

Abstract
Leiomyosarcoma (LMS) is a mesenchymal neoplasm with complex copy number alterations and characteristic loss of tumor suppressor genes without known recurrent activating mutations. Clinical management of advanced LMS relies on chemotherapy and complementary palliative approaches, and research efforts to date have had limited success identifying clinically actionable biomarkers or targeted therapeutic vulnerabilities. To explore the biological underpinning of LMS, we evaluated gene expression patterns of this disease in comparison to diverse sarcomas, non-mesenchymal neoplasms and normal myogenic tissues. We identified a recurrent gene expression program in LMS, with evidence of oncogenic evolution of an underlying smooth muscle lineage-derived program characterized by activation of E2F1 and downstream effectors. Recurrently amplified or highly expressed genes in LMS were identified, including IGF1R and genes involved in retinoid signaling pathways. Though the majority of expressed transcripts were conserved across LMS samples, three separate subtypes were identified that were enriched for muscle-associated transcripts (conventional LMS), immune markers (inflammatory LMS) or a uterine-like gene expression program (uterogenic LMS). Each of these subtypes express a unique subset of genes that may be useful in the management of LMS: IGF1R was enriched in conventional LMS, worse disease-specific survival was observed in inflammatory LMS, and prolactin was elaborated by uterogenic LMS. These results extend our understanding of LMS biology and identify several strategies and challenges for further translational investigation.
Keywords: Leiomyosarcoma, Sarcoma, Gene Expression Profiling, RB1, E2F1, IGF1R, Prolactin
Introduction
Smooth muscle cells exist throughout the body and serve essential functions including contraction of visceral organs, vasoconstriction, bronchoconstriction, piloerection and accommodation. While smooth muscle cells are commonly quiescent and organized into a functional syncytium, upon inflammatory or mitogenic stimuli they can enter an active state capable of proliferation that may be adaptive or pathologic (1). Similar to other mesenchymal-derived tissues, their relative cellular quiescence ostensibly contributes to the low frequency of smooth muscle-derived tumors compared to other more proliferative tissues. Nevertheless, a complex and varied spectrum of benign and malignant smooth muscle tumors exists with diverse anatomic, pathologic and prognostic features (2). Leiomyosarcoma (LMS) is a mesenchymal malignancy derived from smooth muscle cells and represent one of the most common forms of soft tissue sarcoma. Compared to their benign smooth-muscle tumor counterparts, LMS demonstrates nuclear aytpia, mitotic activity and has the propensity for metastasis (3). While LMS may arise in any location, including the large blood vessels, intra-abdominal and retroperitoneal sites (collectively termed extrauterine LMS, or ELMS), the uterus is a common site of origin and uterine LMS (ULMS) helps account for the female gender bias in this disease (4).
LMS has no known recurrent and pathogenic single nucleotide variants, though characteristically bears multiple copy number alterations and loss of tumor suppressor genes including TP53, RB1 and PTEN (5–7). Immunohistochemical markers which support the diagnosis of LMS include smooth muscle actin, desmin and h-caldesmon, though none of these markers are specific for LMS and several histologic variants exist (2). Significant challenges exist in the management of LMS, and for unresectable or metastatic disease standard chemotherapy options are the cornerstone of treatment (8). Insights into the underpinnings of LMS biology are essential to advancing clinical care, as improved means of disease categorization, prognostication and targeted treatment approaches are urgently needed.
Gene expression profiling has been successfully deployed to identify cancer subtypes and their associated oncogenic gene expression programs with unique prognostic and therapeutic characteristics in many malignancies including breast cancer (9), lymphoma (10), colon cancer (11), prostate cancer (12) and others. While these molecular subtypes frequently reflect groups defined by morphologic and immunohistochemical features, they have enabled the identification of high-risk gene expression programs with prognostic and therapeutic implications. Using multi-gene RT-PCR to profile the expression of select genes, these findings have translated into clinical testing that informs treatment decisions in breast, colon and prostate cancer (13,14). Currently, molecular profiling of LMS plays no standard role in clinical management (8), though may be crucial in the future development of molecularly targeted therapies for this disease. Previous efforts at defining gene expression programs and molecular subtypes in LMS, using diverse sequencing methods and sample repositories, have proposed three distinct groups that portend prognostic relevance (5,15,16). However, significant discrepancies exist across the LMS subtypes reported in these studies, and additional characterization and analysis of subtypes is needed in hopes of translating these findings to biological understanding and clinical relevance.
To characterize the distinctive gene expression landscape of LMS and better define its molecular subtypes, we compared LMS to other sarcoma histologies, normal myogenic tissues and non-mesenchymal cancers. We identified a recurrent gene expression program that distinguishes LMS from other sarcomas, activation of oncogenic pathways in comparison to normal myogenic tissues, and genes highly expressed within recurrently amplified regions of the LMS genome. Within LMS, we characterized conventional, inflammatory and uterogenic subtypes, each with a unique and reproducible gene expression profile across independent data sets, which bear prognostic relevance. Individual subtypes express unique markers that may be useful in the diagnosis, surveillance, prognosis and targeted treatment of LMS. Taken together, these findings contribute to establishing the biological framework underlying LMS that will enable translational research into this complex disease.
MATERIALS AND METHODS
RNA-seq.
For novel RNA-seq data, fresh frozen tumor samples were obtained from patients consented to an Institutional Review Board (IRB) approved research protocol and undergoing surgery at the Brigham and Women’s Hospital/Dana-Farber Cancer Institute. Total RNA was isolated using an RNeasy Plus Kit (Qiagen). RNA concentration was measured by Nanodrop (Thermo Scientific) and quality by Bioanalyzer (Agilent). Libraries for Illumina NextSeq 500 sequencing were prepared using TruSeq Stranded mRNA Library Prep Kit (Illumina) and equimolar multiplexed libraries were sequenced with single-end 75 bp reads.
Sequencing data analysis.
Computational methods used for RNA-seq data analysis have been described previously (17). Additional raw data files were obtained from the NCBI’s Database of Genotypes and Phenotypes (18) for normal myogenic tissues, NCI’s Genomic Data Commons Legacy Archive (19) for sarcoma samples, or previously published sarcoma RNA-seq data sets (20,21). All fastq files were aligned to hg19 using STAR (22) with expression quantification using Cufflinks (23) to generate gene expression values in FPKM units. Hierarchical clustering of RNA-seq data was performed using Cluster 3.0 (24) and visualized with Java Treeview (25). Gene set enrichment analysis (GSEA) was performed using Hallmark, KEGG or Reactome gene sets present in the Molecular Signatures Database (software.broadinstitute.org/gsea/), with additional enrichment analysis using Metascape (26). PCA was performed separately on TCGA or novel sequencing data derived from this study where samples were similarly processed. Differential expression analysis of RNA-seq data was performed using edgeR (27), with statistical analyses reporting corrected P-values. Pearson correlation coefficient-based heat map matrices were calculated from log2 mean normalized expression values of the top 100 unique LMS-associated genes, with red indicating strong positive correlation, white neutral and blue negative correlation between samples.
Pan-cancer TCGA RNA-seq expression analysis was performed using the Broad Institute TCGA Genome Data Analysis Center (2016): Firehose stddata_2016_01_28 run, Broad Institute of MIT and Harvard doi: 10.7908/C11G0KM9. LMS samples were included in the SARC TCGA subgroup. The results shown here are in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/. Presented data from cBioPortal (28) was restricted to cancer subtypes with sample size >100, and only the most frequently altered tumor subtypes were shown.
Prolactin Measurements.
Serum samples were obtained from LMS patients consented to an IRB approved research protocol at the Brigham and Women’s Hospital/Dana-Farber Cancer Institute. Prolactin was measured by a sandwich electrochemiluminescence immunoassay on a Roche E modular system (Roche Diagnostics). Samples were mixed with a biotinylated prolactin antibody and a prolactin antibody labeled with ruthenium. Streptavidin-coated magnetic microparticles were used to magnetically entrap the biotinylated antibody, and a chemiluminescent reaction was then electrically stimulated, with resulting photon emission utilized for prolactin quantitation. This assay is approved by the Food and Drug Administration for clinical use.
Cell Culture.
All cell lines tested negative for mycoplasma infection on routine surveillance (MycoAlert mycoplasma detection kit, Lonza Bioscience). Experiments using cell lines were performed within 5 passages from the initial purchased or derived stock without further authentication. Commercially available cell lines include SK-LMS-1 (ATCC Cat# HTB-88, RRID: CVCL_0628; vulvar leiomyosarcoma), SK-UT-1 (ATCC Cat# HTB-114, RRID: CVCL_0533; uterine corpus leiomyosarcoma), SKN (Japanese Collection of Research Bioresources Cell Bank Cat# JCRB0176, RRID: CVCL_3167; uterine corpus leiomyosarcoma), and RKN (Japanese Collection of Research Bioresources Cell Bank Cat# JCRB0173, RRID: CVCL_3156; ovarian leiomyosarcoma). LMS20 was obtained from the Sicinska Laboratory in 2016 and was derived from short-term culture of a surgically resected LMS tumor. Commercial cell lines were cultured in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS, 2 mM L-glutamine, 100 mg/ml penicillin, and 100 mg/ml streptomycin. LMS20 was grown in DMEM/F12 with identical supplementation.
Statistical analysis.
Center values, error bars, P-value cutoffs, number of replicates and statistical tests are identified in the corresponding figure and legend. Error bars are shown for all data points with replicates as a measure of variation within each group. Kaplan-Meier analysis of disease-specific survival was calculated by log rank test from survival data available from the NCI’s Genomic Data Commons (19).
Data availability.
Previously published RNA-seq and 3SEQ data presented in this publication are available through the GEO Publication Reference IDs GSE45510 (16), GSE75885 (20), GSE87581 (21), the NCBI’s Database of Genotypes and Phenotypes (18,29) and the NCI Genomic Data Commons (5,19). Novel RNA-seq data presented in this publication are available online through the GEO Publication Reference ID GSE146360.
