

Summary
Solution-based structural techniques complement high-resolution structural data by providing insight into the oft-missed links between protein structure and function. Here, we present Parallel Chemoselective Profiling, a solution-based structural method for characterizing protein structure and dynamics. Our method utilizes Deep Mutational Scanning saturation mutagenesis data to install amino acid residues with specific chemistries at defined positions on the solvent exposed surface of a protein. Differences in the extent of labeling of installed mutant residues are quantified using targeted mass spectrometry, reporting on each residue’s local environment and structural dynamics. Using our method, we studied how conformation-selective, ATP-competitive inhibitors affect the local and global structure and dynamics of full-length Src kinase. Our results highlight how parallel chemoselective profiling can be used to study a dynamic multi-domain protein, and suggest that our method will be a useful addition to the relatively small toolkit of existing protein footprinting techniques.
Graphical Abstract
eTOC burb
Potter et al. present a solution-based method that leverages saturation mutagenesis data and targeted mass spectrometry to characterize the structure and dynamics of proteins. They demonstrate its utility by characterizing the structural perturbations induced by ATP-competitive, conformation selective inhibitors in full-length Src.
Introduction
Structural characterization techniques are essential tools for studying protein function. Methods such as x-ray crystallography and cryogenic electron microscopy have become standard techniques for providing high resolution protein structure. Despite the general utility of these methods, they only capture part of the picture and are best complemented by techniques that characterize dynamics in solution. The ability to assess protein dynamics in solution yields functional insight beyond static structures and provides information about regions of proteins that cannot be characterized using traditional structural methods. A subset of mass spectrometry-based structural characterization techniques—collectively referred to as protein footprinting methods—profile the solvent exposed surfaces of proteins. These methods take advantage of a relatively simple concept, that the relative solvent accessibility of protein surfaces is a proxy for structure and dynamics. To measure accessibility and report on local residue environment, the solvent exposed residues of a protein are labeled with a chemical modification that can be quantified with downstream analytical methods. Comparative analysis of protein variants or ligand-bound protein complexes can reveal the influence of such perturbations. Advances in mass spectrometer hardware and informatics software have made bottom-up proteomic workflows appealing for detecting and quantifying modified proteins. Two methods that utilize a proteomics-style approach, Hydrogen-Deuterium Exchange mass spectrometry (HDX-MS) and Fast Photochemical Oxidation of Proteins (FPOP) have been developed and employed successfully to map epitope binding sites and characterize the conformational dynamics of proteins, among other applications (Baerga-Ortiz et al., 2002; Chea & Jones, 2018; Hodkinson et al., 2009). These methods, however, rely on relatively specialized liquid chromatography (LC) equipment, which limits their general applicability (Table S1).
Here, we describe the development of a simple and general method that can report on the solvent accessibility of individual residues within a protein of interest (Figure 1). Integral to our method is the use of Deep Mutational Scanning (DMS) data to inform the generation of multiple single amino acid variants of a protein of interest with desired sidechain chemistries at specific locations. Chemoselective labeling of a pool of DMS-guided protein variants under comparative conditions is leveraged to report on protein local environment and dynamics and a Parallel Reaction Monitoring (PRM) assay is used to provide residue-level quantification by liquid chromatography tandem mass spectrometry (LC-MS/MS). Our “Parallel Chemoselective Profiling” method allows rapid characterization of dynamic protein structure under multiple conditions and does not require the use of highly specialized equipment beyond a LC and MS capable of acquiring high resolution tandem mass spectra.
Figure 1. General Schematic for Parallel Chemoselective Profiling.
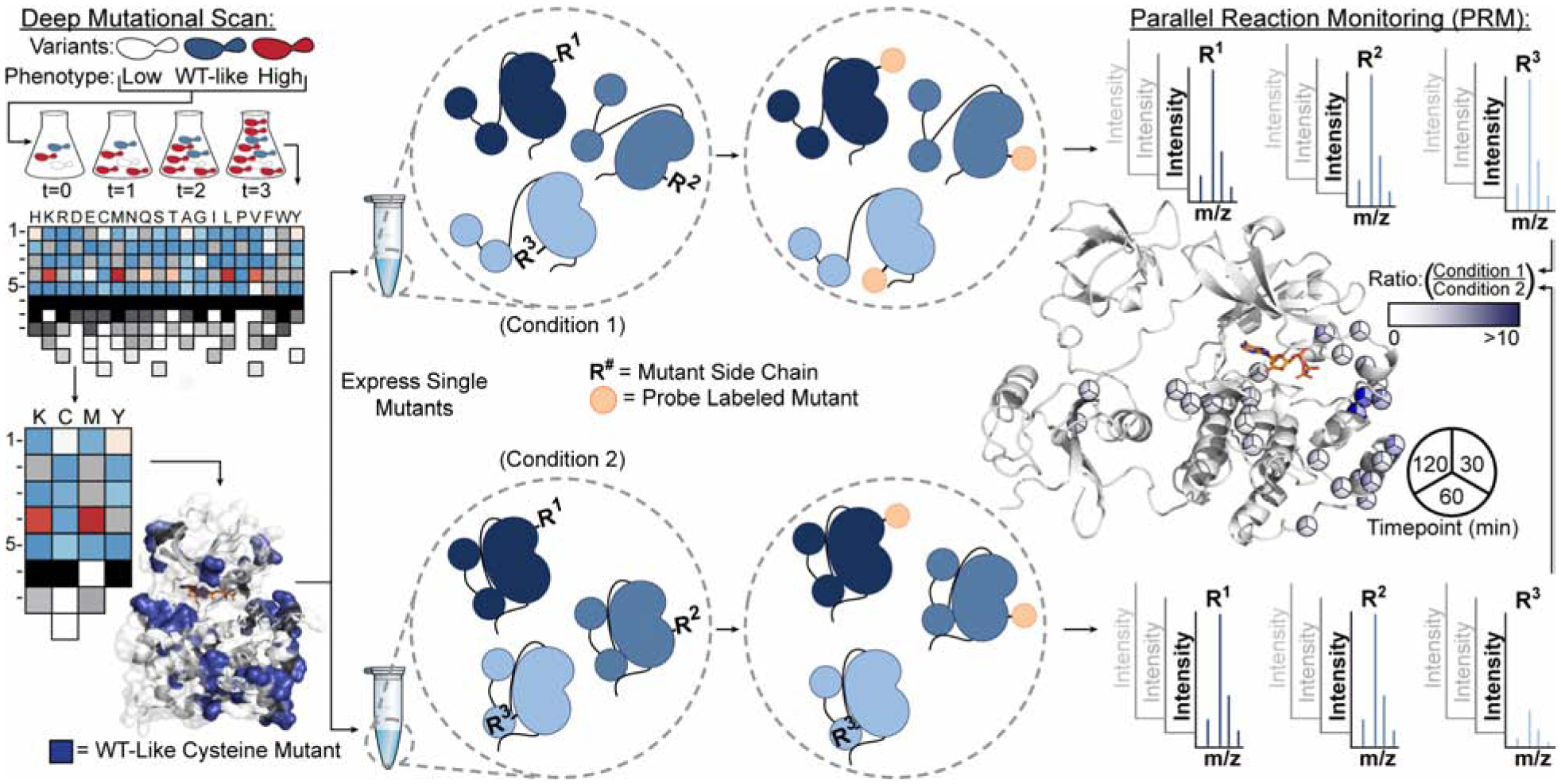
Deep Mutational Scanning is used to determine positions where chemically reactive reporter amino acids can be installed on a protein of interest without perturbing protein function. Individual mutants are expressed and purified in a pooled format and are treated under comparative conditions in vitro. Following treatment, solvent exposed mutant residues are labeled with a chemoselective reagent. Unreacted mutant residues are then capped under denaturing conditions, protein is digested, and labeled residues are quantified using Parallel Reaction Monitoring targeted mass spectrometry. Ratios of labeled residues are compared across timepoints or labeling reagent conditions, which can be visualized and used to infer changes to the local environment of each mutant residue.
To demonstrate the utility of parallel chemoselective profiling, we studied how ATP-binding site occupancy affects the local and global structural dynamics of the tyrosine kinase Src. Using our method, we performed an analysis of how stabilizing the ATP-binding site of full-length Src in two different inactive conformations with inhibitors affects its local and global structure and dynamics. In addition to observing expected changes in local and global conformation, our chemoselective profiling results revealed what appears to be an extended allosteric network within the kinase domain of full-length Src, which links the ATP-binding site to a region on the C-terminal lobe that is more than 20 Å away. These observations corroborate results from HDX-MS experiments that we performed in parallel and elaborate on previously reported NMR studies (Tong, et al. 2017). Together, our findings support the notion of an extended allosteric network within the kinase domain of full-length Src and highlight the utility of parallel chemoselective profiling for characterizing protein structure and dynamics.
Results
DMS data as a resource for identifying mutants appropriate for chemoselective profiling
DMS has emerged as a powerful tool to study the functional effects of thousands of single and multi-amino acid variants in a single experiment (Fowler & Fields, 2014). In contrast with traditional single amino acid-scanning methods, DMS experiments can be used to quantify the effects of all possible amino acid substitutions at each position in a protein. As this field of protein science grows, public repositories—including MaveDB—have been established to distribute data collected from multiplexed assays of variant effects. We posit that these datasets provide a rich source of information which can be leveraged to identify sets of mutants that function similarly to the wild-type (WT) protein but have sidechain chemistries at predefined locations that can be used to report changes in local environment.
We analyzed the DMS datasets of four proteins—ubiquitin, PTEN, Src and BRCA1—that we felt are representative candidates for the application of chemoselective profiling and for which high resolution structural data is available (Table 1). Ubiquitin, PTEN, Src and BRCA1 span a range of molecular weights and vary from single domain to multi-domain architectures. Importantly, these four proteins are representative of a range of biological functions and diverse parameters were used to evaluate the fitness of their variants for the generation of DMS datasets. Ubiquitin is a small, single domain protein that serves as a multi-functional post-translational modification. A yeast growth assay was used to evaluate how mutations affect ubiquitin structure and function. Phosphatase and tensin homolog (PTEN) is a multi-domain phosphoinositide phosphatase. Relative cellular abundance was the parameter used to assess the effects of PTEN mutations. The tyrosine kinase Src is a multi-domain protein that is subject to multiple layers of regulation. Src mutations were scored based on their relative effect on kinase activity. Breast cancer type 1 susceptibility protein (BRCA1) is a tumor suppressor and E3 ubiquitin-protein ligase. While the effects of mutations to the exons encoding the RING and BRCT domains were assessed in HAP1 cells, we only analyzed BCRT domain variants given the more extensive structural characterization of this region. All the DMS experiments we analyzed were performed independently using different assays and analytical techniques, mitigating the chances of a systematic experimental bias.
