

Abstract
Objective
To delineate temporal and spatial dynamics of vascular smooth muscle (SMC) transcriptomic changes during aortic aneurysm development in Marfan syndrome (MFS).
Approach and Results
We performed single-cell RNA sequencing (scRNAseq) to study aortic root/ascending aneurysm tissue from Fbn1C1041G/+ (MFS) mice and healthy controls, identifying all aortic cell types. A distinct cluster of transcriptomically modulated SMCs (modSMCs) was identified in adult Fbn1C1041G/+ mouse aortic aneurysm tissue only. Comparison with atherosclerotic aortic data (ApoE−/− mice) revealed similar patterns of SMC modulation but identified a MFS-specific gene signature including Serpine1 and Klf4. We identified 481 differentially expressed genes between modSMC and SMC subsets; functional annotation highlighted ECM modulation, collagen synthesis, adhesion and proliferation. Pseudotime trajectory analysis of Fbn1C1041G/+ SMC/modSMC transcriptomes identified genes activated differentially throughout the course of phenotype modulation. While modSMCs were not present in young Fbn1C1041G/+ mouse aortas despite small aortic aneurysm, multiple early modSMCs marker genes were enriched, suggesting activation of phenotype modulation. modSMCs were not found in non-dilated adult Fbn1C1041G/+ descending thoracic aortas. scRNAseq from human MFS aortic root aneurysm tissue confirmed analogous SMC modulation in clinical disease. Enhanced expression of TGF-β-responsive genes correlated with SMC modulation in mouse and human datasets.
Conclusions
Dynamic SMC phenotype modulation promotes ECM substrate modulation and aortic aneurysm progression in MFS. We characterize the disease-specific signature of modulated SMCs and provide temporal, transcriptomic context to the current understanding of the role TGF-β plays in MFS aortopathy. Collectively, scRNAseq implicates TGF-β signaling and Klf4 overexpression as potential upstream drivers of SMC modulation.
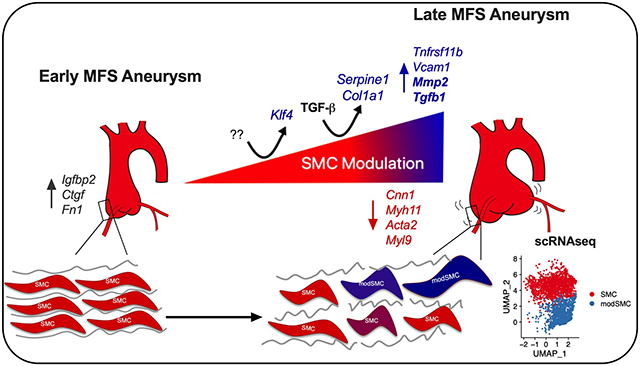
Graphical Abstract
Introduction
Marfan syndrome (MFS) is an inherited connective tissue disorder caused by mutations in the fibrillin-1 gene which affects 1 in 5,000 individuals1,2. This systemic disorder affects extracellular matrix (ECM) composition and leads to highly penetrant aortic root aneurysm development and acute aortic events (dissection or rupture)3. Surgical aneurysm repair remains the only effective treatment for MFS patients. Activation of the transforming growth factor beta (TGF-β) signaling pathway is a known biochemical signature of MFS based on a number of animal model and human studies4–6, but its role in aneurysm initiation and propagation remains highly controversial7,8. Whereas late TGF-β neutralization reduced aneurysm growth in both Fbn1C1041G/+ and Fbn1mgR/mgR mouse model systems9,10, early blockade exacerbated disease in Fbn1mgR/mgR mice. Furthermore, transgenic models of inducible TGF-β type II receptor (Tgfbr2) knockout in SMCs or haploinsufficiency of TGF-β signaling components (Tgfb2 or Smad4) exacerbated aneurysm and dissection in Fbn1C1041G/+ mice7,11–13.
SMCs are the most intensively studied cell type in MFS and are widely held as the culprit cell in aortic aneurysm pathogenesis6,14,15. SMCs maintain tremendous plasticity to modulate their phenotype across a spectrum of functions in response to growth factors, injury and inflammation16–18. Our group and others have reported multiple enhanced contractile genes in human MFS aortic root tissue in parallel with enhanced ECM gene expression compared to control specimens6,19. In vitro experiments confirmed that both of these responses are dampened by TGF-β signaling blockade. Whether changes in ECM are mediated by SMCs or other cell types in vivo remains unknown. Furthermore, the role TGF-β signaling plays during this process is incompletely understood.
Single-cell RNA sequencing (scRNAseq) has added unparalleled resolution to transcriptomic studies, enabling massively parallel investigation of diverse cell types within tissues. This technique has been used to detail SMC gene expression in healthy aortic tissue20, heterogeneity with respect to aortic region21, and phenotype modulation in atherosclerosis16,22. Recently, SMC phenotype profiling using scRNAseq in an aortic aneurysm model resulting from chronic mTOR activation demonstrated deleterious effects of SMC dedifferentiation, proliferation, and reduced ECM synthesis.23 In this study, we apply scRNAseq technology to the Fbn1C1041G/+ murine MFS model and human MFS aortic tissue to define transcriptomic alterations associated with aortic aneurysm development across diverse cell types.
Methods and Materials
Data Availability:
All data and materials have been made publicly available at the gene expression omnibus (GEO) repository under accession ID GSE153534.
Mice
Fbn1C1041G/+ mice (#012885) were purchased from The Jackson Laboratory (Bar Harbor, ME). Mutant mice were backcrossed onto the C57BL/6J background for at least 10 generations by the donating investigator and vendor. The Fbn1C1041G/+ colony was maintained via breeding with littermate control animals. Animals were maintained according to protocols approved by the Institutional Animal Care and Use Committee. Age and sex-matched littermate control animals were used as comparisons. Mice were sacrificed at 4 and 24 weeks for tissue digestion and scRNAseq (n=3 (2 male, 1 female) for each genotype at 4 weeks, n=4 (2 male, 2 female) Fbn1C1041G/+ and n=3 control (2 male, 1 female) at 24 weeks) and histologic studies (n=4–6 for each genotype).
Echocardiography
Mice were anesthetized with inhaled isoflurane. The thorax was prepared with a topical hair removal agent. Images of the aortic root and mid ascending aorta (halfway from the sinotubular junction to the brachiocephalic artery takeoff) were acquired in long axis with an MS700 70MHz MicroScan transducer on the Vevo 2100 system (VisualSonics, Toronto, Canada). Aortic measurements were taken during end diastole. An average of three measurements were taken per animal. Measurements were taken by a technician blinded to animal genotype.
Mouse Aortic dissociation and single cell suspension
Fbn1C1041G/+ mice and littermate controls were anesthetized and sacrificed by cervical dislocation. For aortic root/ascending samples, the aortic root was dissected completely and divided below the aortic valve. The ascending aorta was divided midway between the aortic valve and the takeoff of the brachiocephalic artery (Figure 2A). For descending thoracic aorta, the vessel was transected proximally just beyond the ligamentum arteriosum and distally at the diaphragm. Aortas were transferred to a separate dish and rinsed in PBS. Specimens were combined into groups with age and genotype-matched animals as biologic replicates. The aortas were then digested in enzymatic solution containing 2U/mL Liberase TM (Roche #05401127001) and 2U/mL elastase (Worthington #LS002279) in Hanks buffered saline solution (HBSS) for one hour. The cell suspension was filtered through a 70uM strainer, centrifuged and resuspended in cold HBSS.
Figure 2:

Single cell RNA sequencing (scRNAseq) of Fbn1C1041G/+ and control mouse aortic Root/AS tissue reveals disease-associated cluster. A Experiment workflow for scRNAseq. Aortic root and proximal ascending aorta tissue are digested in enzyme mixture to suspend cells. FACS eliminates debris, cell clumps, and dead cells prior to single cell capture and sequencing. B Uniform Manifold Approximation and Projection (UMAP) dimensional reduction of >9,800 aortic cells from Fbn1C1041G/+ (n=4) and control (n=3) mice. Blue circle denotes major clusters selected for further analysis, red arrows indicate modulated SMC (modSMC) cluster enriched in Fbn1C1041G/+. C Dotplot for highest specificity gene markers of cell clusters. Dot size represents percent of cells expressing denoted gene, color represents average normalized expression level within the denoted cluster. D UMAP projection of modSMC cluster positioned between SMC and fibroblast clusters. E Feature plots demonstrate reduced expression of contractile genes (Acta2, Myl9, Myh11, Tpm2) and enriched expression of fibronectin 1 (Fn1), matrix gla protein (Mgp), nuclear protein 1 (Nupr1) and elastin (Eln) in modSMCs. Color scale indicates range from 5th (Low) to 95th (High) percentile of normalized expression levels by individual cells.
