

SUMMARY
All mammals progress through similar physiological stages throughout life, from early development to puberty, aging, and death. Yet, the extent to which this conserved physiology reflects underlying genomic events is unclear. Here, we map the common methylation changes experienced by mammalian genomes as they age, focusing on comparison of humans with dogs, an emerging model of aging. Using oligo-capture sequencing, we characterize methylomes of 104 Labrador retrievers spanning a 16-year age range, achieving >150× coverage within mammalian syntenic blocks. Comparison with human methylomes reveals a nonlinear relationship that translates dog-to-human years and aligns the timing of major physiological milestones between the two species, with extension to mice. Conserved changes center on developmental gene networks, which are sufficient to translate age and the effects of anti-aging interventions across multiple mammals. These results establish methylation not only as a diagnostic age readout but also as a cross-species translator of physiological aging milestones.
In Brief
Wang et al. create an oligo-capture system to characterize the canine DNA methylome, targeting syntenic regions of the genome conserved across all mammals. Cross-species comparisons reveal a nonlinear epigenetic signature that aligns the progression of life events in dogs, humans, and mice. This conserved signature occurs primarily in modules of developmental genes, leading the team to create a conserved epigenetic clock model of aging that can be trained and operated across different species.
Graphical Abstract
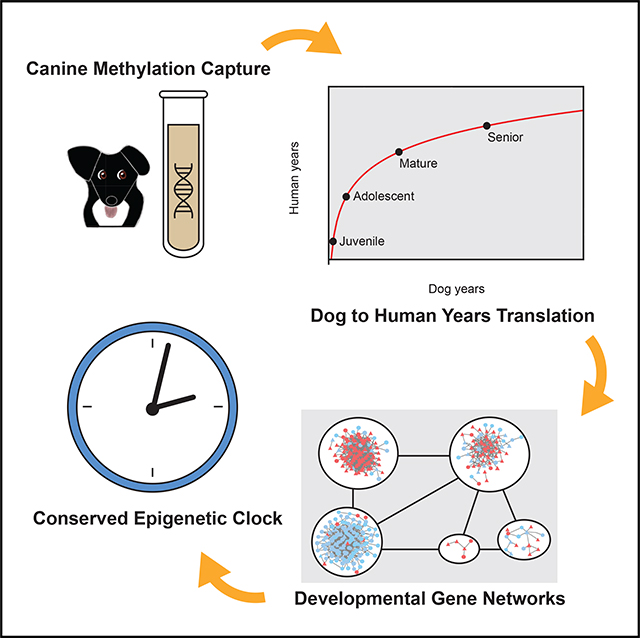
INTRODUCTION
The wisdom that every year in a dog’s life equates to seven human years reflects our deep intuition that development and aging are conserved processes that occur at different rates in different species. All mammals, whether dog, human, or other creature, pass through similar life stages of embryogenesis, birth, infancy, youth, adolescence, maturity, and senescence (Withers, 1992). Although embryonic developmental programs have been relatively well studied, many of the molecular events governing postnatal life stages, including those tied to aging, are still unresolved (Khan et al., 2017). Over the past decade, it has become clear that a prominent molecular alteration during aging is remodeling of the DNA methylome, the pattern of epigenetic modifications whereby methyl groups are present at some cytosine-guanine dinucleotides (methyl-CpGs) but absent from others (Field et al., 2018). The methylation states of tens of thousands of CpGs have been found to change predictably over time, enabling the construction of mathematical models, known as “epigenetic clocks,” that use these shifting patterns to accurately measure the age of an individual (Hannum et al., 2013; Horvath, 2013; Petkovich et al., 2017; Stubbs et al., 2017; Thompson et al., 2017; Wang et al., 2017).
Most questions regarding the relationship between DNA methylation and mammalian life stage remain unanswered (Figure 1). While the rate of methylation change appears to depend on maximal lifespan (Maegawa et al., 2017; Lowe et al., 2018), whether lifespan is the sole factor in determining how the methylome progresses with age, or if CpG states are aligned to specific intermediate milestones in development and aging, remains unknown. It is also unclear if the major epigenetic changes that occur with age involve the same or different (or random) CpG sites in different species. While DNA encoding a highly conserved ribosomal RNA family shows increasing methylation over time at the same conserved CpG sites in mice and humans (Wang and Lemos, 2019), epigenetic clocks trained in humans have not been strong predictors of age in other mammals (Petkovich et al., 2017; Stubbs et al., 2017). Perhaps this is not surprising, as epigenetic clocks are formulated using regression techniques that select only a small subset of the available CpG sites (<500) out of millions of potential subsets that can be used to measure age equivalently (Field et al., 2018).
Figure 1. Physiological versus Epigenetic Change during Development and Aging.

Aging yields similar physiological changes in humans and dogs, yet these changes occur along different time scales. Are these different timescales reflected in the progression of epigenetic changes observed during aging? If so, is this progression consistent with the adage “one dog year equals seven human years,” or does it suggest a different cross-species alignment of time?
Thus far, a major impediment to understanding the relationship between methylation and mammalian life stages has been our inability to characterize conserved epigenetic features in different mammals. Most studies of the human methylome use Illumina methylation arrays, which include oligonucleotide probes designed to explicitly measure the methylation states of >450,000 CpG sites (http://www.illumina.com/). Since a similar array platform has not yet been developed for other species, techniques such as whole-genome bisulfite sequencing (WGBS) have been used, which measure many CpGs without the need for a specific capture system but at relatively low coverage per sequencing run (Chatterjee et al., 2017). Other efforts, such as reduced representation bisulfite sequencing (RRBS), increase coverage for some CpGs but do not guarantee consistent measurements of the same sites across samples (Chatterjee et al., 2017). Therefore, what is needed is (1) to select a suitable model species, and (2) within this species, to develop a CpG capture system that can be more directly aligned with human methylation arrays.
Domestic dogs provide a unique opportunity to address these challenges (Gilmore and Greer, 2015; Kaeberlein et al., 2016). Dogs have been selectively bred by humans for occupation and esthetics (Ostrander et al., 2017), generating over 450 distinct breeds whose members share morphologic and behavioral traits. Most breeds derive from small numbers or popular sires, and dogs have been domesticated for only about 15,000–30,000 years (Vonholdt et al., 2010), leading to strong phenotypic and genetic homogeneity within breeds (Dreger et al., 2016). Dogs share nearly all aspects of their environment with humans, including factors associated with aging such as diet and chemical exposure. They also experience similar levels of health observation and health care intervention as humans (Gilmore and Greer, 2015; Kaeberlein et al., 2016). While the average lifespan differs dramatically across breeds, there is also considerable variability within some breeds, such as the Labrador retriever. Despite extensive phenotypic differences, however, all domestic breeds are members of the same species with a similar developmental, physiological, and pathological trajectory as humans (Gilmore and Greer, 2015; Kaeberlein et al., 2016). Importantly for aging studies, dogs accomplish this progression in many fewer years than humans, generally fewer than 20. Finally, epigenetic clocks have been demonstrated in dogs (Thompson et al., 2017), establishing them as a system for studies of age-related epigenetic remodeling.
Here, we develop the dog as a model system for epigenetic aging, using a specifically designed CpG oligo-capture system to generate high quality methylation data that align directly to human methylomes. Comparison of each dog methylome to its nearest human counterparts reveals a conserved but nonlinear progression of epigenetic changes, with rapid remodeling in puppies, relative to children, which slows markedly in canine adulthood. We show that most of the conserved epigenetic changes occur within specific developmental gene networks, such that a set of 439 conserved CpG sites is sufficient to build a pan-species epigenetic clock of aging. Unlike previous clocks, which are predictive in a single species only, the conserved developmental clock translates to multiple species with relative accuracy.
