

Abstract
Reagents that can selectively recognize specific toxic tau variants associated with onset and progression of AD and other tauopathies can be effective diagnostic and therapeutic tools. We utilized a novel atomic force microscopy (AFM) based biopanning protocol to isolate antibody fragments (scFvs) that selectively bind tau variants present in human AD but not cognitively normal age matched brain tissue. We identified six scFvs (ADT-1 through 6) that readily distinguished between AD and control tissue and sera samples. We utilized three of the scFvs (ADT-2, -4 and -6) to analyze longitudinal plasma samples from 50 human patients, 25 patients which converted to AD during the study and 25 that remained cognitively normal. All three scFvs could distinguish the AD from control samples with higher tau levels in ApoE3,3 AD cases compared to ApoE3,4. Immunohistochemical analyses of human AD brain slices indicated several but not all tau variants overlapping with phosphorylated tau staining. Several of the reagents also showed therapeutic potential, protecting neuronal cells against AD tau induced toxicity.
Keywords: Alzheimer’s Disease, tau, single chain antibody fragment, biomarker
1. Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disease that affects memory and behavior. AD, like many other neurodegenerative diseases, is associated with altered folding of key neuronal proteins, including amyloid-beta (Aβ) and tau, primary components of the hallmark extracellular plaques and intracellular neurofibrillary tangles, respectively. While the plaques and tangles are comprised of fibrillar aggregates of these proteins, many recent studies indicate that small soluble oligomeric aggregates of Aβ and tau play important roles in the pathogenesis and spread of disease. Here we study the role of several key oligomeric tau aggregates in AD using novel reagents that selectively bind variants of tau present in human AD but not cognitively normal brain tissue.
Tau is a natively unfolded microtubule associated protein due to its very low hydrophobic content. The protein contains a projection domain, a basic proline-rich region, and an assembly domain that contains either three or four repeats (Liu and Gong 2008) of a conserved tubulin-binding motif as a result of alternative splicing of exon 10 (Ballatore, Lee et al. 2007, Liu and Gong 2008, Wang and Liu 2008). Tau 4R isoforms have better microtubule binding and stabilizing capabilities compared to the 3R isoforms, and while 3R tau is expressed at the fetal stage, 3R and 4R are present in equal proportions in the adult human brain. Mutations that alter splicing of tau transcript and the ratio of 3R to 4R tau isoforms can lead to neurodegenerative disease (Ballatore, Lee et al. 2007, Wang and Liu 2008). In AD, tau undergoes several post-translational modifications which include aggregation, phosphorylation, glycosylation, glycation, ubiquitination, cleavage or truncation, (reviewed in (Martin, Latypova et al. 2011)). Tau can aberrantly fold into various aggregate morphologies which include β-sheet rich fibrillar forms that result in the formation of paired helical filaments and neurofibrillary tangles (Ghoshal, Garcia-Sierra et al. 2002, Garcia-Sierra, Ghoshal et al. 2003). Hyperphosphorylation of tau decreases the affinity of tau to the microtubules which in turn affects axonal transport (Konzack, Thies et al. 2007, Dubey, Chaudhury et al. 2008). Therefore, tau in human brain tissue can exist in a variety of different lengths and morphologies and with multiple post-translational modifications.
Accumulation of tau is necessary for the development of cognitive deficits in AD models caused by over-expression of Aβ (Marx 2007, Roberson, Scearce-Levie et al. 2007). While neurofibrillary tangles (NFTs) have been implicated in mediating neurodegeneration in AD and tauopathies (Arriagada, Growdon et al. 1992, Bancher, Braak et al. 1993, Guillozet, Weintraub et al. 2003), animal models of tauopathy have shown that memory impairment and neuron loss do not associate well with accumulation of NFT (Brunden, Trojanowski et al. 2008). Animal studies showed that a reduction in neuronal loss and improvement in memory could occur even with accumulation of NFTs (Santacruz, Lewis et al. 2005), and that neuronal loss and NFT pathology did not always co-localize (Meraz-Rios, Lira-De Leon et al., Andorfer, Kress et al. 2003, Polydoro, Acker et al. 2009). Oligomeric tau aggregates correlate well with AD progression (Maeda, Sahara et al. 2006, Berger, Roder et al. 2007, Sahara, Maeda et al. 2008) and high levels of tau oligomers were detected in the frontal lobe cortex (Maeda, Sahara et al. 2006, Yoshiyama, Higuchi et al. 2007) and in CSF during early stages of AD (Vandermeeren, Mercken et al. 1993, Shaw, Vanderstichele et al. 2009). Various oligomeric tau aggregates have been shown to propagate in AD brains through prion-like and receptor mediated mechanisms (Gomez-Ramos, Diaz-Hernandez et al. 2009). For example, tau pathology spreads throughout the brain from early to late stage (Schonheit, Zarski et al. 2004), and seeding with tau aggregates can propagate altered tau folding from the outside to the inside of a cell (Frost, Jacks et al. 2009). Studies have also shown that brain extract from a transgenic mouse with aggregated mutant human tau can transmit tau pathology throughout the brain in mice expressing normal human tau (Clavaguera, Bolmont et al. 2009) and inducing pro-aggregation of human tau induces formation of tau aggregates and tangles composed of both human and normal murine tau (co-aggregation) (Mocanu, Nissen et al. 2008). Therefore, many diverse studies indicate oligomeric tau aggregates are involved in neurodegeneration and so selective AD related tau variants may be excellent early diagnostic and therapeutic targets for AD. Because of the wide diversity of tau forms and the importance of soluble oligomeric tau variants in AD and related dementias, it is critically important to generate reagents that can selectively bind to protein variants implicated in these diseases. Here we develop a panel of antibody-based reagents that specifically bind tau variants that are present in human AD brain samples but not cognitively normal age matched control samples, and show that the reagents have potential diagnostic and therapeutic value for AD.
2. Materials and methods
2.1. Brain Tissue and sera samples:
Human brain tissue, sections and sera samples were obtained from Dr. Thomas Beach, director of the Brain and Body Donation Program at Banner Sun Health Research Institute (BBDP; http://www.brainandbodydonationprogram.org) (Beach 2008, Beach, Adler et al. 2015). Seven different pathologically confirmed AD and two cognitively normal brain tissue samples were obtained from two different brain regions - the superior frontal gyrus and middle temporal gyrus. Brain sections for immunohistochemistry (Kaufman, Thomas et al. 2017) studies were obtained from the superior frontal cortex. Sera samples were obtained from 10 pathologically confirmed AD (Braak stage V and VI) and 10 control cases.
2.2. Brain tissue homogenization:
Frozen brain tissue samples were homogenized as described previously (Kasturirangan, Reasoner et al. 2013). Briefly tissue was sonicated in cold lysis buffer: 25 mM HEPES NaOH (pH 7.9), 150 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 0.5% Triton-X-100, 1 mM dithiothreitol, protease inhibitor cocktail. The homogenized sample was centrifuged, and the supernatant was frozen in −80°C. The presence of fibrillar tau was verified by immunohistochemistry by staining AD and healthy control brain tissue slices with 1:1000 dilution of commercial AT8 antibody (Thermo Fisher Scientific, Catalog # MN1020) against phosphorylated tau Ser202/Thr205.
