

Abstract
A visual cascade detection system has been applied to the detection and analysis of drug-resistance profile of Mycobacterium tuberculosis complex (MTC), a causative agent of tuberculosis. The cascade system utilizes highly selective split RNA-cleaving deoxyribozyme (sDz) sensors. When activated by a complementary nucleic acid, sDz releases the peroxidase-like deoxyribozyme apoenzyme, which, in complex with a hemin cofactor, catalyzes the color changes of the sample’s solution. The excellent selectivity of the cascade has allowed for the detection of point mutations in the sequences of the MTC rpoB, katG, and gyrA genes, which are responsible for resistance to rifampin, isoniazid, and fluoroquinolone, respectively. When combined with isothermal nucleic acid sequence based amplification (NASBA), the assay was able to detect amplicons of 16S rRNA and katG mRNA generated from 0.1 pg and 10 pg total RNA taken for NASBA, respectively, in less than 2 h, producing a signal detectable with the naked eye. The proposed assay may become a prototype for point-of-care diagnosis of drug resistant bacteria with visual signal output.
Keywords: point mutations, Mycobacterium tuberculosis, visual assay, deoxyribozyme cascade, drug susceptibility testing, Nucleic Acid Sequence-Based Amplification (NASBA)
1. Introduction
The widespread occurrence of drug-resistant bacterial infections is a serious healthcare challenge (CDC, 2019). Therefore, diagnostics of bacterial infections should not only reliably identify the causative infection agent, but also determine its drug susceptibility profile in a timely manner (Barenfanger et al., 1999). Molecular diagnostic analysis of the genetic signatures of pathogens is promising for rapid drug-susceptibility testing (DST) that can be administered at the point-of-care (POC) (Wilson, 2011; Schon et al., 2017). They are especially advantageous for the slow-growing Mycobacterium tuberculosis complex (MTC), the cause of tuberculosis (TB), since even rapid phenotypic DST requires at least 1-2 weeks, thus extending the period when patients do not receive proper treatment. The increasing prevalence of multidrug resistant TB (MDR-TB) makes it crucial to analyze drug-susceptibility profiles of MTC pathogens. These infections do not respond to either of the front-line antibiotics – isoniazid (INH) and rifampin (RIF) (Seung et al., 2015). In the cases of extensive drug resistant (XDR) TB, the pathogens are also resistant to fluoroquinolones (FQ) and at least one of the injectable second-line drugs (Seung et al., 2015).
Drug resistance in MTC is mainly caused by point mutations in the genes encoding either drug targets or prodrug-activating enzymes (Almeida Da Silva and Palomino, 2011). It is challenging to discriminate two nucleic acids differing by a single nucleotide using the hybridization probes currently employed in molecular DST. For example, the molecular beacon (MB) probes, which are used in the Xpert MTB/RIF assay endorsed by the World Health organization (WHO) for rapid detection of MDR TB (Horne et al., 2019), may suffer from low affinity and/or selectivity (Kolpashchikov, 2012). Indeed, structurally constrained MB probes are inefficient in the interrogation of highly structured nucleic acids (Kolpashchikov, 2012; Grimes et al., 2010).
Previously, we have addressed these challenges of hybridization analysis by developing split probes (Grimes et al., 2010; Nguyen et al., 2011; Gerasimova et al., 2010a; Gerasimova and Kolpashchikov, 2013a). A split DNA probe utilizing the MB probe as a signal reporter could discriminate single-base substitutions even when the mutations were located in the middle of stable stem structures (Grimes et al., 2010; Nguyen et al., 2011), which is an extremely challenging case for the MB or other non-split hybridization probes (Kolpashchikov, 2010). The MB-based split probes exhibit high discriminatory ability under a broader temperature range than the MB probes do (Stancescu et al., 2016). Split probes with signal amplification capabilities (Gerasimova et al., 2010b, 2015; Gerasimova and Kolpashchikov, 2013b) have been used for the analysis of bacterial RNA, including discrimination of nucleotide substitutions (Gerasimova et al., 2015). This demonstrates the potential of split hybridization probes for POC DST.
An affordable POC test should avoid using sophisticated and expensive equipment (e.g. qPCR instruments or multichannel fluorescence detector). Ideally, the signal should be easily interpreted by the user, for example, by reading a signal observed by the naked eye (Yoo and Lee, 2016). However, visual format often suffers from insufficient sensitivity. Indeed, the human eye senses the color produced by low molecular weight chromophores at concentrations in a micromolar to submicromolar range.1 Such concentrations are unattainable by standard amplification techniques (e.g. PCR). To address the issue, we have developed a post-amplification signal amplification cascade with visual output (Gerasimova et al., 2013; Reed et al., 2019; Wood et al., 2019).
This work combines split probes and a visual signal amplification cascade with isothermal target amplification via the nucleic acid sequence-based amplification (NASBA) technique (Compton, 1991; Kievits et al., 1991; Deiman et al., 2002), and for the first time applies the technology for differentiation of genotypes of drug-resistant and drug-susceptible MTC species. We have demonstrated high selectivity, as well as robust response, with visually detected output in less than 2 h starting from total bacterial RNA. The proposed approach is a step towards POC MTC diagnostics and DST. This approach is also feasible for analysis of drug-resistance profiles of other bacterial pathogens.
