

Abstract
Acute myeloid leukemia (AML) results from the enhanced proliferation and impaired differentiation of hematopoietic stem and progenitor cells. Using an ex vivo functional screening assay, we identified that the combination of the BTK inhibitor ibrutinib and BCL2 inhibitor venetoclax (IBR+VEN), currently in clinical trials for chronic lymphocytic leukemia (CLL), demonstrated enhanced efficacy on primary AML patient specimens, AML cell lines, and in a mouse xenograft model of AML. Expanded analyses among a large cohort of hematologic malignancies (n=651 patients) revealed that IBR+VEN sensitivity associated with select genetic and phenotypic features in both CLL and AML specimens. Among AML samples, 11q23 MLL-rearrangements were highly sensitive to IBR+VEN. Analysis of differentially-expressed genes with respect to IBR+VEN sensitivity indicated pathways preferentially enriched in patient samples with reduced ex vivo sensitivity, including IL-10 signaling. These findings suggest IBR+VEN may represent an effective therapeutic option for patients with AML.
Introduction
The identification of effective therapies based on targeted interventions for human cancers faces the challenges of genetic and epigenetic heterogeneity underlying the disease. Large-scale sequencing efforts have uncovered a spectrum of mutations in many hematologic malignancies, suggesting that combinations of agents will be required to treat these diseases effectively. For patients with acute myeloid leukemia (AML), the long-standing frontline chemotherapy consisting of cytarabine and anthracyclines, has a 5-year overall survival rate of 25%1. Outcomes in older patients, who represent the majority of patients with this disease, are poor with a median survival of 5 to 10 months. Due to their inability to tolerate intensive chemotherapy, many older patients do not receive any anti-leukemic therapy2.
Although molecularly targeted drugs offer substantial promise as treatment options, the effectiveness of individual inhibitors has been limited by resistance mutations and activation of compensatory signaling pathways. Resistance to targeted agents in AML is further complicated by substantial disease heterogeneity and rescue signals from the microenvironment, underscoring the need for combinations of targeted therapies to achieve durable responses.
Recent reports demonstrating the efficacy of combinations of oral, targeted drugs for several adult leukemias establish their potential for improved and durable clinical responses, and shift treatment options away from cytotoxic chemotherapy. The BCL2 inhibitor, venetoclax, has demonstrated single-agent efficacy in patients with AML3, and the combination of venetoclax plus a hypomethylating agent was recently approved as a therapeutic strategy for elderly, treatment-naïve AML patients4, 5. However, responses are of short duration, justifying exploration of additional therapeutic strategies. For chronic lymphocytic leukemia (CLL) and mantle cell lymphoma (MCL), combinations of the BTK inhibitor ibrutinib with the BCL2 inhibitor venetoclax have shown dramatic response in patients with high rates of minimal residual disease negativity6–9. Both ibrutinib and venetoclax as single agents are highly effective in CLL, though their primary mechanisms of action are different; while both drugs induce direct killing, ibrutinib also induces CLL cell egress from the nurturing lymph node microenvironment resulting in redistribution into the peripheral blood10. Tumor cells isolated from patients receiving ibrutinib for mantle cell lymphoma also show venetoclax sensitivity11. Notably, the effectiveness of this combination is anticipated from prior studies showing tumor cells isolated from CLL patients on ibrutinib monotherapy are highly sensitive to venetoclax12, 13.
Efforts to identify new targeted combinations for AML have been aided by the use of ex vivo functional screening of primary patient leukemia cells14. Using this approach, the recent publication of functional and genomic data for a large cohort of AML patient samples described a significant association between samples with FLT3-ITD, NPM1 and DNMT3A mutations and sensitivity to ibrutinib15. This observation prompted us to test the combination of ibrutinib with venetoclax on AML patient samples, given the emerging success of this combination in CLL.
Methods
Patient samples
All patients gave informed consent to participate in this study, which had the approval and guidance of the Institutional Review Boards (IRB) at Oregon Health & Science University (OHSU), University of Utah, University of Texas Medical Center Southwestern, Stanford University, University of Miami, University of Colorado, University of Florida, National Institutes of Health, Fox Chase Cancer Center and University of Kansas. Samples were sent to the coordinating center (OHSU IRB 9570 and 4422), where they were coded and processed. Primary bone marrow aspirates or peripheral blood draws from 651 unique patients with hematologic malignancies were collected and classified according to five general diagnostic groups: AML (n=325), CLL (n=152), acute lymphoblastic leukemia (ALL; n=100), chronic myeloid leukemia (CML; n=27), and myeloproliferative neoplasms or myelodysplastic syndromes (MPN or MDS/MPN; n=47). Samples were assayed for drug sensitivity within 24 hours of receipt. All samples were analyzed for clinical characteristics, with expanded, disease-specific panels of clinical, prognostic, genetic, cytogenetic, and surface antigen characteristics obtained from AML and CLL patient electronic medical records. Genetic characterization of AML samples included results of a clinical deep-sequencing panel of genes commonly mutated in hematologic malignancies.
Cell lines
Human AML patient-derived MOLM14 (FLT3-ITD positive), HL-60, and GDM1 cells were obtained from DSMZ and maintained in RPMI1640 media supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin/100 μg/mL streptomycin, and 2 mmol/L l-glutamine (R10) at 37°C in 5% CO2. Cells were kept in culture no longer than a month at a time, and all cell lines were authenticated by extensive functional and genetic analysis in our lab.
Ex vivo functional screen
Small-molecule inhibitors, purchased from LC Laboratories (Woburn, MA, USA) and Selleck Chemicals (Houston, TX, USA), were reconstituted in DMSO and stored at −80°C. Inhibitors were distributed into 384-well plates prepared with single agents in a 7-point concentration series (10 uM to 0.0137 uM) and the combination of ibrutinib and venetoclax in a 7-point equimolar ratio concentration series identical to those used for single agents. The final concentration of DMSO was ≤0.1% in all wells, and all plates were stored at −20°C and thawed immediately prior to use. Primary mononuclear cells freshly isolated by Ficoll-gradient centrifugation were seeded into 384-well assay plates at 10,000 cells/well in RPMI-1640 media supplemented with fetal bovine serum (10%), L-glutamine, penicillin-streptomycin and b-mercaptoethanol (10−4 M). After three days of culture at 37°C in 5% CO2, methanethiosulfonate (MTS) reagent (CellTiter 96 AQueous One; Promega Madison, WI, USA) was added to each well and absorbance was measured at 490 nm.
Inhibitor dose-response curve analysis and effect-measure calculations
Raw absorbance values were adjusted to a reference blank value (average of positive-control wells containing a drug combination of flavopiridol, staurosporine and velcade), normalized to untreated control wells, and bounded at 0 and 100 to produce cell viability percentages. Normalized viability percentages16 at each dose of single agent or combination 7-point dilution series were analyzed for all patient samples that passed a quality control inspection (based on plate- and profile-specific expectations of drug-induced cell inhibition). A two-parameter probit regression curve was fit to each 7-point log10-transformed dose-response profile using maximum likelihood estimation for the intercept and slope. This parametric model was chosen over a polynomial because the probit’s monotonic shape reflects a dose-response curve typically seen in samples incubated with cytotoxic or inhibitory agents17. From the fitted probit curve for each sample-drug pairing, the half maximum inhibitory concentration (IC50) was defined as the lowest concentration to achieve 50% predicted viability and the area under the curve (AUC) was computed by integration of the curve height across the tested dose range. If the predicted cell viability (i.e., probit curve height) was ≤ 50% at the lowest tested dose or > 50% across the entire dose range, the IC50 was designated as the lowest dose or highest dose, respectively. For sensitivity profiles with 100% normalized viability at all 7 dose points, the IC50 and AUC were designated as the highest tested dose and the maximum possible AUC, respectively. For sensitivity profiles with 0% viability at all 7 dose points, the IC50 and AUC were designated as the lowest tested dose and a value (0.01) just below the minimum probit-derived AUC, respectively.
Mouse xenograft model
NSG mice were purchased from JAX Labs as 5 week old, females and allowed to acclimate form one week prior to the study. MOLM13 cells were injected into tail veins NSG mice (3×105 cells/mouse) and allowed to engraft for 48 hours. Thereafter, mice were treated daily by oral gavage with vehicle, venetoclax (25 mg/kg), ibrutinib (25 mg/kg), or the IBR+VEN combination (5 mice/treatment group) administered sequentially; i.e., venetoclax (25 mg/kg) followed 2 hours later with ibrutinib (25 mg/kg)). On day 17 (15 days of treatment), animals were euthanized and assessed for disease burden. A sample size of 5 animals in the vehicle and 5 animals in each of the drug arms provides >90% power for a comparison of each drug vs. vehicle, and 80% power for comparison of single agent vs. combination with 5% significance level. This study was approved by the Oregon Health & Science University IACUC.
