

Abstract
Under conditions of inherent or induced mitochondrial dysfunction, cancer cells manifest overlapping metabolic phenotypes, suggesting that they may be targeted via a common approach. Here we use multiple oxidative phosphorylation (OXPHOS)-competent and -incompetent cancer cell pairs to demonstrate that treatment with alpha-ketoglutarate (aKG) esters elicits rapid death of OXPHOS-deficient cancer cells by elevating intracellular aKG concentrations, thereby sequestering nitrogen from aspartate through glutamic-oxaloacetic transaminase 1 (GOT1). Exhaustion of aspartate in these cells resulted in immediate depletion of adenylates, which plays a central role in mediating mTOR inactivation and inhibition of glycolysis. aKG esters also conferred cytotoxicity in a variety of cancer types if their cell respiration was obstructed by hypoxia or by chemical inhibition of the electron transport chain (ETC), both of which are known to increase aspartate and GOT1 dependencies. Further, preclinical mouse studies suggested that cell-permeable aKG displays a good biosafety profile, eliminates aspartate only in OXPHOS-incompetent tumors, and prevents their growth and metastasis. This study reveals a novel cytotoxic mechanism for the metabolite aKG and identifies cell-permeable aKG, either by itself or in combination with ETC inhibitors, as a potential anti-cancer approach.
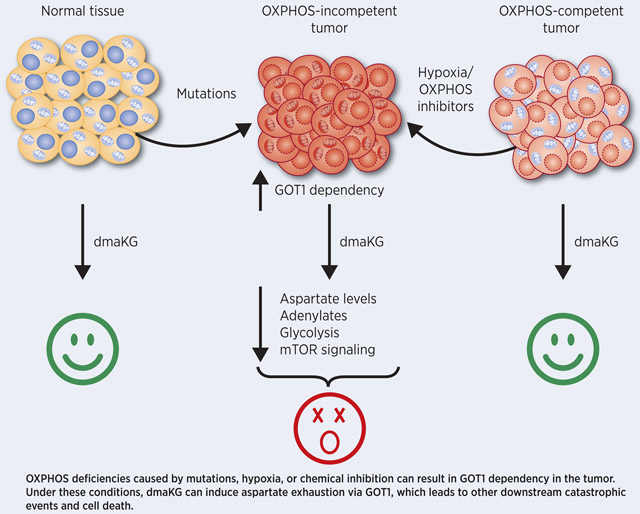
Graphical Abstract
INTRODUCTION
Cellular metabolic reprogramming is an essential step toward tumorigenesis. Cancer metabolism not only has to support the cell’s high anabolic needs but also to respond to various challenges such as low oxygen and nutrient availability pertaining to the tumor environment. Several canonical oncogenes have been shown to regulate cancer cell metabolism (1). The discovery of cancer-associated mutations in the tricarboxylic acid (TCA) cycle enzymes isocitrate dehydrogenase 1 and 2 (IDH1/2), succinate dehydrogenase (SDH) and fumarate hydratase (FH) indicates that significant alterations in metabolic pathways can also drive tumorigenesis (2). Rewiring of metabolism may render cancer cells more dependent than normal cells on specific cellular processes that could be targeted for therapeutic benefit (3,4).
It is important to note that while all cancer cells utilize glucose and secrete lactate in conditions with ample oxygen, a phenomenon termed aerobic glycolysis or the Warburg effect, many cancers also maintain mitochondrial metabolism and require respiratory competency (5,6). However, we now know that OXPHOS defects play a crucial role in a subset of cancers. For example, FH- and SDH-mutant cancers manifest pronounced mitochondrial respiration deficiencies (7-9). Furthermore, pathogenic mitochondrial DNA (mtDNA) mutations occur frequently in a broad range of cancer types (10,11). Additionally, cancer cells that have limited access to oxygen in vivo may exhibit OXPHOS defects (12).
Interestingly, a series of reports have recently demonstrated that cancer cells under hypoxic conditions and cancer cells with TCA cycle or electron transport chain (ETC) mutations display very similar metabolic reprogramming phenotypes. To survive the severe truncation of the OXPHOS pathway, these cells undergo multiple metabolic rearrangements, such as increased glycolysis and utilization of glutamine via reductive instead of oxidative carboxylation to replenish TCA cycle metabolites (13,14). In culture, respiration-incompetent cells are auxotrophic for pyruvate because of its role in maintaining redox balance to support aspartate biosynthesis. Concordantly, aspartate is a common limiting factor for their proliferation (12,15-18). Moreover, it has been found that cytosolic aspartate synthesis via the glutamate oxaloacetate transaminase 1 (GOT1) becomes essential when the ETC is inhibited (15).
Clinically, OXPHOS-defective cancers are often difficult to treat. For example, hypoxia enhances cancer virulence and significantly reduces the efficacy of radiotherapy, chemotherapy and targeted therapy (19). Loss-of-function mutations in FH can cause an aggressive form of kidney cancer called hereditary leiomyomatosis and renal cell carcinoma (HLRCC). HLRCC-associated kidney cancer occurs early in life and can metastasize even when tumors are small (<1 cm) (20,21). In addition, using mitochondrial transfer and cybrid cells, studies have shown that some mtDNA mutations can enhance tumor progression (22-24). The close resemblance in the metabolic phenotypes despite the varying causes of OXPHOS-deficiency suggests that it may be possible to develop a unifying therapeutic approach for such cancers. In the present work, we demonstrate that cell-permeable forms of the TCA cycle metabolite alpha-ketoglutarate (aKG) lead to potent cytotoxicity specifically in OXPHOS-incompetent cancer cells by targeting their dependence on the aspartate biosynthesis pathway.
Materials and Methods
Chemicals
Compounds dmaKG (dimethyl alpha-ketoglutarate, 349631), deaKG (diethyl alpha-ketoglutarate, CDS008208), etaKG (2-oxo-pentanedioic acid 5-ethyl ester 1-(3-trifluoromethyl-benzyl) ester, SML1743), antimycin A (A8674), rotenone (R8875), aspartate (L-aspartic acid potassium salt, A9381), 2-DG (2-deoxy-D-glucose, 25972), adenine (A2786), ATP (adenosine 5’-triphosphate, A7699), ADP (adenosine 5’-diphosphate, A5285), AMP-PCP (β,γ-methyleneadenosine 5’-triphosphate, M7510), , AOA (aminooxyacetic acid hemihydrochloride, C13408), and 3-bromopyruvate (16490), were purchased from Sigma Aldrich. Oligomycin (11341), necrostatin (11658), atpenin (11898), and metformin (13118) were purchased from Cayman Chemical. 3-NPA (3-nitropropionic acid, 4849) were purchased from Tocris. Cyclosporine A (BMLA1950100) were purchased from Enzo Life Sciences. Z-VAD-FMK (A19025) were purchased from Apexbio Technology. MG-132 (S2619) were purchased from Selleck Chemicals. Phenformin (P29690) were purchased from Toronto Research Chemicals.
Cell culture
All of the cell lines were cultured in the standard medium consisting of DMEM (Invitrogen, #11965) supplemented with 10% fetal bovine serum, 1 mM of sodium pyruvate and 1% penicillin-streptomycin at 37 °C with 10% CO2 unless specified otherwise. UOK262 and UOK268 cells were gifts from Drs. Y. Yang and W.M. Linehan at the National Institutes of Health (25,26). HLRCC isogenic cell pairs were generated by infection with viral particles containing pBabe-puro (UOK262-ev and UOK268-ev) or pBabe-puro-FH (UOK262-FH and UOK268-FH) and were maintained in 2 μg/ml of puromycin. UOK262-SLC1A3 and 143B-wt cells expressing SLC1A3 were generated by infection with viruses containing pMXs-SLC1A3 and were maintained in 5 and 4 μg/ml of blasticidin, respectively. 143B cybrid pairs (143B-wt and 143B-CytB) were gifts from Dr. M.G. Vander Heiden (Massachusetts Institute of Technology) with the permission of Dr. N.S. Chandel (Northwestern University), and they were cultured in the standard medium supplemented with 0.1 mg/mL uridine (17). pMXs-GOT1 and pMXs-IRES-blasticidin empty vector were introduced into UOK262, 143B-wt and 786-O by viral transduction to generate stable lines. pMXs-SLC1A3 (Addgene, #72873) and pMXs-GOT1 (Addgene, #72872) were gifts from David Sabatini (15). UMRC2-ev and UMRC2-VHL are human clear cell renal cell carcinoma cell lines carrying pBabe-puro and pBabe-puro-VHL, respectively. These cells were provided by Dr. O. Iliopoulos (Massachusetts General Hospital/MGH) and were maintained in the presence of 2 μg/ml of puromycin. 786-O and UMRC-3 were provided by Dr. O. Iliopoulos (MGH). NCI-H522, DU-145, Cal-12T and HCT116 were gifts from Dr. Cyril Benes (Center for Molecular Therapeutics, MGH). These cells have not been authenticated during this study. Cells were maintained and propagated for two months after thawing. Mycoplasma test was performed routinely using the MycoAlert PLUS Mycoplasma Detection Kit (Lonza, #LT07-701). The FH mutations in HLRCC isogenic cell pairs have been sequence confirmed. Multiple batches of HLRCC isogenic cell pairs were generated and the results were reproducible from different batches.