RESULTS
Characterization of Dog Methylomes with Syntenic Bisulfite Sequencing (SyBS)
Commonly used techniques such as WGBS and RRBS measure large numbers of random CpGs, but such CpGs are often not present in regions conserved across species. To enable high quality evolutionary comparisons of dog methylomes with other mammals, we performed targeted-bisulfite sequencing to systematically characterize CpGs in regions of the dog genome that are syntenic with those measured by Illumina human methylome arrays. Since Illumina arrays have been used to characterize epigenetic aging in many human studies (Alisch et al., 2012; Hannum et al., 2013; Horvath, 2013), our goal was to create a high quality panel of dog methylomes with substantial coverage of CpGs noted in prior human datasets (Figure 2A). Our strategy, henceforth called synteny bisulfite sequencing (SyBS), was designed to capture approximately 90,000 CpGs of the approximately 232,000 conserved CpGs on the Illumina array (STAR Methods).
Figure 2. Interrogating Mammalian Methylomes by Syntenic Bisulfite Sequencing (SyBS).

(A) Strategy used to profile and compare CpG methylation states within blocks of synteny in the mammalian genome. Capture oligonucleotide design: regions of DNA (blue blocks) characterized by the Illumina 450K methylation array in humans are mapped to their syntenic region in dogs using whole-genome alignments between the two species. These regions are used to design oligonucleotides (yellow stars) for capture and enrichment of DNA in the second species. Data generation: A sequencing library is constructed from high quality DNA and bisulfite converted, analogously to WGBS. Syntenic sequences are captured, sequenced, and aligned to the mammalian genome under study. CpG methylation values are called and then filtered to select those conserved with humans for further analysis. For more details see STAR Methods.
(B) Pie charts showing representation of targeted genomic regions. Regions exhibiting significant enrichment (p < 10−10) are indicated using asterisks with * indicating odds ratio > 2.5 and ** odds ratio > 4. UTR, untranslated region; TSS, transcription start site.
(C) Ten dog methylomes were sequenced twice, either with enrichment for syntenic regions (SyBS hybridization) or without enrichment (WGBS). Methylation values (per CpG site per animal) are shown for SyBS (y axis) versus WGBS (x axis). Sites were considered if they were covered by >5 reads with both SyBS and WGBS.
(D) Concordance of SyBS values for one canine DNA sample (S1), for which two independent captures were performed. In (C) and (D) the color captures the density of observations at each point (darker colors represent higher densities), and the r value is the Pearson correlation.
(E) Average coverage of syntenic segments versus total reads in millions, contrasting SyBS with RRBS.
We applied SyBS to characterize the methylomes of 104 dogs, primarily consisting of Labrador retrievers and representing the entire lifespan, from 0.1 to 16 years at the time of blood draw (Figure S1A; Table S1). Libraries were sequenced to an average depth of 163×, with nine dogs removed due to lack of coverage. Captured CpG sites spanned the genome and were enriched for genomic regions that included exons, transcription start sites, and CpG islands (odds ratio >2.5 and p < 10−10 by Fisher’s exact test, Figure 2B). The methylation values associated with captured CpGs were similar to those obtained using WGBS (Pearson correlation r > 0.85, Figure 2C) and showed excellent replication across independent captures from the same samples (r > 0.95, Figures 2D and S1B–S1F). As expected, SyBS achieved substantially higher coverage of syntenic regions compared with non-targeted reduced representation bisulfite methods (~13-fold increase, Figure 2E). For comparison, we obtained previously published methylation profiles from the blood of 320 human individuals aged from 1 to 103 years at the time of sample isolation (Alisch et al., 2012; Hannum et al., 2013). Based on these data, we identified 54,469 well profiled CpGs in both species, thus enabling systematic evolutionary studies of epigenetic changes during life (STAR Methods).
A Concordant but Nonlinear Relationship between Dog and Human Age
We observed the highest methylome similarities (Pearson correlation, STAR Methods) when pairing young dogs with young humans, or aged dogs with older humans. In contrast, the lowest similarities were obtained when pairing young dogs with old humans or vice versa (Figure 3A). The relationship between methylome similarity and age was lost upon permutation (FDR < 0.01; Figure S2A), which indicated that a conserved set of CpG sites are affected during aging in the two mammalian species. Notably, this signal was sufficiently strong to arise in an unsupervised methylome-wide analysis without sub-selection of markers. This result suggested that the conserved methylation changes with age in ribosomal RNA genes, noted previously (Wang and Lemos, 2019), extend more generally to the greater mammalian methylome. It contrasts with previous observations using epigenetic clocks, which did not find strong conservation across species, likely because these clocks are restricted to 80–300 CpGs, selected for optimal age prediction in humans independent of other species (Stubbs et al., 2017).
Figure 3. A Nonlinear Transformation from Dog-to-Human Age.

(A) Dog-human methylome similarities (Pearson correlation, blue-red color range) are shown with dogs and humans ranked from youngest to oldest. Data are lightly smoothed in both dimensions using Gaussian interpolation in matplotlib.
(B) The age of each dog methylome (x axis) is plotted against the average age of the five nearest human methylomes (y axis), 95 dogs are depicted.
(C) Reciprocal plot in which the age of each human methylome (y axis) is plotted against the average age of the five nearest dog methylomes (x axis), 320 humans are depicted.
(D) Logarithmic function for epigenetic translation from dog age (x axis) to human age (y axis). Outlined boxes indicate the approximate age ranges of major life stages as documented qualitatively based on common aging physiology. Juvenile refers to the period after infancy and before puberty, 2–6 months in dogs, 1–12 years in humans; adolescent refers to the period from puberty to completion of growth, 6 months to 2 years in dogs, approximately 12–25 years in humans; Mature refers to the period from 2–7 years in dogs and 25–50 years in humans; Senior refers to the subsequent period until life expectancy, 12 years in dogs, 70 years in humans. Dog life stages are based on veterinary guides and mortality data for dogs (Fleming et al., 2011; Bartges et al., 2012; Inoue et al., 2015). Human life stages are based on literature summarizing life cycle and lifetime expectancy (Bogin and Smith, 1996; CIA, 2013; Arias et al., 2017). Black dots on the curve connect to images of the same yellow Labrador taken at four different ages (courtesy of Sabrina and Michael Mojica, with permission) and to images of a representative human at the equivalent life stages in human years (photos of Tom Hanks drawn from a public machine-learning image repository, Chen et al., 2015).
(E) Mouse-dog methylome similarities shown as in (A).
(F) Data from (E) are summarized by sorting mice according to 0.2-year bins (x axis) and, for each mouse, plotting the average age of the 5 nearest dogs by methylome similarity (y axis). Points illustrate the mean of each bin and bars represent the 95% confidence interval obtained from bootstrapping.
We next investigated whether the conserved methylation changes in dogs and humans follow a constant rate of change with age, with the rate constant depending on the lifespan of each species as suggested previously (Maegawa et al., 2017; Lowe et al., 2018), or whether there was evidence for a more complex trajectory. For this purpose, we assigned the age of each dog to the average age of its nearest humans by methylome-wide similarity (STAR Methods). This analysis revealed a monotonic, time-resolved, nonlinear relationship between dog and human age (Figure 3B considering the k = 5 nearest humans, Figures S2B–S2G considering other k values). Similar results were obtained in a reciprocal analysis assigning each human to its nearest dogs (Figure 3C), as evidenced by the similarity in fitted functions: human_age = 17 ln(dog_age) + 33 (Figure 3B) and human_age = 16 ln(dog_age) + 30 (Figure 3C). Therefore, we combined the reciprocal analyses to generate the single function: human_age = 16 ln(dog_age) + 31 (Figure 3D).
