

Clostridioides difficile infection (CDI) is an urgent public health threat with a high recurrence rate, in part because the causative bacterium has a high rate of antibiotic survival. The (p)ppGpp-mediated bacterial stringent response plays a role in antibiotic tolerance in diverse pathogens and is a potential target for development of new antimicrobials because the enzymes that metabolize (p)ppGpp have no mammalian homologs. We report that stationary-phase onset and antibiotics induce expression of the clostridial ppGpp synthetase RSH and that both chemical inhibition and translational suppression of RSH increase C. difficile antibiotic susceptibility. This demonstrates that development of RSH inhibitors to serve as adjuvants to antibiotic therapy is a potential approach for the development of new strategies to combat CDI.
KEYWORDS: alarmone, antibiotic tolerance, bacterial metabolism, Clostridioides difficile, guanosine tetra- and pentaphosphate, (p)ppGpp, stress response, stringent response
ABSTRACT
The human pathogen Clostridioides difficile is increasingly tolerant of multiple antibiotics and causes infections with a high rate of recurrence, creating an urgent need for new preventative and therapeutic strategies. The stringent response, a universal bacterial response to extracellular stress, governs antibiotic survival and pathogenesis in diverse organisms but has not previously been characterized in C. difficile. Here, we report that the C. difficile (p)ppGpp synthetase RSH is incapable of utilizing GTP or GMP as a substrate but readily synthesizes ppGpp from GDP. The enzyme also utilizes many structurally diverse metal cofactors for reaction catalysis and remains functionally stable at a wide range of environmental pHs. Transcription of rsh is stimulated by stationary-phase onset and by exposure to the antibiotics clindamycin and metronidazole. Chemical inhibition of RSH by the ppGpp analog relacin increases antibiotic susceptibility in epidemic C. difficile R20291, indicating that RSH inhibitors may be a viable strategy for drug development against C. difficile infection. Finally, transcriptional suppression of rsh also increases bacterial antibiotic susceptibility, suggesting that RSH contributes to C. difficile antibiotic tolerance and survival.
IMPORTANCE Clostridioides difficile infection (CDI) is an urgent public health threat with a high recurrence rate, in part because the causative bacterium has a high rate of antibiotic survival. The (p)ppGpp-mediated bacterial stringent response plays a role in antibiotic tolerance in diverse pathogens and is a potential target for development of new antimicrobials because the enzymes that metabolize (p)ppGpp have no mammalian homologs. We report that stationary-phase onset and antibiotics induce expression of the clostridial ppGpp synthetase RSH and that both chemical inhibition and translational suppression of RSH increase C. difficile antibiotic susceptibility. This demonstrates that development of RSH inhibitors to serve as adjuvants to antibiotic therapy is a potential approach for the development of new strategies to combat CDI.
INTRODUCTION
Clostridioides (formerly Clostridium) difficile is an obligate anaerobic Gram-positive bacillus that persists in aerobic environments as metabolically dormant spores (1). C. difficile spores are tolerant to oxygen, heat, and several alcohol-based disinfectants as well as quaternary ammonium-based detergents, making their eradication from contaminated environments extremely challenging (2, 3). Upon ingestion by susceptible hosts and exposure to amino acids and bile salts in the digestive tract, the spores germinate into actively reproducing vegetative cells in the anaerobic mammalian gut (4). The oxygen-intolerant vegetative cells secrete cytotoxins that are internalized by host epithelial cells, leading to actin depolymerization and the collapse of the actin cytoskeleton (5, 6). The subsequent loss of cell-cell contacts compromises the epithelial integrity and results in a massive host inflammatory response (7, 8). This contributes to debilitating clinical presentations of the disease, which range from mild diarrhea to life-threatening pseudomembranous colitis and toxic megacolon (3, 9).
In healthy individuals, the metabolic activity of the commensal gut microbiota provide colonization resistance against C. difficile infection at least in part by limiting the nutrients available to the invading pathogen (10–13). While cases of community-acquired C. difficile infection (CDI) have increased in recent years, CDI predominantly occurs in health care settings such as hospitals and residential elder care facilities, where the gut microbiota of the residents have been disrupted by age or prior antibiotic usage (3, 14–16). Once C. difficile colonizes the host colon, it can survive the host immune system, the regrowth of commensal gut microbes, and antibiotic therapy and cause infections with an extremely high (20 to 30%) rate of recurrence after initial treatment (17, 18).
C. difficile infects over 450,000 patients annually in the United States alone, resulting in an estimated 29,000 deaths and $4.8 billion in associated medical costs (17). The public health impact of CDI has increased in the 21st century due to the emergence of so-called “hypervirulent” epidemic strains with higher levels of sporulation and toxin production, which cause infections with corresponding higher rates of transmission and morbidity (19–23). C. difficile has very high survival rates when treated with multiple classes of antibiotics, including penicillin, cephalosporins, clindamycin, and fluoroquinolones (18, 24, 25). Epidemic strains exhibit lower antibiotic susceptibility than historical strains, contributing to increased CDI recurrence (26, 27). Metronidazole and vancomycin have been the first-line treatments for moderate and severe CDI, respectively, but recent guidelines issued by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology (SHEA) do not recommend the use of metronidazole, as the antibiotic is now losing clinical efficacy (28). Patients treated with vancomycin experience a 10 to 35% rate of recurrence, with at least 40% of recurrent infections manifesting multiple times (29). Patients often experience consecutive relapse episodes, leading to a poor quality of life with limited therapeutic options (29). Fidaxomicin is associated with a lower CDI recurrence rate via the inhibition of C. difficile spore formation and toxin production (30, 31). But fidaxomicin is expensive, preventing its widespread use, and resistance of epidemic strains to the antibiotic has been reported in clinical trials (29–32). As C. difficile demonstrates high survival rates when treated with multiple classes of antibiotics, fecal microbiota transplantation (FMT) is currently the most effective therapy for recurrent CDI (24, 25, 33). The introduction of suppressive bacterial species to C. difficile-colonized gastrointestinal tracts of infected patients has demonstrated a success rate of approximately 90% (34). Unfortunately, the technique carries an inherent risk of transmitting undetected pathogens and/or bacterial agents to the recipient, and the long-term safety and efficacy have yet to be determined (35, 36). Therefore, the development of novel therapies or preventative strategies against CDI is an urgent public health priority.
In addition to competition for nutrients and antibiotic stresses, pathogens are exposed to a number of harsh environmental conditions when colonizing a potential host, including febrile heat and pH changes, reactive oxygen species, and antimicrobial peptides produced by the innate immune system (37, 38). Bacteria have evolved several stress response pathways, including the stringent response (SR). The SR is mediated by the nucleotide alarmones GDP-3′-disphosphate (ppGpp) and GTP-3′-disphosphate (pppGpp), collectively known as (p)ppGpp (39, 40). Cytoplasmic accumulation of (p)ppGpp results in a global halt in cell cycle progression, DNA replication, and rRNA synthesis with a concomitant increase in transcription of biosynthetic and stress response genes that largely favor bacterial survival (39, 40). The SR is further associated with virulence traits such as biofilm formation, sporulation, toxin production, and antibiotic survival in diverse pathogens, including Mycobacterium tuberculosis, Legionella pneumophila, Salmonella enterica, Pseudomonas aeruginosa, Staphylococcus aureus, and Enterococcus faecalis (39, 41–49).
The RelA/SpoT homolog (RSH) proteins that mediate the SR in Gram-positive bacteria are bifunctional (p)ppGpp synthetase/hydrolase enzymes with C-terminal regulatory domains, named for their homology to the RelA and SpoT proteins, which mediate the SR in Gram-negative bacteria (50). When active as synthetases, RSH proteins catalyze pyrophosphate transfer from ATP to the ribose 3′ OH group of GDP or GTP to synthesize ppGpp and pppGpp, respectively (51–53). When active as phosphatases, RSH enzymes transfer pyrophosphate away from (p)ppGpp (51, 52). The synthetase and hydrolase activities of bifunctional RSH enzymes are mutually exclusive; regulation is presumed to occur via protein-protein interactions or small-molecule binding by noncatalytic C-terminal regulatory domains (51, 52, 54–57). RSH enzymes are transcriptionally regulated, with transcription increasing during late exponential growth or stationary phase or upon induced nutrient deprivation by inhibitors of biosynthetic pathways (40, 58–60). The specific nutrients that trigger (p)ppGpp synthesis vary between bacterial species (40, 58, 61–63). The more recently discovered family of small alarmone synthetase (SAS) enzymes consists of monofunctional (p)ppGpp synthetases (64, 65). Gram-positive bacteria typically possess one RSH family enzyme and one or two SASs; Firmicutes bacteria contain two families of SAS, named RelQ and RelP (50). SAS enzymes appear to contribute to the stringent response and also to play an SR-independent role as an allosteric regulator of guanosine and GTP biosynthesis (66–69). Extracellular stresses previously documented to induce SAS gene expression include cell wall stress induced by vancomycin, ethanol, or alkaline stress (46, 59, 64, 70, 71).
Due to the role of (p)ppGpp in bacterial survival and virulence factor production, there has been some pharmaceutical interest in inhibiting the SR by preventing (p)ppGpp synthesis (72–76). The ppGpp structural analog relacin competitively inhibits the activity of RSH family (p)ppGpp synthetases (77). The compound reduces (p)ppGpp accumulation, stationary-phase survival, biofilm formation, and sporulation in Bacillus species (78). Relacin does not affect the activity of SAS family synthetases or inhibit in vivo (p)ppGpp accumulation in Escherichia coli, indicating that SR inhibitors are not universally effective and must be targeted to specific bacterial strains (78, 79).
We have previously confirmed that RSH from C. difficile is a functional ppGpp synthetase (80). Here, we demonstrate that RSH readily synthesizes ppGpp at physiologically relevant concentrations of GDP. In contrast to previously characterized RSH homologs from Gram-positive organisms, C. difficile RSH (RSHCd) interacts poorly with GTP and fails to produce detectable pppGpp under the conditions studied. Utilizing an anaerobic fluorescent transcriptional reporter, we found that stationary-phase onset and exposure to the antibiotics clindamycin and metronidazole stimulate transcription of (p)ppGpp synthetase genes in a strain-specific manner. We further found that inhibition of RSH synthetase activity by relacin along with the knockdown of the rsh gene increases C. difficile antibiotic susceptibility. Conclusively, our findings render the SR pathway an attractive target for the development of new prevention and treatment strategies against CDI.
RESULTS AND DISCUSSION
Clostridioides difficile has conserved (p)ppGpp metabolism genes.
The C. difficile genome contains putative RSH and SAS genes, rsh and relQ, suggesting that the organism could mount a stress-induced SR (Fig. 1B). RSH is a putative bifunctional Rel-Spo homolog with two highly conserved N-terminal synthetase and hydrolase domains and aspartate kinase, chorismate, and TyrA (ACT) and ThrRS, GTPase, and SpoT (TGS) C-terminal regulatory domains (Fig. 1A). The in vitro ppGpp synthetase activity of C. difficile RSH has recently been reported (80). C. difficile RelQ is a putative single-domain small alarmone synthetase (Fig. 1A). The rsh and relQ genes are completely conserved between the historical strain CD630, the erythromycin-susceptible derivative 630Δerm, and the modern epidemic strain R20291 (21, 50, 81, 82). rsh and relQ are both in predicted operons with beta-lactamase-like genes, suggesting a potential role for (p)ppGpp signaling in C. difficile ampicillin tolerance (Fig. 1B). rsh also shares an operon with the predicted d-aminoacyl-tRNA deacylase dtd; in Clostridium perfringens, the rsh homolog and dtd are coexpressed and function together to regulate sporulation (83). A previous microarray analysis of the C. difficile strain 630 transcriptional response to various stresses has shown a 2-fold upregulation of relQ after exposure to ampicillin and clindamycin, further suggesting that (p)ppGpp signaling could play a role in C. difficile antibiotic survival (84).