2.3. Tau protein immunoprecipitation:
Two polyclonal tau antibodies, PA1-18272 against amino-acids 240-450 of tau and PA5-27287 against amino-acid 1-286 (Thermo Fisher Scientific) were used to immunoprecipitate tau. Antibody conjugates were captured using the Pierce Crosslink IP Kit A following the manufacturers’ protocols. The antibody is first bound to the protein A/G agarose which is chemically crosslinked to the resin to prevent the antibody from eluting off the column. AD brain tissue homogenates were pooled into Braak stage III and Braak stage V samples. Tau immunoprecipitation was performed on the pooled AD Braak stage III, Braak stage V and healthy control samples. The integrity of the eluted tau was preserved by neutralizing the low pH elution buffer with 1M Tris, pH 9.5 as recommended by the manufacturer. The immunoprecipitated tau samples were probed with 1:1000 dilution of commercially available AT8 antibody (Thermo Fisher Scientific, Catalog # MN1020) to verify the presence of tau.
2.4. Phage library preparation:
Tomlinson I, J and Sheets phage libraries (Sheets, Amersdorfer et al. 1998) each having a diversity of 108 were amplified as described (http://docplayer.net/30268507-Human-single-fold-scfv-libraries-i-j-tomlinson-i-j.html). Equal phage titers from each library were pooled for biopanning studies.
2.5. Biopanning – AD tau specific morphologies:
A series of negative and positive panning steps were performed as described previously (Williams, Venkataraman et al. 2015) to obtain AD-tau specific reagents. Negative or subtractive selection steps were performed to remove phage particles that bound to non-target sticky protein samples using bovine serum album and aggregated protein morphologies using α-synuclein aggregates. Additional negative panning steps against monomeric tau, healthy tissue samples and healthy tau samples were performed to remove all phage containing antibody fragments that bound to those forms of tau found in healthy individuals. Atomic force microscopy (AFM) imaging was performed after every negative panning step to ensure removal of all antibody fragments binding these off-target antigens. Phage that did not bind to any of the off-target antigens was used for the final positive selection round performed against tau immunoprecipitated from pooled AD Braak stage III and V brain tissue samples. For the positive panning step, the tau preparation was deposited on mica since only nanogram quantities of the antigen are needed and the process can be monitored via AFM imaging. Phage were eluted using trypsin and triethylamine (TEA) and grown on LB – Amp plates overnight at 37°C.
2.6. Phage and scFv purification:
To ensure recovered phage clones code for full length antibody fragments, their sequences were checked for presence of mutations and stop codons. After sequence validation, phage were amplified as described (Sheets, Amersdorfer et al. 1998). Phage titers were performed to verify the concentration of phage (~109 pfu/mL). Soluble scFv protein separate from the phage was also produced for each clone by transforming the plasmid into HB2151 strain of E coli. The scFvs were grown and purified using a protein A Sepharose affinity column (GE Healthcare) as described (Emadi, Barkhordarian et al. 2007, Williams, Venkataraman et al. 2015). Molecular size of the scFvs were checked by western blot using anti c-myc (9E10) monoclonal antibody (1:1000 dilution) which recognizes the c-myc region in the scFv followed by goat anti-mouse HRP (1:2000 dilution). The DNA sequences of the scFvs were also validated using MAFFT, a multiple sequence alignment software.
2.7. Tau monomer phage:
To obtain a detection phage for sandwich ELISA we required a phage particle displaying an scFv that bound all forms of tau including monomeric and oligomeric. To generate this scFv, we used the phage preparation eluted after negative panning with BSA and aggregated α-synuclein, and used this in a single positive panning step using commercially available tau monomer deposited on mica as described (Williams, Venkataraman et al. 2015). The detection phage binds tau variants present in both AD and cognitively normal control samples and does not compete for the same binding sites as the capture scFv in sandwich ELISA.
2.8. ELISA assay:
A phage ELISA was initially performed to check the specificity of each of the phage clones to tau variants in AD brain tissue as described previously (Williams, Venkataraman et al. 2015). Pooled AD and control human brain tissue homogenates were used to coat the plates followed by detection with each of the phage clones. This assay was used to evaluate binding specificity of all the phage clones for AD over cognitively normal control samples.
Soluble antibody fragments (scFv) (ADT-1, ADT-2, ADT-3, ADT-4, ADT-5 and ADT-6) were produced for each of the phage that showed a high ELISA signal with the AD samples. The scFvs were used as the capture antibody in a sandwich ELISA to test reactivity with AD (n=10) and cognitively normal control sera (n=10) samples (Beach 2008, Beach, Adler et al. 2015) as described previously (Williams, Schulz et al. 2017). Briefly, the capture scFv was immobilized to the wells of a high binding 96-well ELISA plate (Costar, USA) and any unbound sites blocked with 2% milk-PBS. A 1/200 v/v dilution of the plasma samples were added to the wells followed by 200 ng/ml of a 40 mM carboxyl biotinylated detection phage. Any bound phages were identified using the avidin-HRP antibody due to the presence of these biotin binding sites on the phage coat proteins. Following addition of the SuperSignal ELISA Femto Maximum Sensitivity Substrate (Thermo Scientific, USA), signal intensities were quantified using the Wallac Victor2 microplate reader. After each incubation step, it was necessary to wash the wells 3-4 times with 0.1% PBS-Tween20 to reduce non-specific binding. Known positive and negative controls were included on each ELISA plate.
2.9. Longitudinal plasma analysis:
Plasma samples were obtained over multiple timepoints from 25 individuals that were initially cognitively normal but converted to AD during the study and 25 individuals that remained cognitively normal throughout the timeline (~10 years). The samples were taken from Mayo Clinic collections obtained by Dr. Steven Younkin and provided with the assistance of Dr. Terrone Rosenberry at the Mayo Clinic College of Medicine in Jacksonville, Florida. Four to five different timepoints were obtained for every individual. Based on clinical diagnoses samples were designated as pre-mild cognitive impairment (pre-MCI), MCI and AD. The individual cases have not been pathologically validated. The results obtained by analyses using the ADT-2, ADT-4 and ADT-6 scFvs were analyzed with respect to gender, genotype and MMSE scores.
2.10. Conversion to IgG and Immunohistochemistry:
Five anti-tau scFvs (ADT-1, ADT-2, ADT-3, ADT-4 and ADT-5) were converted to IgG format, (purchased from MIGS LLC, New Hampshire, USA). Concentration of the purified IgGs were assessed using a BCA kit and ranged between 2-4 mg/mL
Brain sections (Grundman, Petersen et al. 2004) from the superior frontal cortex were treated with 0.1% Triton X-100 and 5% goat serum for 10 minutes. The brain sections were then treated with primary antibodies including either a commercially available antibody against phosphorylated tauSer202/Thr205 (clone: AT8, catalog: MN1020, 1:2000, Thermofisher Scientific, Rockford, IL) or one of the five human anti-tau IgGs at a concentration of 1 ug/ml, 4°C, overnight. The sections were further treated with fluorescently tagged goat anti-mouse IgG (red) and goat anti-human IgG (green) (1:1000) respectively for 1 hour at room temperature and the non-specific background was blocked with 0.03% Sudan black for 5 minutes. The sections were observed and imaged with Leica SP5 (Tian et al., 2015). Commercial AT8 antibody is visualized in red, anti-tau IgG in green and DAPI, which stains the nucleus, in blue.