We found that this function showed strong agreement between the approximate times at which dogs and humans experience common physiological milestones during both development and lifetime aging, i.e., infant, juvenile, adolescent, mature, and senior (Lebeau, 1953; Bogin and Smith, 1996; Bartges et al., 2012) (Figure 3D). The observed agreement between epigenetics and physiology was particularly close for infant/juvenile and senior stages. For instance, the epigenome translated approximately 8 weeks in dogs (0.15 years) to approximately 9 months in humans (0.78 years), corresponding to the infant stage when deciduous teeth develop in both puppies and babies (Bogin and Smith, 1996; Bartges et al., 2012). In seniors, the expected lifespan of Labrador retrievers, 12 years, correctly translated to the worldwide lifetime expectancy of humans, 70 years (Fleming et al., 2011; CIA, 2013). For adolescent and mature stages, the correspondence was more approximate, with the epigenome showing faster changes for dogs, relative to humans, than expected by physiological tables (Inoue et al., 2015; Arias et al., 2017) (Figure 2D). Thus, the canine epigenome progresses through a series of conserved biological states that align with major physiological changes in humans, occurring in the same sequence but at different chronological timepoints during each species’ lifespan.
A conserved nonlinear and weaker epigenetic progression was also observed by comparing the dog methylomes to those measured previously from 133 mice (Petkovich et al., 2017) (Figures 3E and 3F). This weaker effect may be due to the limited number of mice sampled during the developmental period (Table S3). Nevertheless, the ability to translate age among these three diverse mammals indicates that shared physiology may yield conserved molecular transitions in epigenome remodeling with age.
Identification of Genes Exhibiting Conserved Methylation Dynamics with Age
To determine whether the conserved changes were concentrated within particular genes or gene functions, we examined CpG methylation states near 7,942 genes for which orthologs were present in all three species (dogs, humans, and mice, STAR Methods). This analysis identified 394 genes for which methylation values showed conserved time-dependent behavior across species (empirical p < 0.05, Figure S3; Table S2). To understand the underlying gene functions, we mapped them onto the parsimonious composite network (PCNet), a database of approximately 2 106 molecular interactions capturing physical and functional relationships among genes and gene products, in which each interaction has support from multiple sources (Huang et al., 2018). The genes clustered into five highly interconnected network modules (Figure 4), nearly all of which were enriched for developmental functions. Methylation changes at these developmental genes were concentrated near transcription start sites (Figure S4A) and in CpG islands and shores for all three species (Figure S4B). Four modules predominantly increased in methylation with age (FDR < 0.05) and included modules associated with synapse assembly (18 genes) and neuroepithelial cell differentiation (5 genes) and two modules associated with anatomical patterning (117 and 69 genes). These four modules were enriched for polycomb repressor targets, which are predominantly silenced in adult tissues (Xie et al., 2013). A fifth module was enriched in leukocyte differentiation and nucleic-acid metabolism (144 genes) and demonstrated decreasing methylation with age. We also noted that orthologs from all five modules were among the most highly conserved in DNA sequence in the mammalian genome, even accounting for high sequence conservation of developmental genes in general (Figure S5).
Figure 4. Conserved Lifetime Methylation Changes Aggregate in Developmental Networks.

Genes exhibiting conserved age-related methylation behavior were mapped onto a composite molecular interaction network which was subsequently clustered to reveal five major modules, labeled according to enriched Gene Ontology functions (STAR Methods). Colors represent the conserved direction of change with age, with red representing genes that increase in methylation with age and blue representing genes that decrease in methylation with age. Heatmaps show the conserved methylation patterns of a random subset of genes in each module. Columns represent distinct orthologs, while rows represent the average values of all species ranked according to their age in human years and divided into 15 age bins (quantiles). Values are normalized according to the mean and standard deviation of methylation for each ortholog. The fractional species composition of each bin is visualized in the legend.
Further indication of the importance of developmental gene modules was observed when calculating dog-human methylome similarity using CpGs at developmental genes only versus a comparison using all CpGs except those at developmental genes (Figure S6; STAR Methods). This comparison showed that CpGs near developmental genes are both necessary and sufficient to recapitulate the cross-species alignments of age observed earlier (Figure 3). Enrichment of age-related methylation increases in developmental genes has been previously observed in humans (Rakyan et al., 2010) and mice (Maegawa et al., 2010). Our findings extend such observations by highlighting developmental genes as predominant drivers of the ability to align mammalian methylomes and in specifying more precisely where in the developmental gene modules such changes occur.
Translating Age and Aging Effects Using a Conserved Epigenetic Clock
Epigenetic clocks have garnered recent interest due to their ability to translate an individual’s methylome to an accurate prediction of age (Hannum et al., 2013; Horvath, 2013; Petkovich et al., 2017; Stubbs et al., 2017; Thompson et al., 2017; Wang et al., 2017). However, they have typically been applied to study methylome data in one species only, with less success in cross-species application (Stubbs et al., 2017). Therefore, in our final analysis, we examined whether the conserved developmental gene modules could be used to construct a conserved epigenetic clock capable of predicting age in multiple mammalian species.
In particular, we formulated a conserved development clock based on methylation values of the 394 CpGs within the conserved developmental gene modules we had identified earlier (Figure 4; STAR Methods). By training in dogs or alternatively mice, this clock could be used to score the age of a dog or mouse using its methylation profile. Implicit in this analysis was the translation of a dog methylome to its equivalent mouse age or vice versa (Figure 5A). For baseline comparison, we also constructed single species methylome-wide clocks for dogs and mice as per the usual procedure (STAR Methods). In this case, the model was allowed to select the most optimal CpGs for age prediction from the entire methylome of each species (Figure 5A).
Figure 5. A Conserved Development Clock Measuring Age and Physiological Aging.

(A) Construction of epigenetic clocks. Four clocks are constructed, depending on whether the training data are from dogs or mice and whether the input features are from all methylome-wide CpGs or from CpGs in conserved developmental modules only. All four cases yield a regression model for predicting age from CpG markers (STAR Methods).
(B) Scatterplot of predicted versus actual ages for the dog methylome-wide model.
(C) Scatterplot of predicted versus actual ages for the mouse methylome-wide model.
(D) Performance of single species methylome-wide clocks (gray) or conserved developmental clocks (turquoise) as measured by the Spearman correlation between predicted and actual ages within species or across species.
(E) The conserved development clock distinguishes the effects of lifespan-enhancing treatments (orange) from control treatments (gray). For each treatment, mouse epigenetic ages are measured (y axis; conserved development clock trained in mice) and plotted against actual mouse ages binned in 0.1-year bins (x axis). Mean ± 95% confidence intervals shown for each bin and each observation.
(F) As for (E) but training the conserved development clock using data for dogs. For each treatment (orange lifespan-enhancing; gray control), epigenetic ages of each mouse are measured and plotted against actual mouse ages binned in 0.1-year bins (x axis). * denotes p < 0.05 in all panels.
When training and predicting on individuals within a single species, the ages measured by the single species methylome-wide clocks were very accurate (dog rho = 0.99, Figures 5B and 5D; mouse rho = 0.86, Figures 5C and 5D) relative to the conserved development clocks (dog rho = 0.81; mouse rho = 0.78; Figure 5D). However, when applying a clock trained in one species to make predictions in the other, the ages predicted by the conserved development clock (dogs-to-mice rho = 0.73; mice-to-dogs rho = 0.71) were substantially more accurate than those of the single species methylome-wide clocks (dogs-to-mice rho = 0.22; mice-to-dogs rho = 0.32; Figure 5D). In addition, clocks based on the developmental gene modules were more accurate than clocks based on methylation of ribosomal RNA sequences (rho = 0.6 dogs-to-mice and 0.5 mice-to-dogs) (Wang and Lemos, 2019). When applying the conserved development clock to mice treated with lifespan-extending interventions (STAR Methods), the epigenetic ages were 30% less, on average, than those of control mice (p < 10−6, Figure 5E). These same results were observed when using the development clock trained in dogs to predict mouse age (Figure 5F). Together, these results demonstrate that the methylation states of developmental gene modules can be used to construct a conserved model of age that is transferable between mammalian species. In contrast, previous models trained in a single species can appear to achieve extremely high accuracy when testing in that same species, but these models do not readily transfer outside of the original species context.