2. Experimental Section
2.1. Materials and Instruments.
Oligonucleotides were custom made by Integrated DNA Technologies, Inc. (Coralville, IA) and were used without purification, except the IPDz substrate, which was HPLC-purified. The NASBA liquid kit was purchased from Life Sciences Advanced Technologies, Inc. (Saint Petersburg, FL). 2,2’-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS), hemin, triton-X100 and HEPES were purchased from Sigma-Aldrich (St. Louis, MO). DMSO was from Fisher Scientific (Hampton, NH). Hydrogen peroxide solution (3%) was from VWR (Radnor, PA). Non-DEPC treated DNase/RNase-free water was from Boston Bio Products (Ashland, MA). SeaKem® LE agarose was from Lonza (Basel, Switzerland), GelRed® nucleic acid gel stain was from Biotium (Fremont, CA). RiboRuler Low Range RNA Ladder, Middlebrook 7H9 with OADC, and Ambion Turbo DNA-free DNase kit were obtained from ThermoFisher Scientific (Waltham, MA). All other reagents were of analytical grade. Absorption spectra were measured using NanoDrop OneC UV/Vis spectrometer (ThermoFisher Scientific, Waltham, MA). Amplification reactions were carried out using a C1000 Touch Thermo Cycler (Bio-Rad Laboratories, Inc., Hercules, CA). The amplicons were analyzed in 2% agarose gels supplemented with GelRed using a BioRad horizontal gel electrophoresis system. The gel images were acquired and analyzed using a BioRad GelDoc XR Molecular Imager.
2.2. Isolation of Total Bacterial RNA
Cultures (50 mL) of Mycobacterium bovis BCG were grown to log phase (OD600 = 0.5-0.8) in 75 cm2 tissue culture flasks in 7H9-OADC medium. RNA was isolated following the protocol published by Rohde et al. (2007). In brief, addition of guanidine thiocyanate-based lysis buffer stabilized bacterial RNA while leaving mycobacteria intact. Pelleted bacilli were lysed in 65°C Trizol using a BeadBeater and 0.1 mm silicon beads. Total RNA was isolated from Trizol lysates by chloroform extraction and Qiagen RNeasy column purification. DNA contamination was removed using an Ambion Turbo DNA-free DNAse kit according to manufacture protocol. Following DNAse treatment, the concentration of RNA was measured using a NanoDrop spectrophotometer.
2.3. NASBA reaction
Amplification was performed according to the manufacturer’s instruction, with the sample volume decreased to 12 μl, and the reaction time varied (30-90 min). Briefly, samples were prepared by mixing 4 μl of 3×NASBA Reaction Buffer, 2 μl of 6×Nucleotide Mix, 2 μl of 1.5 μM primer mix, and 1 μl of either RNase-free water (for no-target control, NTC) or total bacterial RNA (1-1000 ng/mL). The samples were incubated at 65 °C for 2 min followed by cooling to 41 °C for 10 min. Then, 3 μL of 4×NASBA enzyme cocktail was added, and the samples were incubated at 41 °C for 30-90 min.
2.4. Colorimetric Assay
Samples (30 μL) containing IPDz (1.0 μM), two probe strands (0.1 μM each) in the colorimetric buffer (50 mM HEPES-NaOH, pH 7.4, 50 mM MgCl2, 120 mM NaCl, 20 mM KCl, 0.03% Triton X-100, 1%DMSO) were mixed with either synthetic DNA targets (0-100 nM, Table S1) or the products of NASBA reaction (3.3%) and incubated at 50°C for 15-60 min. After briefly cooling the samples to room temperature, hemin (375 nM), ABTS (1 mM) and H2O2 (1 mM) were added. The stock solutions of hemin and ABTS were prepared daily in DMSO, and H2O2 was diluted daily with water. As soon as the color of the target-containing samples was visible (within 5-15 min after H2O2 addition), the tube images were captured using a smartphone camera, and the absorbance of the samples at 420 nm was measured with a NanoDrop spectrometer. The data was processed in Microsoft Excel and/or Origin.
2.5. Calculation of the Limit of Detection (LOD)
The LOD was calculated using a 3σ/S rule (MacDougall and Crummett, 1980), where σ is the standard deviation for the blank, and S is the slope of the linear trendline for the signal concentration dependence in the linear dynamic range. The LOD values calculated from individual experiments were averaged, and the error was expressed as standard deviation. The data processing was done in Microsoft Excel.
3. Results
3.1. Design of the sDz/PDz system
The cascade of deoxyribozymes (Dz) characterized in this study is based on the consecutive catalytic actions of an RNA-cleaving 10-23 Dz (Santoro and Joyce, 1997) and a peroxidase-like Dz (PDz) (Pavlov et al., 2004). The sequence of 10-23 Dz is split into two fragments, which are elongated with the target-binding fragments (arms) to construct two strands of the split Dz (sDz) (Scheme 1A, top left). The substrate-binding arms are tailored to recognize an inhibited PDz structure (IPDz) with a ribonucleotide cleavage site (Scheme 1A, white triangle). In the presence of a specific nucleic acid target, strands S and U hybridize to the target adjacent to one another. This forms the Dz catalytic core and brings together the IPDz-binding arms to cleave IPDz (Scheme 1A, top middle and right). IPDz cleavage releases PDz, which folds into a G-quadruplex (G4) structure with affinity for hemin. The hemin-PDz holoenzyme facilitates a one-electron transfer reaction to convert the colorless 2,2’-azino-bis(3-ethyl benzothiazoline-6-sulphonate) (ABTS) into the blue-green radical cation (ABTS+.) (Wolfenden and Wilson, 1982), which allows for the visual detection of the nucleic acid target (Scheme 1A, bottom). One specific target molecule triggers release of several PDz molecules, thus amplifying the signal. The target activates the sDz only if it is fully complementary to the target-binding arms of both strand S and U. A point-mutation (single-nucleotide substitution) in the target introduces a destabilizing mismatch in its hybrid with strand S, and arrests signaling despite forming a stable hybrid with strand U (Scheme 1B).
Scheme 1.