Table 1:
Large-scale mutagenesis datasets used in this study.
| Data set | Variant length (#AA) | Mutagenized Residues* | Number of Mutations | Mutational completeness** (%) | Selected phenotype | Citation |
|---|---|---|---|---|---|---|
| Ubiquitin | 76 | 2–76 | 4,800 | 99 | Yeast growth | Roscoe, et al. 2013 |
| PTEN | 403 | 14–351 | 4,112 | 54 | Protein abundance | Matreyek, et al. 2018 |
| SRC | 536 | 270–519 | 3,506 | 73 | Kinase activity | Ahler, et al. 2019 |
| BRCA1 | 1,863 | 1,649–1,859 | 1,317 | 36 | HAP1 cell growth | Findlay, et al. 2018 |
Tolerability and solvent accessibility of Cys substitutions
In the first iteration of our chemoselective profiling method, we analyzed the Cys substitutions contained in the DMS saturation mutagenesis datasets for ubiquitin, PTEN, Src and BRCA1 (Table 1). We chose Cys due to the availability of numerous electrophilic reagents that can be used to selectively label this residue. We extracted the mutational effect scores for the ubiquitin, PTEN, Src and BRCA1 datasets from the original publications. In each of these studies, variants were classified as wild-type like (WT-like) or not wild=type like (Not WT-like) by comparing each variant’s functional score to the distribution of synonymous variants. Specifically, WT-like variants were defined as those variants whose scores were within the distribution of scores of variants with mutant DNA sequences but WT protein sequence. We used these classifications without alteration. Other variants did not have reported scores (Not in DMS). We then mapped these classifications onto the crystal structure of each protein (Figure 2A–D). We quantified each protein’s tolerance of Cys mutations as the proportion of WT-like mutants to the total number of Cys mutants with WT-like and not WT-like classifications and found that in all cases, more than half of all Cys mutants observed were WT-like (Figure 2E,G,I,K). We then used the PyMOL script FindSurfaceResidues to identify the solvent exposed sidechains in each crystal structure. In all cases, a majority of WT-like Cys mutants are located at amino acid positions that have solvent exposed sidechains. Taken together, these analyses suggest that Cys mutations are well tolerated at positions that are generally solvent accessible. Therefore, Cys appears to be a promising choice for developing our chemoselective profiling methodology.
Figure 2. Tolerance and Solvent Accessibility of Cys Substitutions.
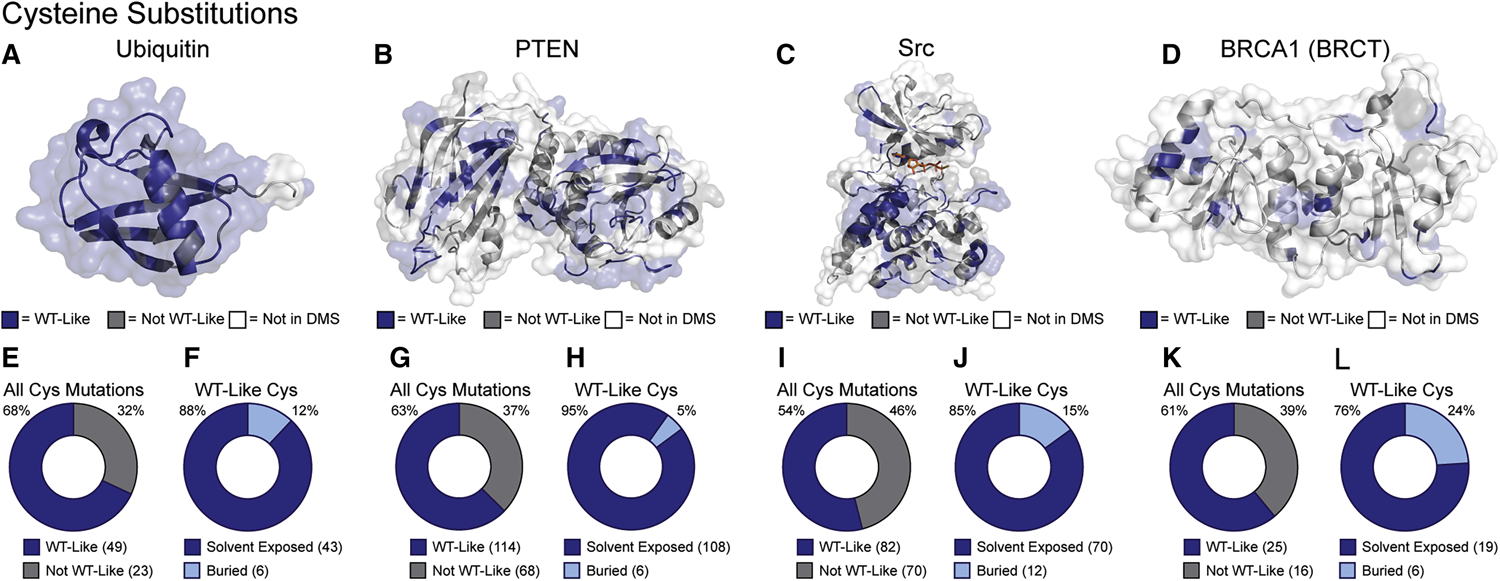
(A-D) Mutational effect scores for Cys substitutions (Table 1) mapped on the crystal structures of ubiquitin, PTEN, Src kinase domain, and BRCA1 BRCT domain (PDB: 1UBQ, 1D5R, 2SRC and 1T29, respectively). (E,G,I,K) Fractions of all Cys mutants classified as WT-like. (F,H,J,L) Fraction of WT-like Cys mutants whose endogenous amino acid contains a solvent-exposed sidechain.
Creation of a Src Cys mutant library for chemoselective profiling
To demonstrate the utility of our method, we chose to apply it to study the conformational dynamics of full-length Src (Src FL). We chose Src as a test case for several reasons. Src has been studied for decades, which has helped define the roles of the SH2 and SH3 domains as allosteric regulators of Src’s phosphotransferase activity (Boggon & Eck, 2004). Additionally, a number of well-characterized point mutants, truncation constructs and small molecule tools for perturbing Src conformational dynamics are available. From a DMS performed on the kinase domain of Src in the context of the full-length enzyme (Figure 3A), which contains 3,506 single amino acid variants, we extracted the mutational effect scores for all Cys mutants (Figure 3B). Of these 152 Cys mutants, 82 of them were classified as WT-like, indicating that Cys mutations at these sites do not significantly affect the catalytic activity of Src FL. From this information, we infer that these WT-like mutations do not disrupt the catalytic machinery or interdomain interactions of Src FL, and therefore we selected these mutants to move forward with for developing our method.
Figure 3. Parallel Reaction Monitoring Assay Development.
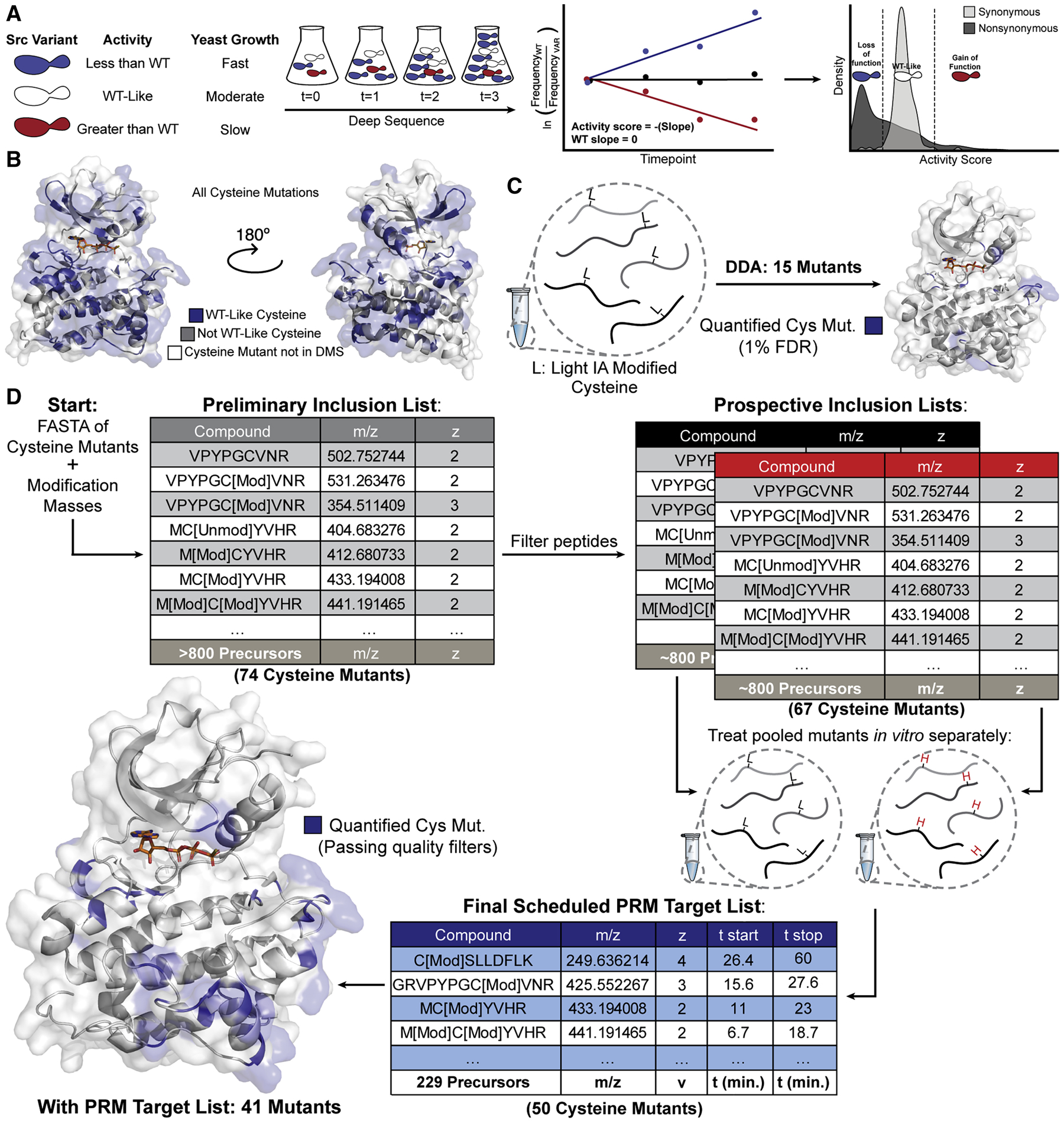
(A) Schematic of the yeast growth-based DMS of Src’s kinase domain (Ahler et al. 2019). (B) Binned activity scores for all Cys substitutions mapped onto Src’s kinase domain (PDB: 3DQW). (C) Initial attempt to identify Cys mutants without enrichment or targeting yielded 15 mutant IDs with mutant peptide FDR<1%. (D) Schematic of PRM assay development. Candidate precursors were identified for all mutant Cys residues by inputting mutant FASTA sequences and modification masses of heavy and light IA into Skyline. Precursors were filtered by removing potentially problematic peptides resulting in prospective inclusion lists for each mass label . Each prospective DDA inclusion list was run in triplicate and resulting spectra were aggregated using Skyline to generate a spectral library and a corresponding Scheduled PRM Target List.
We employed an inverse PCR strategy to generate individual plasmids for each WT-like Cys mutant in mammalian expression vectors (Figure S1A,B). This approach yielded 74 of the 82 possible point mutants in the first pass without optimization. We verified the expression of each construct individually using western blot analysis of transiently transfected HEK293T cells (data not shown). DNA concentrations of the 74 mutant constructs were then normalized and plasmids were pooled and transiently transfected into suspension 293F cells. Resulting recombinant Src FL Cys mutant proteins were dephosphorylated in vitro with the phosphatase YopH and purified as a pool of mutants—hereafter referred to as the Src Cysteine Library (SrcCysLib) (Figure S1C,D).