Results
LMS exhibits a recurrent oncogenic gene expression pattern unique from other sarcomas and normal myogenic tissues.
Several efforts have utilized gene expression profiling to identify diagnostic and prognostic features in soft tissue sarcoma (20,30,31). The most extensive multi-platform analysis of sarcomas was published by the TCGA (5), which compared 206 samples from six sarcoma subtypes, finding that most LMS samples clustered together when compared to other sarcomas. To extend these findings, we performed gene expression comparisons from available (5,20) and novel RNA-seq data sets derived from fresh-frozen tissue. Utilizing transcriptional data sets from TCGA, the French Sarcoma Group (FSG) and Dana-Farber Cancer Institute (DFCI) where a mix of sarcoma subtypes was present, we performed unsupervised hierarchical clustering. In each data set, a majority of LMS samples resided within the same cluster (Fig. 1A–C). Between these data sets, there was overlap in genes expressed at higher levels in LMS compared to other sarcoma subtypes, with gene ontology analysis showing significant enrichment for genes involved in muscle development and function (Fig. 1D–E). The top 500 genes enriched in LMS were expressed at significantly higher levels than non-LMS sarcomas (Fig. 1F), but were expressed at similar levels in normal myogenic tissues derived from the genotype-tissue expression (GTEx) project (Fig. 1G) (29). By contrast, genes enriched in normal smooth muscle compared to cardiac and skeletal muscle were expressed at similar levels in all LMS tumors (Fig. 1H).
Figure 1.
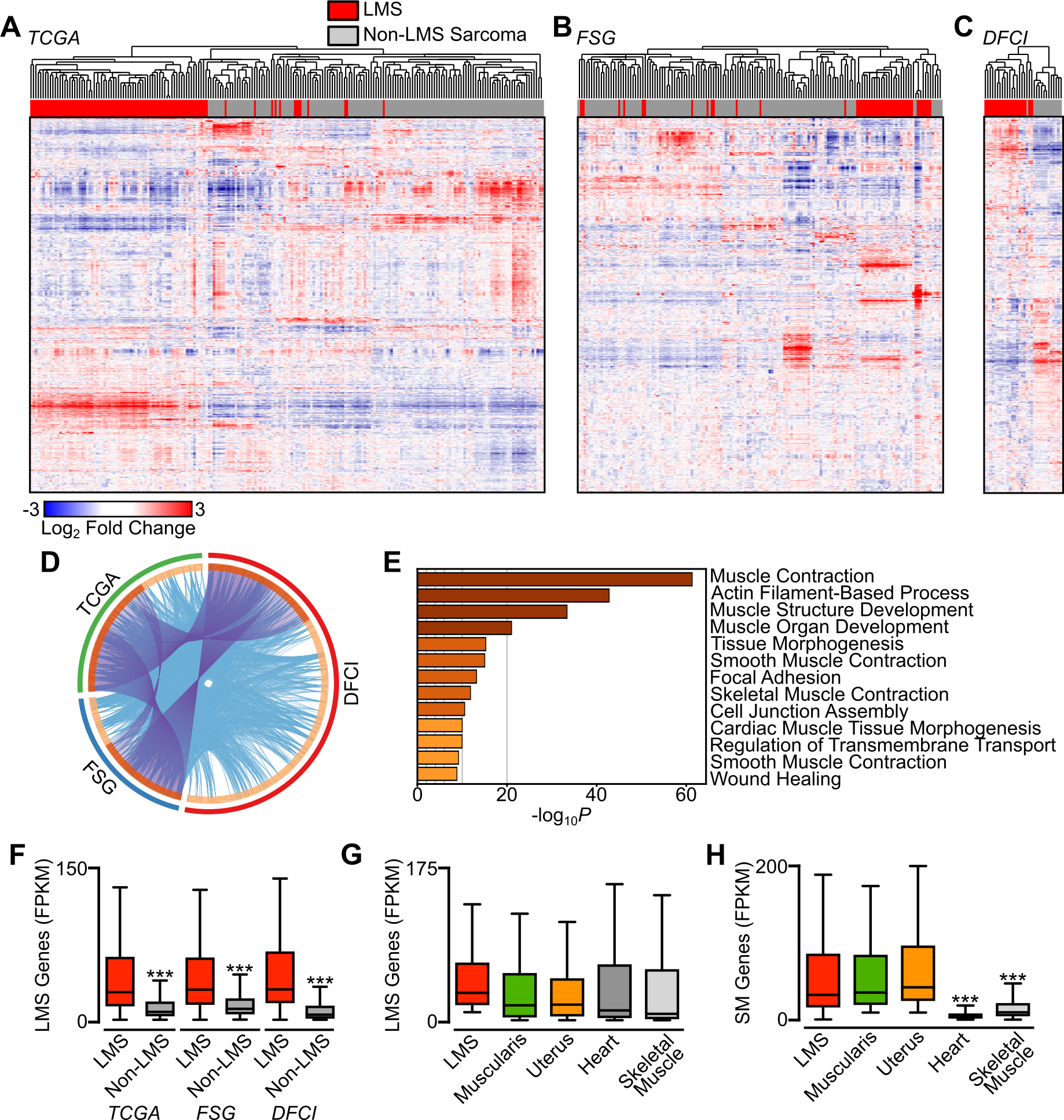
LMS has a recurrent and unique gene expression program among sarcomas. A-C, Unsupervised hierarchical clustering of RNA-seq data from the 10,000 highest expressed genes (rows) and 392 sarcoma samples comprising 10 histologic subtypes (columns). LMS samples are indicated in red and non-LMS sarcoma subtypes is gray. Clustering is divided by data set origin, with The Cancer Genome Atlas in A (TCGA, n=211 with LMS n=88), French Sarcoma Group in B (FSG, n=149 with LMS n=40), and from DFCI in C (Dana-Farber Cancer Institute, n=32 with LMS n=19). D, Circos plot showing overlap of genes enriched in LMS in each data set. The outside arc represents the source data set, the inside arc the individual genes, with darker shading indicating overlap with other data sets. Purple lines indicate shared genes between data sets, and blue lines link genes falling into the same ontologic term. E, Enriched GO terms arising from the top 500 enriched genes from D. Darker coloring indicates higher P-values. F, Box plots displaying expression in FPKM of the top 500 LMS-enriched genes between LMS and non-LMS samples in each sarcoma data set. Data were analyzed by unpaired t-test (compared to LMS; ***, P<0.001). G, Box plots displaying expression in FPKM of the top 500 LMS-enriched genes across normal myogenic tissues (n=85 per tissue) and all LMS samples (n=147). H, Box plots displaying expression in FPKM of the top 500 SM-enriched genes across normal myogenic tissues and all LMS samples. Data were analyzed by one-way ANOVA with Dunnett’s multiple comparisons test (compared to LMS; ***, P<0.001).
To identify pathways activated in LMS compared to normal myogenic tissues, we utilized multiple gene expression pathway databases and gene set enrichment analysis (GSEA). Comparison of smooth muscle-derived tissues, composed of esophageal muscularis and uterus, to all LMS samples showed significant enrichment for cancer-associated pathways including telomere maintenance, cell cycle, and DNA replication in LMS (Fig. 2A). In contrast, there was enrichment in phosphatidylinositol (PI) and FGFR signaling gene sets in normal smooth muscle (Fig. S1A–B). LMS showed significant enrichment for genes involved in myogenesis (Fig. 2B), suggesting that derivation from a myogenic precursor or dedifferentiation from more mature smooth muscle may be relevant in oncogenesis. Notably, gene sets consisting of pathways regulated by E2F and cell cycle regulation were among the most enriched in LMS (Fig. 2A,C–D).
Figure 2.