Fluorescence-Activated Cell Sorting (FACS)
FACS was performed at the Stanford Shared FACS Facility on a Sony SH800 instrument using 7-Aminoactinomycin D (7AAD) as a live/dead cell discrimination marker. The gating strategy shown in Supplemental Figure 1 was used to sort cells from debris and exclude doublets. Single, live cell suspensions were counted to ensure adequate cell density (>150 cells/uL) and immediately used for downstream sequencing applications.
scRNAseq
All portions of the scRNAseq workflow (single cell capture, library preparation, quality control PCR and sequencing) were performed Stanford Genome Sequencing Service Center. Freshly sorted samples were diluted to target 5,000 cells for capture. The cells were processed using a 10× Genomics microfluidics chip to generate barcoded Gel Bead-In Emulsions (GEMs) according to manufacturer protocols. Indexed single cell libraries were then created according to 10× Genomics specifications. Samples were multiplexed and sequenced in pairs on an Illumina HiSeq 4000 device.
scRNAseq Data Analysis
The sequenced data were processed into expression matrices with the ‘Cell Ranger Single cell software suite 3.1.0’ (https://www.10xgenomics.com/) on the Stanford Sherlock High Performance Computing Cluster. Raw base-call files from HiSeq4000 sequencer were demultiplexed to first generate FASTQ files using the cellranger mkfastq pipeline. Subsequently, the reads were aligned to the mouse transcriptome (mm10–3.0.0), cell barcodes and unique molecular identifiers were filtered and corrected using the cellranger count pipeline. The final output filtered expression matrices were imported into the Seurat package in R and built into Seurat objects using the CreateSeuratObject function. Filtering during this step included only genes detected in >3 cells and cells with >200 distinct genes. To exclude doublets/clumps and free RNA, thresholds for individual cell read counts (‘nCount_RNA’) and genes (‘nFeature_RNA’) were determined for each dataset by excluding indices (cells) with counts outside of normal distributions for these variables. Cells with greater than 7.5% mitochondrial gene content were excluded. Representative QC data pre- and post-filtering is shown in Supplemental Figure 2A.
Data normalization, scaling and regression by mitochondrial content were then performed using the SCTransform command under default settings in Seurat. Principal component analysis (PCA) and non-linear dimensional reduction using both Uniform Manifold Approximation and Projection (UMAP) and t-Distributed Stochastic Neighbor Embedding (tSNE) techniques were performed. Cell clustering was then assessed across a range of pre-determined resolution scales to ensure separation of known major aortic cell types without excessively sub-clustering. Example clustering results using low (0.4) and ultra-low (0.2) resolution are presented in Supplemental Figure 3A-B. The FindAllMarkers function in Seurat was applied to performs parallel differential expression testing of all cells within a cluster versus all other cells in the dataset via non-parametric Wilcoxon rank sum test using default parameters.
Dataset integration for human/mouse MFS analysis and comparison between Fbn1C1041G/+ mice and SMC lineage-traced ApoE−/− mice was performed in Seurat using the SCTransform integration workflow24. The SMC lineage-traced scRNAseq dataset for TgMyh11-CreERT2, ROSAtdT/+, ApoE−/− mouse data obtained using published methods22 was generously provided by Dr. Thomas Quertermous.
Bioinformatic Analyses
Functional annotation was performed using the DAVID 6.8 Bioinformatics database25 (https://david.ncifcrf.gov/summary.jsp). The differentially expressed gene set comparing SMC and modSMC clusters was filtered to include genes enriched in modSMCs and input into DAVID. Enriched GO terms classified by biologic process (BP) were identified using default stringency. Significantly enriched KEGG pathways were similarly identified. Benjamini-Hochberg correction was applied to adjust for multiple comparisons. Adjusted p value <0.05 was considered significant.
oPossum (version 3.0, http://opossum.cisreg.ca/oPOSSUM3/) single site analysis (SSA) was used to identify overrepresented transcription factor binding motifs among differentially expressed genes26. Genes enriched in the modSMC (n=216 genes) were input into the SSA tool and compared to all genes in the oPossum database (29,347 genes) using transcription factor binding site matrices from all JASPAR CORE profiles and default binding site parameters. Transcription factors with computed Z-scores greater than two standard deviations above the mean were considered significant. Pseudotime scRNAseq trajectory analysis was performed using Monocle327 in R. Normalized expression data from 24 week Fbn1C1041G/+ SMC and modSMC clusters were exported from Seurat as input, and pseudotime scores for individual cells were incorporated into the original Seurat object as metadata for further analysis.
Aortic histologic tissue preparation
Fbn1C1041G/+ and littermate control animals were sacrificed at 24 weeks of age as previously described. Animals were perfused with 4% paraformaldehyde (Sigma). The aortic root was harvested en bloc with the left ventricular outflow tract. Tissue samples were fixed for 24 hours in 4% PFA then passed through a sucrose gradient and embedded in OCT compound (Fisher). Tissue blocks were then sliced in 7um sections and dried at −80 degrees Celsius. Human tissue was acquired from institutional Surgical Pathology laboratory following formalin fixation, paraffin embedding and sectioning. 7um slices were deparaffinized with xylenes prior to use.
Elastic Van Gieson Stain
Fixed frozen tissue sections were rehydrated in PBS and stained with Accustain Elastin Verhoeff’s Van Gieson (EVG) kit (Sigma Aldrich, St. Louis, MO, USA) according to the manufacturer’s instructions. Briefly, slides were immersed in elastic stain solution (hematoxylin, FeCl, Weigert iodine solution) for 10 minutes, washed, then destained in FeCl solution. Slides were washed, immersed in 95% ethanol, then immersed in Van Gieson stain. Slides were dried with increasing ethanol concentration followed by immersion in xylenes and mounted with perimount.
RNAscope Assay
Slides were processed according to the manufacturer’s instructions (Advanced Cell Diagnostics, Newark CA), and all reagents were obtained from ACD Bio. Dried slides were rinsed in PBS then baked at 60°C for 30 minutes. Slides were then post-fixed in 4% PFA for 15 minutes at 4°C then dehydrated in ethanol. Dehydrated slides were pre-treated with hydrogen peroxide and immersed in 1X Target Retrieval buffer at 100°C for 5 minutes, washed in water followed by 100% ethanol and air dried. Sections were then treated with Protease Plus reagent at 40°C for 30 minutes and washed, followed by hybridization with probes for mouse osteoprotegerin (Tnfrsf11b) and matrix metalloproteinase 2 (Mmp2), Multiplex chromogenic assays were then performed according to manufacturer’s instructions. Human tissue was processed using the same protocol with sequence-specific probes for human plasminogen activator inhibitor-1 (SERPINE1) and TNFRSF11B. Representative photomicrographs were obtained at 5× and 20× magnification.
Human scRNAseq
All studies involving human patient tissue were approved by the Institutional Review Board and informed consent obtained prospectively. Fresh aortic root tissue was collected from the operating room within 30 minutes of excision and kept in cold Hank’s buffered saline solution (HBSS, Sigma). Tissue was dissected to include all vascular layers and divided into four 50mg pieces. Samples were then placed in digestion solution containing Liberase TM (10.4U/mL working concentration, Roche) and elastase (8U/mL, Sigma) and finely minced. Following 60 minutes of digestion at 37°C, digestates were filtered through a 70uM strainer, centrifuged and resuspended with fresh cold HBSS. Samples were sorted with FACS using Calcein green (ThermoFisher) for live/dead discrimination and used fresh for single cell capture and library preparation as with mouse samples.