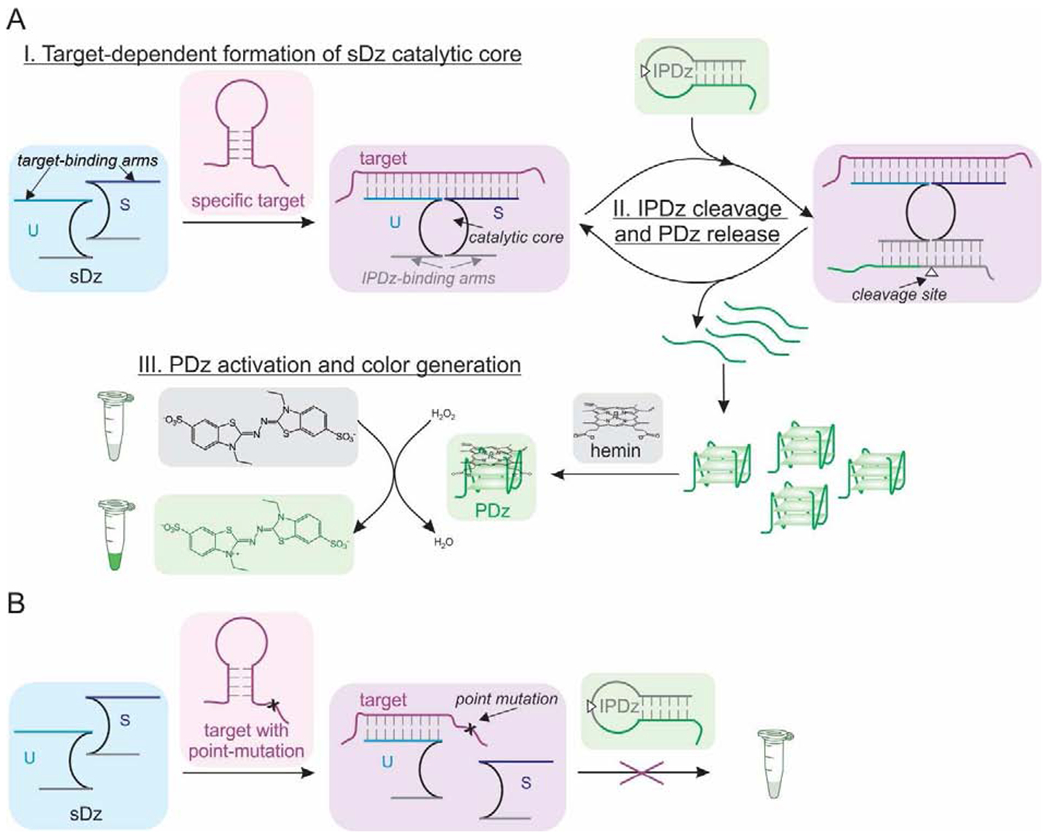
Mechanism of the sDz/PDz cascade system.
The sDz/PDz cascade designed in this work interrogated fragments of the MTC genes conferring bacterial resistance to RIF, INH, and a second-line drug fluoroquinolone (FQ). In addition, 16S rRNA was used as an MTC genetic signature regardless of the bacterial drug-resistance status. To detect the cases of RIF resistance (RIFR), the sDz/PDz cascades were designed to recognize a fragment of the rpoB gene with a site of point mutations in codon 526, which is one of the major point mutations responsible for RIFR (Souglakoff et al., 2004). Along with the sDz/PDz cascade targeting the RIFS genotype, sDz were tailored to recognize either C>T or C>G substitutions, which represent the sequences associated with RIFR (Seifert et al., 2015). In this case, the cascade systems targeting RIFR sequences utilized the same strand U, but contained target-specific strands S. For INHR, the probes interrogated a fragment of the katG gene. In most INHR TB cases, there is a G>C substitution resulting in S315T enzyme mutant (Isakova et al., 2018). For fluoroquinolone-resistance (FQR), a gyrA gene fragment with a site of an A>G substitution in the case of D94G mutant (Pantel et al., 2016) was targeted. Finally, we designed the sDz/PDz cascade targeting a fragment of MTC 16S rRNA, as described earlier (Wood et al., 2019). The 16S rRNA-specific probe along with a set of the point-mutation differentiating probes would enable both MTC detection and DST. The sequences of all the probes and targeted fragments are listed in Table S1.
We optimized the signal-to-background ratios (S/B) and selectivity of eight sDz/PDz cascades. In all the probes, the reporter-containing component – IPDz – was universal. The IPDz structure has been previously optimized to ensure minimal background cleavage and efficient release of PDz for colorimetric signaling after target-induced cleavage (Reed et al., 2019). The only elements of the sDz/PDz cascade that were different between the probes were target-binding arms of strands S and U (Table S1). For the 16S rRNA-specific sDz/PDz probe, the target-binding arms were 19-nt and 22-nt long with a Tm of ~65 °C for the corresponding hybrids. This prevented the cascade to recognize non-specific non-tuberculous mycobacterial species, for example, M. abscessus (Fig. S1).
The stability of the target-probe complex ensures high target-induced signal of the probe. At the same time, the stability is inversely correlated with selectivity of target recognition (Demidov and Frank-Kamenetskii, 2004).To design probes with high affinity to the specific targets and high selectivity against point-mutations, we designed strand U to be long, while keeping strand S short. Strand U was 24-26 nt, with a Tm of 71-78.9 °C (Table S1) for tight binding and unwinding of the target’s secondary structure. Strand S was 13-15 nt to allow stable complex between the probe and only fully complementary target, but not the target with a point-mutation, as can be exemplified for the katG-specific cascade (Fig. S2). The point-mutation site was positioned closer to the middle of the target-binding arm of strands S to ensure themaximal difference between the free energy change values for the matched and mismatched target-probe complex (Table S1).
