

Abstract
Serine hydrolases comprise a large family of enzymes that have diverse roles in key cellular processes, such as lipid metabolism, cell signaling, and regulation of post-translation modifications of proteins. They are also therapeutic targets for multiple human pathologies, including viral infection, diabetes, hypertension, and Alzheimer disease; however, few have well-defined substrates and biological functions. Activity-based probes (ABPs) have been used as effective tools to both profile activity and screen for selective inhibitors of serine hydrolases. One broad-spectrum ABP containing a fluorophosphonate electrophile has been used extensively to advance our understanding of diverse serine hydrolases. Due to the success of this single reagent, several robust chemistries have been developed to further diversify and tune the selectivity of ABPs used to target serine hydrolases. In this review, we highlight approaches to identify selective serine hydrolase ABPs and suggest new synthetic methodologies that could be applied to further advance probe development.
INTRODUCTION
The recent boom in technologies focusing on “-omics” strategies has provided invaluable insights into the genetic, transcriptomic, and proteomic composition of cells. However, these methods are generally unable to identify specific functional regulators of a given cellular state at the protein level. Over the past two decades, activity-based protein profiling (ABPP) has emerged as a valuable strategy for deciphering the functions of many diverse families of enzymes (Adibekian et al., 2011; Bachovchin and Cravatt, 2012). The key component of ABPP is the activity-based probe (ABP), which covalently modifies active site residues in enzymes and thus provides an indirect readout of enzyme activity. Both broad-spectrum and highly selective ABPs have enabled spatiotemporal changes in enzyme activity to be tracked in response to specific cellular perturbations and have thus contributed to our understanding of how specific enzymes drive particular cellular phenotypes. However, a general lack of highly selective chemical probes has prevented the functional characterization of many cellular enzymes. The ability to inhibit and/or readout dynamic changes in activity and localization of specific enzymes in tissues or whole organisms that contain many related enzymes with similar substrate specificities remains a primary goal of contemporary ABPP research. To achieve this, novel approaches are needed to generate probes with enhanced potency and selectivity for specific enzymes. In this review, we highlight strategies commonly used to diversify probe scaffolds and also discuss which new synthetic approaches can be used to advance ABP development in the future. While this review highlights avenues for expanding the chemical space of probes for the serine hydrolase superfamily, the chemical methods discussed here are also generally applicable to ABPs that target other enzyme families and classes.
ABPP and Serine Hydrolases
Serine hydrolases represent one of the most diverse classes of enzymes in eukaryotes, prokaryotes, and viruses. They include proteases, lipases, esterases, peptidases, thioesterases, and amidases (Long and Cravatt, 2011). Due to this extensive diversity, hydrolases have a prominent role in many metabolic functions as well as in disease states, such as cancer and bacterial/viral pathogenesis (Blais et al., 2012; Menendez and Lupu, 2007; Puri and Bogyo, 2013). Consequently, significant effort has been focused on developing chemical tools to identify, classify, and characterize the function of serine hydrolases. One of the original and arguably most successful ABPs ever developed is the broad-spectrum serine hydrolase probe, fluorophosphonate (FP) (Liu et al., 1999). This probe, which was developed more than two decades ago, has become a benchmark reagent for studies of serine hydrolases. Although originally designed to target the lipid-metabolizing enzyme fatty acid amide hydrolase (FAAH), it turned out to be an ideal probe for many diverse serine hydrolases ranging from lipid esterases to proteases. The FP probe relies on the highly selective reactivity of the FP electrophile, which forms an irreversible covalent bond exclusively with serine nucleophiles. Although the FP probe can be derivatized, the core elements of the probe have remained constant with a FP electrophile “warhead” (which covalently modifies the catalytic serine residue in the active site of targeted serine hydrolases), an extended saturated lipid linker, and a tag for either visualization or affinity purification of targets (Figure 1A).
Figure 1. ABP Design and ABPP Workflow.
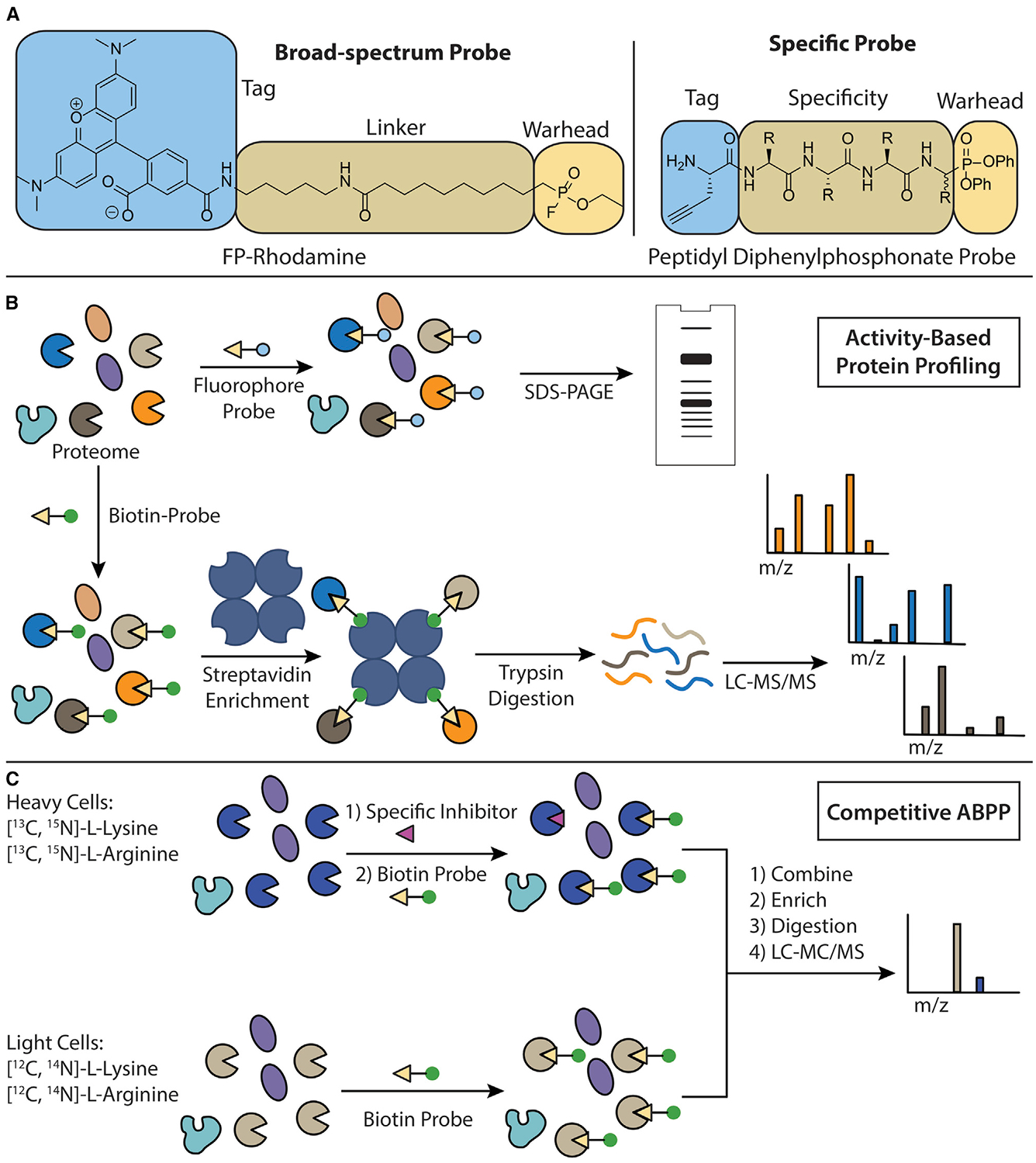
(A) Components of an activity-based probe. The structure of the broad-spectrum FP-rhodamine probe (left). A peptidyl diphenylphosphonate probe scaffold (right). The electrophilic warhead, linker/specificity element, and tag are highlighted.
(B) The workflow of a typical ABPP experiment. Cells or cell lysates are incubated with an ABP. If a fluorescent probe is used, then proteomes can be separated by SDS-PAGE and probe-labeled proteins visualized using in-gel fluorescence. If a biotin-tagged probe is used, then probe-labeled proteins are immobilized on streptavidin resin, digested into tryptic peptides, and subjected to liquid chromatography-tandem mass spectrometry for target identification.
(C) The workflow of a stable isotope competitive ABPP experiment. Cells are grown in the presence of isotopically distinct “heavy” or “light” arginine and lysine. Heavy labeled cells are then pre-treated with an inhibitor before incubation with a broad-spectrum biotinylated ABP. Light labeled cells are pre-treated with a DMSO control before incubation with the same broad-spectrum biotinylated ABP. The heavy and light lysates are mixed together and the same workflow as described in (B) is followed. The heavy-to-light ratio of peptides is then used as a parameter to quantify the selectivity of inhibition of protein targets by the compound of interest.
The FP probe is commonly used in chemical proteomic studies to identify serine hydrolases and also to globally profile how the activity of these enzymes is modulated in a given disease state or cellular condition. Initial profiling studies are often performed using a simple gel-based readout of tagged proteins, which enables rapid and relatively high-throughput analysis of enzyme activity (Wright and Sieber, 2016). However, in many cases, when broad-spectrum probes are used, gel-based methods fail to effectively resolve all targets and a large dynamic range of activities in a given sample can prevent comprehensive analysis by gel. Therefore, probes containing an affinity tag are often used to perform gel-free functional proteomic experiments (Figure 1B). This type of ABPP experiment allows comprehensive mapping of all labeled proteins over a large dynamic range and is often used in combination with isotope labeling to enable highly quantitative readouts of changes in enzyme activity (Figure 1C). There have been many successful applications of the FP probe to ABPP experiments since its inception over 20 years ago. Recent examples include the discovery of a new family of serine hydrolases in S. aureus that are crucial for establishing host infection (Lentz et al., 2018) and the identification of previously uncharacterized serine hydrolases from Vibrio cholerae with important roles in abrogating host expression of intelectin, a protein implicated in the immune response (Hatzios et al., 2016).
A second major application for the FP probe is profiling the specificity and potency of serine hydrolase inhibitors by competitive ABPP (Figure 1C). Competitive ABPP is performed by pre-incubating an inhibitor in a complex biological sample or in vivo and then labeling with a broad-spectrum probe to measure residual enzyme activity. As compounds bind and inhibit their targets, the overall amount of active enzyme available to be labeled by the probe decreases. The limitation of competitive ABPP is that inhibitor specificity can only be judged relative to the enzymes that are effectively labeled by the probe. Thus, broad-spectrum probes, such as the FP probes are ideal for use in competitive ABPP since they label a large number of related enzymes in any given biological sample (Bachovchin et al., 2010). Competitive ABPP is a powerful strategy that has been used extensively to both identify novel small-molecule inhibitors and to confirm the specificity of previously discovered compounds. For example, competitive mass spectrometry-based ABPP with FP-biotin identified the lipase FAAH-4 as the target of the carbamate inhibitor, JZl184, which has previously been shown to regulate lifespan in C. elegans (Chen et al., 2019a). Similarly, competitive gel- and mass spectrometry-based ABPP with FP-rhodamine and FP-biotin, respectively, were used to screen a library of serine-reactive compounds against ABHD12, a lipase that regulates the endocannabinoid system in the brain, resulting in the identification of a potent thiourea inhibitor that demonstrated exquisite selectivity in vivo (Ogasawara et al., 2018). Furthermore, the Cravatt group has reported a competitive ABPP method for measuring the inhibition of serine hydrolases by reversible inhibitors. Key to this approach is the use of kinetically tuned probes that enable competition for target engagement with reversible inhibitors to be measured. For example, an alkyne triazole urea ABP, which has decreased reactivity toward serine hydrolases compared with FP probes, was used to determine the target profiles of various non-covalent piperazine amide inhibitors in living cells and mice (Adibekian et al., 2012).
