

Abstract
Background:
For most of the >2,000 CFTR gene variants reported, neither the associated disease liability nor the underlying basic defect are known, and yet these are essential for disease prognosis and CFTR-based therapeutics. Here we aimed to characterize two ultra-rare mutations - 1717–2A>G (c.1585–2A>G) and S955P (p.Ser955Pro) - as case studies for personalized medicine.
Methods:
Patient-derived rectal biopsies and intestinal organoids from two individuals with each of these mutations and F508del (p.Phe508del) in the other allele were used to assess CFTR function, response to modulators and RNA splicing pattern. In parallel, we used cellular models to further characterize S955P independently of F508del and to assess its response to CFTR modulators.
Results:
Results in both rectal biopsies and intestinal organoids from both patients evidence residual CFTR function. Further characterization shows that 1717–2A>G leads to alternative splicing generating <1% normal CFTR mRNA and that S955P affects CFTR gating. Finally, studies in organoids predict that both patients are responders to VX-770 alone and even more to VX-770 combined with VX-809 or VX-661, although to different levels.
Conclusion:
This study demonstrates the high potential of personalized medicine through theranostics to extend the label of approved drugs to patients with rare mutations.
Keywords: CFTR modulators, theranostics, rare mutations, intestinal organoids, precision medicine
Graphical abstract
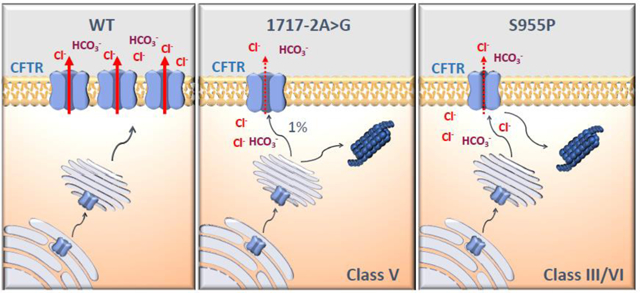
1.1. Introduction
More than 2,000 variants have been reported so far in the CFTR gene which can be grouped into seven functional classes based on their molecular, cellular and functional consequences [1]. However, most of these mutations are still of unknown impact in terms of disease liability and the respective underlying molecular/cellular cause that leads to CFTR dysfunction (https://www.cftr2.org). And yet, with the advent of CFTR modulator drugs, this knowledge is essential as these classes have evolved into theratypes [2]. Therefore, it remains an important challenge to identify and functionally characterize the ~90% of ultra-rare CFTR mutations towards a personalized medicine approach through theranostics [3]. Here, we focused on two ultra-rare mutations as case studies for personalized medicine: c.1585–2A>G (legacy name: 1717–2A>G) [4] and the novel p.Ser955Pro (legacy name: S955P), both identified in two Czech patients with p.Phe508del (F508del - legacy name) on the other allele.
We previously described that 1717–2A>G (an intronic A>G mutation at the second position of IVS11 acceptor) causes aberrant splicing by both retaining six intronic nucleotides and exon 12 skipping [4]. Despite these insights from in vitro studies, the clinical phenotype of this mutation and its effect in patient-derived samples were not described.
S955P (a T>C change at c.2863 in exon 17) is a novel, thus uncharacterized mutation that replaces serine 955 by a proline at the cytoplasmic end of transmembrane helix 8 (TM8) close to the intracytoplasmic loop (ICL) 3 of CFTR, which connects the second nucleotide binding domain (NBD2) to the second transmembrane domain (TMD2) [5].
Here we aimed to characterize RNA splicing pattern and function for these two ultra-rare CFTR mutations in patient-derived rectal biopsies and intestinal organoids from two individuals with CF who had each of these mutations in trans with F508del (p.Phe508del). In parallel, we used a cellular model to characterize S955P-CFTR regarding plasma membrane (PM) trafficking, gating and protein stability independently of F508del-CFTR. Finally, we evaluated the responsiveness of these patients to CFTR modulators.
Results in rectal biopsies and intestinal organoids from both patients evidenced residual CFTR function. Consistently with previous reports, we found that 1717–2A>G leads to two alternatively spliced transcripts and <1% of wt transcripts. In turn, S955P does not affect protein processing but rather channel gating. Finally, we show that intestinal organoids from both individuals respond to VX-770, and even more to VX-809/VX-770 or VX-661/VX-770 combinations, and predict that the latter will be of clinical relevance.
Altogether these data illustrate how complementary in vitro and ex vivo studies can contribute to understand the basic defect of ultra-rare CFTR mutations and to determine their responsiveness to CFTR modulator drugs for possible translation into clinical use.
1.2. Materials and Methods
1.2.1. CF subjects, ethics approval and genomic sequencing
This study was ethically approved at Motol University Hospital and the two patients’ l g l representatives signed informed consents. DNA was collected from both individuals and CFTR gene sequencing was performed by direct Sanger sequencing.
1.2.2. Rectal biopsies and micro-Ussing chamber measurements
Superficial 6–8 rectal mucosa biopsies were obtained by forceps, immediately stored in ice-cold medium and subsequently analyzed in modified micro-Ussing chambers for measurements of equivalent short-circuit current (Ieq-sc) as previously [6].
1.2.3. Intestinal organoids and forskolin-induced swelling (FIS) assay
Crypt isolation from rectal biopsies, organoid culturing and FIS assay were performed as before [7], 3–4 organoids per individual (in duplicate). FIS quantification was done using Cell Profiler and the area under the curve (AUC; t=60; baseline=100%) was calculated using GraphPad Prism 7.0. ANOVA tests with p-value ≤ 0.05 considered as significant.
1.2.4. Organoids immunofluorescence
Organoids immunostaining was performed according to Immunofluorescent Staining Whole-Mount Organoids Protocol by Sigma, using 1 % BSA instead of 5 % horse serum. Organoids were stained for CFTR using the mouse CFTR-specific antibody 528 (1:250, supplied by CFF) followed by incubation with anti-mouse Alexa Fluor 488 - conjugated secondary antibody (1:500, Invitrogen, A-21202). Actin and nuclei were labelled using phalloidin-TRITC (Sigma, P1951) and methyl green (Sigma, 67060), respectively. Images were acquired using Leica SP8 confocal microscope and processed using Fiji software.
1.2.5. mRNA quantification by qRT-PCR
Total RNA isolation from intestinal organoids and reverse transcription was performed as before [16]. RT-PCR (primers in Table S1) was used to assess levels of the three variants of 1717–2A>G transcripts, namely those: i) retaining the last 6 nucleotides of IVS11 (“+6nt”); ii) lacking exon 12 (“Δex12”) [4]; and iii) the normal full-l ngth t nsc ipt (“Nml”). Non-F508del and F508del-transcripts were then separately amplified by qRT-PCR and relative quantification performed as previously for allele specific transcripts [8]. CDX2, CDH17, VIL1, SATB2 relative expression was also assessed by qRT-PCR in 3D intestinal organoids and compared to human nasal epithelial cells (HNEs), using gene specific primers (Harvard Primerbank) and normalization against the housekeeping gene CAP1.
1.2.6. Cell line, Western blotting and micro-Ussing chamber recordings
Lentiviral transduction of the CFBE41o- cell line [9] using S955P-CFTR cDNA plasmid was used after site-directed mutagenesis as previously [10] to generate a novel cell line. CFTR protein was detected in organoids or cells line with anti-CFTR CFF antibodies 450, 570 or 596 as before [10], using calnexin (BD Biosciences antibody) as a loading control. Images were acquired using ChemiDoc XRS+ imaging system BioRad and further processed by Image lab 4.0 software.
CFTR function was assessed in monolayers (transepithelial electrical resistance values above >450 Ω.cm2) mounted in micro-Ussing chambers as previously [11, 12].
1.2.7. Patch-clamp electrophysiological recordings
Patch-clamp was carried out in CHO cells co-transfected with S955P-CFTR-cDNA [pcDNA 3.1 Zeo(+) vector; Invitrogen] as previously [13] being membrane patches excised into an inside-out configuration after the seal resistance >40 GΩ. After the excision, the pipette was perfused with 25 IU of PKA and 2 mM ATP until the current reached a steady-state.