3.2. Selectivity
The optimized cascades recognized both the wild-type and mutant sequences of the katG genes with excellent selectivity (Fig. 1A). In the case of the RIFS-specific cascade, the target with the C>T substitution would form a G-T Wobble base pair with strand S (Table S1), thus complicating differentiation between rB_RIFS and rB_T_RIFR. However, no signal significantly above the background, or a color change, was observed for the RIFS-specific sDz/PDz probe in the presence of rB_T_RIFR (Fig. 1B).
Figure 1. Selectivity of the sDz/PDz cascade in interrogation of katG (A) or rpoB (B) gene sequences.
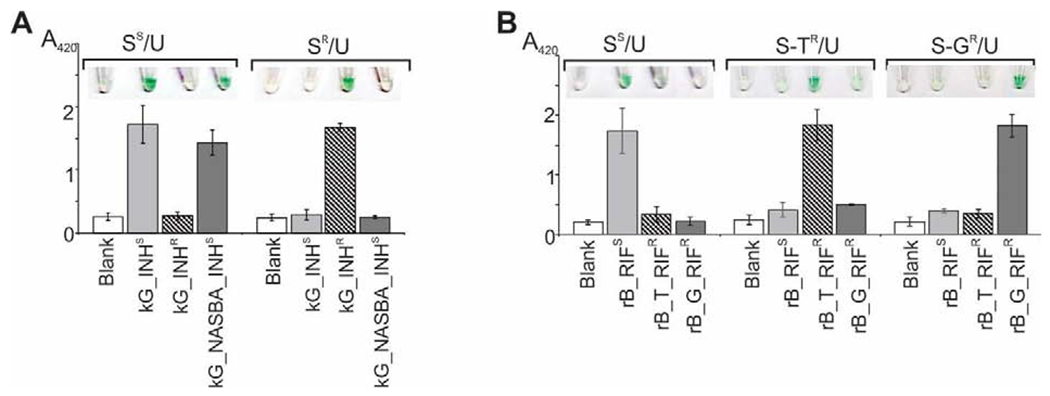
A. The samples contained IPDz, strands U_kG (U) and either S_kG_INHS (SS) or S_kG_INHR (SR) in the absence (“Blank”) or presence of one of the indicated targets. The NASBA sample with the kG_NASBA_INHS amplicon obtained from 83 ng/mL total MTC RNA in a 60-min reaction was added at 3.3% (v/v) to the assay sample. B. The samples containing IPDz, strands U_rB (U) and either S_rB_RIFS (SS), S_rB_T_RIFR (S-TR) or S_rB_G_RIFR (S-GR) in the absence (“Blank”) or presence of one of the indicated targets. All samples in both panels were incubated at 50°C for 45 min. The data is an average of three independent experiments, with relative standard deviations as error bars.
The gyrA-specific cascade system designed according to the same principles failed to discriminate between gA_FQS and gA_FQR. The signal triggered by either target was at the same level, likely due to the T-G Wobble base pair between the target and strand S_gA_FQS (Fig. S3). The discriminatory ability of the split probes can be improved via one of the two approaches (Connelly et al., 2019). In the first approach, the target-binding fragment of strand S was shortened to form strand S2 (Fig. S3, panels B and D, middle). Another approach relied on a conformational constraint in the form of a stem-loop introduced into strand S to form strand s/S (Fig. S3, panels B and D, bottom) (Bonnet et al., 1999). This strategy stabilized the dissociated state, thus disfavoring formation of the mismatched probe-target complex (Kolpashchikov, 2010). Indeed, in the case of FQR-specific probes containing either S2/U2 or s/S/U set of strands, the signal triggered by the mismatched target gA_FQS was at the background level (Fig. S3C). The signal for the FQS-specific probes with S2/U2 or s/S/U strands in the presence of gA_FQR was slightly above the background (Fig. S3A). Still, clear differentiation between gA_FQS and gA_FQR was observed. The signal intensity for the mismatched sample can be mitigated by adjusting the pH (Fig. S4).
3.3. Limit of Detection (LOD) for sDz/PDz cascades
LOD values were determined for all well-performing sDz/PDz cascades (Figs. S5–S8). The values ranged from 1.5 to 13 nM (Table 1). The LODs are close to that reported for the MB probes (Kolpashchikov, 2012), which are currently used as part of the GeneXpert MTB/RIF assay. Similar LOD for the stoichiometric MB probe and the sDz/PDz cascade allowing for signal amplification reflects the higher sensitivity of the fluorescent signal over visual. The latter though has an advantage of the instrument-free observation.
Table 1.
LOD for sDz/PDz Cascadea
| Targeted gene | DrugS | DrugR |
|---|---|---|
| rpoB | 4±2 nM | 3±1 nM (rB_G_RIFR) 5±2 nM (rB_T_RIFR) |
| katG | 1.6±0.8 nM | 5±2 nM |
| gyrA | 13±2 nM (S2/U2) | 13±3 nM (S2/U2) |
| 12±4 nM (s/S/U) | 7.0±0.2 nM (s/S/U) |
DrugS and DrugR indicated drug-susceptible and resistant sequences, respectively. The data is given as average of three independent experiments, with relative standard deviations indicated.
3.4. Interrogation of NASBA amplicons with sDz/PDz probes
For the test to be practically useful, target amplification was needed. Isothermal nucleic acid amplification methods are preferable for POC diagnostics (Li and Macdonald, 2015). We chose Nucleic Sequence-Based Amplification (NASBA), which has been introduced for the detection of RNA-containing viruses, (Compton, 1991; Kievits et al., 1991; Jean et al., 2001; Hibbitts et al., 2003; Pardee et al., 2016; Heo et al., 2019) and applied for amplification of bacterial RNA (Rodriguez-Lazaro et al., 2004). This isothermal technique allows for generation of 2×106-5×107 copies of a single-stranded RNA amplicon (Kievits et al., 1991). We utilized NASBA to amplify a fragment of 16S rRNA for MTC detection. We then amplified a fragment of the katG gene mRNA to demonstrate feasibility of NASBA-assisted DST with the sDz/PDz cascade. This target is also practically significant for MDR-TB analysis, since RIF-monoresistance cases are rare (Sinshaw et al., 2019).