While broad-spectrum probes, such as FP, are highly effective for proteomic discovery of target enzymes and for competitive ABPP, there is still a need for new classes of highly selective probes. Probe specificity is particularly important for imaging applications as there is no opportunity to biochemically resolve labeled proteins. Furthermore, highly selective probes enable rapid and high-throughput strategies to dynamically monitor a specific target enzyme because there is no need for analytic separations of proteins before readout of signals. This interest in probes with a high degree of target selectivity has led to the development of novel chemical strategies to diversify the binding elements of probes as well as the reactive electrophilic warhead used to form the key covalent bond to targets (Shannon and Weerapana, 2015). Therefore, we have chosen to highlight some of these recent chemical strategies to diversify covalent probes with a particular focus on the serine hydrolases due to the large size of this family and the extensive prior efforts applying ABPs to this class of enzymes.
Serine Hydrolase Warheads
Current Electrophilic Warheads.
While the FP probe has proven useful for studies of diverse serine hydrolase targets, its overall general hydrophobic lipid core makes it most effective for enzymes, such as lipases and esterases involved in lipid metabolism. Furthermore, studies using derivatives of the original FP probe containing multiple tags and linkers, confirm that even small changes to the core probe results in altered target profiles (Janssen et al., 2018). These results highlight the possibility of tuning probes for serine hydrolases by making changes to each of the key components of an ABP, including the tag, linker, and electrophile. An obvious strategy for altering the selectivity of a probe is to modify the main reactive electrophile. This group is the key element that forms the covalent bond to the active site nucleophile of a serine hydrolase. The electrophile can be chosen based on its selectivity toward a given class of nucleophile (i.e., phosphonate, which only reacts with serine versus a chloromethyl ketone that reacts with serine and cysteine residues) but also based on its overall intrinsic reactivity toward that nucleophile. By selecting electrophiles with lower chemical reactivity, the resulting probes derive their selectivity predominantly from the binding energy between the target and the linker/specificity elements. However, if this reactivity is too low, labeling will be inefficient, even with strong binding elements attached. On the other hand, if the reactivity is too high, significant off-target labeling becomes a major concern. This was recently demonstrated for inhibitors and probes designed to target the malaria proteasome, where optimized peptide scaffolds designed with a specific electrophile lost most of their target selectivity upon swap of the electrophile from a less reactive to a more reactive group (Yoo et al., 2018).
For serine hydrolases, a variety of covalent electrophiles exist that have been described over the past 40 years. Many of these, including FPs, diphenyl phosphonates, sulfonyl fluorides, β-lactams, carbamates, triazole ureas, and isocoumarins have been used in probe scaffolds to target serine hydrolases (Figure 2A). These electrophiles differ in their nucleophile and enzyme class specificity as well as ease of synthesis, making the choice of an optimal warhead something that must be carefully considered. Electrophilic phosphorous compounds are a common class of serine hydrolase inhibitor and are particularly attractive as ABPs and inhibitors because they possess a high degree of selectivity for the serine nucleophile. Importantly, they have sufficient intrinsic reactivity to yield active site labeling while avoiding non-specific reactions with free hydroxyl groups. This broad class of serine hydrolase inhibitors includes diphenyl phosphonates, FPs, and mixed aryl/alkyl phosphonates. Recently, the Adibekian group used mixed alkyl aryl phosphonate ABPs to target distinct subsets of the serine hydrolase family by varying the phenolate leaving group (Figure 2B) (Wang et al., 2019). These probes offer several advantages over the FP probe, including ease of synthesis and tunable reactivity, as well as increased cell permeability and reduced toxicity, the latter of which permits labeling of serine hydrolases directly in living cells. The usual method for synthesizing mixed alkyl aryl phosphonates involves LiBr-mediated mono de-alkyation of a diethylphosphonate intermediate followed by esterification with a phenol derivative. However, a recent report used a Cu(I) catalyst with diaryliodonium salts to perform a direct aryloxylation resulting in a one-step transformation of the diethylphosphonate to the mixed alkyl aryl phosphonate (Fañanás-Mastral and Feringa, 2014). Similarly, triflic anhydride and pyridine can also be used to activate the phosphonate and enable direct aryloxylation (Huang et al., 2018) (Figure 2B). These advances should enable large libraries of mixed alkyl aryl phosphonates to be synthesized and therefore this scaffold may be used not only for broad-spectrum labeling of serine hydrolases but also for selective inhibition of individual serine hydrolase targets. Interestingly, the mixed alkyl aryl phosphonate warhead has also been used to develop quenched fluorescent ABPs for various serine proteases, suggesting that this class of electrophile can be used to target a diverse range of serine hydrolases (Serim et al., 2015). Unlike the FP electrophile, which labels diverse classes of hydrolases, diphenylphosphonates (DPPs) predominantly target serine proteases and can be further fine-tuned by optimization of a peptide linker. Specifically, a P1 amino acid can be used to target specific serine protease sub-classes (Jackson et al., 1998). For example, a P1 tyrosine or phenylalanine biases selectivity of the diphenyl phosphonate toward chymotrypsin-like proteases, while a P1 arginine results in labeling of trypsin-like proteases (Powers et al., 2002).
Figure 2. Synthetic Strategies for Diversifying Serine Hydrolase-Targeting Electrophilic Warheads.
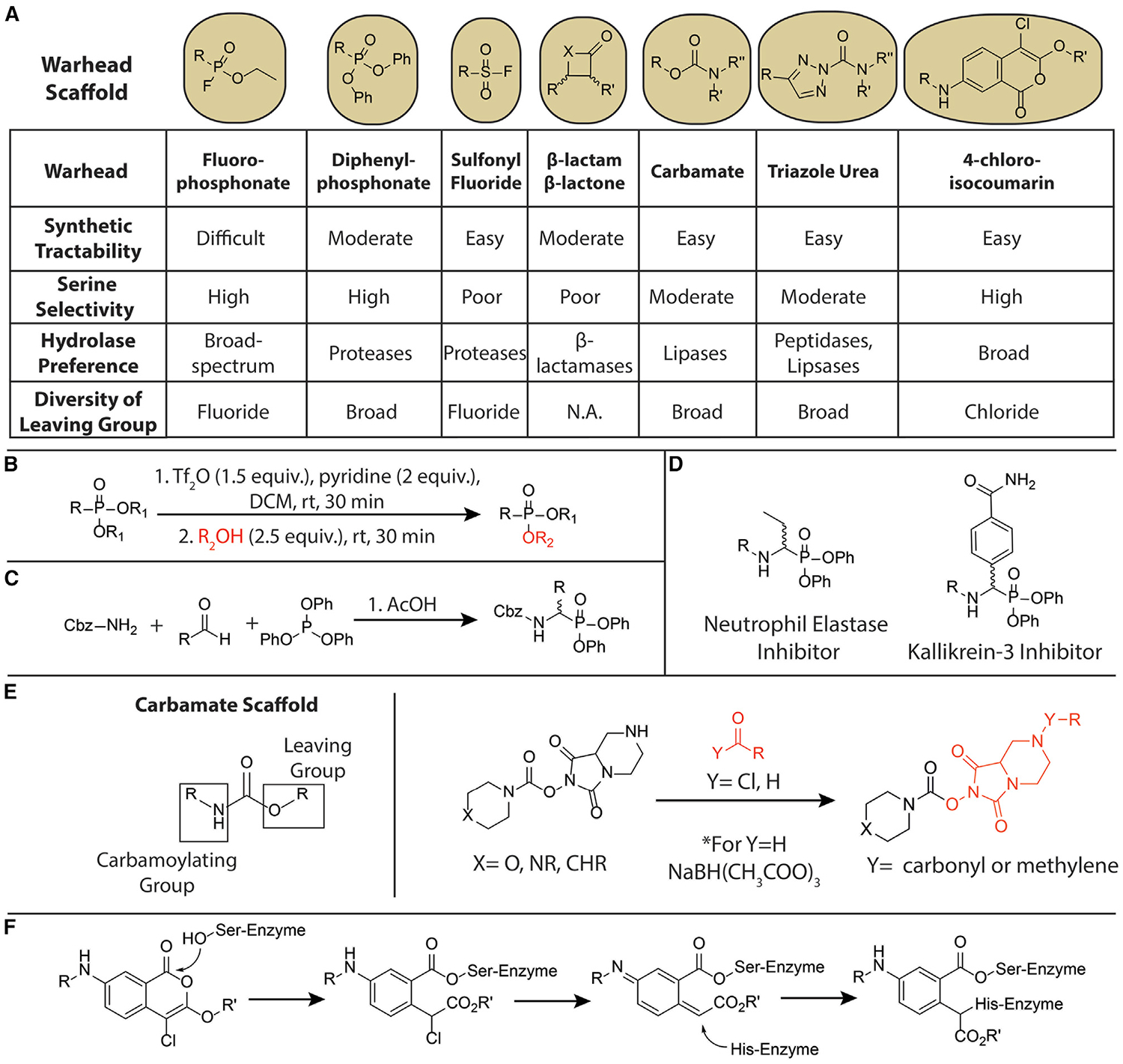
(A) Structures of the commonly used serine hydrolase-targeting electrophilic warheads discussed in this review and key properties to consider for their use in ABPs.
(B) Synthetic route to mixed alkyl/aryl phosphonates described by Huang et al. (2018). The method enables synthesis of diverse mixed alkyl/aryl phosphonates and mixed aryl phosphonates. R = alkyl group. R1 and R2 denote an aryl or alkyl group.
(C) Synthesis of the diphenylphosphonate warhead using an α-amidoalkylation reaction. R = natural or unnatural amino acid.
(D) Examples of tuning the specificity of the DPP electrophile. Examples are a neutrophil elastase DPP inhibitor with a P1 homoalanine and kallikrein 3 DPP inhibitor with a P1 benzamidine.
(E) General structure of the carbamate warhead with the carbamoylating and leaving groups depicted (left). Example of the modification of the leaving group (in red) as a strategy to generate diverse libraries of carbamate inhibitors and probes as described in Cognetta et al. (2015) (right).
(F) The two-hit mechanism of covalent inhibition of serine hydrolases by 4-chloroisocoumarins.
Synthesis of DPPs is accomplished by α-amidoalkylation of triphenyl phosphite with benzyl carbamate and an aldehyde of choice affording racemic Cbz-protected amino diphenyl phosphonates (Figure 2C). Importantly, this chemistry allows installation of an array of natural and non-natural amino acid analogs using aldehydes with diverse R groups. For example, a diphenyl phosphonate analog with a P1 benzamidine was reported as an inhibitor of KLK3, while a P1 homoalanine diphenyl phosphonate showed potent inhibition of neutrophil elastase (Figure 2D) (Kasperkiewicz et al., 2014; Kojtari et al., 2014). The selectivity of the DPP warhead can also be tuned using a range of aryl esters. For instance, Grzywa et al. (2014) developed a panel of diaryl phosphonate probes using an assortment of aryl esters and tested them against human neutrophil elastase, proteinase 3, and cathepsin G. This study found that the p-S-methyl aryl ester greatly increased the potency and selectivity of inhibitors for cathepsin G.