Statistical Analysis
Transthoracic echo data was analyzed in GraphPad. Non-parametric (Mann Whitney U) tests were used to compare aortic root diameter measurements between genotypes. Differential gene expression testing was performed in Seurat as described in the scRNAseq section.
Results
Adult Fbn1C1041G/+ mice display progressive aortopathy
In prior reports, Fbn1C1041G/+ mice demonstrated highly penetrant aortic root and ascending aortic aneurysms associated with low frequency of aortic dissection or rupture before one year of age9,28. We performed transthoracic echocardiography on Fbn1C1041G/+ mice at ages 4 and 24 weeks and sex-matched littermate controls. Mild aneurysmal dilatation of the aortic root was detectable in Fbn1C1041G/+ mice at 4 weeks (1.78 ± 0.15mm versus 1.63 ± 0.07mm, p=0.04, n=6 each genotype) and severely dilated at 24 weeks (2.14 ± 0.30mm versus 1.69±0.10mm, p=0.0006, n=8 each genotype.) Ascending aortic dilatation was not significant at 4 weeks (1.47 ± 0.11mm versus 1.38 ± 0.09mm, p=0.18) but progressed at 24 weeks (1.56 ± 0.18mm versus 1.37 ± 0.16mm p=0.028, Figure 1A-B). Elastic Van Gieson staining of the aortic root demonstrated characteristic elastic lamina breakdown in 24-week Fbn1C1041G/+ animals (Figure 1C).
Figure 1:

Progressive aortopathy in Fbn1C1041G/+ mice. A Long axis echocardiographic aortic root and ascending aorta measurements in 4-week (n=6 mice per genotype) and 24-week old (n=8 per genotype) control and Fbn1C1041G/+ mouse cohorts. B Representative 24-week transthoracic echocardiographic images demonstrating severe dilatation of the aortic root in Fbn1C1041G/+ mice. White arrows depict sinus of Valsalva measurement. Yellow arrow depicts ascending aorta measurement. C Representative elastic Van Gieson (EVG) stain demonstrating severe elastin fiber fragmentation in Fbn1C1041G/+ mice. Box and whisker plots display 95% confidence interval (box), mean (line) and range (whiskers). ***p<0.001, *p<0.05 (Mann-Whitney U non-parametric test).
Aortic tissue scRNAseq in Fbn1C1041G/+ mice identifies aneurysm-associated cell cluster
To precisely define cell-specific transcriptomic changes associated with advanced MFS aortic aneurysms, we performed scRNAseq on isolated aortic root/proximal ascending (‘Root/AS’) tissue from 24-week old Fbn1C1041G/+ mice (n=4 animals) and littermate controls (n=3). Transmural aortic tissue was dissected from the aortic valve to the mid ascending aorta and digested using a validated enzymatic dissociation protocol (see Methods). FACS was used to exclude cell clumps and debris (Figure 2A). Single cell capture and cDNA library preparation were performed using the 10X Genomics Chromium platform followed by library preparation, multiplexed sequencing and quality control and analysis using the Seurat24,29 scRNAseq analysis package in R (see Methods). Our mouse cohort for scRNAseq suggested increased aneurysm size in Fbn1C1041G/+ males compared to females. This was confirmed with echocardiography of a larger cohort of 24-week old mice (2.30 ± 0.24mm versus 1.96 ± 0.18mm, n=9 animals per sex, Supplemental Figure 2C). We therefore initially compared male and female pairs with scRNAseq. We found highly similar cell clustering patterns between the two pairs (Supplemental Figure 2D-E). The male dataset demonstrated increased expression of multiple collagen isoforms in SMCs and fibroblasts (Supplemental Figure 2F) but otherwise few differentially expressed genes within clusters. We therefore combined these samples for comparison to controls.
Fbn1C1041G/+ and control samples (9,815 individual cells) were then jointly analyzed. We identified all major aortic cell types (SMC, multiple fibroblast subsets, endothelial cells, macrophages) within the aorta (Figure 2B). Differential gene expression testing was performed to identify the top gene markers for each cluster (Figure 2C displays top markers for each cluster). The top 100 gene markers for each cluster are listed in Supplemental Table 1. Splitting the analysis by genotype identified a distinct cluster of cells predominantly in Fbn1C1041G/+ samples (14.6% of all cells) and very low frequency in controls (1.5% of all cells). This cluster was positioned in continuity with SMCs and adjacent to the fibroblast clusters in both UMAP and tSNE projections. The transcriptomic profile of these cells was most similar to SMCs, as they preferentially clustered together as a single population under lower resolution conditions (Supplemental Figure 3). These findings suggest this cluster represents a subset of SMCs undergoing phenotypic modulation (modSMCs) toward a fibroblast-like state. To enable transcriptomic analysis across the SMC spectrum, we subset the dataset to isolate the closely associated SMC, modSMC, and fibroblast clusters (90.9% of total cell population, Figure 2D). The modSMC cluster demonstrated intermediate expression levels for SMC cluster markers (Acta2, Myl9, Myh11, Tpm2 Figure 2E), while fibronectin 1 (Fn1), matrix gla protein (Mgp), nuclear protein 1 (Nupr1), and elastin (Eln) were strong markers of the modSMC cluster.
Fbn1C1041G/+ modSMCs co-segregate with analogous cell type in atherosclerosis
SMC phenotype modulation has been extensively studied in murine models of atherosclerosis16,18,22,30,31. To directly compare the transcriptomic profile of SMC modulation in Fbn1C1041G/+ to SMC lineage-traced atherosclerosis-prone (TgMyh11-CreERT2,ROSAtdT/+,ApoE−/−) mice, we performed reference-based dataset integration24 with scRNAseq data from this model (Figure 3A shows mechanism for SMC tracing, adapted from Wirka et al22, integrated dataset shown in Figure 3B). This analysis revealed that 99% of Fbn1C1041G/+ modSMCs co-clustered with SMC-lineage traced cells (87% with the analogous fibromyocyte or ‘FMC’ cluster, 12% with SMC cluster, Figure 3C-D), supporting SMC trans-differentiation into the modSMC identity in Fbn1C1041G/+ mice.
Figure 3:

Fbn1C1041G/+ modSMCs co-cluster with analogous cell type in SMC lineage-traced atherosclerosis model. A Cartoon depiction of SMC lineage-tracing in TgMyh11-CreERT2, ROSAtdT/+, ApoE−/− mouse model. Tamoxifen at 8 weeks induces Cre recombinase-mediated activation of the tandem dimeric Tomato (tdT) in cells expressing Myh11 leading to permanent fluorescence in SMCs. High fat diet induces atherosclerosis prior to scRNAseq. B Integrated scRNAseq datasets from TgMyh11-CreERT2, ROSAtdT/+, ApoE−/− and Fbn1C1041G/+ mice with all cell clusters. C UMAP projection of scRNAseq data split by input sample. Blue oval indicates common SMC sub-clusters in each model. 99% of cells assigned modSMC identity in Fbn1C1041G/+ dataset co-clustered with SMC lineage-traced clusters from the ApoE−/− dataset. D Myh11 expression in integrated dataset. E UMAP projection of SMC/modSMC clusters from both models and feature plots depicting expression of Col1a1, Lum, and Lcn2 (enriched in ApoE−/−) split by model. F Analogous plots for example genes enriched in Fbn1C1041G/+ (‘MFS’) mice (Klf4, Serpine1, Lamc3). Color scale indicates range from 5th (Low) to 95th (High) percentile of normalized expression levels by individual cells.