FIG 1.

(p)ppGpp metabolism in C. difficile. (A) Domain organization of predicted (p)ppGpp metabolism enzymes. RSH contains putative hydrolase, synthetase, and regulatory domains. RelQ is a predicted single domain synthetase. C. difficile does not encode any predicted small alarm hydrolase enzymes. (B) Genomic context of predicted (p)ppGpp metabolism genes (blue). rsh and relQ share predicted operons with beta-lactamase-like proteins (pink). The rsh operon also contains dtd. (C and D) Growth of E. coli carrying empty vector pMMBneo (C) or pMMBneo:rsh (D) in the presence or absence of IPTG. Cells were cultured in LB medium or LB medium supplemented with Casamino Acids (LB-CAA). (E) Growth of E. coli carrying pMMBneo:rshREL in LB medium supplemented with 1% Casamino Acids and/or 1% glucose (GLU). Growth of three independent biologicals with three technical replicates is shown for one assay.
Exogenous expression of full-length C. difficile rsh suppresses the growth of exponentially growing E. coli, consistent with previous observations that intracellular accumulation of (p)ppGpp inhibits bacterial growth (Fig. 1C and D) (39). Supplementation of the E. coli growth medium with Casamino Acids rescued the RSH-dependent growth defect, suggesting that amino acid limitation may trigger the C. difficile stringent response in vivo (Fig. 1D). The presence of the gene fragment encoding the RSHCd synthetase domain (RSH-REL) is extremely toxic to E. coli even in the absence of induction, suggesting that the enzyme is so active that trace translation from uninduced promoter activity results in enough protein to synthesize inhibitory levels of (p)ppGpp. RSH-REL toxicity can be partially rescued by supplementation with 1% Casamino Acids and is further enhanced by supplementation with Casamino Acids and 1% glucose (Fig. 1E). The finding that full-length C. difficile RSH slows E. coli growth while RSH-REL completely arrests it suggests that (p)ppGpp toxicity is modulated by activation of the RSH hydrolase domain, which in homologous proteins is allosterically regulated by the C-terminal regulatory domains (CTD) in response to amino acids, including valine, isoleucine, and leucine (55, 57, 85). The rescue of RSHCd- or RSH-REL-induced growth defects by nutrient supplementation indicates that these enzymes trigger the endogenous E. coli stringent response, which is activated by nutrient limitation (86).
Alignment of the amino acid sequence of RSHCd with those of known proteins belonging to the RSH family of enzymes revealed that the sequences exhibit pronounced similarities to the substrate binding and product coordination regions of the N-terminal catalytic domain (Fig. 2A). Many functionally significant amino acid residues are conserved between the structural template Streptococcus equisimilis RelA (SeqRelA; GenBank accession no. X72832) and RSHCd, suggesting that the enzymes share functional, and likely structural, homology (51, 64). Several hydrophobic amino acid residues found in the synthetase domain of Bacillus subtilis RSH (BsRSH; GenBank accession no. U86377), which are predicted to coordinate binding of inhibitory ppGpp analog relacin, are also well conserved in the RSHCd synthetase domain, suggesting that RSHCd can be inhibited by relacin (64, 78).
FIG 2.

(A) Alignment of the amino acid sequences of the catalytic domains RSHCd and CdRelQ, with those of RSH and RelP/RelQ/SAS enzymes from Bacillus subtilis, Enterococcus faecalis, Staphylococcus aureus, and Streptococcus equisimilis. Residues important for substrate binding, product coordination, and allosteric (p)ppGpp binding in S. aureus RelP are indicated with lines as shown. Highly conserved residues are marked in black, and residues with same polarity are marked in gray. (B) Summary of the sequence alignment of RSHCd catalytic domain with those of RelA and RSH encoded by S. equsimilis and B. subtilis, respectively.
C. difficile RSH exclusively synthesizes ppGpp in vitro.
The in vitro ppGpp synthetase activity of RSHCd using GDP as a substrate has previously been reported (80). While SAS enzymes from E. faecalis and B. subtilis can utilize GMP as a substrate to synthesis pGpp, and RSH homologs from S. equisimilis and M. tuberculosis preferentially utilize GTP to synthesize pppGpp, RSHCd appears to be unable to utilize GMP or GTP and synthesizes ppGpp exclusively under the conditions studied (Fig. 3A) (52, 64, 79, 87–89). GTP does appear to stimulate ATP hydrolysis by RSHCd, suggesting either that this enzyme recognizes GTP but fails to accommodate the triphosphate form of the nucleotide in its active site or that the protein binds GTP without enabling a pyrophosphotransferase reaction (Fig. 3A). The ability of RSHCd to convert GDP and excess radioactive ATP to ppGpp increases in a dose-dependent manner with increasing concentrations of GDP (Fig. 3B). We found that RSHCd binds GDP with a Km of 95 μM (Fig. 3C). As enzymatic substrate affinity is generally close to biologically relevant concentrations of that substrate, this finding is consistent with previous reports that GDP concentrations are 100 to 600 μM in Gram-negative and Gram-positive bacterial cytoplasm during exponential growth and rise during stationary-phase onset (90–93). The finding that RSHCd does not produce pppGpp was further explored by running nucleotide standards separately through an anion exchange high-performance liquid chromatography system (see Fig. S1 in the supplemental material). We found the GTP peak resolved in the chromatogram to be free of contaminants and derivative compounds, suggesting that RSHCd’s inability to utilize GTP is not due to substrate impurity or degradation (Fig. S1).
FIG 3.

In vitro characterization of RSHCd activity. (A) RSH readily transfers radioactive pyrophosphate from [γ-32P]ATP to GDP but cannot utilize either GMP or GTP as a substrate. ATP hydrolysis to inorganic phosphate increases in the presence of GTP. (B) Conversion of [γ-32P]ATP to ppGpp increases with GDP concentration. (C) Lineweaver-Burk plot of RSHCd activity. The Km of RSHCd for GDP is 95 μM. (D) RSHCd activity in the presence of exogenous ppGpp shows no allosteric activation of synthetase activity. Activities at different ppGpp concentrations were compared by one-way analysis of variance (ANOVA). n.s., not significant.
ppGpp is a positive allosteric regulator of ppGpp synthesis by E. coli RelA, and pppGpp has been shown to allosterically activate SAS activity in B. subtilis, raising the possibility that RSHCd is susceptible to feedback regulation by its own product (53, 94). We measured RSHCd synthetase activity in the presence of increasing concentrations of exogenous ppGpp and found no evidence of allosteric regulation (Fig. 3E).
C. difficile RSH activity is unaffected by pH or the identity of the metal ion cofactor.
Like other multidomain RSH family enzymes, RSHCd is a basic enzyme with an isoelectric point of 8.81 (52, 95). RSHCd synthesizes ppGpp but not pppGpp at a buffer pH of 7.5 (Fig. 3A), a value representative of normal colonic perimucosal pH, which ranges between 7.1 and 7.5 (80, 96). To investigate how RSHCd utilizes substrate(s) under various pH conditions, in vitro transferase reactions were conducted separately in buffer pHs ranging from 5 to 10 (Fig. 4). The enzyme synthesizes ppGpp at all buffer conditions studied but does not synthesize pppGpp at any pH, suggesting that the enzyme cannot bind to or accommodate GTP regardless of the titration status of synthetase domain residues (Fig. 4).
FIG 4.

RSHCd synthesizes ppGpp independently of buffer pH. (A) TLC autoradiogram demonstrating the accumulation of ppGpp by RSHCd in the presence of GDP (left) as a result of in vitro transferase reaction at various buffer pHs. RSHCd is incapable of utilizing GTP (right) to produce pppGpp at any given pH value. (B) Graphical representation of the autoradiogram demonstrating that RSHCd utilizes GDP to produce ppGpp without exhibiting pH specificity. ppGpp synthesis at various buffer pH was compared to that at pH 7.5 by one-way ANOVA.
Pyrophosphokinases and nucleic acid-metabolizing enzymes require metal ion cofactors to coordinate negative charge. Magnesium, present in millimolar quantities in bacterial cytoplasm, is the most commonly utilized metal for this purpose and has historically been employed for in vitro (p)ppGpp synthetase assays, although RSH hydrolase domains utilize manganese (51, 52, 97–100). Very recently, Methylobacterium extorquens RSH was shown to synthesize pppGpp more efficiently in the presence of Co2+ than Mg2+ (101). Human DNA polymerase β, whose catalytic palm domain bears structural kinship with the synthetase domain active site of SeqRelA, is capable of utilizing Zn2+, Mn2+, and Co2+, as well as Mg2+, to catalyze primer extension (51, 102). All of these cations have ionic radii similar to that of Mg2+ (102, 103). Ca2+, which has an ionic radius significantly larger than that of Mg2+, does not enable primer extension by DNA polymerase β, confirming that the enzyme is selective for specific metal ion cofactors (102). To investigate the metal cofactor selectivity of RSHCd, in vitro transferase reactions were conducted independently in the presence of different metals (Fig. 5). Of the eight divalent cations that were tested (Mg2+, Mn2+, Co2+, Cu2+, Fe2+, Zn2+, Ni2+, and Ca2+), only Cu2+ reduced RSHCd-driven ppGpp synthesis, suggesting that this enzyme readily employs a wide array of structurally diverse cations for a cofactor (Fig. 5B). As mammalian immune systems sequester metal during bacterial infection and metal starvation can trigger the stringent response in diverse bacteria (62, 63, 104), it is possible that flexible utilization of divalent cations by RSHCd enables (p)ppGpp synthesis under metal-limited conditions.
FIG 5.