DISCUSSION
By using targeted oligonucleotide capture (SyBS), we have produced methylomes of high quality for comparative studies among dogs and other mammals. Analysis of these data shows that multiple mammalian species experience conserved methylation changes during aging, and that the scope of these changes is methylome-wide. Second, the trajectory of changes followed by one species as it ages is not necessarily the same as that followed by another. In particular, dog methylomes remodel very rapidly in early life compared with the methylomes of their human counterparts. Our further analysis in this regard (Figure 3D) suggests that the rate of remodeling is not only determined by the lifespan of a species but also by the timing of key physiological milestones. Previously, CpG methylation states in humans were proposed to exhibit a nonlinear trajectory over time, with non-constant rates of change (Horvath, 2013). The analysis here demonstrates a different point, that methylation changes in one species can be nonlinear with respect to another.
We observe that epigenetic changes during aging center on highly conserved modules of developmental genes, in which methylation generally increases with age. Although the enrichment of developmental pathways has been generally observed in mammals previously (Ciccarone et al., 2018; Field et al., 2018; Horvath and Raj, 2018), our findings show that specific changes at these loci translate the effects of age and anti-aging interventions across species. While the biology of aging has historically been considered as separate from that of development (Miller and Nadon, 2000; Kowald and Kirkwood, 2016), their strong association, demonstrated here, supports a model in which at least some aspects of aging are a continuation of development rather than a distinct process.
Limitations and extensions of our findings are as follows. First, we have used DNA isolated from whole blood, for which age-dependent shifts in leukocyte populations have been described (Jaffe and Irizarry, 2014). In particular, previous studies have found that CD4+ T cells, CD8+ T cells, and B cells decline with age. Although it is possible that such conserved shifts may influence our findings, such decline occurs in both dogs (Greeley et al., 2001) and humans (Jaffe and Irizarry, 2014). Second, our study has focused exclusively on Labrador retrievers, a popular and heterogeneous breed for which we could collect large numbers of unrelated dogs in order to control for population structure. Distinct breeds exhibiting widely varying lifespans (Gilmore and Greer, 2015; Kaeberlein et al., 2016) could yield different age-translation functions.
Further efforts to characterize epigenetic changes across breeds and species may help to address these and broader questions. For example, does the timing of epigenetic changes early in life influence the overall lifespan of a species, or of an individual within that species? Does modulating the timing of developmental events affect lifespan? Again, comparisons of species or sub-species that experience developmental milestones at similar times but with different lifespans (such as distinct dog breeds) may help address these questions, providing critical and complementary data to inform the ongoing cross-species aging studies (Kaeberlein et al., 2016), including clinical trials of aging interventions (Urfer et al., 2017).
Finally, our study has demonstrated that the methylome can be used to quantitatively translate the age-related physiology experienced by one organism (i.e., a model species like dog) to the age at which physiology in a second organism is most similar (i.e., a second model or humans; Figures 3 and 5). These results enable the methylome to act not only as a diagnostic readout of age in a single species, as per the usual epigenetic clock studies, but also for cross-species translation of age and physiological state of aging. Such translation may provide a compelling tool in the quest to understand aging and identify interventions for maximizing healthy lifespan.
STAR★METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Trey Ideker (tideker@ucsd.edu)
Materials Availability
This study did not generate new materials.
Data and Code Availability
The sequencing reads and processed files generated during this study are available at GEO (GSE146920). All original code is freely available for download at 10.5281/zenodo.3864683.
METHOD DETAILS
Annotations
Reference genomes were downloaded from Ensembl for dog (CanFam3.1), mouse (mm10) and human (hg19). Ensembl Biomart version 91 was used for gene, 3′UTR and 5′UTR annotations (Yates et al., 2016). CpG islands, repeat annotations, and chain files were downloaded from the UCSC Genome Browser (Rosenbloom et al., 2015). CpG shores were designated as regions 2 kilobases (kb) outside each CpG island, and CpG shelves were designated as regions 2kb outside of CpG shores. Promoters were designated as regions 2kb upstream and 100 basepairs (bp) downstream of the transcription start sites (TSS) based on gene annotations from Ensembl (Yates et al., 2016). Whole genes were divided into exonic and intronic sequences. Intergenic regions were then defined as the remaining regions of the genome after subtracting all other annotated regions. Definitions of one-to-one orthologs were downloaded from Ensembl Compara (Vilella et al., 2009) for dogs, humans and mice.
Public Datasets
The following datasets were obtained from Gene Expression Omnibus (GEO) or Sequence Read Archives (SRA) [number of individuals included in study in brackets]:
GSE80672 (Petkovich et al., 2017): Methylomes from postnatal mice. Blood, Reduced Representation Bisulfite Sequencing (RRBS) method. [133]
GSE36054 (Alisch et al., 2012): Methylomes from human children. Blood, Infinium 450K array. [35]
GSE40279 (Hannum et al., 2013): Methylomes from human adults. Blood, Infinium 450K array. [285]
SRP065319 (Thompson et al., 2017): Methylomes from dogs and wolves. Blood, RRBS method [92].
Canine Samples
Information on each dog sample used, including age, breed, and source, is given in Table S1, with the age distribution also provided in Figure S1A. For samples sourced from NHGRI, domestic dogs were collected with owners’ signed consents in accordance with standard protocols approved by the NHGRI IACUC committee. Samples were collected at canine-centric events such as dog shows. Alternatively, owners were supplied with a mail-in kit which included instructions, tubes for blood draws and a general information sheet requesting the AKC number (when available), pedigree and date of birth. Blood draws were performed by licensed veterinarians or veterinary technicians. For samples sourced from UC Davis, blood was collected from privately owned dogs through the William R. Pritchard Veterinary Medical Teaching Hospital. Owners specified the breed of each dog. Standard collection protocols were reviewed and approved by the UC Davis IACUC. DNA was extracted either using the Puregene kit (Qiagen) or using the cell lysis protocol described by (Bell et al., 1981), followed by phenol/chloroform extraction with phase separation in 15-mL phase-lock tubes (5-Prime, Inc. Gaithersburg, MD, USA).
SyBS Target Selection
The strategy for syntenic bisulfite sequencing was to base our Illumina Human 450K probe locations were extended 50bp with respect to the strand of each probe. The resulting locations were mapped to the dog genome using liftOver (Rosenbloom et al., 2015) using default parameters. After excluding regions that mapped to sex, mitochondrial and unplaced contigs in the dog genome, we identified approximately 230,000 probes that were syntenic between human and dogs. Hybridization probes were generated to target these regions using the Roche SeqCap-Epi platform. This process produced an 18.8 megabase sequencing library in dogs, containing approximately 90,000 CpGs that were also profiled by the Illumina 450K array in humans.
SyBS Library Preparation and Sequencing
We followed the protocol specified by the Roche SeqCap-Epi platform. Briefly, approximately 500ng of lambda phage DNA (bisulfite-conversion control) was added to 1ug of dog DNA, then sheared to an average of 175bp (Covaris). Sheared DNA was end-repaired, A-tailed and ligated to barcoded adapters. Adapter-ligated libraries were subjected to bisulfite treatment (Zymo EZ DNA methylation lightning kit) following manufacturer’s instructions. Bisulfite-treated libraries were cleaned and amplified using 25 cycles of PCR with a uracil-tolerant enzyme (Kapa). Libraries were pooled equimolarly into 4-plex or 6-plex hybridization capture reactions to a total of 1ug per reaction. Captured product was PCR amplified (10 cycles). Hybridizations were pooled before sequencing and split among 10 lanes on an Illumina HiSeq 4000 in 2×100bp cycles.