2.11. Toxicity Assay:
The ability of the different anti-tau IgGs to block toxicity of tau variants contained in human AD tissue were assessed using cultured neuronal cells. Tau immunoprecipitated from human AD brain was used to induce toxicity in the human neuroblastoma cell line, SH-SY5Y. Once the SH-SY5Y neuronal cells reached confluence they were introduced into a 6-well plate. 100 μg/mL of immunoprecipitated tau from AD or control postmortem brain tissue was introduced into the wells. The cells were then incubated with either a commercial polyclonal tau antibody PA5-27287 (Thermo Fisher), or one of the anti-tau IgGs – ADT-1, ADT-2, ADT-3, ADT-4 or ADT-5. After 12 hours of incubation, toxicity and damage to the cells were measured using a lactate dehydrogenase (LDH) assay kit (Xin et al., 2015).
2.12. Statistical Analysis:
Measurements obtained with the ELISAs are represented as ratios of luminescence measurement for each sample with respect to the PBS background (no antigen) control. Reactivity of each test sample was obtained relative to the average signal of the control group. Any sample with a ratio greater than 1 is considered a positive signal. Statistical significance was assessed using SPSS software (version 24) and one-way ANOVA was performed with LSD post-hoc analyses with significance at p<0.05.
For SH-SY5Y toxicity studies, the LDH values for each IgG antibody were adjusted to a percentage of the AD tau IP+vehicle samples, zeroed to the control tau IP sample, and plotted as log dose-response curves. The log of IC50 was calculated in SPSS by nonlinear regression using the log form of the Hill equation.
To determine the accuracy of anti-tau scFvs in detecting AD over healthy controls, Receiver Operating Characteristic (ROC) curves were generated based on the reactivity of the anti-tau scFvs with postmortem verified AD and healthy control sera. Sensitivity and specificity of the anti-tau scFvs were obtained by setting the cutoff as the average value of the healthy controls. Area Under the Curve (AUC) was calculated as described (Hanley and McNeil 1982). AUC greater than 0.8 is considered a good diagnostic test while 0.5 (straight line) indicates the test cannot significantly differentiate between AD and healthy controls.
3. Results and Discussion
3.1. Tau protein immunoprecipitation:
Tau was immunoprecipitated from pooled AD samples with confirmed tangle pathology and control brain tissue homogenates. When probed with commercial AT8 antibody, AD samples contained high molecular weight phosphorylated tau which was not present in the control samples (Supplemental Figure S1). The immunoprecipitated AD and control tau samples were used for biopanning studies.
3.2. Biopanning:
A series of negative or subtractive panning steps were utilized to remove all phage from the initial pooled phage library that bound to several off-target antigens including BSA, aggregated α-synuclein, healthy control tissue and any forms of tau immunoprecipitated from healthy control tissue. Essentially complete removal of all phage binding these off-target antigens was verified by AFM imaging. After completing the subtractive panning process, a single round of positive panning using the immunoprecipitated tau from AD brain tissue was performed to isolate scFvs selectively binding tau variants present in human AD tissue but not control tissue.
Thirty-five phage clones were recovered from the positive selection step, and 11 complete scFvs showed selective binding to the AD brain tissue samples compared to the control brain tissue samples (Fig 1a) based on ELISA results and sequence analysis for scFv integrity. Six of the eleven phage clones (ADT-1, ADT-2, ADT-3, ADT-4, ADT-5 and ADT-6) with high reactivity and specificity for the pooled AD over control human brain tissue homogenates were selected for further analyses. We expressed soluble scFv from each of the six phage and evaluated reactivity of each scFv with individual AD (n=7) and control (n=2) brain tissue homogenates by sandwich ELISA. All six scFvs showed reactivity with tau variants, present in each of the human AD tissue samples, but not the cognitively normal control brain tissue samples (Fig 1b).
Figure 1: Anti-tau phage characterization with pooled and individual AD and control brain tissue homogenates.
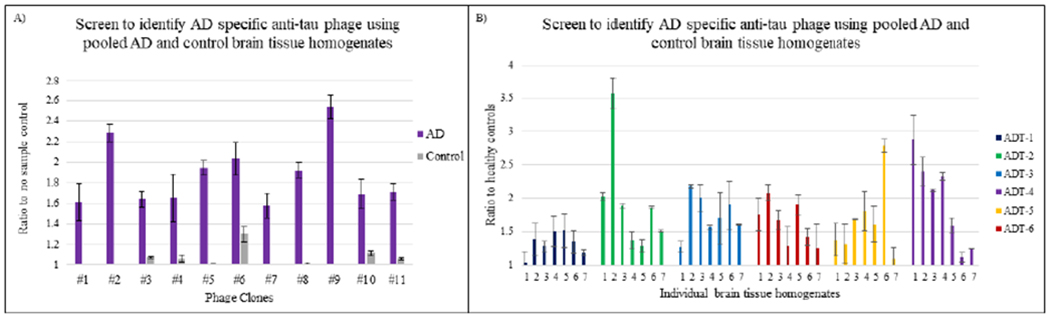
ELISA assay to screen for AD specific anti-tau phages. X axis represents different anti-tau phage and Y axis represents luminescence signal ratio to no sample background control or healthy control. Reactivity of anti-tau phage with: A) pooled AD and control brain tissue homogenates. B) individual AD brain tissue homogenates. All phages have high levels of binding to both pooled and individual AD brain tissue homogenates compared to controls.
3.3. Sera sample analysis:
We determined whether the 6 selected scFvs (ADT-1, ADT-2, ADT-3, ADT-4, ADT-5 and ADT-6) could also recognize tau variants present in human AD (n=10) and controls (n=10) sera samples (Fig 2). All 6 scFvs preferentially bound tau variants present in all 10 AD sera samples compared to cognitively normal controls.
Figure 2: Sandwich ELISA with AD and control sera.