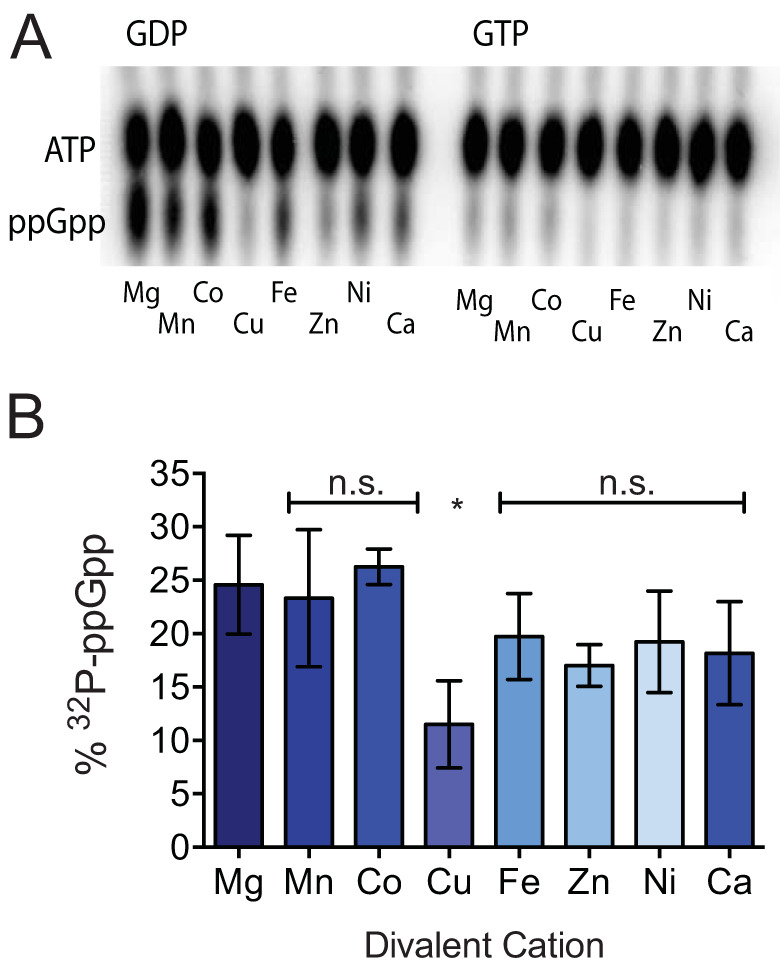
RSHCd utilizes diverse metal cations to synthesize ppGpp. (A) TLC separation of ppGpp produced by RSHCd in vitro (left) in the presence of different divalent cations at buffer pH of 7.5. RSHCd is incapable of producing pppGpp in the presence of any given metal cofactor (right). (B) Graphical representation of the autoradiogram depicting that RSHCd employs many structurally diverse divalent cations to produce ppGpp. Levels of ppGpp synthesis were compared by one-way ANOVA. *, P < 0.01.
C. difficile RSH binds GTP with poor affinity.
To further investigate the lack of pppGpp formation by RSHCd, we examined the enzyme’s interactions with GDP and GTP using isothermal titration calorimetry (ITC). Purified RSHCd at1.2 μM exhibited association constants of 31.9 M−1 and 25.1 M−1 for GDP and GTP, respectively, indicating that RSHCd does bind GTP (Table 1). During exponential growth, bacterial cells maintain GTP concentrations ranging from 900 to 1,100 μM, a 2- to 10-fold excess compared to GDP concentrations (90–92). During stationary-phase onset, both ATP and GTP are rapidly depleted 2- to 4-fold, with ATP levels recovering more quickly due to the “diversion” of guanosine nucleotides into (p)ppGpp (91, 92). GDP levels remain more stable during phase transition (91, 92). Based on our observations, the binding affinity of RSHCd for GTP is lower than that for GDP, which at physiological concentrations during stationary phase would allow the enzyme to preferentially utilize GDP as a substrate. Notably, we found RSHCd to bind GTP at a protein-to-substrate ratio of 1:10. But failure to utilize GTP for subsequent production of pppGpp was found at a protein-to-substrate ratio of 1:200 (Fig. 3A). In vitro transferase reaction combining mixtures of GDP and GTP at a GDP-to-GTP ratio of 1:1 as well as 1:2 also demonstrated RSHCd’s inability to utilize GTP (Fig. S6). These results thus provide further suggestion that while RSHCd transiently binds GTP at different stages of bacterial growth, the enzyme rapidly responds to GDP accessibility and stability, which may also be influenced by intracellular polyphosphate metabolizers.
TABLE 1.
Thermodynamic parameters of RSHCd binding to GXP at 37°Ca
| Substrate | K (M−1) | ΔH (cal/mol) | ΔS (cal/mol/°C) |
|---|---|---|---|
| GDP | 31.9 ± 1.11e7 | −1.916e5 ± 7.235e5 | −611 |
| GTP | 25.1 ± 3.65e6 | −2.165e5 ± 9.176e5 | −692 |
The binding of RSHCd at 0.0012 mM to GDP/GTP at 0.012 mM was measured by ITC. The data were fitted to a single-binding site model. Shown are the values for K, the equilibrium binding constant; ΔH, the enthalpy change associated with protein binding to the ligand; and ΔS, the entropy change associated with binding. Each value is the average from three repeat experiments, with standard deviations.
The “K” value, which represents the association constant calculated from a single site binding model, was found to be slightly lower than the Km of RSHCd calculated for GDP through radiolabeled TLC (Table 1 and Fig. S3). We postulate that this deviation in binding constant may arise from nonspecific interaction of GDP with the CTD of full-length RSHCd (54).
C. difficile rsh transcription is induced by stationary-phase onset.
Transcription of the rsh gene is induced by nutrient limitation during the onset of stationary phase in E. coli and other bacterial species (40, 60, 105). To confirm this in C. difficile, we examined transcript levels of rsh and relQ at culture optical densities at 600 nm (OD600) of 0.8, 1.2, and 1.6, corresponding to exponential growth, transition into stationary phase, and stationary phase, respectively. We found that transcription of rsh increased 3-fold as cells transitioned out of exponential growth and remained elevated as cells entered stationary phase (Fig. 6A). relQ transcription was not growth phase dependent, consistent with previous reports that SAS expression is stimulated by extracellular stress rather than nutrient deprivation or stationary-phase onset (46, 47, 59, 64, 70, 71).
FIG 6.
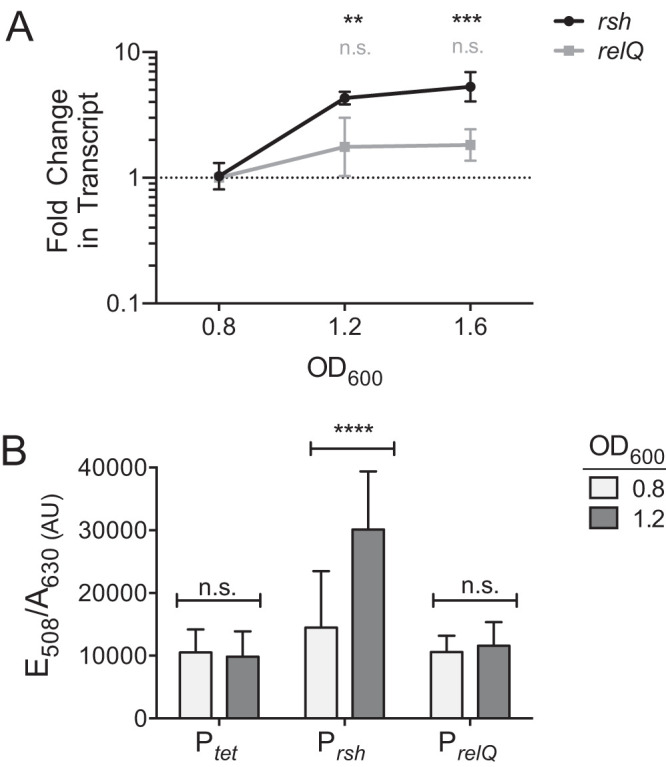
Stationary-phase onset stimulates rsh transcription. (A) qRT-PCR analysis shows that transcription levels of rsh increase 3-fold during the transition from exponential growth to stationary phase, which occurs at OD600 of 1.0 to 1.2, and remain elevated during stationary phase. relQ transcript levels show no significant change. Transcript levels were compared to those of the same gene during exponential growth by two-way ANOVA using Dunnett’s multiple-comparison test. n.s., not significant; **, P < 0.05; ***, P < 0.001. (B) The fluorescent phiLOV2.1 transcriptional reporter also shows a 3-fold increase of rsh promoter activity between exponentially growing cells and cells entering stationary phase. Activities of the relQ promoter and of the tet control promoter are unaffected by growth phase. Transcript levels were compared to those of the same gene during exponential growth by two-way ANOVA using Dunnett’s multiple-comparison test. n.s., not significant; ****, P < 0.0001.
In order to rapidly screen multiple environmental signals as potential regulators of C. difficile SR gene expression, we utilized an anaerobic fluorescent transcriptional reporter incorporating the oxygen-independent flavoprotein phiLOV2.1 (106). We placed the phiLOV2.1 gene under the control of the rsh and relQ promoter regions from C. difficile 630Δerm. To validate our reporter constructs, we repeated the comparison of rsh and relQ transcription at OD600 of 0.8 and 1.2. Our results were consistent with those of the more quantitative reverse transcription-PCR (RT-PCR) experiment. Promoter activity from the tetracycline-inducible control and that from PrelQ were unaffected by growth phase, while the activity of the rsh promoter increased roughly 3-fold upon stationary-phase onset (Fig. 6B).
C. difficile induces rsh and relQ gene transcription in response to antibiotic stress.
Our transcriptional analyses confirmed that stationary-phase onset is a positive regulator of C. difficile rsh transcription. A previous study has demonstrated that antibiotic stress also induces the SR in methicillin-resistant Staphylococcus aureus, resulting in an antibiotic resistance phenotype (47). We used the phiLOV2.1 reporter to monitor transcription of rsh and relQ upon exposure to sublethal concentrations of clindamycin, a medical antibiotic whose use is highly correlated with risk for CDI (107). The rsh promoters in the 630Δerm and R20291 genomes are differentiated by a single purine substitution, while the relQ promoters are identical (Fig. S4). Promoter activity in the tetracycline-inducible control was unaffected by clindamycin in either the 630Δerm or the R20291 genetic background (Fig. 7A). The activity of the (p)ppGpp synthetase promoters was increased by antibiotic stress in a strain-specific manner. The rsh promoter was unaffected by clindamycin in 630Δerm and showed a 50% increase in activity in R20291. Conversely, the relQ promoter was upregulated 2.8-fold in 630Δerm but was unaffected in R20291 (Fig. 7A). To investigate whether (p)ppGpp synthesis could also be stimulated by exposure to an antibiotic that has until recently been clinically effective against C. difficile, we repeated this assay by exposing the bacteria to sublethal amounts of metronidazole (108, 109). The tetracycline-inducible control promoter was not affected by metronidazole exposure, and neither of the (p)ppGpp synthetase promoters displayed any metronidazole response in the 630Δerm background (Fig. 7B). Activity from the rsh promoter increased 80% in R20291, while the relQ promoter showed no response (Fig. 7B).
FIG 7.

Antibiotic-induced transcription of rsh and relQ. PhiLOV2.1 transcriptional reporter activity after 2 h of exposure to 16 μg/ml of clindamycin (A) and 0.3 μg/ml of metronidazole (B) in the C. difficile 630Δerm and R20291 genetic backgrounds. Fluorescence activities normalized by cell density in the presence and absence of antibiotics were compared by two-way ANOVA. *, P < 0.05; **, P < 0.01; ****, P < 0.0001.
Disruption of RSH activity reduces C. difficile antibiotic survival.
Relacin is a structural analog of ppGpp that competitively inhibits Rel/RSH enzymes from E. coli and B. subtilis (78). We found that relacin inhibits the activity of RSHCd in vitro in a dose-dependent manner (Fig. 8A). Relacin independently had no discernible effect on endpoint C. difficile R20291 accumulation in vivo (Fig. 8B). However, the combination of relacin with a sublethal concentration of either clindamycin (Fig. 8C) or metronidazole (Fig. 8D) inhibited final growth yield more than either drug alone. The effect of relacin appeared to be more pronounced with clindamycin, which activated rsh transcription in C. difficile R20291, than with metronidazole.