We designed NASBA primers for amplification of a 95-nt fragment of 16S rRNA and a 143-nt fragment of the katG gene (Table S2, Fig. S9). We varied the concentration of total bacterial RNA taken for amplification, amplification time, and sDz/PDz cascade reaction time (stage II in Scheme 1). For 16S rRNA amplification, either 0.1 pg or 10 pg of total MTC RNA was taken for a 12-μL NASBA reaction. This amount corresponds to 8.3 pg/mL or 833 pg/mL 16S rRNA in the pre-amplified samples, respectively. The amplicon of the correct size was obtained in a 60-min NASBA reaction for both high and low pre-amplified RNA concentration, while 45-min NABSA generated the amplicon only from high RNA concentration (Fig. 2A). When interrogated with the cognate sDz/PDz cascade, the 60-min amplicon obtained with 8.3 pg/mL RNA could be visually detected after as little as 15 min of IPDz cleavage, while a 45-min cleavage step resulted in a signal of maximal intensity (Fig. 2, panels B and C). For 833 pg/mL RNA input, a 15-min incubation with the cascade was sufficient to observe the high intensity color signal (Fig. 2, panels B and D). Incubation of either amplicon with the cascade for longer than 30 min did not improve the S/B much (Table 2). Overall, as little as 0.1 pg of MTC RNA taken into a 12-μL NASBA reaction (8.3 pg/mL pre-amplified RNA), with a 1-μL aliquot taken for the 16S rRNA-specific sDz/PDz cascade, triggered a visually observed signal in less than 2 h, including the hands-on/color development time.
Figure 2. Time-dependence for NASBA/sDz stages of the 16S rRNA-interrogating system.
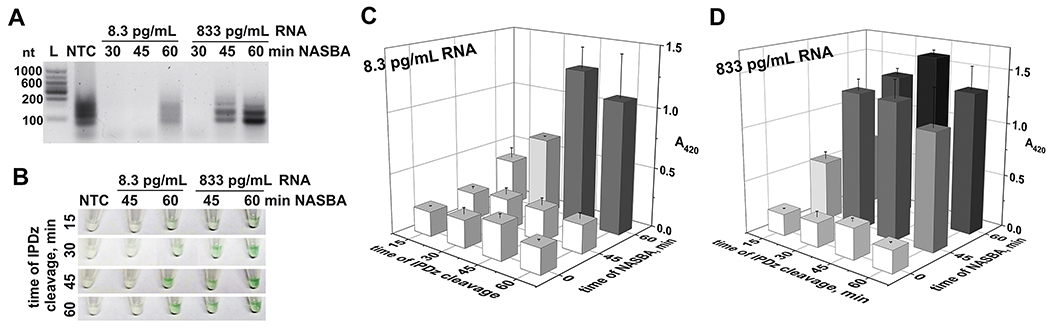
A. Gel electrophoretic analysis of the NASBA products obtained from 8.3 pg/mL or 833 pg/mL total MTC RNA. The NASBA reaction was carried out for 30, 45, or 60 min. NTC – NASBA “no-target control”, incubated for 60 min; L – low-range RNA ladder. B-D. Analysis of the NASBA products from panel A in the colorimetric assay using the16S_MTC-specific sDz/PDz system. The sample indicated as “0 min NASBA” is NTC. The samples containing 3.3% NASBA samples were incubated at 50 °C for 15, 30, 45, or 60 min for IPDz cleavage. B. Images of the sample tubes. C. NASBA samples obtained from 8.3 pg/mL RNA. D. NASBA samples obtained from 833 pg/mL RNA. The data is average of three independent experiments, with errors presented as relative standard deviations.
Table 2.
Signal-to-Background Ratios (S/B) for NASBA/sDz/PDz Cascades Depending on IPDz Cleavage Time (stage II in Scheme 1)a
| gene | [RNA], ng/ml | IPDz cleavage time, min | |||
|---|---|---|---|---|---|
| 15 | 30 | 45 | 60 | ||
| rrs | 0.0083 | 1.9±0.5 | 2.9±0.7 | 3.31±0.01 | 4.26±0.08 |
| 0.83 | 3±1 | 6±2 | 6±1 | 6.3±0.6 | |
| katG | 0.83 | N/D | 4±2 | 4.6±0.5 | N/D |
| 8.3 | 1.8±0.5 | 4±2 | 5±2 | 3±1 | |
| 83 | 2.5±0.9 | 6±3 | 6±1 | 5±2 | |
NASBA reaction was performed for 60 min. The S/B values were calculated as the absorbance of the samples with the RNA amplicon divided by the absorbance of the sample with no-target NASBA control. The data is given as average of three independent experiments, with relative standard deviations indicated. N/D – not determined.
The katG mRNA is present in fewer copies per cell than rRNA. Correspondingly, higher pre-amplified RNA concentration was required to trigger high signal with the cognate sDz/PDz probe. We started with 1 ng of total RNA (83 ng/mL pre-amplified RNA, 104-fold higher than for amplification of 16S rRNA). Addition of 1 μL of the amplification mixture after 60-min NASBA into a 30-μL sample (3.3%) containing the kG_INHS-specific cascade, triggered the color change, which was absent if the same kG_NASBA_INHS NASBA sample was tested with the kG_INHR-specific probe, even though the kG_INHR synthetic target triggered the expected high signal (Fig. 1). This observation confirms that excellent selectivity of the sDz/PDz cascade optimized with synthetic DNA targets is retained when applied to NASBA amplicons with stable secondary structure (predicted free energy of −23.82 kcal/mol, Fig. S9).