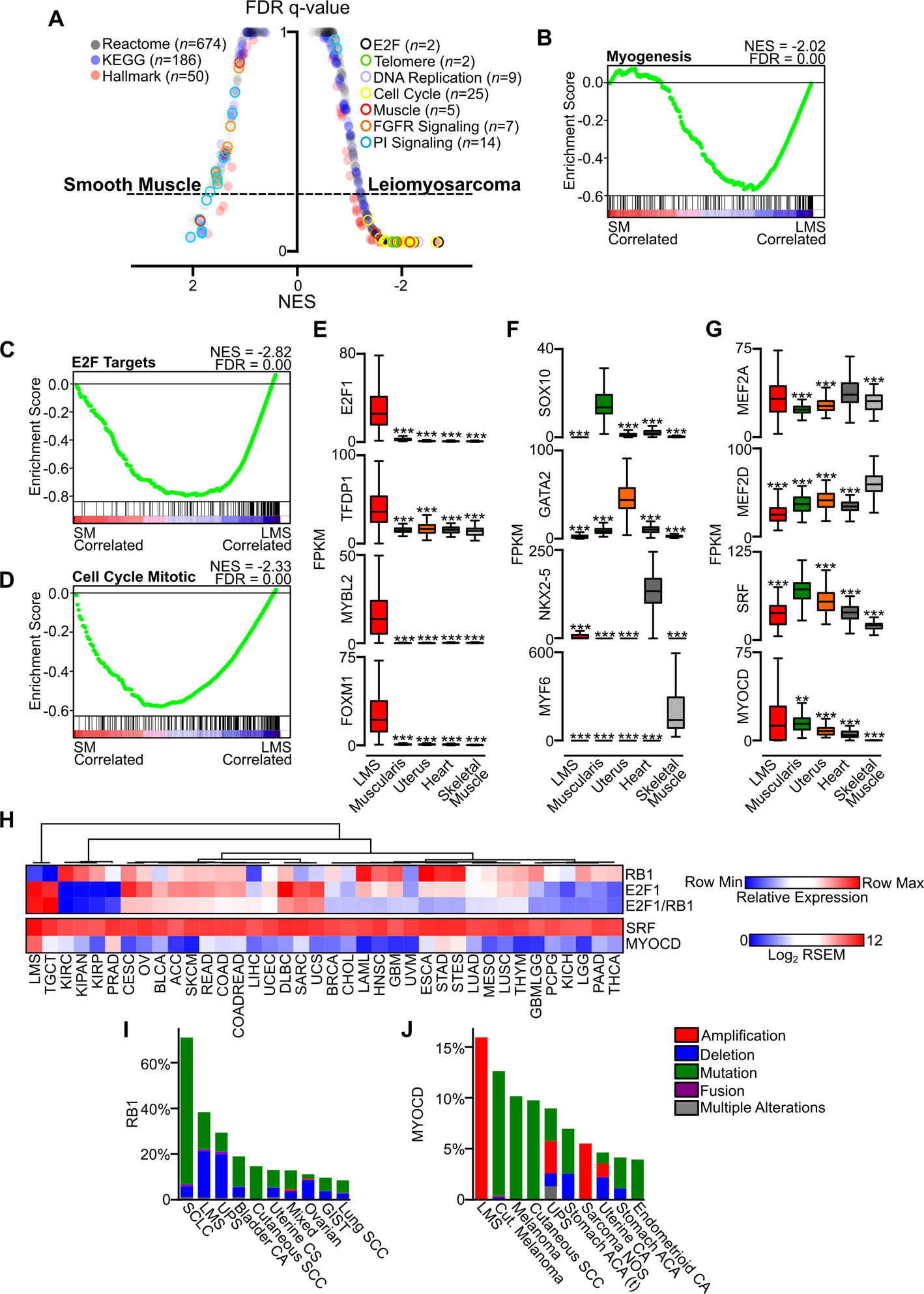
Oncogenic pathways in LMS. A, GSEA plot of false discovery rate (FDR) and normalized enrichment score (NES) in Reactome, KEGG and Hallmark gene sets comparing normal tissue enriched for smooth muscle (SM, combined esophageal muscularis (n=85) and uterus (n=85)) and all LMS samples (n=147). B, Hallmark Myogenesis gene set comparing normal smooth muscle and LMS. C-D, Hallmark E2F targets and Reactome Cell Cycle Mitotic gene sets comparing normal smooth muscle and LMS. E, Enrichment of oncogenic TFs related to E2F1 in LMS (n=147) compared to normal myogenic tissues (n=85 each). F, Select muscle lineage-specific TFs in LMS and normal myogenic tissues. G, Muscle-associated TFs expressed in LMS and normal myogenic tissues. Data were analyzed by one-way ANOVA with Tukey’s post-hoc test (compared to the highest mean expressing group; **, P<0.01; ***, P<0.001). H, Heatmap of TCGA RNA-seq data across cancer subtypes, with LMS samples compiled in their own group. Relative expression of RB1, E2F1 and the ratio of E2F1 to RB1 expression are shown, with heatmap indicating the row minimum and maximum values (upper panel). Samples are arranged by unsupervised hierarchical clustering of the relative expression value. SRF and MYOCD expression in RSEM are indicated across data sets (lower panel, n=14,114). I, Frequency of RB1 alterations in cancer, with the top ten most altered cancer subtypes in cBioPortal shown. J, Frequency of MYOCD alterations in cancer, with the top ten most altered cancer subtypes shown. See Table S1–S2 for cancer subtype abbreviations in TCGA and cBioPortal data.
Given the prominence of cell cycle and E2F-related gene sets, and the characteristic loss of RB1 in LMS which negatively regulates E2F1 (32), we focused on transcriptional regulators in LMS and normal myogenic tissues. Compared to normal muscle, E2F1, it’s binding partner TFDP1, and downstream oncogenic TFs related to E2F1 including MYBL2 (33) and FOXM1 (34) were significantly upregulated in LMS (Fig. 2E). By contrast, other muscle lineage-specific transcription factors associated with tissue differentiation were exclusively expressed in normal myogenic tissues (Fig. 2F) (35–37). Many transcription factors were expressed across LMS and normal myogenic tissues, including members of the myocyte-specific enhancer factor (MEF2) family and serum response factor (SRF) and its transcriptional co-activator MYOCD (Fig. 2G). MYOCD has previously been identified as an amplified and relevant transcription factor in LMS biology (38), and was significantly enriched in LMS compared to normal myogenic tissues.
Given the prominence of RB1 loss and the E2F1-driven transcriptional program in LMS, we sought to compare the expression of these two genes across all cancer types profiled by TCGA, comprising over 14,000 RNA-seq samples. Compared to other TCGA data sets, relative RB1 expression was lower in LMS than 36 of 37 tumor types. By contrast, relative E2F1 expression was higher in LMS than 36 of 37 TCGA tumor types, and the ratio of E2F1 to RB1 was second only to testicular germ cell tumor (Fig. 2H, upper panel). We performed a similar analysis of SRF, which is ubiquitously expressed, and MYOCD, which was substantially enriched in LMS compared to all other cancer subtypes (Fig. 2H, lower panel). These data highlight the unique transcriptional state of LMS, with LMS demonstrating high E2F1 expression with lower RB1, and unique expression of MYOCD among all other cancer types.
To comparatively evaluate for genomic events involving RB1 and MYOCD, we analyzed cancer genomic profiling data from cBioPortal (28) comprising over 74,000 samples. RB1 was altered in 4,139 (6%) of all samples, with LMS ranking second to small cell lung cancer with an alteration frequency of approximately 40% (Fig. 2I). MYOCD was altered in 1,270 samples (2%) of all samples, with LMS comprising the most commonly altered cancer subtype with complete bias towards gene amplification (Fig. 2J). While TP53 and PTEN disruption is also characteristic of LMS, their frequency of alteration is lower in LMS compared to many other cancer types (Fig. S1C–D). Expression of putative tumor suppressors across TCGA cancer subtypes is notable for overall similarity between LMS and all sarcoma samples, with notably higher expression of CDKN2A, CDKN2B and CDKN2C in LMS compared to most other tumor types (Fig. S1E). An inverse correlation of RB1 and CDK inhibitor protein expression has been previously described in several cancer subtypes, and may suggest loss of RB1 and activation of the E2F transcriptional program as a primary driving event in these diseases (6), and further may represent a biomarker of native resistance to CDK 4/6 inhibition (39), though this needs to be formally tested in LMS.
Taken together, these data demonstrate that, among sarcomas, LMS has a unique gene expression profile that differentiates it from other sarcoma subtypes. These data outline an oncogenic platform driving leiomyosarcoma, with initiation of a myogenic program reliant upon RB1 loss and activation of E2F1 and collaborative transcription factors. The genomic locus of one such transcription factor, MYOCD, is recurrently amplified in LMS to support the expression of this myogenic lineage transcriptional regulator. Compared to non-mesenchymal cancer subtypes, LMS demonstrates a higher proportion of copy number alterations in tumor suppressors and putative oncogenes, which is a characteristic shared with other sarcomas (5).
Identification of putative oncogenes in LMS within recurrently amplified genomic regions.
Though there is significant diversity in patterns of aneuploidy across LMS tumors, we and others have previously reported recurrent genomic amplifications in LMS (5–7). These recurrently amplified regions may harbor potential oncogenic dependencies important in LMS biology. Using genes contained within these previously defined amplified genomic loci (7), we evaluated the expression of 1,775 genes in LMS tumors and normal smooth muscle (Fig. 3A). The majority of these genes (n=1,671) were either expressed at low levels (FPKM <10) or not changed between LMS and smooth muscle, and included putative oncogenes such as MYC (5) which contribute to cancer biology in other contexts. By contrast, 104 genes exhibited 2-fold or greater enrichment in LMS, including MYOCD (Table S3). Several of these genes were more highly expressed in LMS compared to other cancer subtypes, and exhibited enrichment similar to that seen for commonly used LMS histologic markers (Fig. 3B).
Figure 3.
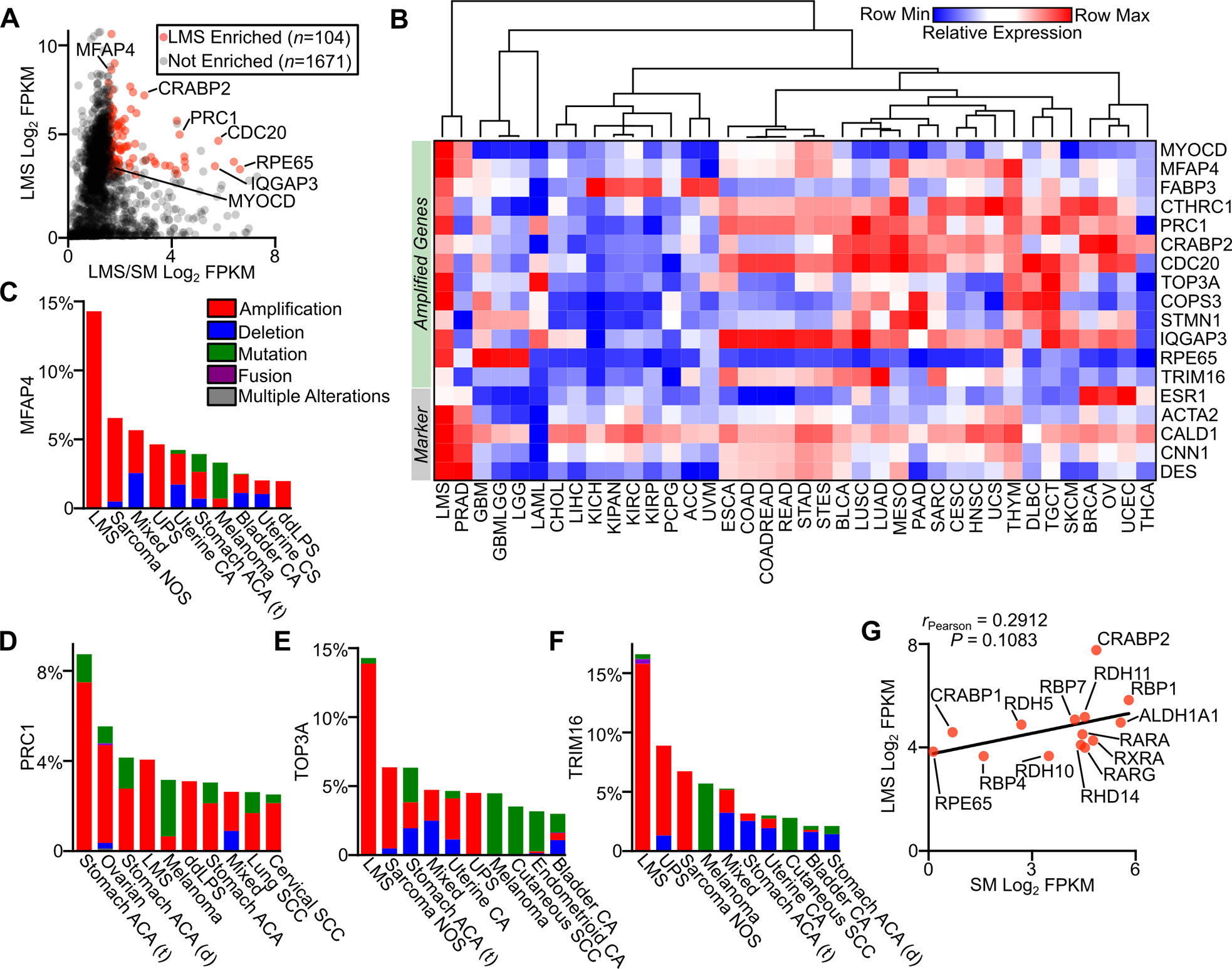
Gene amplification and expression in LMS. A, Plot of LMS log2 FPKM RNA-seq expression versus the log2 ratio of LMS (n=147) to smooth muscle (n=170) FPKM for genes in recurrently amplified regions in LMS. B, Heatmap with unsupervised hierarchical clustering of select amplified genes and standard histologic markers for LMS in TCGA data sets (Table S1). C-F, Frequency of MFAP4, PRC1, TOP3A and TRIM16 alterations across cancer subtypes in cBioPortal, with the top ten most altered cancer subtypes shown (Table S2). G, Plot of expression in FPKM of select components of the retinoic acid signaling pathway in LMS and normal smooth muscle. The Pearson correlation coefficient is shown.
