

Abstract
Tipifarnib is a potent and highly selective inhibitor of farnesyltransferase (FT). FT catalyzes the post-translational attachment of farnesyl groups to signaling proteins that are required for localization to cell membranes. Although all RAS isoforms are FT substrates, only HRAS is exclusively dependent upon farnesylation, raising the possibility that HRAS mutant tumors might be susceptible to tipifarnib-mediated inhibition of FT. Here, we report the characterization of tipifarnib activity in a wide panel of HRAS mutant and wild type HNSCC xenograft models. Tipifarnib treatment displaced both mutant and wild type HRAS from membranes but only inhibited proliferation, survival and spheroid formation of HRAS mutant cells. In vivo, tipifarnib treatment induced tumor stasis or regression in all six HRAS mutant xenografts tested but displayed no activity in six HRAS wild type PDX models. Mechanistically, drug treatment resulted in reduction of MAPK pathway signaling, inhibition of proliferation and induction of apoptosis and robust abrogation of neovascularization, apparently via effects on both tumor cells and endothelial cells. Bioinformatics and quantitative image analysis further revealed that FT inhibition induces progressive squamous cell differentiation in tipifarnib-treated HNSCC PDX. These preclinical findings support that HRAS represents a druggable oncogene in HNSCC through FT inhibition by tipifarnib, thereby identifying a precision therapeutic option for HNSCCs harboring HRAS mutations.
Keywords: HNSCC, HRAS, farnesyltransferase, tipifarnib, precision medicine
Introduction
Squamous cell carcinoma of the head and neck (HNSCC) is the sixth most common cancer worldwide, with an estimated annual incidence of 600,000 patients. In the United States, 55,000 new cases are diagnosed each year, leading to nearly 13,000 deaths annually (1). Early-stage HNSCC disease is treated relatively well with single-modality therapy (either surgery or radiation), but nearly 66% of patients present with advanced disease and fewer than 30% of these patients are cured. Few drugs have proven effective in HNSCC therapy. Systemic, platinum-based chemotherapy is the mainstay of first line treatment, and combination with 5-FU and the anti-EGFR antibody cetuximab has been shown to extend overall survival in the metastatic setting (2). However, despite significant advances in the understanding of the molecular underpinnings of this group of tumors, cetuximab was the only molecularly-targeted drug approved for HNSCC until the arrival of anti-PD-1 antibodies in 2016, and checkpoint inhibitors only produce durable responses in a minority (<15-20%) of patients (3). Thus, identification and exploitation of novel druggable oncogenes in HNSCC is urgently needed to improve patient outcomes.
Farnesyltransferase (FTase) is cytosolic metalloenzyme that catalyzes the transfer of a 15-carbon farnesyl lipid moiety to a group of cellular proteins characterized by a C-terminal CAAX motif (4). Farnesylation is required for cellular membrane insertion and subsequent activity of certain signaling proteins associated with cancer progression (5), spurring the development of several FTase inhibitors (FTIs) in the late 1990’s and early 2000’s. The first selective FTI to enter clinical studies was tipifarnib (R115777), a heterocyclic non peptidomimetic drug that inhibits farnesylation of the canonical FTase substrate lamin A with subnanomolar potency (6). The clinical development of tipifarnib began in 1997 and consisted of more than 70 clinical oncology and hematology studies. Many RAS family proteins are farnesylated at steady state, so FTIs were originally conceived as KRAS inhibitors and tested in high-prevalence KRAS-driven tumors. However, it was subsequently discovered that certain farnesylated proteins, including KRAS and NRAS, can be rescued from membrane displacement in the presence of FTIs by an alternative prenylation pathway mediated by the enzyme geranylgeranyl transferase, so clinical activity of tipifarnib and other FTIs in KRAS- and NRAS-driven cancer was modest. By contrast, the third family member, HRAS, is not a substrate for the geranylgeranylated transferase, and its membrane localization and cellular function are suppressed by FTIs (7).
HRAS was originally identified as an oncogene in chemical carcinogenesis studies of skin squamous cell carcinoma (SCC), and recent genomic analyses reveal that it is the predominant mutated RAS isoform in SCCs of several types, including HNSCC (8). The Cancer Gene Atlas (TCGA) reports that HRAS is mutated in 6% of HNSCC at initial diagnosis (9), and higher frequencies have been reported in some demographic groups associated with exposure to specific oral carcinogens (10). In addition, HRAS mutations have been reported in 15% of patients during acquisition of resistance to cetuximab therapy (11).
Prior to a recent report of tipifarnib activity in HRAS-mutant thyroid carcinoma (12), historical studies of FTIs in HRAS-mutant settings have employed the bladder carcinoma line T24 (6,13), breast cancer lines (14) or recombinant models (6,14,15). Here, we report an in-depth characterization of the antitumor activity of tipifarnib in a series of cell line- and patient-derived HNSCC xenograft models that capture the genomic diversity of this patient subset. Tipifarnib displaced HRAS from cellular membranes and selectively inhibited proliferation and survival of HRAS-mutant HNSCC cells in vitro. In xenograft models, tipifarnib blocked tumor growth and induced regressions in cell line- and early passage patient-derived HNSCCs and was associated with robust inhibition of MAPK pathway signaling downstream of activated HRAS. FTI treatment also blocked neovascularization, in part via HRAS-independent mechanisms, and analysis of gene expression changes following tipifarnib treatment of HRAS-mutant PDX models confirmed a robust G1-S cell cycle block downstream of MAPK pathway inhibition and revealed induction of squamous lineage differentiation in vivo.
Materials and Methods
Cell lines and tissue culture
Human head and neck cancer cell lines CAL27, DETROIT562 (HRAS wild type), HN31 and UMSCC17B (HRAS mutant) were collected as part of the NIDCR Oral and Pharyngeal Cancer Branch cell collection and have been described previously(16,17). The novel cell lines ORL48 (HRAS wild type) and ORL214 (HRAS mutant) were generously provided by Dr. Sok Ching Cheong. To ensure consistency in cell identity, all the cell lines underwent DNA authentication by multiplex STR profiling (Genetica DNA Laboratories, Inc. Burlington, NC, USA) prior to experiments. No mycoplasma was detected through Mycoplasma Detection Kit-QuickTest from Biomake (Houston, TX, USA). See Supplemental Materials for additional details.
Tipifarnib
Tipifarnib was provided by Kura Oncology. See Supplemental Materials for drug storage and preparation in vitro and in vivo.
In vivo mouse experiments and analysis
Studies on cell line-derived HNSCC xenografts were performed at the University of California, San Diego under protocol ASP # S15195, approved by the Institutional Animal Care and Use Committee (IACUC). See Supplemental Materials for additional details. For PDX establishment, fresh surgically removed tumor tissues were obtained by Crown Bio (18) (Crown Bioscience SPF facility) from patients diagnosed as HNSCC with approval by the Institutional Review Boards of the hospital and informed consents from patients, and studies were conducted in accordance with recognized ethical guidelines. The protocol and any amendment(s) or procedures involving the care and use of animals were approved by the Institutional Animal IACUC of CrownBio prior to conduct. During the study, the care and use of animals was conducted in accordance with the regulations of the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC).
RNAi, cell growth assays, 3D spheroids assay, immunoblot analysis, immunohistochemistry and immunofluorescence, microfluidic vasculogenesis assay
See Supplemental Materials for additional details.
HRAS plasma membrane translocation assays
For HRAS-GFP transfection, cells were grown on μslide glass bottom (Ibidi). Cells were transfected with HRAS-GFP and the next day were treated with tipifarnib for 48h and the image acquisition was performed by confocal microscopy.
Mouse Choroidal Explant Assay
Male C57BL/6J mice (age P20) were euthanized and eyes were immediately enucleated for dissection. After removing the cornea and lens, the peripheral choroid-scleral complex was separated from the retina and cut into approximately 1mm x 1mm fragments, then the mouse choroidal explant assay. See Supplemental Materials for additional details.
RNA sequencing and bioinformatic analysis
HN2579 and HN3504 HNSCC PDX tumors were implanted in groups of three animals as described above and allowed to grow to 350-450mm3, treated for four days with vehicle or tipifarnib (80mg/kg BID), excised and snap-frozen. In order to ensure unbiased sampling for each tumor lesion, three fragments in different regions of the tumor were collected by microdissection techniques. See Supplemental Materials for RNA extraction, RNAseq and analysis.
Statistical analysis
GraphPad Prism version 7 for Windows (GraphPad Software, San Diego, CA) was used to perform data analyses, variation estimation and validation of test assumptions. The differences between experimental groups in tumor volume, quantification of immunohistochemical analysis were performed with longitudinal data analysis method, independent t-tests, or ANOVA.