Targeted MS allows quantification of Cys mutant peptides
To generate alkylated, Cys-containing peptides for mass spectrometric analysis, we prepared samples of SrcCysLib under denaturing conditions with a concentration of iodoacetamide (IA) that should yield quantitative alkylation of all Cys residues present. We first attempted to identify the labeled mutant peptides using a shotgun proteomic workflow operating the MS in Data-Dependent Acquisition (DDA) mode, however this yielded few mutant peptide identifications (Figure 3C). Given the low number of Cys mutants identified from the SrcCysLib it became apparent that our parallel chemoselective profiling method would not be amenable to a DDA workflow. We reasoned that in the ideal case, the SrcCysLib contains a pool of mutants expressed in equimolar ratios relative to one another. Therefore, each Src Cys mutant peptide is diluted with 73 copies of the WT Src (Srcwt) peptide. Perfect chromatographic separation of all peptides is not achieved with the run length and consequent organic gradient needed to perform the number of analyses required for this method in a practical amount of LC-MS/MS run time. Therefore, multiple abundant Srcwt peptide analytes, including all non-Cys-containing peptides, compete with the mutant Cys-containing peptides for ionization and identification. This issue is exacerbated by the fact that we anticipate sub-stoichiometric alkylation regimes are required for our profiling experiments. Because we wanted to accurately quantify differentially labeled Cys peptides despite the challenges of low stoichiometry of labeling and the presence of co-eluting Srcwt peptides, we pursued a targeted proteomic workflow.
We implemented a PRM assay to allow rapid quantification of our SrcCysLib peptides (Figure 3D). We began by constructing a preliminary inclusion list using the FASTA sequence of the 74 individual Src mutants with variable modifications specified for oxidation of Met and the products of IA (light) and isotopically-labeled IA (heavy) alkylation on Cys. The list was refined to ~800 precursors by removing short tryptic peptides (<6 amino acids), allowing for up to two missed tryptic cleavages and charge states of +2 through +5, resulting in a list of precursors for 67 mutants for each mass label. We prepared samples by separately alkylating Cys residues under denaturing conditions with light or heavy IA and acquired fragmentation spectra of these Src mutants using our targeted precursor inclusion lists. These results were aggregated in Skyline to produce a list consisting of precursors for each mass label (light and heavy) for 50 Cys mutants. Pierce Peptide Retention Time Standards (PRTC, iRT) were used to schedule the PRM method, reducing the number of targeted precursors at any given time, and increasing the quality of quantification (Figure S2). This final scheduled PRM target list with 229 members allowed us to robustly quantify 41 Cys mutants, 4 Srcwt Cys-containing peptides, 14 Srcwt non-Cys-containing standards and the 15 iRT peptides .
Biochemical modulation and characterization of Src global conformation
We applied our new workflow to map the kinase domain of Src bound to two different conformation-selective, ATP-competitive inhibitors. Numerous co-crystal structures of inhibitor-bound kinase complexes have revealed that inhibitors can stabilize large conformational movements in regions lining the ATP-binding pocket (Tong & Seeliger, 2015; Ung, et al. 2018; Zhao, et al. 2014). In particular, certain pharmacophores promote large displacements of helix αC—which contains a conserved Glu residue that forms a salt bridge with the catalytic Lys residue—or the DFG-motif within the activation loop, from their active conformations in a number of kinases (Figure S3A). Furthermore, we and others have demonstrated that conformation-selective inhibitors are capable of inducing large structural changes that are distal to the ATP-binding site in multi-domain kinases (Aguis et al. 2019; Ahler et al. 2019; Chakraborty et al. 2019; Foda, et al. 2015; Krishnamurty et al. 2013; Kung & Jura, 2016; Kwarcinski et al. 2016; Leonard et al. 2014; Register et al. 2014; Tong et al. 2017). For example, conformation-selective inhibitors modulate the level of SH2 and SH3 domain engagement with the kinase domain of Src by exploiting the allosteric networks that are utilized to control catalytic activity. We felt that a comparison of Cys alkylation rates between two conformation-selective inhibitor-bound Src complexes would provide an ideal test case for determining the utility of parallel chemoselective profiling for mapping protein structure.
For our study, we selected conformation-selective inhibitors 1 and 2, which contain pharmacophores that stabilize the helix αC-out and DFG-out conformations of the ATP-binding site of Src, respectively (Figure 3A). While we have not obtained co-crystal structures of 1 or 2 bound to Src, their interactions with the ATP-binding site can be inferred from structures of electrophilic analogs of these inhibitors complexed to a Cys-containing mutant of Src (Figure S3A). Inhibitor 1 contains a 4-phenoxyphenyl substituent at the C-3 position of the pyrazolopyrimidine scaffold which occupies the hydrophobic pocket created by the outward rotation of helix αC. This promotes the helix αC-out conformation of Src and stabilizes a closed global conformation that is characterized by autoinhibitory engagement of the SH2 and SH3 domains with the backside of the kinase domain. Inhibitor 2 contains a 3-trifluoromethylbenzamide substituent at the C-3 position—connected to the pyrazolopyrimidine scaffold through a rigid (2-methylphenyl)-propargyl linker—that occupies the hydrophobic pocket created by the ~180° “flip” of the DFG-motif at the base of the activation loop from an active conformation. The 3-trifluoromethylbenzamide substituent stabilizes the DFG-out conformation of Src and promotes an open global conformation that is characterized by SH2 and SH3 domains that are disengaged from the kinase domain. Because both inhibitors share an identical scaffold and only vary in the pharmacophore responsible for stabilizing different ATP-binding site conformations, most observed differences in our profiling experiments will likely reflect structural changes in the kinase domain.
Prior to performing parallel chemoselective profiling experiments with inhibitor-bound complexes, we benchmarked the effects of 1 and 2 on the global conformation of Src. First, we characterized how each inhibitor modulates the global conformation of Src with a limited proteolysis assay using a Src construct consisting of the SH3, SH2 and kinase domains (Src 3D). Previously, it has been shown that the protease thermolysin can cleave the linker between the SH2 and kinase domains of Src (SH2-KD-linker), with the rate of cleavage–reported as a half-life–correlating to Src’s global conformation (Agius et al. 2019; Fang et al. 2020; MacAuley & Cooper 1989). Consistent with 2 stabilizing the DFG-out conformation of Src and promoting a more open global conformation, we found that thermolysin more rapidly cleaved the SH2-KD-linker of the inhibitor 2-Src complex than of apo Src (DMSO) (Figure 4A, S4B). We observed the opposite trend for 1, with the inhibitor 1-Src complex showing less susceptibility to cleavage by thermolysin than apo Src. Thus, 1 and 2 promote the expected global conformations of Src.
Figure 4. Biochemical Characterization of Inhibitor-Bound Src 3D Complexes.
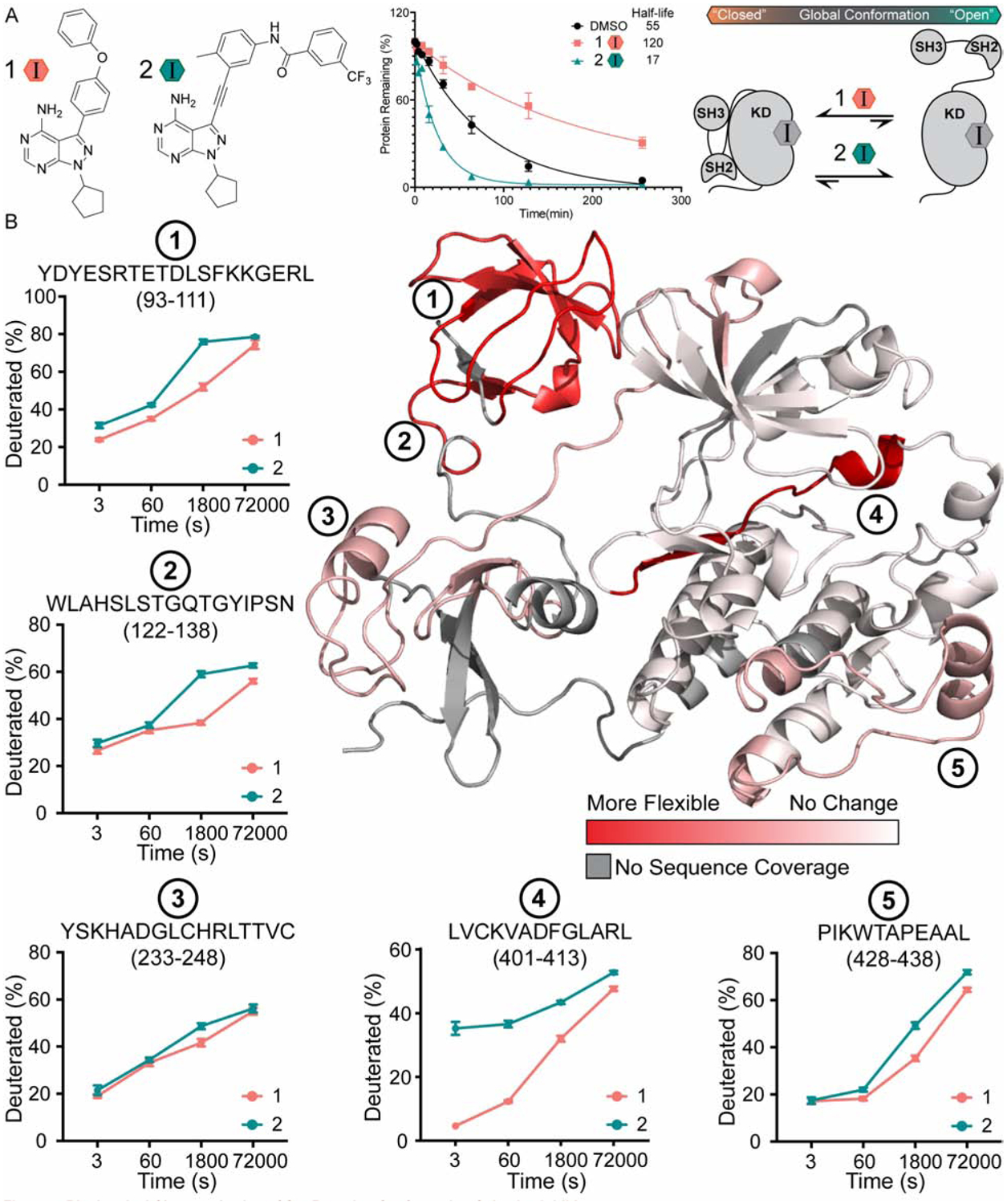
(A) Structures of inhibitors 1 and 2 (Krishnamurty et al., 2013; Fang et al., 2020). Quantification of the limited proteolysis experiments performed with inhibitor-bound complexes of Src 3D. Points represent mean ± SEM. (B) HDX-MS analysis of inhibitor-bound Src 3D complexes. Deuteration differences between the inhibitor 1- and 2-Src 3D complexes are plotted on the crystal structure of Src 3D (PDB: 2SRC) from no change (white) to the largest change (red). Deuterium uptake plots for peptides at regions of interest are shown on the periphery. Points represent mean ± SEM.
To further benchmark how 1 and 2 influence the structure of Src, we performed HDX-MS experiments with the inhibitor 1- and 2-Src 3D complexes. To ensure that quantitively-bound inhibitor-Src 3D complexes were formed, we used saturating concentrations of both inhibitors. Following generation of each inhibitor-Src 3D complex, we initiated deuterium exchange by adding buffered D2O (90% deuterated). At specific timepoints exchange reactions were quenched by lowering the pH to 2.5 and immediately flash freezing samples to lock exchanged deuterium in place and denature the protein. Samples were then processed using standard protocols (see “Methods”). After filtering out peptides showing weak signal or partial overlap, 32 unique peptic peptides remained, which cover 73% of Src 3D’s sequence. In determining which peptic peptides demonstrate a difference between the two inhibitor-bound complexes, we excluded peptides with overlapping sequences that demonstrate different levels of deuteration to reduce the possibility of interpreting noise. This yielded a relatively conservative set of peptides for further analysis (Supplemental file). A summary of the deuteration exchange kinetics from the two inhibitor-bound Src 3D complexes are represented on the crystal structure shown in Figure 4B.