Several of these genes were preferentially amplified in LMS compared to other cancer subtypes in cBioPortal, and may bear relevance to disease pathogenesis. For example, microfibrillar-associated protein 4 (MFAP4) is an extracellular matrix protein involved in intercellular interactions promoting smooth muscle cell proliferation (40), and was found to be highly amplified in LMS compared to other cancers (Fig. 3C). PRC1, TOP3A and TRIM16 were also highly amplified and expressed in LMS compared to other tumor subtypes and normal myogenic tissues (Fig. 3A, D–F), and have been shown to have oncogenic roles and prognostic value in other cancer subtypes (41–43). Like TRIM16, many other transcripts related to the retinoid signaling pathway were expressed in LMS. Compared to smooth muscle, LMS expresses higher levels of many constituents of the retinoid signaling pathway, which includes retinol binding proteins (RBPs), retinol dehydrogenases (RDHs), retinaldehyde dehydrogenases (ALDH1A enzymes), cellular retinoic acid-binding proteins (CRABPs), retinoic acid receptors (RARs), and retinoid X receptors (RXRs) (Fig. 3A,G). Notable is the high expression in LMS of CRABP1 and CRABP2, which are responsible for all-trans-retinoic acid binding and shuttling to metabolic enzymes and nuclear receptors (44). Given the role of retinoic acid signaling in smooth muscle cell homeostasis, proliferation and differentiation (45), these data suggest this pathway may be activated and important in LMS. Taken together, these results highlight a small group of recurrently amplified and highly expressed genes in LMS, and further testing is needed to explore their utility as diagnostic markers or therapeutic vulnerabilities.
Gene expression profiling clusters LMS into conventional, inflammatory and uterogenic subtypes.
Given the clinical diversity seen in LMS and the associated anticipation of LMS subtype-specific oncogenic programs, the wealth of available gene expression datasets, and to build on prior efforts at stratifying disease subtypes, we sought to use gene expression profiling to characterize molecular subtypes of LMS. Unsupervised hierarchical clustering of all LMS samples with known anatomic origin disclosed three distinct gene expression profiles (Fig. 4A). Evaluating genes upregulated in each of these clusters, one showed an abundance of muscle-associated transcripts including CALD1, DMD and MYL9 among others, and was designated conventional LMS (cLMS). The second cluster was enriched in immune-related transcripts including HLA subtypes, interleukins and cytokines, and was designated inflammatory LMS (iLMS). The third subtype was composed predominantly of ULMS, with preservation of genes expressed in the uterus including ESR1, WT1 and HOXA10; this cluster was designated uterogenic LMS (uLMS). Of note, ULMS tumors clustered into each LMS subtype, though all samples outside the uLMS cluster lost expression of the uterogenic transcriptional profile, with stratification being demonstrated by ESR1 expression (Fig. 4A). All subgroups contained both localized and metastatic tumors, and the few tumors with paired primary and metastatic samples clustered within the same subtype (Table S4).
Figure 4.
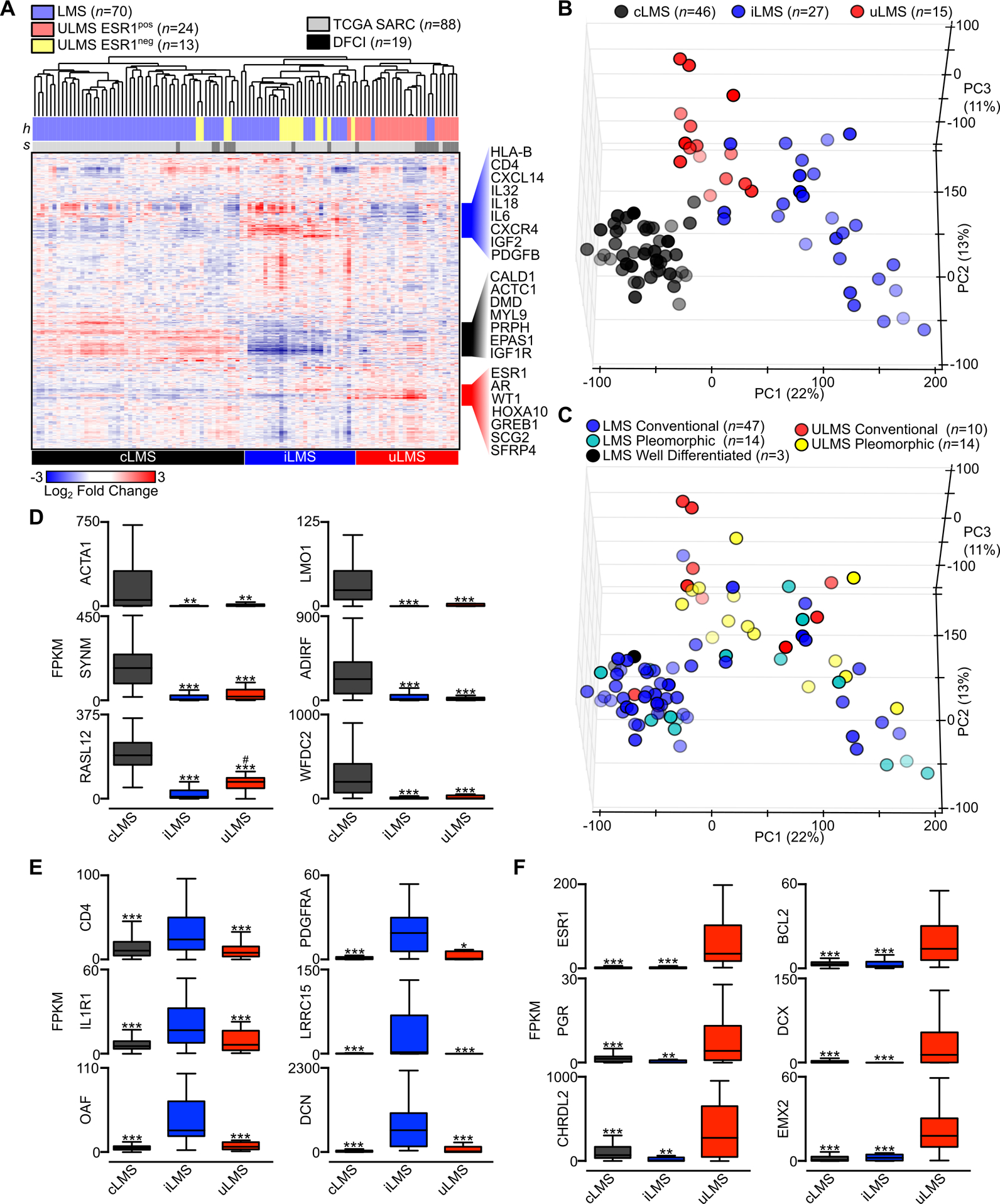
Gene expression profiling defines conventional, inflammatory and uterogenic LMS subtypes. A, Unsupervised hierarchical clustering of RNA-seq data from the 10,000 highest expressed genes (rows) and 107 annotated LMS samples (columns). Histologic tumor subtype (h) and sample origination (s) are indicated with color-coding. An FPKM value of 10 was used as the threshold to determine positive or negative expression of ESR1. Genes listed to the right of the heatmap indicate the identity of select subtype-specific clustered genes. B, PCA of 88 TCGA LMS samples colored by molecular subtype. C, PCA of LMS samples colored by the LMS histologic subtypes well-differentiated, conventional and pleomorphic (inclusive of pleomorphic, undifferentiated and epithelioid histologies). D, Box plots of select cLMS-specific genes. E, Box plots of select iLMS-specific genes. F, Box plots of select uLMS-specific genes. Data were analyzed by one-way ANOVA with Tukey’s post-hoc test (compared to the enriched LMS subtype; *,P<0.05; **, P<0.01; ***, P<0.001).
