

Abstract
Natural killer (NK) cells are potent immune modulators that can quickly lyse tumor cells and elicit inflammatory responses. These characteristics make them ideal candidates for immunotherapy. However, unlike T cells, NK cells do not possess clonotypic receptors capable of specific antigen recognition and cannot expand via activating receptor signals alone. To enable NK cells with these capabilities, we created and have previously described a Tri-specific Killer Engager (TriKE™) platform capable of inducing antigen specificity and cytokine-mediated NK cell expansion. TriKE molecules have three arms: (i) a single chain variable fragment (scFv) against the activating receptor CD16 on NK cells, to trigger NK cell activation; (ii) an scFv against a tumor-associated antigen (CD33 here), to induce specific tumor target recognition; and (iii) an IL15 moiety, to trigger NK cell expansion and priming. Here, we demonstrated that by modifying the anti-CD16 scFv with a humanized single domain antibody against CD16, we improved TriKE functionality. A CD33-targeting second-generation TriKE induced stronger and more specific NK cell proliferation without T-cell stimulation, enhanced in vitro NK cell activation and killing of CD33-expressing targets, and improved tumor control in preclinical mouse models. Given these improved functional characteristics, we propose rapid translation of second-generation TriKEs into the clinic.
Keywords: Natural killer cells, TriKE, Tri-specific antibodies, IL15, Immunotherapy
Introduction
Natural killer (NK) cell-based immunotherapies are quickly gaining traction preclinically and in early phase clinical trials (1). This is due to the ability of NK cells to lyse tumors with minimal priming, their lack of MHC restriction, their capacity to exert antibody-dependent cellular cytotoxicity (ADCC), and to robustly and rapidly produce inflammatory cytokines (2). A number of NK cell immunotherapy modalities are being actively explored, including cellular, cytokine, checkpoint inhibition, and ADCC-driven therapies (3). Developing affordable NK cell-based immunotherapeutic off-the-shelf products will significantly strengthen clinical practice.
Both cytokine- and ADCC-mediated immunotherapies are ideal candidates for off-the-shelf approaches. NK cells express cytokine receptors that modulate NK cell effector function, development, proliferation, and homeostasis. IL2 and IL15 activate a host of downstream signaling molecules that result in enhanced function, proliferation, and survival (4, 5). Because of reduced toxicities and its NK cell–stimulating effects without the augmentation of regulatory T cells (Tregs), IL15 has become a priority cytokine of interest in NK immunotherapy (6, 7). Recombinant human IL15 and an IL15 superagonist complex (N-803, formerly ALT-803) are under investigation in several tumor settings alone and in combination with adoptive transfer in patients with leukemia (8–11). Early clinical data indicate that IL15 can induce NK cell expansion necessary to enhance immunotherapy (12, 13). IL15 also has the ability to prime CD16 signaling on NK cells (14). Amongst these approaches, NK cell–driven function through monoclonal antibodies and through bi- and tri-specific engagers is of significant clinical interest (15–19). Our group previously described a platform, termed Tri-specific Killer Engager (TriKE), which merges the concepts of cytokine signaling and bi-specifics into a single molecule to maximize NK cell immunotherapeutic potential (3).
TriKE molecules are composed of three arms: a single chain variable fragment (scFv) targeting the activating receptor CD16 on NK cells, a scFv targeting a tumor-associated antigen (TAA), and an IL15 moiety to drive NK cell expansion, survival, and priming (3,16). We previously described TriKE molecules against a host of TAAs, including a molecule targeting CD33 meant to enhance NK cell immunotherapy against myeloid malignancies including acute myeloid leukemia (AML), myelodysplastic syndromes (MDS), and other CD33-expressing malignancies such as systemic mastocytosis (20–22). This first-generation TriKE molecule, termed 161533 in previous studies, is called scFv16-m15–33 in this context for clarity and contains a mutant IL15 with an N72D substitution (abbreviated m15)(23), and a phase I/II clinical trial is underway for treatment of refractory AML, high-risk MDS, and systemic mastocytosis (NCT03214666). The present study describes the effect of substituting a “humanized” anti-CD16 single domain, camelid antibody to replace the anti-CD16 scFv, which enabled presentation of wild-type (wt) IL15 without use of m15. The resulting molecule, cam16-wt15–33 TriKE, displays stronger IL15 signaling capabilities, better NK cell activation, and increased NK cell–mediated tumor control both in vitro and in vivo. Thus, we postulate that the second-generation TriKE molecule may prove more efficacious for clinical development.
Materials and Methods
Isolation of immune cells, transplant patient samples, and cell lines
Human healthy donor blood was obtained from Memorial Blood Bank (Minneapolis, MN), and processed to isolate peripheral blood mononuclear cells (PBMCs) using density gradient Ficoll-Paque (GE Healthcare). PBMCs were either cryopreserved in liquid nitrogen with a mix of 90% fetal bovine serum (GIBCO) and 10% DMSO (SIGMA), or were processed fresh to enrich NK cells (90–95%) using the EasySep Human NK Cell Enrichment Kit (Cat. No:19055, STEMCELL Technologies), or were CD3/CD19-depleted using positive-selection kits (Cat. No:17851&17854, STEMCELL Technologies).
All human samples, including AML patient blasts, were obtained after approval from the Institutional Review Board (IRB) at the University of Minnesota and following written informed consent, received in compliance with guidelines by the Committee on the Use of Human Subjects in Research and in accordance with the Declaration of Helsinki. PBMCs collected 28 days after double umbilical cord blood transplant [N=9; are umbilical cord blood transplants but use two units from separate donors to maximize stem cell input] or matched sibling donor transplant [N=9; four bone marrow and five G-CSF–mobilized peripheral blood stem cell grafts] were cryopreserved and stored by Translational Therapy Shared Resource (Masonic Cancer Center, University of Minnesota). For the patient-derived xenograft model, AML blasts were collected between 2012–2016 and cryopreserved from an apheresis of a de novo (no prior chemotherapy) AML patient with normal cytogenetics, which resulted in 87% AML blasts. AML blasts were 98.9% CD34+ and 99.2% CD33+. Samples were stored by Heme Malignancy Tissue Bank at the University of Minnesota, also managed by the Translational Therapy Shared Resource.
The native CD33+ acute promyelocytic leukemia cell line HL-60 was received from American Type Culture Collection (ATCC) on April 2017 for use in this study. HL-60luc cells, obtained from Xianzheng Zhou on 2013 at the University of Minnesota (now at New York Medical College), were authenticated using short tandem repeat (STR) DNA profiling, according to the International Cell Line Authentication Committee (ICLAC) by the University of Arizona Genetics Core on March 2017. HL-60 and HL-60luc cells were used for studies outlined here from 2017–2020. Cells were cultured in RPMI 1640 (Gibco; cat#: 224000–089) supplemented with 10% fetal bovine serum (Gibco cat#: 2614–079) at 37°C, 5% CO2. CTLL-2 cells were received from ATCC in August 2016 and also cultured in RPMI 1640 supplemented with 10% fetal bovine serum and IL2 (Prometheus, 100 U/ml) at 37°C, 5% CO2. All cell lines used in studies were utilized at an early passage (P1-P3) and were mycoplasma negative using the Universal Mycoplasma Detection Kit (ATCC) per the manufacturer’s protocol.
Mice
Mouse experiments were performed after approval from and in accordance with and the guidelines of Institutional Animal Care and Use Committee (IACUC) at the University of Minnesota. NOD scid gamma (NSG) mice and NSG-SGM3 triple transgenic mice, expressing human IL3, GM-CSF, and SCF, were obtained from Jackson Laboratories. Age matched mice, in the 6–8 week old range for the PDX model and 8–10 week old range for the HL-60 model, were used in all studies.
ELISA for in vitro and in vivo detection of IL15
Equimolar concentrations of scFv16-m15–33 and rhIL15 were serially diluted as noted in RPMI media or heat-inactivated Human AB serum (Valley Biomedical, Inc.). The Quantikine ELISA kit (R&D Systems) was used per the manufacturer’s protocol to detect IL15. For in vivo detection of IL15, NSG mice were injected intravenously with equimolar amounts of scFv16-m15–33 (25 μg) or rhIL15 (4.85 μg). Facial vein bleeds were carried out to collect serum at 30, 60, 120, and 240 minutes post-injection for IL15 quantitation. Serum samples were diluted 1:100 and 1:1,000 for the IL15 ELISA assay. An IL15 standard curve, using the manufacturer provided recombinant IL15 within the ELISA KIT, was established to determine the relative concentration. A Tecan Infinite M200Pro plate reader was used to measure absorbance.
Measuring NADP activity of IL15
Prior to beginning of the assay, IL2 was washed off, and 20,000 CTLL-2 cells were incubated in a 96-well plate for 48 hours with indicated concentrations of scFv16-m15–33 and rhIL15. Cells were then incubated for 1 hour at 37°C, 5% CO2 with resazurin (Cat. No:AR002, R&D Systems), a blue reagent that is reduced by metabolically active viable cells to form pink resorufin, identifiable at 570 nm absorbance (24). A Tecan Infinite M200Pro plate reader was used to measure absorbance.
cam16-wt15–33 TriKE construct
Production steps of the second-generation TriKE cam16-wt15–33, including construction, verification, isolation, and purification, were performed as previously described for the first-generation TriKE (20,25). The final construct of the cam16-wt15–33 NcoI/XhoI gene fragment was encoded from a start codon, a humanized camelid anti-CD16 VHH (cam16), a 20 amino acid flanking sequence (PSGQAGAAASESLFVSNHAY), wild-type human IL15, a 7–amino acid flanking sequence (EASGGPE), and anti-CD33 scFv to construct the TriKE sequence (Patent US 2018/0282386 A1).
OCTET
An Octet-based label-free binding assay (OctetRED96, ForteBio, Molecular Devices) was performed to measure binding affinity of the analytes scFv16-m15–33 and cam16-wt15–33 to the ligands CD16a and CD16b. The ligands, rhCD16A or rhCD16B with a c-terminal 6x-HIS tag (R&D Systems cat#: 4325-FC-050 and 1597-FC-050/CF, respectively) were captured on biosensor tips using His tag-specific dip and read biosensors. The biosensor coated with a ligand was dipped into buffer containing an analyte for association and then dipped into assay buffer (PBS). The binding and dissociation was measured using Bio-Layer Interferometry technology. The OCTET assays were carried out by Precision Antibody (Columbia, MD).