Comparison of the HDX-MS results for the inhibitor 1- and 2-Src 3D complexes show that there are five regions, represented by peptides 1 through 5 in Figure 4B, that demonstrate the largest differences in deuterium exchange kinetics. Consistent with 2 generally increasing the solvent accessibility of Src 3D, we found that all five of these regions underwent faster exchange kinetics in the inhibitor 2-Src 3D complex relative to the inhibitor 1-Src 3D complex. The largest overall difference in exchange kinetics was observed for the peptide that contains the DFG-motif in the activation loop (peptide 4), which is consistent with 2’s stabilization of an inactive form of the ATP-binding site where the DFG-motif is “flipped” ~180° relative to the active conformation (DFG-out conformation). Additionally, our HDX-MS experiments recapitulate the observations made using limited proteolysis experiments regarding the global conformation of Src 3D (Figure 4B). We found that peptides 1, 2 and 3, which are located on the regulatory SH2 and SH3 domains, demonstrated faster exchange kinetics in the inhibitor 2-Src 3D complex. This result is congruent with 2 promoting a more open global conformation of Src 3D. Interestingly, deuteration level differences on the complementary surfaces of the kinase domain that directly contact the SH2 and SH3 domains were not observed, possibly because changes in this region occur outside of the temporal range sampled in the HDX-MS experiments (Hamuro, 2017). Finally, in the inhibitor 2-Src 3D complex, we also observed an unexpected increase in deuteration rate for the region represented by peptide 5, which is located at the bottom of the C-terminal lobe of the kinase domain. While the overall measured difference in exchange rate in this region is modest, this structural perturbation, which is likely the result of a long-range allosteric network, is an intriguing result worthy of further inquiry. Together, these HDX-MS experiments provide a basis for comparison with our parallel chemoselective profiling method.
Parallel chemoselective profiling of inhibitor-bound Src complexes
We next performed analogous mapping experiments using our parallel chemoselective profiling workflow. Because our method can be performed with lower concentrations of input protein relative to HDX-MS, these experiments were performed with Src FL, which contains Src’s membrane-interacting N-terminus, rather than Src 3D. To gain residue-level insight into the changes in surface accessibility and dynamics of Src’s kinase domain upon complexation with 1 and 2, we performed chemoselective profiling experiments using our SrcCysLib (Figure 5). In a similar manner that we prepared inhibitor-Src 3D samples for HDX-MS, we separately formed quantitative inhibitor-bound Src FL complexes by incubating SrcCysLib with saturating concentrations of 1 or 2, followed by the addition of three different concentrations of heavy IA. Heavy IA concentrations were selected based on their likelihoods of providing sub-stoichiometric labeling over the timecourse of the experiment, which we reasoned would allow us to measure differences in Cys alkylation rates over multiple timepoints. We then removed and quenched aliquots of each reaction after 30, 60, or 120 min after the addition of heavy IA. Quenching was achieved by denaturing SrcCysLib in guanidinium buffer containing a sufficiently high concentration of light IA to outcompete any background labeling from residual heavy IA. We then heated reactions to 37 °C and digested with mass spectrometric grade trypsin overnight. Following peptide desalting, peptides were analyzed using our PRM workflow and spectra were imported directly into Skyline for quantification.
Figure 5. Parallel Chemoselective Profiling of Inhibitor-Bound Src FL Complexes.
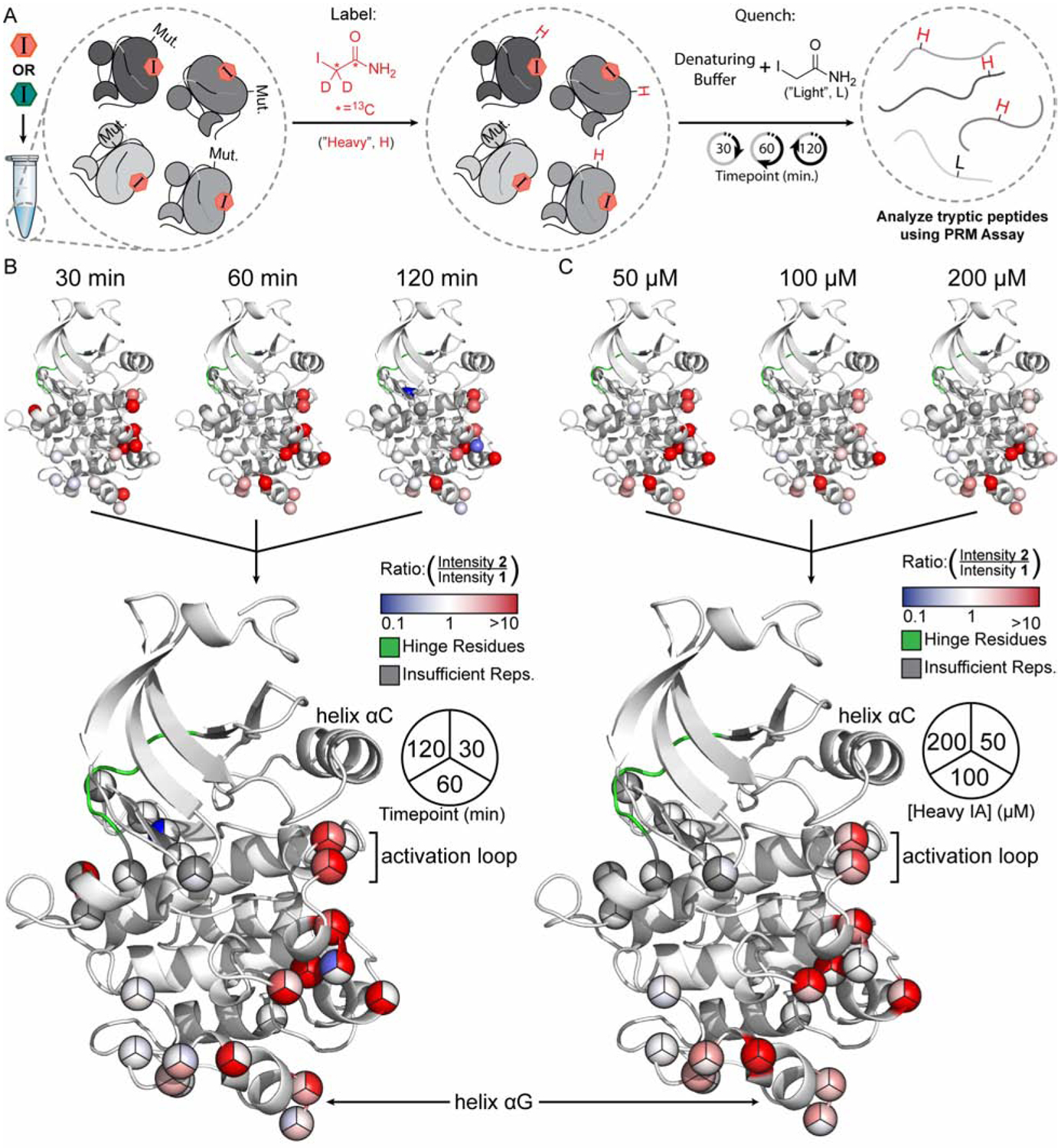
(A) Schematic of the Parallel Chemoselective Profiling experiment. Conformation-selective inhibitors 1 or 2 were added to the SrcCysLib at saturating concentrations to form inhibitor-Src FL complexes. After complexation, heavy IA was added to a final concentration of 50, 100 or 200 μM. At the indicated timepoints, aliquots of the reaction were removed and quenched. Protein was then digested and peptides were analyzed using our PRM assay. (B-C) 2-SrcCysLib/1-SrcCysLib intensity ratios from select conditions (B: 50 μM heavy IA timecourse; C: 60 min timepoint for each heavy IA concentration) mapped on the kinase domain of Src (residues 264–536, human numbering) extracted from PDB ID: 2SRC. Crystal structures are superimposed to generate pie charts.
For our analysis, we only considered residues that had been quantified in two-thirds of our experimental samples, yielding a relatively conservative set of peptides that were robustly quantified in most samples (see “Methods”). This allowed us to utilize 23 Cys mutants and 4 WT Cys residues to probe the structural differences between the inhibitor 1- and 2-SrcCysLib complexes. These Cys residues line a significant portion of the ATP-binding site, including multiple residues on the activation loop, which allowed us to probe local structural differences of the inhibitor-bound Src complexes. Additionally, we quantified a number of residues on the C-terminal lobe of the kinase domain, including in positions that significantly overlap with the region that demonstrated unexpected dynamics in our HDX-MS experiments. While our coverage of residues that line the SH2/SH3 regulatory interface in the kinase domain were poor, we were able to quantify a WT Cys residue that lines the C-terminal tail-binding interface of the SH2 domain. Finally, these are several mutant Cys residues on the kinase domain that are not solvent exposed. These positions served as controls for interpreting our differential alkylation results.
To visualize our parallel chemoselective profiling data, we first generated (2-SrcCysLib/1-SrcCysLib) intensity ratios of the heavy IA labeled peptides for each Cys residue at each timepoint and concentration of heavy IA, which provided nine conditions in total. We then mapped these ratios as a heatmap onto the kinase domain from a crystal structure of Src (Figure 5B–C, S4). To facilitate data interpretation, we superimposed the crystal structures from each condition and represented the heatmaps for each residue as a pie chart. The trends observed in our chemoselective profiling experiments are highly consistent with our HDX-MS results. First, for almost all Cys residues that we observe a difference in labeling between the two inhibitor-bound complexes, the (2-SrcCysLib/1-SrcCysLib) intensity ratios are >1. This is consistent with 2 increasing the overall solvent accessibility of Src FL. Furthermore, we found that the WT Cys residue located in the C-terminal tail-binding interface of the SH2 domain underwent more rapid labeling in the presence of 2, consistent with 2 promoting an open global conformation of Src FL (Figure S5A). We also found high (2-SrcCysLib/1-SrcCysLib) intensity ratios for a number of Cys mutant residues in the activation loop, indicating an increase in solvent exposure for this region when Src FL is bound to 2. While one of these residues (L413C) overlaps with peptic peptide 4 from our HDX-MS experiments, five of these Cys mutations (E415C, T420C, A421C, R422C and Q423C) are C-terminal to the DFG-motif and lie in a region of the activation loop that is highly solvent exposed and dynamic (Figure S5B). Unlike the DFG-motif-containing peptic peptide, this region of the activation loop was highly deuterated at early timepoints and did not show a difference in deuteration levels in our HDX-MS analysis, likely due to the flexibility and intrinsic solvent exposure of this region. Our chemoselective profiling data shows that flipping of the DFG-motif affects the dynamics of distal parts of the activation loop and highlights how our method can complement HDX-MS experiments.