Results
Genomics of HRAS-mutant HNSCC subset
The recent completion of The Cancer Genome Atlas (9) has provided an unprecedented opportunity to perform a pancancer analysis of the genomic alterations in HRAS. We performed a detailed analysis of genomic information in the TCGA database focused on revealing HRAS gene expression levels and mutational status in a broad array of cancer types. This study showed that relatively few cancers harbor HRAS mutations, particularly thyroid cancer, pheochromocytoma and paraganglioma (PCPG), and head & neck squamous cell carcinoma (HNSCC), (Fig. 1A). Among them, the latter also represents the cancer expressing the highest levels of HRAS transcripts, together suggestive of a more prominent role for HRAS in this particular cancer type. The TCGA analysis has also provided a comprehensive genomic characterization of HNSCC (9) (Fig. 1B), supporting that TP53 is one the most mutated genes (71% mutated), followed by FAT1 (23% mutated), NOTCH1 (18% mutated), CASP8 (11% mutated), CDKN2A (22% mutated) genes, and PIK3CA (~18% mutated) (19). In a prior study, we have performed a pathway-specific analysis of the HNSCC oncogenome, which indicated that the PI3K-mTOR signaling pathway is mutated in the highest percentage of the HNSCC lesions (19). Indeed, PIK3CA is the driver oncogene most frequently mutated when considering HPV- and HPV+ HNSCC cases (16.8% and 36.1%, respectively (19). HRAS is mutated at a lower frequency (6.1%), but only in the HPV negative HNSCC group (9), which is often associated with tobacco use and exhibit worse prognosis (9,20).
Figure 1. Genomics of HRAS-mutant HNSCC subset.
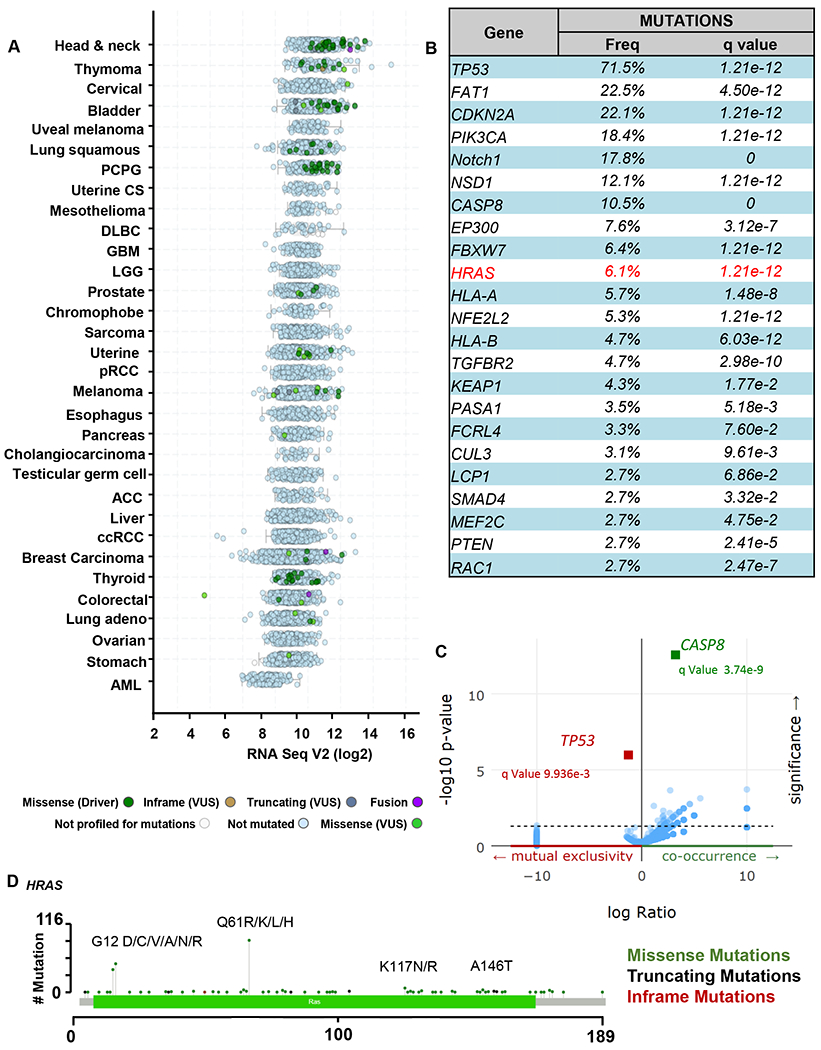
(A) Pan-cancer analysis of the TCGA database focused on HRAS gene expression and mutations. Expression level of HRAS indicated as Log2 TPM (Transcript Count Per Million) and mutation frequency of HRAS (green) across different cancer types in the Cancer Genome Atlas (TCGA) dataset are represented. (B) Percentage of samples with one or more mutations in the major driver-signaling pathways, including HRAS, in HNSCC (TCGA dataset, n=523). Percentage of samples with mutations is indicated, and the corresponding statistical significance (q value). (C) Co-occurrence and mutual exclusivity of HRAS mutations in HNSCC (TCGA dataset, n=523). (D) Mutational Plot representing the analysis of cancer-associated HRAS mutations from TCGA for HNSCC. The frequency of mutations is depicted by the height of the lollipop.
HPV infection has been recently recognized as a viral etiologic agent responsible for HNSCC, more specifically in the oropharynx (21,22). The absence of any HRAS mutations in HPV+ HNSCC prompted us to explore whether there are other significantly altered genomic alterations concomitant with HRAS that may help define better the landscape of HRAS mutant HNSCC. Indeed, aligned with a prior report (23), we found that HRAS mutations define a unique subset of HNSCC, characterized, in most of the cases, by coincident loss of function mutations in caspase 8 (q value 3.74 e−9) and enrichment for absence (nearly mutually exclusive) of TP53 mutations (q value 9.936 e−3) (Fig. 1C). In this regard, HRAS mutant HNSCC cases also exhibit a low overall mutational burden that is associated with poor response to immuno-oncology agents (9), and hence may instead benefit from the development of targeted options disruption HRAS oncogenic signaling. As with the other RAS isoforms, HRAS exhibits mutational hotspots in exons 2 (G12, G13), 3 (Q61) and 4 (K117 and A146), but Q61 mutations are more common than in KRAS and NRAS mutants, where G12 mutations predominate (Fig. 1D).
Tipifarnib inhibits HRAS farnesylation and displaces it from cellular membranes
Ras proteins are synthesized in the cytosol and subsequent post-translational modifications enable their stable association with intracellular membranes, which is required for GTP hydrolysis and RAS signaling (4,5). The rate-limiting modification is farnesylation of the cysteine 186, mediated by farnesyltransferase (Fig. 2A) (24). Since the linkage of a farnesyl moiety to target proteins subtly alters their molecular weight - the so called ‘farnesylation shift’, we used a high concentration gel (18%) to show that tipifarnib treatment deprenylated HRAS in both mutant and WT cell lines (Fig. 2B). To determine whether this de-farnesylation altered the intracellular trafficking of HRAS protein, we expressed GFP-tagged HRAS in 293 cells (Fig. 2C) to be able to follow its localization with and without tipifarnib treatment. HRAS localized to the plasma and nuclear membranes and membranous organelles in control samples as described (25), while tipifarnib treated cells displayed a more cytosolic localization of the protein (Fig. 2D).
Figure 2. Tipifarnib inhibits HRAS farnesylation and displaces it from cellular membranes.