We directly compared the modulated SMC clusters to identify disease-specific signatures. While the modulated SMC clusters co-segregated, over 500 genes were differentially expressed within this cluster between models. Among the strongest markers for atherosclerotic vessels were Col1a1, lumican (Lum), and lipocalin-2 (Lcn2) (Figure 3E), while Kruppel-like factor-4 (Klf4), plasminogen activator inhibitor-1 (Serpine1), and laminin subunit gamma-3 (Lamc3) distinguished MFS modSMCs (Figure 3F).
scRNAseq outlines trajectory of SMC modulation in MFS
Having confirmed that SMC modulation distinguishes murine MFS aortic aneurysm from healthy aortas, we next applied bioinformatic tools to identify biological pathways affected by this process. Differential expression testing to directly compare the SMC and modSMC clusters produced a list of 481 differentially express genes (DEGs) (Figure 4A, complete list in Supplemental Table 2). Genes with enriched expression in modSMCs (n=216, genes) were utilized for functional annotation. These upregulated genes were input into the DAVID bioinformatics database25 to identify gene ontology (GO) terms32 corresponding to biologic processes over-represented in modSMCs (Figure 4B). This analysis highlighted cell adhesion, proliferation and collagen fibril organization among the biologic processes activated during phenotypic modulation. Enhanced DEGs were also analyzed using the Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway database33, identifying ECM-receptor interaction (9–9-fold), Focal adhesion (5.6-fold), and Proteoglycans in cancer (5.4-fold) as the most enriched pathways (adjusted p value <0.05 for all pathways). Functional annotation of downregulated genes in modSMCs compared to SMCs emphasized SMC contraction as the chief biologic process and KEGG pathway affected (not shown). To identify possible transcription factors promoting SMC modulation, we performed transcription factor binding motif overrepresentation analysis using oPOSSUM26. Analyzing the 216 enriched genes in modSMCs identified KLF4 (177/216 genes with predicted binding sites, Z-score 39.7) and SP-1 (162/216 genes, Z-score 29.2) as the top two transcription factors with overrepresented motifs (Figure 4C).
Figure 4:

Dynamic phenotype modulation across SMC spectrum in Fbn1C1041G/+ mice. A Results of dichotomous differential expression test comparing modSMC to SMC clusters from Fbn1C1041G/+ and control mouse merged dataset. B Top 10 significant gene ontology (GO) terms and KEGG pathway terms overrepresented by genes with enhanced expression in modSMCs (n=216 genes). C Transcription factor (TF) motif analysis for genes upregulated in modSMC cluster (n=216 genes). TFs with Z score >2 standard deviations above mean considered significant. D UMAP projections of Fbn1C1041G/+ SMC/modSMC clusters and pseudotime trajectory analysis scores demonstrate dynamic modulation across spectrum. E Scatter plots for example DEGs depicting expression level as a function of pseudotime score. Each point represents a single cell, color scheme depicts Seurat clustering (red=SMC, blue=modSMC).
While dichotomous comparison between SMC sub-clusters is useful to identify DEGs, SMC phenotype change occurs over a continuous spectrum rather than in binary fashion18,31. To model this gradual shift, we performed pseudotime analysis27, which maps a trajectory along a continuous cell state transition and then determines each cell’s progress down this trajectory based upon that cell’s transcriptome. Plotting pseudotime values across the Fbn1C1041G/+ SMC spectrum highlighted gradual transcriptomic evolution (Figure 4D). We analyzed DEG expression levels as a function of pseudotime values, revealing multiple patterns of gene modulation (Figure 4E). Genes downregulated (e.g. Cnn1, Myh11) largely demonstrated gradual downslope and abrupt drop at the SMC/modSMC cluster boundary. Activated genes demonstrated multiple patterns; i) expression gradually increasing within the SMC cluster and peaking relatively early (Serpine1 and Klf4), ii) an abrupt step-up in expression at the transition point between clusters (Igfbp2, Ctgf), or iii) activation later in Pseudotime, peaking at the end of the trajectory in the most modulated subset (Tnfrsf11b and Vcam1).
SMC Modulation occurs temporally and spatially with advanced aneurysm in Fbn1C1041G/+ mice
We performed scRNAseq on aortic Root/AS tissue from 4-week old Fbn1C1041G/+ and control mice to assess for SMC phenotype modulation during early aneurysm development (n=3 animals each genotype, total 7,268 cells). UMAP projection of these data revealed all aortic cell types with a single SMC cluster and no disease-specific clusters (Figure 5A-B). We isolated the SMC cluster and split by Fbn1C1041G/+ (1,462 cells) and control samples (2,860 cells) for direct comparison (Figure 5C). Within the SMC cluster, Fbn1C1041G/+ cells occupied a distinct location in UMAP space, suggesting modest transcriptomic differences between genotypes. While SMC contractile marker (Acta2, Myh11, Cnn1) expression was unchanged between the groups, differential expression analysis comparing diseased to control SMCs identified 72 DEGs including enriched expression of Igfbp2, Ctgf, and Fn1 (Figure 5D).
Figure 5:

scRNAseq from 4-week Fbn1C1041G/+ and control Root/AS tissue. A UMAP dimensional reduction of merged data (>7,200 cells) with clustering of major aortic cell types. Blue circle indicates subsampled SMC cluster. B UMAP plot split by genotype with similar clustering projection and no disease-specific clusters. C UMAP projection of SMC subset color-coded by genotype. D Consistent expression of contractile genes Acta2, Myh11, and Cnn1 across genotypes. Violin plots for markers of Fbn1C1041G/+ SMCs (Igfbp2, Ctgf and Fn1). * denotes adjusted p-value <0.01.
We also performed scRNAseq on non-dilated descending thoracic aortic tissue from 24-week-old Fbn1C1041G/+ and control mice (n=3 animals each genotype, total 4,742 cells) to determine whether SMC modulation is specific to the aneurysmal aorta despite the systemic effects of Fbn1 mutations (Supplemental Figure 4). Similar to young mice, this analysis did not identify disease-specific cell clusters in the descending thoracic aorta.
RNA in-situ hybridization localizes modSMCs within diseased tunica media layer
Elastin fiber fragmentation and ECM degeneration within the aortic tunica media are hallmarks of MFS aortic aneurysm histopathology.34 Matrix metalloproteinases (MMP) including Mmp2 contribute to medial degeneration in MFS pathogenesis35–37; scRNAseq revealed that Mmp2 expression is significantly enriched in modSMCs in adult Fbn1C1041G/+ mice (log2FC = 0.49, p = 1.7E−160, Figure 6A). Osteoprotegerin (Tnfrsf11b) was a strong marker for highly modulated SMCs based on pseudotime analysis and dimensional reduction (Figure 4E, 6A). While both Mmp2 and Tnfrsf11b were detectable in more SMCs in Fbn1C1041G/+ mice at 4 weeks, expression levels did not reach statistical significance (Figure 6B). By 24 weeks, both genes were highly upregulated in Fbn1C1041G/+ mice (SMC/modSMC clusters considered together). We therefore utilized these genes as markers for SMC modulation to histologically localize modSMCs. We performed duplex chromogenic in situ hybridization with the RNAscope assay (Advanced Cell Diagnostics, Newark CA) using probes for Mmp2 (red) and Tnfrsf11b (blue) on aortic root tissue. While 4-week samples demonstrated qualitatively increased Mmp2 expression in the tunica media of Fbn1C1041G/+ mice (representative images of n=3 animals in Figure 6C), minimal Tnfrsf11b was detected in either genotype. In 24-week old samples, duplexed RNA detection revealed diffuse presence of strongly double-positive cells in the tunica media layer of Fbn1C1041G/+ aortic root samples (Figure 6D) while rare positive cells were found in littermate control specimens. In adventitial fibroblasts, diffuse hybridization with Mmp2 probe and minimal Tnfrsf11b positivity was observed in all samples, consistent with scRNAseq findings.
Figure 6:

RNAscope assay for modSMC markers. A UMAP projection of 24-week Fbn1C1041G/+ and control SMC/modSMC clusters with expression plots for matrix metalloproteinase-2 (Mmp2) and osteoprotegerin (Tnfrsf11b) expression enriched in modSMC cluster. B Fraction of SMCs expressing Mmp2 and Tnfrsf11b in 4- and 24-week scRNAseq datasets. Cells with >1 transcript for denoted gene considered positive. *** denotes adjusted p <0.01 (Wilcoxon rank sum test). C Chromogenic amplified in-situ hybridization using sequence-specific probes for Tnfrsf11b (blue) and Mmp2 (red) in 4-week Fbn1C1041G/+ and control aortas identifies qualitatively increased Mmp2 expression with minimal Tnfrsf11b positivity in diseased samples. D 24-week aortic tissue RNAscope. Rare Mmp2-positive cells identified in littermate control animals, double-positive cells in Fbn1C1041G/+ samples (red arrows) represent modSMCs in tunica media. Adventitial fibroblasts display significant Mmp2 and rare Tnfrsf11b staining, confirming efficient hybridization and amplification in all samples. Images shown are representative of results for n=3 animals of each genotype.