1.2.8. Statistical analyses
Data are mean values ± SEM. Statistical analyses were performed on GraphPad Prism 7.0 using two-tailed paired student’s t-tests (unless otherwise stated), with p ≤0.05 considered as significant.
1.3. Results
1.3.1. Clinical characterization of individuals with CF and the 1717–2G>A or S955P mutations
Patient 1 (CF1) is a 4-year-old boy diagnosed with CF at 6 weeks of age by newborn screening (NBS, raised immunoreactive trypsinogen (IRT) of 161 ng/ml), with a sweat chloride value of 78 mM, pancreatic insufficient (PI) (fecal elastase level (FEE) 14 μg/g), good nutritional status (BMI 19; 2,3 SD) and no upper respiratory tract symptoms. Intermittent colonization with P. aeruginosa and chronic colonization with S. aureus were present. Pulmonary lung function tests were difficult to evaluate since he was still too young to perform the spirometry consistently (FVC 1.05 l (pp89%), FEV1 1.05 l/s (pp105%), MEF50 1.72 l/s (pp102%)). Sequencing results showed the presence of a 1717–2A>G/F508del genotype.
Patient 2 (CF2) is a 7-year-old boy with a positive NBS result for CF (IRT: 118 ng/ml), borderline sweat chloride value (51; 55 mM), pancreatic sufficient (PS) (FEE 431 μg/g) and had a good nutritional status (BMI 14; - 0,96 SD). Pathological flora was repeatedly found in the upper respiratory tract, with Haemophilus influenzae, S. aureus and S. pneumoniae colonization, without acute exacerbations nor need of extensive antibiotic treatments. The novel S955P (c.2863T>C, p.Ser955Pro) mutation was identified along with F508del and some non-pathological variants (Table S2). His pulmonary function values were: FVC 1.63 l (pp88%), FEV1 1.60 l/s (pp99%), MEF50 2.40 l/s (pp100%).
1.3.2. Characterization of 1717–2A>G in native tissues
Allele-specific qRT-PCR of transcripts resulting from 1717–2A>G in native tissues demonstrated that these were significantly reduced vs the ones from the F508del-allele, comprising only 6.7±1.2% of total CFTR transcripts (for n=6 independent samples, Fig.1A). This suggests occurrence of a significantly higher rate of nonsense mediated decay (NMD) than seen for the majority of premature termination codon (PTC)-bearing transcripts [8]. Our RT-PCR and qRT-PCR data using primers specific for the predicted alternative transcripts (Fig.1B, C) confirmed the amplification of two alternatively spliced forms derived from the 1717–2A>G allele: one lacking exon 12 (Δex12, 4.7%) and another with 6 intronic nucleotides extra between exons 11 and 12 (+6nt, 1.1%). Interestingly, a very small amount (0.2% of total) of full-length wt CFTR (Nml) transcript derived from the 1717–2A>G allele was also detected (Fig1A,C).
Figure 1. Effect of the 1717–2A>G mutation in native tissues.
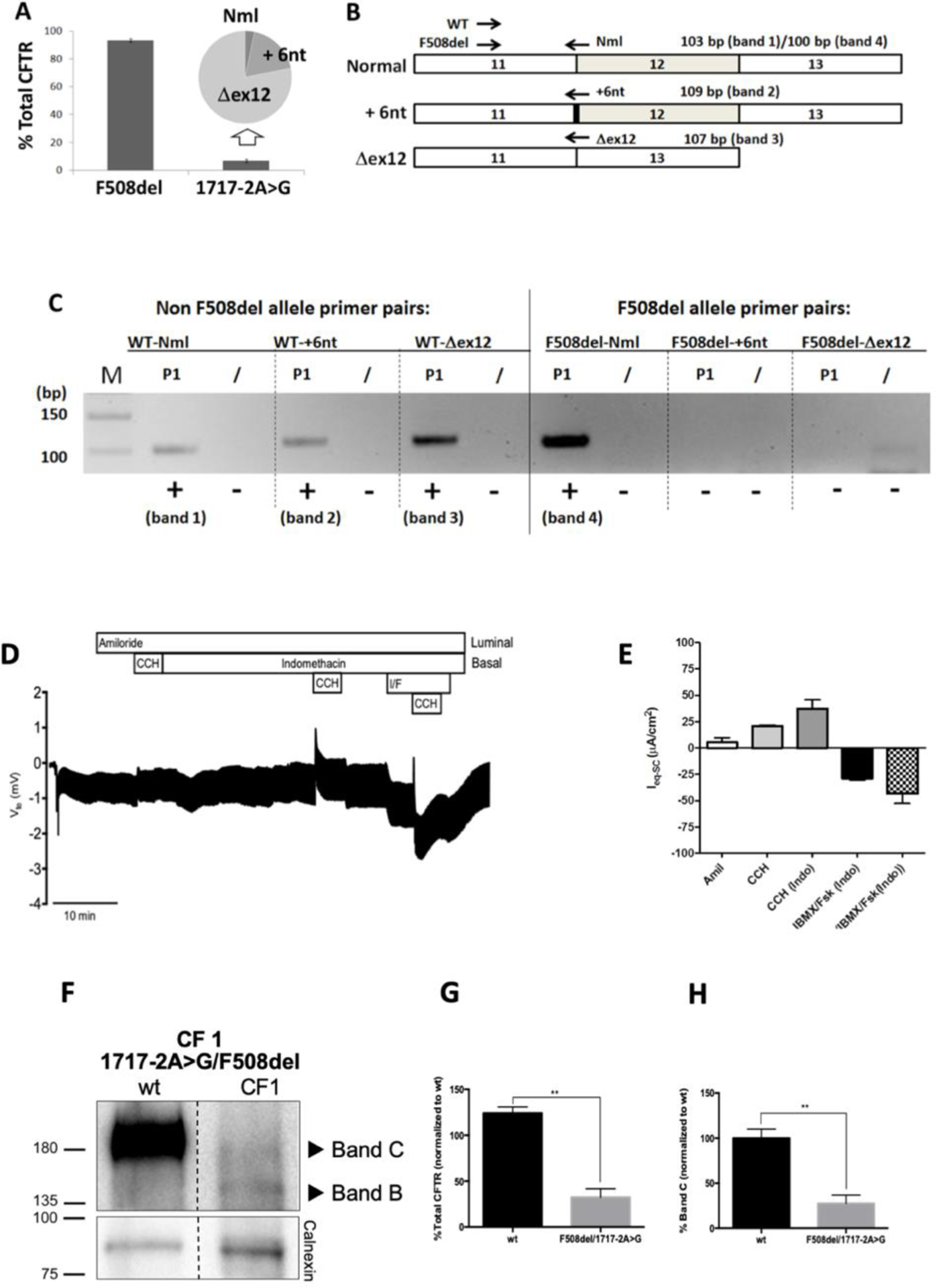
(A) Relative quantification of allele specific CFTR expression in organoids expressing 1717–2A>G/F508del CFTR (CF1): percentage of total CFTR mRNA contributed by each allele is shown (Means ± SEM for n=6 samples). Inset pie chart shows that the 6.7±1.2% of CFTR transcript derived from the 1717–2A>G allele is further divided between three forms (normal spliced product (Nml) − 0.2%, full length product plus retention of 6 intronic nucleotides (+6nt) − 1.1%, and skipping of exon 12 (Dex12) − 4.7%). (B) Scheme for amplification of variant products from intestinal organoid cDNA from CF1 using forward primers specific for non F508del (WT) and F508del alleles, and reverse primers specific for Nml, +6nt, and Dex12 transcripts. Position of primers and expected sizes of products is shown. (C) 4% agarose gel showing products amplified for each combination of primers, for CF1. Bands of the expected size confirming the presence of the full length (band 1) +6nt (band 2) and Dex12 (band 3) products were amplified from the non-F508del allele: only a normally spliced product (band 4) was amplified from the F508del allele. M = DNA marker lane. (D) Representative original recording of the effects of cholinergic (CCH, 100 μM, basolateral) and cAMP-dependent (IBMX/Fsk (I/F), 100 μM/2 μM, basolateral) activation on transepithelial voltage (Vte) in F508del/1717–2A>G rectal biopsies from CF1. Experiments were performed in the presence of Amiloride (Amil, 20 μM, luminal) and/or Indomethacin (Indo, 10 μM basolateral), as indicated in the figure. (E) Summary of activated equivalent short-circuit currents (ΔIeq-sc); data represent the mean of measurements on three rectal biopsies ± SEM. ΔIeq-sc,IBMX/Fsk(Indo) = −28.8 ± 1,7 μA/cm2; ΔIeq-sc,CCH(IBMX/Fsk(Indo)) = −43.3 ± 9.4 μA/cm2. (F) Representative WB analysis of CFTR protein expressed in intestinal organoids from a non-CF control (wt), and from CF1 (n = 3). (G) Quantification of total CFTR expression relative to wt and (H) quantification of CFTR processing (% of Band C related to total CFTR expression). Data are shown as mean ± SEM. Asterisks indicate degree of significant difference calculated by unpaired t-test. ** - p value <0.01.