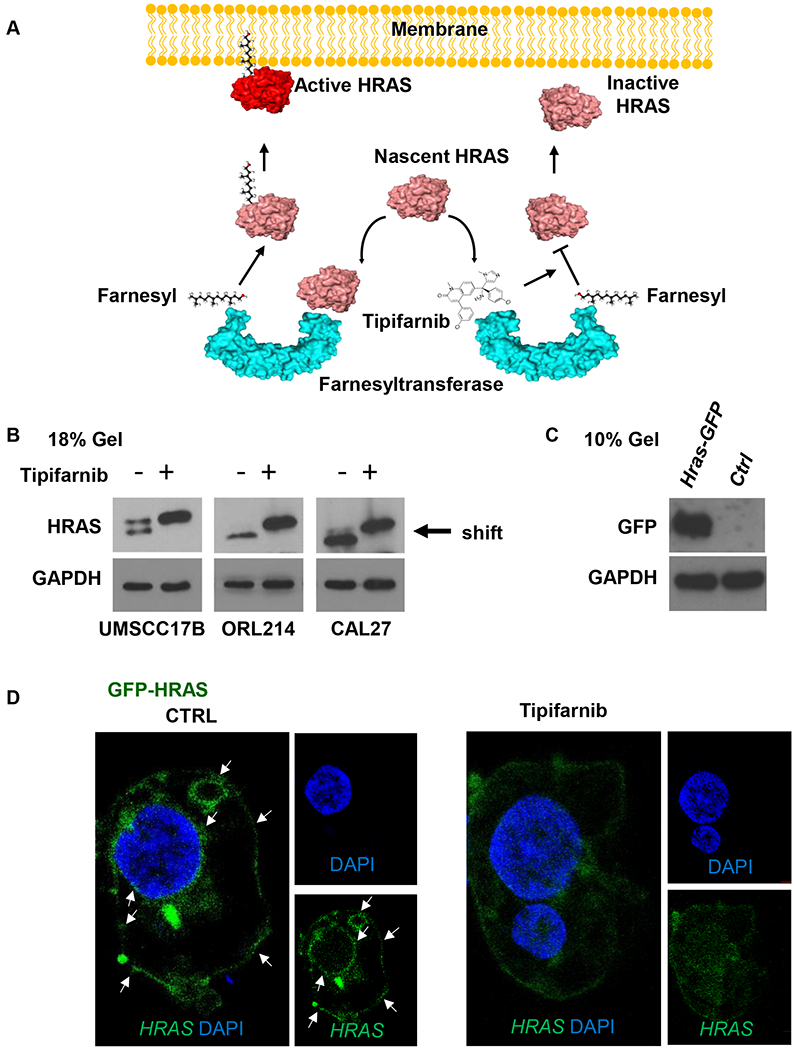
(A) Mechanism of action of tipifarnib. Ras proteins are synthesized in the cytosol and subsequent post-translational modifications enable their stable association with intracellular membranes, which is required for GTP hydrolysis and RAS signaling. HRAS, is only dependent on farnesylation for its membrane localization. The rate-limiting modification is farnesylation of the cysteine 186, mediated by farnesyltransferase. Tipifarnib displaces HRAS from cellular membranes and selectively inhibited proliferation and survival of HRAS-mutant HNSCC cells. (B) Western blot analysis of signaling events in HNSCC cells UMSCC17B, ORL214 (HRAS mut) and CAL27 (HRAS WT). Cells were cultured in 6-well plates and treated with tipifarnib (200nM) or DMSO (0.2 %) as a control for 48h. Western Blot was performed in an 18% gel showing that the HRAS prenylation shift has been abolished by tipifarnib treatment. (C) Western Blot showing HRAS-GFP expression after transfection. (D) HRAS recruitment to the plasma membrane using cells transfected with HRAS-GFP. Transfected cells, were treated with tipifarnib (200nM) for 48h and single plan imaging in the center of the cells has been performed by confocal microscope allowing fluorescent protein localization.
Tipifarnib is selectively cytotoxic to HRAS-mutant HNSCC in vitro
We characterized the effect of tipifarnib in HRAS mutated and WT human HNSCC cell lines to determine whether the drug was selectively active in the HRAS-mutant subset. First, we treated the cells with tipifarnib or, knocked down HRAS by siRNA and the effect was verified by Western blot (Fig. 3A). Further, we compared the proliferation of HRAS mutated HNSCC (UMSCC17B and ORL214) with HRAS WT (CAL27) treated with siRNA or tipifarnib. Although the inhibition of proliferation was incomplete, both tipifarnib and HRAS knockdown significantly reduced growth of the HRAS mutated cell lines, while no significant effects were detected in HRAS WT cells (Fig. 3B). HRAS knockdown with siRNAs and tipifarnib treatment reduced pERK and pMEK levels in HRAS mutant HNSCC cells, consistent with the inhibition of the HRAS-MEK-ERK pathway, but not in HRAS WT cells, using the MEK inhibitor trametinib, which inhibited ERK activation in all cells as a control (Fig. 3C).
Figure 3. Tipifarnib is selectively cytotoxic to HRAS-mutant HNSCC in vitro.
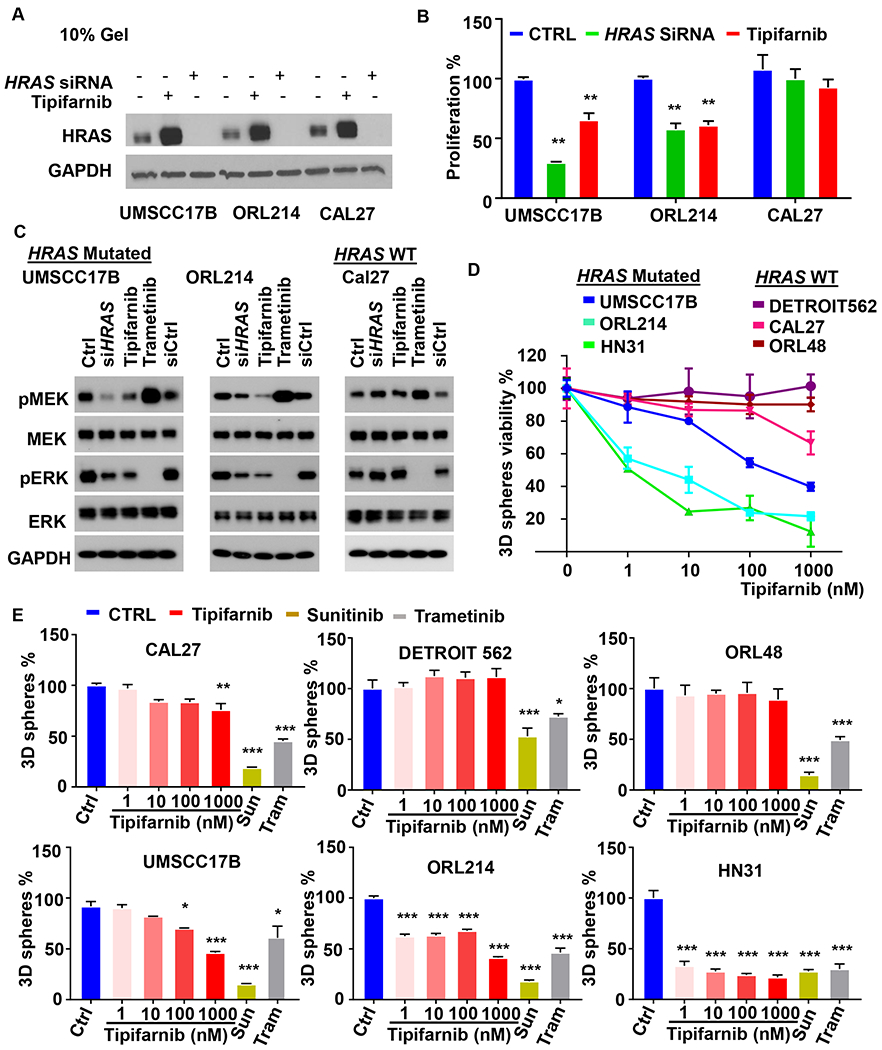
(A) UMSCC17B, ORL214 (HRAS mut) and CAL27 (HRAS WT) were knocked down for HRAS by means of siRNA smart pool, or treated with tipifarnib (200nM) for 48h. SiRNA ctrl was used in the control sample. Cell lysates after treatment were analyzed by Western Blot in a 10% gel showing the HRAS expression in different experimental conditions. (B) Effects of tipifarnib and HRAS siRNA in monolayer culture: viability analysis in the condition described in figure 3A. (*P<0.05, **P< .01, ***P< .001 when compared with the control-treated group, n = 3 per group). (C) Western blot for pMEK/MEK and pERK/ERK on HRAS WT and mutated cell lines, respectively, Cal27 (WT) and UMSCC17B and ORL214 (mutant). Cells were treated with HRAS siRNA (48h), and tipifarnib as above, and using trametinib as a control. (D) 3D spheroids assay for the three dimensional analyses of HRAS inhibition: quantification of the viability of CAL27, DETROIT562 and ORL48 (HRAS WT) UMSCC17B, ORL214, HN31 (HRAS mut) at increasing concentration of (0 to 1000nM) in 3D hydrogel culture. Cells have been treated for 3 weeks. (n=3 per group). (E) With the same setup of the 3D spheroids assay, the stemness potential has been measured quantifying the number of spheroids generated in a 3D controlled hydrogel by CAL27, DETROIT562 and ORL48 (HRAS WT), UMSCC17B, ORL214, HN31 (HRAS mut) at increasing concentration of tipifarnib (o to 1000 nM), using sunitinib and trametinib as positive controls. (*P<0.05, **P< .01, ***P< .001 when compared with the control-treated group, n = 3 per group).
The initial experiments with tipifarnib and HRAS knockdown were performed in monolayer culture, but as several groups have recently reported that this format fails to capture the full potency and selectivity of RAS inhibitors (26). We also tested tipifarnib in a larger panel of HRAS mutant and WT cell lines in 3D spheroid formation assays, measuring effects on viability in terms of metabolic activity (Fig. 3D) and absolute number (Fig. 3E) of 3D tumor spheroids. The broadly active HRAS-non-selective drugs sunitinib and trametinib were employed as controls. 3D assay treatment lasting 3 weeks, evidenced distinct behaviors in different conditions. Tipifarnib displayed dose-dependent inhibition of spheroid viability in HRAS mutant cells only (Fig. 3D) and also selectively reduced the absolute number of colonies (Fig. 3E), suggesting that HRAS inhibition depletes tumor initiating cells. By contrast, sunitinib and trametinib were similarly active in both HRAS mutant and WT HNSCC lines (Fig. 3E).