In general, as little as 0.4% kG_NASBA_INHS amplicon was sufficient to trigger visually observed signals (Fig. S10). This suggests that either amplification time could be shortened, or concentration of pre-amplified RNA can be reduced. Indeed, a 45-min amplification using either 1 ng (83 ng/mL) or 0.1 ng (8.3 ng/mL) of RNA yielded the expected kG_NASBA_INHS amplicon (Fig. 3A). The amplicon obtained with higher RNA concentration triggered the color change after 30 min incubation with the cognate cascade (Fig. S11A), and longer incubation did not significantly affect the signal intensity (Fig. S11B). For lower pre-amplified RNA concentration, the 45-min NASBA product did not trigger significant color change even for prolonged interrogation with the cascade, despite the presence of the product’s band in gel (Fig. 3, panels A-C). Linear correlation between the absorbance and the intensity of the amplicon-containing gel band was noticed (Fig. S11C), and 60-min NABSA with 8.3 ng/mL RNA resulted in a more intense amplicon band in gel (Fig. 3A). Therefore, as expected, the color output after 30-45 min of IPDz cleavage was triggered by 60-min NASBA amplicon obtained from low RNA concentrations (Fig. 3B). The intensity of the color increased slightly with the cleavage time (Fig. 3C), but the S/B was not improved by longer IPDz cleavage (Table 2). Therefore, the NASBA time fixed at 60 min was used to amplify the targeted fragment of the katG mRNA with varying concentration of the pre-amplified bacterial RNA (0.083-83 ng/mL). The generated amplicons were then subjected to the colorimetric assay with kG_INHS-specific sDz/PDz cascade for 30 or 45 min. The signal statistically distinguishable from the no-target amplification control (NTC) was observed with the kG_NASBA_INHS amplicons obtained from 0.83 ng/mL or 83 pg/mL bacterial RNA (Fig. 3D) after 30-min or 45-min IPDz cleavage, respectively (Figure S12). The calculated analytical LODs of the NASBA/sDz/PDz assay were 120 pg/mL and 75 pg/mL for 30- and 45-min IPDz cleavage reaction, respectively (Fig. S12). This value, on average, corresponds to the RNA amount that can be extracted from less than 1000 MTC cells (Hia et al., 2015).
Figure 3. Performance of the sDz/PDz cascade interrogating the katG gene.
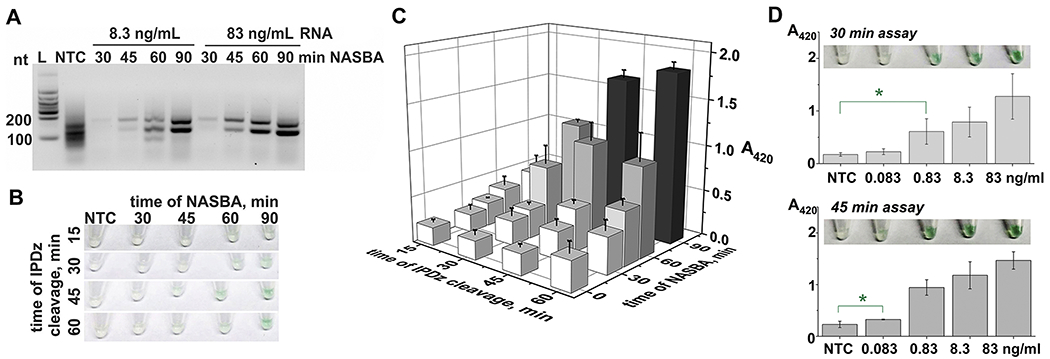
A. Analysis of the NASBA products obtained from 8.3 ng/mL or 83 ng/mL total MTC RNA by 2% agarose gel electrophoresis. The NASBA reaction was carried out for 30, 45, 60, or 90 min. NTC – NASBA “no-target control”, incubated for 90 min; L – low-range RNA ladder. B and C. The NASBA samples from panel A obtained using 8.3 ng/mL RNA were added (3.3% v/v) to the cascade containing strands kG_INHS and kG_U and. The samples’ color change (B) and absorbance at 420 nm (C) was recorded upon 15-, 30-, 45-, or 60-min IPDz cleavage at 50 °C. The sample indicated as “0 min NASBA” is NTC. D. Absorbance at 420 nm and tube images of the samples containing kG_NASBA_INHS amplicon obtained with different pre-amplified RNA concentrations. NASBA was performed for 60 min, and the samples were incubated at 50 °C for 30 min (top) or 45 min (bottom). The data is given as average of three independent experiments, with relative standard deviations indicated.
4. Discussion
Previously, we have designed the sDz/PDz systems reporting flavivirus-specific targets (Reed et al., 2019), and species-typing of mycobacteria (Wood et al., 2019). However, no differentiation of point mutations was shown. In the present work, we achieved discrimination of point mutations by the cascade system and used it for the analysis of nucleic acid sequences associated with drug-resistant MTC species. We interrogated a fragment of MTC katG mRNA obtained via NASBA and demonstrated that the amplicon can be selectively and reliably interrogated, without isolation, by the matched, but not the mismatched, probe. To the best of our knowledge, this study is the first example of NASBA amplification of a bacterial mRNA fragment containing drug-resistance conferring point mutations. We also applied the same methodology for amplification and interrogation of a 16S rRNA fragment as an assay for MTC detection. Both amplicons had relatively stable secondary structures (predicted free energy values were −20.35 kcal/mol and −23.82 kcal/mol for 16S_NASBA_MTC and kG_NASBA_INHS respectively, Fig. S9). A split approach for the probe design allowed target recognition and single mismatch discrimination without the need of an annealing step (Kolpashchikov, 2010). Indeed, the semi-independent interactions of the two sDz strands with the target, and the differential affinities of the strands to the target, ensured unwinding of the structured RNA amplicons.