Next, assessment of CFTR-mediated Cl− secretion in rectal biopsies from CF1 showed that IBMX/Fsk caused a negative transepithelial voltage (Vte) deflection which was further increased by cholinergic co-activation (IBMX/Fsk/CCH), in a biphasic manner (Fig.1D), characteristic of residual CFTR activity [14]. The percentage of CFTR function for this individual is estimated to be ~13% vs non-CF [15].
To evaluate the possible impact of 1717–2A>G on CFTR PM trafficking, total protein was extracted from intestinal organoids and assessed by Western blot (WB). Results show that CFTR was present as some mature form (fully-glycosylated, band C - most likely corresponding to protein from the 1717–2A>G allele) as well as immature form (core-glycosylated, band B), albeit at lower levels (~10%) than that of wt-CFTR (Fig.1F–H). A decrease in processing was also observed, most likely corresponding to protein from the F508del allele. This indicates that CFTR protein resulting from the 1717–2A>G transcripts does not significantly affect CFTR PM trafficking, since F508del (present in the other allele) does not traffic to the PM. However, the data suggest that 1717–2A>G leads to lower CFTR protein levels when compared to non-CF organoids (Fig.1F–H).
1.3.3. Characterization of S955P in native tissues
As expected, S955P (T>C at c.2863) did not alter CFTR splicing nor induce NMD, as confirmed using allele-specific qRT-PCR (data not shown). Analysis of rectal biopsies from CF2 (F508del/S955P) indicates a small negative Vte deflection which was further increased by cholinergic co-activation (IBMX/Fsk/CCH), in a monophasic manner (Fig.2A). This monophasic profile suggests relatively high residual CFTR activity, since lower function typical appears as a biphasic profile [14]. However, the percentage of CFTR function in comparison with non-CF is estimated to be ~18% [15].
Figure 2. Effect of the S955P mutation in native tissues.
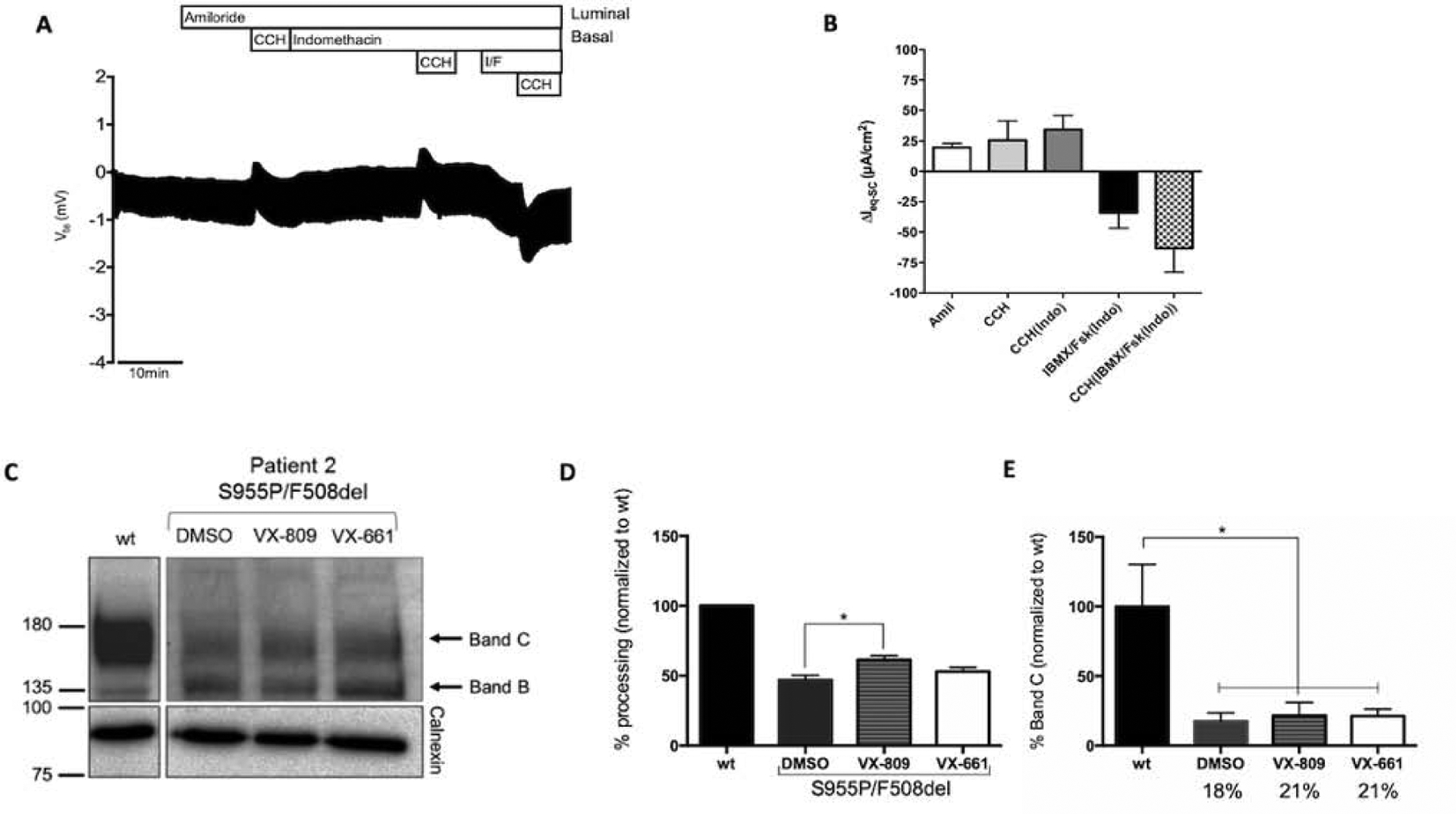
(A) Representative original recording of the effects of cholinergic (CCH, 100 μM, basolateral) and cAMP-dependent (IBMX/Fsk (I/F), 100 μM/2 μM, basolateral) activation on transepithelial voltage (Vte) in rectal biopsies from individual CF2 (F508del/1717–2A>G). Experiments were performed in the presence of Amiloride (Amil, 20μM, luminal) and/or Indomethacin (Indo, 10μM, basolateral), as indicated in the panels. (B) Summary of activated equivalent short-circuit currents (ΔIeq-sc); data represent the mean of measurements on three rectal biopsies ± SEM. CF2 - ΔIeq-sc,IBMX/Fsk(Indo) = −34.1±22.2 μA/cm2; ΔIeq-sc,CCH(IBMX/Fsk(Indo)) = −63.2±34.1 μA/cm2 (18% of WT CFTR function). (C) Representative WB analysis of CFTR protein expressed in intestinal organoids from a non-CF control (wt), and from individual CF2, under basal conditions or following treatment with VX-809 (3μM) or VX-661 (5μM) for 24h (n=3). (D) Quantification of fully-glycosylated protein (band C) for each condition, following densitometry to calculate the percentage of mature CFTR (band C) vs total CFTR expressed (=%processing). (E) Quantification as in D, but the percentage of mature CFTR (band C) was estimated vs mature CFTR (band C) in wt (% band C). Data are shown as mean ± SEM. Asterisks indicate degree of significant difference calculated by unpaired one-way ANOVA using Fisher’s LSD test. * - p value <0.05.