Proliferation assay
The CellTrace Violet Proliferation Kit (Cat. No:C34557, Thermo Fisher) was used to evaluation proliferation (dye dilution) by flow cytometry. 300,000 PBMCs were labeled, treated with indicted proteins (human rhIL15 obtained from R&D systems cat#: 247-ILB) diluted in RPMI 1640 (Gibco; cat#: 224000–089) supplemented with 10% fetal bovine serum (Gibco cat#: 2614–079), harvested 7 days later and analyzed on an LSRII (BD Biosciences). For the characterization of NK and T cells, LIVE/DEAD NEAR IR (Cat. No: L34976, Thermo Fisher), PE-CY7–conjugated anti-CD56 (HCD56, BioLegend), and PE-CF594–conjugated anti-CD3 (UCHT1, BD BioSciences) were used. NK cells and T cells were identified as LiveDead–CD56+CD3– and LiveDead–CD56–CD3+ cells, respectively. Cells were run on an LSRII flow cytometer and analyzed using FlowJo software (BD Biosciences).
Activation assay
Noted human PBMCs (500,000/well) were incubated overnight with noted treatments, harvested, and stained with Live/Dead Fixable Aqua Staining Kit (Cat. No: L-34966, Thermo Fisher), anti-CD56, anti-CD3, PE-conjugated anti-CD69 (FN50, BioLegend), APC-conjugated anti-CD25 (M-A251, BioLegend) to evaluate NK cell activation marker expression with flow cytometry. Cells were run on an LSRII flow cytometer and analyzed using FlowJo software (BD Biosciences).
Function assay measuring CD107a degranulation and IFNγ cytokine production
Flow cytometric assessment of NK cell function was carried out as previously described (25). Briefly, following addition of noted treatments (diluted RPMI 1640 (Gibco; cat#: 224000–089) supplemented with 10% fetal bovine serum (Gibco cat#: 2614–079)) and HL-60 tumor targets (at a 2:1 effector:target ratio), healthy donor, or post-transplant patient PBMCs were stained with FITC-conjugated anti-CD107a (H4A3, BioLegend) at the beginning of a 4-hr incubation. One hour after the addition of anti-CD107a, cells were given Golgi Stop (1:1,500) and Golgi Plug (1:1,000; both from BD Biosciences), and incubated for 3 hrs. Cells were then stained with the Live/Dead Fixable Aqua Staining Kit (Cat. No: L-34966, Thermo Fisher), anti-CD56, and anti-CD3, fixed in 2% paraformaldehyde (Fisher Scientific cat#: 50980487) and permeabilized (eBioscience cat#: 00–8333–56). Permeabilized cells were stained with BV650-conjugated IFNγ (4S.B3, BioLegend), and were evaluated with flow cytometry. Effectors (normal donor or day 28 post-transplant patient PBMCs) were cocultured with targets in each of the experiments. Cells were run on an LSRII flow cytometer and analyzed using FlowJo software (BD Biosciences).
Real-time tumor killing
20,000 HL-60 cells were labeled using CellTrace Far Red Proliferation Kit (Cat. No:C34564, Thermo Fisher). Healthy donor enriched NK cells were incubated at a 2:1 effector:target ratio with CellTrace Far Red–labeled HL-60 targets with noted treatments (30 nM) and with IncuCyte Caspase-3/7 Green Apoptosis Assay reagent (1:250, cat#: 4440, Essen BioScience) in RPMI 1640 (Gibco; cat#: 224000–089) supplemented with 10% fetal bovine serum (Gibco cat#: 2614–079). Plates were placed in an IncuCyte Zoom (Sartorius Inc.) for 24 hours. Readings were taken every 30 minutes. A graph was created representing the percentage of live HL-60 targets (CellTrace Far Red+Caspase3/7–) normalized to live targets alone at the starting time point (0 hr).
In Vivo models
The previously described HL-60luc/human NK cell xenogeneic NSG mouse model was implemented (20). Briefly, mice were irradiated (275 cGy), intravenously (IV) injected with 0.75×106 HL-60luc cells, injected IV with 1×106 CD3/CD19-depleted human NK cells three days later and treated for two weeks intraperitoneally (IP); 5 days a week for two weeks with different concentrations (90, 30, or 10 μg/injection) of the second-generation cam16-wt15–33 TriKE or the first-generation scFv16-m15–33 TriKE. At days 7, 14, and 21, tumor load was assessed after D-luciferin (GoldBio) intraperitoneal injection (150 mg/kg) using bioluminescent imaging (IVIS 100 In Vivo Imaging System). Mice were bled via facial vein on day 21 (100 μl blood per mouse), red blood cells lysed using a quick (45 second) water lysis, then resuspended in PBS and cells were stained with Brilliant Violet 605–conjugated anti-human CD45 (HI30, BioLegend), anti-CD56, and anti-CD3. Data was acquired on an LSRII flow cytometer for 60 secs/sample and analyzed using FlowJo software (BD Biosciences).
An AML patient-derived xenograft (PDX) model was used to compare the activity of cam16-wt15–33 to rhIL-15. 6–8 week old NSG-SGM3 mice were irradiated (125 cGy) and injected IV the next day with 2×106 primary AML cells. Progress of AML engraftment was monitored with weekly facial vein bleeds stained for anti-CD45 and APC-conjugated anti-CD33 (WM53, BioLegend) to determine the proportion of CD45+CD33+ blast cells until detection of at least 1% human AML in blood was achieved (about 6 weeks after AML injection). Mice then received 3×106 CD3/CD19-depleted NK cells and subsequently received IP treatments of either 5 μg rhIL-15 three times weekly or 30 μg cam16-wt15–33 five times weekly for a total of 3 weeks. Mice were euthanized and blood, bone marrow, and spleen was harvested on day 21. Blood and splenocytes were stained with anti-human CD45, anti-CD56, and anti-CD3 to determine CD45+CD56+CD3– NK cell expansion. Bone marrow was stained with anti-human CD45, FITC–conjugated anti-CD34 (561, BioLegend), and anti-CD33 to determine CD45+CD33+ AML blasts.
Statistical analysis
GraphPad Prism (GraphPad Prism Software, Inc, La Jolla, CA) was used to calculate statistics via One-Way ANOVA or Paired T test methods (as indicated) and to generate graphs with error bars showing the mean ± SEM and statistical significance coded as *P<0.05, **P< 0.01, ***P<0.001, and ****P<0.0001.
Results
IL15 potency is diminished in first-generation TriKEs
We previously reported a TriKE composed of a single-chain variable fragment (scFv) against CD16 (scFv16), an N72D mutant IL15 (m15) shown to signal better than wild type IL15, and an scFv against CD33 termed scFv16-m15–33 (20, 23). Because versions of this molecule containing a wild-type IL15 moiety were not functional, the mutant IL15 version of this first-generation TriKE, scFv-m15–33, was selected. Although this molecule displays cytotoxic activity mediated by NK cell engagement and proliferative activity by the IL15 domain, we hypothesized that the activity of the IL15 moiety might be compromised in the first-generation TriKE backbone. To test this, IL15 protein detection in the scFv16-m15–33 TriKE was compared to detection of equimolar amounts of recombinant human IL15 (rhIL15) using an ELISA assay (Figure 1A). Data indicated a significant reduction in IL15 detection when comparing the scFv16-m15–33 TriKE to rhIL15. Results were identical when comparing detection in media (RPMI with 10% FBS) or human serum.
Figure 1. Design of second-generation cam16-wt15–33 TriKE.
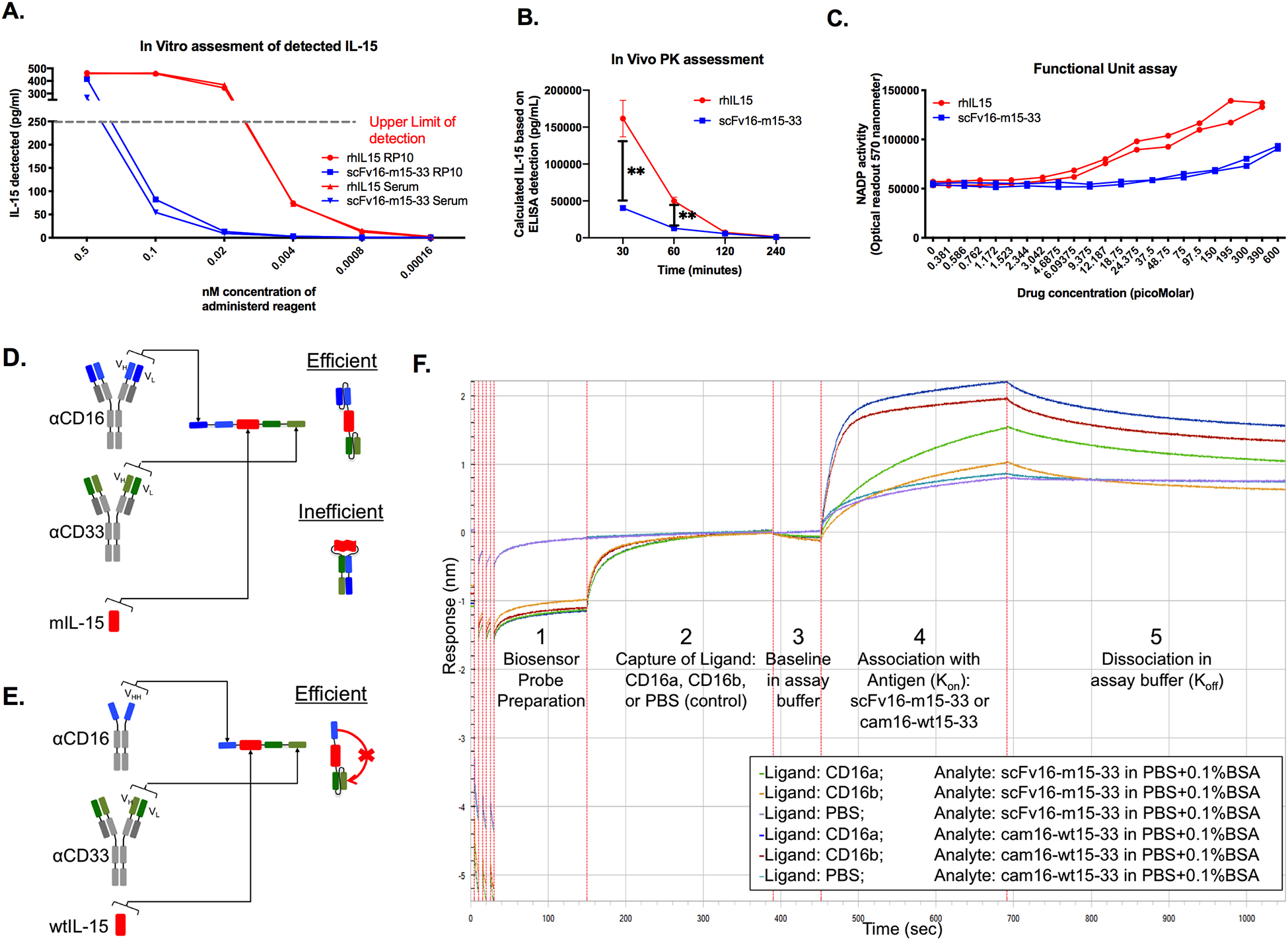
(a) ELISA-based detection of IL15 in equimolar concentrations of scFv16-m15–33 TriKE (in blue) and rhIL-15 (in red); representative of two independent experiments. (b) NSG mice were injected with equimolar amounts of scFv16-m15–33 TriKE or rhIL15, and IL15 concentration in serum were detected by ELISA after 30, 60, 120, and 240 minutes (N=3). Unpaired T tests were used at individual time points to compare IL15 concentration in mice (mean±SEM) with **P<0.01. (c) Activity of IL15 in the scFv16-m15–33 TriKE or rhIL15 was evaluated after a 48 hours in culture with CTLL-2 cells. Representative of two independent experiments. (d) Schematic of theoretic folding inefficiencies between two scFvs in the first-generation scFv16-m15–33 TriKE, causing steric hindrance of IL15. Wt: wild-type; m: mutant. (e) Schematic of the second-generation cam16-wt15–33 TriKE containing a humanized camelid single domain antibody (sdAb/VHH) anti-CD16 arm (in blue), leading to precise folding and reduced steric hindrance of IL15. (f) Octet analysis of binding affinity of scFv16-m15–33 TriKE binding to CD16a (green trace) and CD16b (orange trace) versus cam16-wt15–33 TriKE binding to CD16a (blue trace) and CD16b (red trace). Purple and light blue traces represent unspecific control binding of scFv16-m15–33 and cam16-wt15–33 TriKEs, (respectively). Numbers 1–5 represent different stages (probe preparation, ligand capture, baseline, association, and dissociation) in the Octet assay.
