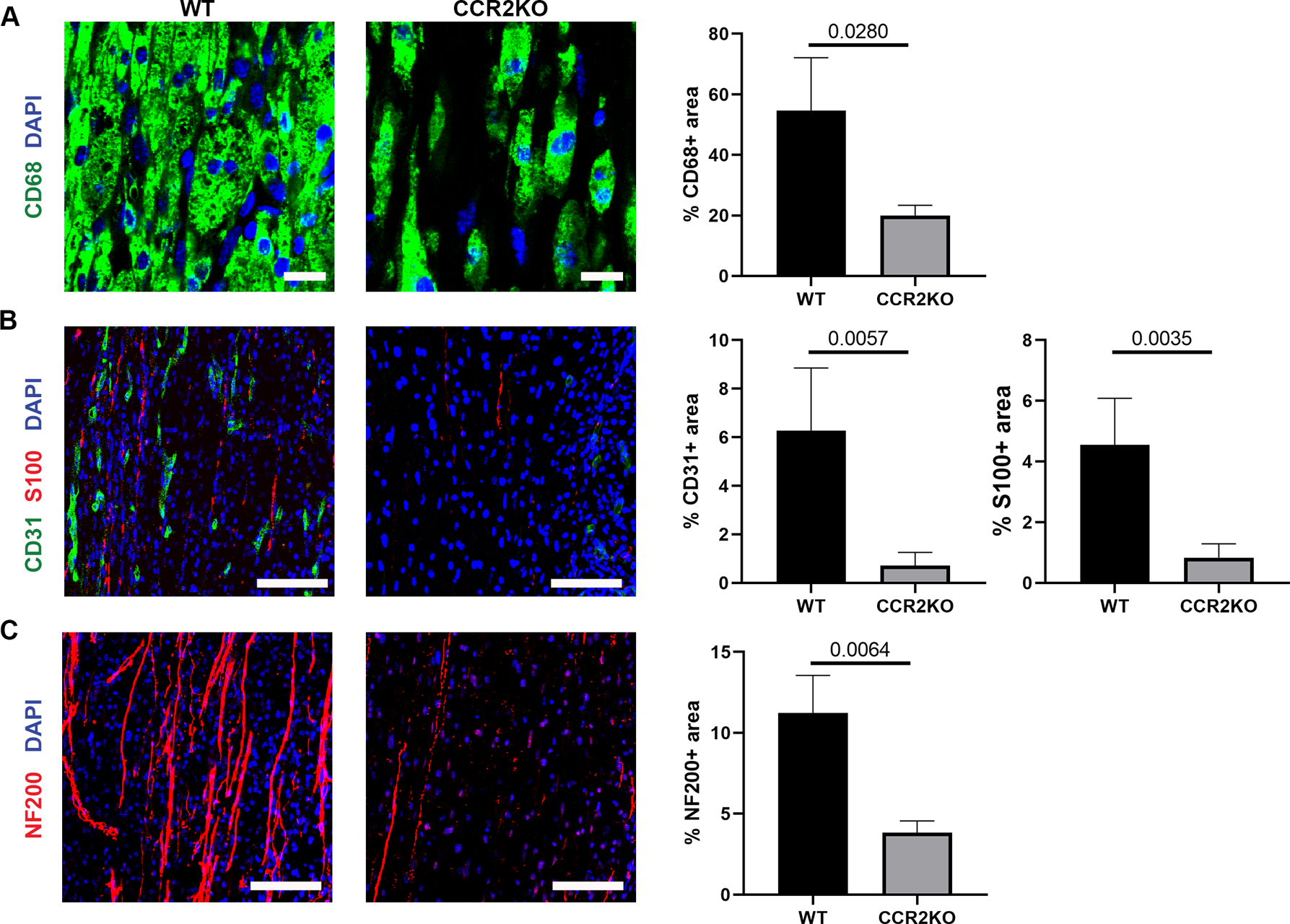
CCR2 promotes angiogenesis and robust regeneration within ANAs. Representative immunofluorescence staining and quantification of macrophages (CD68 and CD206; A), endothelial cells (CD31; B), Schwann cells (S100; B), and axons (NF200; C), within ANAs 14 days post-repair from CCR2 KO and WT mice. Longitudinal sections are presented. Scale bars represent 10 μm (A) or 50 μm (B-C). Mean ± SD, n=4/group; p values shown.
To confirm the consequences of these relatively short-term deficits, a longer-term endpoint (6 weeks) was used to assess the impact to the final quality of nerve regeneration and functional recovery. Compared to WT mice repaired using ANAs, CCR2 KO mice had grossly similar axon regeneration within ANA (NF200), as well as a similar extent of myelination (MBP area; Fig. 7A). And while there was no significant difference in gastrocnemius muscle mass 6 weeks following repair in either mice model, CCR2 KO mice repaired with ANAs experienced a greater proportion of foot faults demonstrating reduced functional recovery compared to WT mice (Fig. 7B). Overall, a CCR2 deficiency impacted angiogenesis and regeneration within the first 2 weeks during regeneration across ANAs, which by 6 weeks had resolved but resulted in impaired functional recovery.
Fig. 7.