WB analysis of intestinal organoids from CF2 showed that CFTR protein is present as both mature CFTR and immature forms, although the former at lower levels (~18%) than that of wt-CFTR (Fig.2C–E). Since the other allele (F508del) does not produce mature CFTR, these results indicate that the mature form results from S955P which does not significantly affect trafficking. However, the observed lower levels suggest that S955P may cause a biogenesis/stability defect.
1.3.4. Biochemical characterization of S955P-CFTR in cell lines
In order to test the individual effect of S955P on PM trafficking, a novel CFBE cell line expressing solely S955P-CFTR was produced (see Methods). Assessment of S955P-CFTR by WB (Fig.3) revealed that it is present as both immature (band B) and mature (band C) forms similarly to wt-CFTR, confirming that S955P does not alter CFTR PM trafficking (Fig.3A,B), as in CF2’s organoids. Also, maturation of S955P-CFTR (band C) was not significantly enhanced by correctors (Fig.3A,B).
Figure 3. Impact of the S955P mutation on protein processing.
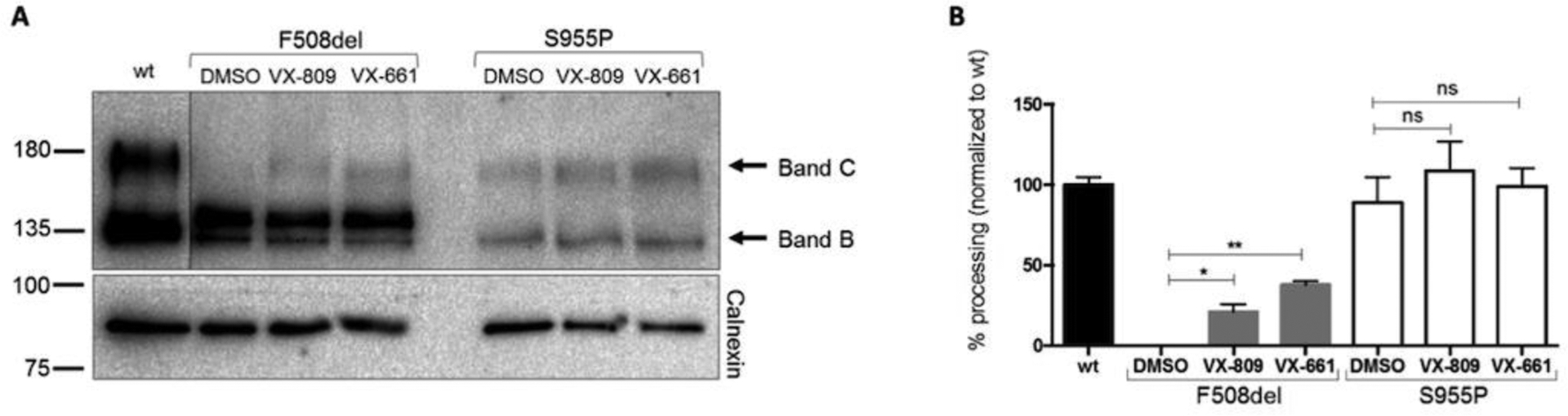
Representative WB analysis of CFTR protein expressed in CFBE cells stably expressing S955P-, F508del or wt-CFTR. CFBE cells were also analyzed following treatment with VX-809 (3 μM) or VX-661 (5μM) for 24h (n=3). For each condition, densitometry was used to calculate the percentage of mature CFTR (band C) vs total CFTR expressed (=%processing). Data were normalized to the efficiency of processing of wt-CFTR and are shown as mean ± SEM. Asterisks indicate degree of significant difference calculated by unpaired t-test. * - p value <0.05, ** - p value <0.01.
1.3.5. Assessment of S955P-CFTR function in cell lines and effect of correctors
To evaluate the function of S955P-CFTR, we assessed transepithelial transport in Ussing chamber of the respective CFBE cells. Fsk/IBMX elicited residual CFTR-mediated Cl− secretion (potentiated by Gen and inhibited by CFTRinh-172) indicating that S955P-CFTR has residual function, consistent with CF2’s rectal biopsies and organoids measurements (Fig.4A,B). Consistently with WB data, treatment with correctors VX-809 or VX-661 did not produce any detectable difference in CFTR-mediated Cl− transport (Fig.4C–E).
Figure 4. Functional characterization of S955P-CFTR in the CFBE cell line.
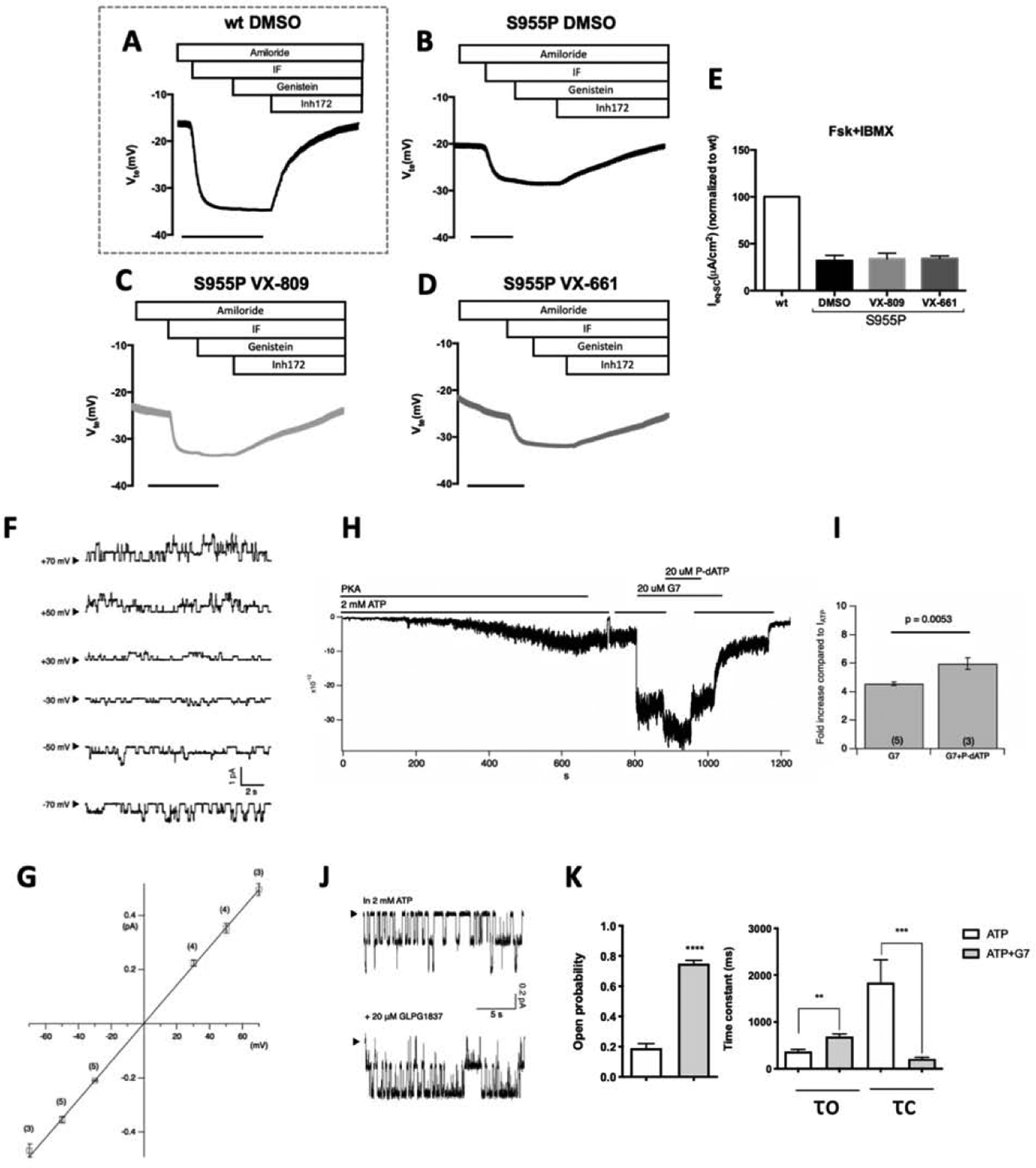
CFBE cells stably expressing (A) wt-CFTR or (B–D) S955P-CFTR, were pre-incubated for 24h with DMSO (0.1% v/v) (A,B) as vehicle control, (C) 3μM VX-809, or (D) 5 μM VX-661 and analyzed under CFTR potentiation with genistein. (E) Graph summarizing equivalent short-circuit currents (Ieq-sc) after apical stimulation with forskolin+IBMX (I/F 2 μM and 100 μM, respectively) obtained in A–D. A low Cl− Ringer solution was used at apical side to stablish a Cl− gradient. Negative transepithelial voltage (Vte) deflections were observed following the addition of apical I/F which were fully reverted by addition of CFTR inhibit Inh172 (30 μM). (F) Representative single-channel CFTR current traces at different membrane potentials in the presence of 2 mM ATP. (G) Single-channel I-V relationships of S955P-CFTR (blue points) vs WT-CFTR (fitting from WT (Yeh et al, 2014)). (H) GLPG1837 further increases macroscopic S955P-CFTR currents in the presence of P-dATP (a high affinity ATP analogue). (I) Comparison of residual currents after incubation with GLPG1837 (G7) shows an increase in 955P-CFTR gating of ~4-fold and ~6-fold in the absence or presence of P-dATP, respectively. (J) Single-channel behavior of GLPG1837-potentiated S955P-CFTR. Comparing these two 20-s traces, longer open events and shorter closed events after potentiation with GLPG1837 were noted. (K) Summary of the additive effect of GLPG1837 on single-channel kinetics of S955P-CFTR. Po, open time (τo), and closed time (τc) were as follows: 0.19±0.03; 368±46ms, and 1841±490 ms for ATP alone (n=4); and 0.75±0.02, 689±56 ms, and 212±35 ms for ATP+GLPG1837 (n=3). Error bars represent the SEM of the mean. Asterisks indicate degree of significant difference calculated by unpaired t-test. ** - p value <0.01, *** - p value <0.001.