Gene expression and pathway enrichment analysis
A differential expression (DE) analysis pipeline was applied to AML patient specimens with ex vivo IBR+VEN sensitivity data and RNA sequencing data available (collected under the Beat AML study; 15). Genes were removed for having >1 HGNC symbol or read counts <10 in at least 90% of samples. Specimens whose median expression value (across all non-filtered genes) was <2 standard deviations below the mean (for this AML sample set) were removed from consideration and the earliest specimen was chosen for patients with multiples. Next, “sensitive” and “resistant” sample groups were identified as the lowest 25% and highest 25%, respectively, of specimens according to combination drug AUC. To account for differences in library size, GC content, and gene length, conditional quantile normalization (CQN)18 was applied to the remaining gene-by-sample matrix of read counts to generate (i) log2 normalized RPKM values for correlation analysis and visual clustering and (ii) normalization factors for DE analysis using the DESeq2 method19. Latent variables representing potential batch effects were identified by count-based surrogate variable analysis20, 21 and entered as covariates into the DESeq2 model to produce gene expression fold-changes (resistant group / sensitive group) and associated Wald test false discovery rate (FDR)-adjusted p-values22. Genes with adjusted p-values <0.01 were considered DE. For pathway topology analysis, reaction-based Reactome pathways23, 24 were converted to gene/protein networks using the “graphite” R/Bioconductor package25 and, after mapping DESeq2 log2 fold changes from Ensembl IDs to Uniprot IDs, tested for significant enrichment of DE gene products using the Pathway Regulation Score (PRS) method26. The PRS algorithm, as implemented by the “ToPASeq” R package, returns normalized (by pathway size) enrichment scores and FDR-adjusted p-values, calculated from null distributions generated by repeatedly permuting the gene labels on the list of fold-changes.
IGHV Mutation status
Following RNA and cDNA isolation, IGHV mutations were analyzed using the IGH Somatic Hypermutation Assay v2.0 (Invivoscribe). Briefly, PCR was performed using the supplied master mixes and Amplitaq Gold DNA polymerase (Applied Biosystems) on a Veriti Thermal Cycler Model #9902 (Applied Biosystems) to amplify the IGH sequence fragment between the leader (VHL) and joining (J) regions as per the manufacturer’s instructions. To confirm amplification of a single clonal product in the expected size range, an aliquot of each sample was run on a 1.5% agarose gel and separated by gel electrophoresis. Samples were then submitted for DNA sequencing using the supplied sequencing primers. The NCBI IgBLAST tool was used to determine the % divergence of each clonal sequence. Samples which showed less than 2% divergence from germline sequence were deemed to have unmutated IGHV.
Assessment of plasma cytokine levels
Plasma isolated and banked from patient samples at the time of ex vivo assay evaluation was assayed for a panel of human inflammatory cytokines using the Luminex platform (R&D Systems) according to the manufacturer’s protocol. Cytokine values were normalized to total protein levels for each sample.
Statistical analysis
IBR+VEN efficacy was compared for all samples (n=651) across a panel of general clinical and disease-specific variables. Subgroups for all mutations, cytogenetic abnormalities, and cell surface antigens were defined as either positive or negative. Combination sensitivity (as measured by % of maximum AUC) for categorical variables was compared using a Mann-Whitney or Kruskal-Wallis test. Comparisons of matched single-agent and combination treatment sensitivity were performed using Friedman test with Dunn’s post-hoc pairwise comparisons. Correlations between continuous clinical variables (e.g. WBC count) and drug sensitivity values were evaluated with Spearman rank correlation coefficients. In vivo response data were compared between treatment groups using a one-way ANOVA test with Bejamini-Hochberg adjustment of p-values for pairwise group comparisons. Plasma cytokine data were compared between IBR+VEN sensitive and resistant samples using a Mann-Whitney test.
Results
AML cells are highly sensitive to ibrutinib combined with venetoclax (IBR+VEN)
Ex vivo screening of primary cells from patients with various hematologic malignancies (Supplemental File S1) including AML (n=325), CLL (n=152), ALL (n=100), MPN or MDS/MPN (n=47), and CML (n=27) revealed several interesting patterns of sensitivity. Sensitivities to the IBR+VEN combination are represented in the radar plot depicting IC50 values across the 5 diagnostic groups (Figure 1A). Consistent with clinical data and previous literature, the IBR+VEN combination was highly effective in CLL specimens (median IC50=0.015 μM). Intriguingly, IBR+VEN also showed similar effectiveness on ALL and AML primary samples (median IC50: 0.018 and 0.054 μM, respectively). By contrast, samples from patients with CML or MDS/MPN were markedly less sensitive to the combination, with median IC50 values near or above 1 μM.
Figure 1. Sensitivity of Ibrutinib + Venetoclax combination on 651 unique leukemia patient samples.
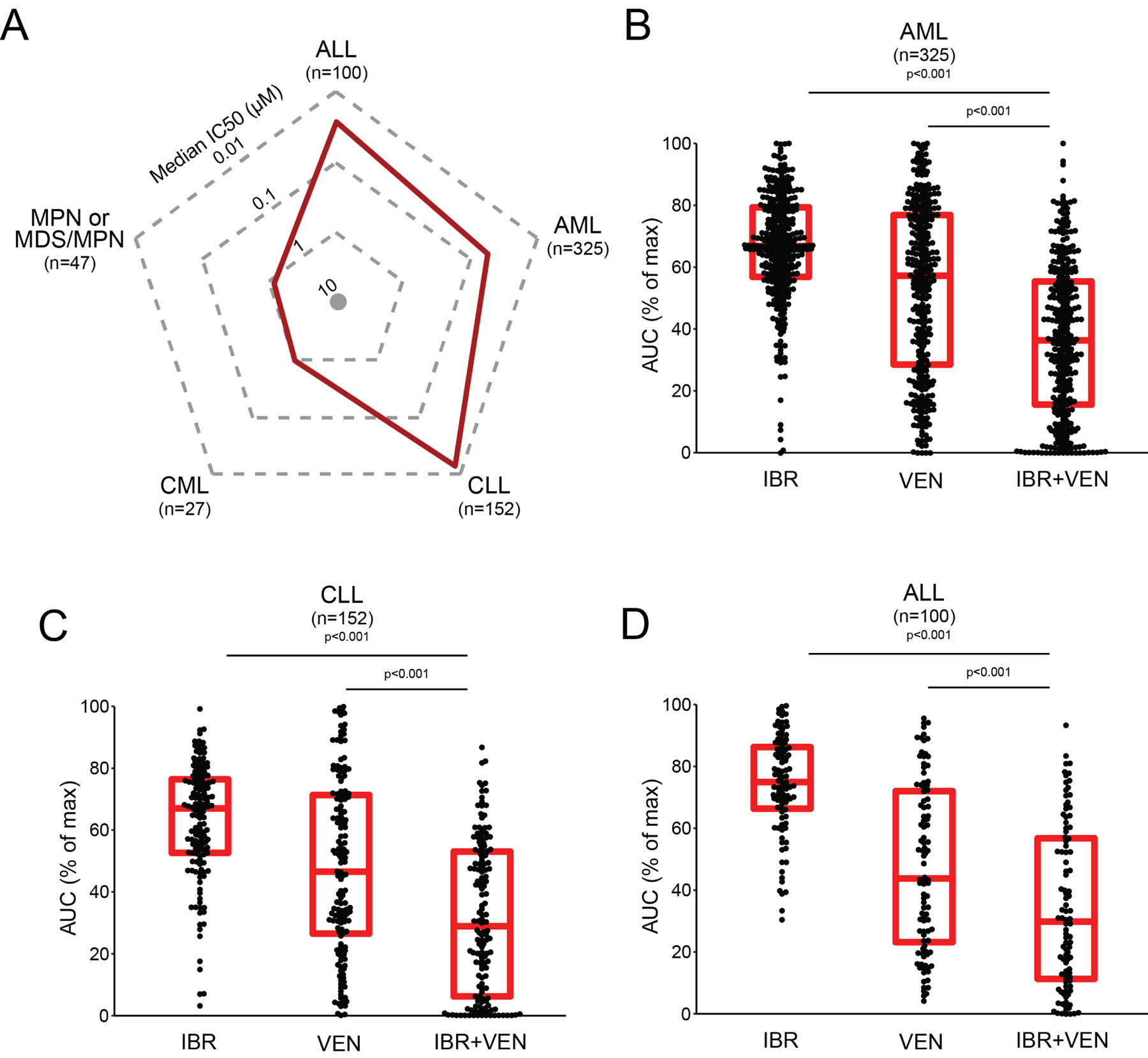
A. Radar plot indicating median IC50 values (red line) for 651 unique patient samples across 5 leukemia subgroups. B-D. Comparisons of Ibrutinib (IBR) and Venetoclax (VEN) sensitivities alone and in combination for AML, CLL and ALL subgroups. Red horizontal bars indicate median and interquartile range for % of maximum AUC. Comparisons were performed with Nemenyi test for the combination to each single agent.