Principal component analysis (PCA) represents an independent mathematical method of representing sample clustering and variation. Applying PCA to the largest subset of annotated LMS samples with identical sequencing methodology performed by the TCGA, three primary clusters again emerge. Samples clustered more closely together by LMS molecular subtype than by anatomic origin (Fig. 4B, Fig. S2A). Histologic descriptions, including well differentiated, conventional and pleomorphic features, poorly separated LMS subtypes, with the exception of well differentiated samples that all clustered with cLMS (Fig. 4C).
We next used differential expression analysis of RNA-seq data from each LMS subtype to identify uniquely expressed genes in cLMS, iLMS and uLMS. In each subgroup comparison, less than 10 percent of all expressed genes were differentially expressed, indicating the overall homogeneity of LMS subtypes (Fig. S2B–D). Nevertheless, numerous genes were recurrently enriched in each subgroup. For cLMS, muscle-associated transcripts including ACTA1, SYNM and LMO1 were unique and highly expressed (Fig. 4D). For iLMS, inflammatory markers, PDGFRA and LRRC15 were enriched among many others (Fig. 4E). For uLMS, uterine and hormone-related transcripts enriched included ESR1, PGR and EMX2 (Fig. 4F). To characterize the immune infiltrate in each LMS subtype from bulk RNA-seq data, we utilized CIBERSORT (46). This analysis found evidence of macrophage, CD4 and CD8 T cell infiltration in each tumor subtype, with M2 macrophage and CD8 T cell estimated leukocyte fraction highest in iLMS (Fig. S2E). These data demonstrate a unique and recurrent gene expression program within the three subtypes of LMS that supersedes anatomic origin, disease state (localized versus metastatic) or histologic characteristics.
Different gene expression programs drive LMS subtype clustering and bear prognostic relevance.
We next sought to characterize LMS subtype-specific transcripts and their prognostic relevance. Selecting from the 100 most highly and differentially expressed transcripts, subtype-specific clustering could be robustly reproduced (Fig. 5A, Table S5). Each subtype-specific gene list was significantly enriched in its associated cluster compared to other subtypes (Fig. 5B). LMS subtype designation was further explored utilizing gene lists from the Molecular Signatures Database. The cLMS subtype showed the most enrichment in genes associated with smooth muscle contraction, though this gene list failed to statistically differentiate cLMS from uLMS (Fig. 5C). iLMS showed enrichment for many gene sets associated with the immune and inflammatory response (Fig. 5D). Comparing all subtype-specific transcripts with normal myogenic tissues, cLMS and iLMS showed significantly increased expression compared to esophageal muscularis, uterus, myocardium and skeletal muscle. In contrast, uLMS-enriched transcripts were not significantly different from expression levels found in normal uterus (Fig. 5E). Compared to the other nine sarcoma subtypes evaluated (Fig. 1A), cLMS and uLMS gene lists were significantly enriched in their respective LMS subgroups, while the iLMS gene list was also highly expressed in other select sarcoma histologies (Fig. 5F), possibly reflecting similar immune system involvement in these other sarcoma subtypes.
Figure 5.
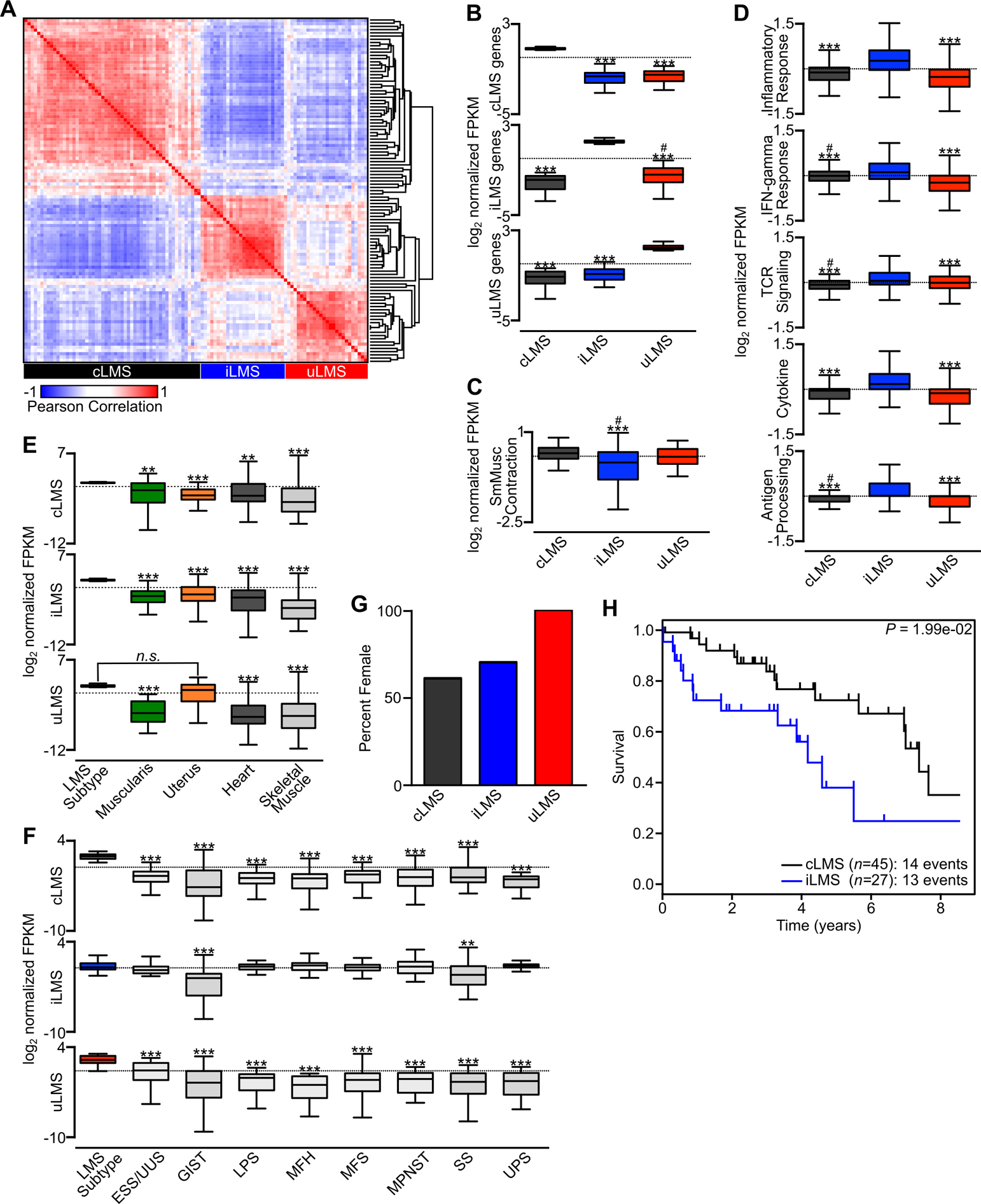
LMS subtype-specific gene expression programs and prognostic relevance. A, Pearson correlation matrix of the top 100 most differentially expressed genes in LMS subtypes. The LMS subtype represented by each cluster is indicated (cLMS n=52, iLMS n=30, uLMS n=25). B, Log2 FPKM values for each LMS subtype normalized to the mean of all LMS samples. For each plot, the indicated subtype-specific gene list was used to compare expression values across subtypes. C, Log2 normalized FPKM values across LMS subtypes for the KEGG Vascular Smooth Muscle Contraction gene list. D, Log2 normalized FPKM values across LMS subtypes for the indicated gene sets including Hallmark Inflammatory Response, Hallmark IFN-gamma Response, KEGG TCR Signaling Pathway, KEGG Cytokine-Cytokine Receptor Interaction and KEGG Antigen Processing and Presentation. E, Log2 normalized FPKM values comparing the indicated LMS subtype and subtype-specific genes to normal myogenic tissues. F, Log2 FPKM values normalized to the mean of all samples comparing the indicated LMS subtype-specific genes to all analyzed sarcoma histologies from Fig. 1A. Data were analyzed by one-way ANOVA with Tukey’s post-hoc test (compared to the indicated LMS subtype; *,P<0.05; **, P<0.01; ***, P<0.001; compared to the non-reference LMS subtype; #,P<0.05). G, Percentage of female patients within each LMS subtype. H, Kaplan-Meier analysis of disease-specific survival comparing cLMS and iLMS subtypes. Survival data were analyzed by log-rank test.
In agreement with this subgroup stratification, a confirmatory RNA-seq data set consisting of 40 LMS tumors could similarly be categorized by this gene list (Fig. S3A) (20). Gene expression signatures for LMS subgroups were similarly enriched in this data set (Fig. S3B–D). These same subgroups were also reproduced in data from 99 LMS formalin-fixed, paraffin-embedded (FFPE) tumors evaluated by 3’ end RNA sequencing (3SEQ) (16), though while significant differences were found between groups the magnitude of changes were less marked in this data set (Fig. S3E–F), perhaps arising from differences in sample source and sequencing technology. Previously identified LMS markers consistent with cLMS were enriched in this subgroup (Fig. S4A–B). ARL4C expression and the CSF1 signature, previously identified as an LMS subtype distinct from the muscle-like subtype (16), were enriched in iLMS, though no consistent enrichment was found in ROR2 or the CINSARC signature (16) (Fig. S4C–F).
All uLMS tumors occurred in women (Fig. 5G). Though sample numbers were smaller in this group, this may indicate that the ELMS tumors clustering within the uLMS subtype originate from a gynecologic cell of origin outside the adult uterus, such as from embryonic remnants of the paramesonephric ducts. Conversely, cLMS or iLMS arising from ULMS may originate from a non-myometrial progenitor cell, such as vascular smooth muscle within the uterus, suggesting a unique cell of origin for each LMS subtype.
Comparing cLMS and iLMS, there was a significantly worse disease-specific survival in the iLMS group (Fig. 5H). In agreement, markers of inflammation, including macrophage infiltration and an elevated CSF1 signature gene expression pattern, have previously been found to portend worse prognosis in LMS (47,48). Other LMS subgroup comparisons of survival were limited by sample size (Fig. S5A–B). Taken together, these data indicate that a defined gene list can distinguish LMS subtypes, that the distinctive gene expression profile of each subtype suggests underlying unique biology and origin, and that this transcriptional program leads to differences in clinical course and outcomes.