FIG 8.

Effect(s) of disrupting RSH-regulated stringent response on C. difficile antibiotic survival. (A) Relacin inhibits the activity of RSHCd in a dose-dependent manner in vitro. Activity at each relacin concentration was compared to that under the 0 mM relacin condition by one-way ANOVA. (B) Relacin alone has no impact on the final growth yield of C. difficile R20291. Cell densities after overnight growth with and without relacin were compared by one-way ANOVA. (C and D) Cell density at 18 h in the presence of sublethal clindamycin (C) and metronidazole (D) with and without relacin. The cell densities of antibiotic-treated samples with and without relacin were compared by unpaired t test. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (E) Knockdown of the rsh gene with anhydrotetracycline inducer (0.5 μg/ml) negatively impacts the final growth yield of the R20291 strain in the presence of metronidazole (0.075 μg/ml). The cell densities of antibiotic-treated samples with and without ATc were compared by ordinary one-way ANOVA. **, P = 0.0042.
Due to the observed modest effect of relacin on a metronidazole-treated epidemic strain, we next confirmed the significance of rsh for C. difficile survival during antibiotic stress by inducing the expression of antisense RNA (asRNA) complementary to the 5′ end of the rsh mRNA, a technique known as gene silencing, which has previously been shown to deplete mRNA transcript levels and “knock down” gene transcription in C. difficile (32). Induction of the asRNA to rsh’s mRNA dramatically suppressed R20291 accumulation when exposed to a sublethal concentration of metronidazole, suggesting that rsh plays a direct role in C. difficile metronidazole tolerance and survival (Fig. 8E). In contrast, proliferation of the empty-vector control strain was unaffected by the inducer (Fig. 8E). Interestingly, cells carrying the pMSPT:as_rsh vector demonstrated partial inhibition even in the absence of the inducer, which was not seen in the empty-vector control strain, which is suggestive of leaky promoter transcription (Fig. 8E).
Conclusions.
The (p)ppGpp-mediated stringent response is ubiquitous among bacteria, so it is unsurprising that it plays a role in the notorious resilience of C. difficile against antibiotic stress. We have previously confirmed that RSH from C. difficile is an active ppGpp synthetase; in this study, we have determined that its ppGpp synthesis activity is robust in a wide range of environmental pHs and can utilize an unexpectedly diverse array of metal cofactors. We have confirmed that RSH binds GTP with lower affinity than GDP but appears to be incapable of utilizing it as a substrate. The failure of RSHCd to utilize GTP at equimolar concentration of GDP and at an excess concentration in the same pool directly indicates that the enzyme is an exclusive ppGpp synthetase. To some degree, it is also likely that pppGpp is unstable or prone to degradation in C. difficile, as suggested in Porphyromonas gingivalis (110). Gram-negative bacteria encode the guanosine pentaphosphate 5′-phosphohydrolase (GPP; GppA) enzyme, which hydrolyzes pppGpp to ppGpp via removal of the 5′-phosphate group (111). In E. coli, conversion of pppGpp to ppGpp is indicated to refine the potency of ppGpp in bacterial physiology (112). However, it remains to be explored whether SR-mounting Gram-positive species encode a protein(s) with identical GPP/GppA functionality (113). Interestingly, pppGpp is a more potent inhibitor of DNA primase in B. subtilis than ppGpp, but degradation of pppGpp for potency refinement has not been reported for the organism (114). This suggests that observations made for relative organisms cannot be extrapolated to the effects of (p)ppGpp potency and metabolism in the C. difficile background. Therefore, future studies are imperative to quantitate intracellular accumulation of (p)ppGpp in C. difficile and to establish proteins involved in polyphosphate breakdown. Furthermore, biochemical characterization of C. difficile RelQ is needed to achieve a more comprehensive understanding of (p)ppGpp metabolism in this organism.
The stringent response governs virulence traits in diverse bacterial pathogens, including biofilm production, spore formation, virulence factor expression, and survival of pharmacological and immune stresses. We have identified stationary-phase onset and antibiotic exposure as environmental stimuli that trigger rsh transcription in C. difficile in a strain- and stress-dependent manner. Interestingly, the historical 630Δerm strain and the epidemic R20291 strain exhibit different transcriptional responses to antibiotics. The epidemic strain increases rsh transcription in response to clindamycin and metronidazole, while the historical strain does not have a transcriptional response to metronidazole and induces transcription of relQ rather than rsh upon exposure to clindamycin. It may be relevant that R20291 contains an intact ermB gene, which implies some cross-resistance to clindamycin as well as erythromycin, while this gene is deleted in 630Δerm, which may affect the perception of antibiotic-induced stress by these bacterial strains (115). While clindamycin and metronidazole are both cytoplasmic rather than extracellular antibiotics, they kill bacteria by distinct mechanisms, targeting protein synthesis and DNA replication, respectively, so it appears that rsh transcription may be a general response to antibiotic stress rather than a response to inhibition of a specific synthetic pathway. Notably, inhibition of RSH activity by relacin amplified the potency of both drugs against the epidemic C. difficile strain R20291. As the effects of relacin on antibiotic efficacy were slight and required millimolar concentrations of the reagent, it is not itself a promising candidate for clinical use against CDI. However, these results serve as an important proof of concept demonstrating that the development of higher-affinity inhibitors against RSHCd could lead either to novel antibiotics or to adjuvants to increase the potency of existing antibiotics against CDI. Lastly, translational suppression of rsh that lead to a more pronounced effect on bacterial antibiotic susceptibility demonstrates the direct role that the stringent response plays in C. difficile antibiotic survival.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
Table S1 in the supplemental material lists the bacterial strains and plasmids used in this study. C. difficile 630Δerm and R20291 were grown at 37°C in tryptone-yeast (TY) medium or in brain heart infusion medium supplemented with 5% yeast extract (BHIS), as indicated (116, 117). The strains were grown in an anaerobic chamber (Coy Laboratory Products, Grass Lake, MI) with an atmosphere of 85% N2, 10% CO2, and 5% H2. E. coli strains were grown in Luria-Bertani broth (LB) at 37°C. Bacterial strains carrying plasmids were maintained using the following antibiotics in the indicated concentrations: 50 or 100 μg/ml of ampicillin (IBI Scientific), 10 or 15 μg/ml of thiamphenicol (Tm; Alfa Aesar), 10 μg/ml of chloramphenicol (VWR), 50 or 100 μg/ml of kanamycin (Kan; Bio Basic Canada Inc.), 0.075 or 0.3 μg/ml of metronidazole (Beantown Chemical), or 16 μg/ml of clindamycin (Tokyo Chemical Industry) as indicated. Relacin (gift from Sigal Ben-Yehuda, Hebrew University of Jerusalem) was added at a 2 mM concentration to C. difficile cultures when cell optical density at 600 nm reached 0.3 to 0.5 (78). Protein overexpression in E. coli was induced with 0.5 mM isopropyl-β-d-thiogalactopyranoside (IPTG).
Plasmid and strain construction.
All restriction enzymes and DNA ligase were purchased from New England BioLabs (NEB). Phusion DNA polymerase was purchased from Thermo Fisher Scientific. rsh (CDR20291_2633) was amplified from C. difficile R20291 genomic DNA using primers (Table S2) that added a C-terminal hexahistidine tag, ligated into the pMMBneo expression vector at the KpnI and PstI restriction sites, and transformed into E. coli BL21 as previously described (80). The plasmid was confirmed by PCR using both gene- and plasmid-specific primers (Table S2). Likewise, rel was amplified from pMMBneo::rsh expression vector using primers (Table S2) that also added a C-terminal hexahistidine tag. The amplicon was subsequently digested and ligated into the pMMBneo vector at the KpnI and PstI cut sites and transformed into an E. coli DH5α background. The plasmid was confirmed by PCR using gene-specific primers (Table S2).
The plasmid pRF185::phiLOV2.12.1 (provided by Gillian Douce, University of Glasgow), containing the phiLOV2.12.1 gene under the control of the tetracycline-inducible promoter Ptet, was digested with BamHI and KpnI to remove the original promoter (106). The predicted promoters upstream of rsh (CD630_27440 and CDR20291_2633) and relQ (CD630_03450 and CDR20291_0350) were amplified from 630Δerm and R20291 genomic DNA using the Prsh and PrelQ forward and reverse primers (Table S2). Prsh was amplified separately from 630Δerm and R20291 genomic DNA, while PrelQ, which is identical between the two strains, was amplified from 630Δerm. The primers introduced cut sites for BamHI and KpnI, which were used to digest the amplified promoters. The promoters were ligated into the digested pRF185_phiLOV2.12.1 plasmid to yield Prsh630Δerm::phiLOV2.12.1, PrshR20291::phiLOV2.12.1, and PrelQ630Δerm::phiLOV2.12.1. The plasmids were subsequently PCR verified for promoter insertion using vector-specific as well as promoter-specific primers (Table S2). The reporter plasmids were then individually transformed into competent E. coli HB101 cells (NEB) carrying the helper plasmid pRK24. HB101 cells carrying pRK24 were mated with C. difficile 630Δerm and R20291 strains as previously described (118). Prsh630Δerm::phiLOV2.12.1 and PrshR20291::phiLOV2.12.1 were mated into their isogenic background strains, and PrelQ630Δerm::phiLOV2.12.1 was mated into both C. difficile strains. Transconjugants were selected on BHIS agar supplemented with 10 μg/ml of thiamphenicol and 100 μg/ml of kanamycin. Finally, transconjugants were verified by PCR using both promoter-specific and phiLOV2.12.1-specific primers (Table S2).
The vector pMSPT, a gift from Julian G. Hurdle (Texas A&M University), was used to clone anhydrotetracycline (ATc)-inducible antisense RNA (asRNA) to the mRNA of C. difficile rsh (32). Vector pMSPT was derived by cloning a paired-terminus region, synthesized by GenScript in pUC57, into the BamHI and SacI sites of pRPF185 (32). An antisense fragment was designed and synthesized to span ∼50 bp upstream and downstream of the start codon to mimic naturally occurring bacterial asRNA that blocks translation by binding to the ribosome binding site (RBS) (32). The antisense fragment synthesized by GenScript was amplified using gene-specific primers (Table S2), digested, and cloned into the SphI and XhoI sites of pMSPT. The derived construct, termed as pMSPT::rsh_as, contained the gene with its own RBS expressed from the Ptet promoter. The construct was subsequently PCR verified for antisense gene insertion using vector-specific primers (Table S2). The RNA interference (RNAi) plasmids, including the empty-vector control, were then individually electroporated into E. coli HB101 cells (NEB) carrying the helper plasmid pRK24. HB101 cells with pRK24 were conjugated with C. difficile R20291 strains as previously described (118). Transconjugants were selected on BHIS agar supplemented with 15 μg/ml of thiamphenicol and 100 μg/ml of kanamycin. Finally, transconjugants were verified by PCR using C. difficile relQ gene-specific and pMSPT vector-specific primers (Table S2).
Growth curves.