Among the three sensitive diagnostic subgroups, in each case the IBR+VEN combination demonstrated superior efficacy compared to either single agent by two different effect measures (IC50 and AUC; Figure 1B–D). For example, IBR+VEN was 38- to 118-fold more potent by AUC across AML patient specimens compared with ibrutinib and venetoclax alone, respectively (adjusted p<0.001).
We also tested this combination on primary cells from two healthy donors to evaluate potential broader toxicity. In contrast to our findings with AML patient specimens, healthy donor mononuclear cells showed little to no sensitivity to IBR+VEN (Figure 2A). To validate our AML findings and to establish whether the efficacy of the IBR+VEN combination represents a synergistic relationship, the combination was tested for sensitivity on the human AML cell lines MOLM14, HL-60 and GDM1 using a dose matrix, which included all possible concentration pairings for each drug’s 7-point dose series. MOLM14 cells were modestly sensitive to both ibrutinib and venetoclax as single agents, but demonstrated enhanced efficacy when used in combination (Figure 2B and Supplemental Figure S1). Synergy scores were calculated using the zero interaction potency (ZIP) model for each dose pair of the 7×7 matrix; a positive score indicates synergy relative to the expected cell inhibition when assuming no interaction27. By this method, the IBR+VEN combination showed synergy in MOLM14, HL60, and GDM1 cells across the surveyed dose matrix (average ZIP score: +10.4, +3.5, and +20.1, respectively; Figure 2C and Supplemental Figure S1).
Figure 2. Ibrutinib + Venetoclax combination is potent and synergistic in AML cells.
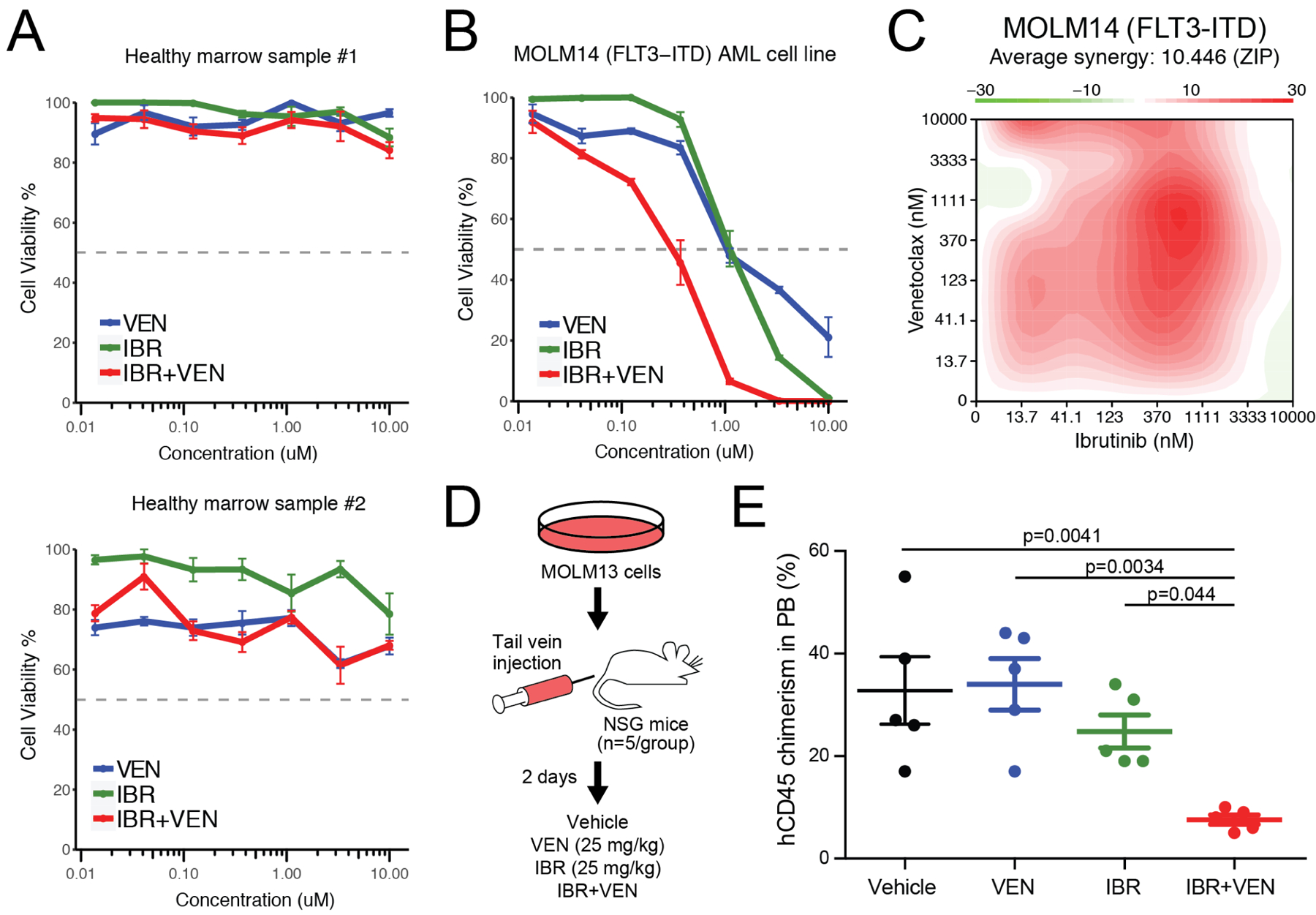
A. Evaluation of IBR and VEN alone and in combination on two healthy donor bone marrow samples. B. Evaluation of IBR and VEN alone and in combination on MOLM14 AML cells. C. Surface plot of IBR+VEN synergy (ZIP score) generated from a 7×7 dose matrix on MOLM14 AML cells. Synergy was calculated with R_SynergyFinder. D. Schematic of in vivo xenograft study. E. Levels of human CD45-positive cells in peripheral blood of NSG mice injected with MOLM13 cells and treated with vehicle, venetoclax (25 mk/kg), ibrutinib (25 mg/kg) or the combination IBR+VEN (25 mg/kg each). FDR-adjusted p-values are indicated for comparisons with the combination.
The IBR+VEN combination was also found to be effective in vivo, where it reduced tumor burden relative to either single agent as assessed by a decrease in human CD45-positive cells in peripheral blood of NSG mice injected with MOLM13 cells (Figure 2D&E). IBR+VEN also reduced spleen weight relative to either single agent, whereas neither hemoglobin levels nor platelet counts were altered across the treatment groups (Supplemental Figure S2). Together, these findings suggest IBR+VEN is well tolerated and demonstrates enhanced efficacy in an in vivo AML xenograft model.
Genetic and clinical features associate with differential sensitivity to IBR+VEN ex vivo
To identify associations between relevant patient characteristics and combination sensitivity ex vivo, IBR+VEN efficacy (as measured by AUC) was broken down according to general clinical and disease-specific features for CLL and AML, our two largest diagnosis categories. For CLL samples, del(11q) was associated with increased sensitivity to IBR+VEN (p=0.008), whereas samples with mutated IGHV had reduced sensitivity (p=0.040) (Figure 3A). Additionally, higher counts for WBCs and lymphocytes were correlated with increased sensitivity to the combination (Spearman r: −0.39 and −0.45, respectively; p<0.001). Conversely, and consistent with the latter, samples with higher percentages of monocytes were less sensitive (Spearman r: 0.35; p<0.001) (Figure 3B).
Figure 3. Select clinical and genetic features in CLL samples associate with differential sensitivity ex vivo to Ibrutinib + Venetoclax.