SyBS Data Processing
Reads obtained from sequencing were demultiplexed and their quality was verified using FastQC (Andrews et al., 2010). Reads were trimmed using TrimGalore (Krueger, 2015) (4bp) then aligned to a bisulfite-converted dog genome (CanFam3.1) using Bismark (v0.14.3) (Krueger and Andrews, 2011), which produced alignments with Bowtie2 (v2.1.0) (Langmead et al., 2009) with parameters “-score_min L,0,−0.2”. Methylation values for CpG sites were determined using MethylDackel (v0.2.1) (Ryan, 2017). Custom Python scripts using BEDtools (v2.25.0) (Quinlan and Hall, 2010) were used to determine on-target reads. Optical PCR duplicates were determined using Picard tools (v1.141) (Picard Toolkit, 2019) and removed using Samtools (v0.1.18) (Li et al., 2009). Coverage of syntenic regions was determined using the number of unique on-target reads that were orthologous to humans, divided by the expected sequencing space. Only CpG sites that were on-target, covered by at least five reads and present across 90% of samples were selected for further analysis. Samples missing more than 30% of CpGs were removed from further analyses resulting in the removal of nine dogs. Missing data for selected CpGs were imputed by performing k-nearest neighbors (k = 10) using fancyimpute in Python. To assess the concordance of methylation values obtained using SyBS with conventional approaches, we also sequenced 10 of the same dogs using whole-genome bisulfite sequencing (libraries prior to enrichment with SyBS probes). Reads were processed and aligned with the canine genome as described above. We saw an average Pearson correlation of r = 0.85 among these 10 samples (range 0.75 – 0.97) (Figure 2C). We also performed independent replicate hybridizations for 6 samples. We saw an average r = 0.97 (range: 0.96 – 0.98) for these technical replicates (Figures 2D and S1B–S1F). We verified that lambda phage DNA exhibited complete conversion (>99.5%). We tested the significance of the enrichment of our captured sequences and genome region annotation using the LOLA package (Sheffield and Bock, 2016) in R (version 3.5.1) (R Core Team, 2018). Enrichment tests are performed using Fisher’s exact tests, with the possible ‘universe’ defined by restriction digestion fragmentation of autosomes in the canine reference genome.
Public RRBS Data Processing
For data generated using Reduced Representation Bisulfite Sequencing (RRBS), methods for alignment and CpG selection were identical to those described above. Since RRBS fragments are generated using restriction enzymes with specific recognition sites, optical PCR duplicates could not be removed and on-target CpGs were not determined. For evolutionary comparative analysis, we included 133 control mice aged between 3 months to 2.5 years (Petkovich et al., 2017). To compare the coverage of syntenic regions between SyBS and non-targeted bisulfite technology, we used a RRBS study in dogs and wolves (Thompson et al., 2017) (Figure 2E).
Human Methylation Array Data Processing
Illumina Infinium 450K methylome array data were quantile normalized using Minfi (Aryee et al., 2014) and missing values were imputed using the Impute package (Hastie et al., 2020) in R. These values were adjusted for cell counts as previously described (Gross et al., 2016). To enable comparisons across different methylation array studies, we implemented beta-mixture quantile dilation (BMIQ) (Horvath, 2013; Teschendorff et al., 2013) and used the median of the (Hannum et al., 2013) dataset as the gold standard. To mitigate residual batch effects, we selected human samples that clustered closely in the first two principal components using scikit-learn v0.19.2 (Pedregosa et al., 2011) and verified that such filtering had little effect on the distribution of ages. We also removed samples for which more than 10% of probes were not adequately detected. This procedure resulted in methylome profiles for 320 humans that could be compared to the SyBS-generated dog methylomes.
Determining Orthologous CpGs
Human Illumina 450K methylation array CpGs were extended by 50bp with respect to the strand using BEDtools and mapped to the target genome (mouse or dog) using liftOver with “-minMatch=0.5”. We verified that the coordinate alignment obtained using 50bp was identical to that obtained using the exact coordinate (1bp) at “-minMatch=0.95”. This procedure allowed us to determine an exact orthologous region for each human CpG and each dog CpG. When multiple dog CpGs were assigned to one human CpG probe region, we took the average methylation value of the aligned CpGs in dogs. This procedure resulted in 54,469 dog-human orthologous CpGs for further analysis. To mitigate batch effects specific to sequencing and/or array platforms, we normalized the sequencing methylation values using BMIQ and performed quantile normalization using the preprocessCore package in R (normalize.quantiles.use.target function) (Bolstad, 2013).
For dog-to-mouse comparisons, CpGs that were separated by 1bp were merged into one region using BEDtools. Each region was then extended by 50bp. The resulting region files were aligned to the target genome using liftOver “-minMatch=0.5”. Only regions that were concordant between the two alignments (i.e., dog to mouse or mouse to dog) were selected for further analysis. CpGs that were assigned to the same aligned regions were averaged to generate 9,404 bins, consisting of 87,915 CpGs from dogs.
QUANTIFICATION AND STATISTICAL ANALYSIS
Dog-Human Pairwise Methylome Similarity
Methylation values of orthologous CpGs were normalized by subtracting the mean and dividing by the standard deviation over individuals (i.e., z-transformed, separately for each species). The resulting z-values represent the tendency to decrease or increase relative to the mean of each CpG within a species. Using these values we calculated the pairwise Pearson correlation between the methylomes of each dog-human pair. Correlation was computed across all orthologous CpG values using the SciPy Python package (Virtanen et al., 2020), forming a 95 × 320 (dog × human) methylome similarity matrix (Figure 3). We also created a coarsened version of this matrix, in which the pairwise similarities were averaged over two-year age windows in both species, forming an 8 × 51 (dog × human) methylome similarity matrix (MSA, Figure S2A).
Given this matrix, we evaluated the significance of association between age and methylome similarity using permutations. Specifically, we generated the following two-by-two contingency table:
| Ages More Different AD(i,j)>AD | Ages More Similar AD(i,j)≤AD | |
|---|---|---|
| Methylomes more different MSA(i,j) ≤ MS | Count1 | Count3 |
| Methylomes more similar MSA(i,j)> MS | Count2 | Count4 |
where MSAis the methylome similarity matrix, AD(i, j) is the age difference computed as | Age bindog –Age binhuman | and Count is the number of occurrences (cells within the MS similarity matrix) for which the table row and column conditions are met. Using these counts, we calculated the p-value using the one tailed Fisher’s exact test and compared this p-value to that obtained when permuting the membership of dogs and humans in two-year age bins across 1000 permutations (Figure S2A).
k-nearest Neighbors Analysis
To achieve a robust assignment of reciprocal nearest neighbors, we used a strategy inspired by Context Likelihood of Relatedness (Madar et al., 2010). Specifically, we z-normalized the MS methylome similarity matrix to form MSZ, as follows:
k-nearest neighbors were assigned to each dog or to each human with respect to MSZ values. This process was implemented in Python using scikit-learn.
Fitting the Epigenetic Age Transfer Function
The nearest neighbor analysis was fit using non-linear regression with the SciPy package in Python. The model fit was specified using the following formula:
Here, “Dog age” was represented by the chronological ages of dogs, and “Human age” was the average age of the nearest human neighbors with respect to methylome similarity. The converse was performed as well, i.e., dog age was represented as the average age of the nearest dog neighbors and human age was the chronological age in humans. For the final age transfer function, the coefficients (A,B) were estimated by bootstrapping an equal number of both dogs and humans. The standard error was estimated using 1000 bootstraps.
Validation Using Mouse Methylomes
Dog-mouse methylome similarity was calculated identically as for dog-human comparisons. A k-nearest neighbors analysis (as described for dogs and humans above) was repeated using the orthologous CpGs for pairwise comparisons involving mice. The mouse methylome data had a highly canalized age distribution which was different from that of the dogs or humans in our study. That is, mice had been sampled at discrete ages, we therefore visualized these data according to 0.2 year bins (Figure 3F).