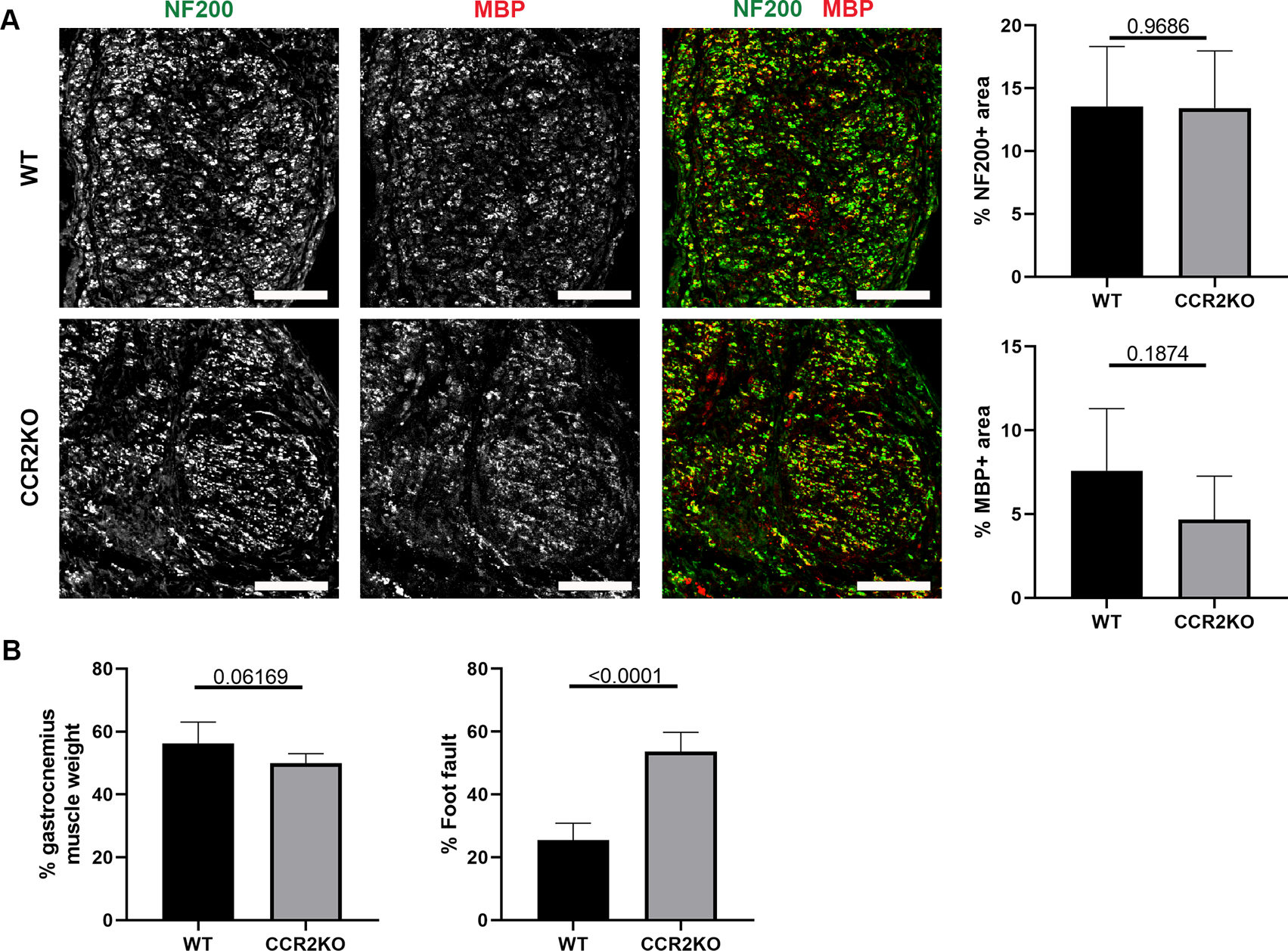
CCR2 deficiency impairs functional recovery across ANAs. A) Representative immunofluorescence staining and quantification of axons and myelin (MBP) within ANAs 6 weeks post-repair from CCR2 KO and WT mice. Cross-sections of nerve are presented. Scale bars represent 50 μm. B) Relative gastrocnemius muscle mass and grid-walk analysis at 6 weeks post-repair from CCR2 KO and WT mice. Mean ± SD, n=5/group; p values shown.
Macrophages are recruited to ANAs via CCL2, which also regulates their phenotype
Given the role of CCR2 in macrophage recruitment and accumulation within ANAs, we assessed if CCL2, the primary chemokine responsible for recruiting CCR2 monocytes, contributed to their recruitment within ANAs. We first verified that SCs were a relative major source of CCL2, where the expression level of CCL2 from SCs was 6-fold that of macrophages’ expression within ANAs (Supplemental Fig. 4). Subsequently, 2 weeks following nerve repair with ANAs, the loss of CCL2 (via CCL2 KO mice) resulted in a reduced accumulation of macrophages in ANAs, as shown via reduced immunohistochemical staining of CD68+ area (Fig. 8A). Given that CCL2 can promote macrophage polarization to an M1 inflammatory phenotype (Carson IV et al., 2017), the proportion of macrophages expressing CD206 (a prominent M2 marker) was also assessed. A greater proportion of CD206+ macrophages relative to total CD68+ macrophages accumulated within ANAs from CCL2 KO vs WT mice, as measured via IHC (Fig. 8A). Similarly, using flow cytometry, there was a reduction of macrophages (CD45+, CD11b+, CD64+, F4/80+) in CCL2 KO mice, both as a proportion of CD11b+ myeloid cells or as the total number of macrophages within ANAs compared to WT mice (Figs. 8B). To determine if difference in macrophage accumulation between ANAs from WT vs CCL2 KO mice was due to macrophage recruitment vs proliferation, we assessed the quantity of macrophages expressing proliferation marker Ki-67, and found a greater proportion, but fewer total number of proliferative macrophages in CCL2 KO mice than WT (Fig. 8C). These data taken together with data derived from clodronate liposome experiments demonstrated that macrophage accumulation within ANAs is largely driven by hematogenous-derived monocytes/macrophages, recruited to the site of injury through CCL2 signaling.