Human aortic root aneurysm scRNAseq confirms analogous SMC modulation associated with enriched TGF-β signaling
To apply our findings from the Fbn1C1041G/+ mouse to clinical disease, we performed scRNAseq on aortic root tissue from a 25-year-old male patient with MFS (pathologic variant FBN1 mutation c.7988G>A) undergoing elective repair of a 4.8cm aortic root aneurysm (Figure 7A). Histologic analysis showed elastin fragmentation and collagen deposition, and in situ hybridization for genes expressed during early (SERPINE1) and late (TNFRSF11B) SMC modulation in mice showed diffuse expression within the tunica media (Figure 7B). Integration of this dataset with Fbn1C1041G/+ male mouse data in Seurat demonstrated a modulated SMC cluster between SMCs and fibroblasts (Figure 7C). We also analyzed the human patient dataset independently and found a similar array of cell types (Figure 7D-E); 81.2% of cells assigned the modSMC identity in the integrated dataset were grouped into an analogous cluster in the human-only analysis. Differential expression testing between human SMCs and modSMCs identified 393 differentially expressed genes (268 upregulated, 125 downregulated. Example DEGs in Figure 7F, complete list in Supplemental Table 3). Collagen expression is a prominent feature of MFS aortic root aneurysm and is inducible by TGF-β in SMC culture6,19. Both mouse and human scRNAseq data demonstrated significantly enhanced Col1a1 in the modulated SMC subset (Figure 8). Furthermore, enhanced expression of connective tissue growth factor (Ctgf) and plasminogen-activator inhibitor 1 (Serpine1), both markers of active TGF-β signaling, was associated with modSMC identity. Expression of Tgfb1 ligand was also significantly enriched in modSMCs within both datasets (Figure 8B-C, E-F).
Figure 7:

Human MFS aortic root aneurysm scRNAseq. A Three-dimensional CTA reconstruction of 4.8cm aortic root aneurysm. B Representative histologic stains and RNAscope in-situ hybridization (20× magnification) for SERPINE1 (blue) and TNFRSF11B (red) from human MFS aortic tissue. ‘L’ indicates aortic lumen. C Results from dataset integration of human and Fbn1C1041G/+ mouse scRNAseq. modSMC cluster (blue) is present within both individual datasets. D Independent dimensional reduction and cell clustering of MFS human scRNAseq data. Similar clustering results are observed when plotted based on integrated dataset clustering (top) or independent clustering (human sample only, bottom). E DotPlot for top cluster markers in human dataset. Dot size denotes percentage of cells expressing noted gene, color scale denotes relative expression values. F Representative expression plots for (DEGs) in human modSMCs compared to SMCs. Reduced genes (top) in red, enriched genes (bottom) in blue. Color scale indicates range from 5th (Low) to 95th (High) percentile of normalized expression levels by individual cells.
Figure 8:

Transcriptomic signature of TGF-β signaling is enhanced in modSMCs. A UMAP dimensional reduction plot of Fbn1C1041G/+ and control adult mice SMC, modSMC and fibroblast clusters. B expression plots and C violin plots demonstrating individual cell transcript levels for canonical TGF-β signaling-responsive genes collagen 1 (Col1a1), connective tissue growth factor (Ctgf), plasminogen activator inhibitor-1 (Serpine1) and TGF-β1 ligand (Tgfb1). D-F Analogous data from human MFS patient aortic root tissue. ***p<0.001 by Wilcoxon rank sum test. Color scale indicates range from 5th (Low) to 95th (High) percentile of normalized expression levels by individual cells.
Discussion
Dynamic alteration of vascular SMC phenotype contributes to many vascular disease states16,22,30,38. While changes in SMC function are dependent on complex signaling processes, the transcriptome provides the most comprehensive view of cellular “programming”. scRNAseq has recently identified heterogeneity in aortic endothelial cells,20 healthy SMCs,21 and dynamic phenotype modulation in murine atherosclerosis and human coronary artery disease22. Although SMC phenotype shift toward a synthetic state and resultant ECM modulation is a core concept in MFS aneurysm formation,6,14,19,39,40 whole transcriptomic assessment to fully this process in vivo is lacking. Using scRNAseq, we 1) characterize dynamic transcriptomic changes across the spectrum of SMC modulation during aneurysm development and progression; 2) identify candidate upstream molecular contributors to phenotype modulation and 3) translate murine scRNAseq findings to clinical human disease using bioinformatic data integration techniques.
Disease-specific signature of SMC phenotype modulation
The principal finding from our initial scRNAseq analysis was the identification of a distinct, disease-specific cell cluster within Fbn1C1041G/+ aortic aneurysm tissue; whether this cluster truly represents a biologically distinct cell type or the most extreme pole along the trajectory of SMC modulation is conceptually debatable. Nevertheless, applying dichotomous (SMC vs. modSMC) comparisons informs the functional changes associated with SMC modulation in broad strokes. Functional annotation of the 216 genes enriched in modSMCs established adhesion, ECM organization and proliferation as the most substantively enhanced pathways. Collagen fibril organization and Positive regulation of collagen biosynthetic process were the most enriched GO biologic process terms (18.3- and 23.8-fold enrichment), driven by enhanced expression of six distinct collagen isoforms and multiple small leucine-rich proteoglycans (Fmod, Lum, Bgn). Furthermore, expression of Tgfb1 ligand, lysyl oxidase-like 1 (Loxl1) and fibrosis-related genes Ctgf, Comp (cartilage oligomeric matrix protein) and Thbs1 (thrombospondin-1) support collagen deposition as a consequence of phenotype modulation41–43. Whether collagen deposition accelerates aneurysm formation or represents a beneficial response to reinforce the aortic wall remains controversial. Clinically, collagen deposition may reduce distensibility and increase aortic stiffness, predisposing to aneurysm and dissection43,44 In contrast, inhibition of mature collagen deposition via lysyl oxidase blockade accelerated aneurysm development in the Fbn1mgR/mgR mouse model45. Thus, while our scRNAseq data suggest that SMC modulation promotes aortic collagen deposition, inhibition of this process will be needed to determine its contribution to aneurysm progression.
Enriched DEGs also included genes related to elastic fiber synthesis (Eln, Fbln2, Fbln5, Fbn1), laminins (Lamc3, Lama2), fibronectin (Fn1) and both enhancers/inhibitors of ECM proteolysis (Mmp2, Timp1, Timp3, Timp4). While the balance of proteolysis/protein deposition in vivo is dependent on activation/inhibition mechanisms downstream of RNA transcription, Mmp2 expression is a central contributor to ECM degradation in MFS pathogenesis, suggesting modSMCs contribute to pathologic ECM remodeling.
Co-clustering of Fbn1C1041G/+ modulated SMCs with an analogous cluster from SMC lineage-traced ApoE−/− mice supported the prevailing model of SMC trans-differentiation in MFS. Nevertheless, these pathologies are quite distinct; whereas phenotypically modulated SMCs hone to the diseased intima and contribute to plaque and fibrous cap formation in atherosclerosis22,31, we observe modSMCs histologically within the tunica media. Furthermore, we identified hundreds of differentially expressed genes between Fbn1C1041G/+ and ApoE−/− models, emphasizing disease-specific mechanisms despite similar relative location in reduced-dimensional single cell transcriptomic space. In this sense, applying broad terms such as “synthetic” or “proliferative” SMCs fails to capture the specificity of SMC contribution to either disease state. A critical question remains whether this pattern is MFS-specific or broadly applicable to thoracic aortic aneurysm in general.