Tipifarnib is highly active in HRAS-mutant HNSCC xenografts
Given that tipifarnib shows selective cytotoxicity to HRAS mutant HNSCC cells in vitro, we next asked whether tipifarnib is sufficient to display tumor suppressive effects in vivo. For these studies, we used UMSCC17B cells exhibiting a HRAS Q61L mutation and ORL214, which has a HRAS G12C mutation. Remarkably, we observed that tipifarnib significantly halted tumor growth from as early as three days after treatment initiation (n=6, p<0.001) (Fig. 4A–C). Tipifarnib reduced pERK in both xenograft models (Fig. 4D). Moreover, IHC for cleaved Caspase 3 and IF for Ki67 showed that, inhibition of HRAS by tipifarnib caused increased apoptosis and a reduction of cell proliferation in HNSCC tumors (Fig. 4D).
Figure 4. Antitumor activity of tipifarnib in cell line-derived HNSCC xenograft models.
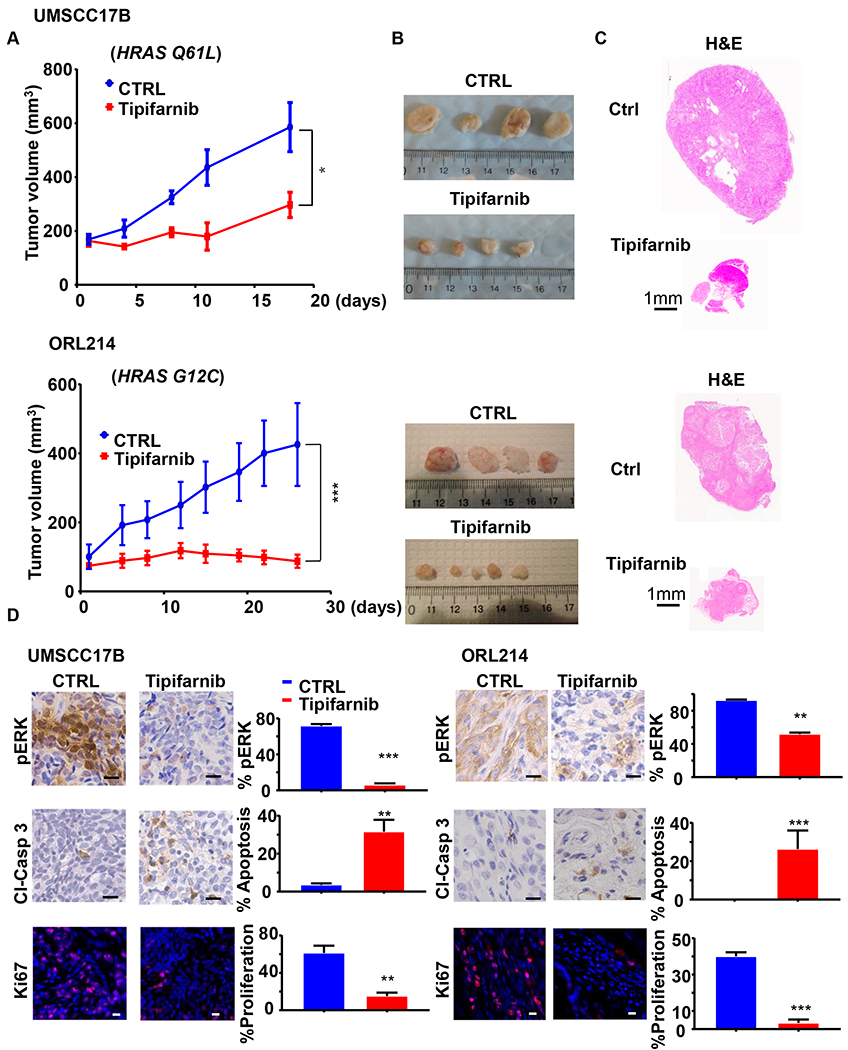
(A) UMSCC17B (top) and ORL214 (bottom) were transplanted into athymic nude mice and NOD-SCID mice respectively and treated with vehicle or tipifarnib (60mg/kg BID) as indicated. (*P<0.05, ***P< .001 when compared with the control-treated group, n = 6 per group). (B) Representative tumor images and (C) histological sections from each treatment group in panel A. (D-E) Left, representative immune-histochemical analysis of pERK and cleaved-Caspase 3, and representative immunofluorescence analysis of Ki67 in tumors from panel A. On the right, quantification from images on the left using Qupath software. (*P<0.05, **P< .01, ***P< .001 when compared with the control-treated group, n = 3 per group).
Activity of Tipifarnib in HRAS mutant is independent of genotype
We next extended our analysis to include a panel of HRAS-mutant and HRAS wild type patient-derived HNSCC xenograft (PDX) models (Fig. S1A), as they may better reflect the complexity of the HNSCC lesions and limit the genetic drift that may occur during establishment and maintenance of HNSCC cell lines in vitro. In addition, because widespread differences exist within individual residues and hotspot gene mutations of amino acids and tumor types (27), the expansion of the study to include PDX models increased the range of hotspot mutations to include A146T (HN1420 model), G12S (HN2579), G13R (HN2581) and K117N (HN3504) (Fig. 1D). Because tipifarnib is likely to be inhibiting the function of other farnesylated proteins in HNSCC models in vivo, it was important to determine if the presence of mutant HRAS was necessary for the robust observed antitumor activity of the drug. As shown in Figure 5, tipifarnib displayed selective antitumor activity in HRAS-mutant HNSCC PDX models. The six HRAS wild type tumors grew progressively while on tipifarnib treatment (Figs. 5A, S1B, S1C) but, in sharp contrast, all four HRAS-mutant tumors were highly sensitive to tipifarnib when compared with the control-treated groups (P<0.01) (Fig. 5A, B), demonstrating that mutant HRAS is required for tumor control by tipifarnib in HNSCC models. Consistent with our previous xenograft data, IHC for pERK (Fig. 5C) and IF for Ki67 (Fig. 5D) showed that tipifarnib caused a reduction of ERK activation and cell proliferation in these PDX models. Moreover, as expected, tipifarnib drastically reduced farneslylated proteins (Fig. 5E), and vessel density by CD31 staining (Fig. 5F), (28).
Figure 5. Antitumor activity of tipifarnib in HRAS mutant patient-derived xenograft models.
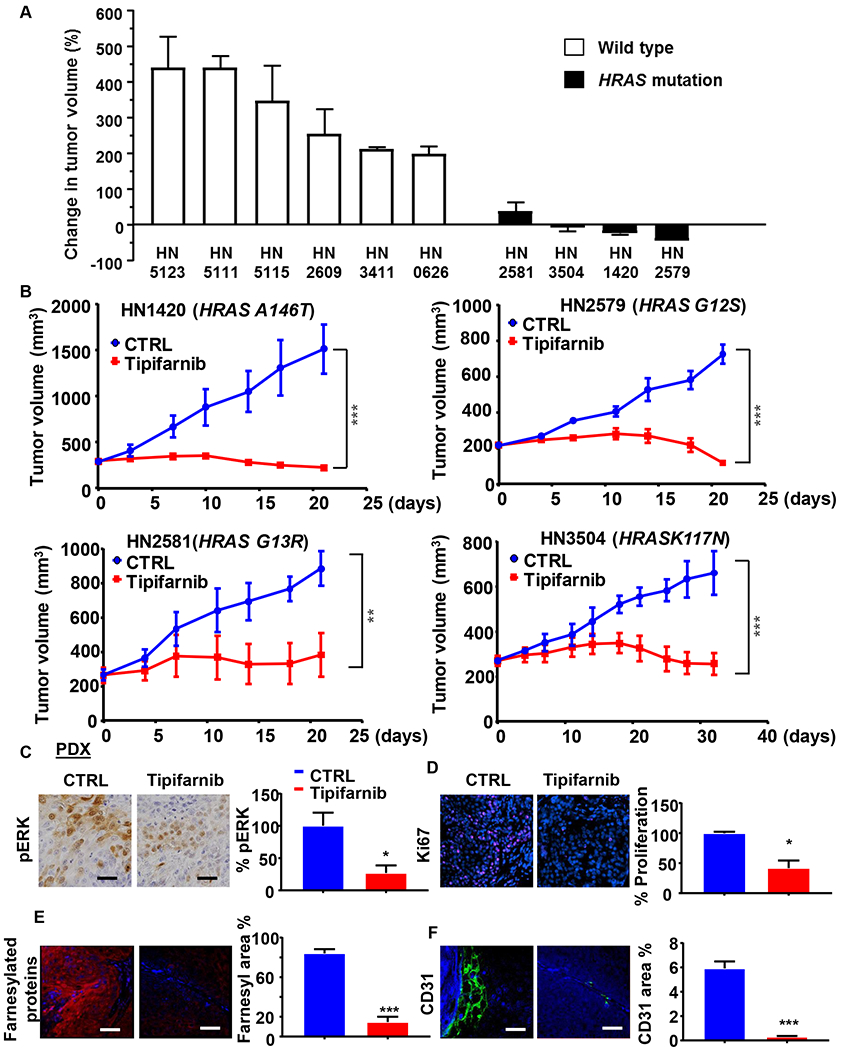
(A) Waterfall plot of tipifarnib antitumor activity in HRAS wild type and mutant PDX models. The columns in the graph represent the volume change comparing before and after treatment with tipifarnib. Athymic nude mice were inoculated subcutaneously with 2–3 mm tumors fragments, the PDX were allowed to establish to 250–350 mm3, than the animals were randomized into groups of three and treated orally with vehicle or tipifarnib (60mg/kg BID) for approximately 20 days. (B) PDX models containing endogenous HRAS mutations. HN1420, HN2579, HN2581 and HN3504 fragments were transplanted into athymic nude mice, treated with vehicle or tipifarnib (60mg/kg BID) as indicated. (*P<0.05, **P< .01, ***P< .001 when compared with the control-treated group, n = 3 per group). (C) Representative immune-histochemical or immunofluorescence analysis (left) and quantification using Qupath (right) of: pERK (top left), Ki67 (top right), farnesylated proteins (bottom left) and CD31 (bottom right) in PDX samples. (*P<0.05, **P< .01, ***P< .001 when compared with the control-treated group, n = 3 per group). The analyzed samples are related to the HN3504 PDX models using the same treatment as in section A.