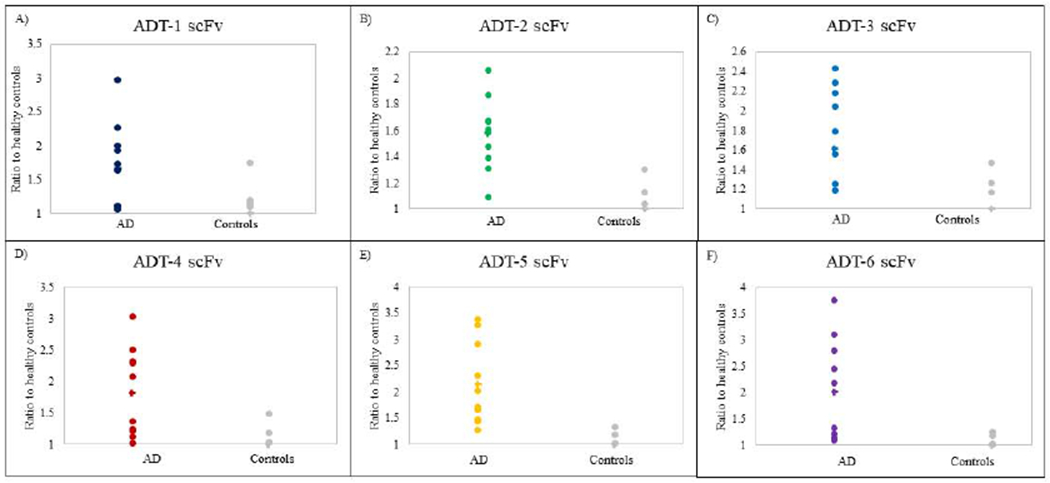
Sandwich ELISA assay testing AD tau scFvs ADT-1, ADT-2, ADT-3, ADT-4, ADT-5, ADT-6 with individual AD brain sera. X axis represents individual AD sera samples and Y axis represents luminescence signal ratio to healthy control. Six AD-tau scFvs have high levels of binding with the 10 AD sera samples compared to controls.
ROC curves for the six anti-tau scFvs were analyzed to determine their ability to select tau variants in AD compared to healthy controls. Five of the six anti-tau scFvs have high sensitivity and specificity to tau variants in AD. The scFvs ADT-2, ADT-4 and ADT-6 recognized tau variants with 80% sensitivity and 90% specificity, 100% sensitivity and 80% specificity and 90% sensitivity and 80% specificity respectively. Together, these three scFvs recognized tau variants with 90% sensitivity and 90% specificity with 0.96 AUC (Table 1, Supplemental Figure S2), so these three scFvs were selected to analyze tau variants in longitudinal AD and control plasma.
Table 1: Specificity and sensitivity of anti-tau scfv against sera:
Six anti-tau scFvs were analyzed to determine their ability to select tau variants in postmortem-verified AD sera compared to healthy control sera. Area Under Curve (AUC), calculated by plotting Receiver Operating curves, represents the combined sensitivity and Specificity scores to select AD samples. The three most effective scFvs (ADT-2, -4, -6) were selected to analyze tau variants in longitudinal AD and control plasma.
| scFv | Sensitivity(%) | Specificity(%) | Area Under Curve (AUC) |
|---|---|---|---|
| ADT-1 | 60 | 80 | 0.69 |
| ADT-2 | 80 | 90 | 0.91 |
| ADT-3 | 70 | 70 | 0.75 |
| ADT-4 | 90 | 80 | 0.96 |
| ADT-5 | 70 | 65 | 0.72 |
| ADT-6 | 90 | 80 | 0.94 |
| ADT2, ADT-4 & ADT-6 | 90 | 90 | 0.96 |
3.4. Longitudinal plasma analysis:
Tau variants in plasma:
Tau variant content in longitudinal plasma samples obtained from 25 individuals that converted to AD over a 10-year timeline (n=119) and individuals that remained cognitively normal (n=121) based on neurological exam were analyzed. Due to limited sample availability, the three most selective scFvs, ADT-2, ADT-4 and ADT-6, were utilized to analyze the longitudinal plasma samples. Plasma samples from individuals that converted to AD showed higher average reactivity toward each of the tau variants tested compared to samples from individuals that did not convert to AD (Fig 3a). Based on one-way ANOVA analysis, all three anti-tau scFvs were able to distinguish the converters from the non-converters. The AD converter and control groups were broken into groups depending on APOE genotype. Both ApoE3/3 and ApoE3/4 genotypes had significantly higher tau levels in the AD converters compared to the controls, and the Apoe3/3 AD cases had higher tau levels than the ApoE3/4 AD cases (Fig 3b).
Figure 3: Longitudinal AD plasma analysis.
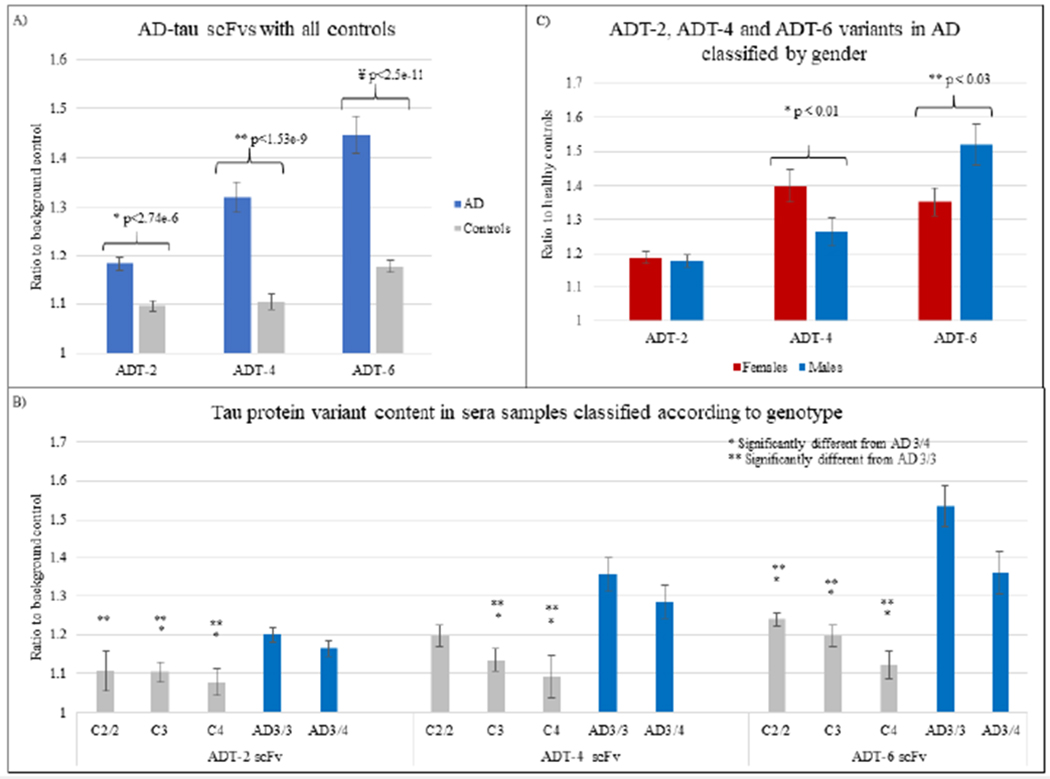
Reactivity of anti tau scFvs ADT-2, ADT-4 and ADT-6 with 241 longitudinal AD and control plasma samples. A) Blue bars represent average of all samples that converted to AD and grey bars represent average of all samples that remained cognitively normal, B) Reactivity to the samples based on ApoE genotype (AD 3/3, AD 3/4, Control 2/2, Controls with at least one copy of ApoE3 (‘C3’), controls with at least one copy of ApoE4(‘C4’)). C) Reactivity of the three anti-tau scFvs classified based on gender. ADT-4 and ADT-6 show significant differences in reactivity between males and females, D) Reactivity of the anti-tau scFvs with the samples classified according to gender and genotype.