More recently, carbamates and ureas have proven to be useful tools for studying individual serine hydrolases (Adibekian et al., 2011; Alexander and Cravatt, 2005). These warheads were introduced to enable rapid synthesis and discovery of selective inhibitors and probes for functionally characterizing serine hydrolases. The two main branches of the core carbamate or urea scaffold are known as the leaving group and the carbamylating (carbamate)/carbamoylating (urea) group (Figure 2E, left). Seminal work by the Cravatt group revealed that carbamates inactivate serine hydrolases by carbamylation of the catalytic serine nucleophile and also demonstrated that carbamate selectivity can be fine-tuned by varying the leaving group. This provides a platform for the development of a wide range of selective urea and carbamate inhibitors. For example, an O-hexafluoroisopropyl carbamate scaffold was leveraged to generate fluorescent ABPs for MAGL and ABDH6 (Chang et al., 2013). Furthermore, a class of N-hydroxyhydantoin (NHH) carbamates have been reported that can be further modified via acylation or reductive amination. Fine-tuning of the NHH leaving group with this approach resulted in the identification of a selective palmitoyl protein thioesterase 1 inhibitor (Figure 2E, right) (Cognetta et al., 2015).
The triazole urea, like the carbamate, has been a more recent addition to the serine hydrolase toolbox and now represents a versatile chemotype for serine hydrolase inhibition due to broad coverage across the enzyme class and the ability to tune selectivity for individual members (Adibekian et al., 2011). A synthetic strategy based on a robust click chemistry approach has enabled rapid generation of triazole urea libraries with diversity present on both the triazole and the carbamoylating group. These libraries have yielded nanomolar and sub-nanomolar inhibitors for several serine hydrolases, including ABHD11, PAFAH2, and acylaminoacyl-peptide hydrolase (APEH) (Adibekian et al., 2011). Another study identified a 1,2,4-triazole urea inhibitor for MAGL with a superior in vivo selectivity profile compared with a previously reported carbamate inhibitor (Aaltonen et al., 2013, 2016). More recently, both N-acyl pyrazoles and benzoxathiazine-3-one, 1,1-dioxides have been developed as selective serine hydrolase inhibitors, thus adding to the growing collection of urea/carbamate inhibitor libraries (Kornahrens et al., 2017; Otrubova et al., 2019).
4-Chloro-isocoumarin (CIC) represents another class of electrophile that is uniquely tuned for selective targeting of serine hydrolases due to its two-hit mechanism of inhibition (Harper et al., 1985). An initial nucleophilic attack by the active site serine opens the heterocyclic ring, creating an acyl enzyme intermediate. While this covalent intermediate can be eliminated by subsequent hydrolysis, the 4-chloro group is rapidly eliminated creating a reactive isoquinonimide methide, which then covalently and irreversibly reacts with the catalytic histidine found in all serine hydrolases (Figure 2F) (Powers et al., 2002). This mechanism makes the CIC electrophile an ideal choice for use in ABPs that are specific for serine hydrolases. CICs have been used to inhibit hydrolases, such as human esterases (Heynekamp et al., 2008), bacterial esterases (Lentz et al., 2018), and depalmitoylases in Toxoplasma gondii (Child et al., 2013; Foe et al., 2018). Furthermore, a recent study used a CIC scaffold to generate a highly selective probe for a depalmitoylating enzyme in T. gondii, further confirming the utility for ABPP (Garland et al., 2018). The selectivity of the CIC scaffold can be tuned by incorporating substituents at the 3 and 7 positions. The Verhelst group synthesized CIC probes containing recognition elements for trypsin-like, elastase-like, and chymotrypsin-like serine proteases attached at various positions on the main aromatic ring. These studies showed that selective labeling and reactivity could be controlled by the location of the substitution (Haedke et al., 2012). In contrast to ureas and carbamates, CIC libraries are limited in scope, with only a few peptide substrates and small-molecule moieties explored to date. It remains to be seen whether CICs with modifications at alternate positions or broader substrate scope will yield useful probes, but this electrophile class undoubtedly represents an unexplored area of chemical diversity.
The use of lactam and lactone electrophiles for the generation of ABPs has mainly focused on targeting bacterial enzymes, especially β-lactamases (Böttcher and Sieber, 2012). Many β-lactamase ABPs are serine hydrolase inhibitors that are derived from penicillin-like antibiotics. Some of the earliest probes in this class were developed by Staub and Sieber (2008) as synthetic lactams for the identification of penicillin binding proteins in various bacterial species. One target of these β-lactam ABPs was discovered to be caseinolytic protease P, a serine protease associated with bacterial virulence (Bhandari et al., 2018). Lactams and lactones have also been used to target lipases, including diacylglycerol lipase-α (DAGL-α), a serine hydrolase that aids in human endocannabinoid synthesis (Baggelaar et al., 2013), and Orlistat, a pan lipase inhibitor used to treat obesity (Heck et al., 2000). It is important to note that, unlike the aforementioned electrophiles, lactams and lactones can also modify catalytic cysteine and threonine residues, and are thus not specific for serine hydrolases. For example, an ABPP study of hepatitis C virus (HCV) infection of human hepatoma cells using three broad-spectrum β-lactam probes of increasing hydro-phobicity resulted in labeling of a broad range of enzymes with different amino acids in their active site, including serine, cysteine, threonine, and glutamic acid (Nasheri et al., 2014). This further supports the reported broad specificity of this compound class. Although many lactam and lactone ABP probe collections do label catalytic residues other than serine, they still represent viable reactive electrophiles that have the potential to be tuned to an acceptable level of overall specificity.
Sulfonyl fluorides were first reported as covalent inhibitors of serine hydrolases as far back as the 1960s (Fahrney and Gold, 1963). Since then, a number of sulfonyl fluoride probes and inhibitors have been prepared to specifically target active site serine residues (Narayanan and Jones, 2015). For example, the aliphatic sulfonyl fluoride inhibitor AM3506 was found to be a selective inhibitor of FAAH, as determined by competitive ABPP (Godlewski et al., 2010). Furthermore, the Verhelst group used a mass spectrometry screening platform to identify peptidyl sulfonyl fluorides as inhibitors against intramembrane rhomboid proteases (Vosyka et al., 2013). More recently, sulfonyl fluorides and fluorosulfates have been used to selectively target individual tyrosine, lysine, and histidine residues on diverse protein targets (Hahm et al., 2020). This unique reactivity profile of sulfonyl fluorides was conceptualized by the Sharpless group in 2014 and is now known as sulfur fluoride exchange chemistry (SuFEx) (Dong et al., 2014). SuFEx reactions occur only when strict requirements are met that enable stabilization of the departing fluoride ion during its transit away from the covalent bond to sulfur. For example, a hydrogen bonding network exists in the binding site of modified proteins that facilitates fluoride extraction. However, the broad reactivity and context-dependent activation of SuFEx, as well as the discovery of more synthetically tractable covalent scaffolds, such as ureas and carbamates, has meant that the sulfonyl fluoride electrophile has been used less frequently in serine hydrolase inhibitors. Despite this, the advent of SuFEx has facilitated the development of new chemistries to robustly install sulfonyl fluoride moieties on diverse chemical scaffolds. Large libraries of small molecules that are amenable to SuFEx chemistry have been synthesized by various groups and are also commercially available (Barrow et al., 2019). Recently, a screen of sulfonyl fluoride compounds against human neutrophil elastase resulted in the identification of 2-(fluorosulfonyl)phenyl fluorosulfate as a potent inhibitor. Importantly, this simple benzenoid also showed significant selectivity over the closely related cathepsin G (Zheng et al., 2019). This finding suggests that, by further expanding SuFEx libraries, it may be possible to identify sulfonyl fluoride inhibitors that selectively react with individual serine hydrolases.
Optimizing Serine Hydrolase Probe Scaffolds
While the selection of a reactive electrophile can help to direct probes toward certain hydrolase families, this functional group alone is usually not sufficient to drive absolute selectivity for individual hydrolases. Thus, the current landscape of probe design tends to focus on developing both novel electrophilic molecules with divergent labeling profiles, and new synthetic methods to derivatize core probe scaffolds. Probe scaffolds can be tuned using diverse methods depending on the serine hydrolase to be targeted. For example, a serine protease can be targeted by conjugating an optimal tetrapeptide substrate to an appropriate electrophile, such as a diphenyl phosphonate. On the other hand, if the target is a lipase or esterase, then broadly reactive serine hydrolase inhibitor scaffolds, such as ureas and carbamates, are likely to be an optimal starting point for probe development. Tuning the specificity of these scaffolds is akin to traditional medicinal chemistry whereby diverse libraries of closely related analogs are synthesized and then screened against the target of interest to identify a lead molecule. In the next section of this review, we highlight recent methods used to optimize the selectivity of probe scaffolds toward an individual serine hydrolase target and also provide suggestions where novel chemistries can be applied to further facilitate this process. Peptidyl Probe Scaffolds. Established Methods. Peptidic specificity elements are often used to tune the selectivity of probe scaffolds, in particular when the serine hydrolase to be targeted is a protease. A range of methods exist for identifying optimal peptide sequences, including positional scanning synthetic combinatorial libraries, phage display, internally quenched fluorescent substrate libraries, and proteomics (Chen et al., 2019b). Short tetrapeptide sequences are routinely incorporated into probe scaffolds in place of non-specific linkers to enable selective inhibition or labeling of individual serine proteases (Schulz-Fincke et al., 2018; Winiarski et al., 2012). This strategy is only applicable to proteases as, unlike lipases and other hydrolases, their specificity pockets are designed to bind to and cleave specific peptide sequences.
Positional scanning libraries (PSLs) allow rapid determination of the specificity of protease active site sub-pockets (Schneider and Craik, 2009). The acquired specificity data can be used to generate optimal peptide substrates for a particular protease, which can then be grafted onto an appropriate probe scaffold to generate a selective ABP (Figure 3A). A potential pitfall of this screening approach is that it does not provide information on binding site cooperativity and thus residues identified as “optimal” from each sub-site screen may not equate to the best substrate when combined into a single peptide sequence. Consequently, it is essential that a small library of substrates is synthesized comprising multiple identified “hit” amino acids from each library sub-screen. The incorporation of non-natural amino acids to PSL substrate libraries, termed hybrid combinatorial substrate libraries (HyCoSuL), has expanded the chemical space that can be explored by this approach and has enabled highly selective substrates and inhibitors to be generated for a range of serine proteases (Poreba et al., 2017). For example, the Drag group used the HyCoSuL approach to identify selective substrates for each of the four main neutrophil serine proteases (neutrophil elastase, cathepsin G, proteinase 3, and neutrophil proteinase 4). Grafting each selective substrate onto a DPP probe scaffold with multiple, spectroscopically unique fluorophore labels, enabled multiplexed imaging of the activity of each protease in live neutrophils using fluorescent microscopy (Figure 3B) (Kasperkiewicz et al., 2017). Similarly, HyCoSuL was used to develop a selective substrate for the “hydrolase important for pathogenesis 1” (Hip1) serine protease in Mycobacterium tuberculosis (Lentz et al., 2016). This sequence was then attached to a diphenyl phosphonate electrophile to generate a selective ABP. Surprisingly, despite being highly potent, the resulting peptidyl DPP demonstrated reversible competitive inhibition kinetics and thus lacked utility as an ABP. Replacement of the DPP moiety with the CIC electrophile enabled the identification of a potent irreversible inhibitor of Hip1 that could be used as a scaffold to generate a selective ABP. This study serves to show the importance of sampling multiple electrophilic warheads when attempting to develop a selective ABP for a hydrolase target.