Tipifarnib inhibits angiogenesis and vasculogenesis
Tumors can increase their blood supply by two recognized mechanisms: de novo formation of vessels by the differentiation of the endothelial progenitor cells, known as vasculogenesis, and angiogenesis, the sprouting of new blood vessels from the existing vasculature. Previous studies indicated that tipifarnib and other FTIs inhibit tumor angiogenesis (13,28), but anti-angiogenic activity can vary between different tumor types, so we sought to expand these observations in the context of HRAS-mutant HNSCC. CD31 immunostaining demonstrated that tipifarnib significantly inhibited vessel formation in both UMSCC17B and ORL214 xenograft tumors (Fig. 6A and B). Moreover, to explore whether tipifarnib may also act on endothelial cells directly, we performed a 3D vasculogenesis assays in a microfluidic model. Briefly, GFP-HUVECS were grown in 3D hydrogel in microfluidic channels for 48 hours with and without tipifarnib. The number of branches was quantified, which revealed the inhibitory effect of tipifarnib on vessels generation (Fig 6C). Choroid sprouting assay can be used as an ex vivo model for studying microvascular angiogenesis. We then tested tipifarnib effects using the choroid sprouting assay. As shown in Figure 6D, tipifarnib robustly inhibited vessel sprouting from mouse choroid in the 3D matrix, suggesting that both pathological and physiological neovascular processes are sensitive to farnesyltransferase inhibition.
Figure 6. Tipifarnib inhibits angiogenesis in vitro, ex vivo and in vivo.
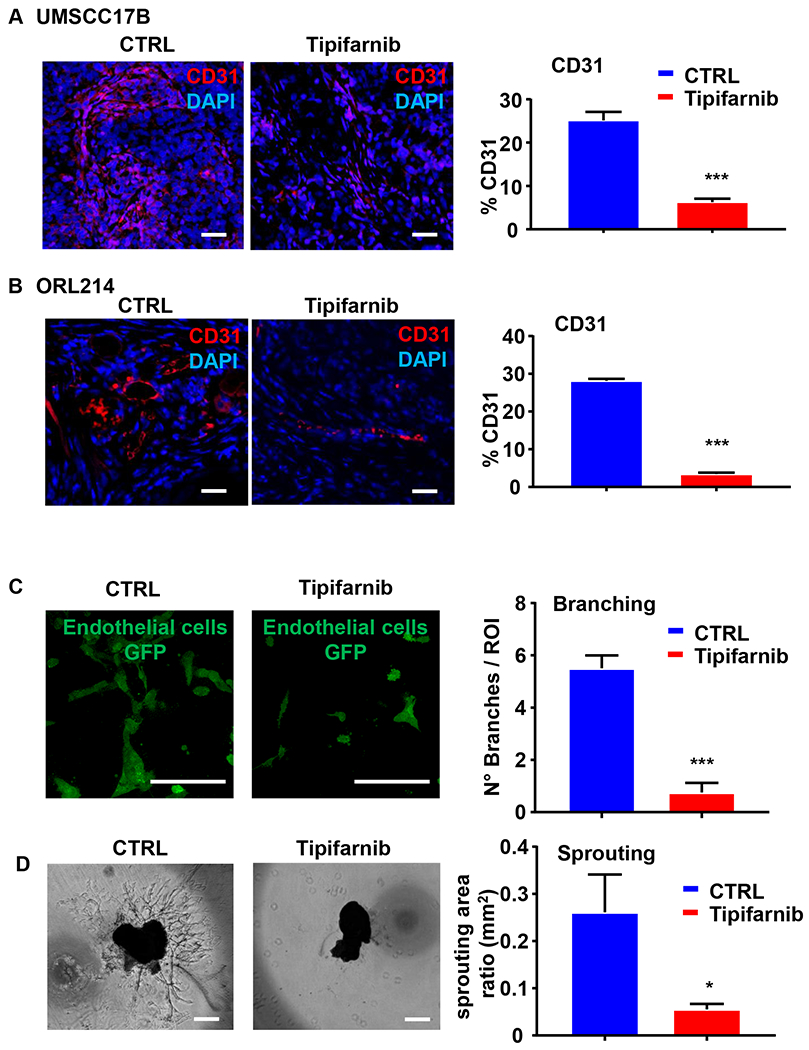
(A-B) UMSCC17B (A) and ORL214 (B) were transplanted into athymic nude mice and NOD-SCID mice respectively and treated with vehicle or tipifarnib (60mg/kg BID) as indicated. Representative immunofluorescence analysis (left) and quantification (right) of CD31 in xenograft models. (C) Vasculogenesis assay in the microfluidic model (left) and quantification (right) of the endothelial cells branching. GFP HUVECs were seeded through microfluidic channels in a 3D environment and treated with tipifarnib 200 nM for 48h. Number of branches were quantified in at least three ROIs for each condition and at least 3microfluidic device for each condition have been cultured. (*P<0.05, **P< .01, ***P< .001 when compared with the control-treated group, n = 3 per group). (D) Mouse Choroidal Explant Assay: vessel outgrowth in a mouse choroid explant model in a 3D environment. Representative images of vessel growth after 6 days of incubation with tipifarnib 200 nM (left) and quantification of the sprouting area (right). (*P<0.05, **P<.01, ***P < .001 when compared with the control-treated group, n = 3 per group).
Tipifarnib induces differentiation in patient-derived tumors
To further elucidate the consequences of interfering with mutant HRAS and other farnesylated targets in HNSCC, we performed bioinformatics analysis of tipifarnib-induced gene expression changes in PDX models. Two tipifarnib-sensitive HRAS-mutant HNSCC PDX models were treated with tipifarnib for four days, at which point the tumors were harvested and processed for RNA sequencing. Data were processed and analyzed using a combination of commercial (Rosalind by OnRamp, Advaita) and Open Source (ENRICHR) resources. As shown in Fig. 7A and E, gene set enrichment analysis (GSEA) revealed two prominent patterns of altered gene expression in tipifarnib-treated tumors. When the two models were analyzed collectively, the predominant changes related to inhibition of cell cycle progression (Fig. 7A, Fig. S2, Fig. S3), as expected from RAS-MAPK biology. Advaita cell pathway analysis showed that the expression of drivers of G2/M progression, such as cyclins A and B, CDK1 and CDC25, and mitotic regulators, including BUB1 and PLK1, was suppressed (Figs. 7B and S2A), consistent with shutdown of RAS-MAPK-cyclin D signaling, the dominant mitogenic pathway in HRAS-mutant cells leading to cell cycle arrest at the G1-S boundary.
Figure 7. Bioinformatics analysis of tipifarnib activity in PDX models.