3.4.1. Gender analysis:
We also analyzed the longitudinal plasma samples to determine if there were any gender differences. ADT-4 recognized tau variants preferentially present in females (n=51) while ADT-6 recognized tau variants preferentially present in males (n=68) (Fig 3c). The ADT-2 scFv did not demonstrate any gender selectivity.
3.4.2. AD Progression:
The longitudinal plasma samples were grouped according to classification based on neurological exam reports. Samples were classified as either pre-MCI, MCI, or AD depending on their neurological exam and performance on mini mental state examination (MMSE). ADT-4 scFv has significantly higher reactivity with samples obtained from patients during the MCI stage compared to samples taken during pre-MCI and AD stages (Fig 4a) suggesting that ADT-4 recognizes a tau variant preferentially present during the MCI phase and present in both females and males (Fig 4b). Since the longitudinal plasma cases were not pathologically validated in post-mortem analyses, the AD cases may include misdiagnosed cases that should be attributed to other AD related dementias or other causes.
Figure 4: Longitudinal plasma analysis: Reactivity based on sample diagnosis and gender.
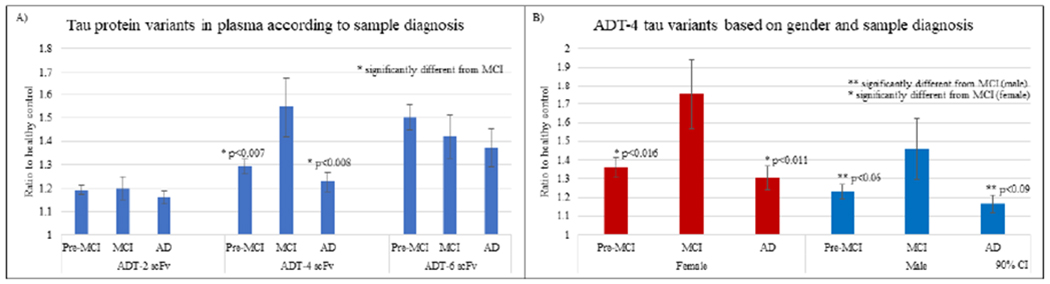
ADT-2, ADT-4 and ADT-6 tau protein variants present in AD plasma samples (n=119) classified based on A) sample diagnosis (Williams, Schulz et al. 2017), B) ADT-4 protein variants by gender. Results indicate that ADT-4 tau variants were significantly higher in samples classified as MCI compared to pre-MCI and AD groups in both females and males.
3.5. Immunohistochemistry:
The location of the different tau variants in human AD brain tissue were analyzed by immunohistochemistry. Human postmortem AD and control brain sections from the superior frontal cortex were probed with five anti-tau IgGs (ADT-1, ADT-2, ADT-3, ADT-4 and ADT-5) and the commercial AT8 anti-phosphorylated tau antibody. As expected, none of the antibodies showed significant reactivity with control brain sections, however widespread positive staining was observed in the AD brain sections (Fig 5). There is extensive extracellular colocalization of commercial AT8 and four of the IgGs (ADT-1, ADT-3, ADT-4 and ADT-5). Interestingly, ADT-2 does not colocalize with AT8 and based on morphology may recognize a tau variant that accumulates along the axons.
Figure 5: Immunofluorescent detection of tau variants in AD and control tissue samples.
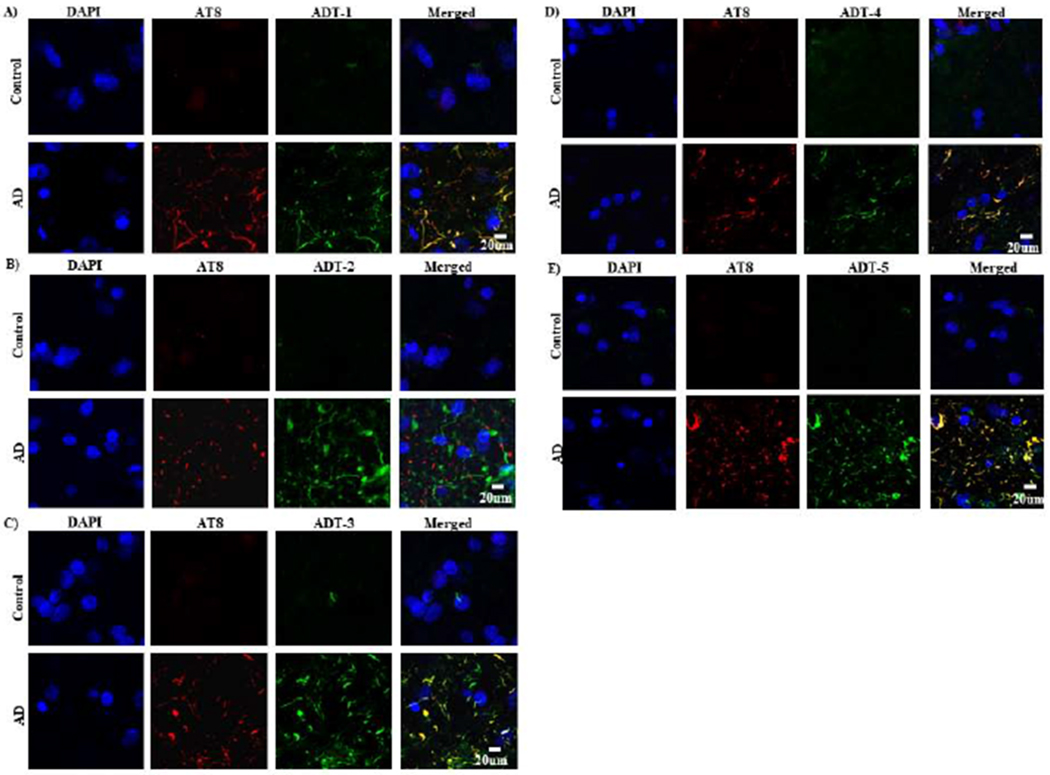
IHC was performed to test reactivity of the anti-tau IgGs with AD and control brain tissue slice. Primary antibodies were applied with mouse against phosphorylated tau (AT8, Covance, 1:2000,) and human against tau variants (clone ADT-1, ADT-2, ADT-3, ADT-4, ADT-5 respectively, 1:100) on a shaking stage overnight, 4°C. Goat anti-mouse IgG(red) and goat anti-human IgG(green) with fluorescence at the concentration of 1:1000 were used respectively as secondary antibodies. The sections were observed and imaged with Leica SP5.
3.6. Therapeutic potential of anti-tau IgGs:
After identifying IgGs (ADT-1, ADT-2, ADT-3, ADT-4 and ADT-5) that preferentially bound to tau forms in AD brain tissue compared to age-matched cognitively normal controls using IHC, we analyzed if the IgGs bound to neurotoxic variants of tau utilizing a human neuroblastoma cell line. We added immunoprecipitated tau from human AD brain tissue to human neuronal SH-SY5Y cells and determined toxicity when co-incubated with different concentrations of either a commercial anti-tau IgG or an IgG version of each of the five scFvs. IP tau from control brain tissue was used as baseline. Three of the IgG (ADT-1, -4, -5) showed improved toxicity-inhibiting IC50 values (4.8nM, 6.4nM, 7.2nM respectively) compared to the commercial antibody PA5-27287 (24.8 nM). IgG ADT-2 showed an equivalent value to the commercial antibody (24.9nM), while ADT-3 showed little protection against toxicity having a relatively high value (IC50 = 13 μM) (Table 2). Individual curves are shown in Supplemental Figure S3. These results indicate selective targeting of tau variants can reduce toxicity, implying potential therapeutic value of selected anti-tau IgGs.