NSG mice were injected with equimolar concentrations of rhIL15 or the scFv16-m15–33 TriKE, and serum was analyzed for 4 hours after administration. IL15 at the 30- and 60-minute post administration time points was significantly greater after rhIL15 injection compared to the scFv16-m15–33 TriKE (Figure 1B). At later time points (2 hours and 4 hours), detection was low for both due to rapid clearance based on size (12.8 kDa for rhIL15 vs. 66.05 kDa for the scFv16-mIL15–33 TriKE). To determine whether functional activity was altered, the scFv16-mIL15–33 TriKE was compared to rhIL15 over a concentration range using an assay that detects NADP activity on CTLL-2 cells that signal through IL15 but do not contain CD16 or CD33 receptors (Figure 1C). Together, results indicated that mIL15 in the TriKE signals with about 25-fold less activity than rhIL15 when presented with the scFv against CD16.
Creation of a second-generation cam16-wt15–33 TriKE
IL15 detection and signaling data indicate that incorporation of IL15 into the TriKE resulted in reduced activity. Because the scFv16-mIL15–33 TriKE contains multiple scFvs, each of which contained a variable heavy (VH) and variable light (VL) chain, the impact on diminished IL15 activity may be mediated by inefficient folding of the scFvs (Figure 1D). Previous studies show that molecules containing multiple scFvs have potential for aggregation and nonspecific folding (26–29). The VH of one scFv aggregates to the VL of the other scFv and vice-versa. To overcome this challenge, a single-domain antibody (sdAb or VHH) sequence was substituted for the scFv anti-CD16 binding domain. Because the sdAb contained only one variable heavy domain that could not aggregate with a VL from an scFv, the TriKE molecule was forced to fold precisely without mispairing (Figure 1E). To accomplish this, the CDR1, CDR2, and CDR3 regions of a camelid (llama) anti-CD16 sdAb were cloned into a humanized sdAb backbone, termed cam16 within this study (30,31). The humanized sdAb backbone, also derived from a camelid, contains four natural amino acid changes generated through evolution that prevent binding of the sdAb to VL fragments. The backbone was humanized by alteration of 12 amino acids to increase the humanness score, as previously described (31). The cam16 was then cloned into a TriKE backbone containing wild-type IL15 (wt15) and an scFv anti-CD33 (cam16-wt15–33 TriKE) or a mutant IL15 and anti-CD33 (cam16-m15–33). Because no difference was detected in proliferative activity between the cam16-wt15–33 TriKE vs. the cam16-m15–33 TriKE, the wild-type version was selected for development (Supplementary Figure S1A-B).
To compare the binding capabilities of the cam16-wt15–33 and scFv16-m15–33 TriKEs, Octet analysis was performed to measure the affinity of each TriKE against the CD16A and CD16B proteins (Figure 1F). Addition of the cam16 arm in the cam16-wt-15–33 TriKE improved binding affinity 28-fold against CD16A compared to the scFv16-m15–33 TriKE (KD=2.32 × 10−8 vs. 6.56 × 10−7 M, respectively). The cam16-wt15–33 TriKE had stronger binding against CD16B than the scFv16-m15–33 TriKE (KD=3.40 × 10−8 vs. 1.44 × 10−6 M, respectively). Both molecules bound CD33 with relatively similar affinity [KD=2.56 × 10−8 M for cam16-wt15–33 vs. 2.21 × 10−8 M for scFv16-m15–33 (Supplementary Figure S1C-D)]. Data indicated that the second-generation cam16-wt15–33 TriKE could bind CD16 with greater affinity than the first-generation scFv16-m15–33 TriKE.
IL15 activity in the cam16-wt15–33 TriKE is greater than the scFv-m15–33 TriKE
To investigate the ability of this new construct to drive IL15 signaling, NK cell proliferation was evaluated on CellTrace-labeled PBMCs incubated with different concentrations of rhIL15, scFv16-m15–33, or cam16-wt15–33. Total NK cell proliferation was highest with cam16-wt15–33 at all concentrations (Figure 2A), and NK cell proliferation beyond three divisions (Figure 2B) was highest in the cam16-wt15–33 group at concentrations of 5 nM or greater. Although we have previously shown that TriKEs can synergize CD16 and IL15 signaling to enhance phospho-STAT5 signaling, this synergy only occurs in the presence of targets needed to crosslink CD16 (21). In the absence of HL-60 targets, IL15 activity increased independently of the CD16 signaling. The IL15 specificity was tested by evaluating T-cell proliferation (Figure 2C–2D). Despite driving the most NK cell proliferation, the cam16-wt15–33 TriKE drove T-cell proliferation significantly less than rhIL15 and the scFv-m15–33 TriKE at the highest concentration tested. Results indicated that the cam16 engager induced delivery of the wild-type IL15 within the TriKE backbone, specifically to NK cells, without cross-stimulation to T cells. Because IL15 signaling can also induce expression of activation markers CD69 and CD25, NK cells were incubated with rhIL15, scFv16-m15–33, or cam16-wt15–33 overnight and expression of activation markers was evaluated. With the exception of the lowest dose, CD69 proportion on NK cells was similar, but the median fluorescence intensity (MFI) for CD69 was higher at all concentrations tested with the cam16-wt15–33 TriKE compared to the scFv16-m15–33 (Figure 2E–2F). The proportion and median florescence of CD25-expressing NK cells was substantially higher with cam16-wt15–33 treatment compared to scFv16-m15–33 (Figure 2G–H). Compared to rhIL15 at the 30 nM concentration, the cam16-wt15–33 TriKE median CD25 expression was higher. These data indicated that IL15 in the cam16-wt15–33 TriKE backbone was stronger than that on the scFv16-m15–33 TriKE and similar to rhIL15.
Figure 2. Second-generation cam16-wt15–33 TriKE demonstrates potent IL15 signaling with enhanced NK cell specificity.