Growth curve assays following the induction of C. difficile rsh in the E. coli DH5α background were performed using a 96-well microtiter plate. Log-phase cultures of E. coli cells were inoculated in LB medium in the presence or absence of 0.5 mM IPTG. The plate was incubated at 37°C with shaking for a total of 12 h. Growth of induced versus uninduced cells was monitored every 30 min.
Endpoint growth assay using RNAi strains.
Growth following the induction of asRNA to the mRNA of C. difficile rsh was performed by a medium dilution technique using a 96-well microtiter plate. Essentially, increasing concentrations of ATc were added to BHIS medium in the absence of antibiotics (Fig. S5). The plate was incubated for 24 h, after which the sublethal concentration of ATc that partially inhibited growth was recorded (Fig. S5). The concentration of ATc (0.5 μg/ml) that moderately inhibited growth was further used to induce rsh_as in assays in which the growth of unstressed versus stressed strains using 0.075 μg/ml of metronidazole was monitored after 12 h of incubation at 37°C.
Purification of Clostridioides difficile RSH.
Expression and purification of native C. difficile RSH from E. coli BL21 cells (NEB) have previously been reported (80). Briefly, the E. coli expression strain BL21 was grown in LB with 50 μg/ml kanamycin at 37°C to an OD600 of 0.16 to 0.25, at which point the temperature was dropped to 30°C and expression was induced using 0.5 mM IPTG for 16 h. Cells were lysed by sonication in the lysis buffer, after which the lysates were clarified by centrifugation and subsequently purified using HisPure nickel-nitrilotriacetic acid (Ni-NTA) resin (G-Biosciences) according to the manufacturer’s protocol. Purified RSHCd was dialyzed overnight at 4°C against dialysis buffer (15.7 mM Tris-HCl [pH 7.6], 471.9 mM NaCl, 15.69 mM MgCl2, 1.57 mM dithiothreitol [DTT], 1.5 mM phenylmethylsulfonyl fluoride [PMSF], and 15.7% glycerol) and stored at – 80°C in 5- to 50-μl aliquots to be used for enzymatic assays.
In vitro measurement of RSH synthetase activity.
ppGpp synthesis by purified RSHCd was carried out as previously described (80). Assays were conducted in buffer containing 10 mM Tris-HCl (pH 7.5), 5 mM ammonium acetate, 2 mM KCl, 0.2 mM DTT, and 0.6 mM ATP. A 5× buffer stock was mixed with the desired concentrations of GDP (0.2 to 0.6 mM), with covarying MgCl2 and 1.0 μCi of [γ-32P]ATP. Reactions were initiated by adding RSH at a final concentration of 0.003 mM and a reaction volume of 10 μl. Reactions were stopped by spotting 2-μl samples on polyethyleneimine (PEI)-cellulose plates, allowing the spots to dry. The plates were subsequently developed in 1.5 M KH2PO4 (pH 3.64 ± 0.03) and autoradiographed using a Storm 860 phosphorimager (GE Healthcare Life Sciences). ppGpp signal was quantitated using ImageJ software (119). RSH activity was expressed as the percentage of [γ-32P]ATP converted into inorganic phosphate at each time point (80). Allosteric feedback regulation of RSHCd by ppGpp was assayed in the same manner in the presence of 0 to 10 μM exogenous ppGpp (TriLink). Relacin inhibition assays were performed under the same reaction conditions using 0.006 mM purified RSHCd enzyme in addition to relacin at concentrations ranging from 0 to 5 mM (78). Finally, the effect(s) of different metal cations as well as buffer pH on RSHCd synthetase activity was evaluated under reaction conditions similar to those mentioned above. Metal cofactors studied included Mg2+, Mn2+, Co2+, Cu2+, Zn2+, Ni2+, Ca2+, and Fe2+ at a concentration of 0.012 mM. Buffer pH studied included pH values of 5.0, 6.0, 7.5, 9.0, and 10.0. The effect(s) of metal cations and buffer pH on (p)ppGpp synthesis was studied at least three times.
Promoter activity analysis using fluorescent phiLOV2.1 reporter constructs.
C. difficile strains containing phiLOV2.1 reporter plasmid were grown anaerobically at 37°C in BHIS-Tm10 for 12 to 16 h. To monitor growth phase-dependent promoter activity, overnight saturated cultures were inoculated 1:20 into fresh BHIS-Tm10 medium and grown with optical density monitored at 600 nm. Samples were collected when cultures reached the OD600 of 0.8 and 1.2. When cultures at the final volume of 3 ml reached the OD600 of 0.8 and 1.2, cell numbers in each sample for fluorescent measurement were normalized to cell culture OD600 of 0.20 at 600 nm. To monitor antibiotic-induced promoter activity, overnight starter cultures were inoculated 1:50 into fresh BHIS-Tm10 containing metronidazole and clindamycin at 1× MIC (Fig. S3) and grown for 2 h. OD600 was recorded for each sample to monitor growth. To minimize discrepancies in fluorescent signal from cellular autofluorescence, cell numbers in each sample were equalized on collection (106). Each sample was collected at a volume that would give a cell count equivalent to 3 ml of the culture with the lowest OD600. After collection, cells were pelleted anaerobically in a microcentrifuge and suspended in 400 μl of anaerobic 1× phosphate-buffered saline (PBS). Duplicate 200-μl samples were aliquoted into a clear-bottomed black 96-well microplate (Thermo Fisher Scientific) and were removed from the anaerobic chamber to measure sample fluorescence intensity. Sample fluorescence using 440-/30-nm excitation and 508-/20-nm emission filters and sample OD630 were measured on a BrandTek plate reader. The instrumental parameters for all fluorescence measurements included a sensitivity limit of 65. Measurements were blanked against 1× PBS and were reported as E508/A630.
RNA isolation and real-time PCR.
Total RNA from C. difficile 630Δerm in BHIS was isolated as previously described (118). Primers were designed using the PrimerQuest tool from IDT DNA Technologies. Samples for RNA isolation were collected when cultures reached OD600 of 0.8 (mid-log phase), 1.2 (stationary-phase onset) and 1.6 (stationary phase). cDNA samples were prepared from 200 ng of RNA using random hexamers and the Tetro cDNA synthesis kit (Bioline). Real-time PCRs were performed with 2 ng of cDNA template using the SensiMix SYBR and fluorescein kit (Bioline). Primers (Table S2) were designed using PrimerQuest tool from IDT DNA Technologies and detected transcripts of rsh and relQ. The rpoC transcript was measured and used as an internal control for normalization. Controls with no reverse transcriptase were also included for all templates and primer sets. The data were analyzed using the threshold cycle (2−ΔΔCT) method with normalization to rpoC and to an of 0.8.
HPLC.
Instrumentation for high-performance liquid chromatography (HPLC) analysis consisted of a dual-pump ultrahigh-performance liquid chromatography system with a UV detector (Schimazdu). The analytical column used was a 4- by 250-mm strong anion-exchange DNAPac PA100 (Thermo Scientific) with a 4- by 50-mm guard column (Thermo Scientific). HPLC buffers A and B consisted of 50 mM KH2PO4 and 500 mM KH2PO4 as well as 500 mM Na2SO4, respectively adjusted to neutral pH. Nucleotide standards, including GDP (Alfa Aesar), GTP (BioBasic), and GMP (BioWorld) at a concentration of 20 μM, were separated using gradient elution at a flow rate of 1.0 ml/min and absorbance at 252 nm as a readout (Fig. S1).
ITC.
Isothermal titration calorimetry (ITC) analysis was conducted on an ITC200 microcalorimeter (Malvern). Purified RSHCd was added onto the cell component (300 μl), and the syringe was filled with GXP ligand (40 μl). Protein and ligand were prepared in the same buffer (10 Mm Tris-HCl, 5 mM AmAce, 1.2 mM MgCl2, 0.2 mM DTT, 0.012 mM ATP, and 2 mM KCl). Concentrations of 0.0012 mM protein and 0.012 mM ligand were used in the experiments in which reactions were carried out at 37°C. ITC experiments were repeated at least three times where the heat of dilution obtained through the titration of ligand into the reference solution was subtracted from the binding curves. The data obtained were fitted using a single-binding-site model by Origin software. Peak integration and the calculation of thermodynamic parameters were also accomplished using Origin (Fig. S2).
Supplementary Material
ACKNOWLEDGMENTS
We declare no financial conflicts of interest.
This work was funded by NIAID 1K22AI118929-01.
We thank Rita Tamayo of UNC Chapel Hill for helpful discussions and for use of the qRT-PCR instrument, Gillian Douce of the University of Glasgow for the pRF185_phiLOV2.12.1 plasmid, Julian G. Hurdle of Texas A&M University for the pMSPT plasmid, Ravi K. R. Marreddy of Texas A&M University for helpful discussions on RNAi, and Alvin Holder of Old Dominion University for use of the ITC instrument. Relacin was a generous gift from Sigal Ben-Yehuda of The Hebrew University of Jerusalem.
Footnotes
Supplemental material is available online only.