Because the S955 residue is close to the TMD2-NBD2 junction, critical for ATP-dependent gating [16], we hypothesized that S955P can affect CFTR gating. To investigate this, S955P-CFTR was expressed in CHO cells and currents were recorded in inside-out patches. The resulting single-channel I-V relationship of S955P-CFTR was nearly identical to that of wt-CFTR (Fig.4F,G), excluding a conductance defect.
To assess S955P-CFTR gating, we first recorded macroscopic currents (Fig.4H) in response to CFTR potentiator GLPG1837 [17] and the high-affinity ATP analogue P-dATP, after phosphorylation with ATP and PKA (in Cl−-based bath). Ten minutes after PKA/ATP exposure, the steady-state current was very small, and the activated current could be abolished by removing ATP (Fig.4H). Application of 20 μM GLPG1837 resulted in a ~4-fold increase (n=3), whereas the addition of P-dATP in the presence of GLPG1837 further enhanced the current by ~20% (~6-fold increase, n=3, Fig.4I).
The 4-fold potentiation of Po by GLPG1837 indicates that the maximal Po without potentiators must be <0.25, which is ~1/2 of wt-CFTR [18], indicating that S955P creates a gating defect. To more accurately estimate Po of S955P-CFTR, membrane patches that yield microscopic single-channel current traces were analysed under ATP or ATP+GLPG1837 (Fig.4I). The high activity of S955P-CFTR under GLPG1837 reassures the accuracy of calculating the number of functional channels in the patch-based on the maximal simultaneous opening steps observed (two in Fig.4J). Single-channel kinetic analysis reveals a Po under ATP of 0.19±0.03 (n=4) with mean open time (τo) of 368±46 ms (n=4) and mean closed time (τc) of 1841±490 ms (n=4), whereas under GLPG1837, Po is 0.75±0.02 (n=3) with τo of 689 ±56 ms and ac of 212±35 ms (Fig.4K), indicating relatively mild gating defects for S955P-CFTR.
1.3.6. Assessment of 1717–2A>G and S955P cellular localization in 3D intestinal organoids
To better decipher the effect of the two rare mutations studied in CFTR cellular localization, 3D-intestinal organoids from CF1 and CF2 were analyzed. qPCR analysis revealed enhanced expression of intestinal markers (CDX2, CDH17, VIL1, SATB2) in 3D intestinal organoids versus human nasal epithelial cells (HNEs), demonstrating the intestinal nature of organoids (Fig.5A).
Fig. 5 – 3D Intestinal organoids characterization.
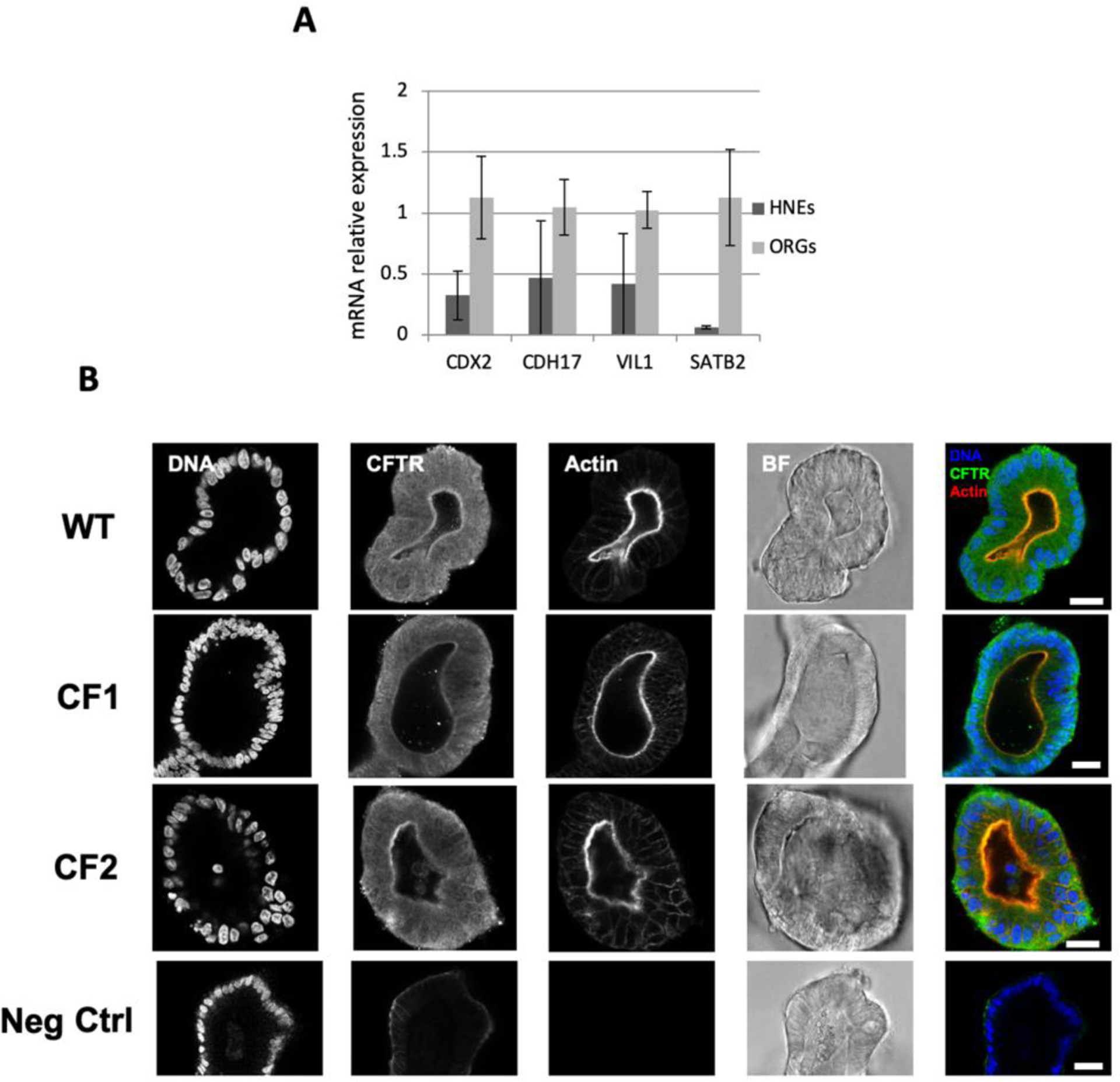
(A) Relative expression of intestinal specific genes (CDX2, CDH17, VIL1, SATB2) was assessed by qRT-PCR using gene specific primers (Harvard Primerbank) in 3D intestinal organoids (ORGs) and human nasal epithelial cells (HNEs). Normalization was performed against the housekeeping gene CAP1. Data is represented as mean ± SEM for n=3. (B) Representative confocal microscopy slices of intestinal organoids (wt, CF1, and CF2). CFTR was immunostained with the 528 antibody (1:250, CFF). Nuclei and F-actin were labelled with Methyl green (1:750) and Phalloidin-TRITC (1:250) respectively, both from Sigma. Corresponding brightfield images (BF) are also shown. Scale bar represents 20μm. Images were acquired with a Leica TCS SP8 confocal microscope.