Table 2: Therapeutic potential of anti-tau IgGs.
SH-SY5Y neuroblastoma cell line was treated with AD or control brain derived tau IP and with different concentrations of either polyclonal anti-tau antibody or IgGs ADT-1, ADT-2, ADT-3, ADT-4 and ADT-5 for 12 hours. The cell damage and toxicity were tested by LDH assay (n=3). IC50 values calculated by nonlinear regression of log dose-response curves. ADT-1, -4 and -5 inhibited toxicity of AD brain derived tau IP more effectively than a polyclonal anti-tau preparation.
| 95% CI (nM) | |||
|---|---|---|---|
| Antibody | IC50 (nM) | Lower | Upper |
| Commercial | 4.96 | 9.49 x10−5 | 2.60 x105 |
| ADT-1 | .961 | 1.60 x10−15 | 5.76 x1014 |
| ADT-2 | 4.97 | 2.09 x10−12 | 1.18 x1013 |
| ADT-3 | > 2500 | N/A | N/A |
| ADT-4 | 1.28 | 1.82 x10−20 | 9.06 x1019 |
| ADT-5 | 1.44 | 8.30 x10−26 | 2.49 x1025 |
3.7. Conclusions
AD is a neurodegenerative disease for which there are currently no effective therapeutics. Both Aβ and tau have been shown to play a critical role in AD pathogenesis (as reviewed in (Ittner and Gotz 2011)). Recent studies have also established the importance of Aβ and tau variants in AD pathogenesis (Roberson, Scearce-Levie et al. 2007, Zempel, Thies et al. 2010, Jin, Shepardson et al. 2011). There are several intermediate variants of tau that exist prior to the formation of intracellular tau tangles which have been hypothesized to play a major role in disease pathogenesis. It has also been shown that these tau variants progressively spread from one cell to another in a neurotoxic manner (Schonheit, Zarski et al. 2004, Clavaguera, Bolmont et al. 2009, Frost, Jacks et al. 2009). Studies have shown these variants to be synaptotoxic and responsible for spread of the disease (Meraz-Rios, Lira-De Leon et al., Marx 2007, Yoshiyama, Higuchi et al. 2007, Brunden, Trojanowski et al. 2008). Reagents that can selectively recognize the different tau variants would provide valuable tools to facilitate diagnosis of AD and could serve as potential novel therapeutics.
Here, we generated a pool of antibody-based reagents that selectively bind different tau variants that are present in human AD but not age-matched control samples (Fig 1–3). The morphology specific reagents recognize tau variants present in both brain tissue homogenate and circulating sera in AD patients, even in pre-symptomatic plasma samples (Fig 3) indicating their potential value as diagnostics for AD. Selected tau variants were preferentially present in either female or male AD patients (Fig 5). The different tau variants were localized to different cell regions where some but not all tau variants correlated with phospho-tau staining (Fig 5). The anti-tau reagents were also capable of countering toxicity induced by tau from pooled AD brain tissue homogenates indicating the potential therapeutic value of these reagents (Table 2). Since tau pathology is not unique to Alzheimer’s Disease and is also observed in other tauopathies such as Frontotemporal Dementia(Lee et al., 2001), these reagents may have value in treating other tauopathies as well. Future studies are planned to determine which tau variants are unique to AD and which ones also are found in other neurodegenerative diseases.
Supplementary Material
Supplemental Figure S1: A) Immunohistochemistry staining of tau: Staining with commercially available anti-phosphorylated tau antibody AT8 showed increased presence of phosphorylated tau fibrils in the human AD brain tissue slice compared to the age matched healthy control; B) Western Blot analysis of immunoprecipitated tau: Staining with the anti-phosphorylated tau antibody, AT8 shows presence of high molecular weight tau species in AD and absence of high molecular weight tau species in control.
Supplemental Figure S2: Specificity and sensitivity of anti-tau scfv against sera: a-f) Receiver Operating Curves (ROC) curves for six anti-tau scFvs were analyzed to determine their ability to select tau variants in postmortem-verified AD compared to healthy control sera. Five of the six anti-tau scFvs have high sensitivity and specificity to tau variants in AD. The scFvs b) ADT-2, d) ADT-4 and f) ADT-6 recognized tau variants with 80% sensitivity and 90% specificity, 100% sensitivity and 80% specificity and 90% sensitivity and 80% specificity respectively. g) combined ADT-2,-4,-6 recognized tau variants with 90% sensitivity and 90% specificity with 0.96 AUC.
Supplemental Figure S3: Log Dose-response curves of anti-tau IgGs. SH-SY5Y neuroblastoma cells were treated with AD or control brain derived tau IP and with different concentrations of either a) polyclonal anti-tau antibody PA5-27287 or IgGs b) ADT-1, c ADT-2, d) ADT-3, e) ADT-4 or f) ADT-5 for 12 hours. The cell damage and toxicity were assessed by LDH assay (n=3). LDH values for each antibody were adjusted to a percentage of the AD tau IP+vehicle samples, zeroed to the control tau IP sample, and plotted as log dose-response curves. ADT-1, -4 and -5 inhibited toxicity of AD brain derived tau IP more effectively than a polyclonal anti-tau preparation.
Identified antibody fragments that selectively bind tau variants present in human postmortem AD brain tissue
Reagents all selectively react with human postmortem AD brain tissue and sera samples but not age-matched cognitively normal control samples
Reagents selectively react with longitudinal human plasma samples from patients that converted to AD indicating early, presymptomatic diagnostic capability
Reagents selectively label tau variants in human brain tissue samples
Reagents block toxicity of tua variants isolated from human AD brain tissue toward a human neuronal cell line
Acknowledgements
We would like to thank Dr. Terrone L. Rosenberry and Dr. Steven Younkin, Mayo Clinic (Florida) for the longitudinal AD and control sera samples. We would also like to thank Dr. Tom Beach for the AD and control brain tissue samples. This work was supported in part by grants from the National Institute on Aging (NIH) 5R21AG04147202 and Department of Defense W81XWH1410467.
Abbreviations
- AD
Alzheimer’s Disease
- Aβ
Amyloid beta
- NFT
Neurofibrillary tangles
- CSF
cerebrospinal fluid
- MTG
Mid temporal gyrus
- MCI
Mild cognitive impairment
- MMSE
Mini Mental State Examination
- scFv
single chain variable fragment
- AFM
Atomic Force Microscopy
- BSA
bovine serum album
- BCA
bicinchoninic acid
- TEA
Triethylamine
- ELISA
Enzyme Linked Immunosorbent Assay
- EDTA
Ethylenediaminetetraacetic Acid
- DAPI
4′,6-diamidino-2-phenylindole
- ROC
Receiver Operating Characteristic
- AUC
Area Under Curve
- One-way ANOVA
One-way Analysis of Variance
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest. M. Sierks is cofounder and CSO of Studio Biotherapeutics. All other authors declare that they have no conflict of interest.