Fig. 8.
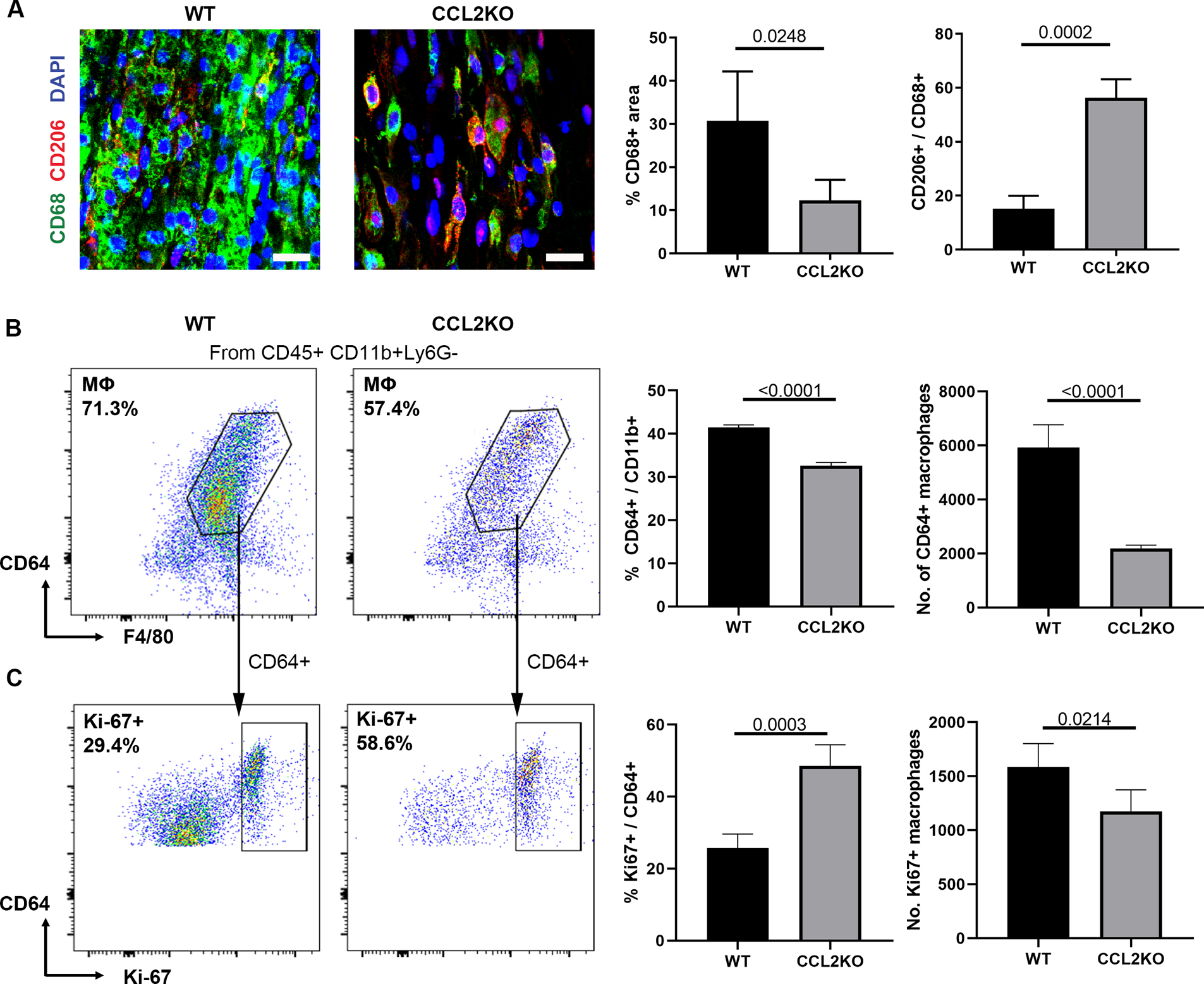
CCL2 promotes robust macrophage accumulation within ANAs. A) Representative immunofluorescence staining and quantification of macrophages (CD68 and CD206) within ANAs 14 days post-repair from CCL2 KO and WT mice. Scale bars represent 10 μm. Flow cytometry analysis for macrophages (CD45+, CD11b+, CD64+, F4/80+; B) among myeloid cells (CD11b+), and proliferative macrophages (CD45+, CD11b+, CD64+, F4/80+, Ki67+; C) among macrophages, within ANAs 14 days post-repair from CCL2 KO and WT mice. For each flow plot, arrows between flow cytometry plots represent further sorting of outlined cell populations for subsequent plots. Mean ± SD, n=4/group; p values shown.
Loss of CCL2 impedes regeneration across ANAs
Given the role of CCL2 in macrophage recruitment within ANAs, we assessed the impact of CCL2 loss on regeneration across nerve repaired ANAs. Fourteen days after nerve transection and repair with ANAs, we found that the ANAs from CCL2 KO mice had reduced ECs (CD31 area) vs WT mice (Fig. 9A). Consistent with the observation that ECs and blood vessels are associated with SC migration and axon extension into an ANA, SC accumulation (S100 area) within ANAs was also reduced among CCL2 KO vs WT mice (Fig. 9A). However, axonal extension (NF200) was not affected (Fig. 9B).
Fig. 9.