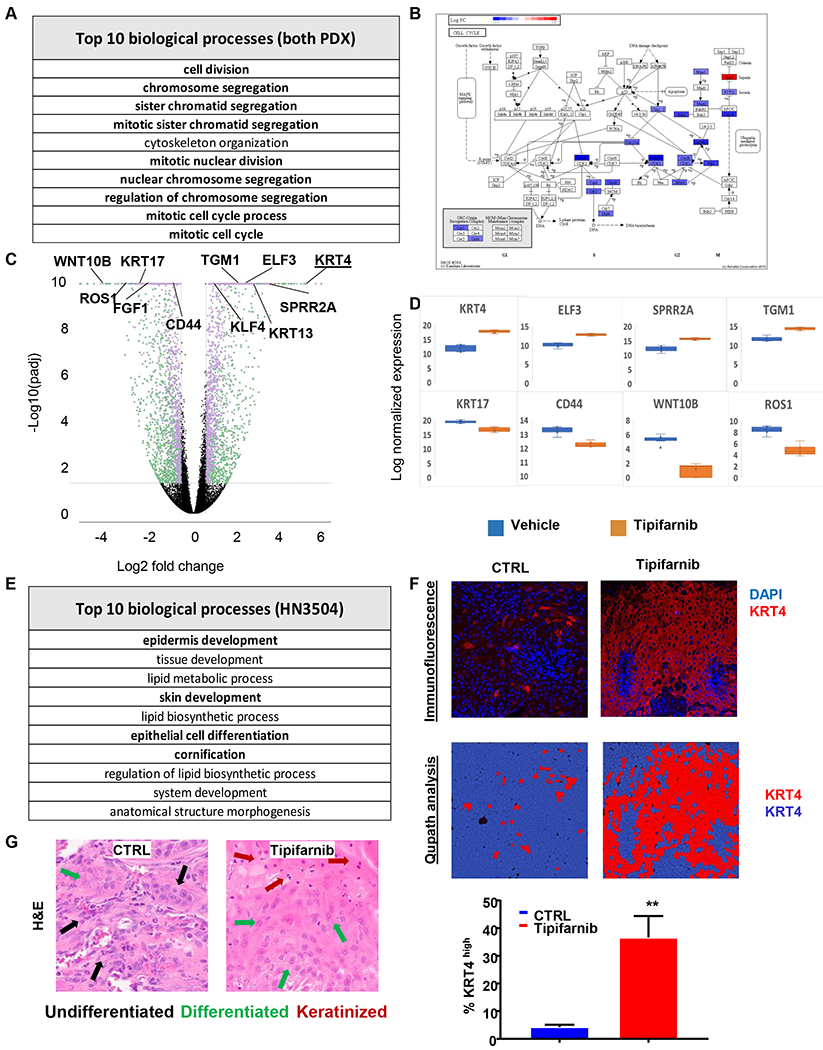
(A) Geneset enrichment analysis of top processes altered by tipifarnib treatment in the combined dataset of HN2579 and HN3504 xenografts. (B) Advaita pathway diagram illustrating the roles of genes suppressed by tipifarnib treatment in G2 and M phases of the cell cycle. (C) Volcano plot of differentially expressed (DE) genes in tipifarnib-treated HN3504 tumors (n = 3794, fold change ≥ 1.5, p-Adj < 0.05). Green: less abundant transcripts, purple: more abundant transcripts. (D) Box-and-whisker plots of representative highly DE genes in tipifarnib-treated HN3504 tumors. Upregulated genes: KRT4 (fold change 35.33, p-Adj 1.01e-54), ELF3 (fold change 6.31, p-Adj 2.41e-37), SPRR2A (fold change 8.97, p-Adj 3.52e-27), TGM1 (fold change 6.16, p-Adj 4.37e-28). Downregulated genes: KRT17 (fold change −5.76, p-Adj 2.75e-20), CD44 (fold change −2.02, p-Adj 2.49e-14), ROS1 (fold change −8.86, p-Adj 4.98e-21), WNT10B (fold change −17.10, p-Adj 9.74e-22). (E) Geneset enrichment analysis of top processes altered by tipifarnib treatment in HN3504 xenografts. (F) Immunofluorescence (upper panel) and quantification analysis using Qupath software (bottom panel) of KRT4 expression in control and tipifarnib-treated HN3504 PDX-tumors. (G) H&E highlighting morphologic evidence of squamous differentiation in control and tipifarnib-treated HN3504 PDX-tumors. BALB/c nu/nu mice were inoculated subcutaneously with 2-3 mm tumors fragments, the PDX were allowed to establish to 250-350 mm3, the animals were randomized into groups of three and treated orally with vehicle or tipifarnib (60mg/kg BID) for approximately 20 days.
However, when the two PDX models were analyzed individually a second prominent transcriptional phenotype emerged in the HN3504 model, where GSEA enriched for processes and pathways associated with differentiation of squamous cells (Fig. 7E), including epidermal development, skin development, epithelial differentiation and cornification. Keratinocytes in stratified squamous epithelia, such as the skin and the lining of the upper aerodigestive tract from which HNSCC is derived, originate as proliferating progenitor cells in the basal layer and progressively differentiate towards specialized post-mitotic cells as they move outward (29). This progression is associated with characteristic alterations in patterns of cytokeratin expression and ultimately enzymatic cross-linking of cellular proteins with barrier function, known as cornification. As shown in the volcano and box and whisker plots of gene expression changes induced by tipifarnib in HN3504 tumors (Figs. 7C–D, S2B, Table S1), the well-characterized squamous differentiation markers cytokeratin 4 (KRT4) and KRT13 (30), the epithelial differentiation transcription factors ELF3 (31) and KLF4 (32), the cornified protein precursor SPRR2A and the cross-linking enzyme transglutaminase-1 (TGM1) were among the most strongly upregulated loci (p-Adj 6.83e-21 – 1.01e-54). By contrast, the basal cytokeratin KRT17, the ‘stemness’ markers CD44 (33) and WNT10B (34) and the HNSCC oncogene ROS1 (35) were all strongly downregulated (p-Adj 2.49e−14 - 9.74e−22). Squamous differentiation in tipifarnib treated HN3504 tumors was confirmed histologically by robust staining for KRT4, and by quantitative analysis of the of the immunofluorescence results. Moreover, the presence of characteristic squamous differentiation morphological features in tipifarnib-treated HN3504 tumors is shown in the H&E staining sections (Fig. 7G).
Discussion
RAS genes are the most common driver oncogenes in human cancer, being mutated in approximately one third of cancer cases, so considerable efforts have been made for decades to develop therapeutics for RAS-driven tumors (36). Despite this significant investment, no drugs directly targeting RAS proteins have been approved, and alternative strategies directed against downstream RAS pathways such as MAPK and PI3K have also proved ineffective, primarily due to feedback reactivation via RAS (37). Direct inhibition of RAS proteins at the catalytic site is impractical due to their picomolar affinity for GTP (37), but a recent breakthrough has enabled direct inhibition of KRASG12C (26).
The only other currently feasible way to disrupt RAS activity directly is through preventing appropriate intracellular localization by interfering with RAS prenylation (36). Several dozen proteins are farnesylated under basal conditions and tipifarnib treatment blocks their prenylation (38), but inhibition of the farnesylation of KRAS and NRAS leads to compensatory geranylgeranylation and the restoration of membrane localization in the presence of tipifarnib (7,38). In contrast, HRAS cannot be geranylgeranylated, and its membrane localization and cellular function may be suppressed by FTIs (38,39). In this study we report that mutant HRAS is a targetable oncogene via farnesyltransferase inhibition in a molecularly-defined subset of HNSCC. Tipifarnib displayed robust and consistent antitumor activity in a series of cell line- and patient-derived xenograft models of HNSCC but, in sharp contrast, tipifarnib was devoid of activity in HRAS wild type HNSCC cell lines and PDX models in vitro and in vivo. Remarkably, tipifarnib displayed significant inhibition of tumor growth in HRAS-mutant xenografts harboring mutations in exon-2 (G12C, G12S, G13R), exon-3 (Q61L) or exon-4 (K117N, A146T), suggesting that all of these mutants are sufficiently oncogenic to drive full malignancy in HNSCC cells, even though exon-2 and exon-3/4 KRAS mutants have been reported to have differing GTPase activities and biologic functions in other cellular contexts (8,40).
HRAS protein was de-prenylated in both HRAS-mutant and wild type HNSCC cells, as indicated by the gel shift and redistribution from intracellular membranes, but tipifarnib only inhibited spheroid growth of the HRAS-mutant UMSCC17B, ORL214 and HN31 cell lines, whereas the cytotoxic multikinase inhibitor sunitinib and the MEK inhibitor trametinib displayed similar activity in both HRAS mutant and HRAS wild type lines. Genetic depletion of HRAS was apparently incompletely effective at inhibiting the proliferation of HRAS-mutant HNSCC cells, but this experiment was performed in monolayer culture, and these growth conditions have recently been shown to partially undermine RAS dependence (26). Tipifarnib activity was also blunted under these conditions, underscoring the importance of using appropriate assay formats to interpret RAS dependence.
The expected effects on mitogenic signaling, cell cycle progression and apoptosis downstream of inhibition of oncogenic HRAS in vivo were observed in both cell-derived xenografts and PDX models. ERK phosphorylation was sharply reduced in UMSCC17B and HN3504, but less so in ORL214, perhaps due to feedback reactivation of the MAP kinase pathway. Indeed, ENRICHR analysis of upregulated gene-sets in PDX models following 4 days of tipifarnib therapy revealed evidence of upregulation of canonical MAPK pathway negative regulators (DUSP1, DUSP3) and activation of collateral epithelial cell oncogenic pathways including EGF/EGFR and HER2/HER3 signaling, PIK3CA and PTPN11 (Fig. S3). Despite this, all models responded well to tipifarnib treatment and proliferation and apoptosis markers were robustly altered at both early and late timepoints during tipifarnib therapy, suggesting that continuous treatment overwhelmed innate tumor resistance mechanisms and maintained sufficient suppression of oncogenic signaling to block tumor growth in these models. Indeed, the antitumor activity of tipifarnib in all HRAS-mutant HNSCC models reported here matches or exceeds that reported with a combination of MAPK and PI3K pathway inhibitors in a HRAS-mutant lung cancer model (41).