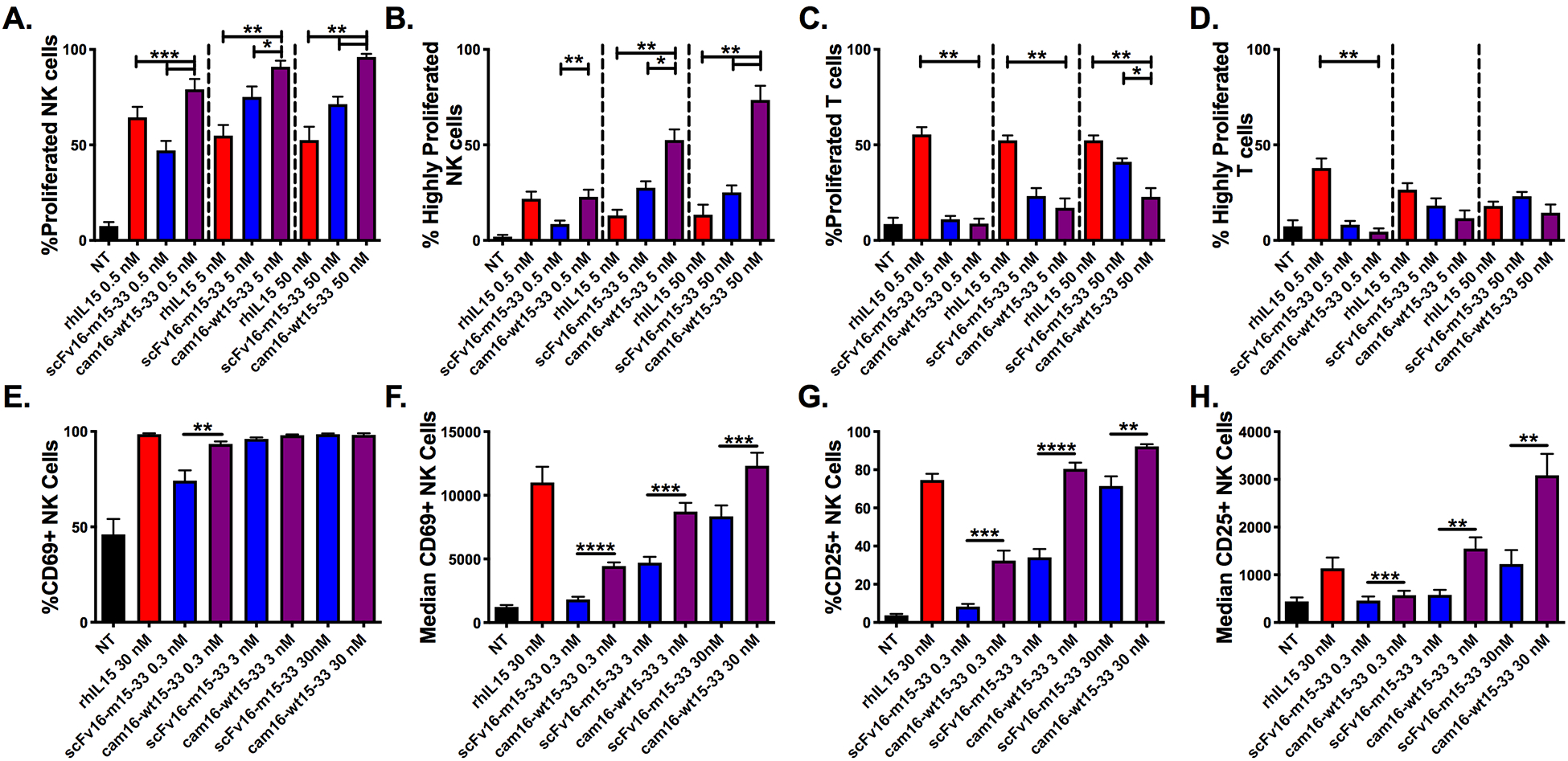
(A-D) PBMCs were CellTrace labeled and incubated for 7 days with noted concentrations of rhIL15 (red), scFv16-m15–33 TriKE (blue), and cam16-wt15–33 TriKE (purple). At the end of the 7 days, cells were harvested, stained, and evaluated by flow cytometry. (a) The proportion of NK cells that proliferated or (b) that highly proliferated (greater than three divisions) with the indicated treatments (N=5). (c) The proportion of T cells that proliferated or (d) that highly proliferated with the indicated treatments (N=5). (A-D) One-way ANOVA with repeated measures was used to calculate differences against the cam16-wt15–33 group within each of the concentrations. Bars show mean±SEM and statistical significance as *P<0.05, **P<0.01, and ***P<0.001. (E-H) NK cells were activated overnight (16 hours) with the noted stimuli or nothing (NT) and activation marker expression was assessed. (e) The CD69+ proportion and (f) median fluorescence intensity (MFI), as well as (g) CD25+ proportion and (h) MFI, was assessed at the time of harvest on NK cells (N=7). Paired T tests between the scFv16-m15–33 and cam16-wt15–33 TriKEs were carried out within each of the concentrations. Bars show mean±SEM and statistical significance as *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
The cam16-wt15–33 TriKE induces NK cell antitumor activity
We next evaluated the ability of the cam16-wt15–33 TriKE to activate NK cell effector functions against CD33-expressing targets. To determine the differential in function between the second-generation cam16-wt15–33 TriKE and the first-generation scFv16-m15–33 TriKE, NK cells were incubated with tumor targets. A gradient of TriKE concentrations (61.7 pM to 15 nM) and degranulation and IFNγ expression was determined after 4 hours (Figure 3A–B). The second-generation cam16-wt15–33 TriKE was functionally superior to the first-generation scFv16-m15–33 TriKE throughout the spectrum of concentrations. Increased functionality was evident at the 30 nM concentration compared to rhIL15 (Figure 3C–D). To evaluate this function over time, we tested the TriKEs in an IncuCyte tumor killing assay (Figure 3E–F, Supplementary Figure S2A). Enriched NK cells were plated with CellTrace Far Red–labeled HL-60 tumor targets, the indicated drugs, and caspase 3/7 green reagent. The number of live tumor cells (CellTrace+caspase 3/7–) was evaluated every 30 minutes for 24 hours. The cam16-wt15–33 TriKE induced more rapid and robust killing than the scFv16-m15–33 TriKE or controls at all time points tested, and increasing the effector to target (E:T) ratio increased killing of the HL-60 cells in the presence of the cam16-wt15–33 TriKE, as expected (Supplementary Figure S2B).
Figure 3. Second-generation cam16-wt15–33 TriKE induces stronger NK cell activation against CD33+ targets.
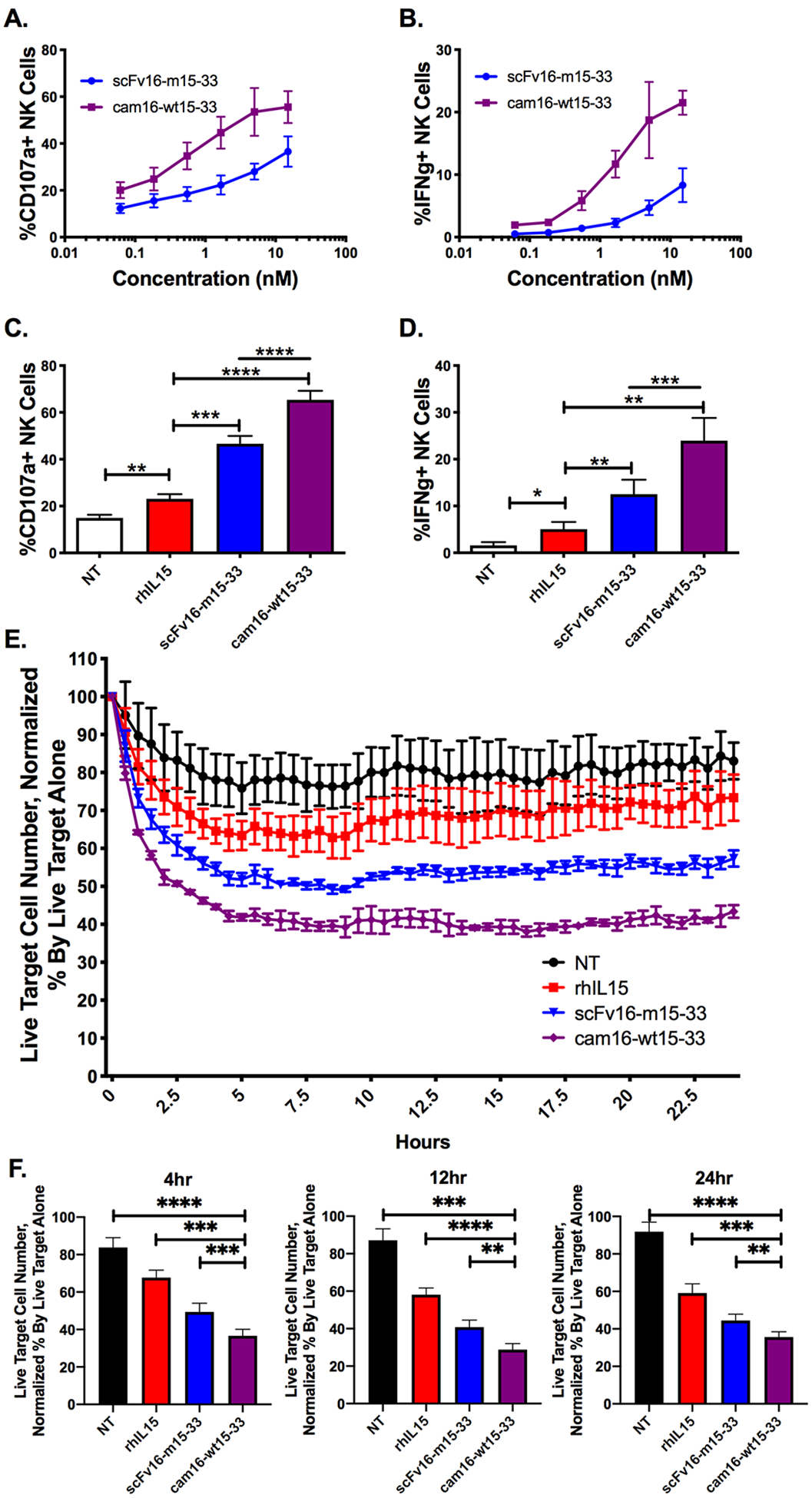
(a) NK cell degranulation (% CD107a+) and (b) IFNγ production were evaluated within PBMCs incubated with CD33+ HL-60 tumor targets at a 2:1 ratio for 4 hours with the indicated concentrations of TriKEs (N=4). (c) Proportion of NK cells expressing CD107a and (d) IFNγ after a 4-hour incubation with 30 nM TriKEs and HL-60 targets (N=8). NT: control (no treatment). (A-D) One-way ANOVA with repeated measures was used to calculate differences against the rhIL15 control group (brackets), and TriKEs were compared with paired T test (lines). (e) A representative experiment of an IncuCyte imaging assay was used to evaluate NK cell–mediated killing of CellTrace Far Red labeled targets over a 24-hour period with the indicated treatments (30 nM). The number of live tumor cells (CellTrace Far Red+Caspase3/7–) was normalized to the number of live tumor targets without effectors to create a percent of remaining cells. (f) Pooled IncuCyte live cell analysis at 4, 12, and 24 hours (N=9). One-way ANOVA with repeated measures was used to calculate differences against cam16-wt15–33. Bars show mean±SEM and statistical significance as *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
The cam16-wt15–33 TriKE enhances NK cell function with short exposure
We compared short exposure to the TriKE molecules to better understand binding characteristics of the two molecules to effectors or targets, which may be important clinically. Effector function (degranulation and cytokine production) was tested after effector or target short-term exposure (15 minutes) to TriKE molecules or rhIL15 followed by coculture of cells. When PBMCs were incubated with TriKE molecules or rhIL15 for 15 minutes, washed, and then cultured with HL-60 tumor targets for 4 hours, the cam16-wt15–33 TriKE induced significantly higher degranulation (CD107a expression) and IFNγ production on the NK cells. In contrast, the scFv16-m15–33 and rhIL15 exhibited weak activity after short-term exposure (Figure 4A). A similar pattern was seen when HL-60 cells were incubated for 15 minutes with TriKEs, washed, and then incubated with PBMCs to evaluate NK cell function (Figure 4B). Because binding to the HL-60 targets via the same anti-CD33 component should be identical between the two TriKEs, the increased activity in this setting was likely due to enhanced signaling mediated by the cam16-wt15–33 once bound. However, it should be pointed out that the short-term cam16-wt15–33 TriKE exposure to PBMCs yielded stronger activation than short-term exposure to HL-60 targets, perhaps highlighting differences in CD16 binding affinity between the two TriKEs. Other exposure times (5 minutes, 30 minutes, 1 hour) were tested and yielded similar results (Supplementary Figure S2C).