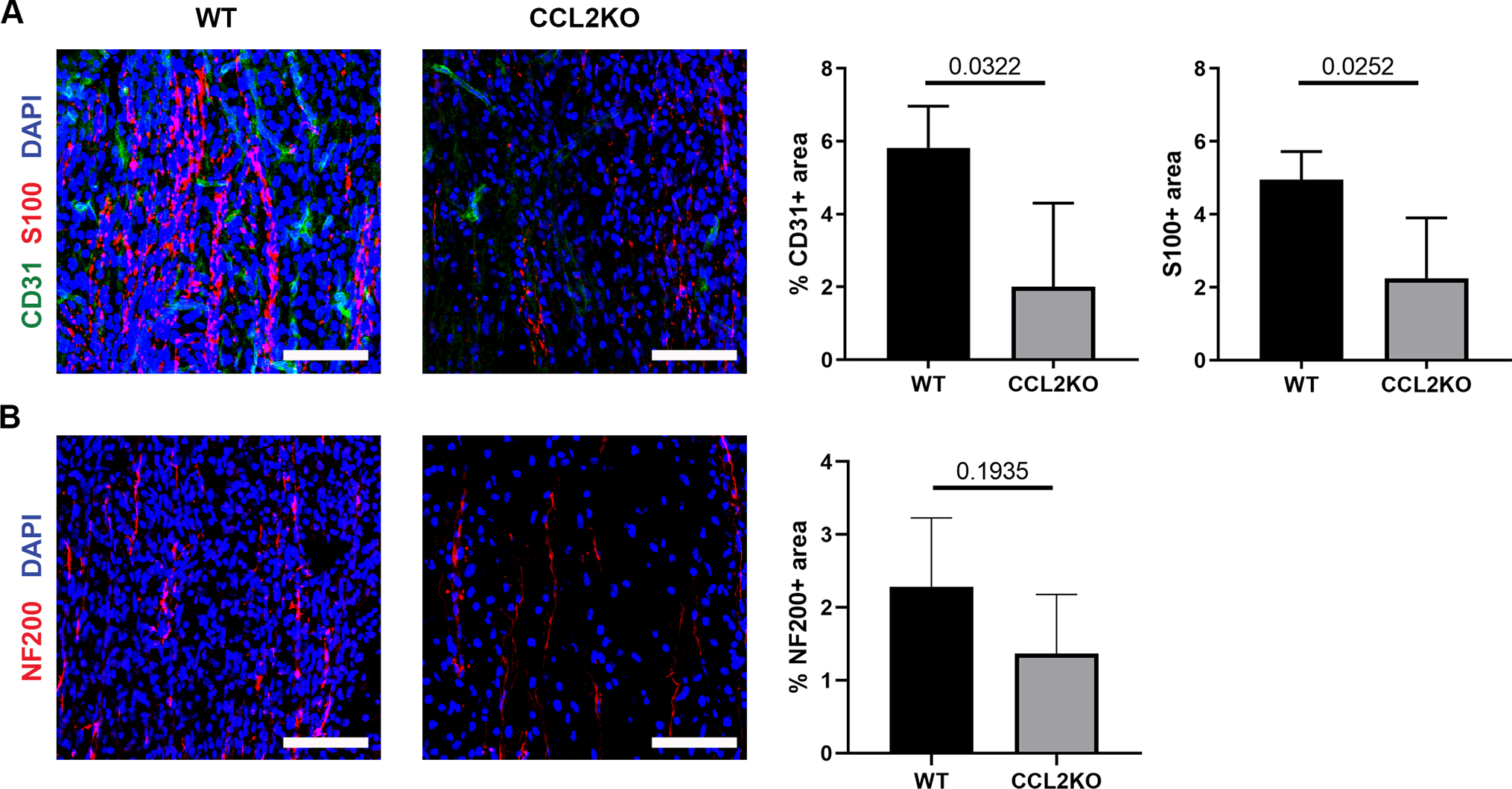
CCL2 promotes angiogenesis and robust regeneration within ANAs. Representative immunofluorescence staining and quantification of endothelial cells (CD31; A), Schwann cells (S100; A), and axons (NF200; B), within ANAs 14 days post-repair from CCL2 KO and WT mice. Longitudinal sections are presented. Scale bars represent 50 μm. Mean ± SD, n=4/group; p values shown.
Similar to the examination of CCR2 KO mice nerves repaired using ANAs, the longer-term impact CCL2 loss was assessed at 6 weeks. Axon regeneration (NF200) and the extent of myelination (MBP) within ANAs were not different among CCL2 KO vs WT mice by this time (Fig. 10A). However, CCL2 KO mice experienced a greater number of foot faults, as well as recovered less muscle mass following nerve denervation with reinnervation (Fig. 10B). Overall, these data demonstrated that CCL2 signaling promotes robust nerve regeneration across an ANA within the first 2 weeks, leading to improved functional recovery across a nerve gap repaired using an ANA as regeneration proceeds.
Fig. 10.