scRNAseq identifies TGF-β and Klf4 as potential drivers of SMC modulation
TGF-β promotes SMC differentiation and contractile gene expression in vitro and in vivo6,19,46,47, yet transcriptomic evidence of enhanced TGF-β signaling (Serpine1, Ctgf, Col1a1) correlated with SMC modulation in 24 week-old mice and human MFS tissue (Figure 8). Serpine1 overexpression also distinguished Fbn1C1041G/+ modulated SMCs from the ApoE−/− subset. While the precise contribution of TGF-β to the genesis and progression of aortic aneurysm remains enigmatic, enhanced TGF-β signaling appears to have deleterious effects later in life across MFS murine models7,8,10,48. Pseudotime trajectory analysis revealed that Serpine1 expression increased gradually along the spectrum of phenotype modulation and peaked with transition to the modSMC cluster (Figure 4E). Accordingly, it is possible that pathologically elevated TGF-β signaling promotes SMC modulation, which may in part explain the harmful role of TGF-β later in the disease process. Tgfb1 ligand overexpression in modSMCs presents the possibility of autocrine and paracrine signaling within the cellular microenvironment
Bioinformatic tools enabled the prediction of upstream transcription factors that may promote SMC modulation, including Klf4 and Sp1, which both potentiate PDGF-mediated SMC phenotype change in vitro.49 Furthermore, Klf4 has been implicated as a mediator of SMC phenotype modulation in murine models of atherosclerosis50 and abdominal aortic aneurysm51. In MFS, KLF4 overexpression was identified in adult aortic tissue specimens and iPSC-derived SMCs52, but reduced Klf4 in very young Fbn1mgR/mgR mice was found to drive deleterious overexpression of contractile markers38. These reports suggest dynamic temporal regulation of both Klf4 expression and SMC phenotype in MFS, though upstream drivers of Klf4 expression in MFS are unknown. In our dataset Klf4 was not only a predicted upstream driver of SMC modulation, but was also overexpressed compared to both control mice and ApoE−/− modulated SMCs. Further, the relatively early rise of Klf4 expression during SMC modulation raises the possibility that it may be involved in the earliest initiating stages of this process. Collectively these data implicate Klf4 as an important target for further study.
Temporal and regional dynamics of SMC transcriptomic changes
We employed two approaches to delineate the temporal dynamics of SMC modulation in MFS aortic aneurysm. First, applying pseudotime analysis to the 24 weeks dataset identified subsets of genes activated differentially throughout the total SMC spectrum. Second, we performed separate scRNAseq analysis of 4 week-old Fbn1C1041G/+ and control mice. We did not identify a distinct disease-specific cell cluster or contractile gene expression reduction despite the development of small aortic aneurysm by this age. Nevertheless, multiple genes activated “early” in modSMCs by pseudotime analysis (Ctgf, Igfbp2) were already enriched at 4 weeks, while “late” markers (Vcam1, Tnfrsf11b) were not. Thus, while global alteration of SMC function may not represent the inciting event in aneurysm development, we propose that SMCs destined to undergo trans-differentiation may be activated early in life. Bunton et al. proposed SMC-mediated elastolysis and synthetic phenotype development as a result of poor adhesion to ECM substrate nearly two decades ago;14 using scRNAseq we add high-resolution, longitudinal transcriptomic context to this concept.
SMC modulation was region-specific; we did not identify a modSMC cluster or enhanced expression of Serpine1, Ctgf, or Col1a1 in SMCs in the descending thoracic aorta from adult Fbn1C1041G/+ mice (Supplemental Figure 4B). This specificity supports the hypothesis that embryonic origin-specific responses to stimuli contribute to focal aortic root aneurysm development despite systemic effects of Fbn1 mutation53,54.
Human aortic tissue scRNAseq complements MFS animal model findings
Human MFS patient aortic root scRNAseq identified a similar pattern of SMC phenotype modulation, augmenting our findings in the murine model. While limited by the availability of only one patient, scRNAseq permits the acquisition of thousands of useful data points from a single aortic tissue sample. We identified osteoprotegerin (TNFRSF11B) as a consistent marker of the most extreme pole of modulated SMCs across species. Elevated osteoprotegerin levels are associated with proteolysis in abdominal aortic aneurysm, and knockout reduced AAA rupture in an experimental murine model. To our knowledge, this is the first report of scRNAseq data from human MFS aortic aneurysm tissue and association of TNFRSF11B overexpression with MFS aneurysm; whether enriched expression contributes to aneurysm progression or merely associates with SMC modulation remains to be determined. Ultimately a larger cohort of human MFS samples and non-aneurysmal controls will be needed to identify the most consistently modulated genes in clinical MFS aortic aneurysm.
Our murine scRNAseq findings did not completely translate to changes in human MFS. For example, expression of angiotensin converting enzyme (Ace) was greatly enriched in modSMCs in the murine dataset (Supplemental Figure 5). Given that enhanced Ace expression could increase local angiotensin II levels and drive promiscuous ERK signaling, our initial theory was that it may contribute to aneurysm formation. This finding did not translate to our human dataset where ACE expression was undetectable in SMCs, modSMCs or fibroblasts. This finding may partially explain the varied results in the losartan clinical trials in MFS patients, which were motivated by losartan efficacy in same murine model system.9,13,55,56 Furthermore, while we and others identified increased contractile gene expression in human MFS compared to controls, murine scRNAseq data did not identify enhanced contractile genes in the SMC cluster. Bulk aortic root tissue RT-PCR of 24-week-old Fbn1C1041G/+ revealed no significant differences in Myh11, Acta2, or Tagln (Supplemental Figure 6). These findings underscore the limitations of small animal models and highlight the importance of further human tissue studies.
This study has several limitations. While scRNAseq permits simultaneous characterization of every cell within the aorta, this data provides a limited view of the true functional changes in MFS pathogenesis that are undoubtedly affected by cellular processes downstream of gene transcription. We inferred that the modSMC cluster trans-differentiates from SMCs based on scRNAseq data and bioinformatic comparisons to SMC lineage-traced mice. However, it is also possible that adventitial stem cells or myofibroblasts may trans-differentiate towards a contractile phenotype and migrate into the tunica media as well. The Fbn1C1041G/+ model faithfully recapitulates MFS aortic root aneurysm pathology, but unlike human disease this model also develops significant dilation of the ascending aorta (Figure 1A). We include the most proximal portion of the ascending aorta in murine scRNAseq experiments to ensure adequate cell yield. These factors limit the specificity of our findings, which would ideally isolate the pathology within the aortic root seen in clinical MFS. Our scRNAseq data failed to explain the significant sexual dimorphism observed in MFS (Supplemental Figure 2). Increased collagen expression in males may partially explain more severe aneurysm progression. Alternatively, while similar expression profiles were found within clusters between sexes, a larger fraction of SMCs may progress towards the modulated phenotype. However, given the large number of biological replicates to confirm such a hypothesis, scRNAseq is not a practical approach for this type of quantification. A more focused, adequately powered study will be needed to definitively address SMC phenotype changes related to sex-related dimorphism. Finally, while we hypothesize that SMC modulation potentiates aortic root aneurysm, we cannot definitively rule out a partially reparative effect of reinforcing the weakened aortic wall via ECM deposition. Further mechanistic studies using pharmacologic or transgenic approaches to inhibit this process will ultimately be needed to answer this important question.
Supplementary Material
Highlights.
Single cell RNA sequencing (scRNAseq) in young and adult Fbn1C1041G/+ Marfan syndrome mice permits high-resolution analysis of smooth muscle cell (SMC) phenotype modulation throughout aortic aneurysm development
Transcriptomic signature of enriched transforming growth factor-beta (TGFβ) signaling is a feature of developed aneurysm and correlates with SMC phenotype change.
Bioinformatic data integration permits direct comparison of human and mouse scRNAseq data, identifying novel markers of SMC modulation in human Marfan syndrome.
Acknowledgements
The authors with to thank the excellent faculty and staff at the Stanford Shared FACS Facility (SSFF) for critical assistance with cell sorting and the Stanford Genome Sequencing Service Center (GSSC) for technical assistance with library preparation and sequencing.
Sources of Funding
This work was supported by NIH grant 1R01AR066629–01A1 (to MPF), NIH grant K08HL152308 and American Heart Association grant AHA18CDA34110206 (to RCW).