Figure 4. Second-generation cam16-wt15–33 TriKE displays stronger activity after short-term exposure.
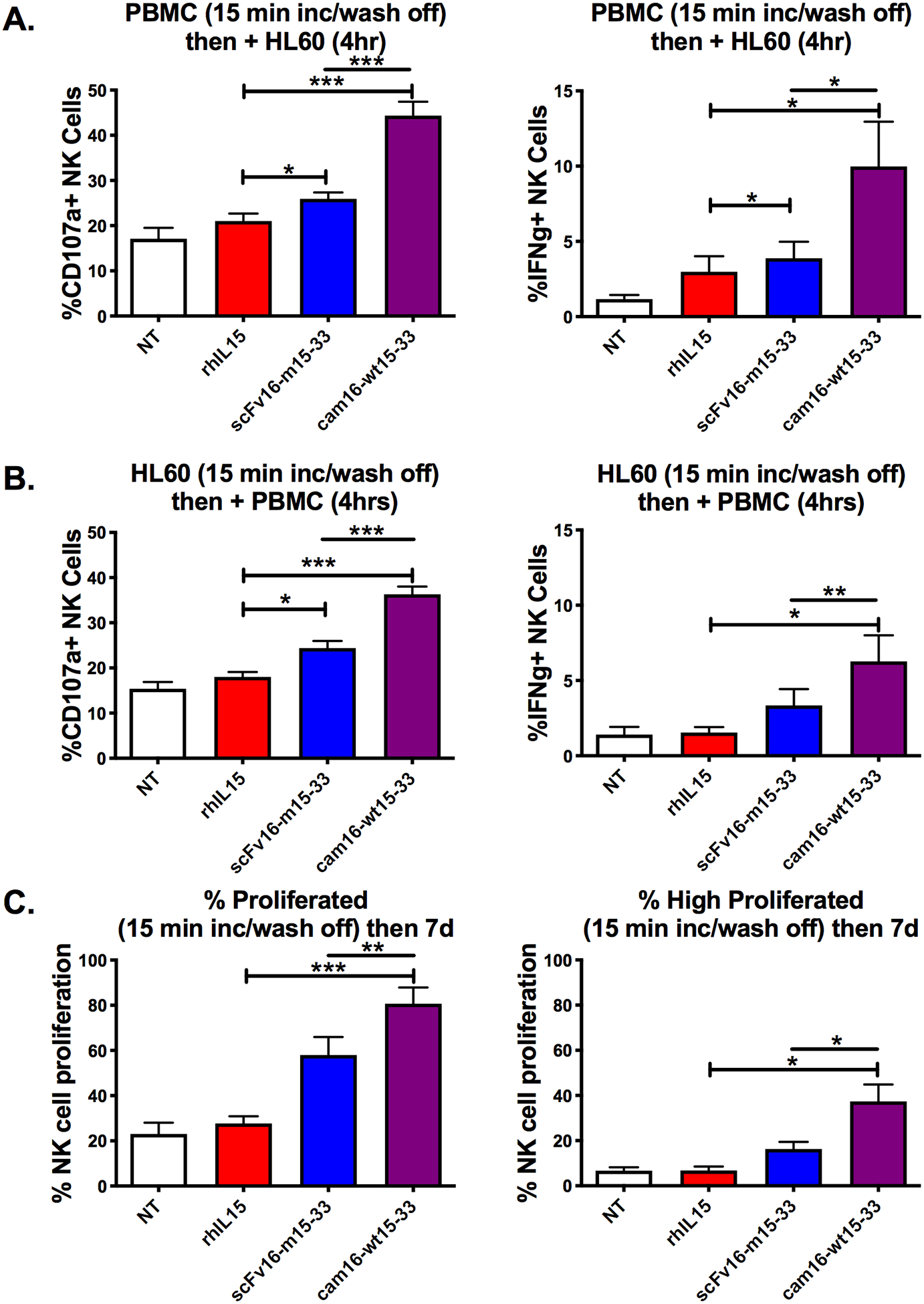
(a) PBMCs were incubated with the indicated treatments (30 nM) for 15 minutes, washed twice, and then incubated with HL-60 targets for 4 hours prior to evaluation of NK cell degranulation and IFNγ production (N=8). (b) HL-60 targets were incubated with the indicated treatments (30 nM) for 15 minutes, washed twice, and then incubated with PBMCs for 4 hours prior to evaluation of NK cell function (N=8). (c) PBMCs were CellTrace labeled, incubated with indicated treatments for 15 minutes, washed twice, and incubated for 7 days in culture with media (no cytokines) to evaluate total NK cell proliferation or those that highly proliferated (N=4). One-way ANOVA with repeated measures was used to calculate differences against the rhIL15 control (brackets), and TriKEs were compared with paired T test (lines). Bars show mean ± SEM and statistical significance as *P<0.05, **P<0.01, and ***P<0.001 (NT: no treatment control).
To evaluate the impact on proliferation, CellTrace-labeled PBMCs were incubated with TriKEs or rhIL15 for 15 minutes, washed, and then incubated for 7 days (Figure 4C). The cam16-wt15–33 TriKE induced the greatest proliferation, followed closely by the scFv16-m15–33 TriKE. rhIL15 alone did not induce proliferation. This differential likely stemmed from differences in binding stability mediated through the cam16 or scFv16, as CD16 signaling alone is not is not active in this setting with no targets present.
cam16-wt15–33 induces enhanced activation of early post-transplant NK cells
To evaluate the impact of TriKE molecules on NK cells post-transplant, patient PBMCs were collected 28 days after transplant, at a time when IFNγ production defects have been shown to occur despite high early engrafting NK cell numbers (32). Eighteen allogeneic hematopoietic cell transplant (alloHCT) recipients, from umbilical cord blood (UCB) and matched sibling donor (MSD) transplants, were studied (Supplementary Table S1). Recipients received a mix of conditioning regimens (myeloablative and reduced intensity), but had high proportions of NK cells (50.70%±8.07% (mean/SEM) for UCB vs. 37.57%±4.44% for MSD). CD16 expression was variable from patient to patient and differed based on transplant source (59.39%±4.30% in UCB recipients vs. 50.01%±6.00% in MSD recipients). AlloHCT samples and targets were treated in a standard 4-hour assay (no priming), or were pre-incubated with treatments overnight, to determine the priming effect of the IL15 moiety in the TriKEs, and then incubated for 4 hours with tumor targets. Under “no priming” conditions, the cam16-wt15–33 TriKE induced higher alloHCT NK cell degranulation than all other groups but little inflammatory cytokine production (Figure 5A–B, Supplementary Figure S3A and S3C). Overnight incubation led to strong degranulation against HL-60 targets with all treatments [Figure 5C, Supplementary Figure S3B and S3D, (top panels)]. In contrast, inflammatory cytokine production of overnight-incubated alloHCT NK cells resulted in similar IFNγ production between the rhIL15 and cam16-wt15–33 groups, which induced superior IFNγ production compared to the scFv16-m15–33 TriKE [Figure 5D, Supplementary Figures S3B and S3D (bottom panels)]. Taken together, the data indicated that short exposure to the cam16-wt15–33 TriKE induced better NK cell degranulation in alloHCT NK cells than all other treatments, and the IL15 moiety within the cam16-wt15–33 TriKE could induce similar priming as rhIL15 with longer exposure.
Figure 5. Second-generation cam16-wt15–33 TriKE enhances function in post-transplant patient samples.
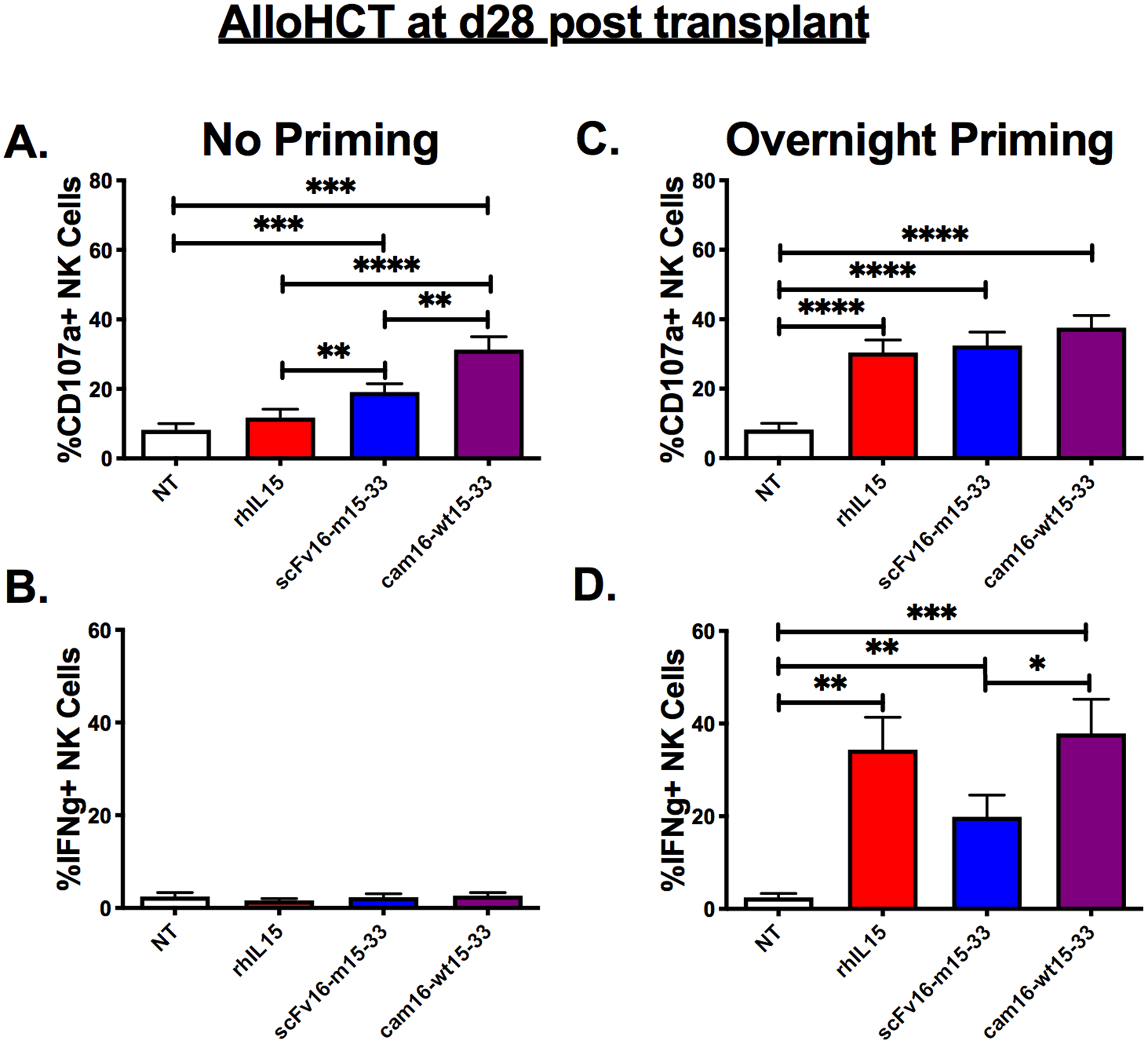
Samples from alloHCT recipients, obtained at day 28 after transplant, were used to evaluate NK cell function with the indicated treatments at a 30 nM concentration (N=18). (a and b) Function was assessed with a standard 4-hour incubation assay (“No Priming”) against HL-60 targets and drug or (c and d) after overnight incubation with the drug, followed by a 4-hour incubation with HL-60 targets and fresh drug. NK cells were assessed for CD107a (a and c) and IFNγ (b and d). One-way ANOVA with repeated measures was used to calculate statistical differences. Bars show mean±SEM and statistical significance as *P<0.05, **P<0.01, ***P<0.001, and ****P<0.0001.