HRAS is among several dozen obligate farnesylated proteins in cells (38), and analysis of treated PDX tumors indicated almost complete disappearance of the farnesyl moiety, raising the possibility that depletion of additional farnesylated target proteins could enhance the antitumor activity of tipifarnib in HRAS-mutant HNSCC. Tipifarnib and other FTIs have previously been shown to possess anti-angiogenic activity (5,28,42) mediated by effects on both tumor (42) and endothelial cells (28,43), and we observed both in this study. The farnesylated proteins involved remain to be identified (44), but the lack of significant effects on tumor growth observed in the panel of HRAS wild type PDX strongly suggests that either (a) inhibition of angiogenesis does not provide sufficient therapeutic value in HNSCC (45) or (b) some or all of the antivascular effects of tipifarnib in vivo are secondary to mutant HRAS blockade (46).
Bioinformatics analysis of tipifarnib-induced gene expression changes in two PDX models further elucidated the multifactorial mechanisms of antitumor activity of the drug in HRAS-mutant HNSCC in vivo. Gene set enrichment analysis (GSEA) of the combined dataset confirmed that FTI treatment induced a robust cell cycle block at the G1/S boundary and also promoted squamous lineage differentiation. Malignant transformation and terminal differentiation are mutually exclusive processes with opposing effects on cellular proliferation. Carcinogenesis in squamous tissues is associated with impairment of differentiation linked to HPV infection or oncogene activation (47,48). Gene set enrichment analysis of HRAS-mutant HN3504 PDX tumors treated for four days with tipifarnib revealed that initiation of epithelial differentiation was a prominent early effect of farnesyl transferase inhibition in this model. HNSCC stem cell markers such as CD44 (33) and WNT10B (34,49) and the basal (proliferative) layer cytokeratin KRT17 (29) were also profoundly suppressed. By contrast, the canonical squamous differentiation markers KRT4 and KRT13 (30), pro-differentiation transcription factors ELF3 (31) and KLF4 (32), and cornification markers like SPRR2A, all of which have been reported to be downregulated in HNSCC, were among the most strongly upregulated genes. This suggests that oncogenic HRAS may suppress squamous differentiation in HNSCC and that this can be reversed by tipifarnib treatment.
In the present study, we have characterized the antitumor activity and mechanisms of action of tipifarnib in a large series of HNSCC CDX and PDX models. Tipifarnib displayed robust and selective activity in HRAS mutant models harboring all of the known hotspot loci. Collectively, these data demonstrate that mutant HRAS represents an actionable oncogene in HNSCC that can be targeted with tipifarnib via inhibition of proliferation and angiogenesis and induction of apoptosis and terminal squamous cell differentiation, resulting in consistent stasis or tumor regression in vivo.
Tipifarnib was previously studied in an extensive development campaign consisting of more than 70 clinical trials in a variety of tumor types in the late 1990’s and early 2000’s without the benefit of methods to enrich for clinical activity such as the use of next-generation sequencing to identify patients with specific driver mutations. Although durable responses were achieved in several cancers, response rates were insufficient to support registrational studies in unselected patient populations. Since its reintroduction into the clinic in 2015, several cohorts of HRAS mutant patients have been treated in a single-arm Phase 2 trial (NCT02383927), with encouraging preliminary findings. As reported in 2018 (45,50), among 7 evaluable HNSCC patients, 5 (71%) achieved a confirmed partial response with a median duration of response of 14.1 months. Importantly, no HRAS mutant HNSCC patient experienced an objective response on his last therapy prior to receiving tipifarnib (including platinum, immunotherapy and cetuximab +/− chemotherapy regimens). Based on these initial encouraging clinical responses, and our current findings, an international, multicenter, open-label, single arm study of tipifarnib after failure of platinum-based therapy in recurrent or metastatic HNSCC with HRAS mutations with registrational intent, AIM-HN, is currently underway (NCT03719690). Indeed, we expect that our experimental studies in genetically-defined HNSCC systems harboring HRAS mutations may support the rationale for selectively enrolling HRAS mutant HNSCC patients in future tipifarnib trials, this representing as a novel precision therapeutic approach for HNSCC based on their oncogenomic landscape.
Supplementary Material
Acknowledgements
This project was supported by National Institute of Dental and Craniofacial Research (NIH/NIDCR) grant 1R01DE026870 to M. Gilardi, Z. Wang, Y. Goto, and J.S. Gutkind; and the NIH grant S10OD021831 to Z. Mikulski. Mara Gilardi was supported by FIRC-AIRC fellowship for abroad (Italian Foundation for cancer research). We thank the Staff of La Jolla Institute Microscopy Core Facility, in particular to Drs. Mcardle, Kiosses and Marcovecchio.
Footnotes
Disclosures: MJ is an employee and stockholder of Kumquat Biosciences, AG and FB are employees and stockholders of Kura Oncology. JSG is a member of the advisory board of Oncoceutics, Domain Therapeutics, and Vividion.
References:
- 1.Cohen EE, LaMonte SJ, Erb NL, Beckman KL, Sadeghi N, Hutcheson KA, et al. American Cancer Society Head and Neck Cancer Survivorship Care Guideline. CA Cancer J Clin 2016;66:203–39 [DOI] [PubMed] [Google Scholar]
- 2.Vermorken JB, Mesia R, Rivera F, Remenar E, Kawecki A, Rottey S, et al. Platinum-based chemotherapy plus cetuximab in head and neck cancer. N Engl J Med 2008;359:1116–27 [DOI] [PubMed] [Google Scholar]
- 3.Chow LQM, Haddad R, Gupta S, Mahipal A, Mehra R, Tahara M, et al. Antitumor Activity of Pembrolizumab in Biomarker-Unselected Patients With Recurrent and/or Metastatic Head and Neck Squamous Cell Carcinoma: Results From the Phase Ib KEYNOTE-012 Expansion Cohort. J Clin Oncol 2016;34:3838–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sebti SM. Protein farnesylation: implications for normal physiology, malignant transformation, and cancer therapy. Cancer Cell 2005;7:297–300 [DOI] [PubMed] [Google Scholar]
- 5.Rowinsky EK, Windle JJ, Von Hoff DD. Ras protein farnesyltransferase: A strategic target for anticancer therapeutic development. J Clin Oncol 1999;17:3631–52 [DOI] [PubMed] [Google Scholar]
- 6.End DW, Smets G, Todd AV, Applegate TL, Fuery CJ, Angibaud P, et al. Characterization of the antitumor effects of the selective farnesyl protein transferase inhibitor R115777 in vivo and in vitro. Cancer Res 2001;61:131–7 [PubMed] [Google Scholar]
- 7.Whyte DB, Kirschmeier P, Hockenberry TN, Nunez-Oliva I, James L, Catino JJ, et al. K- and N-Ras are geranylgeranylated in cells treated with farnesyl protein transferase inhibitors. J Biol Chem 1997;272:14459–64 [DOI] [PubMed] [Google Scholar]
- 8.Li S, Balmain A, Counter CM. A model for RAS mutation patterns in cancers: finding the sweet spot. Nat Rev Cancer 2018;18:767–77 [DOI] [PubMed] [Google Scholar]
- 9.Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018;173:291–304 e6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Su SC, Lin CW, Liu YF, Fan WL, Chen MK, Yu CP, et al. Exome Sequencing of Oral Squamous Cell Carcinoma Reveals Molecular Subgroups and Novel Therapeutic Opportunities. Theranostics 2017;7:1088–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Braig F, Voigtlaender M, Schieferdecker A, Busch CJ, Laban S, Grob T, et al. Liquid biopsy monitoring uncovers acquired RAS-mediated resistance to cetuximab in a substantial proportion of patients with head and neck squamous cell carcinoma. Oncotarget 2016;7:42988–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Untch BR, Dos Anjos V, Garcia-Rendueles MER, Knauf JA, Krishnamoorthy GP, Saqcena M, et al. Tipifarnib Inhibits HRAS-Driven Dedifferentiated Thyroid Cancers. Cancer Res 2018;78:4642–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cohen-Jonathan E, Evans SM, Koch CJ, Muschel RJ, McKenna WG, Wu J, et al. The farnesyltransferase inhibitor L744,832 reduces hypoxia in tumors expressing activated H-ras. Cancer Res 2001;61:2289–93 [PubMed] [Google Scholar]