Conserved Methylation Changes in Orthologs
We considered 14,652 one-to-one orthologs in dogs, humans and mice that were within 2.5kb of orthologous CpGs. Among these, we identified 7,934 orthologous genes for which methylation values were available. Methylation values were then logit-transformed; multiple CpGs assigned to one gene were represented by the average methylation value. We assigned to each ortholog a ‘methylation conservation score’ using the following procedure. First, the age of each dog or mouse individual was translated to the equivalent human age using the epigenetic age translation functions built using the k-nearest neighbors analysis. We ranked all individuals according to their age in human years and divided this ranking into 15 quantile bins. Logit-transformed methylation values were averaged within each bin and species. For each gene and species we calculated the Spearman correlation between the gene’s methylation values and age. Genes were then ranked by sign(correlation) ∗ −log10(correlationp value) within each of the three species. We computed the Euclidean norm of the three ranks and sought genes with very low norms (for which methylation was consistently among the most increasing with age across species) or with very high norms (for which methylation was consistently among the most decreasing with age across species). Significance was determined using a two-sided empirical p-value < 0.05, yielding 394 genes.
We examined methylation within gene bodies by calculating the distance of each CpG relative to the transcription start site and normalized these distances by the gene length. We then grouped CpGs into 10 bins and calculated the average methylation using a rolling window (window = 3) among genes grouped according to their conservation status: conserved increasing or decreasing methylation with age and not conserved. We then calculated the difference between the oldest 20% and youngest 20% for each species. We repeated this analysis using all one-to-one orthologs or grouped according to their developmental gene status (Figure S4A). To test enrichment of genomic regions for conserved developmental genes, for each developmental gene, we annotated its underlying CpGs according to their genomic region (TSS, intergenic, 5′UTR, repeats, exonic, 3′UTR, intronic, CpG shelves, CpG shores, CpG islands). We then tested for enrichment using the Fisher’s exact test between conserved and not conserved developmental genes using a p-value cutoff of < 0.005 for each species (Figure S4B).
Network Analysis
We downloaded the PCNet parsimonious composite human functional interaction network from (Huang et al., 2018) and subselected gene orthologs with significantly conserved methylation trajectories (see above) resulting in a subnetwork with 355 nodes and 2003 edges. We visualized the network using Cytoscape (Shannon et al., 2003) (version 3.7) and performed community detection using clusterMaker2 (Shannon et al., 2003). To annotate modules, we performed functional enrichment using a hypergeometric test for each term within the Biological Process branch of the human Gene Ontology (GO) (Ashburner et al., 2000) and adjusted for false-discovery rate using a very strict Benjamini-Hochberg procedure (FDR <0.001) implemented using statsmodel in python. Significant GO terms were clustered according to gene-set similarity using Enrichment Map (Merico et al., 2011), and gene modules were clustered according to their Jaccard overlap, revealing high-level functional categories (Figure 4).
Developmental Genes Analysis
Genes were ranked according to their methylation conservation score (see above) and subdivided into 25 evenly spaced bins, separating genes with significantly conserved decreases or increases in methylation for a total of 27 bins. We then obtained PhyloP (Siepel et al., 2006) sequence conservation scores according to the orthologous CpGs assigned to each gene. Finally, we averaged the PhyloP scores in each methylation conservation score bin, estimating the 95% confidence interval by bootstrapping (Figure S5). We assessed the significance of the interaction between methylation conservation score and developmental gene status using ANOVA.
We restricted to orthologous CpGs profiled across dogs, humans and mice (6,906 CpGs) that were within 2.5kb of the gene bodies of all orthologous genes (‘all CpGs’). From this set, we identified CpGs near development genes (‘devCpGs’); we also controlled for the number of CpGs with 100 randomly-sampled subsets of CpGs that were equal in size from those not near developmental genes (‘not devCpGs’). We calculated the methylome similarity (as described above) based on these CpG subsets for pairwise comparisons of species (dog and human, dog and mouse). For each pairwise comparison (Species 1, Species 2), we identified the 5-nearest neighbors in Species 2 for each individual of Species 1, then binned the actual age of Species 1 into five discrete bins and calculated the average neighbor age for each bin with the 95% confidence interval estimated by bootstrapping (Figure S6).
Conserved Development Clock Analysis
Dog and mouse epigenetic clocks were built with Elastic net (scikit-learn in Python) using either methylome-wide CpG values (~90000 CpGs across both species) or 394 CpG values associated with developmental gene modules (Figure 5). We refer to the ages predicted from this model as “epigenetic ages”. Hyperparameters were tuned using five-fold cross validation in the dog data. Since some other clocks in the literature use ten-fold cross validation, we also tested the parameters selected using a ten-fold cross-validation procedure. We found an increase in median absolute error (MAE) in this case, thus we proceeded with fivefold. Performance of the final model was assessed by Spearman correlation of actual versus epigenetic age (output of the Elastic net model) for 11 dogs which had not been used for training, and for the control mice described above. As controls in Figures 5E and 5F, we built 100 control clocks using 100 randomly sampled sets of 394 CpGs that had been profiled in dogs and mouse but were not in developmental gene modules. For analysis involving lifespan-enhancing intervention mice, we obtained DNA methylation data profiled from whole blood from (Petkovich et al., 2017), processed as described above. We removed GHRKO from further analysis, as principal component analysis using the 394 conserved CpGs revealed clustering due to treatment. All remaining mice used in this analysis are described in Table S3. We applied the epigenetic clock, trained in mice or trained in dogs, and evaluated the effect of longevity-enhancing interventions using a log-likelihood ratio test.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological Samples | ||
| Labrador retrievers whole blood samples | UC Davis | N/A |
| Labrador retrievers whole blood samples | NHGRI | N/A |
| Critical Commercial Assays | ||
| Kapa HTP Library Prep Illumina | Roche | KK8234 |
| SeqCap Adapter kit A | Roche | 7141530001 |
| SeqCap Adapter kit B | Roche | 7141548001 |
| SeqCap Epi Developer S | Roche | 7139071001 |
| SeqCap Epi Developer Reagent | Roche | 6684335001 |
| SeqCap EZ HE-Oligo Kit A | Roche | 6777287001 |
| SeqCap EZ HE-Oligo Kit B | Roche | 6777317001 |
| VK SeqCap Epi Reagent Kit plus | Roche | 7185723001 |
| Seqcap EZ Pure Capture Bead Kit | Roche | 6977952001 |
| Zymo EZ DNA methylation | Zymo | 11-334 |
| Kapa Library qPCR quantification | Roche | KK4824 |
| Bioanalyzer (DNA 1000) | Agilent | 5067-1504 |
| Bionalyzer machine | Agilent | 2938C |
| Kapa HiFi HotStart Uracil Polymerase | Roche | KK2801 |
| Covaris microTUBES | Covaris | 520166 |
| Ampure beads | Beckman Coulter | A63881 |
| HiSeq 4000 Reagent kit | Illumina | PE-410-1001 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE146920 |
| Code and associated files | This paper | https://doi.org/10.5281/zenodo.3864683 |
| Dog genome (CanFam 3.1) | Yates et al., 2016 | http://dec2017.archive.ensembl.org/ |
| Mouse genome (mm10) | Yates et al., 2016 | http://dec2017.archive.ensembl.org/ |
| Human genome (hg19) | Yates et al., 2016 | http://dec2017.archive.ensembl.org/ |
| CpG Islands & repeat regions | Rosenbloom et al., 2015 | https://hgdownload.soe.ucsc.edu/downloads.html |
| Gene & ortholog annotations | Vilella et al., 2009 | http://dec2017.archive.ensembl.org/ |
| Mouse RRBS data | Petkovich et al., 2017 | GEO: GSE80672 |
| Human (children) 450K array | Alisch et al., 2012 | GEO: GSE36054 |
| Human (adults) 450K array. | Hannum et al., 2013 | GEO: GSE40279 |
| Dog and wolf RRBS | Thompson et al., 2017 | SRA: SRP065319 |
| Gene Ontology | Ashburner et al., 2000 | http://geneontology.org/docs/download-ontology/ |
| Software and Algorithms | ||
| LiftOver | Rosenbloom et al., 2015 | http://hgdownload.soe.ucsc.edu/downloads.html#utilities_downloads |
| FASTQC | N/A | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| TrimGalore (v0.4.5) | N/A | http://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ |
| Bismark (v0.14.3) | Krueger and Andrews, 2011 | http://www.bioinformatics.babraham.ac.uk/projects/bismark/ |
| MethylDackel | N/A | https://github.com/dpryan79/MethylDackel |
| Picard Tools (v1.141) | N/A | https://broadinstitute.github.io/picard/ |
| SAMtools | Li et al., 2009 | http://www.htslib.org/ |
| BEDtools (v2.25.0) | Quinlan and Hall, 2010 | https://bedtools.readthedocs.io/en/latest/ |
| fancyimpute | N/A | https://pypi.org/project/fancyimpute/ |
| Minfi (R) | Aryee et al., 2014 | https://bioconductor.org/packages/release/bioc/html/minfi.html |
| Impute (R) | Hastie et al., 2020 | https://www.bioconductor.org/packages/release/bioc/html/impute.html |
| Modified beta-mixture quantile dilation (BMIQ, R) | Horvath, 2013; Teschendorff et al., 2013 | PMID: 24138928; Additional File 24 |
| LOLA (R) | Sheffield and Bock, 2016 | https://bioconductor.org/packages/release/bioc/html/LOLA.html |
| preprocessCore (R) | Bolstad, 2013 | http://bioconductor.org/packages/release/bioc/html/preprocessCore.html |
| statsmodel (v0.8.0 Python) | Perktold et al. | http://www.statsmodels.org/stable/index.html |
| SciPy (v1.1.1 Python) | Virtanen et al., 2020 | https://www.scipy.org/ |
| scikit-learn (v0.19.2 Python) | Pedregosa et al., 2011 | https://scikit-learn.org/stable/ |
| Cytoscape (v3.7) | Shannon et al., 2003 | https://cytoscape.org/download.html |
| clusterMaker2 | Shannon et al., 2003 | http://apps.cytoscape.org/apps/clustermaker2 |
| Enrichment Map | Merico et al., 2011 | http://apps.cytoscape.org/apps/enrichmentmap |
Highlights.