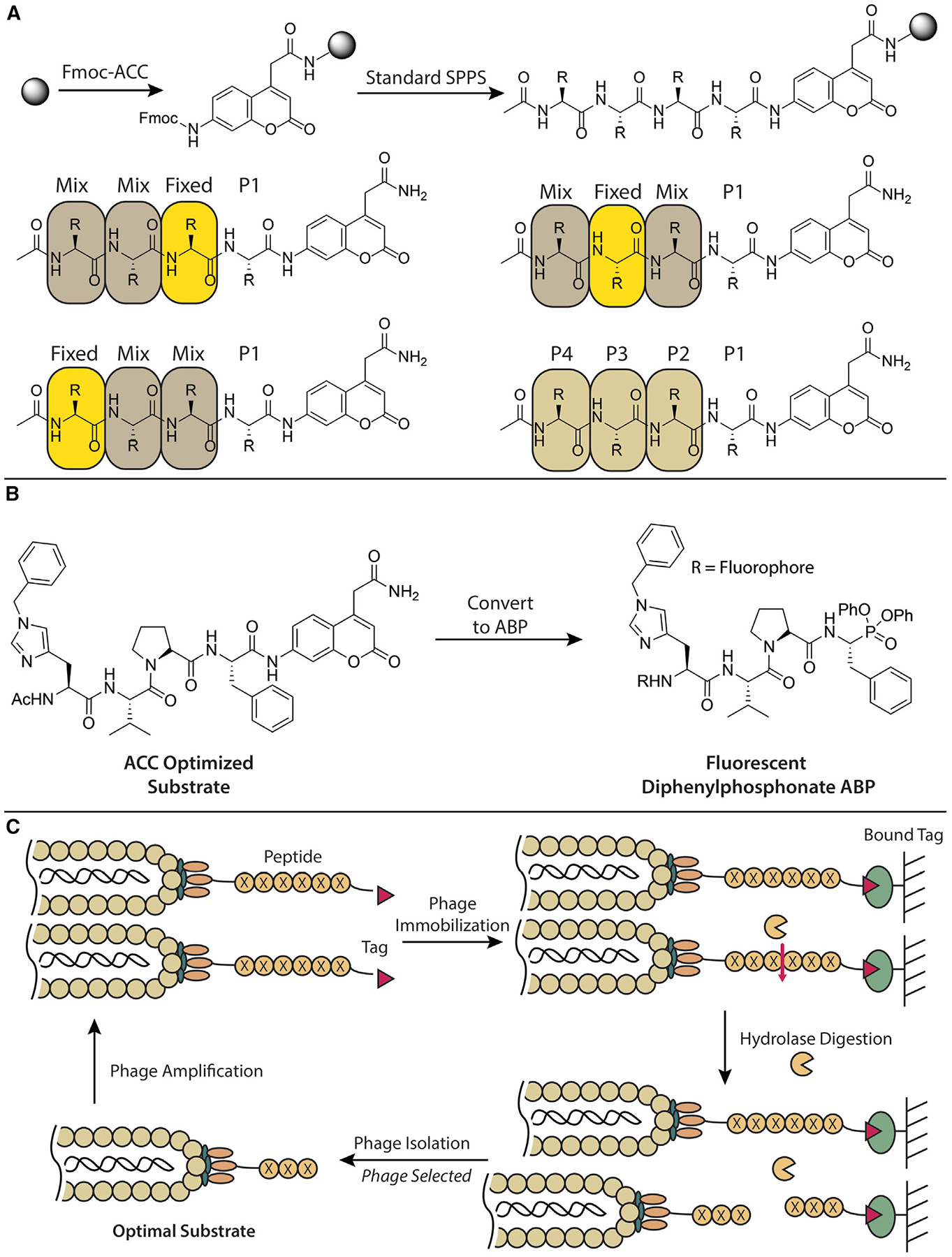
Figure 3. Strategies to Generate Selective ABPs Using Peptide Probe Scaffolds.

(A) Schematic that shows the synthesis of ACC peptides (top). Examples of positional scanning libraries of peptide substrates (bottom). Positional scanning is performed by holding one position fixed as any natural or unnatural amino acid (yellow) while the remaining positions contain an isokinetic mixture of natural amino acids (brown). Screening of each sub-library enables identification of optimal side-chain residues for each protease sub-pocket. The most effectively processed amino acids (beige) for each position are then combined to generate an optimized tetrapeptide substrate (bottom right).
(B) Conversion of an optimized ACC substrate to an ABP. The ACC reporter group in the optimized substrate is replaced with an electrophile to generate a selective ABP. This example shows the structure of an ABP selective for cathepsin G (Kasperkiewicz et al., 2017).
(C) Schematic showing the use of phage display to identify optimal protease substrates. Phage libraries displaying biotinylated peptides are tethered to streptavidin resin and then incubated with a protease of interest. Phage displaying optimal peptide sequences are cleaved from the resin and collected. The phage are then amplified, and the process can be repeated until the consensus sequence of an optimal substrate is identified.
Phage display is an alternative technique that can be used to identify optimal peptide sequences for protease targets. An advantage of phage display is that extended peptide sequences can be screened, and the resulting hits often form interactions with the target protein that extend outside of the active site. Such “exo-site” interactions are often unique to a particular hydrolase which increases the chance of identifying a selective substrate and covalent probe (Ramirez et al., 2018). In phage display, degenerate DNA sequences encoding random peptides (up to 109 unique sequences) are fused to a phage coat protein to enable presentation on the surface of the phage. The most commonly used bacteriophage is the M13 filamentous phage. In this system, the peptide library DNA is ligated into the pIII gene, which codes for the minor coat protein. This approach enables billions of peptides to be rapidly screened against a protein of interest. Identification of optimal protease substrates using phage display was reported as far back as 1993 (Matthews and Wells, 1993). In the most recent description of this approach, each presented peptide displays an N-terminal avi-Tag to permit quantitative biotinylation and immobilization on streptavidin magnetic beads. The immobilized phage library is then incubated with a protease, and phage that display favorable substrates are cleaved into solution and can be subsequently amplified in bacteria (Figure 3C). This process can be repeated for multiple rounds, resulting in an enrichment of optimal substrates (Deperthes, 2002; Zhou et al., 2020). To date, phage display has been used to profile the active site specificity of many serine proteases, including thrombin (Kretz et al., 2018), mannose-binding lectin-associated serine protease (Kocsis et al., 2010), and kallikrein-related peptidase 14 (Felber et al., 2005). Phage display identifies cooperativity between many residues and thus remains a powerful tool for identifying highly optimized protease substrates that can be grafted onto different probe scaffolds to enable development of selective ABPs.
Future Opportunities.
To date, the use of phage display and PSLs has been restricted to identification of linear substrate sequences. Consequently, the ABPs generated from these peptides are generally limited to use in vitro due to rapid in vivo degradation and/or clearance. One possibility for addressing this shortcoming is to use cyclic peptides for ABP design. Despite being relatively unexplored in the field of ABPP, the global market for cyclic peptide therapeutics is currently estimated to be over $20 billion (Henninot et al., 2018). Cyclic peptides are less prone to non-specific proteolysis and tend to have enhanced membrane permeability, greater oral availability, and improved binding characteristics compared with linear peptides (Lau et al., 2015; Nielsen et al., 2017). The latter fact is due to their constrained form, which results in a smaller loss of conformational entropy upon binding (Driggers et al., 2008). Importantly, cyclic peptides have been widely used as selective inhibitors of serine hydrolases (Driggers et al., 2008). For example, sunflower trypsin inhibitor-1, a 14-amino acid cyclic peptide that is found in sunflower seeds, was tuned to be selective for individual kallikrein-related peptidases by grafting preferred P1–P4 substrate sequences into the native scaffold (Shariff et al., 2014).
Over the last decade, the groups of Heinis and Derda have revolutionized phage display by introducing a range of chemical “linchpins” that facilitate peptide cyclization by covalent modification of two or more fixed cysteine residues present in the displayed linear peptides (Derda and Jafari, 2018; Heinis et al., 2009). This has enabled large libraries of monocyclic/bicyclic peptides to be generated and resulted in highly selective binders for diverse proteins, especially inhibitors of serine hydrolase targets (Deyle et al., 2017). For example, a phage screen led to the identification of a highly specific bicyclic inhibitor of matrix metalloproteinase 2, a zinc-dependent endopeptidase that belongs to a family of more than 20 enzymes with structurally conserved catalytic domains and shallow substrate binding sites (Maola et al., 2019). That work used tris-(bromomethyl)benzene to form a bicyclic library by covalently modifying three cysteine residues present in each displayed linear peptide. After identification of an optimal binding bicyclic peptide, the addition of a metal binding hydroxamate functional group further increased the potency of the resulting inhibitor. Such an approach could also be applied to other classes of protease targets by installing class-specific electrophiles after identification of optimal cyclic or bicyclic peptide scaffolds. More recently, phage display was used to screen large libraries of genetically encoded double-bridged peptides against coagulation Factor Xia. This resulted in the identification of a highly selective inhibitor, which was resistant to degradation by gastrointestinal proteases and could thus be administered orally to mice (Kong et al., 2020). Importantly, there are a number of ways in which cyclic peptide phage libraries could be used to aid ABP development. For example, a chemical linchpin could be used to both form cyclic peptides and introduce a tag, such as biotin, to permit immobilization on streptavidin resin. This would enable a library of cyclic peptide substrates to be screened against proteases, and the enriched cyclic peptides could be grafted onto probe scaffolds to generate ABPs with improved in vivo properties. Alternatively, phage display could be used for de novo development of highly selective covalent inhibitors, which could be transformed into ABPs by conjugation to an appropriate tag. Although rare, there are a few examples of using genetically encoded libraries for identifying selective covalent peptide inhibitors. For example, the Schultz group used orthogonal aminoacyl-tRNA synthetase/ tRNACUA pairs, together with a split intein system to biosynthesize a library of ribosomal peptides containing an expanded set of amino acid building blocks. Screening of this library against the HIV protease resulted in the identification of a lead peptide containing the keto amino acid p-benzoylphenylalanine, which covalently modified the ε-amino group of Lys 14 on the protease leading to formation of a Schiff base adduct (Young et al., 2011). Alternatively, the Taki group used a T7 bacteriophage display library to identify linear peptide binders for glutathione S-transferase. These hits were then transformed into covalent inhibitors by modifying an internal cysteine residue with ethenesulfonyl fluoride (Uematsu et al., 2018). More recently, our lab has developed a general, unbiased approach to directly screen diverse pools of covalent peptide inhibitors using phage display. This was enabled by modifying peptides displayed on M13 bacteriophage with a 1,3-dichloroacetone linker that both induced peptide cyclization and allowed for introduction of a weak electrophile (Figure 4A). Using this approach, we were able to identify cyclic peptides that irreversibly inhibited tobacco etch virus cysteine protease (Chen and Bogyo, 2019) and highly selective inhibitors with nanomolar potency for FphF, a serine hydrolase found in S. aureus (unpublished data). The successful optimization of this latter inhibitor suggests that phage display is not only limited to targeting serine proteases but can also be used for identifying selective covalent cyclic peptides for a diverse range of serine hydrolases. This next-generation screening approach for identifying cyclic ABPs is potentially transformative as it combines the benefits of phage display (high diversity, direct screening, successive amplification, and selection) with the strengths of synthetic probe libraries (addition of diverse warheads). By using the various electrophilic warheads discussed throughout this review it should be possible to develop ultrapotent and selective probes for a diverse range of serine hydrolases.