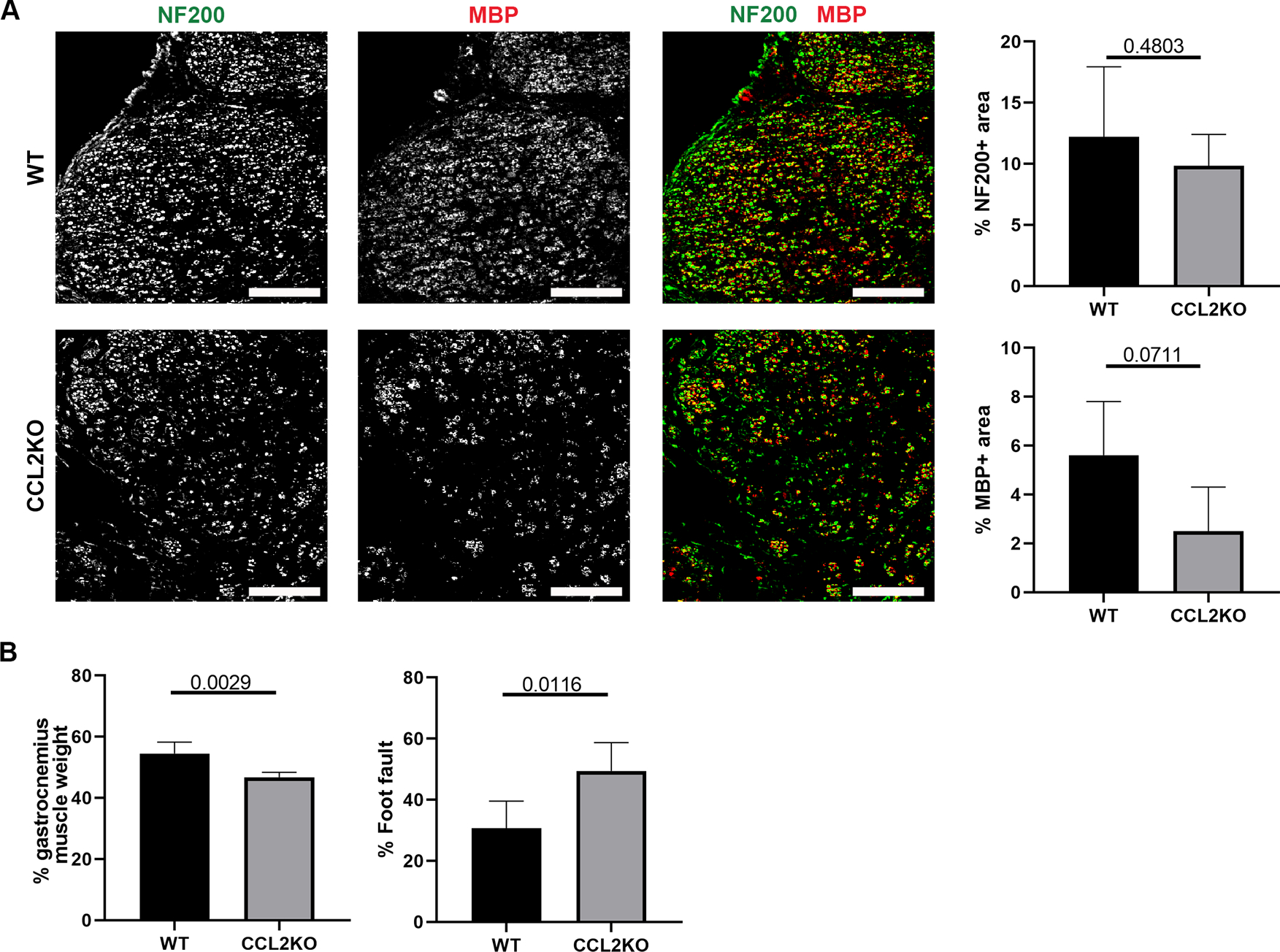
CCL2 deficiency impairs functional recovery across ANAs. A) Representative immunofluorescence staining and quantification of axons and myelin (MBP) within ANAs 6 weeks post-repair from CCL2 KO and WT mice. Cross-sections of nerve are presented. Scale bars represent 50 μm. B) Relative gastrocnemius muscle weight and grid-walk analysis at 6 weeks post-repair from CCL2 KO and WT mice. Mean ± SD, n=5/group; p values shown.
Discussion
While nerve gaps repaired using initially acellular biomaterials and scaffolds, such as ANAs, require angiogenesis to promote regeneration, how angiogenesis drives cell repopulation and the sources that promote angiogenesis have had limited studies. Furthermore, there has been increasing realization of the role of macrophages during peripheral nerve regeneration, which extends the roles of macrophages beyond promoting myelin clearance and SC maturation, but also orchestrating essential angiogenesis (Cattin et al., 2015; Lindborg et al., 2017; Stratton et al., 2018). Our work demonstrates that macrophages are important for angiogenesis across acellular nerve scaffolds, where the macrophages that promote angiogenesis within ANAs are primarily hematogenous-derived macrophages with essential signaling via the CCL2/CCR2 axis. Furthermore, angiogenesis within ANAs driven by macrophages promotes efficient SC and T cell repopulation and subsequent nerve regeneration that promotes functional recovery.
Following nerve repair with ANAs, we observed a drastic increase in angiogenesis within the nerve graft. Surprisingly, this is a relatively delayed process as there was limited angiogenesis even 10 days after injury, and as well, angiogenesis continues for at least another 8 days concurrent with the accumulation of key regenerative cells, such as SCs. Thus, angiogenesis is tightly linked to nerve regeneration across ANAs. Furthermore, unlike small nerve gap injuries (~3 mm) where angiogenesis occurs rapidly (<4 days) (Cattin et al., 2015), larger nerve gaps repaired by ANAs (~10 mm) require substantially more time to vascularize.
Unsurprisingly, angiogenesis was necessary for nerve regeneration across ANAs, but the cells that are driven to repopulate the ANAs was unclear. Previously, it was shown that in a nerve transection model resulting in a small nerve gap (~3 mm), ECs provide a substrate for SC migration (Cattin et al., 2015). In our studies, inhibition of angiogenesis and EC migration almost completely abolished SC cell migration and accumulation within an ANA, despite the well-organized ECM matrix provided by an ANA bridging the nerve gap, which could have potentially served as substrate for SC migration despite a lack of blood vessels. Furthermore, T cells, which contribute to axon regeneration in ANA repaired nerves (Pan et al., 2019a), were absent in ANAs without angiogenesis, suggesting that blood vessels are required for T cells to accumulate. Finally, while the inhibition of angiogenesis via a VEGFR inhibitor increased the proportion of macrophages within ANAs – likely due to the decrease in number of Schwann cells and T cells – their total numbers were decreased compared to models with robust angiogenesis. Therefore, it is possible that macrophages that first arrive within ANAs are independent of blood vessels, but subsequent recruitment of additional macrophages may require blood vessels.