REFERENCES
- 1.Gerding DN, Muto CA, Owens RC Jr. 2008. Measures to control and prevent Clostridium difficile infection. Clin Infect Dis 46:S43–S49. doi: 10.1086/521861. [DOI] [PubMed] [Google Scholar]
- 2.Rodriguez-Palacios A, LeJeune JT. 2011. Moist-heat resistance, spore aging, and superdormancy in Clostridium difficile. Appl Environ Microbiol 77:3085–3091. doi: 10.1128/AEM.01589-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Curry SR. 2017. Clostridium difficile. Clin Lab Med 37:341–369. doi: 10.1016/j.cll.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 4.Howerton A, Ramirez N, Abel-Santos E. 2011. Mapping interactions between germinants and Clostridium difficile spores. J Bacteriol 193:274–282. doi: 10.1128/JB.00980-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Voth DE, Ballard JD. 2005. Clostridium difficile toxins: mechanism of action and role in disease. Clin Microbiol Rev 18:247–263. doi: 10.1128/CMR.18.2.247-263.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Bella S, Ascenzi P, Siarakas S, Petrosillo N, di Masi A. 2016. Clostridium difficile toxins A and B: insights into pathogenic properties and extraintestinal effects. Toxins 8:134. doi: 10.3390/toxins8050134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ng J, Hirota SA, Gross O, Li Y, Ulke-Lemee A, Potentier MS, Schenck LP, Vilaysane A, Seamone ME, Feng H, Armstrong GD, Tschopp J, Macdonald JA, Muruve DA, Beck PL. 2010. Clostridium difficile toxin-induced inflammation and intestinal injury are mediated by the inflammasome. Gastroenterology 139:542–552.e5523. doi: 10.1053/j.gastro.2010.04.005. [DOI] [PubMed] [Google Scholar]
- 8.Jones AM, Kuijper EJ, Wilcox MH. 2013. Clostridium difficile: a European perspective. J Infect 66:115–128. doi: 10.1016/j.jinf.2012.10.019. [DOI] [PubMed] [Google Scholar]
- 9.Oldfield EI, Oldfield EC III, Johnson DA. 2014. Clinical update for the diagnosis and treatment of Clostridium difficile infection. World J Gastrointest Pharmacol Ther 5:1–26. doi: 10.4292/wjgpt.v5.i1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, Littmann E, van den Brink MR, Jenq RR, Taur Y, Sander C, Cross JR, Toussaint NC, Xavier JB, Pamer EG. 2015. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517:205–208. doi: 10.1038/nature13828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ng SC, Lam EF, Lam TT, Chan Y, Law W, Tse PC, Kamm MA, Sung JJ, Chan FK, Wu JC. 2013. Effect of probiotic bacteria on the intestinal microbiota in irritable bowel syndrome. J Gastroenterol Hepatol 28:1624–1631. doi: 10.1111/jgh.12306. [DOI] [PubMed] [Google Scholar]
- 12.Claesson MJ, Cusack S, O’Sullivan O, Greene-Diniz R, de Weerd H, Flannery E, Marchesi JR, Falush D, Dinan T, Fitzgerald G, Stanton C, van Sinderen D, O’Connor M, Harnedy N, O’Connor K, Henry C, O’Mahony D, Fitzgerald AP, Shanahan F, Twomey C, Hill C, Ross RP, O’Toole PW. 2011. Composition, variability, and temporal stability of the intestinal microbiota of the elderly. Proc Natl Acad Sci U S A 108(Suppl 1):4586–4591. doi: 10.1073/pnas.1000097107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Britton RA, Young VB. 2014. Role of the intestinal microbiota in resistance to colonization by Clostridium difficile. Gastroenterology 146:1547–1553. doi: 10.1053/j.gastro.2014.01.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Crogan NL, Evans BC. 2007. Clostridium difficile: an emerging epidemic in nursing homes. Geriatr Nurs 28:161–164. doi: 10.1016/j.gerinurse.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 15.Cohen SHMD, Gerding DNMD, Stuart Johnson MD, Kelly CPMD, Loo VGMD, McDonald LCMD, Pepin JMD, Wilcox MHMD, Infectious Diseases Society of America. 2010. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the Society for Healthcare Epidemiology of America (SHEA) and the Infectious Diseases Society of America (IDSA). Infect Control Hosp Epidemiol 31:431–455. doi: 10.1086/651706. [DOI] [PubMed] [Google Scholar]
- 16.Chitnis AS, Holzbauer SM, Belflower RM, Winston LG, Bamberg WM, Lyons C, Farley MM, Dumyati GK, Wilson LE, Beldavs ZG, Dunn JR, Gould LH, MacCannell DR, Gerding DN, McDonald LC, Lessa FC. 2013. Epidemiology of community-associated Clostridium difficile infection, 2009 through 2011. JAMA Intern Med 173:1359–1367. doi: 10.1001/jamainternmed.2013.7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lessa FC, Mu Y, Bamberg WM, Beldavs ZG, Dumyati GK, Dunn JR, Farley MM, Holzbauer SM, Meek JI, Phipps EC, Wilson LE, Winston LG, Cohen JA, Limbago BM, Fridkin SK, Gerding DN, McDonald LC. 2015. Burden of Clostridium difficile infection in the United States. N Engl J Med 372:825–834. doi: 10.1056/NEJMoa1408913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cole SA, Stahl TJ. 2015. Persistent and recurrent Clostridium difficile colitis. Clin Colon Rectal Surg 28:65–69. doi: 10.1055/s-0035-1547333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Denève C, Janoir C, Poilane I, Fantinato C, Collignon A. 2009. New trends in Clostridium difficile virulence and pathogenesis. Int J Antimicrobial Agents 33:S24–S28. doi: 10.1016/S0924-8579(09)70012-3. [DOI] [PubMed] [Google Scholar]
- 20.He M, Miyajima F, Roberts P, Ellison L, Pickard DJ, Martin MJ, Connor TR, Harris SR, Fairley D, Bamford KB, D'Arc S, Brazier J, Brown D, Coia JE, Douce G, Gerding D, Kim HJ, Koh TH, Kato H, Senoh M, Louie T, Michell S, Butt E, Peacock SJ, Brown NM, Riley T, Songer G, Wilcox M, Pirmohamed M, Kuijper E, Hawkey P, Wren BW, Dougan G, Parkhill J, Lawley TD. 2013. Emergence and global spread of epidemic healthcare-associated Clostridium difficile. Nat Genet 45:109–113. doi: 10.1038/ng.2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Stabler RA, He M, Dawson L, Martin M, Valiente E, Corton C, Lawley TD, Sebaihia M, Quail MA, Rose G, Gerding DN, Gibert M, Popoff MR, Parkhill J, Dougan G, Wren BW. 2009. Comparative genome and phenotypic analysis of Clostridium difficile 027 strains provides insight into the evolution of a hypervirulent bacterium. Genome Biol 10:R102. doi: 10.1186/gb-2009-10-9-r102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kelly CP, LaMont JT. 2008. Clostridium difficile—more difficult than ever. N Engl J Med 359:1932–1940. doi: 10.1056/NEJMra0707500. [DOI] [PubMed] [Google Scholar]
- 23.Bartlett JG. 2010. Clostridium difficile: progress and challenges. Ann N Y Acad Sci 1213:62–69. doi: 10.1111/j.1749-6632.2010.05863.x. [DOI] [PubMed] [Google Scholar]
- 24.Kuijper EJ, Coignard B, Tull P, ESCMID Study Group for Clostridium difficile, EU Member States, European Centre for Disease Prevention and Control. 2006. Emergence of Clostridium difficile-associated disease in North America and Europe. Clin Microbiol Infect 12(Suppl 6):2–18. doi: 10.1111/j.1469-0691.2006.01580.x. [DOI] [PubMed] [Google Scholar]
- 25.McFarland LV. 2008. Antibiotic-associated diarrhea: epidemiology, trends and treatment. Future Microbiol 3:563–678. doi: 10.2217/17460913.3.5.563. [DOI] [PubMed] [Google Scholar]
- 26.Peng Z, Jin D, Kim HB, Stratton CW, Wu B, Tang YW, Sun X. 2017. Update on antimicrobial resistance in Clostridium difficile: resistance mechanisms and antimicrobial susceptibility testing. J Clin Microbiol 55:1998–2008. doi: 10.1128/JCM.02250-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Banawas SS. 2018. Clostridium difficile infections: a global overview of drug sensitivity and resistance mechanisms. BioMed Res Int 2018:8414257. doi: 10.1155/2018/8414257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khanna S, Gerding DN. 2019. Current and future trends in Clostridioides (Clostridium) difficile infection management. Anaerobe 58:95–102. doi: 10.1016/j.anaerobe.2019.04.010. [DOI] [PubMed] [Google Scholar]
- 29.Al-Jashaami LS, DuPont HL. 2016. Management of Clostridium difficile infection. Gastroenterol Hepatol (N Y) 12:609–616. [PMC free article] [PubMed] [Google Scholar]
- 30.Babakhani F, Bouillaut L, Gomez A, Sears P, Nguyen L, Sonenshein AL. 2012. Fidaxomicin inhibits spore production in Clostridium difficile. Clin Infect Dis 55:S162–S169. doi: 10.1093/cid/cis453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Babakhani F, Bouillaut L, Sears P, Sims C, Gomez A, Sonenshein AL. 2013. Fidaxomicin inhibits toxin production in Clostridium difficile. J Antimicrob Chemother 68:515–522. doi: 10.1093/jac/dks450. [DOI] [PubMed] [Google Scholar]
- 32.Marreddy RK, Wu X, Sapkota M, Prior AM, Jones JA, Sun D, Hevener KE, Hurdle JG. 2019. The fatty acid synthesis protein enoyl-ACP reductase II (FabK) is a target for narrow-spectrum antibacterials for Clostridium difficile infection. ACS Infect Dis 5:208–217. doi: 10.1021/acsinfecdis.8b00205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Corrigan RM, Bellows LE, Wood A, Gründling A. 2016. ppGpp negatively impacts ribosome assembly affecting growth and antimicrobial tolerance in Gram-positive bacteria. Proc Natl Acad Sci U S A 113:E1710–E1719. doi: 10.1073/pnas.1522179113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shogbesan O, Poudel DR, Victor S, Jehangir A, Fadahunsi O, Shogbesan G, Donato A. 2018. A systematic review of the efficacy and safety of fecal microbiota transplant for Clostridium difficile infection in immunocompromised patients. Can J Gastroenterol Hepatol 2018:1394379. doi: 10.1155/2018/1394379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McDonald LC, Gerding DN, Johnson S, Bakken JS, Carroll KC, Coffin SE, Dubberke ER, Garey KW, Gould CV, Kelly C, Loo V, Shaklee Sammons J, Sandora TJ, Wilcox MH. 2018. Clinical practice guidelines for Clostridium difficile infection in adults and children: 2017 update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin Infect Dis 66:987–994. doi: 10.1093/cid/ciy149. [DOI] [PubMed] [Google Scholar]
- 36.Boyle ML, Ruth-Sahd LA, Zhou Z. 2015. Fecal microbiota transplant to treat recurrent Clostridium difficile infections. Crit Care Nurse 35:51–64. doi: 10.4037/ccn2015356. [DOI] [PubMed] [Google Scholar]
- 37.LeGrand EK, Day JD. 2016. Self-harm to preferentially harm the pathogens within: non-specific stressors in innate immunity. Proc R Soc B 283:20160266. doi: 10.1098/rspb.2016.0266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Martins D, McKay G, Sampathkumar G, Khakimova M, English AM, Nguyen D. 2018. Superoxide dismutase activity confers (p)ppGpp-mediated antibiotic tolerance to stationary-phase Pseudomonas aeruginosa. Proc Natl Acad Sci U S A 115:9797–9802. doi: 10.1073/pnas.1804525115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Potrykus K, Cashel M. 2008. (p)ppGpp: still magical? Annu Rev Microbiol 62:35–51. doi: 10.1146/annurev.micro.62.081307.162903. [DOI] [PubMed] [Google Scholar]
- 40.Irving SE, Corrigan RM. 2018. Triggering the stringent response: signals responsible for activating (p)ppGpp synthesis in bacteria. Microbiology 164:268–276. doi: 10.1099/mic.0.000621. [DOI] [PubMed] [Google Scholar]