Oligo-capture sequencing of methylomes from 104 Labradors, 0–16 years old
Methylome similarity translates dog years to human years logarithmically
Conserved age-related changes predominately impact developmental gene networks
Formulation of a conserved epigenetic clock transferable across mammals
ACKNOWLEDGMENTS
This work is supported by the following: the California Institute for Regenerative Medicine (CIRM Epigenome Center to T.I.), the National Institute for Environmental Health Sciences (T.I., ES014811), the National Institute of General Medical Sciences (A.R.C., GM108865 and T.W., GM008666), the National Institute on Aging (P.D.A., AG031862), the National Institute of Dental and Craniofacial Research (D.L.B., DE022532), the Maxine Adler Endowed Chair Funds (D.L.B.) and the Intramural Program of the National Human Genome Research Institute (A.H. and E.A.O.). We also gratefully acknowledge Sabrina and Michael Mojica for providing images that document the lifespan of their beloved Labrador, Cosmo
Footnotes
DECLARATION OF INTERESTS
T.I. is co-founder of Data4Cure Inc, is on the Scientific Advisory Board, and has an equity interest. T.I. is on the Scientific Advisory Board of Ideaya Biosciences, has an equity interest, and receives sponsored research funding. The terms of these arrangements have been reviewed and approved by the University of California San Diego in accordance with its conflict of interest policies. T.I. and T.W. hold a patent entitled “Methylome profiling of animals and uses thereof,” international patent application #PCT/US18/49103, US application #16/638,454.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.cels.2020.06.006.
REFERENCES
- Alisch RS, Barwick BG, Chopra P, Myrick LK, Satten GA, Conneely KN, and Warren ST (2012). Age-associated DNA methylation in pediatric populations. Genome Res. 22, 623–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S, et al. (2010). FastQC: a quality control tool for high throughput sequence data. [Google Scholar]
- Arias E, Heron M, and Xu J (2017). United States life tables, 2013. Natl Vital Stat. Rep. 66, 1–64. [PubMed] [Google Scholar]
- Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, and Irizarry RA (2014). Minfi: a flexible and comprehensive Bioconductor package for the analysis of infinium DNA methylation microarrays. Bioinformatics 30, 1363–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, et al. (2000). Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartges J, Boynton B, Vogt AH, Krauter E, Lambrecht K, Svec R, and Thompson S (2012). AAHA canine life stage guidelines. J. Am. Anim. Hosp. Assoc. 48, 1–11. [DOI] [PubMed] [Google Scholar]
- Bell GI, Karam JH, and Rutter WJ (1981). Polymorphic DNA region adjacent to the 5′ end of the human insulin gene. Proc. Natl. Acad. Sci. USA 78, 5759–5763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogin B, and Smith BH (1996). Evolution of the human life cycle. Am. J. Hum. Biol. 8, 703–716. [DOI] [PubMed] [Google Scholar]
- Bolstad BM (2013). ‘preprocessCore: a collection of pre-processing functions’, R package version, 1. https://github.com/bmbolstad/preprocessCore.
- Chatterjee A, Rodger EJ, Morison IM, Eccles MR, and Stockwell PA (2017). Tools and strategies for analysis of genome-wide and gene-specific DNA methylation patterns. Methods Mol. Biol. 1537, 249–277. [DOI] [PubMed] [Google Scholar]
- Chen B, Chen C, and Hsu WH (2015). Face recognition and retrieval using cross-age reference coding With cross-age celebrity dataset. IEEE Trans. Multimedia 17, 804–815. [Google Scholar]
- CIA (2013). The World Factbook 2013–14 (Central Intelligence Agency). [Google Scholar]
- Ciccarone F,Tagliatesta S, Caiafa P, and Zampieri M (2018). DNA methylation dynamics in aging: how far are we from understanding the mechanisms? Mech. Ageing Dev. 174, 3–17. [DOI] [PubMed] [Google Scholar]
- Dreger DL, Rimbault M, Davis BW, Bhatnagar A, Parker HG, and Ostrander EA (2016). Whole-genome sequence, SNP chips and pedigree structure: building demographic profiles in domestic dog breeds to optimize genetic-trait mapping. Dis. Models Mech. 9, 1445–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Field AE, Robertson NA, Wang T, Havas A, Ideker T, and Adams PD (2018). DNA methylation clocks in aging: categories, causes, and consequences. Mol. Cell 71, 882–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming JM, Creevy KE, and Promislow DEL (2011). Mortality in North American dogs from 1984 to 2004: an investigation into age-, size-, and breed-related causes of death. J. Vet. Intern. Med. 25, 187–198. [DOI] [PubMed] [Google Scholar]
- Gilmore KM, and Greer KA (2015). Why is the dog an ideal model for aging research? Exp. Gerontol. 71, 14–20. [DOI] [PubMed] [Google Scholar]
- Greeley EH, Ballam JM, Harrison JM, Kealy RD, Lawler DF, and Segre M (2001). The influence of age and gender on the immune system: a longitudinal study in Labrador Retriever dogs. Vet. Immunol. Immunopathol. 82, 57–71. [DOI] [PubMed] [Google Scholar]
- Gross AM, Jaeger PA, Kreisberg JF, Licon K, Jepsen KL, Khosroheidari M, Morsey BM, Swindells S, Shen H, Ng CT, et al. (2016). Methylome-wide analysis of chronic HIV infection reveals five-year increase in biological age and epigenetic targeting of HLA. Mol. Cell 62, 157–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hannum G, Guinney J, Zhao L, Zhang L, Hughes G, Sadda S, Klotzle B, Bibikova M, Fan JB, Gao Y, et al. (2013). Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol. Cell 49, 359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hastie T, Tibshirani R, Narasimhan B, and Chu G (2020). impute: impute: Imputation for microarray data. R package version 1.62.0. https://www.bioconductor.org/packages/release/bioc/html/impute.html.