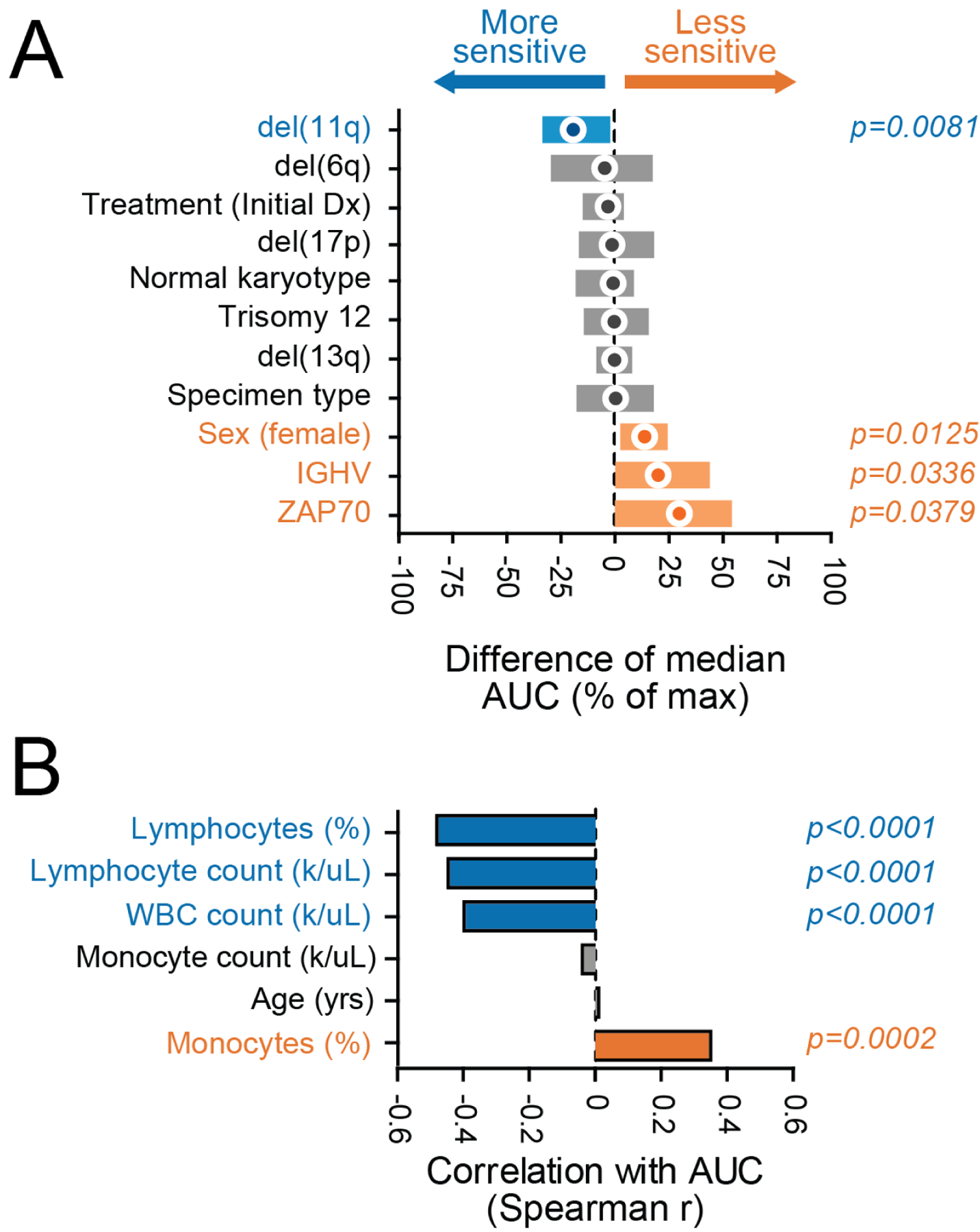
Comparisons of IBR+VEN sensitivities (% of max AUC) with respect to categorical variables (panel A) and continuous variables (panel B). Categorical variables were compared by Mann-Whitney test; circles indicate difference of median AUC and bars indicate 95% confidence interval. Continuous variables were correlated by Spearman rank test.
Our AML cohort (n=325) included patients with a wide range of disease subtypes representing both newly diagnosed and relapsed/refractory status. Among AML specimens, increased sensitivity to the combination of IBR+VEN was significantly associated with MLL-rearrangement (t(v;11)(v;q23); p=0.035), PML-RARA translocation (t(15;17); p=0.019), FLT3-ITD (p<0.001), and NPM1 mutations (p<0.001) (Figure 4A). Other clinical/genetic features including inv(16), prior MDS, loss of chromosome 7, and RUNX1-RUNXT1 (t(8;21)) translocations were associated with reduced sensitivity to the combination (range of p-values: <0.001 to 0.026). Moreover, higher percentages of bone marrow and peripheral blasts were correlated with increased sensitivity to the combination (Spearman r: −0.39 and −0.37, respectively; p<0.001) (Figure 4B). ; a similar trend has been reported fror venetoclax as a single agent.28
Figure 4. Genetic abnormalities in AML samples associate with differential sensitivity ex vivo to Ibrutinib + Venetoclax.
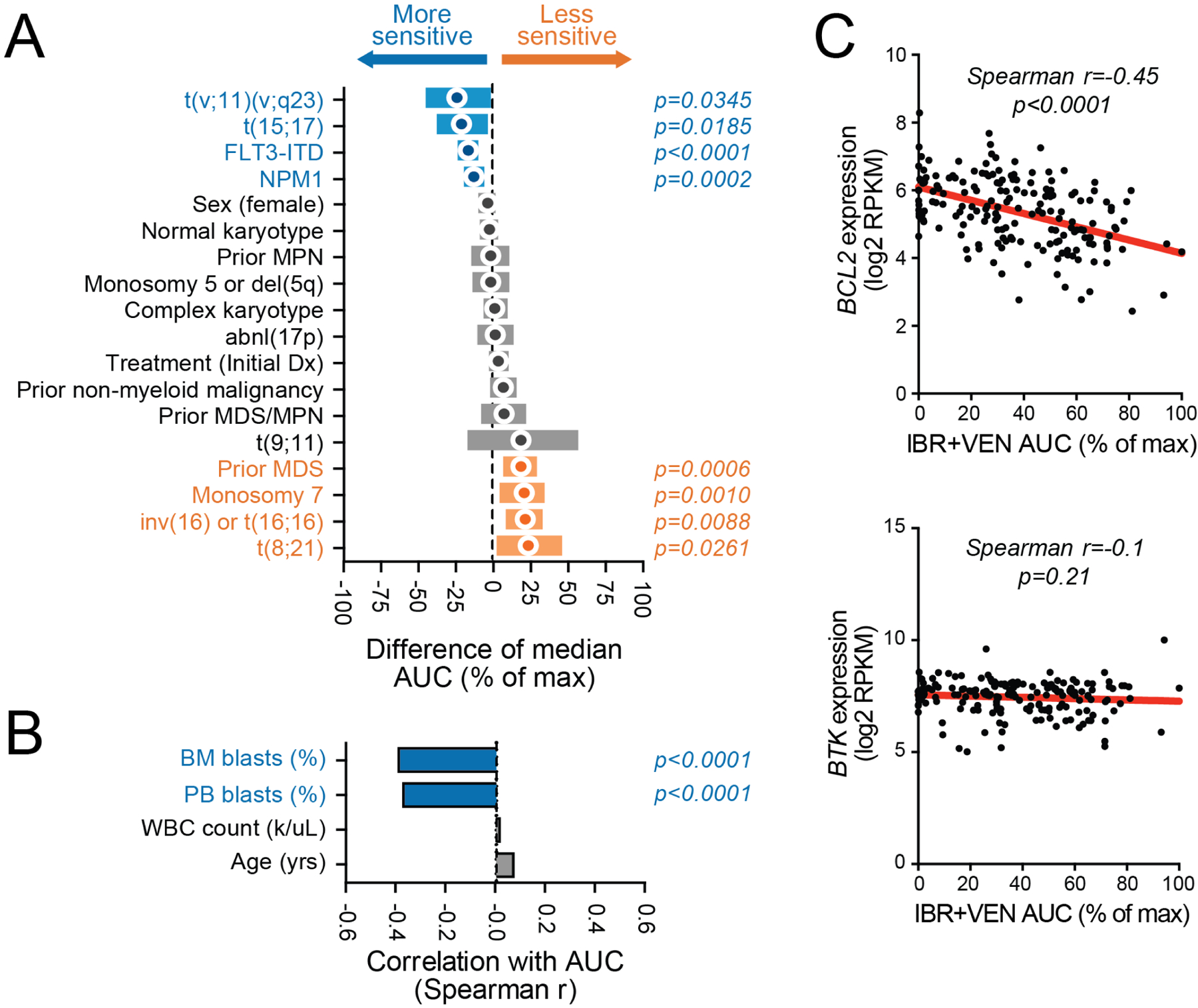
Comparisons of IBR+VEN sensitivities (% of max AUC) with respect to categorical variables (panel A) and continuous variables (panel B). Categorical variables were compared by Mann-Whitney test; circles indicate difference of median AUC and bars indicate 95% confidence interval. Continuous variables were correlated by Spearman rank test. C. Scatter plots of IBR+VEN AUC with expression levels of their respective canonical drug target: BTK and BCL2.