CFTR was immunostained in 3D intestinal organoids from CF1 and CF2. Along with the previous results from the WB analysis, confocal microscopy images also confirmed the presence of 1717–2A>G (CF1) and S955P (CF2) -CFTR in the luminal membrane of the organoids (actin) (Fig.5B). CFTR expression in CF1 organoids is not as clear as in CF2; this might be explained by the lower expression detected in this individual, that might be too low to be properly detected by this technique.
1.3.6. Function of 1717–2A>G- and S955P-CFTR in organoids and response to modulators
Since CFTR-mediated Cl− secretion measurements in rectal biopsies cannot be used to assess response to CFTR modulators due to poor compound penetration into native tissue, next we performed the forskolin (Fsk)-induced swelling (FIS) assay in organoids, as described [19]. We thus pre-incubated intestinal organoids from CF1 (1717–2A>G/F508del) and CF2 (S955P/F508del) individuals with potentiator VX-770 alone or in combination with correctors VX-809 or VX-661, and stimulated CFTR channel function with Fsk, in parallel with F508del/F508del organoids (Fig.6).
Fig. 6 – Results from forskolin-induced swelling (FIS) assay on intestinal organoids from CF1 (F508del/1717–2A>G genotype) and CF2 (F508del/S955P genotype).
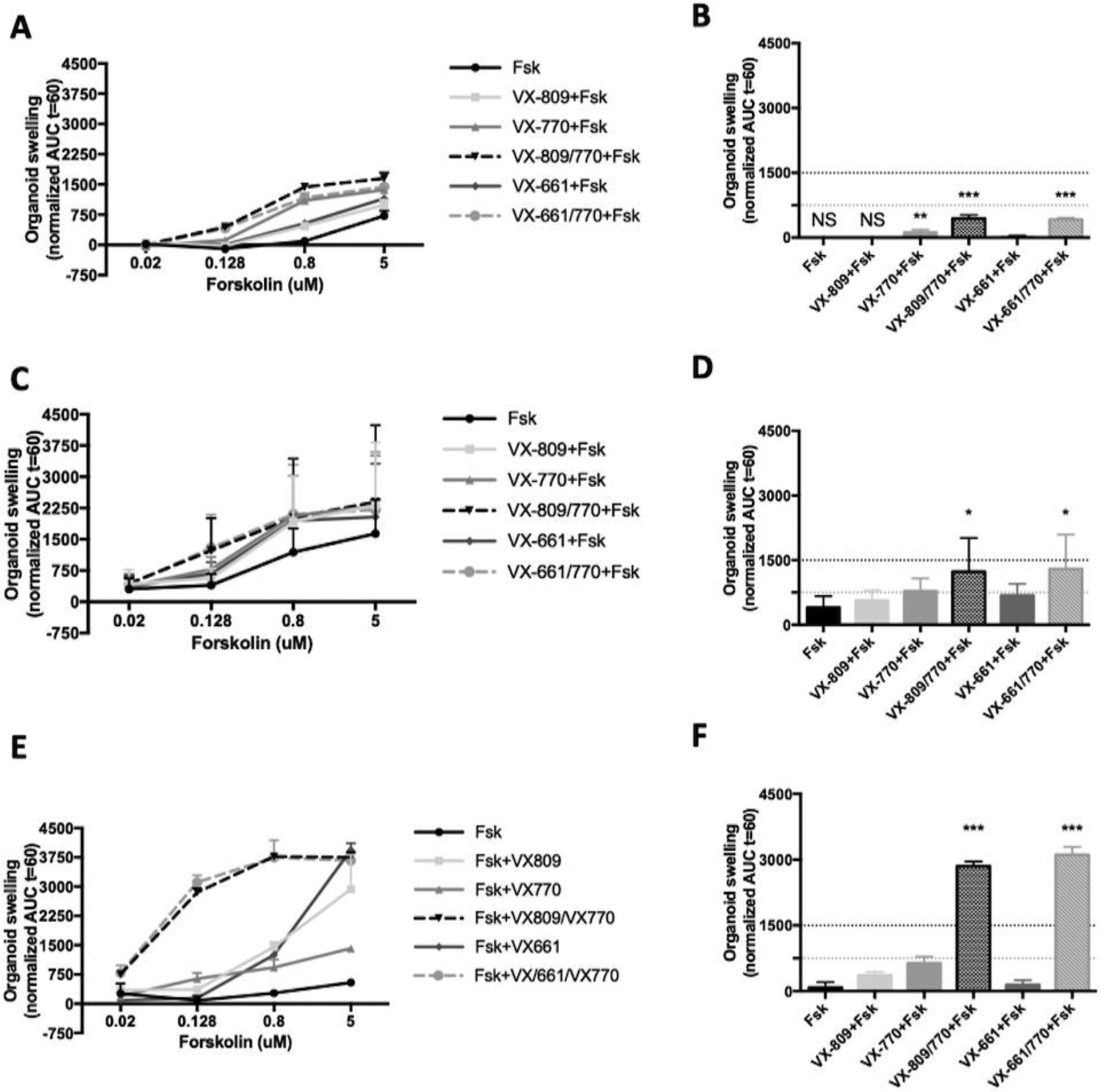
Quantification of FIS in organoids from (A) CF1, (C) CF2 and (E) F508del homozygous control for all treatments at forskolin (Fsk) concentrations of 0.02, 0.128, 0.8 and 5 μM, expressed as the AUC of organoid surface area increase (baseline = 100%, t = 60 min). Quantification of organoid swelling for all treatments at [Fsk] = 0.128 μM for (B) CF1, (D) CF2 and (F) F508del homozygous control. The dashed yellow and green lines represent the established thresholds for medium and high clinical benefit potential for treatments, respectively. NS - No swelling. Data represent the mean of measurements on 5–8 replicate wells per condition ± SEM. Asterisks indicate degree of significant difference calculated by unpaired one-w y ANOVA using Fisher’s LSD test. * - p value <0.05, ** - p value <0.01, *** - p value <0.001.