Cytotoxicity assay
All of the cytotoxicity results shown as % dead cells were determined using the trypan blue dye exclusion assay. For this assay, 20,000 cells per well were seeded in 24-well plates, and treatments started the next day. 24 hr after treatment, both floating and adherent cells were harvested to determine % cell death using trypan blue at 0.2 w/v% final concentration.
Annexin-V apoptosis assay
Apoptosis was measured using FITC Annexin V Apoptosis Detection Kit with PI (BioLegend, 640906). Cells were treated with dmaKG for indicated duration and were harvested by trypsinization, washed with ice-cold phosphate buffered saline (PBS), and finally resuspended in binding buffer. The staining was performed as recommended by the manufacturer. After 20 min of incubation in the dark, samples were analyzed on a Fortessa X-20 flow cytometer (BD Biosciences). The apoptotic cell fraction was measured based on the fraction of cells that are FITC+ and PI- (early apoptotic) or that are FITC+ and PI+ (late apoptotic) in a dot plot. At least 10,000 events were assessed per measurement.
Proliferation assay
10,000 cells/well were seeded in 12-well plates, and dmaKG was added the following day. Cells were grown for 4 days under the indicated conditions. Cells were then washed twice with PBS and incubated with 0.2% filtered crystal violet solution for 15 min. Cells were washed with water approximately three times until all extracellular crystal violet was removed. Intracellular crystal violet was then dissolved in methanol. Absorbance of the solution was measured at 570 nm using the FLUOstar Omega Microplate Reader (BMG Labtech).
Measurement of mitochondrial transmembrane potential (φm)
2 × 105 cells/well were plated in 12-well plate and were treated with dmaKG (6.6 mM) for 8 h. 20 minutes before termination, tetramethylrhodamine, ethyl ester (TMRE) was added to cells at a final concentration of 0.2 μM and incubated at 37 0C. Cells were trypsinized, washed and suspended in 0.5 mL PBS. Trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP) (20 μM) was used as a negative control. The cell fluorescence was measured by flow cytometry with appropriate channels.
Extracellular flux analysis
The cells were plated in a XF96 cell culture microplate at 25,000 cells/well and maintained in regular cell growth conditions for at least 12 hr prior to the experiments. On the day of the assay, cells were treated with indicated compounds for indicated times. Oxygen consumption rate (OCR), extracellular acidification rate (ECAR) and proton efflux rate (PER) were measured according to the manufacturer’s instructions for the Seahorse XF Glycolysis Stress Test, the XF Cell Mito Stress Test, and the XF Real-Time ATP Rate Assay. Data analysis was performed using the Seahorse Wave software.
ATP measurement
The ATP content was measured using the CellTiter-Glo® Luminescent Cell Viability Assay (Promega, G7570). Briefly, 5000 cells/well were plated in a 96-well plate. On the next day, cells were treated with compounds as indicated in the figure legends and then lysed using the assay reagent followed by the luminescence measurement using the FLUOstar Omega Microplate Reader (BMG Labtech). Relative ATP levels was calculated as compared to the control of the same cell types.
Measurement of the adenine nucleotide pool
2 × 105 cells/well were plated in a 12-well plate. On the next day, cells were treated as indicated in the figure legends. At the end of the experiments, cells were washed with cold saline followed by ATP extraction with the boiling method and sample processing per the manufacturer’s protocol. Cryopreserved mouse organs or xenograft tumors (20 mg) from in vivo efficacy studies were processed through trichloroacetic acid deproteination followed by extraction into 10 mM Tris-EDTA (TE) buffer using ether that is pre-saturated in TE buffer. The levels of the total adenine nucleotide pool or the levels of individual adenine nucleotides (ATP, ADP or AMP) were measured using the ATP/ADP/AMP Assay kit (Biomedical Research Service, Department of Biochemistry, University at Buffalo, A-125). The luminescence was measured using the FLUOstar Omega Microplate Reader (BMG Labtech).
Western blotting
Cell lysates were prepared in Laemmli sample buffer (Bio-Rad, #1610737) supplemented with Halt™ Protease and Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific, #78440). The proteins were separated by 12% SDS-PAGE and transferred onto polyvinylidene fluoride (PVDF) membranes. After blocking with 5% non-fat milk in Tris-buffered saline and 0.1% Tween 20, membranes were incubated with primary antibodies at 4°C overnight, followed by 1:20000 horseradish peroxidase (HRP)-conjugated secondary antibody (Cell Signaling Technologies) for 2 hr. Bands were visualized using Clarity ECL Western Blotting Substrate (Bio-Rad, #1705060) and ChemiDoc Touch Imaging System (Bio-Rad Laboratories). Antibodies were purchased from Cell Signaling Technologies for prolylhydroxy-HIF-1α (34345), phospho-p70S6K1 (9234S), p70S6K1 (2708S), phospho-S6 (5364S), S6 (2317S), phospho-4EBP1 (9451S), and 4EBP1 (9644S) and from BD Biosciences for HIF-1α (610958).
Animal studies
All animal experiments were performed with approval of the Institutional Animal Care and Use Committee (IACUC) at the Massachusetts General Hospital.
For UOK262 xenograft tumor model, athymic nude mice were injected subcutaneously in the right flank with 7.5 × 106 UOK262 cells. In approximately two months, palpable tumors were formed, and treatment was started. dmaKG was diluted in phosphate buffered saline (75 mg/ml) daily before the injection. The solution was administered via intraperitoneal injection based on body weight (200 μL/20 g) twice a day with a 2-h interval, which equals to 750 mg/kg per injection. Blind measurements of tumor mass were performed twice a week using a caliper and volume was calculated using 0.5*(short dimension)2*(long dimension). Body weight was measured once a week.
For 143B-CytB and 143B-wt xenograft tumor models, athymic nude mice were injected subcutaneously on the right side of abdominal region with 5 × 106 143B-CytB or 143B-wt cells. Tumor growth was monitored at regular intervals and once the tumor volume was above or equal to 200 mm3, mice were randomly assigned into groups and treatment was started. All mice were injected intraperitoneally (100 μL/20 g) twice a day with a 2-h interval between two injections. The control group received phosphate buffered saline (PBS). The dmaKG group received dmaKG dissolved in PBS (1000 mg/kg per injection). The phenformin group received phenformin dissolved in PBS (25 mg/kg per injection). The co-treatment group receive PBS containing both dmaKG (1000 mg/kg per injection) and phenformin (25 mg/kg per injection). Tumor dimensions and body weights were measured every third day and volume was measured using the above-mentioned formula.
At the conclusion of the experiments, some tumors and organs were harvested and fixed in 4% paraformaldehyde/PBS, pH 7.4. The tissues were then embedded in paraffin and serially sectioned. For routine histological screening, tissue sections were stained with hematoxylin and eosin by standard methods. In addition, some tumors and organs were harvested and snap frozen in liquid N2 for additional downstream analyses. Moreover, blood was collected by cardiac puncture after the animal was euthanized and was submitted to MGH CCM Clinical Pathology Lab for chem panel and serum analysis.
Statistical analysis
All statistical analyses were performed in GraphPad Prism. Statistical methods used for each experiment are as indicated in the figure legends.
Additional information can be found in Supplementary Methods.
Results
Cell-permeable aKG induces rapid death of OXPHOS-incompetent cancer cells
To uncover metabolic vulnerabilities unique to OXPHOS-incompetent cancer cells, we created isogenic cell pairs carrying an empty vector (ev) or an expression vector of wild-type FH (FH) from two FH-mutant HLRCC cell lines - UOK268 derived from a primary kidney tumor and UOK262 derived from a metastasis of a kidney tumor (25,26). It has been shown that FH mutation leads to accumulation of fumarate and that fumarate may act as an oncometabolite by impinging on several pathways associated with cancer (8,27-29). As expected, we found that repletion of FH into HLRCC cells results in normalization of fumarate levels, increases in oxygen consumption rate (OCR), and decreases in extracellular acidification rate (ECAR). In addition, the ev cells rely more on glucose, pyruvate and glutamine for proliferation as compared to their FH-replete counterparts (Supplementary Fig. S1A-F). These results indicate that reconstitution of FH in HLRCC cells restores mitochondrial respiration at least partially.
Because fumarate can compete with aKG for binding to aKG-dependent dioxygenases, we initially surmised that addition of dimethyl alpha-ketoglutarate (dmaKG), a cell-permeable form of aKG, might reverse some of the phenotypes observed in FH-mutant cells attributed to fumarate accumulation. Serendipitously, we noticed that dmaKG at low millimolar concentrations caused rapid and massive cell death within 24 hours (hr) in both UOK262 and UOK268 ev cells but not in their FH-replete counterparts (Fig. 1A). Even at sub-toxic concentrations, dmaKG displayed anti-proliferative effects more potently on the ev cells as compared to the FH-replete cells. Of note, we have found that cells become more sensitive or resistant to dmaKG at a lower or higher pH, respectively. Moreover, removing pyruvate or supplementing additional pyruvate in the medium has no significant impact on dmaKG sensitivity (Supplementary Fig. S2A-C).