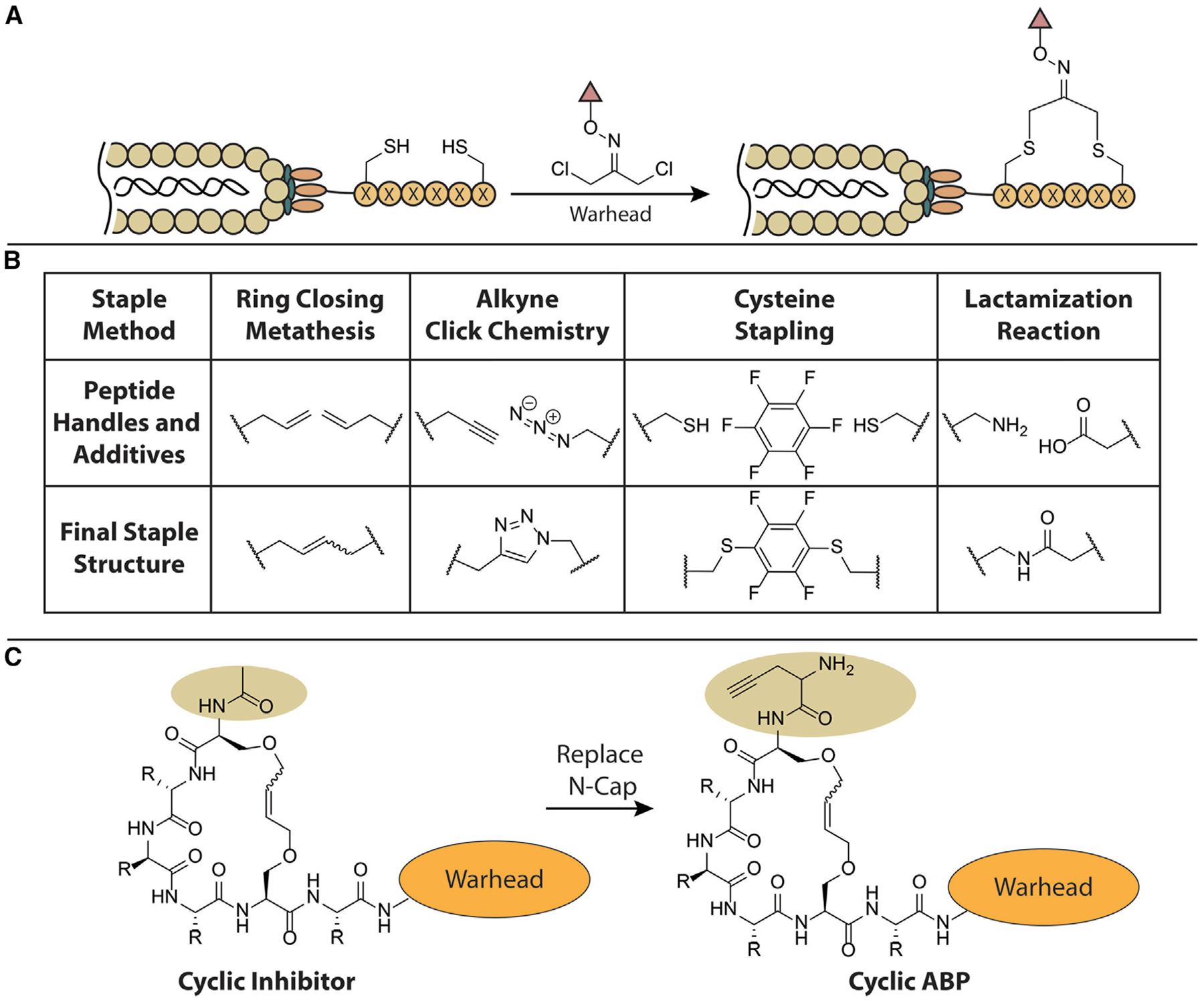
Figure 4. Cyclic Peptide Linkages and Their Incorporation into ABP Scaffolds.
(A) The general strategy to covalently inhibit target proteins by peptides cyclized with dichloro-oxime linkers that contain an appropriate electrophilic warhead.
(B) Structures of several diverse peptide staples.
(C) Schematic of a stapled peptide inhibitor and its conversion to a cyclic peptide ABP. The i, i+4 linkage is shown but in principle can vary. The peptide staple and warhead may also be varied.
In addition to the use of genetically encoded libraries, chemical stapling of linear peptides generated by solid-phase peptide synthesis is another powerful method for forming cyclic peptides. A broad range of peptide-stapling technologies exist and may be used for generating large libraries of cyclic substrates and/or covalent inhibitors (Figure 4B). The introduction of a peptide staple to a linear peptide sequence results in an increase in helicity and/or hydrophobicity, which has been shown to improve membrane permeability, target specificity, and overall stability (Dougherty et al., 2019). For example, the cyclic peptide clinical candidate Ciluprevir, an HCV NS3 serine protease inhibitor, was optimized from a linear hexapeptide using ring-closing metathesis to form the cyclic structure (Rosenquist et al., 2014). This candidate ultimately led to the approved HCV medication, Simeprevir. It is possible that the favorable properties of stapled peptides may be conferred to an ABP by incorporating a carefully designed stapled substrate into a probe scaffold (Figure 4C).
Overall, the research area of covalent cyclic peptide inhibitor development is in its infancy, but recent advances in peptide display technologies and chemical stapling methods, as well as the wide variety of electrophilic warheads available, mean that this class of inhibitor could be used routinely in the future for the development of ultrapotent and exquisitely selective ABPs for diverse serine hydrolase targets.
Small-Molecule Probe Scaffolds. Established Methods.
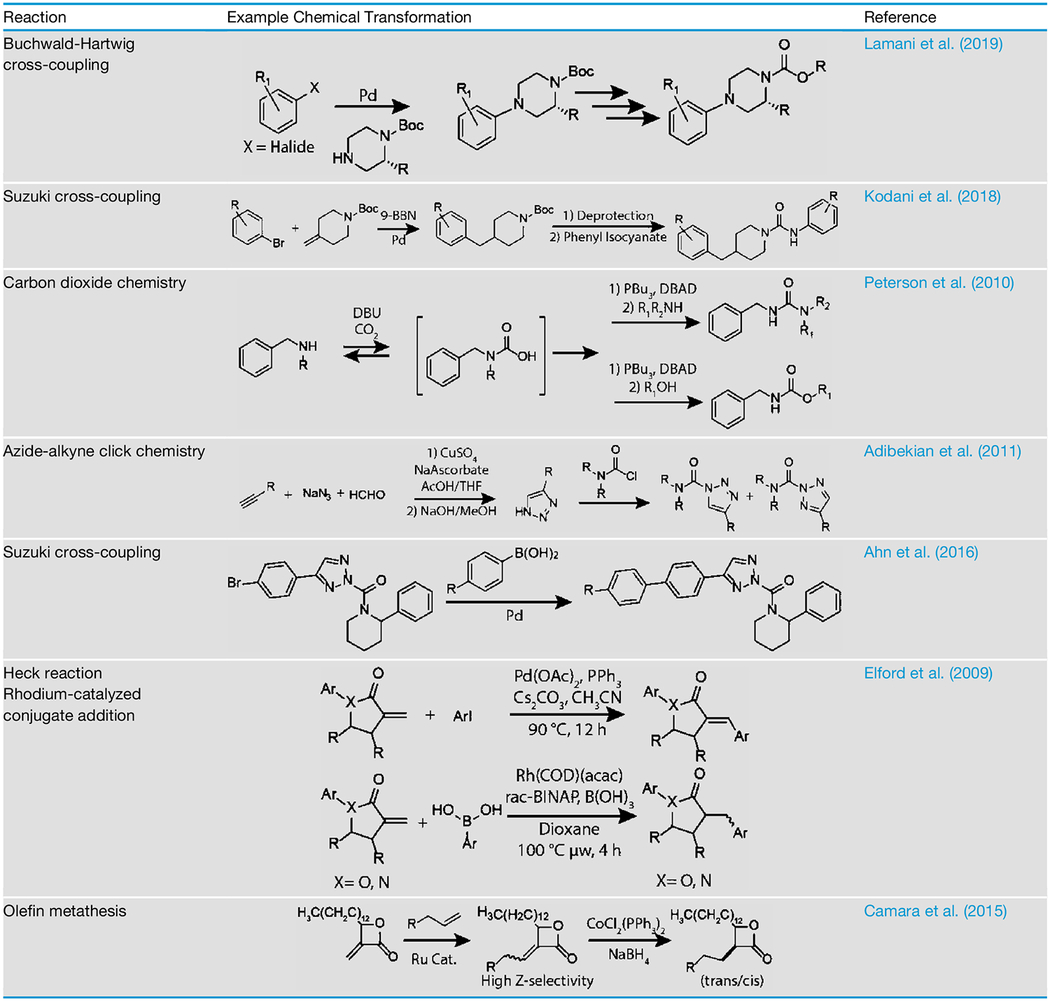
In this section, we highlight current methods used to synthesize diverse libraries of non-peptidic serine hydrolase-targeting probe scaffolds. Of the small-molecule probe scaffolds identified to date, carbamates and ureas have been the most widely used as they can be quickly diversified via relatively simple chemistries. For example, a recent report focused on introducing bulky groups onto the carbamoylating moiety of the carbamate scaffold using Buchwald-Hartwig cross-coupling to increase selectivity for FAAH (Table 1) (Lamani et al., 2019). Similarly, palladium cross-coupling chemistry has been used to generate secondary amine derivatives of t-butyl-methylenepiperidine-1-carboxylate, which were then used as carbamoylating moieties (Kodani et al., 2018). The generated amines were reacted with phenyl isocyanates to generate novel ureas (Table 1). Using these chemistries, it was possible to iteratively change the carbamoylating and leaving group and ultimately identify a lead molecule with dual potency for FAAH and soluble epoxide hydrolase.
Table 1.
Methods for Diversifying Small-Molecule Serine Hydrolase-Targeting Probe Scaffolds
|
Although these strategies can quickly generate diverse libraries, other methods are still needed to synthesize ABPs from accessible starting materials. Another possible route for generating ureas and carbamates is through the use of carbon dioxide. Under basic conditions, amines can react with carbon dioxide to form isocyanates in situ, which can then be reacted with a variety of primary and secondary amines or alcohols to form either ureas or carbamates under Mitsunobu-like conditions (Table 1) (Peterson et al., 2010).This strategy could be used to produce a diverse set of isocyanates using readily available amines and alcohols.
The triazole urea scaffold is synthesized using a two-step procedure. Substituted alkynes are first reacted with in-situ-formed azido methanol to yield 4-substituted triazoles, which can then be carbamoylated by use of triphosphgene and a carbamoyl chloride to afford triazole urea products (Table 1) (Adibekian et al., 2011; Kalisiak et al., 2008). This strategy was initially used to find a sub-nanomolar inhibitor for APEH. Selective inhibition of APEH with the optimized triazole urea enabled identification of its substrates by quantitative proteomics (Adibekian et al., 2011). Further work has created additional diversity in the triazole urea scaffold. For example, incorporation of an aryl halide on the triazole provided a chemical handle for derivatization through the use of palladium cross-coupling reactions. This approach was used for developing DAGL-β and lysophospholipase-like 1 inhibitors (Table 1) (Ahn et al., 2016; Hsu et al., 2013). Triazole ureas have also been transformed into imaging probes. For example, conjugation of a BODIPY dye to the carbamoylating group of an optimized triazole urea enabled single-cell imaging of FphE in S. aureus (Chen et al., 2019a, 2019b). In addition, the van der Stelt group converted a triazole urea inhibitor for DAGL-α into a quenched fluorescent probe by attaching a fluorophore on the carbamoylating moiety and a quencher on the triazole leaving group (van Rooden et al., 2018). These examples serve to highlight how synthetic chemistry has enabled the discovery of diverse probes using the triazole urea scaffold.