LMS subtypes have unique expression of putative oncogenic programs.
In evaluating for possible subtype-specific pathway or oncogene activation, we noted that IGF1R was significantly enriched in the cLMS group (Fig. 6A). Despite the overall group enrichment, there was within-group variability in expression levels (Fig. 6B). Stratifying IGF1R expression by the median value, significant differences in survival were observed, with higher IGF1R expression associated with worse outcomes in cLMS (Fig. 6C). These data implicate IGF1R signaling in cLMS pathogenesis, and further studies are warranted to evaluate its utility as a predictive marker of disease course and in future development of IGF1R-directed therapies for LMS (49).
Figure 6.
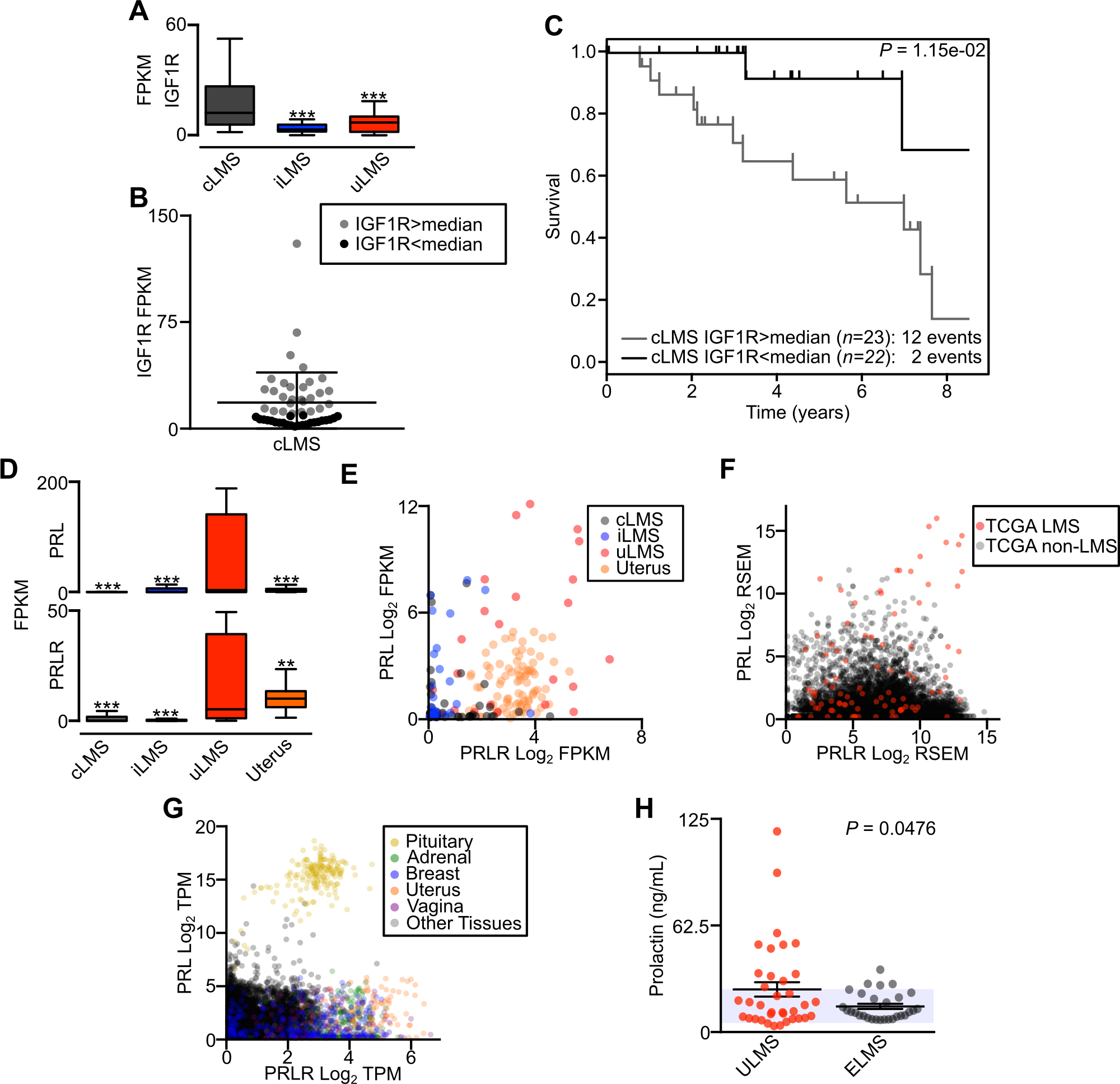
LMS subtype-specific oncogene-associated pathways. A, Box plot displaying FPKM of IGF1R between LMS subtypes (cLMS n=52, iLMS n=30, uLMS n=25). B, IGF1R expression level in cLMS samples, with color differentiating samples above and below the median value. C, Kaplan-Meier analysis of disease-specific survival comparing cLMS stratified by IGF1R expression above or below the median. Survival data were analyzed by log-rank test. D, Box plot displaying FPKM of PRL (top) and PRLR (bottom) between LMS subtypes. Data were analyzed by one-way ANOVA with Tukey’s post-hoc test (compared to the indicated LMS subtype; **, P<0.01; ***, P<0.001). E, Log2 FPKM scatterplot of PRL and PRLR comparing LMS subtypes and normal uterus (n=85). F, Scatterplot of PRL and PRLR log2 RSEM in all TCGA tumors (n=14,114). LMS tumors are indicated in red. G, Scatterplot of PRL and PRLR log2 TPM in GTEx normal tissues (n=10,788). Select tissues are differentiated by color. H, ELISA of serum prolactin levels in patients with metastatic ULMS (n=36) or ELMS (n=29). The shaded area indicates reference values for normal female prolactin levels; P -value indicates unpaired t-test.
Among the most differentially expressed transcripts in the uLMS group were prolactin (PRL) and the prolactin receptor (PRLR) (Fig. 6D). While PRLR was expressed in normal uterus, a subset of uLMS tumors exhibited PRL and PRLR upregulation (Fig. 6E). Evaluating PRL and PRLR expression across all TCGA samples, a subset of LMS had distinctively high expression of both transcripts (Fig. 6F). Though PRLR is expressed in many normal tissues including the uterus, breast, vagina and adrenal glands, PRL expression is normally restricted to the pituitary (Fig. 6G). To determine if elevated levels of circulating prolactin could be detected in LMS patients, we performed a prolactin ELISA on serum from patients with metastatic ULMS or ELMS. Though tumors in these patients could not be further stratified by molecular subtype for comparisons, plasma from patients with ULMS had significantly higher levels of prolactin compared to ELMS, with a subset of patients having high levels of circulating prolactin (Fig. 6H). Formal studies are needed to determine if prolactin could be a useful circulating tumor marker in some patients with uLMS, or may act in an autocrine loop to stimulate tumor growth.
In an attempt to validate potential LMS oncogenic pathways, we profiled five LMS cell lines by RNA-seq. Using PCA, the global gene expression program of these cell lines did not resemble that from LMS tumors (Fig. S6A). Further, many distinctive transcripts enriched in LMS and associated with smooth muscle lineage are not expressed in cell lines, with the exception of those related to cell cycle activation, and cell lines express genes that may support growth programs unrelated to LMS biology (Fig. S6B–E, Table S6). Taken together with prior studies on LMS cell lines (50), these data argue for the broad loss of fidelity of LMS cell lines to their tumor of origin. This may owe in part to the characteristic loss of tumor suppressors and lack of defined oncogene activation in LMS, enabling the evolution of divergent growth programs in vitro, and underscores the urgent need to develop valid pre-clinical models of this disease to enable translational research.
Discussion
In the present study we explore the gene expression profile of LMS through comparisons to other sarcoma subtypes, normal myogenic tissues and non-mesenchymal cancers. In comparisons to sarcomas, the vast majority of LMS tumor samples cluster together, suggesting the oncogenic evolution of an underlying smooth muscle lineage-derived gene expression program. Among cancer subtypes, LMS is notable for loss of RB1 and associated activation of E2F1 and related downstream pathways, possibly representing a common and early oncogenic event. Supporting the preeminence of RB1 loss in the development of mesenchymal neoplasms, and LMS in particular, patients with hereditary retinoblastoma are at high risk of developing sarcomas. In survivors of hereditary retinoblastoma treated with radiation, sarcomas account for the majority of cancer diagnoses (51), with LMS representing the most common histology (52). E2F1 is a transcription factor that is negatively regulated by the Rb family of proteins with complex and context-dependent roles in driving cell cycle, cellular growth and survival (53). With the commonality of RB1 alterations in LMS (5,54), E2F1 proceeds with an unchecked oncogenic program, and targeting this pathway represents a challenging but promising strategy in this disease. Beyond this prominent means of oncogenesis, these data have identified or reinforced several additional cancer-associated pathways that may represent diagnostic opportunities or therapeutic vulnerabilities in LMS. In addition the to loss of common tumor suppressors (e.g. TP53, PTEN), potentially oncogenic pathways and tumor biology in LMS that merit further evaluation include those involving MYOCD, IGF1R and retinoid signaling. Identifications of such transcripts present within recurrently amplified regions of LMS may suggest their selection during tumor evolution.
The majority of expressed genes were conserved across LMS samples. However, through hierarchical clustering and PCA, three separate LMS subtypes were distinguished by a subset of uniquely expressed genes. Among subtypes, cLMS was found to have enrichment for smooth-muscle associated proteins, iLMS for markers of immune cell activity and uLMS a uterine-like gene expression program. The genes that best distinguished these subtypes were conserved across different cohorts, sample preparations, sequencing methods and between localized and metastatic disease, arguing for the generalizability of these markers, the biology giving rise to their expression and the utility of this experimental approach.