- 41.Hammer BK, Tateda ES, Swanson MS. 2002. A two-component regulator induces the transmission phenotype of stationary-phase Legionella pneumophila. Mol Microbiol 44:107–118. doi: 10.1046/j.1365-2958.2002.02884.x. [DOI] [PubMed] [Google Scholar]
- 42.Pizarro-Cerdá J, Tedin K. 2004. The bacterial signal molecule, ppGpp, regulates Salmonella virulence gene expression. Mol Microbiol 52:1827–1844. doi: 10.1111/j.1365-2958.2004.04122.x. [DOI] [PubMed] [Google Scholar]
- 43.Vogt SL, Green C, Stevens KM, Day B, Erickson DL, Woods DE, Storey DG. 2011. The stringent response is essential for Pseudomonas aeruginosa virulence in the rat lung agar bead and Drosophila melanogaster feeding models of infection. Infect Immun 79:4094–4104. doi: 10.1128/IAI.00193-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.van Delden C, Comte R, Bally AM. 2001. Stringent response activates quorum sensing and modulates cell density-dependent gene expression in Pseudomonas aeruginosa. J Bacteriol 183:5376–5384. doi: 10.1128/jb.183.18.5376-5384.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Geiger T, Goerke C, Fritz M, Schäfer T, Ohlsen K, Liebeke M, Lalk M, Wolz C. 2010. Role of the (p)ppGpp synthase RSH, a RelA/SpoT homolog, in stringent response and virulence of Staphylococcus aureus. Infect Immun 78:1873–1883. doi: 10.1128/IAI.01439-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Abranches J, Martinez AR, Kajfasz JK, Chávez V, Garsin DA, Lemos JA. 2009. The molecular alarmone (p)ppGpp mediates stress responses, vancomycin tolerance, and virulence in Enterococcus faecalis. J Bacteriol 191:2248–2256. doi: 10.1128/JB.01726-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aedo S, Tomasz A. 2016. Role of the stringent stress response in the antibiotic resistance phenotype of methicillin-resistant Staphylococcus aureus. Antimicrob Agents Chemother 60:2311–2317. doi: 10.1128/AAC.02697-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Primm TP, Andersen SJ, Mizrahi V, Avarbock D, Rubin H, Barry CE III. 2000. The stringent response of Mycobacterium tuberculosis is required for long-term survival. J Bacteriol 182:4889–4898. doi: 10.1128/jb.182.17.4889-4898.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weiss LA, Stallings CL. 2013. Essential roles for Mycobacterium tuberculosis Rel beyond the production of (p)ppGpp. J Bacteriol 195:5629–5638. doi: 10.1128/JB.00759-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Atkinson GC, Tenson T, Hauryliuk V. 2011. The RelA/SpoT homolog (RSH) superfamily: distribution and functional evolution of ppGpp synthetases and hydrolases across the tree of life. PLoS One 6:e23479. doi: 10.1371/journal.pone.0023479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hogg T, Mechold U, Malke H, Cashel M, Hilgenfeld R. 2004. Conformational antagonism between opposing active sites in a bifunctional RelA/SpoT homolog modulates (p)ppGpp metabolism during the stringent response [corrected]. Cell 117:57–68. doi: 10.1016/S0092-8674(04)00260-0. [DOI] [PubMed] [Google Scholar]
- 52.Mechold U, Murphy H, Brown L, Cashel M. 2002. Intramolecular regulation of the opposing (p)ppGpp catalytic activities of Rel(Seq), the Rel/Spo enzyme from Streptococcus equisimilis. J Bacteriol 184:2878–2888. doi: 10.1128/jb.184.11.2878-2888.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Steinchen W, Schuhmacher JS, Altegoer F, Fage CD, Srinivasan V, Linne U, Marahiel MA, Bange G. 2015. Catalytic mechanism and allosteric regulation of an oligomeric (p)ppGpp synthetase by an alarmone. Proc Natl Acad Sci U S A 112:13348–13353. doi: 10.1073/pnas.1505271112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Syal K, Joshi H, Chatterji D, Jain V. 2015. Novel pppGpp binding site at the C-terminal region of the Rel enzyme from Mycobacterium smegmatis. FEBS J 282:3773–3785. doi: 10.1111/febs.13373. [DOI] [PubMed] [Google Scholar]
- 55.Gratani FL, Horvatek P, Geiger T, Borisova M, Mayer C, Grin I, Wagner S, Steinchen W, Bange G, Velic A, Macek B, Wolz C. 2018. Regulation of the opposing (p)ppGpp synthetase and hydrolase activities in a bifunctional RelA/SpoT homologue from Staphylococcus aureus. PLoS Genet 14:e1007514. doi: 10.1371/journal.pgen.1007514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ronneau S, Caballero-Montes J, Coppine J, Mayard A, Garcia-Pino A, Hallez R. 2018. Regulation of (p)ppGpp hydrolysis by a conserved archetypal regulatory domain. Nucleic Acids Res 47:843–854. doi: 10.1093/nar/gky1201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fang M, Bauer CE. 2018. Regulation of stringent factor by branched-chain amino acids. Proc Natl Acad Sci U S A 115:6446–6451. doi: 10.1073/pnas.1803220115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Boutte CC, Crosson S. 2013. Bacterial lifestyle shapes stringent response activation. Trends Microbiol 21:174–180. doi: 10.1016/j.tim.2013.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anderson KL, Roberts C, Disz T, Vonstein V, Hwang K, Overbeek R, Olson PD, Projan SJ, Dunman PM. 2006. Characterization of the Staphylococcus aureus heat shock, cold shock, stringent, and SOS responses and their effects on log-phase mRNA turnover. J Bacteriol 188:6739–6756. doi: 10.1128/JB.00609-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang T, Zhu J, Wei S, Luo Q, Li L, Li S, Tucker A, Shao H, Zhou R. 2016. The roles of RelA/(p) ppGpp in glucose-starvation induced adaptive response in the zoonotic Streptococcus suis. Sci Rep 6:27169. doi: 10.1038/srep27169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Pulschen AA, Sastre DE, Machinandiarena F, Crotta Asis A, Albanesi D, de Mendoza D, Gueiros-Filho FJ. 2017. The stringent response plays a key role in Bacillus subtilis survival of fatty acid starvation. Mol Microbiol 103:698–712. doi: 10.1111/mmi.13582. [DOI] [PubMed] [Google Scholar]
- 62.Miethke M, Westers H, Blom E-J, Kuipers OP, Marahiel MA. 2006. Iron starvation triggers the stringent response and induces amino acid biosynthesis for bacillibactin production in Bacillus subtilis. J Bacteriol 188:8655–8657. doi: 10.1128/JB.01049-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Colomer-Winter C, Gaca A, Lemos J. 2017. Association of metal homeostasis and (p)ppGpp regulation in the pathophysiology of Enterococcus faecalis. Infect Immun 85:e00260-17. doi: 10.1128/IAI.00260-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nanamiya H, Kasai K, Nozawa A, Yun C-S, Narisawa T, Murakami K, Natori Y, Kawamura F, Tozawa Y. 2008. Identification and functional analysis of novel (p)ppGpp synthetase genes in Bacillus subtilis. Mol Microbiol 67:291–304. doi: 10.1111/j.1365-2958.2007.06018.x. [DOI] [PubMed] [Google Scholar]
- 65.Lemos JA, Lin VK, Nascimento MM, Abranches J, Burne RA. 2007. Three gene products govern (p)ppGpp production by Streptococcus mutans. Mol Microbiol 65:1568–1581. doi: 10.1111/j.1365-2958.2007.05897.x. [DOI] [PubMed] [Google Scholar]
- 66.Gaca AO, Kajfasz JK, Miller JH, Liu K, Wang JD, Abranches J, Lemos JA. 2013. Basal Levels of (p)ppGpp in Enterococcus faecalis: the magic beyond the stringent response. mBio 4:e00646-13. doi: 10.1128/mBio.00646-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kriel A, Bittner AN, Kim SH, Liu K, Tehranchi AK, Zou WY, Rendon S, Chen R, Tu BP, Wang JD. 2012. Direct regulation of GTP homeostasis by (p)ppGpp: a critical component of viability and stress resistance. Mol Cell 48:231–241. doi: 10.1016/j.molcel.2012.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kriel A, Brinsmade SR, Tse JL, Tehranchi AK, Bittner AN, Sonenshein AL, Wang JD. 2014. GTP dysregulation in Bacillus subtilis cells lacking (p)ppGpp results in phenotypic amino acid auxotrophy and failure to adapt to nutrient downshift and regulate biosynthesis genes. J Bacteriol 196:189–201. doi: 10.1128/JB.00918-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bittner AN, Kriel A, Wang JD. 2014. Lowering GTP level increases survival of amino acid starvation but slows growth rate for Bacillus subtilis cells lacking (p)ppGpp. J Bacteriol 196:2067–2076. doi: 10.1128/JB.01471-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Geiger T, Kastle B, Gratani FL, Goerke C, Wolz C. 2014. Two small (p)ppGpp synthases in Staphylococcus aureus mediate tolerance against cell envelope stress conditions. J Bacteriol 196:894–902. doi: 10.1128/JB.01201-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pando JM, Pfeltz RF, Cuaron JA, Nagarajan V, Mishra MN, Torres NJ, Elasri MO, Wilkinson BJ, Gustafson JE. 2017. Ethanol-induced stress response of Staphylococcus aureus. Can J Microbiol 63:745–757. doi: 10.1139/cjm-2017-0221. [DOI] [PubMed] [Google Scholar]
- 72.Beljantseva J, Kudrin P, Jimmy S, Ehn M, Pohl R, Varik V, Tozawa Y, Shingler V, Tenson T, Rejman D, Hauryliuk V. 2017. Molecular mutagenesis of ppGpp: turning a RelA activator into an inhibitor. Sci Rep 7:41839. doi: 10.1038/srep41839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Andresen L, Varik V, Tozawa Y, Jimmy S, Lindberg S, Tenson T, Hauryliuk V. 2016. Auxotrophy-based high throughput screening assay for the identification of Bacillus subtilis stringent response inhibitors. Sci Rep 6:35824. doi: 10.1038/srep35824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.de la Fuente-Núñez C, Reffuveille F, Haney EF, Straus SK, Hancock REW. 2014. Broad-spectrum anti-biofilm peptide that targets a cellular stress response. PLoS Pathog 10:e1004152. doi: 10.1371/journal.ppat.1004152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dutta NK, Klinkenberg LG, Vazquez M-J, Segura-Carro D, Colmenarejo G, Ramon F, Rodriguez-Miquel B, Mata-Cantero L, Porras-De Francisco E, Chuang Y-M, Rubin H, Lee JJ, Eoh H, Bader JS, Perez-Herran E, Mendoza-Losana A, Karakousis PC. 2019. Inhibiting the stringent response blocks Mycobacterium tuberculosis entry into quiescence and reduces persistence. Sci Adv 5:eaav2104. doi: 10.1126/sciadv.aav2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Syal K, Flentie K, Bhardwaj N, Maiti K, Jayaraman N, Stallings CL, Chatterji D. 2017. Synthetic (p)ppGpp analogue is an inhibitor of stringent response in mycobacteria. Antimicrob Agents Chemother 61:e00443-17. doi: 10.1128/AAC.00443-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wexselblatt E, Kaspy I, Glaser G, Katzhendler J, Yavin E. 2013. Design, synthesis and structure-activity relationship of novel Relacin analogs as inhibitors of Rel proteins. Eur J Med Chem 70:497–504. doi: 10.1016/j.ejmech.2013.10.036. [DOI] [PubMed] [Google Scholar]
- 78.Wexselblatt E, Oppenheimer-Shaanan Y, Kaspy I, London N, Schueler-Furman O, Yavin E, Glaser G, Katzhendler J, Ben-Yehuda S. 2012. Relacin, a novel antibacterial agent targeting the stringent response. PLoS Pathog 8:e1002925. doi: 10.1371/journal.ppat.1002925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gaca AO, Colomer-Winter C, Lemos JA. 2015. Many means to a common end: the intricacies of (p)ppGpp metabolism and its control of bacterial homeostasis. J Bacteriol 197:1146–1156. doi: 10.1128/JB.02577-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Pokhrel A, Poudel A, Purcell EB. 2018. A purification and in vitro activity assay for a (p)ppGpp synthetase from Clostridium difficile. J Vis Exp 2018(141):e58547. doi: 10.3791/58547. [DOI] [PubMed] [Google Scholar]