In addition to characterizing local and global structural differences between inhibitor-bound Src FL complexes, we also observed high (2-SrcCysLib/1-SrcCysLib) intensity ratios in regions of the kinase domain distal to the ATP-binding site. Most notably, we found that 2 led to a dramatic increase in the labeling of mutant Cys residues that lie in an area of the kinase domain’s C-terminal lobe that overlaps with the region represented by peptic peptide 5 from our HDX-MS experiments relative to 1. This region of the C-terminal lobe includes residues R472C, E478C and R479C, which reside on the αG helix. It is intriguing that one of these C-terminal lobe residues (R472C) demonstrates intensity ratios that are comparable to a residue (L413C) that is directly adjacent to the flipped DFG-motif in the activation loop, despite being more than 20 Å away from the ATP-binding site. R472C shows higher (2-SrcCysLib/1-SrcCysLib) intensity ratios than the two residues at the opposite end of the helix αG, suggesting that the solvent accessibility of this region of helix αG is most influenced by ATP-binding site occupancy. Both our HDX-MS and chemoselective profiling results are consistent with a hypothesized allosteric coupling between the ATP-binding site and the helix αG of Src (Tong, et al. 2017), a structural element that is conserved in other eukaryotic protein kinases (Kornev & Taylor, 2015; McClendon et al., 2014; Taylor et al., 2019). Given the spatial connectivity of the residues that show increased labeling in the presence of 2 from the activation loop to helix αG, our profiling method suggests the potential composition of the allosteric network that connects these two spatially separated regions. Our ability to obtain residue level information into what appears to be an extended allosteric network provides complementary insight into an intriguing trend that we observed with HDX-MS.
Application of parallel chemoselective profiling beyond Cys
We anticipate that parallel chemoselective profiling will be amenable to amino acid substitutions beyond Cys. To explore this possibility, we considered other amino acids that can be selectively labeled. Recently, a chemoselective labeling reagent for Met was reported (Li et al., 2017). We analyzed the mutational effect scores of Met substitutions for the same DMS datasets listed in Table 1. We applied the same three bins for Met mutational effect scores that were used when analyzing the Cys mutational effect scores and mapped them to the crystal structures of ubiquitin, PTEN, Src and BRCA1 (Figure 6A–D). We also performed the same tolerance analysis, which revealed trends like those observed for Cys. With the exception of Src, more than half of all Met substitutions surveyed in the DMS data were classified as WT-like (Figure 6E,G,I,K). Additionally, the solvent exposure analysis conducted with PyMOL revealed that a majority of WT-like substitutions are on solvent exposed sidechains (Figure 6F,H,J,L). Interestingly, in the case of BRCA1, Met is especially well tolerated. This observation highlights the importance of selecting a suitable amino acid and chemoselective labeling reagent pair, which maximizes the potential solvent exposed surface available for profiling. Our analysis suggests that Met is also a promising candidate for use in parallel chemoselective profiling.
Figure 6. Parallel Chemoselective Profiling Beyond Cys.
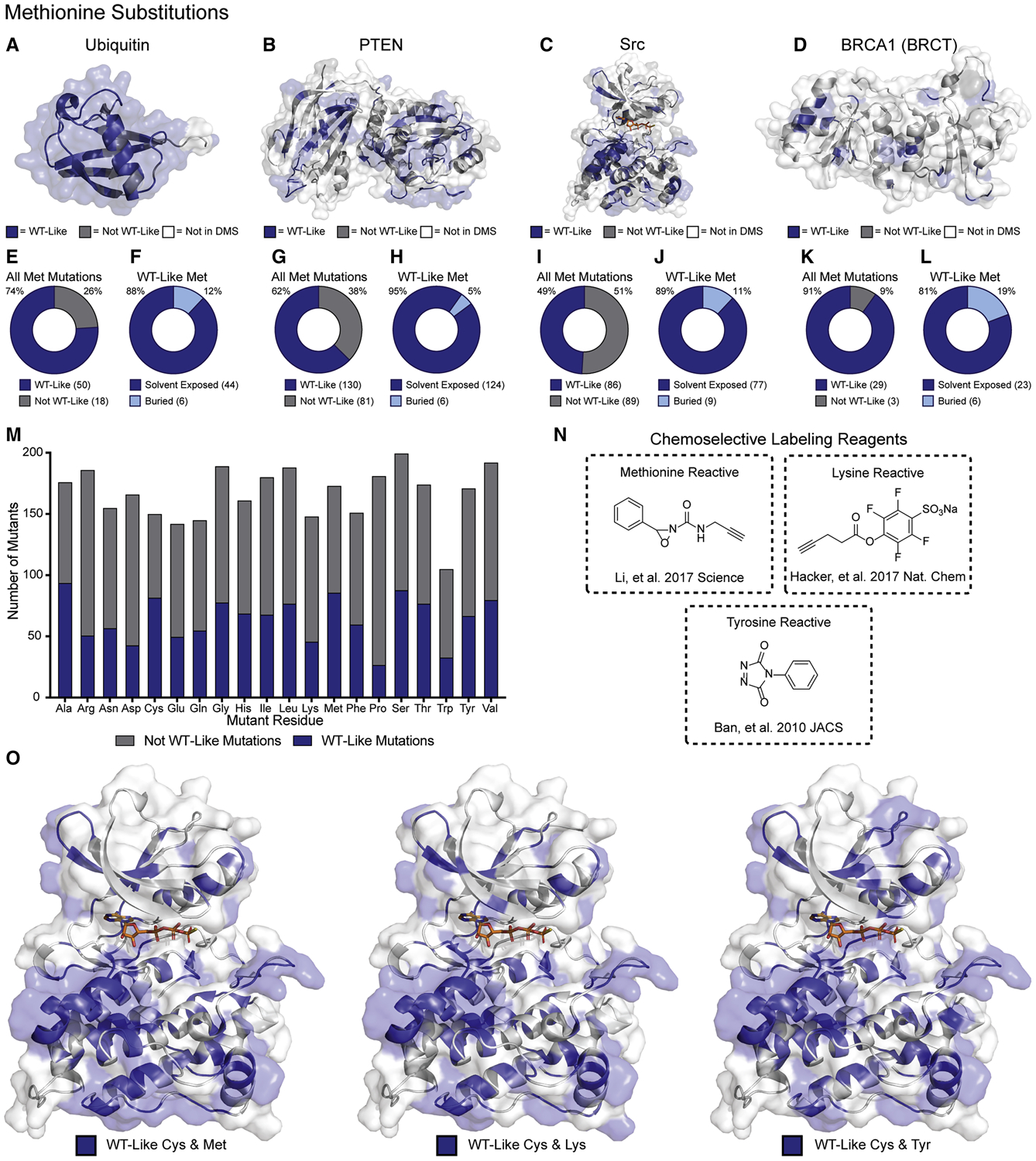
(A-D) Mutational effect scores for Cys substitutions (Table 1) mapped on the crystal structures of ubiquitin, PTEN, Src kinase domain, and BRCA1 BRCT domain (PDB: 1UBQ, 1D5R, 2SRC and 1T29, respectively). (E,G,I,K) Fractions of all Met mutants classified as WT-like. (F,H,J,L) Fraction of all WT-like Met substitutions that are solvent exposed in each crystal structure. (M) All single amino acid substitutions from the DMS of Src kinase domain and the fraction of those that result in WT-like substitutions. (N) Chemoselective labeling reagents for Met, Tyr, and Lys. (O) WT-like Met, Lys or Tyr residues mapped with WT-like Cys residues.
Met does not appear to be a special case. For example, if we consider all 3,506 variants from the Src kinase domain DMS dataset, there are other amino acid substitutions that are well tolerated as indicated by the fraction of each amino acid substitution characterized as WT-like (Figure 6M). Similar ratios of WT-like substitutions relative to the total number of substitutions quantified are observed for the other DMS datasets contained in Table 1 (Figure S6). Among these well-tolerated amino acid substitutions are several examples where chemoselective labeling reagents are available (Figure 6N).
Discussion
In this work we present parallel chemoselective profiling, a solution-based method for studying protein structure and dynamics. Chemoselective profiling leverages DMS data to install amino acids with desired chemistries at defined locations on a protein surface and differential labeling of mutant residues—measured in parallel using targeted mass spectrometry—is used to infer structural changes. Our method has the potential to be generalized to any protein target of interest and, importantly, does not require specialized instrumentation beyond a LC and MS capable of obtaining high-resolution tandem mass spectra. To demonstrate the utility of our method, we applied it to the tyrosine kinase Src. By performing a comparative chemoselective profiling experiment with inhibitor-bound Src complexes, we were able to characterize how ATP-binding site occupancy influences the structure and dynamics of full-length Src. By combining our chemoselective profiling results with complementary HDX-MS experiments, we were able to observe what appears to be an extended allosteric network within the kinase domain of Src FL, linking the ATP-binding site, activation loop and helix αG, which resides more than 20 Å away from the ATP-binding site. This structural feature is conserved amongst eukaryotic protein kinases (Taylor et al., 2012) and is thought to contain inter- and intramolecular regulatory binding interfaces (McClendon et al., 2014), although the exact role of the helix αG has not been explored explicitly for Src.
A majority of our parallel chemoselective profiling data significantly overlapped with the HDX-MS experiments that we performed, allowing us to cross-validate the insight we obtained into how conformation-selective, ATP-competitive inhibitors affect Src’s structure and dynamics. Our experiments highlight the intrinsic complementarity of HDX-MS—which probes backbone amide flexibility—and covalent labeling approaches which probe side-chain solvent accessibility. The lower concentration of input protein required for chemoselective profiling compared to HDX-MS allowed us to profile a library of Src FL constructs—which contain a membrane-interacting N-terminal region—rather than the solubility-optimized truncation variant Src 3D that was used in our HDX-MS experiments and all prior structural characterizations of Src. Furthermore, chemoselective profiling provided residue-level insight into Src’s structure and dynamics in some areas of Src’s kinase domain where HDX-MS showed more modest effects. We did, however, find regions in the kinase domain that we were not able to profile using our method but that were probed with HDX-MS. This leaves room for future improvements in our workflow, which can be tuned for specific applications. Two regions that were particularly under-represented in our chemoselective profiling data were the αF pocket and N-terminal lobe of Src’s kinase domain. We speculate that despite these areas possessing WT-like and solvent exposed Cys mutations that poor coverage can be attributed to unfavorable properties of the tryptic peptides in this region (e.g. high charge state and/or nonoptimal length). One potentially useful avenue for increasing the coverage of these areas would be to redistribute the trypsin cut-sites on peptides that contain WT-like Cys mutations. The addition or elimination of trypsin cut-sites can be guided by DMS data, which can provide suitable Lys and Arg replacements or suggest positions where WT-like Lys or Arg mutations can be introduced. The optimal combination of trypsin cut-sites could then be determined empirically. Similarly, alternative proteases with different cut-site specificity such as GluC or chymotrypsin could be employed. Either of these modifications to the general approach could dramatically increase the coverage of regions of interest that are poorly characterized in first pass experiments.
Another potential strategy for increasing the solvent accessible surface available for profiling is the use of multiple single amino acid-mutant libraries in combination. For example, if we consider the mutational effect scores for Met, Lys and Tyr—which all can be selectively labeled with suitable reagents—in addition to Cys, we can increase the solvent-exposed surface of Src’s kinase domain sampled by WT-like mutants (Figure 6O). For any protein of interest, it should be possible to find complementary combinations of WT-like amino acids that provide maximal coverage of solvent exposed surfaces.