In vivo activity of the second-generation cam16-wt15–33 TriKE
Two different models were used to evaluate the efficacy of the cam16-wt15–33 TriKE in vivo. One employed intravenous (IV) injection of a luciferase-labeled AML cell line (HL-60luc) into NSG mice to track tumor development using live imaging. Three days after tumor inoculation, NK cells were injected IV, and mice were treated 5 days a week for two weeks with different concentrations (90, 30, or 10 μg/injection) of the second-generation cam16-wt15–33 TriKE or the first-generation scFv16-m15–33 TriKE (Figure 6A). The mice were evaluated for tumor burden using bioluminescence over the course of the three weeks (Figure 6B, Supplementary Figure 4A-B). Data showed that at 90 and 30 μg doses, the cam16-wt15–33 TriKE treatment induced significantly better tumor control (130-fold and 12-fold, respectively) than the scFv16-m15–33 TriKE (Figure 6C). The differential in proliferative activity of the cam16-wt15–33 TriKE with the scFv16-m15–33 TriKE is illustrated by higher NK cell counts in the blood at day 21 at all doses tested (Figure 6D).
Figure 6. Enhanced function of second-generation cam16-wt15–33 TriKE against AML targets in vivo.
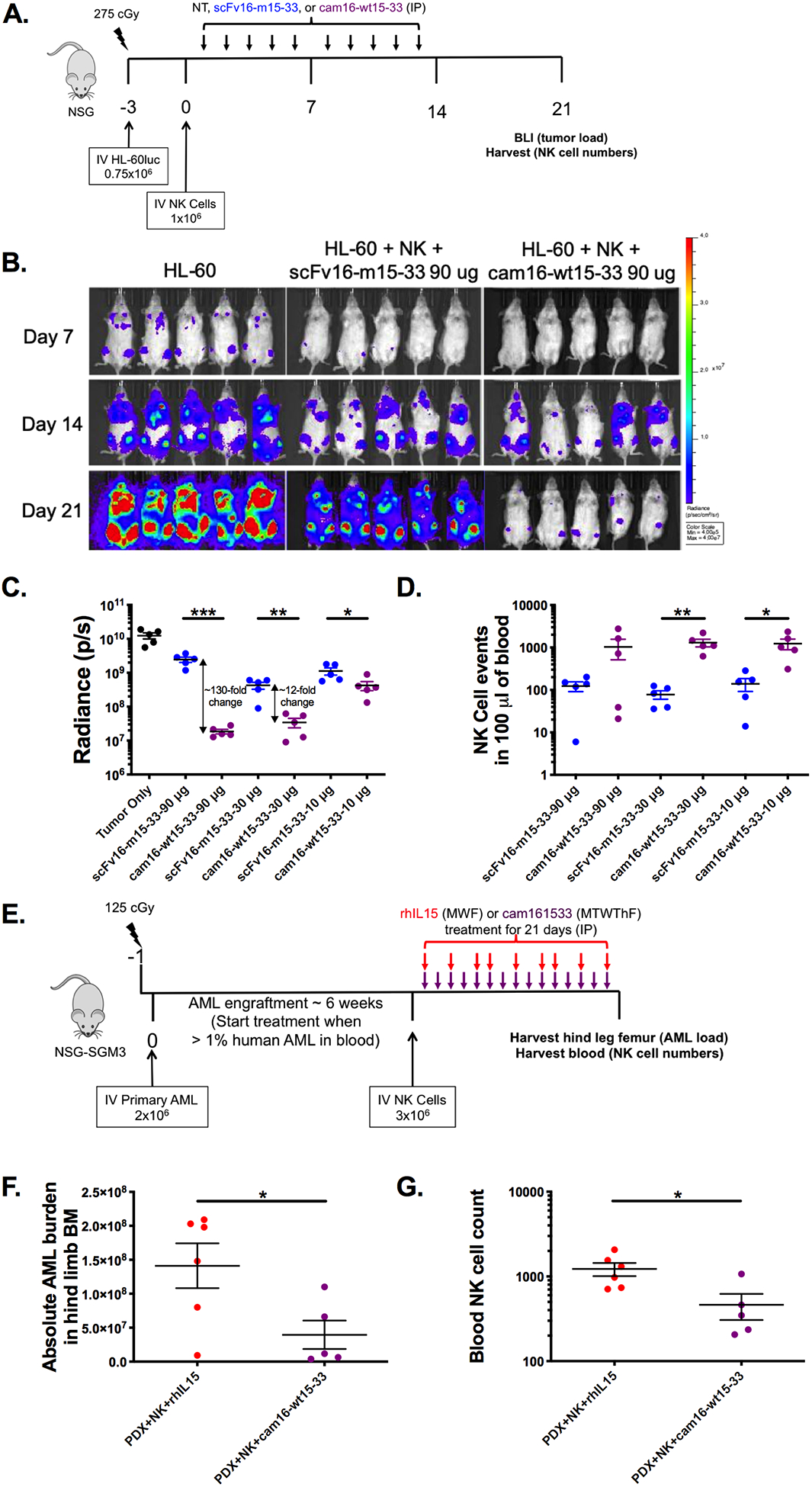
(a) Diagram of HL-60/human NK cell xenogneic NSG model for evaluation of TriKE activity. Mice were treated IP with noted concentrations of scFv16-m15–33 (blue) or cam16-wt15–33 (purple) 5 times weekly for 2 weeks. At days 7, 14, and 21, tumor load was assessed by bioluminescent imaging, and blood collected to evaluate NK cell expansion at day 21. (b) Image of individual mouse radiance on 90 μg treatment group over the course of the experiment. (c) Summary of tumor radiance on all treatment groups (90, 30, and 10 μg per injection groups), and (d) NK cell numbers at day 21 (N=5/group, representative of two experiments). (e) Diagram of AML patient-derived xenograft (PDX) model used to compare the second-generation cam16-wt15–33 TriKE to rhIL15. Upon AML engraftment, mice were injected with NK cells and treatments, and tissues were harvested 3 weeks later for evaluation of (f) AML blasts (CD45+CD33+) numbers in bone marrow and (g) NK cell numbers in a 100 μL of blood (N=5–6, representative of two experiments). Unpaired T test was used to calculate statistical differences. Bars show mean±SEM and statistical significance as *P<0.05, **P< 0.01, ***P<0.001, and ****P<0.0001.
The second in vivo system used a patient-derived xenograft (PDX) tumor model in which primary acute myeloid leukemia (AML) blasts from a cancer patient were injected into NSG-SGM3 mice (blast characteristics found in Supplementary Figure S4C), and tumors were allowed to grow for about 6 weeks. Once the tumor was established, NK cells were IV injected, and mice were treated for three weeks with cam16-wt15–33 TriKE or rhIL15 (IP) to evaluate therapeutic efficacy (Figure 6E). Absolute leukemia burden in the bone marrow and NK cell numbers in the blood were then assessed. cam16-wt15–33 TriKE treatment resulted in less tumors in the bone marrow than with rhIL-15 treatment (Figure 6F), despite lower NK cell numbers found in the blood (Figure 6G). NK cell numbers did not differ in the spleen, indicating that there might be differences in localization/effect of rhIL15 vs. cam16-wt15–33 TriKE (Supplementary Figure S4D). In vivo data indicated that the cam16-wt15–33 TriKE induced better AML tumor control.
Discussion
The TriKE platform is unique among NK engagers because it incorporates cytokine signaling, specific tumor targeting, and activation of ADCC within a single molecule (16–19). TriKEs not only drive specific tumor killing but also induce NK cell proliferation and survival via the IL15 moiety. Unlike T cells (e.g. CD3 stimulation in bispecific T-cell engagers [BiTEs]), which can be induced to expand through crosslinking of activating receptors, NK cells require a cytokine signal to induce proliferation. Although activating receptor signals may synergize with cytokines to enhance proliferation, NK cell activating receptors, including CD16, do not induce proliferation on their own (33). The first-generation TriKE, scFv16-m15–33 (161533 in previous studies), provides both cytokine and activating signals (20). Our findings indicated that IL15 functionality within this molecule is limiting. The IL15 moiety within the first-generation TriKE showed a 25-fold less function compared to rhIL15. This decrease may be influenced by steric hindrance mediated by mispairing of the two scFvs (34). We therefore substituted in a humanized single domain antibody (sdAb) against CD16 incapable of pairing with the light chain of the anti-CD33 scFv, resulting in improved function of the IL15 moiety within the second-generation cam16-wt15–33 TriKE. An alternative possibility to decreased mispairing is that the smaller size of the sdAb may also reduce steric hindrance to present IL15 more efficiently. Regardless of the mechanism, the IL15 activity was altered by its flanking engaging sequences. This allowed use of wild-type IL15 within this platform (23) and no longer required the use of enhanced IL15 mutants used previously (20). In all aspects, including proliferation, degranulation, inflammatory cytokine secretion, tumor killing, and in vivo function, the second-generation cam16-wt15–33 was more potent than first-generation scFv16-m15–33. This differential in functionality was particularly evident in the HL-60luc xenogeneic mouse experiments. The cam16-wt15–33 TriKE induced increases in tumor control and NK cell numbers compared to the scFv16-m15–33 TriKE.