Data show that for CF1 organoids, CFTR residual function was detected only at Fsk concentration of 5μM (Fig.6A, black line) and that incubation with VX-770 alone already showed response at 0.128μM Fsk, which was statistically different from control (Fig.6B). However, either combination of VX-809/VX-770 or VX-661/VX-770 induced a significantly higher extent of swelling (Fig.6B, red bar vs. blue/brown bar).
For CF2, swelling was detected without any CFTR modulator already at 0.02μM Fsk (Fig.6C, black line), thus confirming residual activity, as in rectal biopsies from this individual. Nevertheless, at this Fsk concentration, potentiator VX-770 already induced modest but significant swelling (Fig.6C,D, red vs. black bar), while the combinations VX-809/VX-770 or VX-661/VX-770 induced further swelling vs control (Fig.6C,D, blue and brown bars).
1.4. Discussion
Results shown here provide the molecular and functional characterization of the S955P and 1717–2A>G CFTR mutations in patient-derived materials and in a bronchial epithelial cell line (S955P) so as to determine the respective basic defect and their responsiveness to CFTR modulators.
So far, 1717–2A>G was only reported in two patients worldwide, CF1 in this study and in another individual with CF from Romania, both in trans with F508del [20]. An in vitro study also described a 1717–2A>G-CFTR minigene [4] originating two alternatively spliced forms, namely: one with six intronic extra nucleotides between exons 11 and 12, and another with exon 12 skipped; both introducing PTCs. Here we confirmed in native tissues the presence of both these forms (Fig.1), albeit at very low abundance which can be ascribed to degradation via NMD. Using CF1 native tissue mRNA we were also able to measure very low abundance (0.2% of total CFTR) of full-length (wt) transcripts derived from the 1717–2A>G allele, thus explaining the residual function measured in both CF1 rectal biopsies (Fig.1D) and organoids (Fig.5A). Since this minor amount of wt-CFTR mRNA produced normally processed CFTR protein (albeit at <10% of wt-CFTR) confirmed by WB and immunostaining, 1717–2A>G should be considered as a class V mutation, which describes those causing a major reduction in the levels of normal CFTR protein.
As S955P is a novel mutation, here reported for the first time (CF2), we first aimed to characterize it. By WB of both organoids and S955P-CFTR CFBE cells we showed that it does not affect CFTR PM trafficking (Figs.2C,3A), since it expresses CFTR band C, consistent with the relatively high residual function in rectal biopsies (~18% vs non-CF), in CFBE cells and intestinal organoids (Figs.2A,4B,5C). The presence of S955P in the apical membrane was also confirmed by confocal microscopy of 3D intestinal organoids from CF2 (Fig.5B).
Single-channel recordings of S955P-CFTR showed normal channel conductance (Fig.4F,G) but an open-probability of 50% vs wt-CFTR (Fig.4H). Altogether, these data are consistent with S955P, being a class III (gating) mutation. The fact that in organoids it also appears at much lower levels than wt-CFTR (Fig.2C) suggests that S955P-CFTR is unstable, thus possibly also being a class VI mutation [6]. Of note, another example of a mixed class III/VI mutations is Q1412X [21].
Comparing the clinical phenotypes of both individuals CF1 and CF2 with the characterization of their respective ultra-rare mutations (1717–2A>G and S955P, respectively), we can conclude that they are consistent. Indeed, the more severe phenotype observed for CF1 (higher sweat [Cl−] values, PI and P. aeruginosa colonization) vs CF2 (who had a milder clinical phenotype) is in agreement with a smaller residual function measured in his intestinal organoids and rectal biopsies vs CF2.
Finally, we also assessed the effect of CFTR modulators on these two ultra-rare mutations. Our results in the intestinal organoids from CF1 (1717–2A>G/F508del) evidenced that VX-770 alone was able to partially rescue CFTR function but the VX-809/VX-770 or VX661/770 combinations had an even more significant effect (Fig.5B), but still below the threshold for a possible clinical benefit [22]. Since the potentiator alone already had a positive effect on rescuing CFTR function, it is very likely it acted on the protein resulting from the 1717–2A>G-allele, as F508del is not responsive to this potentiator alone. Indeed, besides class III and IV mutations, which have been listed as responding to potentiator monotherapy, class V splicing mutations also have the potential to benefit from this therapy [23].
Analysis of intestinal organoids from the patient with the S955P mutation showed that VX-770 appears to have a minor significant effect which is different from the control (Fig.5D). However, the combination of correctors and potentiator resulted in a significantly higher response (Fig.5D) which is in the range of possible clinical benefit [22].
Clearly, the amount of residual CFTR function measured in rectal biopsies, as well as in intestinal organoids (Fsk only), combined with our results in the S955P-CFTR CFBE cell line, lead us to conclude that S955P generates a relatively functional CFTR protein (~18% of wt). However, the effect of the potentiator is not as striking as we might expect. This could be due to the high residual function already measured in the presence of only Fsk which might mask the swelling effect of the potentiator alone.
Altogether, this work evidences the importance of analyzing patient-derived materials ex vivo, complemented by in vitro cell line to characterize rare CFTR mutations. As these cell models recapitulate several features of the parental organ, they are useful to understand the impact of genetic factors on the disease and predict clinical efficacy of therapies. Also, as cell background may significantly influence the pharmacological rescue of CFTR mutants the most promising therapies should be further validated in stablished cell models, in order to identify effective compounds that might result in optimal therapeutic benefits in the clinical scenario. Furthermore, we find a good correlation between the results in the airways cell line and intestinal organoids, thus further validating this patient-derived ex vivo system as a good model to predict clinical efficacy of therapies.
Furthermore, with this work, and by testing these patient-derived materials ex vivo, we managed to predict that one of these individuals will likely get clinical benefit from treatment with current CF-approved drugs. This appears as a good way forward for label extension of these approved drugs for individuals with CF carrying ultra-rare mutations, since costly therapeutic efforts are not directed at developing new drugs to rescue CFTR in those patients.
Supplementary Material
Highlights:
Characterization of CFTR mutations is key to adequately choose CFTR modulator drugs
1717–2A>G causes alternative splicing generating <1% normal CFTR mRNA
S955P leads to a defect in channel gating
Organoid theranostics predicts that both patients will respond to approved drugs
Acknowledgements
Work supported by UIDB/04046/2020 and UIDP/04046/2020 centre grants from FCT/MCTES Portugal (to BioISI), and research grants (to MDA): “HIT-CF” (H2020-SC1-2017-755021) from EU; SRC 013 from CF Trust-UK; and Orphan Mutations (Ref. AMARAL16I0) and “PTSense” (AMARAL19G0), both from CFF-USA; Charles University Grant Agency GA UK No. 412217 and by Ministry of Health, Czech Republic - conceptual development of research organisation, Motol University Hospital, Prague, Czech Republic 00064203 (both to TD). TCH is supported by an NIHR01 (DK55835) and a grant from CFF (Hwang19G0). Faculty of Sciences of the University of Lisbon’s Microscopy Facility is a node of the Portuguese Platform of BioImaging (PPBI), reference PPBI-POCI-01-0145-FEDER-022122 from FCT/MCTES. SR was recipient of fellowship SFRH/BD/142857/2018 from BioSys PhD programme PD/00065/2012 from FCT (Portugal).