Figure 1. Esters of aKG are selectively toxic to cancer cells with mitochondrial dysfunction.

(A) Cytotoxicity of α-ketoglutarate esters (dmaKG, dimethyl alpha-ketoglutarate; deaKG, diethyl alpha-ketoglutarate; etaKG, 5-ethyl 1-(3-(trifluoromethyl)benzyl) alpha-ketoglutarate) in fumarate hydratase (FH)-deficient HLRCC cells carrying either an empty vector (UOK262-ev, UOK268-ev) or a FH expression vector (UOK262-FH, UOK268-FH). Chemical structures of these esters are shown in the respective cytotoxicity plots. Unless otherwise mentioned, % Dead cells throughout the manuscript denotes % trypan blue-positive cells after 24 hr of treatment.
(B) Flow cytometric analysis of mitochondrial membrane potential (MMP or ΔΨm) following staining with potentiometric fluorescent probe (TMRE, 200 nM) in UOK262 cell pairs. dmaKG, 6.6 mM for 8 hr.
(C) Flow cytometric analysis of apoptosis following staining with FITC-conjugated annexin-V and propidium iodide (PI) in UOK262 and UOK268 cell pairs with or without dmaKG treatment until the indicated time. Early apoptosis, annexin-V-positive and PI-negative; Late apoptosis, annexin-V-positive and PI-positive.
(D) Cytotoxicity of dmaKG, deaKG, and etaKG in 143B-wt and 143B-CytB cybrid cell pair.
(E) Cytotoxicity of dmaKG in combination with chemical inhibitors of ETC Complex I (Metformin 5 mM, Phenformin 300 μM, Rotenone 0.2 μM), Complex II (Atpenin 0.5 μM, 3-NPA 0.2 mM), Complex III (Antimycin A 50 μM), and Complex V (Oligomycin 1 μM) in UOK262-FH and 143B-wt cells.
(F) Cytotoxicity of dmaKG in various cancer cell types cultured in normoxia (21% O2) or hypoxia (1% O2). dmaKG, 3.3 mM for NCI-H522, 13.2 mM for MDA-MB-231 and 786-O, 6.6 mM for VHL-deficient UMRC2 renal cell carcinoma cells (UMRC2-ev) and their VHL-reconstituted counterparts (UMRC2-pVHL).
All data are shown as mean with individual data points or mean ± SD except in (B). Statistical analysis was performed for (A) and (D) (two-way ANOVA with Tukey's multiple comparisons, ns p>0.5, *p≤0.05, **p≤ 0.01, ***p≤0.001, ****p≤0.0001).
Similar to dmaKG, other cell-permeable forms of aKG were also differentially toxic between the cell pairs, indicating that aKG maybe the active moiety (Fig. 1A). Alpha-ketoglutaric acid without a cell-permeable moiety was not cytotoxic up to 13.2 mM, indicating that entry of aKG into the cell is critical for toxicity. Furthermore, we did not find any differential toxicity with cell-permeable forms of other TCA cycle metabolites, highlighting the special property of aKG esters in targeting FH-mutant cancer cells (Supplementary Fig. S3A-B).
To characterize the mechanisms leading to cell death, we found that dmaKG caused a significant decrease in the mitochondrial membrane potential and an increase in annexin V-positive cells within 6 hr in the ev cells (Fig. 1B-C). Accordingly, we found that cyclosporine A (CsA), which prevents the opening of the mitochondrial permeability transition pore, and the pan-caspase inhibitor Z-VAD-FMK suppressed dmaKG-induced cell death, albeit less effectively with higher concentrations of dmaKG (Supplementary Fig. S4A-D). These results suggest that cell death in the presence of dmaKG involves a mitochondria-regulated pathway and may occur via both caspase-dependent and caspase-independent mechanisms.
To investigate whether dmaKG exerts cytotoxicity under other OXPHOS-incompetent cancer conditions, we employed an isogenic cybrid pair of human osteosarcoma 143B cells containing either wild-type mtDNA (143B-wt) or mtDNA with a mutation in the ETC complex III cytochrome b gene (143B-CytB) (14,17). The data showed that various aKG esters induced pronounced death of the respiration-deficient 143B-CytB cells but not the respiration-proficient 143B-wt cells (Fig. 1D). However, chemical inhibition of the ETC complex I, II, III or V in either UOK262-FH or 143B-wt cells elicited dmaKG sensitivity in these cells (Fig. 1E). Likewise, treatment with phenformin, an ETC complex I inhibitor, induced dmaKG toxicity in a variety of cancer cell types (Supplementary Fig. S5). Changing the culture condition from 21% to 1% O2 also induced dmaKG cytotoxicity in a spectrum of cancer cell lines, though its potency varied depending on the cancer cell types (Fig. 1F). We additionally tested an isogenic cell pair of UMRC2, which is a VHL-deficient renal cancer cell line that expresses both HIF-1α and HIF-2α (30). UMRC2 carrying an empty vector (ev) and UMRC2 VHL-replete (VHL) cells were both insensitive to dmaKG in normoxia but became equally sensitive in hypoxia, suggesting that a strong suppression of oxidative metabolism, not the pseudohypoxia phenotype, is associated with dmaKG sensitivity (Fig. 1F). In sum, the findings strongly suggest that cell-permeable aKG confers potent cytotoxicity selectively towards OXPHOS-incompetent cancer cells.
dmaKG increases both aKG and L-2HG
We initially hypothesized that, upon dmaKG treatment, OXPHOS-deficient cells died because they accumulated a higher level of aKG compared to OXPHOS-proficient cells. Using mass spectrometry, we found that intracellular aKG levels increased 5 to 25 fold within 0.5 hour (hr) of treatment. After 5 hr of incubation, aKG levels were higher in OXPHOS-deficient conditions compared to OXPHOS-proficient conditions in both UOK268 and 143B cell pairs, whereas in UOK262 pair the levels were similar (Supplementary Fig. S6A). Hence, OXPHOS deficiency may increase the amount of aKG accumulating in the cells. Nevertheless, even when the intracellular aKG levels are similar as in the UOK262 pair, the OXPHOS-deficient cells die more than the OXPHOS-proficient cells, implying an inherent difference in their responses to high levels of aKG.
In addition, we considered the possibility that aKG was converted to 2-hydroxyglutarate (2-HG) inside the cell and that 2-HG might cause cell death. 2-HG has two enantiomers. While D-2HG is known as an oncometabolite of the IDH1/2 mutations, L-2HG elevates naturally under hypoxia or acidic conditions (31,32). 2-HG may also be converted to aKG via the enzymes D-2HGDH and L-2HGDH. We found that dmaKG significantly increased L-2-HG levels in UOK262, UOK268 and 143B cell pairs except 143B-wt. However, dimethyl L-2HG (dmHG) up to 13.2 mM was not toxic to UOK262-ev or UOK268-ev cells, and yet it potentiated the toxicity of dmaKG possibly due to a further increase in the intracellular aKG concentrations (Supplementary Fig. S6A-D). These results suggest that aKG but not 2-HG accumulation elicits cytotoxicity in OXPHOS-defective cells.
dmaKG inhibits glycolysis in OXPHOS-incompetent cells
aKG may inhibit mitochondrial ATP synthase (33), which may decrease ATP level leading to cell death. Interestingly, we observed that dmaKG decreased ATP levels acutely in less than 4 hr. Moreover, the reduction was significantly greater in the ev cells than in the FH-replete cells (Fig. 2A). However, oligomycin, a well-characterized inhibitor of ATP synthase, did not reduce ATP levels. Rather, inhibition of glycolysis using 2-deoxy-D-glucose (2DG) rapidly decreased ATP levels in the ev cells within the same timeframe and to a similar extent as did dmaKG. Nonetheless, 2DG reduced ATP levels indiscriminately between ev and FH-replete cells. Furthermore, 2DG was not cytotoxic even after 24 hr treatment (Fig. 2B). Although these results do not clarify the mechanism by which dmaKG decreases ATP levels in HLRCC cells, they indicate that lowering of ATP levels is not sufficient to cause HLRCC cell death.
Figure 2. Glycolysis and HIF-1α levels are differentially affected in OXPHOS-competent versus OXPHOS-incompetent cells by dmaKG.

(A) ATP levels as measured by CellTiter-Glo® Luminescent Cell Viability Assay in UOK262 (4 hr) and UOK268 (2.5 hr) cell pairs after treatment with the indicated compounds. dmaKG, 6.6 mM; 2-deoxy-D-glucose (2-DG), 20 mM; oligomycin A (OliA), 20 μM.
(B) Cytotoxicity of compounds shown in (A) after 24 hr of treatment.
(C) Real-Time ATP Rate Assay using Seahorse XFe96 Analyzer on UOK262 cell pair. Cells were treated with dmaKG (6.6 mM) for the indicated times. mitoATP, ATP production rate via mitochondrial respiration; glycoATP, ATP production rate via glycolysis.
(D) Seahorse XF Glycolysis Stress Test assay on UOK262 cell pair. Cells were treated with dmaKG (6.6 mM) for the indicated times and the effects on glycolysis and glycolytic capacity were measured.