Abbreviations
- scRNAseq
Single-cell RNA sequencing
- Fbn1
Fibrillin-1
- MFS
Marfan Syndrome
- Root/AS
Aortic root/ascending aorta
- SMC
Smooth muscle cell
- modSMC
Modulated smooth muscle cell
- ECM
Extracellular matrix
- DEG
Differentially express gene
- TGF-β
Transforming growth factor-beta
- Klf4
Kruppel-like factor 4
- Ctgf
Connective tissue growth factor
- Serpine1
Plasminogen activator inhibitor-1
- Col1a1
Collagen type 1 alpha 1
- Mmp2
Matrix metalloproteinase-2
- Tnfrsf11b
Tumor necrosis factor receptor super family member 11b (Osteoprotegerin)
- Myh11
Myosin heavy chain 11
Footnotes
Disclosures
The authors confirm that there are no financial disclosures or conflicts of interest.
References
- 1.Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352(6333):337–339. doi: 10.1038/352337a0 [DOI] [PubMed] [Google Scholar]
- 2.Judge DP, Dietz HC. Marfan’s syndrome. Lancet. 2005;366(9501):1965–1976. doi: 10.1016/S0140-6736(05)67789-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saeyeldin A, Zafar MA, Velasquez CA, Ip K, Gryaznov A, Brownstein AJ, Li Y, Rizzo JA, Erben Y, Ziganshin BA, Elefteriades JA. Natural history of aortic root aneurysms in Marfan syndrome. Ann Cardiothorac Surg. 2017;6(6):625–632. doi: 10.21037/acs.2017.11.10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Judge DP, Dietz HC. Therapy of Marfan Syndrome. Annu Rev Med. Published online 2008. doi: 10.1146/annurev.med.59.103106.103801 [DOI] [PubMed] [Google Scholar]
- 5.Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science (80- ). 2006;312(5770):117–121. doi: 10.1126/science.1124287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Crosas-Molist E, Meirelles T, López-Luque J, et al. Vascular smooth muscle cell phenotypic changes in patients with marfan syndrome. Arterioscler Thromb Vasc Biol. 2015;35(4):960–972. doi: 10.1161/ATVBAHA.114.304412 [DOI] [PubMed] [Google Scholar]
- 7.Wei H, Hu JH, Angelov SN, Fox K, Yan J, Enstrom R, Smith A, Dichek DA. Aortopathy in a Mouse Model of Marfan Syndrome Is Not Mediated by Altered Transforming Growth Factor beta Signaling. J Am Hear Assoc. 2017;6(1). doi: 10.1161/JAHA.116.004968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tellides G Further Evidence Supporting a Protective Role of Transforming Growth Factor-β (TGFβ) in Aortic Aneurysm and Dissection. Arterioscler Thromb Vasc Biol. 2017;37(11):1983–1986. doi: 10.1161/ATVBAHA.117.310031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Habashi JP, Judge DP, Holm TM, et al. Losartan, an AT1 antagonist, prevents aortic aneurysm in a mouse model of Marfan syndrome. Science (80- ). Published online 2006. doi: 10.1126/science.1124287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook JR, Clayton NP, Carta L, Galatioto J, Chiu E, Smaldone S, Nelson CA, Cheng SH, Wentworth BM, Ramirez F. Dimorphic effects of transforming growth factor-beta signaling during aortic aneurysm progression in mice suggest a combinatorial therapy for Marfan syndrome. Arter Thromb Vasc Biol. 2015;35(4):911–917. doi: 10.1161/ATVBAHA.114.305150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lindsay ME, Schepers D, Bolar NA, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44(8):922–927. doi: 10.1038/ng.2349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, Offermanns S, Humphrey JD, Tellides G. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014;124:755–767. doi: 10.1172/JCI69942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Holm TM, Habashi JP, Doyle JJ, et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in Marfan syndrome mice. Science. 2011;332(6027):358–361. doi: 10.1126/science.1192149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bunton TE, Jensen Biery N, Myers L, Gayraud B, Ramirez F, Dietz HC. Phenotypic alteration of vascular smooth muscle cells precedes elastolysis in a mouse model of Marfan syndrome. Circ Res. Published online 2001. doi: 10.1161/01.RES.88.1.37 [DOI] [PubMed] [Google Scholar]
- 15.Dale M, Fitzgerald MP, Liu Z, et al. Premature aortic smooth muscle cell differentiation contributes to matrix dysregulation in Marfan Syndrome. Pereira LV, ed. PLoS One. 2017;12(10):e0186603. doi: 10.1371/journal.pone.0186603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu M, Gomez D . Smooth Muscle Cell Phenotypic Diversity At the Crossroads of Lineage Tracing and Single-Cell Transcriptomics. Arter Thromb Vasc Biol. Published online 2019. doi: 10.1161/ATVBAHA.119.312131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Riches K, Clark E, Helliwell RJ, Angelini TG, Hemmings KE, Bailey MA, Bridge KI, Scott DJA, Porter KE. Progressive Development of Aberrant Smooth Muscle Cell Phenotype in Abdominal Aortic Aneurysm Disease. J Vasc Res. 2018;55(1):35–46. doi: 10.1159/000484088 [DOI] [PubMed] [Google Scholar]
- 18.Lacolley P, Regnault V, Nicoletti A, Li Z, Michel JB. The vascular smooth muscle cell in arterial pathology: A cell that can take on multiple roles. Cardiovasc Res. Published online 2012. doi: 10.1093/cvr/cvs135 [DOI] [PubMed] [Google Scholar]
- 19.Pedroza AJ, Koyano T, Trojan J, Rubin A, Palmon I, Jaatinen K, Burdon G, Chang P, Tashima Y, Cui JZ, Berry G, Iosef C, Fischbein MP. Divergent effects of canonical and non‐canonical TGF‐β signalling on mixed contractile‐synthetic smooth muscle cell phenotype in human Marfan syndrome aortic root aneurysms. J Cell Mol Med. 2019;(December):1–15. doi: 10.1111/jcmm.14921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kalluri AS, Vellarikkal SK, Edelman ER, Nguyen L, Subramanian A, Ellinor PT, Regev A, Kathiresan S, Gupta RM. Single-Cell Analysis of the Normal Mouse Aorta Reveals Functionally Distinct Endothelial Cell Populations. Circulation. Published online 2019. doi: 10.1161/CIRCULATIONAHA.118.038362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dobnikar L, Taylor AL, Chappell J, Oldach P, Harman JL, Oerton E, Dzierzak E, Bennett MR, Spivakov M, Jørgensen HF. Disease-relevant transcriptional signatures identified in individual smooth muscle cells from healthy mouse vessels. Nat Commun. Published online 2018. doi: 10.1038/s41467-018-06891-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wirka RC, Wagh D, Paik DT, et al. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nat Med. Published online 2019. doi: 10.1038/s41591-019-0512-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li G, Humphrey JD, Tellides G, et al. Chronic mTOR activation induces a degradative smooth muscle cell phenotype Graphical abstract Find the latest version : Chronic mTOR activation induces a degradative smooth muscle cell phenotype. J Clin Invest. Published online 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R. Comprehensive Integration of Single-Cell Data. Cell. Published online 2019. doi: 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. Published online 2009. doi: 10.1038/nprot.2008.211 [DOI] [PubMed] [Google Scholar]
- 26.Kwon AT, Arenillas DJ, Hunt RW, Wasserman WW. Opossum-3: Advanced analysis of regulatory motif over-representation across genes or chip-seq datasets. G3 Genes, Genomes, Genet. Published online 2012. doi: 10.1534/g3.112.003202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. Published online 2014. doi: 10.1038/nbt.2859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Judge DP, Biery NJ, Keene DR, Geubtner J, Myers L, Huso DL, Sakai LY, Dietz HC. Evidence for a critical contribution of haploinsufficiency in the complex pathogenesis of Marfan syndrome. J Clin Invest. 2004;114(2):172–181. doi: 10.1172/JCI20641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol. Published online 2018. doi: 10.1038/nbt.4096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gomez D, Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovasc Res. Published online 2012. doi: 10.1093/cvr/cvs115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Basatemur GL, Jørgensen HF, Clarke MCH, Bennett MR, Mallat Z. Vascular smooth muscle cells in atherosclerosis. Nat Rev Cardiol. Published online 2019. doi: 10.1038/s41569-019-0227-9 [DOI] [PubMed] [Google Scholar]