Several previous reports have evaluated LMS molecular subtypes by various methodologies. Initial microarray profiling and later 3SEQ identified three groups consisting of (1) a “muscle-enriched” subtype, (2) “subtype II” enriched for the CSF1 gene signature and ARL4C which had a worse prognostic outcome, and (3) “subtype III” defined as cases that did not fit into the other subtypes (15,16). More recently, primary analysis of TCGA LMS samples also noted three distinct LMS subtypes by mRNA expression (5). However, there has been both limited analysis of drivers of LMS clustering and discrepancies between studies. Differences may be attributed to sequencing methodology (microarray, 3SEQ and RNA-seq), sample type (FFPE, fresh frozen, disease state) and variable exclusion of samples in clustering analysis. In the present investigation, we have utilized differential expression analysis of RNA-seq data to identify uniform signatures for cLMS, iLMS and uLMS that stratify tumors across multiple data sets. Further, we have described distinguishing biological processes and pathways enriched in these subtypes that may explain their cellular origin and give translational insight into LMS.
A subset of cLMS expresses IGF1R, which is associated with worse prognosis in this subtype and may represent an informative prognostic marker. Further, in early clinical trials utilizing combination treatment with mTOR and IGF1R inhibition, some patients with LMS derived benefit (49,55). Based on early clinical experience and these data, additional preclinical studies with combination therapy utilizing IGF1R inhibition in this LMS subtype are warranted. As evidenced by the iLMS subtype and in several prior reports, LMS with evidence of immune infiltration has a worse prognosis (16,47,48). Though isolated cases of successful treatment with checkpoint blockade have been reported, larger trials have shown no clear benefit in ULMS (56). Further efforts at histologic characterization of immune infiltrates in LMS subtypes are warranted, as this iLMS subset may most benefit from treatment with checkpoint inhibition or other immune-based therapies (57).
The uLMS subtype maintains components of a myometrium-derived gene expression program, including expression of estrogen, progesterone and prolactin receptors. There is limited evidence that targeting the estrogen receptor with aromatase inhibition may have clinical benefit in select ULMS patients (58), suggesting functional dependency upon myometrial lineage-related hormonal receptors (59). Whether the ectopic expression of prolactin in a subset of uLMS supports tumor growth in an autocrine loop is uncertain. However, evidence of oncogenic activity from autocrine prolactin signaling, and the therapeutic utility of blocking the prolactin receptor, has previously been reported in breast cancer (60). These data suggest that prolactin signaling may be an uncommon means of acquired autocrine stimulation in several cancer subtypes of reproductive origin.
Translational opportunities in cancer research rely upon the use of preclinical models of disease. In attempting to model features of LMS biology in vitro, we evaluated five LMS cell lines and found little relationship between their gene expression program and that of LMS tumors, either in their global transcriptional program or transcripts enriched in LMS or of smooth muscle lineage. This may have arisen from an incorrect diagnosis of the malignancy these cells were derived from, in vitro selection for alternative growth programs in the setting of tumor suppressor loss common in LMS or a combination of these and other factors. Nevertheless, development of preclinical models of LMS is essential to furthering our understanding of LMS biology and attempts at future translational investigation, and these findings warrant additional efforts in developing such models.
Together with other unbiased sequencing efforts, these data highlight the complexity and diversity of LMS biology, including the common loss of tumor suppressors, recurrently amplified genes and evidence of a smooth muscle lineage. Given the limited outcomes data available for this study, additional research regarding prognostic markers in larger case series is warranted. The emerging understanding of this disease and its subtypes will facilitate additional translational research. Given the frequently limited benefit of currently available systemic therapies, there is an urgent need for the development of targeted therapeutic approaches in LMS and an appreciation for which patients they may benefit most.
Supplementary Material
Implications:
Leiomyosarcoma has a recurrent oncogenic transcriptional program and consists of molecular subtypes with biological and possible clinical implications.
ACKNOWLEDGEMENTS
Support for this work was provided by the following sources: Erica’s Entourage Sarcoma Epigenome Project Fund (G.D.D.), the Leiomyosarcoma Support & Direct Research Foundation (M.L.H.), the Catherine England Leiomyosarcoma Fund (S.G.), the Jill Effect (S.G.), the Spivak Faculty Advancement Fund (M.L.H.), the Sarcoma Foundation of America research grant (M.L.H.), the Shore Fellowship Program (M.L.H.) and the Harvard Catalyst Medical Research Investigator Training Program, NIH Award UL 1TR002541 (M.L.H.).
We thank the Dana-Farber Cancer Institute Molecular Biology Core Facilities, Boston Children’s Hospital Department of Laboratory Medicine and the Brigham and Women’s Hospital tissue bank. We thank the Dana-Farber Cancer Institute Center for Sarcoma and Bone Oncology for support. Brian Crompton, Joanna Przybyl and Matt van de Rijn provided valuable feedback on the data and manuscript. We are indebted to the patients and families who donated clinical samples that enabled this research, and prior research efforts that made these annotated data sets publically available.
Footnotes
Competing Financial Interests
G.D.D. reports financial relationships with Ariad, AstraZeneca, Bayer, Blueprint Medicines, Kolltan Pharmaceuticals and Pfizer. S.A.A. consults for Imago Biosciences, Cyteir Therapeutics, C4 Therapeutics, Syros Pharmaceuticals, OxStem Oncology and Accent Therapeutics. S.A.A. has received research support from Janssen, Novartis, and AstraZeneca. S.G. reports financial relationships with Blueprint Medicines, Deciphera Pharmaceuticals, ARIAD, Pfizer, Bayer, Exelixis, Eli Lilly, AstraZeneca, Research to Practice, MORE Health, Huron Consulting, Daiichi Sankyo, Novartis, Wolter Kluwers Health, Abbvei, Abbott Labs and Allergan. None of these relationships constitute a conflict of interest for the present work. The remaining authors declare no conflict of interest.
REFERENCES
- 1.Newby AC, Zaltsman AB. Molecular mechanisms in intimal hyperplasia. J Pathol. 2000;190:300–9. [DOI] [PubMed] [Google Scholar]
- 2.Fletcher CD. World Health Organization Classification of Tumours. 2002;:1–415. [Google Scholar]
- 3.Miettinen M Smooth muscle tumors of soft tissue and non-uterine viscera: biology and prognosis. Modern Pathology. Nature Publishing Group; 2014;27:S17–S29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ducimetière F, Lurkin A, Ranchère-Vince D, Decouvelaere A-V, Péoc’h M, Istier L, et al. Incidence of sarcoma histotypes and molecular subtypes in a prospective epidemiological study with central pathology review and molecular testing. Najbauer J, editor. PLoS ONE. Public Library of Science; 2011;6:e20294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.The Cancer Genome Atlas Research Network. Comprehensive and Integrated Genomic Characterization of Adult Soft Tissue Sarcomas. Cell. 2017;171:950–965.e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chudasama P, Mughal SS, Sanders MA, Hübschmann D, Chung I, Deeg KI, et al. Integrative genomic and transcriptomic analysis of leiomyosarcoma. Nat Comms. Springer US; 2018;:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hemming ML, Klega K, Rhoades J, Ha G, Acker KE, Andersen JL, et al. Detection of Circulating Tumor DNA in Patients With Leiomyosarcoma With Progressive Disease. JCO Precision Oncology. 2019;:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.George S, Serrano C, Hensley ML, Ray-Coquard I. Soft Tissue and Uterine Leiomyosarcoma. J Clin Oncol. 2018;36:144–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Perou CM, Sørlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–52. [DOI] [PubMed] [Google Scholar]
- 10.Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–11. [DOI] [PubMed] [Google Scholar]
- 11.Marisa L, de Reyniès A, Duval A, Selves J, Gaub MP, Vescovo L, et al. Gene Expression Classification of Colon Cancer into Molecular Subtypes: Characterization, Validation, and Prognostic Value. Kemp C, editor. PLoS Med. 2013;10:e1001453–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lapointe J, Li C, Higgins JP, van de Rijn M, Bair E, Montgomery K, et al. Gene expression profiling identifies clinically relevant subtypes of prostate cancer. Proc Natl Acad Sci USA. 2004;101:811–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Paik S, Tang G, Shak S, Kim C, Baker J, Kim W, et al. Gene Expression and Benefit of Chemotherapy in Women With Node-Negative, Estrogen Receptor–Positive Breast Cancer. Journal of Clinical Oncology. 2006;24:3726–34. [DOI] [PubMed] [Google Scholar]