(E-G) Immunoblotting of HIF-1α. UOK262 and UOK268 cell pairs were treated with or without 6.6 mM dmaKG (E) or 9.9 mM deaKG (F). 143B cell pairs were treated with dmaKG (9.9 mM) (G). β-actin is used as the loading control.
In (A-D), bar graphs are shown as mean with individual data points or mean ± SD. Statistical analysis was performed using two-way ANOVA with Tukey’s multiple comparisons (ns p>0.5, *p≤0.05, **p≤0.01, ***p≤0.001, ****p≤0.0001).
Next, we examined whether dmaKG might inhibit glycolysis using the Seahorse Real-Time ATP Rate Assay. In both UOK262-ev and -FH cells, glycolysis contributed more towards overall ATP production than did mitochondrial respiration (Fig. 2C). Meanwhile, we found that dmaKG reduced glycolytic ATP production in UOK262-ev but not in UOK262-FH. To confirm these findings, we measured the extracellular acidification rate (ECAR) using the Glycolysis Stress Assay. The results showed that dmaKG reduced both glycolysis and glycolytic capacity preferentially in the ev cells (Fig. 2D). Similarly, dmaKG lowered ECAR in UOK268-ev and 143B-CytB but not in UOK268-FH and 143B-wt. Moreover, we found that glucose uptake in UOK262-ev was unaffected in the presence of dmaKG (Supplementary Fig. S7A-C). Although ECAR is an indirect measurement of glycolysis, these results suggest that dmaKG inhibits the glycolytic pathway at the steps downstream of glucose transport and only in OXPHOS-incompetent cells.
dmaKG exerts opposing effects on HIF-1α levels in OXPHOS-competent versus -incompetent conditions
It is known that HLRCC cells exhibit a pseudohypoxic phenotype due to normoxic stabilization of hypoxia-inducible factor-1α (HIF-1α), a key regulator of glycolysis (28). Since aKG is a cofactor of HIF prolyl hydroxylases (PHDs), enhancing PHD activities may lead to HIF-1α degradation. Unexpectedly, we observed that, upon dmaKG treatment, HIF-1α protein was decreased in UOK262-ev and UOK268-ev, whereas it was increased in UOK262-FH and UOK268-FH (Fig. 2E). The related compound deaKG conferred similar effects as dmaKG (Fig. 2F). Cell-permeable aKG derivatives have been shown to both increase and decrease HIF-1α protein stability, suggesting that the effects may depend on cell type, culture conditions, and the aKG derivatives used (34,35). Hou et al. recently reported that dmaKG causes HIF-1α stabilization in various cancer cell lines by inhibiting PHD2 (34). In UOK262-FH, our data also suggest that dmaKG stabilizes HIF-1α by inhibiting its prolyl hydroxylation (Supplementary Fig. S8A). Therefore, it is puzzling why HIF-1α does not accumulate in OXPHOS-incompetent cells as in OXPHOS-competent cells in response to cell-permeable aKG. In UOK262-ev, the decreases in HIF-1α protein levels were not due to changes in its mRNA levels or protein degradation (Supplementary Fig. S8A). Results in the 143B cell pair were consistent with those in the HLRCC cell pairs (Fig. 2G, Supplementary Fig. S8B). Collectively, these data indicate that dmaKG may increase the stability of HIF-1α in OXPHOS-competent conditions. However, in OXPHOS-incompetent conditions, dmaKG may downregulate HIF-1α through translation via mechanisms such as the mTOR pathway (36).
dmaKG abrogates aspartate synthesis in OXPHOS-incompetent conditions
Although the pseudohypoxia phenotype may play some roles in FH-mutant cancers, previous studies have shown that reductions in HIF-1α level do not cause HLRCC cell death (37,38). To explore alternative cytotoxic mechanisms, we performed LC-MS based metabolic profiling of key and relevant metabolites following 0.5 and 5 hr of dmaKG treatment in the UOK262 cell pair (Fig. 3A). Not surprisingly, we found that dmaKG increased citrate levels implying reductive carboxylation in both ev and FH-replete cells (14). Nonetheless, these cells showed clear differences in their oxidative metabolism in response to dmaKG. In FH-replete cells, dmaKG increased all TCA cycle metabolites downstream of aKG including succinate, fumarate and malate. In ev cells, although succinate was increased, there was a decrease in fumarate and no increase in malate. We also found that dmaKG significantly reduced aspartate levels within 0.5 hr in both UOK262-ev and UOK262-FH cells. However, while aspartate level gradually recovered in UOK262-FH, it did not recover in UOK262-ev. Meanwhile, dmaKG caused only modest changes in other amino acids and the changes were largely similar between ev and FH-replete cells (Fig. 3B).
Figure 3. dmaKG treatment depletes aspartate levels in respiration-deficient cells via GOT1.

(A) Mass spectrometry analysis of UOK262 cell pairs indicating the relative abundances of TCA cycle intermediates (citrate, succinate, fumarate, malate) and aspartate in control or with 0.5 or 5 hr of dmaKG (6.6 mM) treatment. α-KG, alpha-ketoglutarate; OAA, oxaloacetate.
(B) The log2 fold change in amino acids in UOK262 cell pairs after 5 hr of dmaKG (6.6 mM) treatment as compared to untreated control.
(C) Mass spectrometry analysis of 143B cell pairs in control or with 3 or 6 hr of dmaKG (9.9 mM) treatment.
(D) The log2 fold change in amino acids in 143B cell pairs after 6 hr of dmaKG (9.9 mM) treatment as compared to untreated control.
(E) Depiction of the enzymatic function of GOT1/2.
(F) Mass spectrometry analysis of aspartate levels in UOK262 cell pairs. Cells were treated with or without 6.6 mM dmaKG (left panel) or 2 mM aminooxyacetic acid (AOA) (right panel) for 6 hr. AOA potently decreases aspartate levels in both UOK262-ev and UOK262-FH, whereas dmaKG causes a stronger reduction in aspartate in UOK262-ev than in UOK262-FH.
(G) Targeted metabolic flux analysis using 13C4,15N-aspartate in 143B-wt cells expressing SLC1A3. The 15N-glutamate/13C4,15N-aspartate ratio increased upon dmaKG treatment. There is an approximately 8-fold increase in the labelled glutamate/aspartate ratio upon treatment with dmaKG but a 40-fold increase with co-treatment of dmaKG and phenformin as compared to their respective controls without dmaKG.
(H) Cytotoxicity of dmaKG in UOK262-ev and 143B-CytB cells. These cells additionally carried pMXs empty vector (ev) or pMXs-GOT1 (GOT1). Cells were treated with dmaKG (4.4 mM for UOK262-ev and 6.6 mM for 143B-CytB) for 24 hr.
(I)The effects of various compounds on cell proliferation as determined by crystal violet staining. UOK262 cells carried pMXs empty vector (ev) or pMXs-GOT1 (GOT1) were treated with the vehicle (Control), 3.3 mM dmaKG, or 2 mM AOA for 72 hr.
Data are shown as mean and individual data points or mean ± SD. Statistical analysis was performed in (A, C, H-I) (n=4, two-way ANOVA with Tukey’s multiple comparisons) and in (F-G) (n=4, unpaired two-tailed t test with Welch’s correction, ns p>0.5, *p≤0.05, **p≤ 0.01, ***p≤0.001, ****p≤0.0001).
Metabolic profiling of the 143B cell pair following 3 and 6 hr of dmaKG treatment showed similar results (Fig. 3C-D). In 143B-CytB, dmaKG increased citrate levels due to reductive metabolism of aKG (14). Moreover, dmaKG caused significant increases in succinate but decreases in fumarate and malate, suggesting that oxidative metabolism downstream of succinate may be inefficient in these cells. In comparison, in 143B-wt cells in which oxidative metabolism is efficient, dmaKG did not increase citrate or succinate. Notably, dmaKG increased malate levels in 143B-wt and UOK262-FH but not in 143B-CytB or UOK262-ev. In both UOK262 and 143B cell pairs, basal aspartate levels were lower in OXPHOS-incompetent conditions compared to OXPHOS-competent conditions. Treatment with dmaKG caused a further 80-90% reduction in aspartate levels in UOK262-ev and 143B-CytB, but only 40-60% reduction in UOK262-FH and 143B-wt. However, in 143B-wt cells treated with phenformin, dmaKG again reduced aspartate level to only 10% of the control level (Supplementary Fig. S9A-B). These results indicate that dmaKG induces severe aspartate deficiency selectively in OXPHOS-incompetent conditions.