We also explored which myeloid cell populations are required for angiogenesis. Neutrophils, monocytes, and macrophages are major leukocytes within ANAs following repair, but their appearance during the regenerative process was later previously reported for nerve injury (Lindborg et al., 2017). As ANAs initially lack any cells, the accumulation of neutrophils and macrophages were delayed compared to normal injured nerves as reflected in our data. And, the higher proportion of neutrophils and macrophages within ANAs compared to nerve injury may represent a greater need for leukocytes in ANAs due to their acellular nature (Lindborg et al., 2017). Neutrophils and macrophages have both been suggested to contribute to angiogenesis, either independently or in conjunction (Cattin and Lloyd, 2016; Lin et al., 2006; Shojaei et al., 2008). However, their contribution to angiogenesis in ANAs or other scaffold repaired nerves has not been assessed. We found that loss of neutrophils had a limited impact on angiogenesis within ANAs, while loss of macrophages drastically impacted angiogenesis within ANAs. These findings are consistent with previous findings following nerve injury, that despite neutrophil infiltration, their impact on regeneration was limited (Nadeau et al., 2011). On the other hand, macrophage-derived angiogenic factors have been shown to be a major contributor to angiogenesis across a small (~3 mm), unrepaired nerve gap (Cattin et al., 2015), suggesting that mechanisms for spontaneous nerve regeneration are closely recapitulated during nerve regeneration across a gap repaired using biomaterials.
Macrophages that accumulate within injured nerve can be derived via local proliferation of tissue resident macrophages, or via the recruitment of monocytes that mature into macrophages (hematogenous-derived). Both sources of macrophages are robust in numbers within injured nerves (Mueller et al., 2003) suggesting that either source could contribute to regeneration across a nerve gap. We found that hematogenous-derived macrophages are the main initial contributing source of macrophages promoting angiogenesis and regeneration within ANAs repairing a nerve gap. These findings are well-supported for several reasons. Firstly, previous studies found that by day 7, a majority of macrophages within an injured nerve are hematogenous-derived macrophages (Mueller et al., 2003). Secondly, we found that liposomes preferentially targeted hematogenous macrophages since Dil encapsulated liposomes have higher uptake in macrophages from ANAs of WT mice than that of CCR2KO mice. Thirdly, liposomal clodronate treatment did not have an effect on the number of macrophages in uninjured nerves, suggesting that it is inefficient at depleting tissue-resident macrophages.
Further evidence for the role of hematogenous-derived macrophages was provided by studies using CCR2 KO mice. Additionally, as various chemokines signal via CCR2, among them, CCL2 is expressed by SC, as well as neurons and their axons, making it a likely candidate for macrophage recruitment within injured nerve (Kwon et al., 2015; Niemi et al., 2016; Van Steenwinckel et al., 2015). Indeed, we found that loss of CCL2 reduced macrophage accumulation within ANAs but had no impact on the proliferation of macrophages, suggesting that it promotes the recruitment of hematogenous-derived macrophages. Furthermore, loss of CCL2 resulted in reduced macrophage recruitment, angiogenesis, and SC accumulation, which is consistent with our other data that loss of macrophage accumulation within ANAs contributes to reduced nerve regeneration.
While we have focused on the accumulation of macrophages within ANA repairing injured nerve, macrophages are also known to accumulate both proximally and distally to the site of nerve injury. Proximally, macrophages accumulate around neuron cell bodies in the DRG and spinal cord and are known to promote nerve regeneration (Kwon et al., 2015). Distally, macrophages accumulate around the NMJs previously innervated by the damaged axons from the nerve (Vannucci et al., 2019). Previous studies have shown that loss of CCL2/CCR2 impacts macrophage accumulation at DRGs, and loss of CCL2 delays regeneration (Niemi et al., 2016, 2013). Thus, the effects measured in our studies from the loss of macrophage in CCL2KO or CCR2KO mice are complicated by the potential loss of macrophages within DRG and NMJ. Further studies will be required to determine if locally depleted macrophages within sites of injury, like at only the ANA, also still affect nerve regeneration.
Despite the profound impact that loss of hematogenous-derived macrophages had on early angiogenesis and regeneration within ANAs, we found that by 6 weeks following repair with ANAs, there were few obvious differences in axon regeneration or myelination. Thus, there is likely compensation within the nerves of CCR2 KO and CCL2 KO mice to mitigate the loss of hematogenous-derived macrophages during nerve regeneration. One possibility is the increase in proliferation of tissue-resident macrophages within nerve. Indeed, we found that in CCL2 KO mice, while there were fewer macrophages recruited within ANAs, there was increased proliferation of macrophages within the ANAs, which may partially compensate for the loss of hematogenous-derived macrophage. While this compensation delayed nerve regeneration across a 10 mm ANA, eventually regeneration proceeded, but it resulted in impaired functional recovery. Therefore, exploring this concept for longer and critically-sized ANAs (i.e. >3 cm) could be enlightening. As critically-sized ANAs (>3 cm) fail to support nerve regeneration and undergo deficient angiogenesis compared to shorter ANAs (<3 cm) (Pan et al., 2019a), understanding how CCR2 macrophage accumulation, how CCL2 expression is regulated, and whether tissue resident macrophages can contribute to angiogenesis and regeneration within these critically-sized ANAs could reveal major insights as to why they are functionally limited by length.