β-lactam and β -lactone ABPs have been widely used to profile the targets of antibiotic compounds (Böttcher and Sieber, 2012). Various studies have shown that substitution on the β-lactam and β-lactone ring can alter the reactivity and selectivity of these core scaffolds (Sharifzadeh et al., 2017). This unique reactivity of β-lactams has been leveraged to enable profiling of the rhomboid intramembrane proteases, which is a class of protease that has been difficult to characterize due to the lack of effective warheads (Pierrat et al., 2011). Due to the prevalence of β-lactams in natural products, there is an expansive body of literature describing their synthesis (Kamath and Ojima, 2012). Libraries of lactones have largely focused on the use of diversity-oriented synthesis. One method has used a combination of Heck cross-couplings and rhodium-catalyzed conjugate additions to develop a library of lactones that cover a large range of chemical space (Table 1) (Elford et al., 2009). Synthetic libraries also include bicyclic lactams and lactones, further broadening the chemical space of these scaffolds. Olefin cross-metathesis has also been used to develop a wide variety of lactone libraries (Table 1) (Camara et al., 2015).
Future Opportunities.
Overall, a wide range of synthetic methodologies exist to enable rapid diversification of small-molecule probe scaffolds that target serine hydrolases. However, some electrophiles, such as mixed alkyl aryl phosphonates and chloroisocoumarins remain relatively underrepresented in the literature despite showing great promise as selective inhibitors. Generally, probe scaffold libraries are limited to a few hundred compounds due to the need for multiple synthetic steps, as well as a need for chromatographic purification. Therefore, covalent inhibitor libraries remain modest in size and consequently we lack selective inhibitors for many members of the serine hydrolase family. Therefore, highly robust chemistries that can be performed without the need for purification, such as “click chemistry” approaches could be used to address these shortcomings.
To date, click chemistry has been only sparingly used for developing optimal ABPs. The properties of these reactions are compatible with high-throughput screening and would enable rapid diversification of probe scaffolds to generate libraries made up of thousands of analogs. Copper(I)-catalyzed azide-alkyne cycloaddition (CuAAC) has been used by medicinal chemists to develop libraries of bioactive molecules, but has yet to be harnessed for the development of diverse libraries of covalent inhibitors (Thirumurugan et al., 2013). The major advantage of CuAAC is that the reaction can take place in aqueous buffers and the resulting crude reaction mixtures can be screened directly in biochemical assays (Rostovtsev et al., 2002). A recent study demonstrated the quantitative conversion of over 1,200 amines to azides using fluorosulfuryl azide. Furthermore, crude azide mixtures were directly reacted with alkynes using CuAAC conditions resulting in near quantitative conversion to triazoles (Meng et al., 2019). This landmark study opens up the possibility of generating vast libraries of covalent serine hydrolase inhibitors simply by ligating azides, which are made from readily available primary amines, to alkyne-tagged versions of probe scaffolds (Figure 5A). Importantly, this could facilitate the identification of selective covalent inhibitors based on underutilized scaffolds, such as mixed alkyl aryl phosphonates and chloroisocoumarins, which can be readily modified with an alkyne handle. Crude reaction mixtures can be tested directly against the serine hydrolase of interest and thus the entire screening process, from synthesis to activity readout, can be performed in a microwell plate potentially resulting in a significant decrease in the time taken to identify an optimal ABP.
Figure 5. Click Chemistry and the Generation of Synthetic Libraries.
(A) General strategy for developing selective ABPs for serine hydrolase targets by derivatizing small-molecule probe scaffolds with diverse libraries of readily available azides. R = aryl or alkyl group.
(B) Example of the optimization of an inhibitor for the cysteine protease SpeB using SuFEx chemistry. Left: structures of the lead SpeB inhibitor and the resulting iminosulfur oxydifluoride analogs. Right: representative optimized SpeB inhibitors (Kitamura et al., 2019).
(C) Optimization of a serine hydrolase probe scaffold using a SuFEx library screening approach.
Similarly, SuFEx click chemistry, developed by the Sharpless group, also presents new avenues for synthesizing diverse ABPs and serine hydrolase inhibitors (Dong et al., 2014). This method enables metal-free coupling of amines, aromatic alcohols, carbon nucleophiles, and aryl silyl ethers to multi S-F bond-containing “hubs” to generate sulfonamides, sulfates, and sulfonates (Barrow et al., 2019). Importantly, these transfor mations occur in metal-free, biocompatible reaction conditions, meaning that crude reaction mixtures can be screened directly against protein targets. Unlike CuAAC, the sulfur(VI)-containing motifs resulting from SuFEx reactions often possess 3D structure, multiple hydrogen-bond donors/acceptors, and drug-like lipophilicity. In a recent paper by the Wolan group, a modest nitrile inhibitor for SpeB, a bacterial cysteine protease, was modified with an iminosulfur oxydifluoride motif and rapidly diversified with 460 diverse amines (Kitamura et al., 2019). The resulting library was directly screened against SpeB and yielded several drug-like inhibitors with up to 300-fold higher potency (Figure 5B). In addition, the synthesis of the library was repeated on a miniaturized scale using an Echo Acoustic liquid handler. A strong correlation in inhibitory potency was observed between the picomole scale and nanomole scale syntheses, thus suggesting that SuFEx libraries can be readily generated using robotic screening systems. Using a similar method to diversify serine hydrolase probe scaffolds could result in potent, selective, and in vivo compatible ABPs (Figure 5C). Furthermore, given that iminosulfur oxydifluoride analogs contain two S-F bonds, and can be readily synthesized using thionyl tetrafluoride gas, it is foreseeable that probe libraries of even greater diversity could be synthesized by sequentially modifying probe scaffolds with two different amine moieties. Overall, given that library synthesis is likely to be carried out in microplates using liquid handling systems, and that crude mixtures can be screened directly against serine hydrolase targets, both CuAAC and SuFEx click chemistry are likely to become powerful methods for rapidly diversifying probe scaffolds and identifying optimal ABPs.
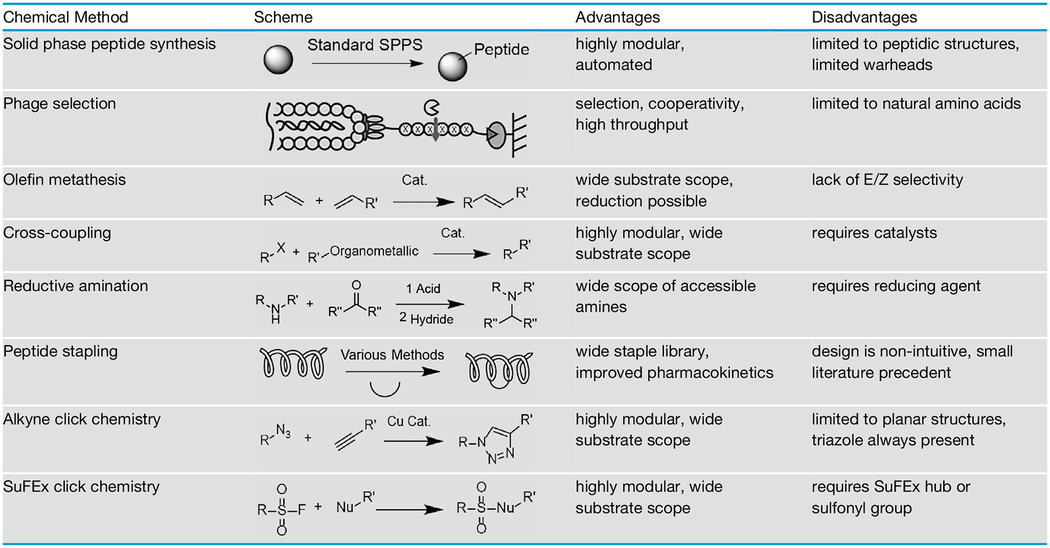
Table 2.
Chemical Methods for Tuning the Selectivity of Serine Hydrolase Probe Scaffolds
|
SIGNIFICANCE.
For a full understanding of the role of serine hydrolases in human health and disease, selective inhibitors and active site probes need to be developed. In this review, we describe the diverse range of electrophilic warheads available to selectively target serine hydrolases. Moreover, we highlight the various methods available to tune the selectivity of probe scaffolds. A summary of these methods can be found in Table 2. For optimizing peptide scaffolds, positional scanning libraries and phage display are powerful approaches, but these tend to yield linear peptide sequences that generally cannot be applied to in vivo models. There is a wide selection of chemical linchpins available that facilitate peptide cyclization and we hypothesize that generating large libraries of covalent cyclic peptides for screening against targets will facilitate the identification of potent, selective, and in vivo compatible ABPs. Similarly, there are a variety of synthetic methods described in the literature that enable optimization of various small-molecule probe scaffolds. However, many of the described approaches are based on multi-step syntheses and require purification, thus limiting library sizes to a few hundred compounds. We suggest that the use of CuAAC and SuFEx click chemistries will enable rapid diversification of a wide variety of probe scaffolds in one step and enable direct screening of crude reaction mixtures against hydrolase targets. We envision that this review will help to focus future efforts to expand the scope of ABPs for the study of this clinically and biologically important family of enzymes.
ACKNOWLEDGMENTS
This work was supported by the National Science Foundation Graduate Research Fellowship under grant no. DGE-1656518 and Stanford ChEM-H O’Leary-Thiry Graduate Fellowship, (awarded to F.F). IT was also funded by a Stanford Graduate Fellowship under the William R. and Sara Hart Kimball Fellowship (awarded to J.B.) and the Enhancing Diversity in Graduate Education Doctoral Fellowship Program (awarded to F.F. and J.B.).