IBR+VEN sensitivity relative to the expression of their respective canonical targets
Expression levels (normalized RPKM) of BTK and BCL2, the respective canonical targets of ibrutinib and venetoclax, were compared with sensitivity to the combination for a subset of the AML cohort (56%; 181/325) with available RNA-seq data (Figure 4C). Higher expression levels of BCL2 correlated with sensitivity to the combination as evaluated by AUC (Spearman r=−0.46; p<0.001) whereas expression levels of BTK were not significantly correlated with sensitivity to the combination (Spearman r=−0.10; p=0.191).
Distinct patterns of differentially expressed genes associate with IBR+VEN sensitivity
DE analysis of the 25% most sensitive (AUC (% of max) ≤18.5; n=45) versus the 25% most resistant (AUC (% of max) >55.5; n=45) AML samples to IBR+VEN yielded 7769 genes with an FDR-adjusted p-value < 0.01 (DESeq2 method; Supplemental File S2). Unsupervised hierarchical clustering of the top 1000 most differentially-expressed genes revealed marked differences between sensitive and resistant samples (Figure 5A). Analysis across these two samples sets indicated higher BCL2 expression levels associated with the IBR+VEN sensitive samples (FDR-adjusted p=1.18E-10). Within the BCL2 family we and others28, 29 observed higher levels of BCL2A1 associated with IBR+VEN resistant samples (FDR-adjusted p=2.55E-16) (Figure 5B). Consistent with a lack of correlation between BTK expression levels and IBR+VEN sensitivity (Figure 4C), BTK was not differentially expressed between IBR+VEN sensitive and resistant samples. However, among other reported kinase targets of ibrutinib featuring the conserved cysteine aligning with C481 in BTK30, two other TEC family members (TEC, TXK; Figure 5B) and two ERBB family kinases (EGFR, ERBB2; Supplemental Figure S3) showed significantly elevated expression levels in IBR+VEN sensitive samples (FDR-adjusted p value range: 2.56E-4 to 2.67E-7).
Figure 5. Patterns of differentially-expressed genes associate with sensitivity or resistance to IBR+VEN.
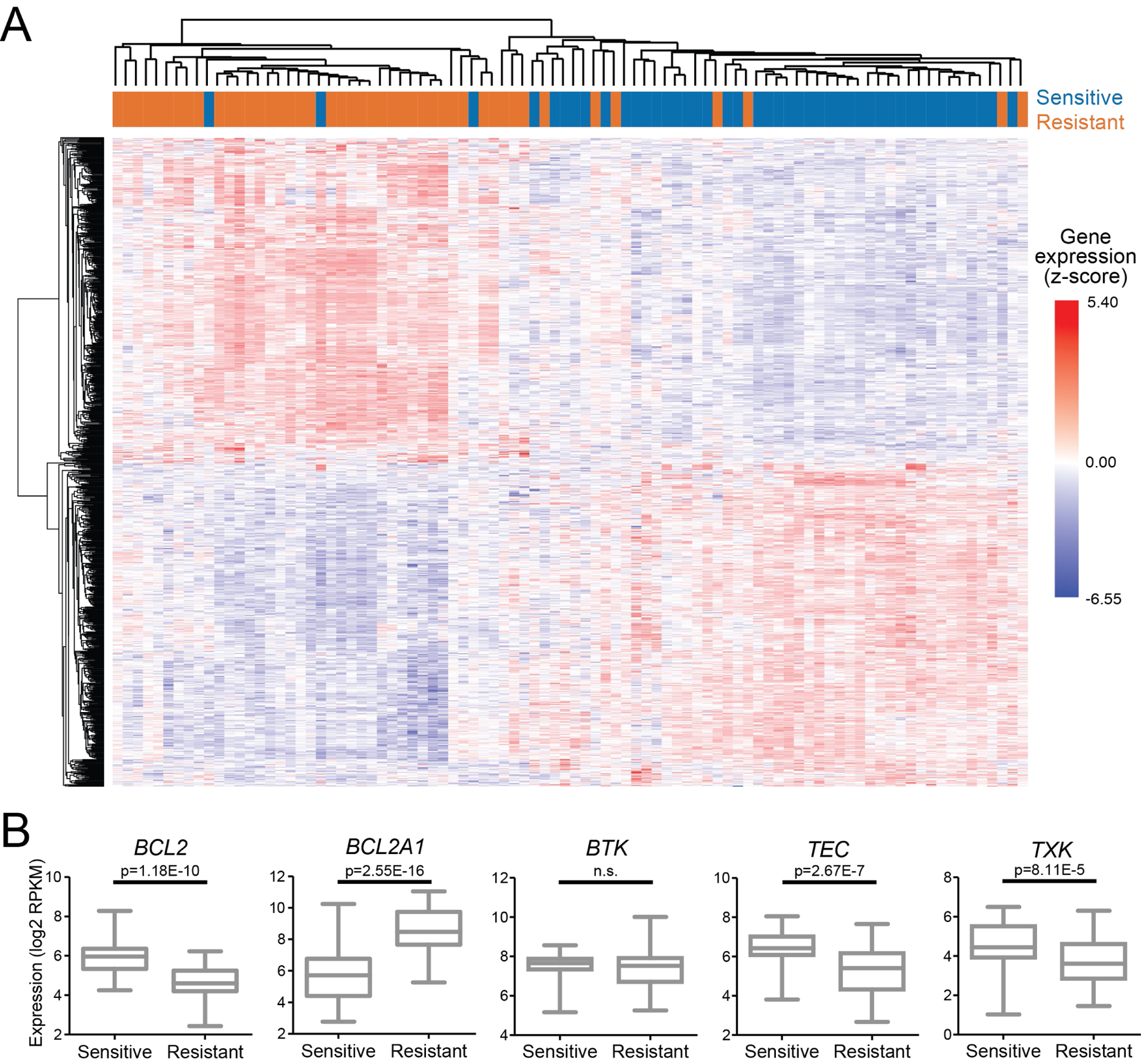
A. Hierarchical clustering of top 1000 DE genes between sensitive and resistant AML samples (lowest and highest quartile, respectively) by % of max AUC for IBR+VEN. Sensitive and resistant samples are designated in blue and orange, respectively and expression values are represented as z-score. B. Dot plot comparisons of expression for select DE genes.
IL-10 signaling pathway genes are overexpressed in samples with ex vivo resistance to IBR+VEN
The fold-changes of differentially expressed genes (defined as genes with FDR-adjusted p<0.01) and the functional connections between their protein products in annotated biological pathways were combined via the PRS method26 to identify significantly enriched Reactome pathways (Supplemental File S3). The IL-10 signaling pathway had one of the top enrichment scores, with 28 DE genes among the 39 genes belonging to this pathway. All but 2 of these 28 DE genes were upregulated in IBR+VEN resistant samples (Figure 6A), suggesting that IL-10-dependent inflammatory cytokine pathways may contribute to decreased sensitivity to IBR+VEN. Notably, IL-10-dependent, NF-kB-mediated resistance mechanisms have been previously identified in mantle cell lymphomas for the IBR+VEN combination31. Concordantly, we detected elevated expression of IL-10, CD40LG, NFKB1/2, and BIRC5 in IBR+VEN resistant patient samples (FDR-adjusted p<0.001; Figure 6B and Supplemental File S2).
Figure 6. Pathway analysis indicates IL-10 signaling is enriched in IBR+VEN resistant samples.