It has been previously shown that aspartate is synthesized by cytosolic GOT1 under OXPHOS-incompetent conditions in contrast to mitochondrial GOT2 under OXPHOS-competent conditions (15). GOT1/2 are transaminases that carry out the reversible reactions from glutamate and oxaloacetate to aKG and aspartate (Fig. 3E). Hence, increasing intracellular aKG concentrations may be incompatible with aspartate synthesis. Moreover, our data suggest that dmaKG may preferentially affect GOT1 as compared to GOT2. Concordantly, a potent, non-selective transaminase inhibitor aminooxyacetic acid (AOA) suppressed aspartate levels to similar levels in UOK262-ev and UOK262-FH (Fig. 3F). We noticed that dmaKG reduced aspartate level even more than AOA did in UOK262-ev, suggesting that aKG may deplete aspartate further by trapping its nitrogen through GOT1 and yield glutamate. To test this hypothesis, we investigated the transfer efficiency of the 15N atom from isotope-labelled aspartate to aKG, which would produce 15N-labelled glutamate. Because most cells do not take up aspartate efficiently, we expressed the glutamate-aspartate transporter (SLC1A3) in 143B-wt cells to enhance the uptake of labelled aspartate. The data showed that dmaKG significantly elevated the ratio of labelled glutamate to labelled aspartate in the presence of phenformin, but the effect was less in the absence of phenformin (Fig. 3G). Moreover, overexpression of GOT1 enhanced dmaKG toxicity in both UOK262-ev and 143B-CytB cells (Fig. 3H). As compared to dmaKG, AOA exerted a cytostatic effect in UOK262-ev cells and GOT1 overexpression decreased this effect (Fig. 3I). Similarly, overexpression of GOT1 augmented dmaKG toxicity in 143B-wt and 786-O, a renal cancer cell line, under hypoxia or phenformin-treated conditions, whereas it suppressed AOA and phenformin mediated cytotoxic effect (Supplementary Fig. S10A-B). These results highlight the differences in the actions of dmaKG and AOA, and they strongly suggest that dmaKG consumes aspartate and causes cell death in OXPHOS-incompetent cells through GOT1 activity.
Aspartate and adenylate depletions are the main causes of dmaKG cytotoxicity
As previously reported, we found that GOT1 expression became essential under respiration-deficient conditions (15). To determine whether aspartate depletion might be the cause of dmaKG cytotoxicity in OXPHOS-incompetent cells, we supplemented aspartate in the culture media. The results showed that adding 10 mM of aspartate almost completely rescued both HLRCC cell lines and 143B-CytB cells from dmaKG’s toxic and anti-proliferative effects (Fig. 4A-B, Supplementary Fig. S11A-B). Moreover, in UOK262 cells expressing SLC1A3 ectopically, 150 μM of aspartate effectively prevented dmaKG-induced cell death (Fig. 4C). Thus, the data support that dmaKG mediates cytotoxicity by inducing aspartate deficiency in these cells.
Figure 4. Cell death mediated by dmaKG is caused by aspartate and adenine nucleotide depletion.

(A) Cytotoxicity in UOK262-ev, UOK268-ev, and 143B-CytB following 24 hr treatment with dmaKG at the indicated concentrations with or without 10 mM aspartate.
(B) Cell proliferation as measured by crystal violet staining in UOK262-ev, UOK268-ev, and 143B-CytB following 72 hr of dmaKG treatment at the indicated concentrations with or without 10 mM aspartate.
(C) Cytotoxicity in UOK262 cells expressing SLC1A3 (UOK262-SLC1A3). Cells were treated with 6.6 mM dmaKG with or without 150 μM of aspartate.
(D) Measurement of adenine nucleotide pool (AMP+ADP+ATP) using a biochemical assay in UOK262 cell pair following dmaKG (6.6 mM) treatment at the indicated times. dmaKG caused a rapid and stable decline of adenylates in UOK262-ev. In UOK262-FH, after an initial decrease, adenylates reverts back to baseline levels. All treatment samples were compared to the mean of the respective control group.
(E) Adenine nucleotide pool levels in dmaKG-treated UOK262-ev (6.6 mM, 6 hr) and 143B-CytB (9.9 mM, 6 hr). Cells were treated in media with or without 10 mM aspartate supplementation.
(F) Cytotoxicity in UOK262-ev, UOK268-ev, and 143B-CytB following 24 hr treatment with dmaKG at the indicated concentrations with or without 110 μM adenine, 1.2 mM ATP, or a cell-permeable ATP analog AMP-PCP (100 μM).
All data are shown as mean and individual data points (two-way ANOVA with Tukey’s multiple comparisons, ns p>0.5, *p≤0.05, **p≤ 0.01, ***p≤0.001, ****p≤0.0001).
Because a major role of aspartate in proliferating cells is to participate in nucleotide synthesis, we assessed whether the dmaKG-mediated decrease in ATP might be in part due to adenylate depletion. Using a biochemical method, we found that adenylates (ATP/ADP/AMP) exhausted quickly upon dmaKG treatment in UOK262-ev but not in UOK262-FH cells (Fig. 4D). Moreover, aspartate supplementation restored adenylate levels in both UOK262-ev and 143B-CytB treated with dmaKG, indicating that the exhaustion was caused by aspartate deficiency (Fig. 4E). In another experiment using the UOK262 pair, we analyzed ATP, ADP and AMP levels individually and found that dmaKG reduced all of them in ev but not FH-replete cells. In contrast, the glycolysis inhibitors 2-DG and 3-bromopyrvate (3-BrPy) reduced only ATP but not ADP or AMP in both UOK262-ev and -FH cells (Supplementary Fig. S12). To determine the role of adenylate deficiencies, we added adenine or ATP to the media and observed significant rescue of dmaKG cytotoxicity, except that rescue with ATP was not seen in 143B-CytB (Fig. 4F). Uptake of ATP has been shown to vary amongst different cell types (39). We tested whether the ATP analog β,γ-methyleneadenosine 5’-triphosphate (AMP-PCP) might permeate cell membrane and found that it substantially reduced dmaKG sensitivity in 143B-CytB. We confirmed that adenylate levels were restored by ATP addition in UOK262-ev but not in 143B-CytB and by AMP-PCP in 143B-CytB (Supplementary Fig. S13A-B). In sum, these findings demonstrate that in OXPHOS-incompetent cells, adenylates are rapidly depleted when aspartate is limited. This depletion may lead to not only cessation of DNA synthesis and cell proliferation but also other downstream consequences that cumulatively contribute to cell death.
Aspartate and adenylate deficiencies lead to inhibition of glycolysis
We sought to investigate further the downstream consequences of aspartate and adenylate deficiencies and to explore the mechanism by which dmaKG suppressed glycolysis in OXPHOS-incompetent cells. Surprisingly, supplementation of aspartate in UOK262-SLC1A3 as well as ADP or ATP in UOK262-ev nullified the effects of dmaKG on glycolysis and glycolytic capacity (Fig. 5A). Supplementation of aspartate and AMP-PCP also restored glycolysis and glycolytic capacity in dmaKG-treated 143B-CytB cells (Fig. 5B). These data imply that glycolysis might be halted due to the lack of adenylates. In addition to ADP and ATP, NAD+ is also an essential co-factor for glycolysis. We measured NAD+ and NADH in the UOK262 pair and found that the NAD+/NADH ratios were not significantly changed by dmaKG. However, total NAD+ and NADH level decreased in the presence of dmaKG, possibly because biosynthesis of NAD+ via both de novo and salvage pathways requires ATP (Supplementary Figure S14A-B). Likewise, we observed that NAD+ and NADH declined more slowly compared to the adenylates in UOK262-ev. Collectively these findings suggest that adenylates are essential for glycolysis. Therefore, adenylate depletion mediated by dmaKG and aspartate deficiency can inhibit glycolysis.
Figure 5. dmaKG shuts down glycolysis and the mTOR pathway in OXPHOS-deficient cells.

(A-B) ECAR values indicating glycolysis and glycolytic capacity as determined using the Seahorse XF Glycolysis Stress assay. Cell types and treatment conditions are as indicated in each panel. dmaKG, 6.6 mM in (A) and 9.9 mM in (B); ADP, 1.2 mM; ATP, 1.2 mM; AMP-PCP, 100 μM; aspartate (Asp), 150 μM in (A) and 10 mM in (B).
(C) Immunoblotting for components of mTOR pathway in UOK262, UOK268, and 143B cell pairs. Cells were treated with dmaKG (6.6 mM for UOK262 and UOK268 cell pairs and 9.9 mM for 143B cell pair) for the indicated times.
(D) Immunoblotting for components of mTOR pathway in UOK262-ev and 143B-CytB. Cells were treated with dmaKG (6.6 mM for UOK262-ev and 9.9 mM for 143B-CytB) for the indicated times. Media was additionally supplemented with or without aspartate (Asp, 10 mM), ATP (1.2 mM) or AMP-PCP (100 μM).
Data in bar graphs are shown as mean and individual data points (two-way ANOVA with Tukey’s multiple comparisons, ns p>0.5, *p≤0.05, **p≤ 0.01, ***p≤0.001, ****p≤0.0001).