- 14.Lee KH, Koh M, Moon A. Farnesyl transferase inhibitor FTI-277 inhibits breast cell invasion and migration by blocking H-Ras activation. Oncol Lett 2016;12:2222–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kohl NE, Omer CA, Conner MW, Anthony NJ, Davide JP, deSolms SJ, et al. Inhibition of farnesyltransferase induces regression of mammary and salivary carcinomas in ras transgenic mice. Nat Med 1995;1:792–7 [DOI] [PubMed] [Google Scholar]
- 16.Amornphimoltham P, Patel V, Sodhi A, Nikitakis NG, Sauk JJ, Sausville EA, et al. Mammalian target of rapamycin, a molecular target in squamous cell carcinomas of the head and neck. Cancer Res 2005;65:9953–61 [DOI] [PubMed] [Google Scholar]
- 17.Wang Z, Martin D, Molinolo AA, Patel V, Iglesias-Bartolome R, Degese MS, et al. mTOR co-targeting in cetuximab resistance in head and neck cancers harboring PIK3CA and RAS mutations. J Natl Cancer Inst 2014;106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Guo S, Qian W, Cai J, Zhang L, Wery JP, Li QX. Molecular Pathology of Patient Tumors, Patient-Derived Xenografts, and Cancer Cell Lines. Cancer Res 2016;76:4619–26 [DOI] [PubMed] [Google Scholar]
- 19.Iglesias-Bartolome R, Martin D, Gutkind JS. Exploiting the head and neck cancer oncogenome: widespread PI3K-mTOR pathway alterations and novel molecular targets. Cancer Discov 2013;3:722–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cancer Genome Atlas N Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015;517:576–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.D’Souza G, Kreimer AR, Viscidi R, Pawlita M, Fakhry C, Koch WM, et al. Case-control study of human papillomavirus and oropharyngeal cancer. The New England Journal of Medicine 2007;356:1944–56 [DOI] [PubMed] [Google Scholar]
- 22.Gillison ML, Shah KV. Human papillomavirus-associated head and neck squamous cell carcinoma: mounting evidence for an etiologic role for human papillomavirus in a subset of head and neck cancers. Curr Opin Oncol 2001;13:183–8 [DOI] [PubMed] [Google Scholar]
- 23.Cancer Genome Atlas Research N. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012;489:519–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sacco E, Spinelli M, Vanoni M. Approaches to Ras signaling modulation and treatment of Ras-dependent disorders: a patent review (2007--present). Expert Opin Ther Pat 2012;22:1263–87 [DOI] [PubMed] [Google Scholar]
- 25.Santra T, Herrero A, Rodriguez J, von Kriegsheim A, Iglesias-Martinez LF, Schwarzl T, et al. An Integrated Global Analysis of Compartmentalized HRAS Signaling. Cell Rep 2019;26:3100–15 e7 [DOI] [PubMed] [Google Scholar]
- 26.Janes MR, Zhang J, Li LS, Hansen R, Peters U, Guo X, et al. Targeting KRAS Mutant Cancers with a Covalent G12C-Specific Inhibitor. Cell 2018;172:578–89 e17 [DOI] [PubMed] [Google Scholar]
- 27.Chang MT, Asthana S, Gao SP, Lee BH, Chapman JS, Kandoth C, et al. Identifying recurrent mutations in cancer reveals widespread lineage diversity and mutational specificity. Nat Biotechnol 2016;34:155–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Scott AN, Hetheridge C, Reynolds AR, Nayak V, Hodivala-Dilke K, Mellor H. Farnesyltransferase inhibitors target multiple endothelial cell functions in angiogenesis. Angiogenesis 2008;11:337–46 [DOI] [PubMed] [Google Scholar]
- 29.Koster MI, Roop DR. Mechanisms regulating epithelial stratification. Annu Rev Cell Dev Biol 2007;23:93–113 [DOI] [PubMed] [Google Scholar]
- 30.Sakamoto K, Aragaki T, Morita K, Kawachi H, Kayamori K, Nakanishi S, et al. Down-regulation of keratin 4 and keratin 13 expression in oral squamous cell carcinoma and epithelial dysplasia: a clue for histopathogenesis. Histopathology 2011;58:531–42 [DOI] [PubMed] [Google Scholar]
- 31.Luk IY, Reehorst CM, Mariadason JM. ELF3, ELF5, EHF and SPDEF Transcription Factors in Tissue Homeostasis and Cancer. Molecules 2018;23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Abrigo M, Alvarez R, Paparella ML, Calb DE, Bal de Kier Joffe E, Gutkind JS, et al. Impairing squamous differentiation by Klf4 deletion is sufficient to initiate tongue carcinoma development upon K-Ras activation in mice. Carcinogenesis 2014;35:662–9 [DOI] [PubMed] [Google Scholar]
- 33.Baillie R, Tan ST, Itinteang T. Cancer Stem Cells in Oral Cavity Squamous Cell Carcinoma: A Review. Front Oncol 2017;7:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rhee CS, Sen M, Lu D, Wu C, Leoni L, Rubin J, et al. Wnt and frizzled receptors as potential targets for immunotherapy in head and neck squamous cell carcinomas. Oncogene 2002;21:6598–605 [DOI] [PubMed] [Google Scholar]
- 35.Shih CH, Chang YJ, Huang WC, Jang TH, Kung HJ, Wang WC, et al. EZH2-mediated upregulation of ROS1 oncogene promotes oral cancer metastasis. Oncogene 2017;36:6542–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cox AD, Fesik SW, Kimmelman AC, Luo J, Der CJ. Drugging the undruggable RAS: Mission possible? Nat Rev Drug Discov 2014;13:828–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stephen AG, Esposito D, Bagni RK, McCormick F. Dragging ras back in the ring. Cancer Cell 2014;25:272–81 [DOI] [PubMed] [Google Scholar]
- 38.Storck EM, Morales-Sanfrutos J, Serwa RA, Panyain N, Lanyon-Hogg T, Tolmachova T, et al. Dual chemical probes enable quantitative system-wide analysis of protein prenylation and prenylation dynamics. Nat Chem 2019;11:552–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Berndt N, Hamilton AD, Sebti SM. Targeting protein prenylation for cancer therapy. Nat Rev Cancer 2011;11:775–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stolze B, Reinhart S, Bulllinger L, Frohling S, Scholl C. Comparative analysis of KRAS codon 12, 13, 18, 61, and 117 mutations using human MCF10A isogenic cell lines. Sci Rep 2015;5:8535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kiessling MK, Curioni-Fontecedro A, Samaras P, Atrott K, Cosin-Roger J, Lang S, et al. Mutant HRAS as novel target for MEK and mTOR inhibitors. Oncotarget 2015;6:42183–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Han JY, Oh SH, Morgillo F, Myers JN, Kim E, Hong WK, et al. Hypoxia-inducible factor 1alpha and antiangiogenic activity of farnesyltransferase inhibitor SCH66336 in human aerodigestive tract cancer. J Natl Cancer Inst 2005;97:1272–86 [DOI] [PubMed] [Google Scholar]
- 43.Oh SH, Kim WY, Kim JH, Younes MN, El-Naggar AK, Myers JN, et al. Identification of insulin-like growth factor binding protein-3 as a farnesyl transferase inhibitor SCH66336-induced negative regulator of angiogenesis in head and neck squamous cell carcinoma. Clin Cancer Res 2006;12:653–61 [DOI] [PubMed] [Google Scholar]
- 44.Yue X, Lin X, Yang T, Yang X, Yi X, Jiang X, et al. Rnd3/RhoE Modulates Hypoxia-Inducible Factor 1alpha/Vascular Endothelial Growth Factor Signaling by Stabilizing Hypoxia-Inducible Factor 1alpha and Regulates Responsive Cardiac Angiogenesis. Hypertension 2016;67:597–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Saada-Bouzid E, Le Tourneau C. Beyond EGFR Targeting in SCCHN: Angiogenesis, PI3K, and Other Molecular Targets. Front Oncol 2019;9:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Charvat S, Duchesne M, Parvaz P, Chignol MC, Schmitt D, Serres M. The up-regulation of vascular endothelial growth factor in mutated Ha-ras HaCaT cell lines is reduced by a farnesyl transferase inhibitor. Anticancer Res 1999;19:557–61 [PubMed] [Google Scholar]
- 47.White AC, Tran K, Khuu J, Dang C, Cui Y, Binder SW, et al. Defining the origins of Ras/p53-mediated squamous cell carcinoma. Proc Natl Acad Sci U S A 2011;108:7425–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He F, Melamed J, Tang MS, Huang C, Wu XR. Oncogenic HRAS Activates Epithelial-to-Mesenchymal Transition and Confers Stemness to p53-Deficient Urothelial Cells to Drive Muscle Invasion of Basal Subtype Carcinomas. Cancer Res 2015;75:2017–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wend P, Wend K, Krum SA, Miranda-Carboni GA. The role of WNT10B in physiology and disease. Acta Physiol (Oxf) 2012;204:34–51 [DOI] [PubMed] [Google Scholar]
- 50.Ho AL, Chau N, Bauman J, Bible K, Chintakuntlawar A, Cabanillas ME, et al. 1046OPreliminary results from a phase II trial of tipifarnib in squamous cell carcinomas (SCCs) with HRAS mutations. Annals of Oncology 2018;29 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.