Besides delivering a targeted ADCC signal, TriKEs specifically deliver IL15 to NK cells mediated by anti-CD16 engagement (20,25). The second-generation cam16-wt15–33 TriKE bound to CD16 on NK cells 28-fold better than scFv16-m15–33. This differential in binding was also reflected in the delivery of IL15 to NK cells vs. T cells. Despite delivering a stronger IL15 signal to NK cells, the cam16-wt15–33 TriKE, compared to rhIL-15 and scFv16-m15–33, induced minimal stimulation of T cells, presumably by specific delivery to CD16-expressing cells. This targeted IL15 may be particularly important for a better safety profile, which will need to be tested clinically. Cytokines have been reported in the development of leukemias and solid tumors. Targeted delivery would minimize this potential risk (35–38). IL15 may exacerbate autoimmune disorders such as rheumatoid arthritis, systemic lupus erythematous, inflammatory bowel disease and multiple sclerosis (39). Thus, targeted delivery of IL15 via the cam16-wt15–33 TriKE may limit NK-independent autoimmune effects.
In the hematopoietic transplantation setting, specifically targeting IL15 to NK cells via the cam16-wt15–33, may enhance donor graft function to promote a graft-versus-leukemia (GVL) responses (1,2,40–43). Targeted delivery of IL15 could amplify NK cell expansion and function without off-target stimulation of CD8+ T cells that are known to exacerbate potentially lethal graft-versus-host disease (44). The targeted delivery of IL15 to NK cells, coupled with the ability to potently drive ADCC in vitro and in vivo, makes cam16-wt15–33 TriKE immunotherapeutic candidate for ongoing research. NK cell engagers have the potential to work alone on endogenous NK cells, in settings were NK cells are diminished or dysfunctional, and in combination with NK cell adoptive transfer.
Supplementary Material
Acknowledgements
We would like to acknowledge the Translational Therapy, Flow Cytometry, and Imaging cores at the University of Minnesota for their excellent services. This work was supported in part by NCI P30 CA077598, Minnesota Masonic Charities and the Killebrew-Thompson Memorial Fund. We would also like to thank Xianzheng Zhou, at New York Medical College, for use of his HL-60luc cells.
Financial Support:
This work was supported in part by DoD CA150085 (MF), NIH U01HL127479 (DAV), NCI P01 CA111412 (MF and JSM), P01 CA65493 (MF and JSM), R35 CA197292 (MF and JSM), and the Randy Shaver Cancer Research and Community Foundation (DAV). TriKE™ is a trademark of GT Biopharma, Inc. and this work was supported in part by their research funding (MF, DAV and JSM).
Footnotes
Conflict of Interest Statement:
MF, JSM and DAV receive consulting honorary from GT Biopharma, which has licensed the TriKE platform from the University of Minnesota.
References
- 1.Miller JS, Lanier LL. Natural Killer Cells in Cancer Immunotherapy. Annu Rev Canc Biol. 2019;3:77–103. doi: 10.1146/annurev-cancerbio-030518-055653. [DOI] [Google Scholar]
- 2.Foley B, Felices M, Cichocki F, Cooley S, Verneris MR, Miller JS. The biology of NK cells and their receptors affects clinical outcomes after hematopoietic cell transplantation (HCT). Immunol Rev. 2014;258(1):45–63. doi: 10.1111/imr.12157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davis ZB, Vallera DA, Miller JS, Felices M. Natural killer cells unleashed: Checkpoint receptor blockade and BiKE/TriKE utilization in NK-mediated anti-tumor immunotherapy. Semin Immunol. 2017;31:64–75. doi: 10.1016/j.smim.2017.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fehniger TA, Cooper MA, Caligiuri MA. Interleukin-2 and interleukin-15: immunotherapy for cancer. Cytokine Growth Factor Rev. 2002;13(2):169–83. [DOI] [PubMed] [Google Scholar]
- 5.Waldmann TA. The biology of IL-15: implications for cancer therapy and the treatment of autoimmune disorders. J Investig Dermatol Symp Proc. 2013;16(1):S28–30. doi: 10.1038/jidsymp.2013.8. [DOI] [PubMed] [Google Scholar]
- 6.Waldmann TA. The biology of interleukin-2 and interleukin-15: implications for cancer therapy and vaccine design. Nat Rev Immunol. 2006;6(8):595–601. doi: 10.1038/nri1901. [DOI] [PubMed] [Google Scholar]
- 7.Kennedy-Nasser AA, Ku S, Castillo-Caro P, Hazrat Y, Wu MF, Liu H, Melenhorst J, Barrett AJ, Ito S, Foster A, Savoldo B, Yvon E, Carrum G, Ramos CA, Krance RA, Leung K, Heslop HE, Brenner MK, Bollard CM. Ultra low-dose IL-2 for GVHD prophylaxis after allogeneic hematopoietic stem cell transplantation mediates expansion of regulatory T cells without diminishing antiviral and antileukemic activity. Clin Cancer Res. 2014;20(8):2215–25. doi: 10.1158/1078-0432.CCR-13-3205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cooley S, He F, Bachanova V, Vercellotti GM, DeFor TE, Curtsinger JM, Robertson P, Grzywacz B, Conlon KC, Waldmann TA, McKenna DH, Blazar BR, Weisdorf DJ, Miller JS. First-in-human trial of rhIL-15 and haploidentical natural killer cell therapy for advanced acute myeloid leukemia. Blood Adv. 2019;3(13):1970–80. doi: 10.1182/bloodadvances.2018028332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wrangle JM, Velcheti V, Patel MR, Garrett-Mayer E, Hill EG, Ravenel JG, Miller JS, Farhad M, Anderton K, Lindsey K, Taffaro-Neskey M, Sherman C, Suriano S, Swiderska-Syn M, Sion A, Harris J, Edwards AR, Rytlewski JA, Sanders CM, Yusko EC, Robinson MD, Krieg C, Redmond WL, Egan JO, Rhode PR, Jeng EK, Rock AD, Wong HC, Rubinstein MP. ALT-803, an IL-15 superagonist, in combination with nivolumab in patients with metastatic non-small cell lung cancer: a non-randomised, open-label, phase 1b trial. Lancet Oncol. 2018;19(5):694–704. doi: 10.1016/S1470-2045(18)30148-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Romee R, Cooley S, Berrien-Elliott MM, Westervelt P, Verneris MR, Wagner JE, Weisdorf DJ, Blazar BR, Ustun C, DeFor TE, Vivek S, Peck L, DiPersio JF, Cashen AF, Kyllo R, Musiek A, Schaffer A, Anadkat MJ, Rosman I, Miller D, Egan JO, Jeng EK, Rock A, Wong HC, Fehniger TA, Miller JS. First-in-human phase 1 clinical study of the IL-15 superagonist complex ALT-803 to treat relapse after transplantation. Blood. 2018;131(23):2515–27. doi: 10.1182/blood-2017-12-823757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller JS, Morishima C, McNeel DG, Patel MR, Kohrt HE, Thompson JA, Sondel PM, Wakelee HA, Disis ML, Kaiser JC, Cheever MA, Streicher H, Creekmore SP, Waldmann TA, Conlon KC. A First-in-Human Phase 1 Study of Subcutaneous Outpatient Recombinant Human IL-15 (rhIL-15) in Adults with Advanced Solid Tumors. Clin Cancer Res. 2017. doi: 10.1158/1078-0432.CCR-17-2451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dubois S, Conlon KC, Muller JR, Hsu-Albert J, Beltran N, Bryant BR, Waldmann TA. IL15 Infusion of Cancer Patients Expands the Subpopulation of Cytotoxic CD56(bright) NK Cells and Increases NK-Cell Cytokine Release Capabilities. Cancer Immunol Res. 2017;5(10):929–38. doi: 10.1158/2326-6066.CIR-17-0279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Conlon KC, Potter EL, Pittaluga S, Lee CR, Miljkovic MD, Fleisher TA, Dubois S, Bryant BR, Petrus M, Perera LP, Hsu J, Figg WD, Peer CJ, Shih JH, Yovandich JL, Creekmore SP, Roederer M, Waldmann TA. IL15 by Continuous Intravenous Infusion to Adult Patients with Solid Tumors in a Phase I Trial Induced Dramatic NK-Cell Subset Expansion. Clin Cancer Res. 2019. doi: 10.1158/1078-0432.CCR-18-3468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nguyen QH, Roberts RL, Ank BJ, Lin SJ, Thomas EK, Stiehm ER . Interleukin (IL)-15 enhances antibody-dependent cellular cytotoxicity and natural killer activity in neonatal cells. Cell Immunol. 1998;185(2):83–92. doi: 10.1006/cimm.1998.1286. [DOI] [PubMed] [Google Scholar]
- 15.Chames P, Van Regenmortel M, Weiss E, Baty D. Therapeutic antibodies: successes, limitations and hopes for the future. Br J Pharmacol. 2009;157(2):220–33. doi: 10.1111/j.1476-5381.2009.00190.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Felices M, Lenvik TR, Davis ZB, Miller JS, Vallera DA. Generation of BiKEs and TriKEs to Improve NK Cell-Mediated Targeting of Tumor Cells. Methods Mol Biol. 2016;1441:333–46. doi: 10.1007/978-1-4939-3684-7_28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rothe A, Sasse S, Topp MS, Eichenauer DA, Hummel H, Reiners KS, Dietlein M, Kuhnert G, Kessler J, Buerkle C, Ravic M, Knackmuss S, Marschner JP, Pogge von Strandmann E, Borchmann P, Engert A. A phase 1 study of the bispecific anti-CD30/CD16A antibody construct AFM13 in patients with relapsed or refractory Hodgkin lymphoma. Blood. 2015;125(26):4024–31. doi: 10.1182/blood-2014-12-614636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gauthier L, Morel A, Anceriz N, Rossi B, Blanchard-Alvarez A, Grondin G, Trichard S, Cesari C, Sapet M, Bosco F, Rispaud-Blanc H, Guillot F, Cornen S, Roussel A, Amigues B, Habif G, Caraguel F, Arrufat S, Remark R, Romagne F, Morel Y, Narni-Mancinelli E, Vivier E. Multifunctional Natural Killer Cell Engagers Targeting NKp46 Trigger Protective Tumor Immunity. Cell. 2019;177(7):1701–13 e16. doi: 10.1016/j.cell.2019.04.041. [DOI] [PubMed] [Google Scholar]