Abbreviations:
- CFTR
Cystic fibrosis transmembrane conductance regulator
- CF
Cystic fibrosis
- FIS
Forskolin induced swelling assay
- VX-661
Tezacaftor
- VX-809
Lumacaftor
- VX-770
Ivacaftor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References
- 1.Boeck K De, Amaral MD. Rapid Review Progress in therapies for cystic fi brosis. Lancet Respir Med 2016;2006:1–13. [DOI] [PubMed] [Google Scholar]
- 2.Clancy JP, Cotton CU, Donaldson SH, Solomon GM, VanDevanter DR, Boyle MP, Gentzsch M, Nick JA, Illek B, Wallenburg JC, Sorscher EJ, Amaral MD, Beekman JM, Naren AP, Bridges RJ, Thomas PJ, Cutting G, Rowe S, Durmowicz AG, Mense M, Boeck KD, Skach W, Penland C, Joseloff E, Bihler H, Mahoney J, Borowitz D, Tuggle KL. CFTR modulator theratyping: Current status, gaps and future directions. J. Cyst. Fibros 2019;18:22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Davies JC, Drevinek P, Elborn JS, Kerem E, Lee T, Amaral MD, de Boeck K. Speeding up access to new drugs for CF: Considerations for clinical trial design and delivery. J Cyst Fibros 2019;18:677–84. [DOI] [PubMed] [Google Scholar]
- 4.Sharma N, Sosnay PR, Ramalho AS, Douville C, Franca A, Gottschalk LB, Park J, Lee M, Vecchio-Pagan B, Raraigh KS, Amaral MD, Karchin R, Cutting GR. Experimental Assessment of Splicing Variants Using Expression Minigenes and Comparison with In Silico Predictions. Hum Mutat 2014;35:1249–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hwang TC, Yeh JT, Zhang J, Yu YC, Yeh HI, Destefano S. Structural mechanisms of CFTR function and dysfunction. J. Gen. Physiol 2018;150:539–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hirtz S, Gonska T, Seydewitz HH, Thomas J, Greiner P, Kuehr J, Brandis M, Eichler I, Rocha H, Lopes AI, Barreto C, Ramalho A, Amaral MD, Kunzelmann K, Mall M. CFTR Cl - channel function in native human colon correlates with the genotype and phenotype in cystic fibrosis. Gastroenterology 2004;127:1085–95. [DOI] [PubMed] [Google Scholar]
- 7.Berkers G, van Mourik P, Vonk AM, Kruisselbrink E, Dekkers JF, de Winter-de Groot KM, Arets HGM, Marck-van der Wilt REP, Dijkema JS, Vanderschuren MM, Houwen RHJ, Heijerman HGM, van de Graaf EA, Elias SG, Majoor CJ, Koppelman GH, Roukema J, Bakker M, Janssens HM, van der Meer R, Vries RGJ, Clevers HC, de Jonge HR, Beekman JM, van der Ent CK. Rectal Organoids Enable Personalized Treatment of Cystic Fibrosis. Cell Rep 2019;26:1701–1708.e3. [DOI] [PubMed] [Google Scholar]
- 8.Clarke LA, Awatade NT, Felício VM, Silva IA, Calucho M, Pereira L, Azevedo P, Cavaco J, Barreto C, Bertuzzo C, Gartner S, Beekman J, Amaral MD. The effect of premature termination codon mutations on CFTR mRNA abundance in human nasal epithelium and intestinal organoids: a basis for read-through therapies in cystic fibrosis. Hum Mutat 2019;40:326–34. [DOI] [PubMed] [Google Scholar]
- 9.Canato S, Santos JD, Carvalho AS, Aloria K, Amaral MD, Matthiesen R, Falcao AO, Farinha CM. Proteomic interaction profiling reveals KIFC1 as a factor involved in early targeting of F508del-CFTR to degradation. Cell Mol Life Sci 2018;75:4495–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Awatade NT, Ramalho S, Silva IAL, Felício V, Botelho HM, de Poel E, Vonk A, Beekman JM, Farinha CM, Amaral MD. R560S: A class II CFTR mutation that is not rescued by current modulators. J Cyst Fibros 2019;18:182–9. [DOI] [PubMed] [Google Scholar]
- 11.Farinha CM, Sousa M, Canato S, Schmidt A, Uliyakina I, Amaral MD. Increased efficacy of VX-809 in different cellular systems results from an early stabilization effect of F508del-CFTR. Pharmacol Res Perspect 2015;3:e00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tian Y, Schreiber R, Wanitchakool P, Kongsuphol P, Sousa M, Uliyakina I, Palma M, Faria D, Traynor-Kaplan AE, Fragata JI, Amaral MD, Kunzelmann K. Control of TMEM16A by INO-4995 and other inositolphosphates. Br J Pharmacol 2013;168:253–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yeh J, Hwang T. Positional effects of premature termination codons on the biochemical and biophysical properties of CFTR. J Physiol 2019;:Accepted article, ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mall M, Wissner A, Seydewitz HH, Kuehr J, Brandis M, Greger R, Kunzelmann K. Defective cholinergic Cl− secretion and detection of K+ secretion in rectal biopsies from cystic fibrosis patients. Am J Physiol Liver Physiol 2000;278:G617–24. [DOI] [PubMed] [Google Scholar]
- 15.Sousa M, Servidoni MF, Vinagre AM, Ramalho AS, Bonadia LC, Felício V, Ribeiro MA, Uliyakina I, Marson FA, Kmit A, Cardoso SR, Ribeiro JD, Bertuzzo CS, Sousa L, Kunzelmann K, Ribeiro AF, Amaral MD. Measurements of CFTR-Mediated Cl- Secretion in Human Rectal Biopsies Constitute a Robust Biomarker for Cystic Fibrosis Diagnosis and Prognosis. PLoS One 2012;7. doi: 10.1371/journal.pone.0047708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Billet A, Mornon JP, Jollivet M, Lehn P, Callebaut I, Becq F. CFTR: Effect of ICL2 and ICL4 amino acids in close spatial proximity on the current properties of the channel. J Cyst Fibros 2013;12:737–45. [DOI] [PubMed] [Google Scholar]
- 17.Davies JC, Van de Steen O, van Koningsbruggen-Rietschel S, Drevinek P, Derichs N, McKone EF, Kanters D, Allamassey L, Namour F, de Kock H, Conrath K. GLPG1837, a CFTR potentiator, in p.Gly551Asp (G551D)-CF patients: An open-label, single-arm, phase 2a study (SAPHIRA1). J Cyst Fibros 2019;18:693–9. [DOI] [PubMed] [Google Scholar]
- 18.Yeh HI, Yeh JT, Hwang TC. Modulation of CFTR gating by permeant ions. J Gen Physiol 2015;145:47–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dekkers JF, Wiegerinck CL, de Jonge HR, Bronsveld I, Janssens HM, de Winter-de Groot KM, Brandsma AM, de Jong NWM, Bijvelds MJC, Scholte BJ, Nieuwenhuis EES, van den Brink S, Clevers H, van der Ent CK, Middendorp S, Beekman JM. A functional CFTR assay using primary cystic fibrosis intestinal organoids. Nat Med 2013;19:939–45. [DOI] [PubMed] [Google Scholar]
- 20.Popa I, Pop L, Popa Z, Schwarz MJ, Hambleton G, Malone GM, Haworth A, Super M. Cystic fibrosis mutations in Romania. Eur J Pediatr 1997;156:212–3. [DOI] [PubMed] [Google Scholar]
- 21.Haardt M, Benharouga M, Lechardeur D, Kartner N, Lukacs GL. C-terminal truncations destabilize the cystic fibrosis transmembrane conductance regulator without impairing its biogenesis. A novel class of mutation. J Biol Chem 1999;274:21873–7. [DOI] [PubMed] [Google Scholar]
- 22.Dekkers JF, Berkers G, Kruisselbrink E, Vonk A, Jonge HR De, Janssens HM, Bronsveld I, Graaf EA Van De, Nieuwenhuis EES, Houwen RHJ, Vleggaar FP, Escher JC, Rijke YB De, Majoor CJ, Heijerman HGM, Groot KMDW De, Clevers H, Ent CK Van Der, Beekman JM. Characterizing responses to CFTR-modulating drugs using rectal organoids derived from subjects with cystic fibrosis. Sci Transl Med 2016;8:13. [DOI] [PubMed] [Google Scholar]
- 23.Van Goor F, Yu H, Burton B, Hoffman BJ. Effect of ivacaftor on CFTR forms with missense mutations associated with defects in protein processing or function. J Cyst Fibros 2014;13:29–36. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.