- 14.Knezevic D, Goddard AD, Natraj N, Cherbavaz DB, Clark-Langone KM, Snable J, et al. Analytical validation of the Oncotype DX prostate cancer assay - a clinical RT-PCR assay optimized for prostate needle biopsies. BMC Genomics. 2013;14:690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beck AH, Lee C-H, Witten DM, Gleason BC, Edris B, Espinosa I, et al. Discovery of molecular subtypes in leiomyosarcoma through integrative molecular profiling. Oncogene. Nature Publishing Group; 2009;29:845–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Guo X, Jo VY, Mills AM, Zhu SX, Lee C-H, Espinosa I, et al. Clinically Relevant Molecular Subtypes in Leiomyosarcoma. Clin Cancer Res. 2015;21:3501–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hemming ML, Lawlor MA, Zeid R, Lesluyes T, Fletcher JA, Raut CP, et al. Gastrointestinal stromal tumor enhancers support a transcription factor network predictive of clinical outcome. Proc Natl Acad Sci USA. 2018;115:E5746–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mailman MD, Feolo M, Jin Y, Kimura M, Tryka K, Bagoutdinov R, et al. The NCBI dbGaP database of genotypes and phenotypes. Nat Genet. 2007;39:1181–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Grossman RL, Heath AP, Ferretti V, Varmus HE, Lowy DR, Kibbe WA, et al. Toward a Shared Vision for Cancer Genomic Data. N Engl J Med. 2016;375:1109–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lesluyes T, Pérot G, Largeau MR, Brulard C, Lagarde P, Dapremont V, et al. RNA sequencing validation of the Complexity INdex in SARComas prognostic signature. Eur J Cancer. 2016;57:104–11. [DOI] [PubMed] [Google Scholar]
- 21.Przybyl J, Kowalewska M, Quattrone A, Dewaele B, Vanspauwen V, Varma S, et al. Macrophage infiltration and genetic landscape of undifferentiated uterine sarcomas. JCI Insight. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2012;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28:511–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.de Hoon MJL, Imoto S, Nolan J, Miyano S. Open source clustering software. Bioinformatics. 2004;20:1453–4. [DOI] [PubMed] [Google Scholar]
- 25.Saldanha AJ. Java Treeview--extensible visualization of microarray data. Bioinformatics. 2004;20:3246–8. [DOI] [PubMed] [Google Scholar]
- 26.Zhou Y, Bin Zhou, Pache L, Chang M, Khodabakhshi AH, Tanaseichuk O, et al. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat Comms. Springer US; 2019;:1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2009;26:139–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio Cancer Genomics Portal: An Open Platform for Exploring Multidimensional Cancer Genomics Data: Figure 1. Cancer Discov. 2012;2:401–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, et al. The Genotype-Tissue Expression (GTEx) project. Nat Genet. Nature Publishing Group; 2013;45:580–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takahashi A, Nakayama R, Ishibashi N, Doi A, Ichinohe R, Ikuyo Y, et al. Analysis of Gene Expression Profiles of Soft Tissue Sarcoma Using a Combination of Knowledge-Based Filtering with Integration of Multiple Statistics. Brock G, editor. PLoS ONE. 2014;9:e106801–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Beck AH, West RB, van de Rijn M. Gene expression profiling for the investigation of soft tissue sarcoma pathogenesis and the identification of diagnostic, prognostic, and predictive biomarkers. Virchows Arch. 5 ed 2009;456:141–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Biswas AK, Johnson DG. Transcriptional and Nontranscriptional Functions of E2F1 in Response to DNA Damage. Cancer Research. 2012;72:13–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Musa J, Aynaud M-M, Mirabeau O, Delattre O, Grünewald TG. MYBL2 (B-Myb): a central regulator of cell proliferation, cell survival and differentiation involved in tumorigenesis. Nature Publishing Group. Nature Publishing Group; 2017;:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zona S, Bella L, Burton MJ, de Moraes GN, Lam EWF. FOXM1: An emerging master regulator of DNA damage response and genotoxic agent resistance. BBA - Gene Regulatory Mechanisms. Elsevier B.V; 2014;1839:1316–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kohli S, Ahuja S, Rani V. Transcription factors in heart: promising therapeutic targets in cardiac hypertrophy. Curr Cardiol Rev. 2011;7:262–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Andreucci JJ, Grant D, Cox DM, Tomc LK, Prywes R, Goldhamer DJ, et al. Composition and Function of AP-1 Transcription Complexes during Muscle Cell Differentiation. Journal of Biological Chemistry. 2002;277:16426–32. [DOI] [PubMed] [Google Scholar]
- 37.Bentzinger CF, Wang YX, Rudnicki MA. Building Muscle: Molecular Regulation of Myogenesis. Cold Spring Harbor Perspectives in Biology. 2012;4:a008342–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pérot G, Derré J, Coindre J-M, Tirode F, Lucchesi C, Mariani O, et al. Strong smooth muscle differentiation is dependent on myocardin gene amplification in most human retroperitoneal leiomyosarcomas. Cancer Research. 2009;69:2269–78. [DOI] [PubMed] [Google Scholar]
- 39.Condorelli R, Spring L, O’Shaughnessy J, Lacroix L, Bailleux C, Scott V, et al. Polyclonal RB1 mutations and acquired resistance to CDK 4/6 inhibitors in patients with metastatic breast cancer. Annals of Oncology. 2017;29:640–5. [DOI] [PubMed] [Google Scholar]
- 40.Pilecki B, Schlosser A, Wulf-Johansson H, Trian T, Moeller JB, Marcussen N, et al. Microfibrillar-associated protein 4 modulates airway smooth muscle cell phenotype in experimental asthma. Thorax. 2015;70:862–72. [DOI] [PubMed] [Google Scholar]
- 41.Chen J, Rajasekaran M, Xia H, Zhang X, Kong SN, Sekar K, et al. The microtubule-associated protein PRC1 promotes early recurrence of hepatocellular carcinoma in association with the Wnt/β-catenin signalling pathway. Gut. 2016;65:1522–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hou G-X, Liu P, Yang J, Wen S. Mining expression and prognosis of topoisomerase isoforms in non-small-cell lung cancer by using Oncomine and Kaplan–Meier plotter. Franco R, editor. PLoS ONE. 2017;12:e0174515–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Marshall GM, Bell JL, Koach J, Tan O, Kim P, Malyukova A, et al. TRIM16 acts as a tumour suppressor by inhibitory effects on cytoplasmic vimentin and nuclear E2F1 in neuroblastoma cells. Oncogene. 2010;29:6172–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rhinn M, Dolle P. Retinoic acid signalling during development. Development. 2012;139:843–58. [DOI] [PubMed] [Google Scholar]
- 45.Chen F, Shao F, Hinds A, Yao S, Ram-Mohan S, Norman TA, et al. Retinoic acid signaling is essential for airway smooth muscle homeostasis. JCI Insight. 2018;3:393–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee C-H, Espinosa I, Vrijaldenhoven S, Subramanian S, Montgomery KD, Zhu S, et al. Prognostic Significance of Macrophage Infiltration in Leiomyosarcomas. Clin Cancer Res. 2008;14:1423–30. [DOI] [PubMed] [Google Scholar]
- 48.Espinosa I, Beck AH, Lee C-H, Zhu S, Montgomery KD, Marinelli RJ, et al. Coordinate expression of colony-stimulating factor-1 and colony-stimulating factor-1-related proteins is associated with poor prognosis in gynecological and nongynecological leiomyosarcoma. Am J Pathol. 2009;174:2347–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Schwartz GK, Tap WD, Qin L-X, Livingston MB, Undevia SD, Chmielowski B, et al. Cixutumumab and temsirolimus for patients with bone and soft-tissue sarcoma: a multicentre, open-label, phase 2 trial. Lancet Oncol. 2013;14:371–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Miyata T, Sonoda K, Tomikawa J, Tayama C, Okamura K, Maehara K, et al. Genomic, Epigenomic, and Transcriptomic Profiling towards Identifying Omics Features and Specific Biomarkers That Distinguish Uterine Leiomyosarcoma and Leiomyoma at Molecular Levels. Sarcoma. 2015;2015:412068–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kleinerman RA, Tucker MA, Tarone RE, Abramson DH, Seddon JM, Stovall M, et al. Risk of New Cancers After Radiotherapy in Long-Term Survivors of Retinoblastoma: An Extended Follow-Up. Journal of Clinical Oncology. 2005;23:2272–9. [DOI] [PubMed] [Google Scholar]
- 52.Kleinerman RA, Tucker MA, Abramson DH, Seddon JM, Tarone RE, Fraumeni JF Jr. Risk of Soft Tissue Sarcomas by Individual Subtype in Survivors of Hereditary Retinoblastoma. JNCI Journal of the National Cancer Institute. 2007;99:24–31. [DOI] [PubMed] [Google Scholar]
- 53.Wu Z, Zheng S, Yu Q. The E2F family and the role of E2F1 in apoptosis. Int J Biochem Cell Biol. 2009;41:2389–97. [DOI] [PubMed] [Google Scholar]
- 54.Cuppens T, Moisse M, Depreeuw J, Annibali D, Colas E, Gil-Moreno A, et al. Integrated genome analysis of uterine leiomyosarcoma to identify novel driver genes and targetable pathways. Int J Cancer. 2017;142:1230–43. [DOI] [PubMed] [Google Scholar]
- 55.Quek R, Wang Q, Morgan JA, Shapiro GI, Butrynski JE, Ramaiya N, et al. Combination mTOR and IGF-1R Inhibition: Phase I Trial of Everolimus and Figitumumab in Patients with Advanced Sarcomas and Other Solid Tumors. Clin Cancer Res. 2011;17:871–9. [DOI] [PubMed] [Google Scholar]
- 56.Ben-Ami E, Barysauskas CM, Solomon S, Tahlil K, Malley R, Hohos M, et al. Immunotherapy with single agent nivolumab for advanced leiomyosarcoma of the uterus: Results of a phase 2 study. Cancer. 2017;123:3285–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pollack SM, He Q, Yearley JH, Emerson R, Vignali M, Zhang Y, et al. T-cell infiltration and clonality correlate with programmed cell death protein 1 and programmed death-ligand 1 expression in patients with soft tissue sarcomas. Cancer. 2017;123:3291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.George S, Feng Y, Manola J, Nucci MR, Butrynski JE, Morgan JA, et al. Phase 2 trial of aromatase inhibition with letrozole in patients with uterine leiomyosarcomas expressing estrogen and/or progesterone receptors. Cancer. 2013;120:738–43. [DOI] [PubMed] [Google Scholar]
- 59.Baek M-H, Park J-Y, Park Y, Kim K-R, Kim D-Y, Suh D-S, et al. Androgen receptor as a prognostic biomarker and therapeutic target in uterine leiomyosarcoma. J Gynecol Oncol. 2018;29:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Howell SJ, Anderson E, Hunter T, Farnie G, Clarke RB. Prolactin receptor antagonism reduces the clonogenic capacity of breast cancer cells and potentiates doxorubicin and paclitaxel cytotoxicity. Breast Cancer Res. 2008;10:R68. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Previously published RNA-seq and 3SEQ data presented in this publication are available through the GEO Publication Reference IDs GSE45510 (16), GSE75885 (20), GSE87581 (21), the NCBI’s Database of Genotypes and Phenotypes (18,29) and the NCI Genomic Data Commons (5,19). Novel RNA-seq data presented in this publication are available online through the GEO Publication Reference ID GSE146360.