References
- Andorfer C, Kress Y, Espinoza M, de Silva R, Tucker KL, Barde YA, Duff K and Davies P (2003). “Hyperphosphorylation and aggregation of tau in mice expressing normal human tau isoforms.” J Neurochem 86(3): 582–590. [DOI] [PubMed] [Google Scholar]
- Arriagada PV, Growdon JH, Hedley-Whyte ET and Hyman BT (1992). “Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer’s disease.” Neurology 42(3 Pt 1): 631–639. [DOI] [PubMed] [Google Scholar]
- Ballatore C, Lee VM and Trojanowski JQ (2007). “Tau-mediated neurodegeneration in Alzheimer’s disease and related disorders.” Nat Rev Neurosci 8(9): 663–672. [DOI] [PubMed] [Google Scholar]
- Bancher C, Braak H, Fischer P and Jellinger KA (1993). “Neuropathological staging of Alzheimer lesions and intellectual status in Alzheimer’s and Parkinson’s disease patients.” Neurosci Lett 162(1-2): 179–182. [DOI] [PubMed] [Google Scholar]
- Beach TG (2008). “Physiologic origins of age-related beta-amyloid deposition.” Neurodegener Dis 5(3-4): 143–145. [DOI] [PubMed] [Google Scholar]
- Beach TG, Adler CH, Sue LI, Serrano G, Shill HA, Walker DG, Lue L, Roher AE, Dugger BN, Maarouf C, Birdsill AC, Intorcia A, Saxon-Labelle M, Pullen J, Scroggins A, Filon J, Scott S, Hoffman B, Garcia A, Caviness JN, Hentz JG, Driver-Dunckley E, Jacobson SA, Davis KJ, Belden CM, Long KE, Malek-Ahmadi M, Powell JJ, Gale LD, Nicholson LR, Caselli RJ, Woodruff BK, Rapscak SZ, Ahern GL, Shi J, Burke AD, Reiman EM and Sabbagh MN (2015). “Arizona Study of Aging and Neurodegenerative Disorders and Brain and Body Donation Program.” Neuropathology 35(4): 354–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger Z, Roder H, Hanna A, Carlson A, Rangachari V, Yue M, Wszolek Z, Ashe K, Knight J, Dickson D, Andorfer C, Rosenberry TL, Lewis J, Hutton M and Janus C (2007). “Accumulation of pathological tau species and memory loss in a conditional model of tauopathy.” J Neurosci 27(14): 3650–3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunden KR, Trojanowski JQ and Lee VM (2008). “Evidence that non-fibrillar tau causes pathology linked to neurodegeneration and behavioral impairments.” J Alzheimers Dis 14(4): 393–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavaguera F, Bolmont T, Crowther RA, Abramowski D, Frank S, Probst A, Fraser G, Stalder AK, Beibel M, Staufenbiel M, Jucker M, Goedert M and Tolnay M (2009). “Transmission and spreading of tauopathy in transgenic mouse brain.” Nat Cell Biol 11(7): 909–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubey M, Chaudhury P, Kabiru H and Shea TB (2008). “Tau inhibits anterograde axonal transport and perturbs stability in growing axonal neurites in part by displacing kinesin cargo: neurofilaments attenuate tau-mediated neurite instability.” Cell Motil Cytoskeleton 65(2): 89–99. [DOI] [PubMed] [Google Scholar]
- Emadi S, Barkhordarian H, Wang MS, Schulz P and Sierks MR (2007). “Isolation of a human single chain antibody fragment against oligomeric alpha-synuclein that inhibits aggregation and prevents alpha-synuclein-induced toxicity.” J Mol Biol 368(4): 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Jacks RL and Diamond MI (2009). “Propagation of tau misfolding from the outside to the inside of a cell.” J Biol Chem 284(19): 12845–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia-Sierra F, Ghoshal N, Quinn B, Berry RW and Binder LI (2003). “Conformational changes and truncation of tau protein during tangle evolution in Alzheimer’s disease.” J Alzheimers Dis 5(2): 65–77. [DOI] [PubMed] [Google Scholar]
- Ghoshal N, Garcia-Sierra F, Wuu J, Leurgans S, Bennett DA, Berry RW and Binder LI (2002). “Tau conformational changes correspond to impairments of episodic memory in mild cognitive impairment and Alzheimer’s disease.” Exp Neurol 177(2): 475–493. [DOI] [PubMed] [Google Scholar]
- Gomez-Ramos A, Diaz-Hernandez M, Rubio A, Diaz-Hernandez JI, Miras-Portugal MT and Avila J (2009). “Characteristics and consequences of muscarinic receptor activation by tau protein.” Eur Neuropsychopharmacol 19(10): 708–717. [DOI] [PubMed] [Google Scholar]
- Grundman M, Petersen RC, Ferris SH, Thomas RG, Aisen PS, Bennett DA, Foster NL, Jack CR Jr., Galasko DR, Doody R, Kaye J, Sano M, Mohs R, Gauthier S, Kim HT, Jin S, Schultz AN, Schafer K, Mulnard R, van Dyck CH, Mintzer J, Zamrini EY, Cahn-Weiner D and Thal LJ (2004). “Mild cognitive impairment can be distinguished from Alzheimer disease and normal aging for clinical trials.” Arch Neurol 61(1): 59–66. [DOI] [PubMed] [Google Scholar]
- Guillozet AL, Weintraub S, Mash DC and Mesulam MM (2003). “Neurofibrillary tangles, amyloid, and memory in aging and mild cognitive impairment.” Arch Neurol 60(5): 729–736. [DOI] [PubMed] [Google Scholar]
- Hanley JA and McNeil BJ (1982). “The meaning and use of the area under a receiver operating characteristic (ROC) curve.” Radiology 143(1): 29–36. [DOI] [PubMed] [Google Scholar]
- Ittner LM and Gotz J (2011). “Amyloid-beta and tau--a toxic pas de deux in Alzheimer’s disease.” Nat Rev Neurosci 12(2): 65–72. [DOI] [PubMed] [Google Scholar]
- Jin M, Shepardson N, Yang T, Chen G, Walsh D and Selkoe DJ (2011). “Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration.” Proc Natl Acad Sci U S A 108(14): 5819–5824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasturirangan S, Reasoner T, Schulz P, Boddapati S, Emadi S, Valla J and Sierks MR (2013). “Isolation and characterization of antibody fragments selective for specific protein morphologies from nanogram antigen samples.” Biotechnol Prog 29(2): 463–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman SK, Thomas TL, Del Tredici K, Braak H and Diamond MI (2017). “Characterization of tau prion seeding activity and strains from formaldehyde-fixed tissue.” Acta Neuropathol Commun 5(1): 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konzack S, Thies E, Marx A, Mandelkow EM and Mandelkow E (2007). “Swimming against the tide: mobility of the microtubule-associated protein tau in neurons.” J Neurosci 27(37): 9916–9927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F and Gong CX (2008). “Tau exon 10 alternative splicing and tauopathies.” Mol Neurodegener 3: 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda S, Sahara N, Saito Y, Murayama S, Ikai A and Takashima A (2006). “Increased levels of granular tau oligomers: an early sign of brain aging and Alzheimer’s disease.” Neurosci Res 54(3): 197–201. [DOI] [PubMed] [Google Scholar]