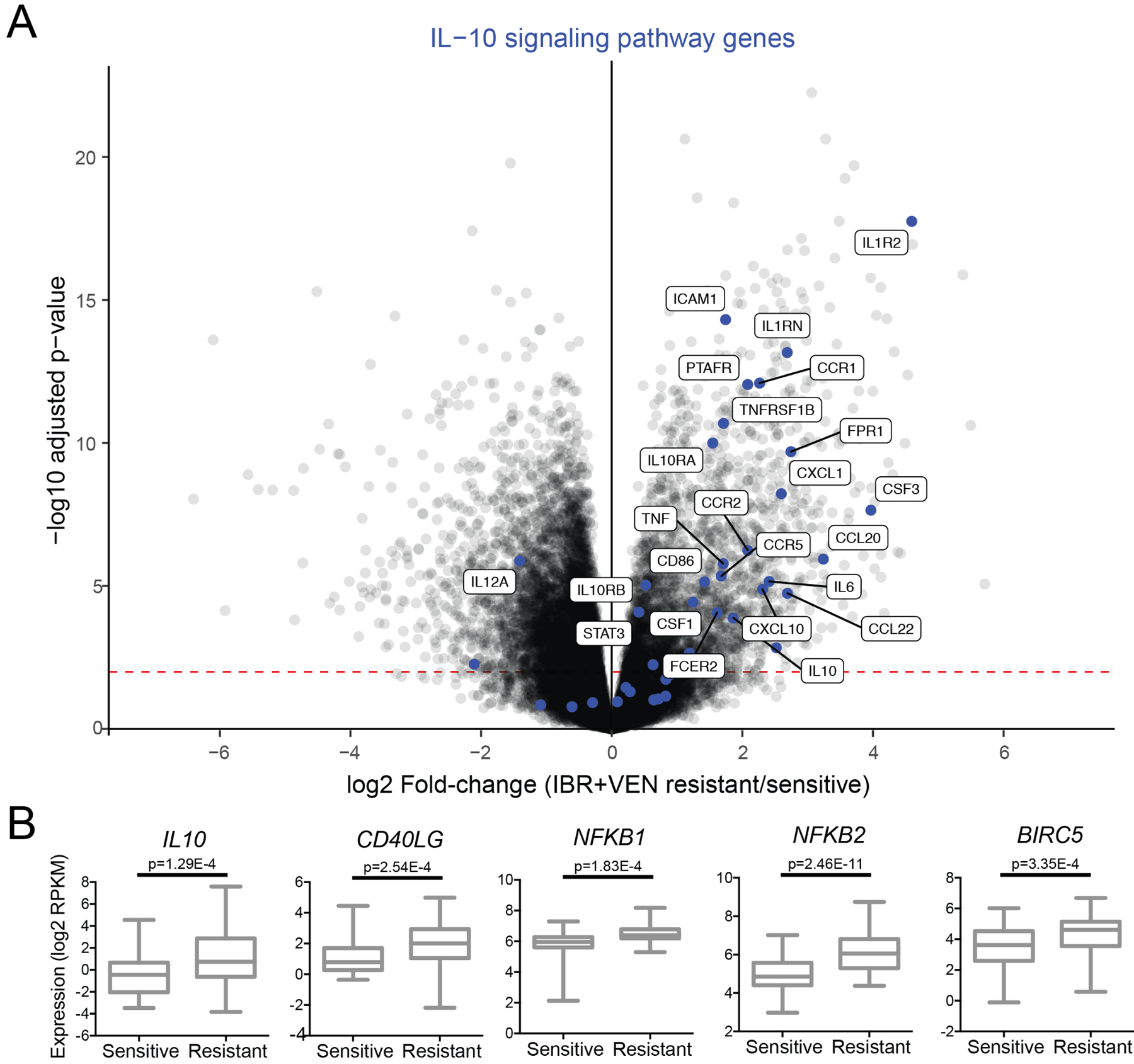
A. The IL-10 signaling pathway is enriched in IBR+VEN resistant samples as represented in the volcano plot depicting the relative expression of ~22,000 genes. Genes involved in the IL-10 signaling pathway are highlighted by blue dots; differentially expressed genes with an adjusted p-value < 0.001 are labeled. Red dashed line indicates p-value of 0.01. B. Dot plot comparisons of expression for select DE inflammatory signaling pathway genes.
We also evaluated levels of inflammatory cytokines in plasma from a subset of the most and least IBR+VEN sensitive samples in our AML cohort. We observed increased levels of the pro-inflammatory cytokines TNF-alpha and IFN-gamma in IBR+VEN-resistant samples compared to sensitive samples (Supplemental Figure S4). Correspondingly, RNAseq-based expression levels of both of these genes were significantly increased in the resistant samples.
Discussion
The combination of IBR+VEN was previously identified as a promising therapeutic strategy for CLL and mantle cell lymphoma (MCL)31–33. These observations have prompted numerous clinical trials in which the combination has shown significant efficacy. Results presented from the CLARITY trial showed after 12 months on IBR+VEN combination treatment an absence of morphological evidence of CLL in the marrow biopsy and achievement of MRD-negative remission in 87% and 41% of patients, respectively6. Similarly high response rates were observed in the CAPTIVATE trial in cohorts of CLL patients with either relapsed/refractory or untreated high-risk disease7, 9. These promising outcomes, coupled with confirmation that the two drugs can be given in combination without obvious additional toxicity, are likely to alter the landscape of CLL therapy.
Our ex vivo drug testing process surveys patient samples across the full spectrum of hematologic malignancies, where we expectedly detected potent IBR+VEN sensitivity in primary CLL samples. Unexpectedly, our data revealed that the therapeutic potential of IBR+VEN may extend to AML, as demonstrated by the impressive sensitivity to this combination in our large patient sample set (median IC50: 0.054 μM, n=325). The IBR+VEN combination was important more effective than either single agent at reducing tumor burden in vivo in a xenograft model of AML. Previously reported levels of each drug in plasma (ibrutinib Cmax for 140 mg QD: 0.084 μM 34; venetoclax Cmax for 400 mg QD: 2.51 μM35) suggest effective combination concentrations would be achievable in patients. The most recent approval for AML has been for use of venetoclax in combination with hypomethylating agents in newly diagnosed, elderly patients not fit for standard chemotherapy36. In this setting, the initial response rates of 60–80% are highly encouraging; however, 1-year survival rates of 30–40% along with significantly lower activity in relapsed/refractory patients indicate that alternative venetoclax combinations may be beneficial.
Different clinical and genetic features of AML guide the selection of treatment options and/or confer altered risk stratification and prognosis. The most extreme example is that of the PML-RARA rearrangement (t(15;17)), which has an excellent prognosis when treated with the combination of arsenic trioxide and retinoic acid regimen37. By contrast, complex karyotype, monosomy 7/del(7q), monosomy 5/del(5q), TP53 mutations, and del(17p) are features of adverse risk patients with more limited treatment options. Among the panel of clinical and genetic features of AML for which we had available annotations, we found select features showing either increased (11q23 MLL and PML-RARA rearrangements, FLT3 and NPM1 mutations) or decreased (inv(16), monosomy 7, RUNX1-RUNXT1 rearrangement, prior MDS) sensitivity to this combination. Importantly, there were no significant differences in sensitivity to IBR+VEN observed for several features typically associated with adverse risk, including complex karyotype, abnormal 17p, prior MPN, and deletion of chr5 or 5q. With respect to CLL patient samples, the characteristics most associated with increased sensitivity to IBR+VEN were del(11q), male gender, and non-mutated IGHV. Our clinical record mining for mutated genes in CLL patients was restricted to IGHV, as this gene has prognostic impact for treatment. It is formally possible that unsurveyed genes or mutations may contribute to lower sensitivity to the combination.