- 32.Blake JA, Christie KR, Dolan ME, et al. Gene ontology consortium: Going forward. Nucleic Acids Res. Published online 2015. doi: 10.1093/nar/gku1179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. Published online 2017. doi: 10.1093/nar/gkw1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Grewal N, Gittenberger-de Groot AC. Pathogenesis of aortic wall complications in Marfan syndrome. Cardiovasc Pathol Published online 2018. doi: 10.1016/j.carpath.2018.01.005 [DOI] [PubMed] [Google Scholar]
- 35.Xiong W, Meisinger T, Knispel R, Worth JM, Baxter BT. MMP-2 Regulates Erk1/2 phosphorylation and aortic dilatation in marfan syndrome. Circ Res. Published online 2012. doi: 10.1161/CIRCRESAHA.112.268268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Merk DR, Chin JT, Dake BA, Maegdefessel L, Miller MO, Kimura N, Tsao PS, Iosef C, Berry GJ, Mohr FW, Spin JM, Alvira CM, Robbins RC, Fischbein MP. MiR-29b participates in early aneurysm development in Marfan syndrome. Circ Res Published online 2012. doi: 10.1161/CIRCRESAHA.111.253740 [DOI] [PubMed] [Google Scholar]
- 37.Chung a WY, Au Yeung K, Cortes SF, Sandor GGS, Judge DP, Dietz HC, van Breemen C. Endothelial dysfunction and compromised eNOS/Akt signaling in the thoracic aorta during the progression of Marfan syndrome. Br J Pharmacol. 2007;150(8):1075–1083. doi: 10.1038/sj.bjp.0707181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dale M, Fitzgerald MP, Liu Z, et al. Premature aortic smooth muscle cell differentiation contributes to matrix dysregulation in Marfan Syndrome. PLoS One Published online 2017. doi: 10.1371/journal.pone.0186603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Milewicz DM, Guo D-C, Tran-Fadulu V, Lafont AL, Papke CL, Inamoto S, Kwartler CS, Pannu H. Genetic Basis of Thoracic Aortic Aneurysms and Dissections: Focus on Smooth Muscle Cell Contractile Dysfunction. Annu Rev Genomics Hum Genet. 2008;9(1):283–302. doi: 10.1146/annurev.genom.8.080706.092303 [DOI] [PubMed] [Google Scholar]
- 40.Perrucci GL, Rurali E, Gowran A, Pini A, Antona C, Chiesa R, Pompilio G, Nigro P. Vascular smooth muscle cells in Marfan syndrome aneurysm: the broken bricks in the aortic wall. Cell Mol Life Sci. Published online 2016. doi: 10.1007/s00018-016-2324-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Halper J, Kjaer M. Basic Components of Connective Tissues and Extracellular Matrix: Elastin, Fibrillin, Fibulins, Fibrinogen, Fibronectin, Laminin, Tenascins and Thrombospondins. In: 2014. doi: 10.1007/978-94-007-7893-1_3 [DOI] [PubMed] [Google Scholar]
- 42.Brigstock DR. Connective tissue growth factor (CCN2, CTGF) and organ fibrosis: Lessons from transgenic animals. J Cell Commun Signal Published online 2010. doi: 10.1007/s12079-009-0071-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang X, LeMaire SA, Chen L, Shen YH, Gan Y, Bartsch H, Carter SA, Utama B, Ou H, Coselli JS, Wang XL. Increased collagen deposition and elevated expression of connective tissue growth factor in human thoracic aortic dissection. Circulation. 2006;114(1 Suppl):I200–5. doi: 10.1161/CIRCULATIONAHA.105.000240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Baumgartner D, Baumgartner C, Mátyás G, Steinmann B, Löffler-Ragg J, Schermer E, Schweigmann U, Baldissera I, Frischhut B, Hess J, Hammerer I. Diagnostic power of aortic elastic properties in young patients with Marfan syndrome. J Thorac Cardiovasc Surg. 2005;129(4):730–739. doi: 10.1016/j.jtcvs.2004.07.019 [DOI] [PubMed] [Google Scholar]
- 45.Busnadiego O, Gorbenko del Blanco D, González-Santamaría J, et al. Elevated expression levels of lysyl oxidases protect against aortic aneurysm progression in Marfan syndrome. J Mol Cell Cardiol. 2015;85:48–57. doi: 10.1016/j.yjmcc.2015.05.008 [DOI] [PubMed] [Google Scholar]
- 46.Hu JH, Wei H, Jaffe M, Airhart N, Du L, Angelov SN, Yan J, Allen JK, Kang I, Wight TN, Fox K, Smith A, Enstrom R, Dichek D a. Postnatal Deletion of the Type II Transforming Growth Factor-β Receptor in Smooth Muscle Cells Causes Severe Aortopathy in Mice. Arterioscler Thromb Vasc Biol. 2015;35(12):2647–2656. doi: 10.1161/ATVBAHA.115.306573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Li W, Li Q, Jiao Y, Qin L, Ali R, Zhou J, Ferruzzi J, Kim RW, Geirsson A, Dietz HC, Offermanns S, Humphrey JD, Tellides G. Tgfbr2 disruption in postnatal smooth muscle impairs aortic wall homeostasis. J Clin Invest. 2014;124(2):755–767. doi: 10.1172/JCI69942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holm TM, Habashi JP, Doyle JJ, et al. Noncanonical TGFβ signaling contributes to aortic aneurysm progression in marfan syndrome mice. Science (80- ). 2011;332(6027):358–361. doi: 10.1126/science.1192149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Deaton RA, Gan Q, Owens GK. Spl-dependent activation of KLF4 is required for PDGF-BB-induced phenotypic modulation of smooth muscle. Am J Physiol - Hear Circ Physiol. Published online 2009. doi: 10.1152/ajpheart.01230.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shankman LS, Gomez D, Cherepanova OA, Salmon M, Alencar GF, Haskins RM, Swiatlowska P, Newman AAC, Greene ES, Straub AC, Isakson B, Randolph GJ, Owens GK. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nat Med. Published online 2015. doi: 10.1038/nm.3866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Salmon M, Johnston WF, Woo A, Pope NH, Su G, Upchurch GR, Owens GK, Ailawadi G. KLF4 regulates abdominal aortic aneurysm morphology and deletion attenuates aneurysm formation. Circulation. Published online 2013. doi: 10.1161/CIRCULATIONAHA.112.000238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Granata A, Serrano F, Bernard WG, McNamara M, Low L, Sastry P, Sinha S. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet. Published online 2017. doi: 10.1038/ng.3723 [DOI] [PubMed] [Google Scholar]
- 53.Sinha S, Iyer D, Granata A. Embryonic origins of human vascular smooth muscle cells: Implications for in vitro modeling and clinical application. Cell Mol Life Sci. 2014;71(12):2271–2288. doi: 10.1007/s00018-013-1554-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Granata A, Serrano F, Bernard WG, McNamara M, Low L, Sastry P, Sinha S. An iPSC-derived vascular model of Marfan syndrome identifies key mediators of smooth muscle cell death. Nat Genet. 2017;49(1):97–109. doi: 10.1038/ng.3723 [DOI] [PubMed] [Google Scholar]
- 55.Groenink M, Den Hartog AW, Franken R, Radonic T, De Waard V, Timmermans J, Scholte AJ, Van Den Berg MP, Spijkerboer AM, Marquering HA, Zwinderman AH, Mulder BJM. Losartan reduces aortic dilatation rate in adults with Marfan syndrome: A randomized controlled trial. Eur Heart J. Published online 2013. doi: 10.1093/eurheartj/eht334 [DOI] [PubMed] [Google Scholar]
- 56.Teixido-Tura G, Forteza A, Rodríguez-Palomares J, González Mirelis J, Gutiérrez L, Sánchez V, Ibáñez B, García-Dorado D, Evangelista A. Losartan Versus Atenolol for Prevention of Aortic Dilation in Patients With Marfan Syndrome. J Am Coll Cardiol. Published online 2018. doi: 10.1016/j.jacc.2018.07.052 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data and materials have been made publicly available at the gene expression omnibus (GEO) repository under accession ID GSE153534.