REFERENCES
- Aaltonen N, Savinainen JR, Ribas CR, Rönkkö J, Kuusisto A, Korhonen J, Navia-Paldanius D, Häyrinen J, Takabe P, Käsnänen H, et al. (2013). Piperazine and piperidine triazole ureas as ultrapotent and highly selective inhibitors of monoacylglycerol lipase. Chem. Biol 20, 379–390. [DOI] [PubMed] [Google Scholar]
- Aaltonen N, Kedzierska E, Orzelska-Gorka J, Lehtonen M, Navia-Paldanius D, Jakupovic H, Savinainen JR, Nevalainen T, Laitinen JT, Park-kari T, et al. (2016). In vivo characterization of the ultrapotent monoacylglycerol lipase inhibitor (1H-1,2,4-triazol-1-yl)methanone (JJKK-048). J. Pharmacol. Exp. Ther 359, 62–72. [DOI] [PubMed] [Google Scholar]
- Adibekian A, Martin BR, Wang C, Hsu K-L, Bachovchin DA, Niessen S, Hoover H, and Cravatt BF (2011). Click-generated triazole ureas as ultrapotent in vivo-active serine hydrolase inhibitors. Nat. Chem. Biol 7, 469–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adibekian A, Martin BR, Chang JW, Hsu K-L, Tsuboi K, Bachovchin DA, Speers AE, Brown SJ, Spicer T, Fernandez-Vega V, et al. (2012). Confirming target engagement for reversible inhibitors in vivo by kinetically tuned activity-based probes. J. Am. Chem. Soc 134, 10345–10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahn K, Boehm M, Brown MF, Calloway J, Che Y, Chen J, Fennell KF, Geoghegan KF, Gilbert AM, Gutierrez JA, et al. (2016). Discovery of a selective covalent inhibitor of lysophospholipase-like 1 (LYPLAL1) as a tool to evaluate the role of this serine hydrolase in metabolism. ACS Chem. Biol 11, 2529–2540. [DOI] [PubMed] [Google Scholar]
- Alexander JP, and Cravatt BF (2005). Mechanism of carbamate inactivation of FAAH: implications for the design of covalent inhibitors and in vivo functional probes for enzymes. Chem. Biol 12, 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachovchin DA, and Cravatt BF (2012). The pharmacological landscape and therapeutic potential of serine hydrolases. Nat. Rev. Drug Discov 11, 52–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachovchin DA, Ji T, Li W, Simon GM, Blankman JL, Adibekian A, Hoover H, Niessen S, and Cravatt BF (2010). Superfamily-wide portrait of serine hydrolase inhibition achieved by library-versus-library screening. Proc. Natl. Acad. Sci. U S A 107, 20941–20946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baggelaar MP, Janssen FJ, van Esbroeck ACM, den Dulk H, Allarà M, Hoogendoorn S, McGuire R, Florea BI, Meeuwenoord N, van den Elst H, et al. (2013). Development of an activity-based probe and in silico design reveal highly selective inhibitors for diacylglycerol lipase-α in brain. Angew. Chem. Int. Ed 52, 12081–12085. [DOI] [PubMed] [Google Scholar]
- Barrow AS, Smedley CJ, Zheng Q, Li S, Dong J, and Moses JE (2019). The growing applications of SuFEx click chemistry. Chem. Soc. Rev 48, 4731–4758. [DOI] [PubMed] [Google Scholar]
- Bhandari V, Wong KS, Zhou JL, Mabanglo MF, Batey RA, and Houry WA (2018). The role of ClpP protease in bacterial pathogenesis and human diseases. ACS Chem. Biol 13, 1413–1425. [DOI] [PubMed] [Google Scholar]
- Blais DR, Nasheri N, McKay CS, Legault MCB, and Pezacki JP (2012). Activity-based protein profiling of host–virus interactions. Trends Biotechnol 30, 89–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher T, and Sieber SA (2012). β-lactams and β-lactones as activity-based probes in chemical biology. MedChemComm 3, 408–417. [Google Scholar]
- Camara K, Kamat SS, Lasota CC, Cravatt BF, and Howell AR (2015). Combining cross-metathesis and activity-based protein profiling: new β-lactone motifs for targeting serine hydrolases. Bioorg. Med. Chem. Lett 25, 317–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JW, Cognetta AB, Niphakis MJ, and Cravatt BF (2013). Proteome-wide reactivity profiling identifies diverse carbamate chemotypes tuned for serine hydrolase inhibition. ACS Chem. Biol 8, 1590–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, and Bogyo M (2019). A phage display approach to identify highly selective covalent binders. bioRxiv 10.1101/791533. [DOI] [Google Scholar]
- Chen AL, Lum KM, Lara-Gonzalez P, Ogasawara D, Cognetta AB, To A, Parsons WH, Simon GM, Desai A, Petrascheck, et al. (2019a). Pharmacological convergence reveals a lipid pathway that regulates C. elegans lifespan. Nat. Chem. Biol 15, 453–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Yim JJ, and Bogyo M (2019b). Synthetic and biological approaches to map substrate specificities of proteases. Biol. Chem 401, 165–182. [DOI] [PubMed] [Google Scholar]
- Child MA, Hall CI, Beck JR, Ofori LO, Albrow VE, Garland M, Bowyer PW, Bradley PJ, Powers JC, Boothroyd JC, et al. (2013). Small-molecule inhibition of a depalmitoylase enhances Toxoplasma host-cell invasion. Nat. Chem. Biol 9, 651–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cognetta AB, Niphakis MJ, Lee H-C, Martini ML, Hulce JJ, and Cravatt BF (2015). Selective N-hydroxyhydantoin carbamate inhibitors of mammalian serine hydrolases. Chem. Biol 22, 928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deperthes D (2002). Phage display substrate: a blind method for determining protease specificity. Biol. Chem 383, 1107–1112. [DOI] [PubMed] [Google Scholar]
- Derda R, and Jafari MR (2018). Synthetic cross-linking of peptides: molecular linchpins for peptide cyclization. Protein Pept. Lett 25, 1051–1075. [DOI] [PubMed] [Google Scholar]
- Deyle K, Kong XD, and Heinis C (2017). Phage selection of cyclic peptides for application in research and drug development. Acc. Chem. Res 50, 1866–1874. [DOI] [PubMed] [Google Scholar]
- Dong J, Krasnova L, Finn MG, and Sharpless KB (2014). Sulfur(VI) fluoride exchange (SuFEx): another good reaction for click chemistry. Angew. Chem. Int. Ed 53, 9430–9448. [DOI] [PubMed] [Google Scholar]
- Driggers EM, Hale SP, Lee J, and Terrett NK (2008). The exploration of macrocycles for drug discovery—an underexploited structural class. Nat. Rev. Drug Discov 7, 608–624. [DOI] [PubMed] [Google Scholar]
- Elford TG, Ulaczyk-Lesanko A, De Pascale G, Wright GD, and Hall DG (2009). Diversity-oriented synthesis and preliminary biological screening of highly substituted five-membered lactones and lactams originating from an allyboration of aldehydes and imines. J. Comb. Chem 11, 155–168. [DOI] [PubMed] [Google Scholar]
- Fahrney DE, and Gold AM (1963). Sulfonyl fluorides as inhibitors of esterases. I. Rates of reaction with acetylcholinesterase, chymotrypsin, and trypsin. J. Am. Chem. Soc 85, 997–1000. [Google Scholar]
- Fañanás-Mastral M, and Feringa BL (2014). Copper-catalyzed synthesis of mixed alkyl aryl phosphonates. J. Am. Chem. Soc 136, 9894–9897. [DOI] [PubMed] [Google Scholar]
- Felber LM, Borgoño CA, Cloutier SM, Kündig C, Kishi T, Chagas JR, Jichlinski P, Gygi CM, Leisinger H-J, Diamandis EP, et al. (2005). Enzymatic profiling of human kallikrein 14 using phage-display substrate technology. Biol. Chem 386, 291–298. [DOI] [PubMed] [Google Scholar]
- Foe IT, Onguka O, Amberg-Johnson K, Garner RM, Amara N, Beatty W, Yeh E, and Bogyo M (2018). The Toxoplasma gondii active serine hydrolase 4 regulates parasite division and intravacuolar parasite architecture. mSphere 3, e00393–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland M, Schulze CJ, Foe IT, van der Linden WA, Child MA, and Bogyo M (2018). Development of an activity-based probe for acyl-protein thioesterases. PLoS One 13, e0190255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godlewski G, Alapafuja SO, Bátkai S, Nikas SP, Cinar R, Offertáler L, Osei-Hyiaman D, Liu J, Mukhopadhyay B, Harvey-White J, et al. (2010). Inhibitor of fatty acid amide hydrolase normalizes cardiovascular function in hypertension without adverse metabolic effects. Chem. Biol 17, 1256–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grzywa R, Burchacka E, Łęcka M, Winiarski Ł, Walczak M, Łupicka-Słowik A, Wysocka M, Burster T, Bobrek K, Csencsits-Smith K, et al. (2014). Synthesis of novel phosphonic-type activity-based probes for neutrophil serine proteases and their application in spleen lysates of different organisms. ChemBioChem 15, 2605–2612. [DOI] [PubMed] [Google Scholar]
- Haedke U, Götz M, Baer P, and Verhelst SHL (2012). Alkyne derivatives of isocoumarins as clickable activity-based probes for serine proteases. Bioorg. Med. Chem 20, 633–640. [DOI] [PubMed] [Google Scholar]
- Hahm HS, Toroitich EK, Borne AL, Brulet JW, Libby AH, Yuan K, Ware TB, McCloud RL, Ciancone AM, and Hsu KL (2020). Global targeting of functional tyrosines using sulfur-triazole exchange chemistry. Nat. Chem. Biol 16, 150–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harper JW, Hemmi K, and Powers JC (1985). Reaction of serine proteases with substituted isocoumarins: discovery of 3,4-dichloroisocoumarin, a new general mechanism based serine protease inhibitor. Biochemistry 24, 1831–1841. [DOI] [PubMed] [Google Scholar]
- Hatzios SK, Abel S, Martell J, Hubbard T, Sasabe J, Munera D, Clark L, Bachovchin DA, Qadri F, Ryan ET, et al. (2016). Chemoproteomic profiling of host and pathogen enzymes active in cholera. Nat. Chem. Biol 12, 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heck AM, Yanovski JA, and Calis KA (2000). Orlistat, a new lipase inhibitor for the management of obesity. Pharmacotherapy 20, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinis C, Rutherford T, Freund S, and Winter G (2009). Phage-encoded combinatorial chemical libraries based on bicyclic peptides. Nat. Chem. Biol 5, 502–507. [DOI] [PubMed] [Google Scholar]
- Henninot A, Collins JC, and Nuss JM (2018). The current state of peptide drug discovery: back to the future? J. Med. Chem 61, 1382–1414. [DOI] [PubMed] [Google Scholar]
- Heynekamp JJ, Hunsaker LA, Vander Jagt TA, Royer RE, Deck LM, and Vander Jagt DL (2008). Isocoumarin-based inhibitors of pancreatic cholesterol esterase. Bioorg. Med. Chem 16, 5285–5294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu K-L, Tsuboi K, Whitby LR, Speers AE, Pugh H, Inloes J, and Cravatt BF (2013). Development and optimization of piperidyl-1,2,3-triazole ureas as selective chemical probes of endocannabinoid biosynthesis. J. Med. Chem 56, 8257–8269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Denne J, Yang C, Wang H, and Kang JY (2018). Direct aryloxylation/alkyloxylation of dialkyl phosphonates for the synthesis of mixed phosphonates. Angew. Chem. Int. Ed 57, 6624–6628. [DOI] [PubMed] [Google Scholar]
- Jackson DS, Fraser SA, Ni LM, Kam CM, Winkler U, Johnson DA, Froelich CJ, Hudig D, and Powers JC (1998). Synthesis and evaluation of diphenyl phosphonate esters as inhibitors of the trypsin-like granzymes A and K and mast cell tryptase. J. Med. Chem 41, 2289–2301. [DOI] [PubMed] [Google Scholar]
- Janssen APA, van der Vliet D, Bakker AT, Jiang M, Grimm SH, Campiani G, Butini S, and van der Stelt M (2018). Development of a multiplexed activity-based protein profiling assay to evaluate activity of endocannabinoid hydrolase inhibitors. ACS Chem. Biol 13, 2406–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalisiak J, Sharpless KB, and Fokin VV (2008). Efficient synthesis of 2-substituted-1,2,3-triazoles. Org. Lett 10, 3171–3174. [DOI] [PubMed] [Google Scholar]