- 19.Ellwanger K, Reusch U, Fucek I, Wingert S, Ross T, Muller T, Schniegler-Mattox U, Haneke T, Rajkovic E, Koch J, Treder M, Tesar M. Redirected optimized cell killing (ROCK(R)): A highly versatile multispecific fit-for-purpose antibody platform for engaging innate immunity. MAbs. 2019;11(5):899–918. doi: 10.1080/19420862.2019.1616506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vallera DA, Felices M, McElmurry R, McCullar V, Zhou X, Schmohl JU, Zhang B, Lenvik AJ, Panoskaltsis-Mortari A, Verneris MR, Tolar J, Cooley S, Weisdorf DJ, Blazar BR, Miller JS. IL15 Trispecific Killer Engagers (TriKE) Make Natural Killer Cells Specific to CD33+ Targets While Also Inducing Persistence, In Vivo Expansion, and Enhanced Function. Clin Cancer Res. 2016;22(14):3440–50. doi: 10.1158/1078-0432.CCR-15-2710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sarhan D, Brandt L, Felices M, Guldevall K, Lenvik T, Hinderlie P, Curtsinger J, Warlick E, Spellman SR, Blazar BR, Weisdorf DJ, Cooley S, Vallera DA, Onfelt B, Miller JS. 161533 TriKE stimulates NK-cell function to overcome myeloid-derived suppressor cells in MDS. Blood Adv. 2018;2(12):1459–69. doi: 10.1182/bloodadvances.2017012369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Don Yun H, Felices M, Vallera DA, Hinderlie P, Cooley S, Arock M, Gotlib J, Ustun C, Miller JS. Trispecific killer engager CD16xIL15xCD33 potently induces NK cell activation and cytotoxicity against neoplastic mast cells. Blood Adv. 2018;2(13):1580–4. doi: 10.1182/bloodadvances.2018018176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhu X, Marcus WD, Xu W, Lee HI, Han K, Egan JO, Yovandich JL, Rhode PR, Wong HC. Novel human interleukin-15 agonists. J Immunol. 2009;183(6):3598–607. doi: 10.4049/jimmunol.0901244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Prabst K, Engelhardt H, Ringgeler S, Hubner H. Basic Colorimetric Proliferation Assays: MTT, WST, and Resazurin. Methods Mol Biol. 2017;1601:1–17. doi: 10.1007/978-1-4939-6960-9_1. [DOI] [PubMed] [Google Scholar]
- 25.Felices M, Kodal B, Hinderlie P, Kaminski MF, Cooley S, Weisdorf DJ, Vallera DA, Miller JS, & Bachanova V. Novel CD19-targeted TriKE restores NK cell function and proliferative capacity in CLL. Blood Adv. 2019;3(6):897–907. doi: 10.1182/bloodadvances.2018029371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Glaven RH, Anderson GP, Zabetakis D, Liu JL, Long NC, Goldman ER. Linking Single Domain Antibodies that Recognize Different Epitopes on the Same Target. Biosensors (Basel). 2012;2(1):43–56. doi: 10.3390/bios2010043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Iezzi ME, Policastro L, Werbajh S, Podhajcer O, Canziani GA. Single-Domain Antibodies and the Promise of Modular Targeting in Cancer Imaging and Treatment. Front Immunol. 2018;9:273. doi: 10.3389/fimmu.2018.00273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hassanzadeh-Ghassabeh G, Devoogdt N, De Pauw P, Vincke C, Muyldermans S. Nanobodies and their potential applications. Nanomedicine (Lond). 2013;8(6):1013–26. doi: 10.2217/nnm.13.86. [DOI] [PubMed] [Google Scholar]
- 29.Rozan C, Cornillon A, Petiard C, Chartier M, Behar G, Boix C, Kerfelec B, Robert B, Pelegrin A, Chames P, Teillaud JL, Baty D. Single-domain antibody-based and linker-free bispecific antibodies targeting FcgammaRIII induce potent antitumor activity without recruiting regulatory T cells. Mol Cancer Ther. 2013;12(8):1481–91. doi: 10.1158/1535-7163.MCT-12-1012. [DOI] [PubMed] [Google Scholar]
- 30.Behar G, Siberil S, Groulet A, Chames P, Pugniere M, Boix C, Sautes-Fridman C, Teillaud JL, Baty D. Isolation and characterization of anti-FcgammaRIII (CD16) llama single-domain antibodies that activate natural killer cells. Protein Eng Des Sel. 2008;21(1):1–10. doi: 10.1093/protein/gzm064. [DOI] [PubMed] [Google Scholar]
- 31.Vincke C, Loris R, Saerens D, Martinez-Rodriguez S, Muyldermans S, Conrath K. General strategy to humanize a camelid single-domain antibody and identification of a universal humanized nanobody scaffold. J Biol Chem. 2009;284(5):3273–84. doi: 10.1074/jbc.M806889200. [DOI] [PubMed] [Google Scholar]
- 32.Foley B, Cooley S, Verneris MR, Curtsinger J, Luo X, Waller EK, Weisdorf DJ, Miller JS. NK cell education after allogeneic transplantation: dissociation between recovery of cytokine-producing and cytotoxic functions. Blood. 2011;118(10):2784–92. doi: 10.1182/blood-2011-04-347070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ali AK, Nandagopal N, Lee SH. IL-15-PI3K-AKT-mTOR: A Critical Pathway in the Life Journey of Natural Killer Cells. Front Immunol. 2015;6:355. doi: 10.3389/fimmu.2015.00355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dietrich S, Gross AW, Becker S, Hock B, Stadlmayr G, Ruker F, Wozniak-Knopp G. Constant domain-exchanged Fab enables specific light chain pairing in heterodimeric bispecific SEED-antibodies. Biochim Biophys Acta Proteins Proteom. 2019. doi: 10.1016/j.bbapap.2019.07.003. [DOI] [PubMed] [Google Scholar]
- 35.Hodge DL, Yang J, Buschman MD, Schaughency PM, Dang H, Bere W, Yang Y, Savan R, Subleski JJ, Yin XM, Loughran TP Jr., Young HA. Interleukin-15 enhances proteasomal degradation of bid in normal lymphocytes: implications for large granular lymphocyte leukemias. Cancer Res. 2009;69(9):3986–94. doi: 10.1158/0008-5472.CAN-08-3735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wu S, Fischer L, Gokbuget N, Schwartz S, Burmeister T, Notter M, Hoelzer D, Fuchs H, Blau IW, Hofmann WK, Thiel E. Expression of interleukin 15 in primary adult acute lymphoblastic leukemia. Cancer-Am Cancer Soc. 2010;116(2):387–92. doi: 10.1002/cncr.24729. [DOI] [PubMed] [Google Scholar]
- 37.Kuniyasu H, Ohmori H, Sasaki T, Sasahira T, Yoshida K, Kitadai Y, Fidler IJ. Production of interleukin 15 by human colon cancer cells is associated with induction of mucosal hyperplasia, angiogenesis, and metastasis. Clin Cancer Res. 2003;9(13):4802–10. [PubMed] [Google Scholar]
- 38.Khawam K, Giron-Michel J, Gu Y, Perier A, Giuliani M, Caignard A, Devocelle A, Ferrini S, Fabbi M, Charpentier B, Ludwig A, Chouaib S, Azzarone B, Eid P. Human renal cancer cells express a novel membrane-bound interleukin-15 that induces, in response to the soluble interleukin-15 receptor alpha chain, epithelial-to-mesenchymal transition. Cancer Res. 2009;69(4):1561–9. doi: 10.1158/0008-5472.CAN-08-3198. [DOI] [PubMed] [Google Scholar]
- 39.Di Sabatino A, Calarota SA, Vidali F, Macdonald TT, Corazza GR. Role of IL-15 in immune-mediated and infectious diseases. Cytokine Growth Factor Rev. 2011;22(1):19–33. doi: 10.1016/j.cytogfr.2010.09.003. [DOI] [PubMed] [Google Scholar]
- 40.Cooley S, Trachtenberg E, Bergemann TL, Saeteurn K, Klein J, Le CT, Marsh SG, Guethlein LA, Parham P, Miller JS, Weisdorf DJ. Donors with group B KIR haplotypes improve relapse-free survival after unrelated hematopoietic cell transplantation for acute myelogenous leukemia. Blood. 2009;113(3):726–32. doi: 10.1182/blood-2008-07-171926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cooley S, Weisdorf DJ, Guethlein LA, Klein JP, Wang T, Le CT, Marsh SG, Geraghty D, Spellman S, Haagenson MD, Ladner M, Trachtenberg E, Parham P, Miller JS. Donor selection for natural killer cell receptor genes leads to superior survival after unrelated transplantation for acute myelogenous leukemia. Blood. 2010;116(14):2411–9. doi: 10.1182/blood-2010-05-283051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Davis ZB, Felices M, Verneris MR, Miller JS. Natural Killer Cell Adoptive Transfer Therapy: Exploiting the First Line of Defense Against Cancer. Cancer J. 2015;21(6):486–91. doi: 10.1097/PPO.0000000000000156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Grzywacz B, Moench L, McKenna D Jr., Tessier KM, Bachanova V, Cooley S, Miller JS, Courville EL. Natural Killer Cell Homing and Persistence in the Bone Marrow After Adoptive Immunotherapy Correlates With Better Leukemia Control. J Immunother. 2019;42(2):65–72. doi: 10.1097/CJI.0000000000000250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Perkey E, Maillard I. New Insights into Graft-Versus-Host Disease and Graft Rejection. Annu Rev Pathol. 2018;13:219–45. doi: 10.1146/annurev-pathol-020117-043720. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.