The NASBA/sDz/PDz system reported here allows for MTC detection and DST within 2 h. Specifically, amplification of 10 pg of MTC RNA in a 12-μL NASBA reaction within 60 min produces enough amplicon of either 16S rRNA or katG mRNA to trigger the color change after 30-min IPDz cleavage (Table 2), plus 10-15 min hands-on and color development time. This turnaround time is similar to that reported for the state-of-the art Xpert MTB/RIF assay (Kwak et al., 2013). Taking into account similar LOD for the hybridization probes used in Xpert MTB/RIF and the assay reported here, as well as similar or higher amplification efficiency of NASBA in comparison with PCR (Jean et al., 2001), one can speculate that the reported assay may exhibit sensitivity comparable with the Xpert MTB/RIF assay.
The Xpert MTB/RIF assay is based on qPCR, so it requires a specialized device for temperature cycling and fluorescent detection. The reliance on a self-contained device and a single-use cartridge is advantageous with respect to minimizing cross-contamination and biohazardous risk, but contributes to high cost of the assay for low-resource high-incident settings. The NASBA/sDz/PDz assay reported herein relies on isothermal amplification without the need of precise temperature control and fast temperature cycling. In addition, it offers a color change as an easily readable signal without instrumentation (e.g. with the naked eye). This latter feature of the assay makes it similar to the line probe assays (LPA) (Ninan et al., 2016), which represent the second WHO-endorsed methods for MTC molecular DST. Somewhat low specificity has been reported for the detection of INHR by the LPA assays (Ninan et al., 2016). This may be attributable to insufficient ability of the conventional hybridization probes to differentiate bacterial genotypes corresponding to INHR and INHS phenotypes. The split approach we have developed ensures excellent discrimination of point mutations (Kolpashchikov, 2010). This lays the foundation for reliable DST of the pathogen. If combined with a self-enclosed device and/or strip-based format, the reported assay may be promising for quick and inexpensive POC analysis for MTC, as well as other bacterial pathogens.
5. Conclusions
In this work, we have demonstrated for the first time that the sDz/PDz cascade exhibits excellent selectivity for discrimination of point mutations in both synthetic targets and single-stranded NASBA amplicons of bacterial RNA. As low as 10 pg of bacterial RNA in a 12-μL NASBA reaction (830 pg/mL) could be used post-amplification to visually detect the absence of a point mutation in the katG gene, which would otherwise indicate isoniazid resistance. In the case of a 16S rRNA NASBA amplicon, even 100-fold lower RNA concentration was sufficient to trigger high color intensity with the cognate sDz/PDz probe. For both amplicons, the complete analysis, starting from total bacterial RNA, could be done in 1.5-2 h. The proposed assay can be intergraded in POC MTC DST or adopted for determination of drug resistance status of other clinically important bacteria.
Supplementary Material
Visual discrimination of point mutations by split deoxyribozyme cascade probes;
In situ interrogation of NASBA amplicons from mycobacterial RNA;
Analysis of an isoniazid-resistant genotype of M. tuberculosis from 10 pg total RNA;
The assay time including RNA amplification and analysis is less with 2 h.
Acknowledgments
This work was supported by the National Institutes of Health (grant R21AI123876). The authors would like to acknowledge Ms. Sandra Geden and Ms. Lauren Brumsey for preparation of mycobacterial strains and nucleic acids. The authors are grateful to Dr. Kolpashchikov for helpful discussions and critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The extinction coefficient of a typical chromophore is ~50-100 mM−1cm−1, so the absorbance of ~0.1 o.u, which is at threshold of what human eye can detect, corresponds to ~1-2 μM chromophore solution at a 1-cm path.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- Almeida Da Silva PE, Palomino JCJ 2011. Antimicrob. Chemother 66, 1417–1430. [DOI] [PubMed] [Google Scholar]
- Barenfanger J, Drake C, Kacich G 1999. J. Clin. Microbiol 37, 1415–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonnet G, Tyagi S, Libchaber A, Kramer FR 1999Proc. Natl. Acad. Sci. U.S.A 96, 6171–6176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan D, Coughlan H, Clancy E, Dimov N, Barry T, Kinahan D, Ducree J, Smith TJ, Galvin P 2017. Sens. Actuators B Chem 239, 235–242. [Google Scholar]
- CDC. Antibiotic Resistance Threats in the United States, 2019. Atlanta, GA: U.S. Department of Health and Human Services, CDC; 2019. [Google Scholar]
- Compton J 1991. Nature 350, 91–92. [DOI] [PubMed] [Google Scholar]
- Connelly RP, Verduzco C, Farnell S, Yishay T, Gerasimova YV 2019. ACS Chem. Biol 14, 2701–2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deiman B, van Aarle P, Sillekens P 2002. Mol. Biotechnol 20, 163–179. [DOI] [PubMed] [Google Scholar]
- Demidov VV, Frank-Kamenetskii MD 2004. Trends Biochem. Sci 29, 62–71. [DOI] [PubMed] [Google Scholar]