Combined inhibition of BTK and BCL2, the respective canonical targets of ibrutinib and venetoclax, represents a possible mechanism of IBR+VEN efficacy. BTK has been shown to contribute to proliferation, survival, and migration in AML blast cells38, 39 and it is possible that ibrutinib acts predominately on this kinase. It is also possible that the sensitivity of AML specimens to IBR+VEN represents an “overlapping” leukemia40, although such bi-phenotypic leukemias are uncommon among adults (<1% of all acute leukemias) and the median age among AML patient samples in our cohort was 60.9 years. Furthermore, a majority of tested AML patient specimens showed sensitivity to the IBR+VEN combination, suggesting that the mechanism of efficacy of this combination is dramatically broader than a rare subset of AML. Importantly, while we observed that ex vivo sensitivity to IBR+VEN associated with higher levels of BCL2 gene expression, we observed no significant association between IBR+VEN sensitivity and BTK expression. It remains possible that post-translational modification of BTK could contribute to differences in sensitivity to IBR+VEN, though this finding may also be indicative of involvement of other ibrutinib targets in AML15. Beyond BTK, ibrutinib also potently inhibits several other kinases, including FLT3 and multiple TEC and SRC family members30, which may provide therapeutic opportunities in other malignancies41. It is formally possible that ibrutinib acts on multiple targets in AML cells to achieve its enhanced sensitivity when combined with venetoclax. Notably, we identified increased expression in IBR+VEN sensitive samples for several ibrutinib targets (TEC, TXK, EGFR, ERBB2) that contain a cysteine residue aligning with C481 in BTK, which is the covalent binding site for ibrutinib30. Each of these four kinases as well as FLT3 are reported to have in vitro IC50 values for ibrutinib below 100 nM42, indicating that the effective concentrations of IBR+VEN could inhibit these targets and suggesting additional complexity with respect to ibrutinib sensitivity. With respect to FLT3, the association of FLT3-ITD mutations with sensitivity to ibrutinib has been established15, 38, 39. Moreover, elevated levels of TXK have been described in Behcet’s disease, whose pathogenesis is associated with excessive Th1 cytokine production and inflammatory signaling43.
Within the BCL2 family, higher expression levels of BCL2A1 and BCL2L11 (BIM) were detected in IBR+VEN resistant samples. This observation is consistent with prior studies that found elevated expression levels of BCL2A1, a BCR-regulated gene, are indicative of resistance to apoptosis inducers44–46. Many genes involved in cytokine signaling, including members of the IL-10 signaling pathway (as annotated by Reactome), were significantly upregulated in AML samples resistant to IBR+VEN, in a manner reminiscent of resistance mechanisms previously reported in mantle cell lymphomas31. Moreover, IL-6, an IL-10 signaling pathway member, has been shown to upregulate MCL1 and BCL-XL in myeloma cells47 or modulate MCL1:BIM priming48, thereby promoting resistance to BCL2 family inhibitors. While these mechanisms may provide insight into IBR+VEN resistance, the majority of AML patient samples are sensitive to IBR+VEN, thus warranting consideration of this combination for AML therapy.
Supplementary Material
Acknowledgements
Supported by grants from the National Cancer Institute (1U01CA217862, 1U54CA224019, 3P30CA069533). J.W.T. received grants from the V Foundation for Cancer Research, the Gabrielle’s Angel Foundation for Cancer Research, and the National Cancer Institute (1R01CA183947). A.V.D. is a Leukemia and Lymphoma Society Scholar in Clinical Research. Disclosures: J.W.T. has received research support from Agios, Aptose, Array, AstraZeneca, Constellation, Genentech, Gilead, Incyte, Janssen, Petra, Seattle Genetics, Syros, Takeda. B.J.D. serves on the advisory boards for Gilead, Aptose, and Blueprint Medicines. B.J.D. is principal investigator or coinvestigator on Novartis and BMS clinical trials. His institution, OHSU, has contracts with these companies to pay for patient costs, nurse and data manager salaries, and institutional overhead. He does not derive salary, nor does his laboratory receive funds from these contracts. The authors certify that the drugs tested in this study were chosen independently and without input from any of our industry partners. A.V.D. received research support from Gilead Sciences, Genentech, Verastem Oncology, Bayer Oncology, Takeda Oncology, Bristol-Myers-Squibb, MEI, Aptose Bioscences, Astra Zeneca; honoraria and consulting fees from Abbvie, Pharmacyclics, Gilead Sciences, Verastem Oncology, TG Therapeutics, Celgene, Teva Oncology, Astra Zeneca, Curis, Seattle Genetics, not relevant to this work.
Footnotes
Conflict of Interest Statement
This manuscript contains original research, has not been previously published, and is not under consideration for publication elsewhere. A.V. Danilov has received commercial research grants from Gilead Sciences, Verastem, Takeda, AstraZeneca, and Verastem Oncology, and is a consultant/advisory board member for AbbVie, AstraZeneca, Genentech, Verastem Oncology, Seattle Genetics, TG Therapeutics, Curis, Celgene, Teva Oncology, and Gilead Sciences. B.J. Druker serves on the board of directors of Amgen, Burroughs Wellcome Fund, and CureOne; reports receiving other commercial research support from Novartis, Bristol-Myers Squibb, and Pfizer (institutional funding—PI or co-investigator on clinical trials funded via contract with OHSU); has ownership interest (including stock, patents, etc.) in Amgen, Blueprint Medicines, MolecularMD (inactive—acquired by ICON Laboratories), GRAIL, Patent 6958335 (exclusively licensed to Novartis), Henry Stewart Talks, Merck via Dana-Farber Cancer Institute (royalty payments); is a consultant/advisory board member for Aileron Therapeutics, ALLCRON, Third Coast Therapeutics, Monojul (inactive), Baxalta (inactive), CTI Biopharma (inactive), Aptose, Beta Cat, Blueprint Medicines, Celgene, Cepheid, GRAIL (former), Gilead (former), and Patient True Talk; and is an uncompensated joint steering committee member for Beat AML LLC. J.W. Tyner has received commercial research grants from Agios, Aptose, Array, AstraZeneca, Constellation, Genentech, Gilead, Incyte, Janssen, Petra, Seattle Genetics, Syros, and Takeda; has received honoraria from the speakers bureaus of Therapeutic Advances in Childhood Leukemia and Hermedicus - Acute Leukemia Forum. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Dohner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. The New England journal of medicine 2015. September 17; 373(12): 1136–1152. [DOI] [PubMed] [Google Scholar]
- 2.Medeiros BC, Satram-Hoang S, Hurst D, Hoang KQ, Momin F, Reyes C. Big data analysis of treatment patterns and outcomes among elderly acute myeloid leukemia patients in the United States. Ann Hematol 2015. July; 94(7): 1127–1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Konopleva M, Pollyea DA, Potluri J, Chyla B, Hogdal L, Busman T, et al. Efficacy and Biological Correlates of Response in a Phase II Study of Venetoclax Monotherapy in Patients with Acute Myelogenous Leukemia. Cancer discovery 2016. October; 6(10): 1106–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2018. October 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pollyea DA, Stevens BM, Jones CL, Winters A, Pei S, Minhajuddin M, et al. Venetoclax with azacitidine disrupts energy metabolism and targets leukemia stem cells in patients with acute myeloid leukemia. Nat Med 2018. December; 24(12): 1859–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hillmen P, Rawstron A, Brock K, Vicente SM, Yates F, Bishop R, et al. Ibrutinib Plus Venetoclax in Relapsed/Refractory CLL: Results of teh Bloodwise TAP Clarity Study. Blood 2018; 132: 182. [Google Scholar]
- 7.Jain N, Keating MJ. Thompson PA, Ferrajoli A, Burger JA, Borthakur G, et al. Combined Ibrutinib and Venetoclax in Patients with Treatment-Naive High-Risk Chronic Lympocytic Leukemia. Blood 2018; 132: 696. [Google Scholar]
- 8.Rogers KA, Huang Y, Ruppert AS, Awan FT, Heerema NA, Hoffman C, et al. Phase 1b study of obinutuzumab, ibrutinib, and venetoclax in relapsed and refractory chronic lymphocytic leukemia. Blood 2018. October 11; 132(15): 1568–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tam CS, Anderson MA, Pott C, Agarwal R, Handunnetti S, Hicks RJ, et al. Ibrutinib plus Venetoclax for the Treatment of Mantle-Cell Lymphoma. The New England journal of medicine 2018. March 29; 378(13): 1211–1223. [DOI] [PubMed] [Google Scholar]