Despite the number of residues available for chemoselective labeling, there are several considerations to make when selecting a residue and labeling reagent pair. First, it is important to analyze the frequency and solvent accessibility of the selected amino acid in the WT protein. For example, if Lys were selected as the residue for a parallel chemoselective profiling experiment and there happens to be a number of solvent exposed Lys residues in the WT protein, it is possible that labeling of these residues may influence the structural dynamics of the protein. This is a critical consideration for any covalent labeling strategy. Having abundant and accessible reactive sidechains on a single protein may complicate the interpretation of a profiling experiment. Importantly, parallel chemoselective profiling can potentially address this issue by “scrubbing” problematic endogenous residues from a protein of interest by installing a chemically inert WT-like mutation at a problematic position. It is worth emphasizing the inherently comparative nature of parallel chemoselective profiling experiments which makes the analysis of WT-like mutants possible. While we expect individual WT-like mutants to behave like the WT protein with respect to structure and function, we cannot exclude the possibility that some mutations that are classified as WT-like in a DMS experiment may exhibit altered structural dynamics. However, given the comparative nature of parallel profiling experiments, we do not envision this being a systematic problem. Additionally, the ability to make parallel measurements on multiple neighboring residues increases our overall confidence in the observations made for the dynamics of a particular structural feature. Taken together, the parallel chemoselctive profiling method presented here introduces a useful addition to the relatively small toolkit available for studying protein structure and dynamics in solution.
STAR Methods
RESOURCE AVAILABILITY
LEAD CONTACT
Further information and requests for resources and reagents should be directed to the Lead Contact, Dustin J. Maly (djmaly@uw.edu).
MATERIALS AVAILABILITY
All plasmids generated in this study are freely available without restriction.
DATA AND CODE AVAILABILITY
This study did not generate large datasets or code.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian cell culture
FreeStyle™ 293F cells were used for production of SrcCysLib protein. Cells were maintained in FreeStyle™ 293 Expression Medium (Gibco) with constant orbital shaking (125 RPM) and an 8% CO2 environment. FreeStyle™ 293F cells are transformed and adapted from female human embryonic kidney cells (Life Technologies). Cells were transfected a 1×106 cells/mL with a 3:1 ratio of PEI Max to plasmid DNA.
METHOD DETAILS
Src WT-like Cysteine Library (SrcCysLib)
Positions of cysteine mutants were informed by the deep mutation scanning data previously reported (Ahler et al., 2019). Briefly, interactions between the regulatory domains of Src and the catalytic kinase domain of Src serve negative regulatory roles. Disruption of these interfaces releases this inhibition and increases Src kinase activity. Site saturation mutagenesis was employed to generate single amino acid variants of Src kinase domain in the context of full-length enzyme (3,506, ~70% of possible mutants were made and assayed). This mutant library was adapted to a yeast growth assay (Kritzer et al. 2018). Once transformed into S. Cerv. samples were taken at different timepoints and deep-sequenced to quantify each mutant. “WT-like” mutations were defined as those mutants with activity scores within two standard deviations of the activity scores for all synonymous wt Src variants.
There are 82 individual WT-like cysteine mutants in the DMS dataset, of which 74 were constructed using an inverse PCR approach with no optimization (Figure S1A). Expression and purification of full-length Src from bacterial expression systems has proven to be inefficient in our hands compared to yields of recombinant protein from truncated variants of Src. To address this, we made several modifications to previously used expression protocols. First, we selected the FreeStyle 293F (ThermoScientific) as our protein production host to take advantage of the endogenous co-factors and chaperones to increase the yield of folded protein. Because the N and C termini of Src are known to be important for regulating the conformational equilibria of Src, we desired recombinant Src cysteine mutants to have near-native termini. To achieve this, we expressed Src cysteine mutants as His6-SUMO* fusions for two reasons. First, expressing proteins as SUMO fusions has been shown to increase fusion protein solubility and expression levels (Butt et al., 2005). Endogenous mammalian SUMO proteases are able to cleave SUMO fusions, so we employed the complementary SUMO protease Ulp1* which, like wild-type Ulp1 cleaves C-terminal to a Gly-Gly motif releasing the fused protein with a “scarless” or native N-terminus (Figure S1C) (Liu et al. 2008).
Mutagenesis primers were designed by centering each primer at the mutagenic position (Figure S1B). The TGC codon was used to encode the cysteine mutation at each position. Selections with Tm 55 °C were selected 5’ and 3’ of each mutation. The combined sequence constituted the forward mutagenic primer. The reverse primer is the reverse complement of the 5’ Tm 55 °C portion of the forward primer.
PCR and thermocycler conditions were as follows:
| 7.5 μL 2X Q5 Master Mix (New England Bioscience) |
| 0.45 μL 10 μM Forward primer |
| 0.45 μL 10 μM Reverse Primer |
| 2 μL 10ng/μL template |
| 5.05 μL H2O |
| Temperature | Time |
|---|---|
| 98 °C | 20 sec |
| 98 °C | 20 sec |
| 55 °C | 30 sec |
| 72 °C | 1 min/kb |
| Go to Step 2 | X10 |
| 72 °C | 5 min |
After amplification, digest template by adding 1 μL FastDigest Dpn1 (ThermoScientific) to each reaction, incubate at 37 °C for 30 min. Transform 1 μL reaction mixture into 10 μL high efficiency NEB5α using heat shock, 120 μL SOC, 30 min outgrowth at 37 °C, 300 RPM shaking. The entire reaction was plated on LB+Carb. plates and incubated overnight at 37 °C. The full gene was sequenced and point mutations were observed for 74 of the 82 constructs attempted.
Transfection and purification of SrcCysLib
With the 74 successful constructs in hand, DNA concentration was normalized to 6.66 μg per construct and transfected into 500 mL Freestyle 293F cells (1×106 cells/mL) using polyethyleneimine ‘MAX’ (MW 40,000, PEI; Polysciences, Inc.) at 3 μL PEI/1 μg DNA. After 48 h outgrowth, cells were pelleted and stored at −80 °C. Cells were thawed on ice and lysed in lysis buffer containing protease inhibitors (Pierce, A32963) as previously described (Ahler, et al. 2019). The only modification to this procedure was the use of ULP1* (Brigham, et al. 2013) instead of ULP1. 20 μL aliquots of each step of the protein purification were added to 10 μL 3X SDS-PAGE loading buffer. Western blot analysis was performed on these samples using phospho- and non-phospho activation loop antibodies (Figure S1D).
Solvent exposure analysis of Ubiquitin, PTEN, Src (kinase domain) and BRCA1 (BRCT domain)
Crystal structures for each protein were imported into PyMOL for analysis. The script FindSurfaceResidues was downloaded from the PyMOL Wiki (https://pymolwiki.org/index.php/FindSurfaceResidues) and used with its default settings (2.5 Ang2 cutoff). After residues were identified by the script, they were visually inspected to ensure that the sidechain and not the backbone of each residue were solvent exposed. Numbers of residues were tabulated and reported as fractions of the total length of crystalized protein.
HDX-MS of 3D Src in presence of conformation selective inhibitors
0.2 mg/mL 3D Src was preincubated with 20 μM conformation selective inhibitor 1 or 2 in protein dilution buffer (50 mM HEPES, pH 7.8, 150 mM NaCl, 1 mM DTT, 5% glycerol) at room temperature for 30 min to prepare a kinase-ligand complex. 10 μL of this was then added to 90 μL of buffered D2O (prepared 5 mL with 4.5 mL of D20 and 0.5 mL of 10X protein dilution buffer and 0.2 μg/mL of Gu-1-Fibrino peptide) to initiate deuteration at 22 °C. Deuterium exchange was quenched after 3 s, 1 min, 30 min, and 20 h by adding the reaction to 100 μL of ice-cold quench buffer (0.2% formic acid, 8 M Urea, 0.1% trifluoroacetic acid) for a final pH of 2.5. All time points for each inhibitor were collected in triplicate. Samples were immediately frozen in a dry ice/ethanol and stored at −80 °C. Undeuterated samples were prepared the same except using buffered H2O instead of D2O.
Frozen samples were thawed on a 5 °C block for 4 min prior to injection onto a loading loop. The loaded sample was passed over a custom packed pepsin column (Porcine pepsin immobilized on POROS 20-AL resin; 2.1 × 50 mm column) (Wang, et al. 2002) kept at 12 °C with a flow of 0.1% trifluoroacetic acid (TFA) and 2% acetonitrile (ACN) at 200 μL/min.
Digested peptic fragments were trapped onto a Waters XSelect CSH C18 XP VanGuard Cartridge (2.1 × 5 mm, 2.5 μm). After 5 minutes of loading, digestion, and trapping, peptides were resolved on an analytical column (Waters BEH 1 × 100 mm, 1.7μm, 130Å) using a gradient of 3% to 40% solvent B for 9 minutes (A: 0.1% FA, 0.025% TFA, 2% ACN; B) 0.1% FA in ACN). The LC system was coupled to a Thermo Orbitrap performing full scans over the m/z range of 300 to 1500 at a resolution of 30,000. The MS source conditions were set to minimize loss (Walters, et al. 2012). Undeuterated samples were run prior to and at the end of all the LCMS runs.
During the analytical separation step, a series of 250 μL injections were used to clean the pepsin column: 1) 0.1% Fos-12 with 0.1% TFA; 2) 2 M guanidinium HCl in 0.1% TFA; 3) 10% acetic acid, 10% acetonitrile, 5% isopropanol (Majumdar, et al. 2012; Hamuro and Coales, 2018). After each gradient the trapping column was washed with a series of 250 μL injections: 1) 10% FA; 2) 30% trifluoroethanol; 3) 80% methanol; 4) 66% isopropanol, 34% ACN; 5) 80% ACN. During the trap washes the analytical column was cleaned with three rapid gradients (Fang, J., et al. 2011).
Peptic peptides were identified from data-dependent acquisition (DDA) experiments on undeuterated samples by exact mass and tandem mass spectrometry (MS/MS) spectra using Protein Prospector (Baker and Chalkley, 2014) filtering with a score cutoff of 15. Mass shifts were determined using HD-Examiner v2 (Sierra Analytics). The GluFib (CAS: 103213-49-6, Sigma) internal standard peptide was checked in all samples to verify that back-exchange levels were consistent in all experiments (Zhang, et al. 2012).
We binned peptides based on their fractional deuteration levels. We assigned the deepest shade of red to the peptide KAVDFGLARL which showed the maximal differential fractional deuteration level of 50.02% between the two inhibitors. We then normalized color intensity from red to white for the rest of the peptides and mapped these to the crystal structure of 3D Src (Figure 4B).
Half-life characterization of 3D Src conformation selective inhibitor bound complexes
Limited proteolysis of Src in presence of inhibitors was modified from (Aguis, et al. 2019). Briefly, 1 μM of Src was preincubated with 20 μM of the inhibitors 1, 2 or DMSO (4%) in proteolysis buffer (50 mM Tris-HCl pH 8.0, 100 mM NaCl, 0.5 mM CaCl2) at room temperature for 30 min to prepare a kinase-ligand complex. Proteolysis was initiated by adding 3.8 μM Thermolysin (Promega, catalog number: V4001) stock solution to the kinase-ligand complex to a final concentration of 60 nM. 20 μL of this mixture was then added to 10 μL of 50 mM EDTA to terminate proteolysis at various time points (0, 2, 4, 8, 16, 32, 64, 128, 256 min). The quenched samples were analyzed by SDS-PAGE (12% Bis-Tris gel in SDS running buffer, and stained with SYPRO Ruby ThermoFisher Scientific: catalog number S12000). Band intensities were analyzed by ImageStudioLite imaging software. Percent protein remaining was computed based on band intensity at 0 min and was plotted against time on GraphPad Prism 8. The curve was fit to an exponential decay equation using GraphPad Prism 8 software to obtain half-lives of each kinase-ligand complex.