- Heo S, Kim HR, Lee HJ 2019. Plant Pathol. J 35, 164–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hia F, Chionh YH, Pang YL, DeMott MS, McBee ME, Dedon PC 2015. Nucleic Acids Res. 43, e32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hibbitts S, Rahman A, John R, Westmoreland D, Fox JD 2003. J. Virol. Methods 108, 145–155. [DOI] [PubMed] [Google Scholar]
- Horne DJ, Kohli M; Zifodya JS, Schiller I; Dendukuri N, Tollefson D, Schumacher SG, Ochodo EA, Pai M, Steingart KR 2019. Cochrane Database Syst. Rev 2019, CD009593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimova YV, Hayson A, Ballantyne J, DM Kolpashchikov DM 2010a. ChemBioChem 11, 1762–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimova YV, Peck S, Kolpashchikov DM 2010b. Chem. Commun 46, 8761–8763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimova YV, Cornett EM, Edwards E, Su X, Rohde KH, Kolpashchikov DM 2013. ChemBioChem 14, 2087–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimova YV, Kolpashchikov DM 2013a. Biosens. Bioelectron 41, 386–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimova YV, Kolpashchikov DM 2013b. Angew. Chem. Int. Ed 52, 10586–10588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerasimova YV, Yakovchuk P, Dedkova LM, Hecht SM, Kolpashchikov DM 2015. RNA 21, 1834–1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimes J, Gerasimova YV, Kolpashchikov DM 2010. Angew. Chem. Int. Ed 49, 8950–8953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isakova J, Sovkhozova N, Vinnikov D, Goncharova Z, Talaibekova E, Aldasheva N, Aldashev A 2018. BMC Microbiol. 18, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jean J, Blais B, Darveau A, Fliss I 2001. Appl. Environ. Microbiol 67, 5593–5600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kao M-C, Durst RA 2010. Anal. Lett 43, 1756–1769. [Google Scholar]
- Kievits T, van Gemen B, van Strijp D, Schukkink R, Dircks M, Adriaanse H, Malek L, Sooknanan R, Lens P 1991. J. Virol. Methods 35, 273–286. [DOI] [PubMed] [Google Scholar]
- Kolpashchikov DM 2010. Chem. Rev 110, 4709–4723. [DOI] [PubMed] [Google Scholar]
- Kolpashchikov DM 2012. Scientifica 2012, 928783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak N, Choi SM, Lee J, Park YS, Lee C-H, Lee S-M, Yoo C-G, Kim YW, Han SK, Yim J-J 2013. PLoS One 8, e77456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Macdonald J 2015. Biosens. Bioelectron 64, 196–211. [DOI] [PubMed] [Google Scholar]
- MacDougall D, Crummett WB 1980. Anal. Chem 52, 2242–2249. [Google Scholar]
- Nadal A, Coll A, Cook N, Pla M 2007. J. Microbiol. Methods 68, 623–632. [DOI] [PubMed] [Google Scholar]
- Nguyen C, Grimes J, Gerasimova YV, Kolpashchikov DM 2011. Chem. Eur. J 17, 13052–13058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninan MM, Gowri M, Christopher DJ, Rupali P, Michael JS 2016. Pathog. Glob. Health 110, 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantel A, Petrella S, Veziris N, Matrat S, Bouige A, Ferrand H, Sougakoff W, Mayer C, Aubry AJ 2016. Antimicrob. Chemother 71, 2428–2431. [DOI] [PubMed] [Google Scholar]
- Pardee K, Green AA, Takahashi MK, Braff D, Lambert G, Lee JW, Ferrante T, Ma D, Donghia N, Fan M, Daringer NM, Bosch I, Dudley DM, O’Connor DH, Gehrke L, Collins JJ 2016. Cell 165, 1255–1266. [DOI] [PubMed] [Google Scholar]
- Pavlov V, Xiao Y, Gill R, Dishon A, Kotler M, Willner I 2004. Anal. Chem 76, 2152–2156. [DOI] [PubMed] [Google Scholar]
- Reed AJ, Connelly RP, Williams A, Tran M, Shim BS, Choe H, Gerasimova YV 2019. Sens. Actuators B Chem 282, 945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Lazaro D, Lloyd J, Herrewegh A, Ikonomopoulos J, D’Agostino M, Pla M, Cook N 2004. FEMS Microbiol. Let 237, 119–126. [DOI] [PubMed] [Google Scholar]
- Rohde KH, Abramovitch RB, Russell DG 2007. Cell Host Microbe 2, 352–364. [DOI] [PubMed] [Google Scholar]
- Santoro SW, Joyce GF 1997. Proc. Natl. Acad. Sci. U.S.A 94, 4262–4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schon T, Miotto P, Koser CU, Viveiros M, Bottger E, Cambau E 2017. Clin. Microbiol. Infect 23, 154–160. [DOI] [PubMed] [Google Scholar]
- Seifert M, Catanzaro D, Catanzaro A, Rodwell TC 2015. PLos One 10, e0119628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seung KJ, Keshavjee S, Rich ML 2015. Cold Spring Harb. Perspect. Med 5, a017863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinshaw W, Kebede A, Bitew A,, Tesfaye E, Tadesse M, Mehamed Z, Yenew B, Amare M, Dagne B, Diriba G, Alemu A, Getahun M, Fikadu D, Desta K, Tola HH 2019. BMC Infect. Dis 19, 641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sougakoff W, Rodrigue M, Truffot-Pernot C, Renard M, Durin N, Szpytma M, Vachon R, Troesch A, Jarlier V 2004. Clin. Microbiol. Infect 10, 289–294. [DOI] [PubMed] [Google Scholar]
- Stancescu M, Fedotova TA, Hooyberghs J, Balaeff A, Kolpashchikov DM 2016. J. Am. Chem. Soc 138, 13465–13468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson ML 2011. Clin. Infect. Dis 52, 1350–1355. [DOI] [PubMed] [Google Scholar]
- Wolfenden BS, Willson RL 1982. J. Chem. Soc. Perkin Trans 2, 805–812. [Google Scholar]
- Wood HN, Sidders AE, Brumsey LE, Morozkin ES, Gerasimova YV, Rohde KH 2019. Clin. Chem 65, 333–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoo SM, Lee SY 2016. Trends Biotechnol. 34, 7–25. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.