- 81.Sebaihia M, Wren BW, Mullany P, Fairweather NF, Minton N, Stabler R, Thomson NR, Roberts AP, Cerdeno-Tarraga AM, Wang H, Holden MT, Wright A, Churcher C, Quail MA, Baker S, Bason N, Brooks K, Chillingworth T, Cronin A, Davis P, Dowd L, Fraser A, Feltwell T, Hance Z, Holroyd S, Jagels K, Moule S, Mungall K, Price C, Rabbinowitsch E, Sharp S, Simmonds M, Stevens K, Unwin L, Whithead S, Dupuy B, Dougan G, Barrell B, Parkhill J. 2006. The multidrug-resistant human pathogen Clostridium difficile has a highly mobile, mosaic genome. Nat Genet 38:779–786. doi: 10.1038/ng1830. [DOI] [PubMed] [Google Scholar]
- 82.Hussain HA, Roberts AP, Mullany P. 2005. Generation of an erythromycin-sensitive derivative of Clostridium difficile strain 630 (630Deltaerm) and demonstration that the conjugative transposon Tn916DeltaE enters the genome of this strain at multiple sites. J Med Microbiol 54:137–141. doi: 10.1099/jmm.0.45790-0. [DOI] [PubMed] [Google Scholar]
- 83.Saito R, Talukdar P, Alanazi S, Sarker M. 2018. RelA/DTD-mediated regulation of spore formation and toxin production by Clostridium perfringens type A strain SM101. Microbiology 164:835–847. doi: 10.1099/mic.0.000655. [DOI] [PubMed] [Google Scholar]
- 84.Emerson JE, Stabler RA, Wren BW, Fairweather NF. 2008. Microarray analysis of the transcriptional responses of Clostridium difficile to environmental and antibiotic stress. J Med Microbiol 57:757–764. doi: 10.1099/jmm.0.47657-0. [DOI] [PubMed] [Google Scholar]
- 85.Grant GA. 2006. The ACT domain: a small molecule binding domain and its role as a common regulatory element. J Biol Chem 281:33825–33829. doi: 10.1074/jbc.R600024200. [DOI] [PubMed] [Google Scholar]
- 86.Traxler MF, Summers SM, Nguyen HT, Zacharia VM, Hightower GA, Smith JT, Conway T. 2008. The global, ppGpp‐mediated stringent response to amino acid starvation in Escherichia coli. Mol Microbiol 68:1128–1148. doi: 10.1111/j.1365-2958.2008.06229.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sajish M, Tiwari D, Rananaware D, Nandicoori VK, Prakash B. 2007. A charge reversal differentiates (p)ppGpp synthesis by monofunctional and bifunctional Rel proteins. J Biol Chem 282:34977–34983. doi: 10.1074/jbc.M704828200. [DOI] [PubMed] [Google Scholar]
- 88.Gaca AO, Abranches J, Kajfasz JK, Lemos JA. 2012. Global transcriptional analysis of the stringent response in Enterococcus faecalis. Microbiology 158:1994–2004. doi: 10.1099/mic.0.060236-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Corrigan RM, Bowman L, Willis AR, Kaever V, Gründling A. 2015. Cross-talk between two nucleotide-signaling pathways in Staphylococcus aureus. J Biol Chem 290:5826–5839. doi: 10.1074/jbc.M114.598300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Park JO, Rubin SA, Xu Y-F, Amador-Noguez D, Fan J, Shlomi T, Rabinowitz JD. 2016. Metabolite concentrations, fluxes and free energies imply efficient enzyme usage. Nat Chem Biol 12:482–489. doi: 10.1038/nchembio.2077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Buckstein MH, He J, Rubin H. 2008. Characterization of nucleotide pools as a function of physiological state in Escherichia coli. J Bacteriol 190:718–726. doi: 10.1128/JB.01020-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Varik V, Oliveira SRA, Hauryliuk V, Tenson T. 2017. HPLC-based quantification of bacterial housekeeping nucleotides and alarmone messengers ppGpp and pppGpp. Sci Rep 7:11022. doi: 10.1038/s41598-017-10988-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Chubukov V, Sauer U. 2014. Environmental dependence of stationary-phase metabolism in Bacillus subtilis and Escherichia coli. Appl Environ Microbiol 80:2901–2909. doi: 10.1128/AEM.00061-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shyp V, Tankov S, Ermakov A, Kudrin P, English BP, Ehrenberg M, Tenson T, Elf J, Hauryliuk V. 2012. Positive allosteric feedback regulation of the stringent response enzyme RelA by its product. EMBO Rep 13:835–839. doi: 10.1038/embor.2012.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gasteiger E, Gattiker A, Hoogland C, Ivanyi I, Appel RD, Bairoch A. 2003. ExPASy: the proteomics server for in-depth protein knowledge and analysis. Nucleic Acids Res 31:3784–3788. doi: 10.1093/nar/gkg563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Nugent S, Kumar D, Rampton D, Evans D. 2001. Intestinal luminal pH in inflammatory bowel disease: possible determinants and implications for therapy with aminosalicylates and other drugs. Gut 48:571–577. doi: 10.1136/gut.48.4.571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Trachsel E, Redder P, Linder P, Armitano J. 2019. Genetic screens reveal novel major and minor players in magnesium homeostasis of Staphylococcus aureus. PLoS Genet 15:e1008336. doi: 10.1371/journal.pgen.1008336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Avarbock D, Avarbock A, Rubin H. 2000. Differential regulation of opposing RelMtb activities by the aminoacylation state of a tRNA·ribosome·mRNA·RelMtb complex. Biochemistry 39:11640–11648. doi: 10.1021/bi001256k. [DOI] [PubMed] [Google Scholar]
- 99.Avarbock A, Avarbock D, Teh J-S, Buckstein M, Wang Z-m, Rubin H. 2005. Functional regulation of the opposing (p) ppGpp synthetase/hydrolase activities of RelMtb from Mycobacterium tuberculosis. Biochemistry 44:9913–9923. doi: 10.1021/bi0505316. [DOI] [PubMed] [Google Scholar]
- 100.Gaca AO, Kudrin P, Colomer-Winter C, Beljantseva J, Liu K, Anderson B, Wang JD, Rejman D, Potrykus K, Cashel M, Hauryliuk V, Lemos JA. 2015. From (p)ppGpp to (pp)pGpp: characterization of regulatory effects of pGpp synthesized by the small alarmone synthetase of Enterococcus faecalis. J Bacteriol 197:2908–2919. doi: 10.1128/JB.00324-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sobala M, Bruhn-Olszewska B, Cashel M, Potrykus K. 2019. Methylobacterium extorquens RSH enzyme synthesizes (p)ppGpp and pppApp in vitro and in vivo, and leads to discovery of pppApp synthesis in Escherichia coli. Front Microbiol 10:859. doi: 10.3389/fmicb.2019.00859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Vashishtha AK, Wang J, Konigsberg WH. 2016. Different divalent cations alter the kinetics and fidelity of DNA polymerases. J Biol Chem 291:20869–20875. doi: 10.1074/jbc.R116.742494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Vashishtha AK, Konigsberg WH. 2016. Effect of different divalent cations on the kinetics and fidelity of RB69 DNA polymerase. Biochemistry 55:2661–2670. doi: 10.1021/acs.biochem.5b01350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Becker KW, Skaar EP. 2014. Metal limitation and toxicity at the interface between host and pathogen. FEMS Microbiol Rev 38:1235–1249. doi: 10.1111/1574-6976.12087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hanna N, Ouahrani-Bettache S, Drake KL, Adams LG, Köhler S, Occhialini A. 2013. Global Rsh-dependent transcription profile of Brucella suis during stringent response unravels adaptation to nutrient starvation and cross-talk with other stress responses. BMC Genomics 14:459. doi: 10.1186/1471-2164-14-459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Buckley AM, Jukes C, Candlish D, Irvine JJ, Spencer J, Fagan RP, Roe AJ, Christie JM, Fairweather NF, Douce GR. 2016. Lighting up Clostridium difficile: reporting gene expression using fluorescent Lov domains. Sci Rep 6:23463. doi: 10.1038/srep23463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Deneve C, Delomenie C, Barc MC, Collignon A, Janoir C. 2008. Antibiotics involved in Clostridium difficile-associated disease increase colonization factor gene expression. J Med Microbiol 57:732–738. doi: 10.1099/jmm.0.47676-0. [DOI] [PubMed] [Google Scholar]
- 108.Kuijper EJ, Wilcox MH. 2008. Decreased effectiveness of metronidazole for the treatment of Clostridium difficile infection? Clin Infect Dis 47:63–65. doi: 10.1086/588294. [DOI] [PubMed] [Google Scholar]
- 109.Vardakas KZ, Polyzos KA, Patouni K, Rafailidis PI, Samonis G, Falagas ME. 2012. Treatment failure and recurrence of Clostridium difficile infection following treatment with vancomycin or metronidazole: a systematic review of the evidence. Int J Antimicrob Agents 40:1–8. doi: 10.1016/j.ijantimicag.2012.01.004. [DOI] [PubMed] [Google Scholar]
- 110.Kim H-M, Davey ME. 2020. Synthesis of ppGpp impacts type IX secretion and biofilm matrix formation in Porphyromonas gingivalis. NPJ Biofilms Microbiomes 6:5. doi: 10.1038/s41522-020-0115-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Keasling J, Bertsch L, Kornberg A. 1993. Guanosine pentaphosphate phosphohydrolase of Escherichia coli is a long-chain exopolyphosphatase. Proc Natl Acad Sci U S A 90:7029–7033. doi: 10.1073/pnas.90.15.7029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Mechold U, Potrykus K, Murphy H, Murakami KS, Cashel M. 2013. Differential regulation by ppGpp versus pppGpp in Escherichia coli. Nucleic Acids Res 41:6175–6189. doi: 10.1093/nar/gkt302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Choi MY, Wang Y, Wong LL, Lu B-t, Chen W-y, Huang J-D, Tanner JA, Watt RM. 2012. The two PPX-GppA homologues from Mycobacterium tuberculosis have distinct biochemical activities. PLoS One 7:e42561. doi: 10.1371/journal.pone.0042561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang JD, Sanders GM, Grossman AD. 2007. Nutritional control of elongation of DNA replication by (p)ppGpp. Cell 128:865–875. doi: 10.1016/j.cell.2006.12.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Baines SD, Wilcox MH. 2015. Antimicrobial resistance and reduced susceptibility in Clostridium difficile: potential consequences for induction, treatment, and recurrence of C. difficile Infection. Antibiotics (Basel) 4:267–298. doi: 10.3390/antibiotics4030267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sorg JA, Dineen SS. 2009. Laboratory maintenance of Clostridium difficile. Curr Protoc Microbiol 12:9A.1.1–9A.1.10. doi: 10.1002/9780471729259.mc09a01s12. [DOI] [PubMed] [Google Scholar]
- 117.Smith CJ, Markowitz SM, Macrina FL. 1981. Transferable tetracycline resistance in Clostridium difficile. Antimicrob Agents Chemother 19:997–1003. doi: 10.1128/AAC.19.6.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Purcell EB, McKee RW, McBride SM, Waters CM, Tamayo R. 2012. Cyclic diguanylate inversely regulates motility and aggregation in Clostridium difficile. J Bacteriol 194:3307–3316. doi: 10.1128/JB.00100-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Schneider CA, Rasband WS, Eliceiri KW. 2012. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.