Parallel Reaction Monitoring (PRM) of Src Cysteine Mutants
Samples were prepared for spectral libraries by denaturing SrcCysLib in buffer containing isotopically light or heavy (Sigma 721328) IA; final concentrations: 860 nM pooled cysteine mutant protein, 100 mM Tris pH 7, 1.6 M guanidinium HCl, 1 mM CaCl2, 1.3% DMSO, 10 mM IA in 75 μL. Samples were heated to 37 °C for 10 min, then 1 μL of 0.5 μg/μL MS grade trypsin was added. pH was adjusted to 8.5 using 1 N NaOH and samples were incubated overnight at 37 °C with 500 RPM orbital shaking. Samples were acidified with 15 μL 88% formic acid and 2 μL of 1.5 pmol/μL Pierce retention time calibration mix was added to each sample (ThermoScientific 88321). Samples were desalted on C-18 StageTips made in house by washing the StageTips with 50 μL Buffer B (80% acetonitrile in water, 0.1% TFA), equilibrating the StageTips with 50 μL Buffer A (5% acetonitrile in water, 0.1% TFA), applying the sample, washing with 50 μL Buffer A and eluting into a Lo-Bind microcentrifuge tube using 50 μL Buffer B. Peptides were then loaded on a self-pulled 360 μm OD × 100 μm ID 15 cm column with a 7 μm tip packed with 3 μm Reprosil C18 resin (Dr. Maisch, Germany). Peptides were analyzed by nanoLC-MS in a 60 min linear gradient from 10% to 35% buffer B (buffer A: 0.1% acetic acid; buffer B: 0.1% acetic acid, 80% acetonitrile) (Thermo EASY nLC 1200) on an Orbitrap Fusion™ Lumos™ Tribrid™ Mass Spectrometer. Top Speed data-dependent acquisition with 3 sec cycle time was used with each inclusion list. The method consisted of Orbitrap FTMS spectra (R = 60 000 at 200 m/z; m/z 350–1600; 7e5 target; max 20 ms ion injection time) for MS1 and HCD MS/MS spectra (R = 30,000 at 200 m/z; 29% CE; 5e4 target; max 100 ms injection time) were collected with an intensity filter set at 2.5e4 and dynamic exclusion for 15 sec. Inclusion lists for each spectral library (heavy or light) were generated by importing a FASTA sequence for each of the 74 cysteine mutant full length Src sequences into Skyline. PRM precursor masses were selected for unique tryptic peptides ≥6 amino acids, allowing up to two missed cleavages and charge states +2 through +5 with variable modifications on cysteine for each mass label, and variable oxidation of methionine. To generate spectral libraries, resultant DDA .RAW files were converted to the .mzML file format using MSConvert and search using MSGF+ against the combined Src cysteine mutant FASTA database which contained the Pierce iRT peptide sequences. The resulting .mzID files were imported into Skyline for analysis and generation of spectral libraries.
PRM assay samples were prepared as master mixes in triplicate for each heavy IA concentration—50, 100 and 200 μM. First, SrcCysLib was diluted to 1.3 μM in 100 mM Tris pH 7.6, 2 mM MgCl2, 100 mM NaCl with 20 μM of 1 or 2 added from a DMSO stock (2% DMSO final). Inhibitor-SrcCysLib complexes were formed at RT for 30 mins after which heavy IA (Sigma 721328) was added from a 50X stock. At the indicated timepoints (30, 60 or 120 min) 50 μL was taken from each master mix and quenched in 25 μL 3X denaturing buffer: 4.5 M guanidinium HCl, 3 mM CaCl2, 15 mM TCEP, 30 mM IA. Samples were then processed like library samples (above). Spectra were collected for each sample as described above with the following modifications. The Orbitrap spectra were collected using the following settings: HCD CE at 29%, R = 15,000 at 200 m/z, m/z 350–1100, 5e4 target, max injection time at 22 ms, with the scheduled PRM target list
After collecting data, raw spectra were imported into Skyline for peptide-centric analysis and intensity values for each peptide were exported for further processing in Microsoft Excel. We analyzed peptides that contained intensity values in either heavy or light channel in two thirds of experimental samples. This included several peptides that did not have heavy intensity values in the experimental samples. These cysteines did not have an appreciable amount of heavy alkylation product and we therefore imputed intensity values of 1. We normalized the heavy intensity values to internal standards (iRT and Srcwt peptides). Ratios of heavy labeled peptides from inhibitor 2/1 were then calculated and mapped to the crystal structure of Src (PDB IS: 2SRC). The crystal structures were then overlaid in Photoshop to generate pie charts for each residue.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data fitting and statistical analysis was performed using GraphPad Prism version 8. Statistical values including exact n and statistical significance are reported in the figure legends. Thermolysin characterization of Src 3D is plotted as mean ± SEM (n = 2 per condition) (Figure 4A). Deuteration levels of selected peptic peptides are plotted as mean ± SEM (n = 4 per inhibitor condition) (Figure 4B). Ratios of intensity values—normalized to internal standards—from PRM assay are represented as means (n = 2 or 3) and transformed to a linear heatmap, represented as wedges of pie charts (Figure 5B and 5C).
Supplementary Material
DNA_Oligos_and_Constructs.xlsx contains all DNA oligonucleotides used and all of the protein constructs contained in the SrcCysLib. Relates to Figures 3 and 5.
Export_HDX_Data_20200515.xlsx contains deuteration levels of peptic peptides from HDX-MS experiments of conformation-selective inhibitor-bound Src 3D complexes. Relates to Figure 4.
Parallel_Chemoselective_Profiling_Data.xlsx contains normalized intensity values of heavy and light IA labeled cysteine-containing peptides. Relates to Figure 5.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Phospho-Src Family (Tyr416) | Cell Signaling Technology | Cat#2101 |
| Non-phospho-Src (Tyr416) | Cell Signaling Technology | Cat#2102 |
| Bacterial and Virus Strains | ||
| E. coli (BL21 DE3 +GroEL + YopH) | Ahler, et al. 2019 | |
| NEB 5-alpha Competent E. coli (high efficiency) | N.E.B. | Cat#C2987H |
| Biological Samples | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Pierce retention time calibration mix | ThermoFisher Scientific | Cat#88321 |
| Isotopically heavy iodoacetamide | Sigma | Cat#721328 |
| Thermolysin | Promega | Cat#V4001 |
| Glu-1-Fibrino (GluFib) peptide | Sigma | CAS#103213-49-6 |
| ULP1* | Brigham, et al. 2013 | |
| Critical Commercial Assays | ||
| Deposited Data | ||
| Ubiquitin crystal structure | Vijay-Kumar, et al. 1987 | PDB: 1UBQ |
| PTEN crystal structure | Lee, et al. 1999 | PDB: 1D5R |
| Src Kinase Domain crystal structure | Azam, et al. 2008 | PDB: 3DQW |
| Src 3D crystal structure | Xu, et al. 1999 | PDB: 2SRC |
| BRCA1 BRCT domain crystal structure | Shiozaki, et al. 2004 | PDB: 1T29 |
| Experimental Models: Cell Lines | ||
| FreeStyle 293F | ThermoFisher Scientific | Cat#R79007 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| DNA oligos | This paper | See Supplemental Item DNA_Oligos_and_Constructs.xlsx |
| Recombinant DNA | ||
| Various point mutants in pCDNA5 | This paper | See Supplemental Item DNA_Oligos_and_Constructs.xlsx |
| Src3D (residues 87–536) in pMCSG7 | Ahler, et al. 2019 | |
| Software and Algorithms | ||
| PyMOL | Schrödinger | https://pymol.org/ |
| GraphPad Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Image Studio Light | LiCor | https://www.licor.com/bio/products/software/image_studio_lite/ |
| msConvertGUI | ProteoWizard | http://proteowizard.sourceforge.net/tools.shtml |
| Skyline | MacCoss Lab (UW) | http://skyline.ms |
| Other | ||
Significance.
Our goal was to develop a general targeted mass spectrometry-based method that can be used to characterize protein structure and dynamics without the use of highly specialized equipment. We present Parallel Chemoselective Profiling, a protein footprinting method that leverages Deep Mutational Scanning data to install reactive amino acids on a protein’s surface that can be labeled using chemoselective reagents. Differences in residue labeling are then compared and used to make inferences into protein structure and dynamics. We used our method to profile the conformational dynamics of full-length Src upon complexation of its kinase domain with two conformation-selective, ATP-competitive inhibitors. Our study revealed that ATP-binding site occupancy induces multiple local and distal changes to Src’s structure and dynamics. We demonstrate that our method complements Hydrogen-Deuterium Exchange Mass Spectrometry experiments and provides residue-level insight into an unexplored extended allosteric network intrinsic to the kinase domain of Src.
Highlights.
Deep mutational scanning data is used to guide the generation of protein variants
Parallel chemoselective labeling of variants allows mapping of solvent accessibility
Differences in quantified labeling rates are used to infer structure and dynamics
Parallel analysis reveals an extended allosteric network in Src’s kinase domain
Acknowledgements
This work used an EASY-nLC1200 UHPLC and Thermo Scientific Orbitrap Fusion Lumos Tribrid mass spectrometer purchased with funding from a National Institutes of Health SIG grant S10OD021502.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Authors declare no competing interests.