Aspartate and adenylate deficiencies lead to inhibition of the mTOR pathway
The deficiencies in aspartate and adenylates also prompted us to investigate the mTOR pathway, which is a major nutrient-sensing mechanism in the cell (40). Both amino acid and purine nucleotide shortages can abrogate mTOR activity, which normally stimulates protein synthesis via phosphorylation of P70S6K1 and 4E-BP1 (41-43). In HLRCC and 143B cell pairs, we found that dmaKG suppressed phosphorylation of P70S6K1, 4E-BP1 and S6 more strongly in the OXPHOS-deficient cells than in the OXPHOS-proficient cells (Fig. 5C). Supplementation of aspartate and ATP in UOK262-ev or aspartate and AMP-PCP in 143B-CytB reversed these effects, indicating that dmaKG-mediated aspartate and adenylate deficiencies can lead to mTOR inhibition (Fig. 5D). Inhibition of mTOR will lead to growth arrest and may contribute to cell death (40). Meanwhile, since HIF-1α is known to be translationally regulated by mTOR, these findings suggest that dmaKG downregulates HIF-1α in HLRCC cells but not in their FH-replete cell pairs due to its selective inhibition of mTOR in OXPHOS-incompetent cells (36).
In vivo anti-tumor activity of dmaKG
To gain insight into dmaKG’s effects on OXPHOS-deficient tumors in vivo, we generated xenograft tumors in mice using the 143B cell pair. In the 143B-CytB tumor model, tumor growth curves began to diverge between the control group and the dmaKG group after treatment started (Fig. 6A, left panel). By Day 6, numerous metastatic tumor nodules appeared in two out of six mice in the control group, hence their tumor volumes were not counted thereafter. On the other hand, in the dmaKG group, tumor metastasis was not found in any of the six mice (Fig. 6B) and tumor growth was impeded. In contrast, in the 143B-wt xenograft tumor model, treatment with dmaKG or phenformin did not affect tumor growth, whereas dmaKG and phenformin co-treatment effectively abrogated tumor progression (Fig. 6A, right panel). No tumor metastasis was found in any of the 143B-wt xenografts, consistent with a previous report implicating an inverse relationship between OXPHOS competency and metastatic potential of 143B tumors (23). In addition, both aspartate and adenylate levels were significantly reduced by dmaKG treatment in 143B-CytB tumors and by dmaKG and phenformin co-treatment in 143B-wt tumors (Fig. 6C-D). Accordingly, Western blot analysis showed that mTOR signaling was inhibited by dmaKG treatment in 143B-CytB tumors and by dmaKG and phenformin co-treatment in 143B-wt tumors (Supplementary Fig. S15A-B). We also extracted brain, heart, kidney, liver, lung, and spleen from the mice bearing 143B-wt tumors and found that neither aspartate nor adenine nucleotide pool levels were significantly affected in these organs by any of the treatments (Fig. 6E-F). The mouse body weight, a complete blood count (CBC) analysis and a panel of blood chemistry assays measured at the conclusion of the experiment also did not find any toxicity caused by the treatments (Supplementary Fig. S16A-C).
Figure 6. dmaKG exerts antitumor effects with high specificity in mouse xenograft tumor models.

(A) Tumor volume of 143B-CytB (left panel) and 143B-wt (right panel) xenograft tumor after treatment with indicated drugs for the length of time as specified. Data are shown as mean and individual data points (n=6 except that two of the 143B-CytB tumors in the saline group were not taken into account from 6 days after treatment started due to metastases).
(B) Bar graph indicating the percentage of metastatic tumors in 143B-CytB xenografts for the duration of the treatment.
(C) Mass spectrometry analysis of aspartate levels in 143B-CytB (left panel) and in 143B-wt (right panel) tumors from the above experiments.
(D) Bar graphs plotted with the relative luminescence units (RLU) indicating the abundances of adenylates (AMP, ADP, ATP) in 143B-CytB (left panel) and 143B-wt (right panel) tumors. Statistical analysis was performed to compare the abundances of total adenine nucleotide pool (ATP+ADP+AMP) in each treatment group versus the saline-injected groups. Statistical significances are indicated above a horizontal bar of each group.
(E) Mass spectrometry analysis of aspartate levels in 143B-wt xenograft tumors.
(F) Adenylate levels in the various vital organs of mice harboring 143B-wt tumors in each treatment group.
Statistical analyses were performed for 143B-CytB data (A, C-D) using unpaired two-tailed t test with Welch’s correction and for 143B-wt data (A, C-F) using one-way ANOVA with Brown-Forsythe and Welch correction (ns p>0.5, *p≤0.05, **p≤ 0.01, ***p≤0.001, ****p≤0.0001).
We additionally investigated dmaKG’s effects using UOK262 xenograft tumor model. It has been previously shown that while UOK262-ev can form xenograft tumors in mice, UOK262-FH cannot, suggesting that the FH mutation is crucial for the tumorigenicity of HLRCC cells (38). Similar to its effect on 143B-CytB tumors, dmaKG significantly reduced the progression, aspartate level, and mTOR signaling in UOK262 tumors (Supplementary Fig. S17A-C). In tumor sections from dmaKG-treated mice, regions of dead cells were clearly visible, whereas such regions were seldom observed in tumor sections from saline-treated animals (Supplementary Fig. S17D-E). Meanwhile, no tissue damage was seen in sections of heart, liver and kidneys after 3 weeks of treatment and mouse weight changes between control and dmaKG-treated groups were similar (Supplementary Fig. S18A-B). These data indicate that treatment with dmaKG is well tolerated and depletes aspartate in OXPHOS-impaired tumors selectively, providing important evidence of dmaKG’s anti-cancer efficacy in vivo.
Discussion
Recent studies have shown that aspartate, especially under the conditions of impaired mitochondrial metabolism, is a limiting factor for tumor growth (12,18). However, a strategy to target aspartate availability in cancers with mitochondrial dysfunction had yet to be identified. Meanwhile, it was unclear whether one could selectively deplete aspartate in cancer without affecting normal cells in vivo. In this report, we identify that cell-permeable aKG esters, such as dmaKG, are toxic to cancers exhibiting mitochondrial metabolism deficiency by promoting aspartate utilization and depletion via GOT1, therefore rendering aspartate inaccessible to alternative biosynthetic usage (Fig. 7). Moreover, the current study provides encouraging in vivo evidence that indicates anticancer efficacy and specificity of this approach, warranting its future clinical investigations.
Figure 7. The cytotoxic mechanism and the targeting specificity of dmaKG.

(Left panel) Under OXPHOS-competent conditions, aspartate is normally produced by GOT2 in the mitochondria. dmaKG can increase intracellular aKG levels and promote mitochondrial metabolism to maintain aspartate levels. (Right panel) Under OXPHOS-incompetent conditions, cells rely on GOT1 in the cytosol for aspartate synthesis. Aberrant elevation in aKG levels can result in nitrogen trapping (converting aKG to Glu) via GOT1, leading to aspartate exhaustion. Blue arrows depict the mode of action of dmaKG. Glu, glutamate; OAA, oxaloacetate.
Notably, the data suggest that there are several differences between respiration-proficient and respiration-deficient cells that contribute to their differential sensitivities to dmaKG (Fig. 7). First, after entering the cell, dmaKG is converted into aKG by esterases, causing a surge of intracellular aKG level in both conditions. However, competency in mitochondrial metabolism can cause aKG levels to normalize quickly, presumably through its oxidative utilization by the mitochondrial TCA cycle. Thus, OXPHOS-incompetent cells may experience a higher amount of aKG accumulation for a longer period of time compared to OXPHOS-competent cells due to their mitochondrial metabolism defects. Second, the increase in aKG level is likely to be higher in the cytosol than in the mitochondria, causing a stronger effect on GOT1 as compared to GOT2. It is known that GOT1 is the main aspartate producer in OXPHOS-incompetent conditions in contrast to GOT2 in OXPHOS-competent conditions (15). Third, aKG can increase TCA cycle metabolites such as malate inside the mitochondria of OXPHOS-competent cells. Although we cannot reliably detect oxaloacetate levels by mass spectrometry, increases in malate may increase oxaloacetate, thereby boosting aspartate production via GOT2 in OXPHOS-competent conditions. All of these mechanisms combined may account for the selective depletion of aspartate in cancer cells with mitochondrial dysfunction. As compared to the GOT1/2 inhibitor AOA, which inhibits aspartate synthesis and reduces aspartate levels equally between OXPHOS-competent and -incompetent conditions, dmaKG converts GOT1 from an aspartate producing enzyme to an aspartate utilizing enzyme in OXPHOS-incompetent conditions (Fig. 7). These findings highlight the unique therapeutic specificity of dmaKG for OXPHOS-incompetent tumors, which will reduce the likelihood of adverse effects on normal cells.
While dmaKG induces potent cytotoxicity in a variety of cancer cell types under OXPHOS-incompetent conditions, the concentration required for its toxicity varies. This may be due to differences in the endogenous metabolite levels and the rates of metabolism in different cell types. However, even at non-toxic concentrations, dmaKG also inhibits cell proliferation preferentially in OXPHOS-incompetent conditions. Similarly, using GOT1 knockout cells, Birsoy et al. showed that the level of aspartate depletion determines whether cells die or become growth arrested (15). In addition to OXPHOS deficiency, it is possible that some genetic mutations which alter the aspartate synthesis pathways, aspartate utilization, or the cell’s reliance on redox balance mediated by the aspartate-malate shuttle may also induce dmaKG sensitivities (44,45). Thus, cell-permeable aKG derivatives may confer anti-cancer effects in cancer types other than those with TCA cycle or mtDNA mutations.