- Kamath A, and Ojima I (2012). Advances in the chemistry of β-lactam and its medicinal applications. Tetrahedron 68, 10640–10664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasperkiewicz P, Altman Y, D’Angelo M, Salvesen GS, and Drag M (2017). Toolbox of fluorescent probes for parallel imaging reveals uneven location of serine proteases in neutrophils. J. Am. Chem. Soc 139, 10115–10125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasperkiewicz P, Poreba M, Snipas SJ, Parker H, Winterbourn CC, Salvesen GS, and Drag M (2014). Design of ultrasensitive probes for human neutrophil elastase through hybrid combinatorial substrate library profiling. Proc. Natl. Acad. Sci. U S A 111, 2518–2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura S, Zheng Q, Woehl JL, Solan A, Chen E, Dillon N, Hull M, Kotaniguchi M, Kitamura S, Nizet V, et al. (2019). SuFEx-enabled high-throughput medicinal chemistry. ChemRxiv 10.26434/chemrxiv.11385906.v1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocsis A, Kékesi KA, Szász R, Végh BM, Balczer J, Dobó J, Závodszky P, Gál P, and Pál G (2010). Selective inhibition of the lectin pathway of complement with phage display selected peptides against mannose-binding lectin-associated serine protease (MASP)-1 and −2: significant contribution of MASP-1 to lectin pathway activation. J. Immunol 185, 4169–4178. [DOI] [PubMed] [Google Scholar]
- Kodani SD, Wan D, Wagner KM, Hwang SH, Morisseau C, and Hammock BD (2018). Design and potency of dual soluble epoxide hydrolase/fatty acid amide hydrolase inhibitors. ACS Omega 3, 14076–14086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kojtari A, Shah V, Babinec JS, Yang C, and Ji H-F (2014). Structure-based drug design of diphenyl α-aminoalkylphosphonates as prostate-specific antigen antagonists. J. Chem. Inf. Model 54, 2967–2979. [DOI] [PubMed] [Google Scholar]
- Kong X-D, Moriya J, Carle V, Pojer F, Abriata LA, Deyle K, and Heinis C (2020). De novo development of proteolytically resistant therapeutic peptides for oral administration. Nat. Biomed. Eng 4, 560–571. [DOI] [PubMed] [Google Scholar]
- Kornahrens AF, Cognetta AB, Brody DM, Matthews ML, Cravatt BF, and Boger DL (2017). Design of benzoxathiazin-3-one 1,1-dioxides as a new class of irreversible serine hydrolase inhibitors: discovery of a uniquely selective PNPLA4 inhibitor. J. Am. Chem. Soc 139, 7052–7061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretz CA, Tomberg K, Van Esbroeck A, Yee A, and Ginsburg D (2018). High throughput protease profiling comprehensively defines active site specificity for thrombin and ADAMTS13. Sci. Rep 8, 2788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamani M, Malamas MS, Farah SI, Shukla VG, Almeida MF, Weerts CM, Anderson J, Wood JT, Farizatto KLG, Bahr BA, et al. (2019). Piperidine and piperazine inhibitors of fatty acid amide hydrolase targeting excitotoxic pathology. Bioorg. Med. Chem 27, 115096. [DOI] [PubMed] [Google Scholar]
- Lau YH, de Andrade P, Wu Y, and Spring DR (2015). Peptide stapling techniques based on different macrocyclisation chemistries. Chem. Soc. Rev 44, 91–102. [DOI] [PubMed] [Google Scholar]
- Lentz CS, Ordonez AA, Kasperkiewicz P, La Greca F, O’Donoghue AJ, Schulze CJ, Powers JC, Craik CS, Drag M, Jain SK, et al. (2016). Design of selective substrates and activity-based probes for hydrolase important for pathogenesis 1 (HIP1) from Mycobacterium tuberculosis. ACS Infect. Dis 2, 807–815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lentz CS, Sheldon JR, Crawford LA, Cooper R, Garland M, Amieva MR, Weerapana E, Skaar EP, and Bogyo M (2018). Identification of a S. aureus virulence factor by activity-based protein profiling (ABPP). Nat. Chem. Biol 14, 609–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Patricelli MP, and Cravatt BF (1999). Activity-based protein profiling: the serine hydrolases. Proc. Natl. Acad. Sci. U S A 96, 14694–14699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Long JZ, and Cravatt BF (2011). The metabolic serine hydrolases and their functions in mammalian physiology and disease. Chem. Rev 111, 6022–6063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maola K, Wilbs J, Touati J, Sabisz M, Kong X, Baumann A, Deyle K, and Heinis C (2019). Engineered peptide macrocycles can inhibit matrix metalloproteinases with high selectivity. Angew. Chem. Int. Ed 58, 11801–11805. [DOI] [PubMed] [Google Scholar]
- Matthews D, and Wells J (1993). Substrate phage: selection of protease substrates by monovalent phage display. Science 260, 1113–1117. [DOI] [PubMed] [Google Scholar]
- Menendez JA, and Lupu R (2007). Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat. Rev. Cancer 7, 763–777. [DOI] [PubMed] [Google Scholar]
- Meng G, Guo T, Ma T, Zhang J, Shen Y, Sharpless KB, and Dong J (2019). Modular click chemistry libraries for functional screens using a diazotizing reagent. Nature 574, 86–89. [DOI] [PubMed] [Google Scholar]
- Narayanan A, and Jones LH (2015). Sulfonyl fluorides as privileged warheads in chemical biology. Chem. Sci 6, 2650–2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasheri N, McKay CS, Fulton K, Twine S, Powdrill MH, Sherratt AR, and Pezacki JP (2014). Hydrophobic triaryl-substituted β-lactams as activity-based probes for profiling eukaryotic enzymes and host-pathogen interactions. ChemBioChem 15, 2195–2200. [DOI] [PubMed] [Google Scholar]
- Nielsen DS, Shepherd NE, Xu W, Lucke AJ, Stoermer MJ, and Fairlie DP (2017). Orally absorbed cyclic peptides. Chem. Rev 117, 8094–8128. [DOI] [PubMed] [Google Scholar]
- Ogasawara D, Ichu T-A, Vartabedian VF, Benthuysen J, Jing H, Reed A, Ulanovskaya OA, Hulce JJ, Roberts A, Brown S, et al. (2018). Selective blockade of the lyso-PS lipase ABHD12 stimulates immune responses in vivo. Nat. Chem. Biol 14, 1099–1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otrubova K, Chatterjee S, Ghimire S, Cravatt BF, and Boger DL (2019). N-Acyl pyrazoles: effective and tunable inhibitors of serine hydrolases. Bioorg. Med. Chem 27, 1693–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson SL, Stucka SM, and Dinsmore CJ (2010). Parallel synthesis of ureas and carbamates from amines and CO2 under mild conditions. Org. Lett 12, 1340–1343. [DOI] [PubMed] [Google Scholar]
- Pierrat OA, Strisovsky K, Christova Y, Large J, Ansell K, Bouloc N, Smiljanic E, and Freeman M (2011). Monocyclic β-lactams are selective, mechanism-based inhibitors of rhomboid intramembrane proteases. ACS Chem. Biol 6, 325–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poreba M, Salvesen GS, and Drag M (2017). Synthesis of a HyCoSuL peptide substrate library to dissect protease substrate specificity. Nat. Protoc 12, 2189–2214. [DOI] [PubMed] [Google Scholar]
- Powers JC, Asgian JL, Ekici ÖD, and James KE (2002). Irreversible inhibitors of serine, cysteine, and threonine proteases. Chem. Rev 102, 4639–4750. [DOI] [PubMed] [Google Scholar]
- Puri AW, and Bogyo M (2013). Applications of small molecule probes in dissecting mechanisms of bacterial virulence and host responses. Biochemistry 52, 5985–5996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez MLG, Poreba M, Snipas SJ, Groborz K, Drag M, and Salvesen GS (2018). Extensive peptide and natural protein substrate screens reveal that mouse caspase-11 has much narrower substrate specificity than caspase-1. J. Biol. Chem 293, 7058–7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenquist Å, Samuelsson B, Johansson P-O, Cummings MD, Lenz O, Raboisson P, Simmen K, Vendeville S, de Kock H, Nilsson, et al. (2014). Discovery and development of simeprevir (TMC435), a HCV NS3/4A protease inhibitor. J. Med. Chem 57, 1673–1693. [DOI] [PubMed] [Google Scholar]
- Rostovtsev VV, Green LG, Fokin VV, and Sharpless KB (2002). A stepwise Huisgen cycloaddition process: copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes 4. Angew. Chem 114, 2708–2711. [DOI] [PubMed] [Google Scholar]
- Schulz-Fincke A-C, Blaut M, Braune A, and Gütschow M (2018). A BODIPY-tagged phosphono peptide as activity-based probe for human leukocyte elastase. ACS Med. Chem. Lett 9, 345–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Serim S, Baer P, and Verhelst SHL (2015). Mixed alkyl aryl phosphonate esters as quenched fluorescent activity-based probes for serine proteases. Org. Biomol. Chem 13, 2293–2299. [DOI] [PubMed] [Google Scholar]
- Shannon DA, and Weerapana E (2015). Covalent protein modification: the current landscape of residue-specific electrophiles. Curr. Opin. Chem. Biol 24, 18–26. [DOI] [PubMed] [Google Scholar]
- Shariff L, Zhu Y, Cowper B, Di W-L, and Macmillan D (2014). Sunflower trypsin inhibitor (SFTI-1) analogues of synthetic and biological origin via N/S acyl transfer: potential inhibitors of human kallikrein-5 (KLK5). Tetrahedron 70, 7675–7680. [Google Scholar]
- Sharifzadeh S, Boersma MJ, Kocaoglu O, Shokri A, Brown CL, Shirley JD, Winkler ME, and Carlson EE (2017). Novel electrophilic scaffold for imaging of essential penicillin-binding proteins in Streptococcus pneumoniae. ACS Chem. Biol 12, 2849–2857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Staub I, and Sieber SA (2008). β-lactams as selective chemical probes for the in vivo labeling of bacterial enzymes involved in cell wall biosynthesis, antibiotic resistance, and virulence. J. Am. Chem. Soc 130, 13400–13409. [DOI] [PubMed] [Google Scholar]
- Thirumurugan P, Matosiuk D, and Jozwiak K (2013). Click chemistry for drug development and diverse chemical–biology applications. Chem. Rev 113, 4905–4979. [DOI] [PubMed] [Google Scholar]
- Uematsu S, Tabuchi Y, Ito Y, and Taki M (2018). Combinatorially screened peptide as targeted covalent binder: alteration of bait-conjugated peptide to reactive modifier. Bioconjug. Chem 29, 1866–1871. [DOI] [PubMed] [Google Scholar]
- van Rooden EJ, Kohsiek M, Kreekel R, van Esbroeck ACM, van den Nieuwendijk AMCH, Janssen APA, van den Berg RJBHN, Overkleeft HS, and van der Stelt M (2018). Design and synthesis of quenched activity-based probes for diacylglycerol lipase and α,β-hydrolase domain containing protein 6. Chem. Asian J 13, 3491–3500. [DOI] [PubMed] [Google Scholar]
- Vosyka O, Vinothkumar KR, Wolf EV, Brouwer AJ, Liskamp RMJ, and Verhelst SHL (2013). Activity-based probes for rhomboid proteases discovered in a mass spectrometry-based assay. Proc. Natl. Acad. Sci. U S A 110, 2472–2477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Abegg D, Dwyer BG, and Adibekian A (2019). Discovery and evaluation of new activity-based probes for serine hydrolases. ChemBioChem 20, 2212–2216. [DOI] [PubMed] [Google Scholar]
- Winiarski Ł, Oleksyszyn J, and Sieńczyk M (2012). Human neutrophil elastase phosphonic inhibitors with improved potency of action. J. Med. Chem 55, 6541–6553. [DOI] [PubMed] [Google Scholar]
- Wright MH, and Sieber SA (2016). Chemical proteomics approaches for identifying the cellular targets of natural products. Nat. Prod. Rep 33, 681–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo E, Stokes BH, de Jong H, Vanaerschot M, Kumar T, Lawrence N, Njoroge M, Garcia A, Van der Westhuyzen R, Momper JD, et al. (2018). Defining the determinants of specificity of Plasmodium proteasome inhibitors. J. Am. Chem. Soc 140, 11424–11437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young TS, Young DD, Ahmad I, Louis JM, Benkovic SJ, and Schultz PG (2011). Evolution of cyclic peptide protease inhibitors. Proc. Natl. Acad. Sci. U S A 108, 11052–11056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Woehl JL, Kitamura S, Santos-Martins D, Smedley CJ, Li G, Forli S, Moses JE, Wolan DW, and Sharpless KB (2019). SuFEx-enabled, agnostic discovery of covalent inhibitors of human neutrophil elastase. Proc. Natl. Acad. Sci. U S A 116, 18808–18814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Li S, Leung KK, O’Donovan B, Zou JY, DeRisi JL, and Wells JA (2020). Deep profiling of protease substrate specificity enabled by dual random and scanned human proteome substrate phage libraries. Biochemistry 10.1101/2020.05.09.086264. [DOI] [PMC free article] [PubMed] [Google Scholar]