Conclusions
Our data demonstrate that the CCL2/CCR2 axis promotes the recruitment of hematogenous-derived macrophages within ANAs. These macrophages are important for promoting angiogenesis and subsequent accumulation of non-neuronal support cells within ANAs that promote axon regeneration, such as SCs and T cells. These findings suggest therapies to promote robust angiogenesis within biomaterials and scaffolds used to repair a nerve gap could target hematogenous-derived macrophages.
Supplementary Material
Acknowledgements
We thank Daniel Hunter for helpful discussion and Lauren Schellhardt for help with animal care. This work was supported in part by the National Institutes of Neurological Disorders and Stroke of the National Institutes of Health (NIH) under award numbers R01 NS086773 (SEM), R01 NS115960 (MDW), and K08 NS096232 (AKSW) to Washington University, an award from the McDonnell Center for Cellular and Molecular Neuroscience (MDW), and by a Pilot Project Award from the Hope Center for Neurological Disorders at Washington University (MDW). The content is solely the responsibility of the authors and does not represent the views of the NIH or Washington University.
Funding:
This work was supported in part by the National Institutes of Neurological Disorders and Stroke of the National Institutes of Health (NIH) under award numbers R01 NS086773, R01 NS115960, and K08 NS096232 to Washington University.
M.D.W. has consulted for The Foundry, LLC and Foundry Therapeutics, LLC and been a recipient of a sponsored research agreement with Checkpoint Surgical, Inc. All other authors have no competing interests to declare.
Footnotes
Data Availability
The experimental data required to reproduce the findings from this study will be made available to interested investigators upon request.
Bibliography
- Beck-Schimmer B, Schwendener R, Pasch T, Reyes L, Booy C, Schimmer RC, 2005. Alveolar macrophages regulate neutrophil recruitment in endotoxin-induced lung injury. Respir. Res 6, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boring L, Gosling J, Chensue SW, Kunkel SL, Farese RV, Broxmeyer HE, Charo IF, 1997. Impaired monocyte migration and reduced type 1 (Th1) cytokine responses in C-C chemokine receptor 2 knockout mice. J. Clin. Invest 100, 2552–61. 10.1172/JCI119798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carson IV WF, Salter-Green SE, Scola MM, Joshi A, Gallagher KA, Kunkel SL, 2017. Enhancement of macrophage inflammatory responses by CCL2 is correlated with increased miR-9 expression and downregulation of the ERK1/2 phosphatase Dusp6. Cell. Immunol 314, 63–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattin A-L, Burden JJ, Van Emmenis L, Mackenzie FE, Hoving JJA, Garcia Calavia N, Guo Y, McLaughlin M, Rosenberg LH, Quereda V, Jamecna D, Napoli I, Parrinello S, Enver T, Ruhrberg C, Lloyd AC, 2015. Macrophage-Induced Blood Vessels Guide Schwann Cell-Mediated Regeneration of Peripheral Nerves. Cell 162, 1127–1139. 10.1016/j.cell.2015.07.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cattin A-L, Lloyd AC, 2016. The multicellular complexity of peripheral nerve regeneration. Curr. Opin. Neurobiol 39, 38–46. 10.1016/j.conb.2016.04.005 [DOI] [PubMed] [Google Scholar]
- Crown SE, Yu Y, Sweeney MD, Leary JA, Handel TM, 2006. Heterodimerization of CCR2 chemokines and regulation by glycosaminoglycan binding. J. Biol. Chem 281, 25438–25446. [DOI] [PubMed] [Google Scholar]
- Hankenson KD, Dishowitz M, Gray C, Schenker M, 2011. Angiogenesis in bone regeneration. Injury 42, 556–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ijkema-Paassen J, Jansen K, Gramsbergen A, Meek MF, 2004. Transection of peripheral nerves, bridging strategies and effect evaluation. Biomaterials. 10.1016/S0142-9612(03)00504-0 [DOI] [PubMed] [Google Scholar]
- Kwon MJ, Shin HY, Cui Y, Kim H, Le Thi AH, Choi JY, Kim EY, Hwang DH, Kim BG, 2015. CCL2 mediates neuron–macrophage interactions to drive proregenerative macrophage activation following preconditioning injury. J. Neurosci 35, 15934–15947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leckenby JI, Furrer C, Haug L, Personeni BJ, Vögelin E, 2020. A retrospective case series reporting the outcomes of Avance nerve allografts in the treatment of peripheral nerve injuries. Plast. Reconstr. Surg 145, 368e–381e. [DOI] [PubMed] [Google Scholar]
- Lin Y-J, Lai M-D, Lei H-Y, Wing L-YC, 2006. Neutrophils and macrophages promote angiogenesis in the early stage of endometriosis in a mouse model. Endocrinology 147, 1278–1286. [DOI] [PubMed] [Google Scholar]
- Lindborg JA, Mack M, Zigmond RE, 2017. Neutrophils are critical for myelin removal in a peripheral nerve injury model of Wallerian degeneration. J. Neurosci 37, 10258–10277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu B, Rutledge BJ, Gu L, Fiorillo J, Lukacs NW, Kunkel SL, North R, Gerard C, Rollins BJ, 1998. Abnormalities in monocyte recruitment and cytokine expression in monocyte chemoattractant protein 1-deficient mice. J. Exp. Med 187, 601–8. 10.1084/jem.187.4.601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeddu P, 2005. Therapeutic angiogenesis and vasculogenesis for tissue regeneration. Exp. Physiol 90, 315–326. [DOI] [PubMed] [Google Scholar]