Aspartate is a substrate for biosynthesis from phosphoribosyl pyrophosphate (PRPP) to inosine monophosphate (IMP) and from IMP to AMP. We show that dmaKG leads to a rapid decline of adenylates in OXPHOS-incompetent cells due to aspartate deficiency. This is consistent with the results of Garcia-Bermudez et al., which show that the rate of aspartate-mediated nucleotide synthesis increases under OXPHOS-incompetent conditions (12). Moreover, dmaKG mediates suppression of glycolysis and mTOR inhibition. Unexpectedly, we found that restoration of adenylate levels restores glycolysis. Interestingly, mature human erythrocytes are devoid of de novo nucleotide biosynthesis. It has been reported that restoration of adenylates in aged erythrocytes also restores glycolytic activities in these cells (46). Taken together, these results demonstrate that adenine nucleotides can play a limiting role in glycolysis. In addition, we show that supplementation of either aspartate or adenylates restore mTOR activity. Although mTOR is known to integrate a plethora of signals ranging from nutrients to growth factors to stress, these data suggest that at least one of the early signals that leads to mTOR inactivation in dmaKG-treated cells is adenylate depletion. Previously, two groups independently reported that mTOR complex 1 (mTORC1) is distinctly sensitive to purine but not pyrimidine nucleotide levels (41,43). Hoxhaj et al. show that inactivation of mTORC1 by purine deficiency is not mediated by the energy sensor AMP kinase (AMPK) (43). Collectively, the evidence indicates that dmaKG mediates cytotoxicity by depleting aspartate and adenylates in respiration-incompetent cancer cells. This leads to cessation of glycolysis, the major energy producing mechanism in these cells, as well as to inactivation of mTOR, the indispensable cell growth signaling pathway.
Using mouse xenograft tumor models, we found that in vivo administration of dmaKG replicates its effects observed in the cell culture system. It hampers the progression of UOK262 and 143B-CytB tumors that have intrinsic mitochondrial dysfunction, greatly reduces aspartate and adenine nucleotide levels, and inhibits mTOR signaling in them. These effects are not seen in 143B-wt tumors that have functional mitochondrial metabolism, indicating the specificity of this therapeutic strategy. Treatment of 143B-CytB-implanted animals with dmaKG also completely abrogated metastatic nodules. Additionally, our cell culture experiments indicate that dmaKG sensitivity can be enhanced by hypoxia or by co-treatment with an ETC inhibitor in cancers that do not have intrinsic mitochondrial defects. Since cancer cells in vivo often reside in a variable degree of hypoxic environment, it will be informative to investigate the potential added benefits of dmaKG in conjunction with other standard therapeutic approaches. We demonstrate that dmaKG and phenformin in combination, but not individually, impair the progression of 143B-wt tumors. While we were pursuing these experiments, Sica et al. reported that ETC complex I inhibitor BAY87-2243 elicits synthetic lethality with dmaKG and concluded that cell death is caused by energy failure due to inhibition of OXPHOS and a MDM2-dependent inhibition of glycolysis (47). Although their report did not investigate the roles of GOT1, it independently confirms the therapeutic synergy between dmaKG and OXPHOS inhibition. Interestingly, using a number of analyses in mouse blood and various organs, we have not found any significant toxicity in the mice treated with dmaKG or co-treated with dmaKG and phenformin for a short period (15 - 21 days). Moreover, aspartate and adenylate levels in various mouse organs are unaffected, in contrast to their significant decreases in the tumors. These data may imply that cancer cells and normal cells are differentially sensitive to ETC inhibitors in vivo, which could be due to some intrinsic differences in their mitochondrial metabolism efficiencies or extrinsic differences in the oxygen levels of their environments. In other studies, dmaKG has been shown to confer cardiac protection in a mouse model of thoracic aortic constriction and to extend lifespan in nematodes (33,48,49). Together, these findings suggest that aKG derivatives may have positive safety profiles, although a thorough preclinical evaluation of toxicity for a long-term treatment with dmaKG is needed. In conclusion, this study identifies a new and promising therapeutic strategy, which may form the basis for the development of various broad-spectrum anti-cancer approaches.
Supplementary Material
Significance.
Findings demonstrate that OXPHOS deficiency caused by either hypoxia or mutations, which can significantly increase cancer virulence, renders tumors sensitive to alpha-ketoglutarate esters by targeting their dependence upon GOT1 for aspartate synthesis.
Acknowledgements
The authors thank Drs. Cyril Benes, Matthew Vander Heiden, Lucas Sullivan, Navdeep Chandel, Youfeng Yang, and Marston Linehan for providing human cancer cell lines, Dr. Miguel Rivera for assistance on interpreting tumor sections, and Dr. Mohammed Mahamdeh for assistance on microscope image acquisition and processing. This work is supported by the Hassenfeld Scholar Award (to J.-R.J.Y.), NIH R01 CA215410 (to J.-R.J.Y.), NIH R01 CA160458 (to O.I.), and the Claudia Adams Barr Program (to E.T.C). A.M.I. is supported by the NIH/NCI (R37 CA251543, K08 CA201483, P30 CA008748).
Footnotes
Conflict of interest: none
References
- 1.Pavlova NN, Thompson CB. The Emerging Hallmarks of Cancer Metabolism. Cell Metab 2016;23(1):27–47 doi 10.1016/j.cmet.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mullen AR, DeBerardinis RJ. Genetically-defined metabolic reprogramming in cancer. Trends Endocrinol Metab 2012;23(11):552–9 doi 10.1016/j.tem.2012.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luengo A, Gui DY, Vander Heiden MG. Targeting Metabolism for Cancer Therapy. Cell Chem Biol 2017;24(9):1161–80 doi 10.1016/j.chembiol.2017.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vander Heiden MG, DeBerardinis RJ. Understanding the Intersections between Metabolism and Cancer Biology. Cell 2017;168(4):657–69 doi 10.1016/j.cell.2016.12.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nat Chem Biol 2015;11(1):9–15 doi 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vyas S, Zaganjor E, Haigis MC. Mitochondria and Cancer. Cell 2016;166(3):555–66 doi 10.1016/j.cell.2016.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lorendeau D, Rinaldi G, Boon R, Spincemaille P, Metzger K, Jager C, et al. Dual loss of succinate dehydrogenase (SDH) and complex I activity is necessary to recapitulate the metabolic phenotype of SDH mutant tumors. Metab Eng 2016. doi 10.1016/j.ymben.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Sullivan LB, Martinez-Garcia E, Nguyen H, Mullen AR, Dufour E, Sudarshan S, et al. The proto-oncometabolite fumarate binds glutathione to amplify ROS-dependent signaling. Mol Cell 2013;51(2):236–48 doi 10.1016/j.molcel.2013.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tyrakis PA, Yurkovich ME, Sciacovelli M, Papachristou EK, Bridges HR, Gaude E, et al. Fumarate Hydratase Loss Causes Combined Respiratory Chain Defects. Cell Rep 2017;21(4):1036–47 doi 10.1016/j.celrep.2017.09.092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chatterjee A, Mambo E, Sidransky D. Mitochondrial DNA mutations in human cancer. Oncogene 2006;25(34):4663–74 doi 10.1038/sj.onc.1209604. [DOI] [PubMed] [Google Scholar]
- 11.Larman TC, DePalma SR, Hadjipanayis AG, Cancer Genome Atlas Research N, Protopopov A, Zhang J, et al. Spectrum of somatic mitochondrial mutations in five cancers. Proc Natl Acad Sci U S A 2012;109(35):14087–91 doi 10.1073/pnas.1211502109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Garcia-Bermudez J, Baudrier L, La K, Zhu XG, Fidelin J, Sviderskiy VO, et al. Aspartate is a limiting metabolite for cancer cell proliferation under hypoxia and in tumours. Nat Cell Biol 2018;20(7):775–81 doi 10.1038/s41556-018-0118-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, et al. Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 2011;481(7381):380–4 doi 10.1038/nature10602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mullen AR, Wheaton WW, Jin ES, Chen PH, Sullivan LB, Cheng T, et al. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2011;481(7381):385–8 doi 10.1038/nature10642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, Sabatini DM. An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 2015;162(3):540–51 doi 10.1016/j.cell.2015.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gui DY, Sullivan LB, Luengo A, Hosios AM, Bush LN, Gitego N, et al. Environment Dictates Dependence on Mitochondrial Complex I for NAD+ and Aspartate Production and Determines Cancer Cell Sensitivity to Metformin. Cell Metab 2016;24(5):716–27 doi 10.1016/j.cmet.2016.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, Vander Heiden MG. Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 2015;162(3):552–63 doi 10.1016/j.cell.2015.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sullivan LB, Luengo A, Danai LV, Bush LN, Diehl FF, Hosios AM, et al. Aspartate is an endogenous metabolic limitation for tumour growth. Nat Cell Biol 2018;20(7):782–8 doi 10.1038/s41556-018-0125-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wilson WR, Hay MP. Targeting hypoxia in cancer therapy. Nat Rev Cancer 2011;11(6):393–410 doi 10.1038/nrc3064. [DOI] [PubMed] [Google Scholar]