- 10.Byrd JC, Furman RR, Coutre SE, Flinn IW, Burger JA, Blum KA, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. The New England journal of medicine 2013. July 4; 369(1): 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiron D, Dousset C, Brosseau C, Touzeau C, Maiga S, Moreau P, et al. Biological rational for sequential targeting of Bruton tyrosine kinase and Bcl-2 to overcome CD40-induced ABT-199 resistance in mantle cell lymphoma. Oncotarget 2015. April 20; 6(11): 8750–8759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cervantes-Gomez F, Lamothe B, Woyach JA, Wierda WG, Keating MJ, Balakrishnan K, et al. Pharmacological and Protein Profiling Suggests Venetoclax (ABT-199) as Optimal Partner with Ibrutinib in Chronic Lymphocytic Leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research 2015. August 15; 21(16): 3705–3715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Deng J, Isik E, Fernandes SM, Brown JR, Letai A, Davids MS. Bruton’s tyrosine kinase inhibition increases BCL-2 dependence and enhances sensitivity to venetoclax in chronic lymphocytic leukemia. Leukemia 2017. October; 31(10): 2075–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Friedman AA, Letai A, Fisher DE, Flaherty KT. Precision medicine for cancer with next-generation functional diagnostics. Nat Rev Cancer 2015. December; 15(12): 747–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tyner JW, Tognon CE, Bottomly D, Wilmot B, Kurtz SE, Savage SL, et al. Functional genomic landscape of acute myeloid leukaemia. Nature 2018. October 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tyner JW, Yang WF, Bankhead A 3rd, Fan, Fletcher LB, Bryant J, et al. Kinase pathway dependence in primary human leukemias determined by rapid inhibitor screening. Cancer Res 2013. January 1; 73(1): 285–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altshuler B Modeling of dose-response relationships. Environmental Health Perspectives 1981; 42: 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hansen KD, Irizarry RA, Wu Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics 2012. April; 13(2): 204–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 2014; 15(12): 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Leek JT, Storey JD. Capturing heterogeneity in gene expression studies by surrogate variable analysis. PLoS Genet 2007. September; 3(9): 1724–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Buja A, Eyuboglu N. Remarks on Parallel Analysis. Multivariate behavioral research 1992. October 1; 27(4): 509–540. [DOI] [PubMed] [Google Scholar]
- 22.Benjamini Y, Hochberg Y. On the Adaptive Control of the False Discovery Rate in Multiple Testing With Independent Statistics. Journal of Educational and Behavioral Statistics 2000. March 20, 2000; 25(1): 60–83. [Google Scholar]
- 23.Matthews L, Gopinath G, Gillespie M, Caudy M, Croft D, de Bono B, et al. Reactome knowledgebase of human biological pathways and processes. Nucleic acids research 2009. January; 37(Database issue): D619–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G, et al. The Reactome pathway knowledgebase. Nucleic acids research 2014. January; 42(Database issue): D472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sales G, Calura E, Cavalieri D, Romualdi C. graphite - a Bioconductor package to convert pathway topology to gene network. BMC Bioinformatics 2012. January 31; 13: 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ibrahim MA, Jassim S, Cawthorne MA, Langlands K. A topology-based score for pathway enrichment. J Comput Biol 2012. May; 19(5): 563–573. [DOI] [PubMed] [Google Scholar]
- 27.Yadav B, Wennerberg K, Aittokallio T, Tang J. Searching for Drug Synergy in Complex Dose-Response Landscapes Using an Interaction Potency Model. Comput Struct Biotechnol J 2015; 13: 504–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang HW B; Bottomly D; Kurtz SE; Eide CA; Damnernsawad A; Romine K, et al. Biomarkers predicting venetoclax sensitivity and strategies for venetoclax combination treatment. Blood 2018; 132: 175. [Google Scholar]
- 29.Bisaillon R, Moison C, Thiollier C, Krosl J, Bordeleau ME, Lehnertz B, et al. Genetic characterization of ABT-199 sensitivity in human AML. Leukemia 2020. January; 34(1): 63–74. [DOI] [PubMed] [Google Scholar]
- 30.Pan Z, Scheerens H, Li SJ, Schultz BE, Sprengeler PA, Burrill LC, et al. Discovery of selective irreversible inhibitors for Bruton’s tyrosine kinase. ChemMedChem 2007. January; 2(1): 58–61. [DOI] [PubMed] [Google Scholar]
- 31.Jayappa KD, Portell CA, Gordon VL, Capaldo BJ, Bekiranov S, Axelrod MJ, et al. Microenvironmental agonists generate de novo phenotypic resistance to combined ibrutinib plus venetoclax in CLL and MCL. Blood Adv 2017. June 13; 1(14): 933–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Axelrod M, Ou Z, Brett LK, Zhang L, Lopez ER, Tamayo AT, et al. Combinatorial drug screening identifies synergistic co-targeting of Bruton’s tyrosine kinase and the proteasome in mantle cell lymphoma. Leukemia 2014. February; 28(2): 407–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao X, Bodo J, Sun D, Durkin L, Lin J, Smith MR, et al. Combination of ibrutinib with ABT-199: synergistic effects on proliferation inhibition and apoptosis in mantle cell lymphoma cells through perturbation of BTK, AKT and BCL2 pathways. British journal of haematology 2015. March; 168(5): 765–768. [DOI] [PubMed] [Google Scholar]
- 34.Scheers E, Leclercq L, de Jong J, Bode N, Bockx M, Laenen A, et al. Absorption, metabolism, and excretion of oral (1)(4)C radiolabeled ibrutinib: an open-label, phase I, single-dose study in healthy men. Drug Metab Dispos 2015. February; 43(2): 289–297. [DOI] [PubMed] [Google Scholar]
- 35.Roberts AW, Davids MS, Pagel JM, Kahl BS, Puvvada SD, Gerecitano JF, et al. Targeting BCL2 with Venetoclax in Relapsed Chronic Lymphocytic Leukemia. The New England journal of medicine 2016. January 28; 374(4): 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DiNardo CD, Pratz K, Pullarkat V, Jonas BA, Arellano M, Becker PS, et al. Venetoclax combined with decitabine or azacitidine in treatment-naive, elderly patients with acute myeloid leukemia. Blood 2019. January 3; 133(1): 7–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lo-Coco F, Orlando SM, Platzbecker U. Treatment of acute promyelocytic leukemia. The New England journal of medicine 2013. October 10; 369(15): 1472. [DOI] [PubMed] [Google Scholar]
- 38.Rushworth SA, Murray MY, Zaitseva L, Bowles KM, MacEwan DJ. Identification of Bruton’s tyrosine kinase as a therapeutic target in acute myeloid leukemia. Blood 2014. February 20; 123(8): 1229–1238. [DOI] [PubMed] [Google Scholar]
- 39.Oellerich T, Mohr S, Corso J, Beck J, Dobele C, Braun H, et al. FLT3-ITD and TLR9 use Bruton tyrosine kinase to activate distinct transcriptional programs mediating AML cell survival and proliferation. Blood 2015. March 19; 125(12): 1936–1947. [DOI] [PubMed] [Google Scholar]
- 40.Gutierrez A, Kentsis A. Acute myeloid/T-lymphoblastic leukaemia (AMTL): a distinct category of acute leukaemias with common pathogenesis in need of improved therapy. British journal of haematology 2018. March; 180(6): 919–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Berglof A, Hamasy A, Meinke S, Palma M, Krstic A, Mansson R, et al. Targets for Ibrutinib Beyond B Cell Malignancies. Scandinavian journal of immunology 2015. September; 82(3): 208–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Honigberg LA, Smith AM, Sirisawad M, Verner E, Loury D, Chang B, et al. The Bruton tyrosine kinase inhibitor PCI-32765 blocks B-cell activation and is efficacious in models of autoimmune disease and B-cell malignancy. Proceedings of the National Academy of Sciences of the United States of America 2010. July 20; 107(29): 13075–13080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Suzuki N, Nara K, Suzuki T. Skewed Th1 responses caused by excessive expression of Txk, a member of the Tec family of tyrosine kinases, in patients with Behcet’s disease. Clinical medicine & research 2006. June; 4(2): 147–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vogler M BCL2A1: the underdog in the BCL2 family. Cell Death Differ 2012. January; 19(1): 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vogler M, Butterworth M, Majid A, Walewska RJ, Sun XM, Dyer MJ, et al. Concurrent up-regulation of BCL-XL and BCL2A1 induces approximately 1000-fold resistance to ABT-737 in chronic lymphocytic leukemia. Blood 2009. April 30; 113(18): 4403–4413. [DOI] [PubMed] [Google Scholar]
- 46.Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood 2010. April 22; 115(16): 3304–3313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Puthier D, Derenne S, Barille S, Moreau P, Harousseau JL, Bataille R, et al. Mcl-1 and Bcl-xL are co-regulated by IL-6 in human myeloma cells. British journal of haematology 1999. November; 107(2): 392–395. [DOI] [PubMed] [Google Scholar]
- 48.Gupta VA, Matulis SM, Conage-Pough JE, Nooka AK, Kaufman JL, Lonial S, et al. Bone marrow microenvironment-derived signals induce Mcl-1 dependence in multiple myeloma. Blood 2017. April 6; 129(14): 1969–1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.