- Martini R, Fischer S, López‐Vales R, David S, 2008. Interactions between Schwann cells and macrophages in injury and inherited demyelinating disease. Glia 56, 1566–1577. [DOI] [PubMed] [Google Scholar]
- Mokarram N, Dymanus K, Srinivasan A, Lyon JG, Tipton J, Chu J, English AW, Bellamkonda RV, 2017. Immunoengineering nerve repair. Proc. Natl. Acad. Sci 114, E5077–E5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller M, Leonhard C, Wacker K, Ringelstein EB, Okabe M, Hickey WF, Kiefer R, 2003. Macrophage Response to Peripheral Nerve Injury: The Quantitative Contribution of Resident and Hematogenous Macrophages. Lab. Investig 83, 175. [DOI] [PubMed] [Google Scholar]
- Nadeau S, Filali M, Zhang J, Kerr BJ, Rivest S, Soulet D, Iwakura Y, de Rivero Vaccari JP, Keane RW, Lacroix S, 2011. Functional recovery after peripheral nerve injury is dependent on the pro-inflammatory cytokines IL-1β and TNF: implications for neuropathic pain. J. Neurosci 31, 12533–12542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi JP, DeFrancesco-Lisowitz A, Cregg JM, Howarth M, Zigmond RE, 2016. Overexpression of the monocyte chemokine CCL2 in dorsal root ganglion neurons causes a conditioning-like increase in neurite outgrowth and does so via a STAT3 dependent mechanism. Exp. Neurol 275, 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niemi JP, DeFrancesco-Lisowitz A, Roldán-Hernández L, Lindborg JA, Mandell D, Zigmond RE, 2013. A critical role for macrophages near axotomized neuronal cell bodies in stimulating nerve regeneration. J. Neurosci 33, 16236–16248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Hunter DA, Schellhardt L, Jo S, Santosa KB, Larson EL, Fuchs AG, Snyder-Warwick AK, Mackinnon SE, Wood MD, 2019a. The accumulation of T cells within acellular nerve allografts is length-dependent and critical for nerve regeneration. Exp. Neurol 318, 216–231. 10.1016/j.expneurol.2019.05.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D, Mackinnon SE, Wood MD, 2019b. Advances in the repair of segmental nerve injuries and trends in reconstruction. Muscle Nerve mus 26797 10.1002/mus.26797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poppler LH, Ee X, Schellhardt L, Hoben GM, Pan D, Hunter DA, Yan Y, Moore AM, Snyder-Warwick AK, Stewart SA, 2016. Axonal growth arrests after an increased accumulation of Schwann cells expressing senescence markers and stromal cells in acellular nerve allografts. Tissue Eng. Part A 22, 949–961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rbia N, Shin AY, 2017. The role of nerve graft substitutes in motor and mixed motor/sensory peripheral nerve injuries. J. Hand Surg. Am 42, 367–377. [DOI] [PubMed] [Google Scholar]
- Robinson LR, 2000. Traumatic injury to peripheral nerves. Muscle Nerve Off. J. Am. Assoc. Electrodiagn. Med 23, 863–873. [DOI] [PubMed] [Google Scholar]
- Sager HB, Hulsmans M, Lavine KJ, Moreira MB, Heidt T, Courties G, Sun Y, Iwamoto Y, Tricot B, Khan OF, 2016. Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circ. Res 119, 853–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saheb-Al-Zamani M, Yan Y, Farber SJ, Hunter DA, Newton P, Wood MD, Stewart SA, Johnson PJ, Mackinnon SE, 2013. Limited regeneration in long acellular nerve allografts is associated with increased Schwann cell senescence. Exp. Neurol 247, 165–177. 10.1016/j.expneurol.2013.04.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shojaei F, Singh M, Thompson JD, Ferrara N, 2008. Role of Bv8 in neutrophil-dependent angiogenesis in a transgenic model of cancer progression. Proc. Natl. Acad. Sci 105, 2640–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E, 1992. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature 359, 843–845. [DOI] [PubMed] [Google Scholar]
- Siebert H, Sachse A, Kuziel WA, Maeda N, Brück W, 2000. The chemokine receptor CCR2 is involved in macrophage recruitment to the injured peripheral nervous system. J. Neuroimmunol 110, 177–185. [DOI] [PubMed] [Google Scholar]
- Stratton JA, Holmes A, Rosin NL, Sinha S, Vohra M, Burma NE, Trang T, Midha R, Biernaskie J, 2018. Macrophages Regulate Schwann Cell Maturation after Nerve Injury. Cell Rep. 24, 2561–2572. [DOI] [PubMed] [Google Scholar]
- Van Steenwinckel J, Auvynet C, Sapienza A, Reaux-Le Goazigo A, Combadiere C, Parsadaniantz SM, 2015. Stromal cell-derived CCL2 drives neuropathic pain states through myeloid cell infiltration in injured nerve. Brain. Behav. Immun 45, 198–210. [DOI] [PubMed] [Google Scholar]
- Vannucci B, Santosa KB, Keane AM, Jablonka‐Shariff A, Lu C, Yan Y, MacEwan M, Snyder‐Warwick AK, 2019. What is Normal? Neuromuscular junction reinnervation after nerve injury. Muscle Nerve 60, 604–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnatsch A, Ioannou M, Wang Q, Papayannopoulos V, 2015. Neutrophil extracellular traps license macrophages for cytokine production in atherosclerosis. Science (80-. ) 349, 316–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.