- 20.Linehan WM, Rouault TA. Molecular pathways: Fumarate hydratase-deficient kidney cancer--targeting the Warburg effect in cancer. Clin Cancer Res 2013;19(13):3345–52 doi 10.1158/1078-0432.CCR-13-0304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tomlinson IP, Alam NA, Rowan AJ, Barclay E, Jaeger EE, Kelsell D, et al. Germline mutations in FH predispose to dominantly inherited uterine fibroids, skin leiomyomata and papillary renal cell cancer. Nat Genet 2002;30(4):406–10 doi 10.1038/ng849. [DOI] [PubMed] [Google Scholar]
- 22.Ishikawa K, Takenaga K, Akimoto M, Koshikawa N, Yamaguchi A, Imanishi H, et al. ROS-generating mitochondrial DNA mutations can regulate tumor cell metastasis. Science 2008;320(5876):661–4 doi 10.1126/science.1156906. [DOI] [PubMed] [Google Scholar]
- 23.Nunes JB, Peixoto J, Soares P, Maximo V, Carvalho S, Pinho SS, et al. OXPHOS dysfunction regulates integrin-beta1 modifications and enhances cell motility and migration. Hum Mol Genet 2015;24(7):1977–90 doi 10.1093/hmg/ddu612. [DOI] [PubMed] [Google Scholar]
- 24.Petros JA, Baumann AK, Ruiz-Pesini E, Amin MB, Sun CQ, Hall J, et al. mtDNA mutations increase tumorigenicity in prostate cancer. Proc Natl Acad Sci U S A 2005;102(3):719–24 doi 10.1073/pnas.0408894102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang Y, Valera V, Sourbier C, Vocke CD, Wei M, Pike L, et al. A novel fumarate hydratase-deficient HLRCC kidney cancer cell line, UOK268: a model of the Warburg effect in cancer. Cancer Genet 2012;205(7–8):377–90 doi 10.1016/j.cancergen.2012.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang Y, Valera VA, Padilla-Nash HM, Sourbier C, Vocke CD, Vira MA, et al. UOK 262 cell line, fumarate hydratase deficient (FH-/FH-) hereditary leiomyomatosis renal cell carcinoma: in vitro and in vivo model of an aberrant energy metabolic pathway in human cancer. Cancer Genet Cytogenet 2010;196(1):45–55 doi 10.1016/j.cancergencyto.2009.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adam J, Hatipoglu E, O’Flaherty L, Ternette N, Sahgal N, Lockstone H, et al. Renal cyst formation in Fh1-deficient mice is independent of the Hif/Phd pathway: roles for fumarate in KEAP1 succination and Nrf2 signaling. Cancer Cell 2011;20(4):524–37 doi 10.1016/j.ccr.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Isaacs JS, Jung YJ, Mole DR, Lee S, Torres-Cabala C, Chung YL, et al. HIF overexpression correlates with biallelic loss of fumarate hydratase in renal cancer: novel role of fumarate in regulation of HIF stability. Cancer Cell 2005;8(2):143–53 doi 10.1016/j.ccr.2005.06.017. [DOI] [PubMed] [Google Scholar]
- 29.Sciacovelli M, Goncalves E, Johnson TI, Zecchini VR, da Costa AS, Gaude E, et al. Fumarate is an epigenetic modifier that elicits epithelial-to-mesenchymal transition. Nature 2016;537(7621):544–7 doi 10.1038/nature19353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gameiro PA, Yang J, Metelo AM, Perez-Carro R, Baker R, Wang Z, et al. In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab 2013;17(3):372–85 doi 10.1016/j.cmet.2013.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, et al. Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab 2015;22(2):304–11 doi 10.1016/j.cmet.2015.06.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Intlekofer AM, Wang B, Liu H, Shah H, Carmona-Fontaine C, Rustenburg AS, et al. L-2-Hydroxyglutarate production arises from noncanonical enzyme function at acidic pH. Nat Chem Biol 2017;13(5):494–500 doi 10.1038/nchembio.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, Deng G, et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014;510(7505):397–401 doi 10.1038/nature13264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hou P, Kuo CY, Cheng CT, Liou JP, Ann DK, Chen Q. Intermediary metabolite precursor dimethyl-2-ketoglutarate stabilizes hypoxia-inducible factor-1alpha by inhibiting prolyl-4-hydroxylase PHD2. PLoS One 2014;9(11):e113865 doi 10.1371/journal.pone.0113865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.MacKenzie ED, Selak MA, Tennant DA, Payne LJ, Crosby S, Frederiksen CM, et al. Cell-permeating alpha-ketoglutarate derivatives alleviate pseudohypoxia in succinate dehydrogenase-deficient cells. Mol Cell Biol 2007;27(9):3282–9 doi 10.1128/MCB.01927-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hudson CC, Liu M, Chiang GG, Otterness DM, Loomis DC, Kaper F, et al. Regulation of hypoxia-inducible factor 1alpha expression and function by the mammalian target of rapamycin. Mol Cell Biol 2002;22(20):7004–14 doi 10.1128/mcb.22.20.7004-7014.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sourbier C, Ricketts CJ, Matsumoto S, Crooks DR, Liao PJ, Mannes PZ, et al. Targeting ABL1-mediated oxidative stress adaptation in fumarate hydratase-deficient cancer. Cancer Cell 2014;26(6):840–50 doi 10.1016/j.ccell.2014.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tong WH, Sourbier C, Kovtunovych G, Jeong SY, Vira M, Ghosh M, et al. The glycolytic shift in fumarate-hydratase-deficient kidney cancer lowers AMPK levels, increases anabolic propensities and lowers cellular iron levels. Cancer Cell 2011;20(3):315–27 doi 10.1016/j.ccr.2011.07.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qian Y, Wang X, Li Y, Cao Y, Chen X. Extracellular ATP a New Player in Cancer Metabolism: NSCLC Cells Internalize ATP In Vitro and In Vivo Using Multiple Endocytic Mechanisms. Mol Cancer Res 2016;14(11):1087–96 doi 10.1158/1541-7786.MCR-16-0118. [DOI] [PubMed] [Google Scholar]
- 40.Kim J, Guan KL. mTOR as a central hub of nutrient signalling and cell growth. Nat Cell Biol 2019;21(1):63–71 doi 10.1038/s41556-018-0205-1. [DOI] [PubMed] [Google Scholar]
- 41.Emmanuel N, Ragunathan S, Shan Q, Wang F, Giannakou A, Huser N, et al. Purine Nucleotide Availability Regulates mTORC1 Activity through the Rheb GTPase. Cell Rep 2017;19(13):2665–80 doi 10.1016/j.celrep.2017.05.043. [DOI] [PubMed] [Google Scholar]
- 42.Goberdhan DC, Wilson C, Harris AL. Amino Acid Sensing by mTORC1: Intracellular Transporters Mark the Spot. Cell Metab 2016;23(4):580–9 doi 10.1016/j.cmet.2016.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoxhaj G, Hughes-Hallett J, Timson RC, Ilagan E, Yuan M, Asara JM, et al. The mTORC1 Signaling Network Senses Changes in Cellular Purine Nucleotide Levels. Cell Rep 2017;21(5):1331–46 doi 10.1016/j.celrep.2017.10.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Allen EL, Ulanet DB, Pirman D, Mahoney CE, Coco J, Si Y, et al. Differential Aspartate Usage Identifies a Subset of Cancer Cells Particularly Dependent on OGDH. Cell Rep 2016;17(3):876–90 doi 10.1016/j.celrep.2016.09.052. [DOI] [PubMed] [Google Scholar]
- 45.Ilic N, Birsoy K, Aguirre AJ, Kory N, Pacold ME, Singh S, et al. PIK3CA mutant tumors depend on oxoglutarate dehydrogenase. Proc Natl Acad Sci U S A 2017;114(17):E3434–E43 doi 10.1073/pnas.1617922114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tsuboi KK. Limiting Role of Adenine Nucleotide in the Glycolysis of the Human Erythrocyte. J Biol Chem 1965;240:582–7. [PubMed] [Google Scholar]
- 47.Sica V, Bravo-San Pedro JM, Izzo V, Pol J, Pierredon S, Enot D, et al. Lethal Poisoning of Cancer Cells by Respiratory Chain Inhibition plus Dimethyl alpha-Ketoglutarate. Cell Rep 2019;27(3):820–34 e9 doi 10.1016/j.celrep.2019.03.058. [DOI] [PubMed] [Google Scholar]
- 48.Marino G, Pietrocola F, Eisenberg T, Kong Y, Malik SA, Andryushkova A, et al. Regulation of autophagy by cytosolic acetyl-coenzyme A. Mol Cell 2014;53(5):710–25 doi 10.1016/j.molcel.2014.01.016. [DOI] [PubMed] [Google Scholar]
- 49.Marino G, Pietrocola F, Kong Y, Eisenberg T, Hill JA, Madeo F, et al. Dimethyl alpha-ketoglutarate inhibits maladaptive autophagy in pressure overload-induced cardiomyopathy. Autophagy 2014;10(5):930–2 doi 10.4161/auto.28235. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.