References
- Abo M, Li C, Weerapana E. (2018). Isotopically-labeled iodoacetamide-alkyne probes for quantitative cysteine-reactivity profiling. Mol Pharm. 15, 743–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agius MP, Ko KS, Johnson TK, Kwarcinski FE, Phadke S, Lachacz EJ, Soellner MB (2019). Selective proteolysis to study the global conformation and regulatory mechanisms of c-Src kinase. ACS Chem Biol 14 7, 1556–1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahler E Register AC, Chakraborty S, Fang L, Dieter EM, Sitko KA, Vidadala RSR, Trevillian BM, Golkowski M, Gelman H, Stephany JJ, Rubin AF, Merritt EA, Fowler DM, Maly DJ (2019). A combined approach reveals a regulatory mechanism coupling Src’s kinase activity, localization and phosphotransferase-independent functions. Mol. Cell74, 2, 393–408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam M, Seeliger MA, Gray N, Kuriyan J, Daley GQ (2008). c-Src kinase domain Thr338Ile mutant in complex with ATPgS Nat. Struct. Mol. Biol 15: 1109–1118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker PR, Chalkley RJ (2014). MS-viewer: A web-based spectral viewer for proteomics results. Mol. Cell. Proteomics 13(5) 1392–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ban H, Gavrilyuk J, Barbas CF (2010). Tyrosine bioconjugation through aqueous ene-type reactions: A click-like reaction for tyrosine. JACS 132, 5, 1523–1525 [DOI] [PubMed] [Google Scholar]
- Boggon TJ, Eck MJ (2004). Structure and regulation of Src family kinases. Oncogene 23, 7918–7927 [DOI] [PubMed] [Google Scholar]
- Brigham JL, Perera BG, Maly DJ (2013). A hexylchloride-based catch-and-release system for chemical proteomic applications. ACS Chem. Biol 8(4): 691–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt TR, Edavettal SC, Hall JP, Mattern MR (2005). SUMO fusion technology for difficult-to-express proteins. Protein Expression and Purification 43, 1, 1–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakraborty S, Inukai T, Fang L, Golkowski M, Maly DJ (2019). Targeting dynanmic ATP-binding site features allows discrimination between highly homologous protein kinases. ACS Chem Biol 14 6, 1249–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chea EE, and Jones LM (2018). Modifications generated by fast photochemical oxidation of proteins reflect the native conformations of proteins. Protein Science 27, 1047–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito D, Weile J, Shendure J, Starita LM, Papenfuss AT, Roth FP, Fowler DM, Rubin AF (2019). MaveDB: An open-source platform to distribute and interpret data from multiplexed assays of variant effect. Genome Biol 20, 223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang J, Rand KD, Beuning PJ, Engen JR (2011). False EX1 signatures caused by samples carryover during HX MS analyses. Int. J. Mass Spectrom 302 (1–3) 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Vilas-Boas J, Chakraborty S, Potter ZE, Register A, Seeliger M, Maly DJ (2020). How ATP-Competitive Inhibitors Allosterically Modulate Tyrosine Kinases That Contain a Src-like Regulatory Architecture. ChemRxiv, 10.26434/chemrxiv.12148524.v2 [DOI] [PMC free article] [PubMed]
- Findlay GM, Daza RM, Martin B, Zhang MD, Leith AP, Gasperini M, Janizek JD, Huang X, Starita LM, Shendure J(2019). Accurate classification of BRCA1 variants with saturation genome editing. Nature 562, 217–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foda ZH, Shan Y, Kim ET, Shaw DE, Seeliger MA (2015). A dynamically coupled allosteric network underlies bunding cooperativity in Src kinase. Nat. Comm 6, 5939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler DM, Fields S (2014). Deep mutational scanning: A new style of protein science. Nat. Meth 11, 801–807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gau BC, Sharp JS, Rempel D, Gross ML (2009). Fast photochemical oxidation of protein footprints faster than protein unfolding. Anal. Chem 81, 16, 6563–6571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacker SM, Backus KM, Lazear MR, Forli S, Correia BE, Cravatt BF (2017). Global profiling of lysine reactivity and ligandability in the human proteome. Nat. Chem 9(12):1181–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambly DM, Gross ML (2005). Laser flash photolysis of hydrogen peroxide to oxidize protein solvent-accessible residues on the microsecond timescale. J. Am. Soc. Mass Spectrom 16, 2057–2063 [DOI] [PubMed] [Google Scholar]
- Hamuro Y (2017). Determination of equine cytochrome c backbone amide hydrogen/deuterium exchange rates by mass spectrometry using a wider time window and isotope envelope. J. Am. Soc. Mass Spectrom 28, 3, 486–497 [DOI] [PubMed] [Google Scholar]
- Hamuro Y and Coales SJ (2018). Optimization of feasibility stage for hydrogen/deuterium exchange mass spectrometry. J. Am. Soc. Mass Spectrom 29(3) 623–629. [DOI] [PubMed] [Google Scholar]
- Hietpas RT, Jensen JD, Bolon DN (2011). Experimental illumination of a fitness landscape. Proc. Natl. Acad. Sci 108, 19, 7896–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodkinson JP, Jahn TR, Radford SE, and Ashcroft AE (2009). HDX-ESI-MS reveals enhanced conformational dynamics of the amyloidogenic protein β2- microglobulin upon release from the MHC-1. Journal of the American Society for Mass Spectrometry 20, 278–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houde D, Arndt J, Domeier W, Berkowitz S, and Engen JR (2009). Characterization of IgG1 Conformation and Conformational Dynamics by Hydrogen/Deuterium Exchange Mass Spectrometry. Analytical Chemistry 81, 2644–2651 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung JE, Jura N (2016). Structural basis for the non-catalytic functions of protein kinases. Structure. 24, 1, 7–2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur U, Johnson DT, Chea EE, Deredge DJ, Espino JA, Jones LM (2018). Evolution of structural biology through the lens of mass spectrometry. Anal. Chem 91, 142–155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornev AP, Taylor SS (2016). Dynamics driven allostery in protein kinases. Trends Biochem. Sci 40(11): 628–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamurty R, Brigham JL, Leonard SE, Ranjitkar P, Larson ET, Dale EJ, Merritt EA, Maly DJ (2013). Active site profiling reveals coupling between domains in Src-family kinases. Nat. Chem. Biol 9 1, 43–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kritzer JA, Freyzon Y, Lindquist S (2018). Yeast can accommodate phosphotyrosine: v-Src toxicity in yeast arises from a single disrupted pathway. FEMS Yeast Res. 18, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyan J, Eisenberg D (2007). The origin of protein interactions and allostery in colocalization. Nature12, 450 (7172), 982–990 [DOI] [PubMed] [Google Scholar]
- Kwarcinski FE, Brandvold KR, Phadke S, Beleh OM, Johnson TK, Meagher JL, Seeliger MA, Stuckey JA, Soellner MB (2016). Conformation-selective analogues of dasatinib reveal insight into kinase inhibitor binding and selectivity. ACS Chem Biol 11 5, 1296–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard SE, Register AC, Krishnamurty R, Brighty GJ, Maly DJ (2014). Divergent modulation of Src-family kinase regulatory interactions with ATP-competitive inhibitors. ACS Chem Biol 9 (8), 1894–905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JO, Yang H, Georgescu MM, Di Cristofano A, Maehama T, Shi Y, Dixon JE, Pandolfi P, Pavletich NP (1999). Crystal structure of the PTEN tumor suppressor: implications for its phosphoinositide phosphatase activity and membrane association. Cell 99: 323–334 [DOI] [PubMed] [Google Scholar]
- Li S, Yang X, Jia S, Weeks AM, Hornsby M, Peter SL, Nichiporuk RV, Iavarone AT, Wells JA, Toste FD, Chang CJ (2017). Redox-based reagents for chemoselective methionine bioconjugation. Science 355,6325, 597–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majumdar R, Manikwar R, Hickey JM, Arora J, Middaugh CR, Volkin DB, Weis DD (2012). Minimizing carry-over in an online pepsin digestion system used for the H/D exchange mass spectrometric analysis of an IgG1 monoclonal antibody. J. Am. Soc. Mass Spectrom 23(12) 2140–8. [DOI] [PubMed] [Google Scholar]
- Manning G, Whyte DB, Martinez R, Hunter T, Sudarsanam S (2002). The protein kinase complement of the human genome. Science 298, 5600, 1912–1934 [DOI] [PubMed] [Google Scholar]
- Matreyek KA, Stariata LM, Stephany JJ, Martin B, Chiasson MA, Gray VE, Kircher M, Dines JN, Hause RJ, Bhatia S, Evans WE, Relling MV, Yang W, Shendure J, Dowler DM (2018). Multiplex assessment of protein variant abundance by massively parallel sequencing. Nat. Genet 50, 874–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClendon CL, Kornev AP, Gilson MK, Taylor SS (2014). Dynamic architecture of a protein kinase. Proc. Natl. Acad. Sci. USA 111(43): E4623–E4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellicena P, Kuriyan J (2006). Protein-protein interactions in the allosteric regulation of protein kinases. Curr. Opin. Struct. Biol 16, 6, 702–709 [DOI] [PubMed] [Google Scholar]
- Register AC, Leonard SE, Maly DJ (2014). SH2-catalutic domain linker heterogeneity influences allosteric coupling across the SFK family. Biochemistry 53 (44), 6910–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscoe B, Thayer KM, Zeldovish KB, Fushman D, Bolon DN (2013). Analyses of the effects of all ubiquitin point mutants on yeast growth rate. J. Mol. Biol 425(8) 1363–1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiozaki EN, Gu L, Yan N, Shi Y (2004). Structure of the BRCT repeats of BRCA1 bound to a BACH1 phosphopeptide: implications for signaling. Mol. Cell 14: 405–412 [DOI] [PubMed] [Google Scholar]
- Taylor SS, Meharena HS, Kornev AP (2019). Evolution of a dynamic molecular switch. IUBMB Life 71, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SS, Keshwani MM, Steichen JM, Kornev AP (2012). Evolution of the eukatyotic protein kinases as dynamic molecular switches. Philos. Trans. R. Soc. Long. B. Biol. Sci 367(1602): 2517–2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong M, Pelton JG, Gill ML, Zhang W, Picart F, Seeliger MA (2017). Survey of solution dynamics in Src kinase reveals allosteric cross talk between the ligand binding and regulatory sites. Nat.. Commun 8 1, 2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong M, Seeliger MA (2015). Targeting conformation plasticity of protein kinases. ACS Chem. Biol 10, 1, 190–200 [DOI] [PubMed] [Google Scholar]
- Ung PM, Rahman R, Schlessinger A (2018). Redefining the protein kinase conformation space with machine learning. Cell Chem. Biol 25, 7, 916–924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vahihi S and Konermann L (2016). Probing the time scale of FPOP: Radical reactions over tens of milliseconds. J. Am. Soc. Mass Spectrom 27, 7, 1156–1164 [DOI] [PubMed] [Google Scholar]
- Vijay-Kumar S, Bugg CE, Cook WJ (1987). Structure of ubiquitin refined at 1.8 A resolution. J. Mol. Biol 194: 531–544 [DOI] [PubMed] [Google Scholar]
- Walters BT, Ricciuti A, Mayne L, Englander SW (2012). Minimizing back exhange in the hydrogen exchange-mass spectrometry experiment. J. Am. Soc. Mass Spectrom 23(12) 2132–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Pan H, Smith DL (2002). Hydrogen Exchange-Mass Spectrometry: Optimization of Digestion Conditions. Mol. Cell. Proteomics 1(2) 132–8 [DOI] [PubMed] [Google Scholar]
- Weerapana E#, Wang C#, Simon GM, Richter F, Khare S, Dillon BDM, Bachovchin DA, Mowen K, Baker D, Cravatt BF. (2010). Quantitative reactivity profiling predicts functional cysteines in proteomes. Nature 468, 790–795. #These authors contributed equally to this work. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Doshi A, Lei M, Eck MJ, Harrison SC (1999). Crystal structures of c-Src reveal features of its autoinhibitory mechanism. Mol. Cell 3: 629–638 [DOI] [PubMed] [Google Scholar]
- Zhang H, Wen J, Huang R, Blankenship RE, Gross ML (2012). Mass spectrometry-based carboxyl footprinting of proteins: method evaluation. Int. J. Mass Spectrom 312: 78–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhang A, Xiao G (2012). Improved protein hydrogen/deuterium exchange mass spectrometry platform with fully automated data processing. Anal. Chem 84(11) 4942–9. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Wu H, Wang L, Liu Y, Knapp S, Liu Q, Gray NS (2014). Exploration of type II binding mode: A privileged approach for kinase inhibitor focused drug discovery? ACS Chem. Biol 9, 6, 1230–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
DNA_Oligos_and_Constructs.xlsx contains all DNA oligonucleotides used and all of the protein constructs contained in the SrcCysLib. Relates to Figures 3 and 5.
Export_HDX_Data_20200515.xlsx contains deuteration levels of peptic peptides from HDX-MS experiments of conformation-selective inhibitor-bound Src 3D complexes. Relates to Figure 4.
Parallel_Chemoselective_Profiling_Data.xlsx contains normalized intensity values of heavy and light IA labeled cysteine-containing peptides. Relates to Figure 5.
Data Availability Statement
This study did not generate large datasets or code.