- Martin L, Latypova X and Terro F (2011). “Post-translational modifications of tau protein: implications for Alzheimer’s disease.” Neurochem Int 58(4): 458–471. [DOI] [PubMed] [Google Scholar]
- Marx J (2007). “Alzheimer’s disease. A new take on tau.” Science 316(5830): 1416–1417. [DOI] [PubMed] [Google Scholar]
- Meraz-Rios MA, Lira-De Leon KI, Campos-Pena V, De Anda-Hernandez MA and Mena-Lopez R “Tau oligomers and aggregation in Alzheimer’s disease.” J Neurochem 112(6): 1353–1367. [DOI] [PubMed] [Google Scholar]
- Mocanu MM, Nissen A, Eckermann K, Khlistunova I, Biernat J, Drexler D, Petrova O, Schonig K, Bujard H, Mandelkow E, Zhou L, Rune G and Mandelkow EM (2008). “The potential for beta-structure in the repeat domain of tau protein determines aggregation, synaptic decay, neuronal loss, and coassembly with endogenous Tau in inducible mouse models of tauopathy.” J Neurosci 28(3): 737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polydoro M, Acker CM, Duff K, Castillo PE and Davies P (2009). “Age-dependent impairment of cognitive and synaptic function in the htau mouse model of tau pathology.” J Neurosci 29(34): 10741–10749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberson ED, Scearce-Levie K, Palop JJ, Yan F, Cheng IH, Wu T, Gerstein H, Yu GQ and Mucke L (2007). “Reducing endogenous tau ameliorates amyloid beta-induced deficits in an Alzheimer’s disease mouse model.” Science 316(5825): 750–754. [DOI] [PubMed] [Google Scholar]
- Sahara N, Maeda S and Takashima A (2008). “Tau oligomerization: a role for tau aggregation intermediates linked to neurodegeneration.” Curr Alzheimer Res 5(6): 591–598. [DOI] [PubMed] [Google Scholar]
- Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M and Ashe KH (2005). “Tau suppression in a neurodegenerative mouse model improves memory function.” Science 309(5733): 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schonheit B, Zarski R and Ohm TG (2004). “Spatial and temporal relationships between plaques and tangles in Alzheimer-pathology.” Neurobiol Aging 25(6): 697–711. [DOI] [PubMed] [Google Scholar]
- Shaw LM, Vanderstichele H, Knapik-Czajka M, Clark CM, Aisen PS, Petersen RC, Blennow K, Soares H, Simon A, Lewczuk P, Dean R, Siemers E, Potter W, Lee VM and Trojanowski JQ (2009). “Cerebrospinal fluid biomarker signature in Alzheimer’s disease neuroimaging initiative subjects.” Ann Neurol 65(4): 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheets MD, Amersdorfer P, Finnern R, Sargent P, Lindquist E, Schier R, Hemingsen G, Wong C, Gerhart JC and Marks JD (1998). “Efficient construction of a large nonimmune phage antibody library: the production of high-affinity human single-chain antibodies to protein antigens.” Proc Natl Acad Sci U S A 95(11): 6157–6162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandermeeren M, Mercken M, Vanmechelen E, Six J, van de Voorde A, Martin JJ and Cras P (1993). “Detection of tau proteins in normal and Alzheimer’s disease cerebrospinal fluid with a sensitive sandwich enzyme-linked immunosorbent assay.” J Neurochem 61(5): 1828–1834. [DOI] [PubMed] [Google Scholar]
- Wang JZ and Liu F (2008). “Microtubule-associated protein tau in development, degeneration and protection of neurons.” Prog Neurobiol 85(2): 148–175. [DOI] [PubMed] [Google Scholar]
- Williams SM, Schulz P, Rosenberry TL, Caselli RJ and Sierks MR (2017). “Blood-Based Oligomeric and Other Protein Variant Biomarkers to Facilitate Pre-Symptomatic Diagnosis and Staging of Alzheimer’s Disease.” J Alzheimers Dis. [DOI] [PubMed] [Google Scholar]
- Williams SM, Venkataraman L, Tian H, Khan G, Harris BT and Sierks MR (2015). “Novel Atomic Force Microscopy Based Biopanning for Isolation of Morphology Specific Reagents against TDP-43 Variants in Amyotrophic Lateral Sclerosis.” (96): e52584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshiyama Y, Higuchi M, Zhang B, Huang SM, Iwata N, Saido TC, Maeda J, Suhara T, Trojanowski JQ and Lee VM (2007). “Synapse Loss and Microglial Activation Precede Tangles in a P301S Tauopathy Mouse Model.” Neuron 53(3): 337–351. [DOI] [PubMed] [Google Scholar]
- Zempel H, Thies E, Mandelkow E and Mandelkow EM (2010). “Abeta oligomers cause localized Ca(2+) elevation, missorting of endogenous Tau into dendrites, Tau phosphorylation, and destruction of microtubules and spines.” J Neurosci 30(36): 11938–11950. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1: A) Immunohistochemistry staining of tau: Staining with commercially available anti-phosphorylated tau antibody AT8 showed increased presence of phosphorylated tau fibrils in the human AD brain tissue slice compared to the age matched healthy control; B) Western Blot analysis of immunoprecipitated tau: Staining with the anti-phosphorylated tau antibody, AT8 shows presence of high molecular weight tau species in AD and absence of high molecular weight tau species in control.
Supplemental Figure S2: Specificity and sensitivity of anti-tau scfv against sera: a-f) Receiver Operating Curves (ROC) curves for six anti-tau scFvs were analyzed to determine their ability to select tau variants in postmortem-verified AD compared to healthy control sera. Five of the six anti-tau scFvs have high sensitivity and specificity to tau variants in AD. The scFvs b) ADT-2, d) ADT-4 and f) ADT-6 recognized tau variants with 80% sensitivity and 90% specificity, 100% sensitivity and 80% specificity and 90% sensitivity and 80% specificity respectively. g) combined ADT-2,-4,-6 recognized tau variants with 90% sensitivity and 90% specificity with 0.96 AUC.
Supplemental Figure S3: Log Dose-response curves of anti-tau IgGs. SH-SY5Y neuroblastoma cells were treated with AD or control brain derived tau IP and with different concentrations of either a) polyclonal anti-tau antibody PA5-27287 or IgGs b) ADT-1, c ADT-2, d) ADT-3, e) ADT-4 or f) ADT-5 for 12 hours. The cell damage and toxicity were assessed by LDH assay (n=3). LDH values for each antibody were adjusted to a percentage of the AD tau IP+vehicle samples, zeroed to the control tau IP sample, and plotted as log dose-response curves. ADT-1, -4 and -5 inhibited toxicity of AD brain derived tau IP more effectively than a polyclonal anti-tau preparation.