- Horvath S (2013). DNA methylation age of human tissues and cell types. Genome Biol. 14, R115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath S, and Raj K (2018). DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat. Rev. Genet. 19, 371–384. [DOI] [PubMed] [Google Scholar]
- Huang JK, Carlin DE, Yu MK, Zhang W, Kreisberg JF, Tamayo P, and Ideker T (2018). Systematic evaluation of molecular networks for discovery of disease genes. Cell Syst. 6, 484–495.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue M, Hasegawa A, Hosoi Y, and Sugiura K (2015). A current life table and causes of death for insured dogs in Japan. Prev. Vet. Med. 120, 210–218. [DOI] [PubMed] [Google Scholar]
- Jaffe AE, and Irizarry RA (2014). Accounting for cellular heterogeneity is critical in epigenome-wide association studies. Genome Biol. 15, R31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeberlein M, Creevy KE, and Promislow DEL (2016). The dog aging project: translational geroscience in companion animals. Mamm. Genome 27, 279–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan SS, Singer BD, and Vaughan DE (2017). Molecular and physiological manifestations and measurement of aging in humans. Aging Cell 16, 624–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowald A, and Kirkwood TBL (2016). Can aging be programmed? A critical literature review. Aging Cell 15, 986–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, et al. ; SciPy 1.0 Contributors (2020). SciPy 1.0: Fundamental Algorithms for Scientific Computing in Python. Nat. Methods 17, 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F (2015). https://github.com/FelixKrueger/TrimGalore. [Google Scholar]
- Krueger F, and Andrews SR (2011). Bismark: a flexible aligner and methylation caller for bisulfite-Seq applications. Bioinformatics 27, 1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebeau A (1953). L’âge du chien et celui de l’homme Essai de statistique sur la mortalité canine. Bul. de l’Ac. Vét. de France 26, 229–232. [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe R, Barton C, Jenkins CA, Ernst C, Forman O, Fernandez-Twinn DS, Bock C, Rossiter SJ, Faulkes CG, Ozanne SE, et al. (2018). Ageing-associated DNA methylation dynamics are a molecular readout of lifespan variation among mammalian species. Genome Biol. 19, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madar A, Greenfield A, Vanden-Eijnden E, and Bonneau R (2010). DREAM3: network inference using dynamic context likelihood of relatedness and the inferelator. PLoS One 5, e9803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, Hinkal G, Kim HS, Shen L, Zhang L, Zhang J, Zhang N, Liang S, Donehower LA, and Issa JP (2010). Widespread and tissue specific age-related DNA methylation changes in mice. Genome Res. 20, 332–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maegawa S, Lu Y, Tahara T, Lee JT, Madzo J, Liang S, Jelinek J, Colman RJ, and Issa JJ (2017). Caloric restriction delays age-related methylation drift. Nat. Commun. 8, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merico D, Isserlin R, and Bader GD (2011). Visualizing gene-set enrichment results using the cytoscape plug-in enrichment map. Methods Mol. Biol. 781, 257–277. [DOI] [PubMed] [Google Scholar]
- Miller RA, and Nadon NL (2000). Principles of animal use for gerontological research. J. Gerontol. A Biol. Sci. Med. Sci. 55, B117–B123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostrander EA, Wayne RK, Freedman AH, and Davis BW (2017). Demographic history, selection and functional diversity of the canine genome. Nat. Rev. Genet. 18, 705–720. [DOI] [PubMed] [Google Scholar]
- Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Blondel M, Prettenhofer P, Weiss R, Dubourg V, Vanderplas JT, et al. (2011). Scikit-learn: machine learning in Python. Journal of machine learning research 12, 2825–2830. [Google Scholar]
- Petkovich DA, Podolskiy DI, Lobanov AV, Lee SG, Miller RA, and Gladyshev VN (2017). Using DNA methylation profiling to evaluate biological age and longevity interventions. Cell Metab. 25, 954–960.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2018). R: a language and environment for statistical computing (R Foundation for Statistical Computing). https://www.R-project.org.
- Rakyan VK, Down TA, Maslau S, Andrew T, Yang TP, Beyan H, Whittaker P, McCann OT, Finer S, Valdes AM, et al. (2010). Human aging-associated DNA hypermethylation occurs preferentially at bivalent chromatin domains. Genome Res. 20, 434–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom KR, Armstrong J, Barber GP, Casper J, Clawson H, Diekhans M, Dreszer TR, Fujita PA, Guruvadoo L, Haeussler M, et al. (2015). The UCSC Genome Browser database: 2015 update. Nucleic Acids Res. 43, D670–D681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan D (2017). MethylDackel: a (mostly) universal methylation extractor for BS-seq experiments, 2017. https://github.com/dpryan79/MethylDackel.
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheffield NC, and Bock C (2016). LOLA: enrichment analysis for genomic region sets and regulatory elements in R and bioconductor. Bioinformatics 32, 587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Pollard KS, and Haussler D (2006). New methods for detecting lineage-specific selection. In Lecture Notes in Computer Science (Springer), pp.190–205. [Google Scholar]
- Stubbs TM, Bonder MJ, Stark AK, Krueger F, BI Ageing Clock Team, von Meyenn F, Stegle O, and Reik W. (2017). Multi-tissue DNA methylation age predictor in mouse. Genome Biol. 18, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teschendorff AE, Marabita F, Lechner M, Bartlett T, Tegner J, Gomez-Cabrero D, and Beck S (2013). A beta-mixture quantile normalization method for correcting probe design bias in Illumina Infinium 450 k DNA methylation data. Bioinformatics 29, 189–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson MJ, vonHoldt B, Horvath S, and Pellegrini M (2017). An epigenetic aging clock for dogs and wolves. Aging 9, 1055–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toolkit Picard (2019). Broad Institute, GitHub Repository. http://broadinstitute.github.io/picard/. [Google Scholar]
- Urfer SR, Kaeberlein TL, Mailheau S, Bergman PJ, Creevy KE, Promislow DEL, and Kaeberlein M (2017). A randomized controlled trial to establish effects of short-term rapamycin treatment in 24 middle-aged companion dogs. GeroScience 39, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilella AJ, Severin J, Ureta-Vidal A, Heng L, Durbin R, and Birney E (2009). EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 19, 327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vonholdt BM, Pollinger JP, Lohmueller KE, Han E, Parker HG, Quignon P, Degenhardt JD, Boyko AR, Earl DA, Auton A, et al. (2010). Genome-wide SNP and haplotype analyses reveal a rich history underlying dog domestication. Nature 464, 898–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, and Lemos B (2019). Ribosomal DNA harbors an evolutionarily conserved clock of biological aging. Genome Res. 29, 325–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Tsui B, Kreisberg JF, Robertson NA, Gross AM, Yu MK, Carter H, Brown-Borg HM, Adams PD, and Ideker T (2017). Epigenetic aging signatures in mice livers are slowed by dwarfism, calorie restriction and rapamycin treatment. Genome Biol. 18, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Withers PC (1992). Comparative Animal Physiology (Cole Publishing Company). https://books.google.com/books/about/Comparative_Animal_Physiology.html?hl=&id=Ba_wAAAAMAAJ.
- Xie W, Schultz MD, Lister R, Hou Z, Rajagopal N, Ray P, Whitaker JW, Tian S, Hawkins RD, Leung D, et al. (2013). Epigenomic analysis of multilineage differentiation of human embryonic stem cells. Cell 153, 1134–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates A, Akanni W, Amode MR, Barrell D, Billis K, Carvalho-Silva D, Cummins C, Clapham P, Fitzgerald S, Gil L, et al. (2016). Ensembl 2016. Nucleic Acids Res. 44, D710–D716. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing reads and processed files generated during this study are available at GEO (GSE146920). All original code is freely available for download at 10.5281/zenodo.3864683.