

Abstract
Aim:
Up-regulation of inflammasome proteins was reported in dystrophin-deficient muscles. However, it remains to be determined whether inflammasome activation plays a role in the pathogenesis of Duchenne muscular dystrophy. This study was therefore set out to investigate whether genetic disruption of the inflammasome pathway impacts the disease progression in mdx mice.
Main methods:
Mice deficient in both dystrophin and ASC (encoded by Pycard [PYD And CARD Domain Containing]) were generated. The impact of ASC deficiency on muscular dystrophy of mdx mice were assessed by measurements of serum cytokines, Western blot, real-time PCR and histopathological staining.
Key findings:
The pro-inflammatory cytokines such as TNF-α, IL-6, KC/GRO and IL-10 were markedly increased in the sera of 8-week-old mdx mice compared to WT. Western blotting showed that P2X7, caspase-1, ASC and IL-18 were upregulated. Disruption of ASC and dystrophin expression in the mdx/ASC−/− mice was verified by Western blot analysis. Histopathological analysis did not find significant alterations in the muscular dystrophy phenotype in mdx/ASC−/− mice as compared to mdx mice.
Significance:
Taken together, our results show that disruption of the central adaptor ASC of the inflammasome is insufficient to alleviate muscular dystrophy phenotype in mdx mice.
Keywords: ASC, DMD, mdx mice, inflammasome, inflammation, muscle regeneration
1. Introduction
Duchenne muscular dystrophy (DMD) is an X-linked inherited neuromuscular disorder1. It is the most common type of muscular dystrophy, caused by genetic mutations in DMD, the gene encoding dystrophin protein2. Dystrophin is involved in forming a mechanical link between the cytoskeleton and the extracellular matrix, and thereby stabilizes the plasma membrane integrity through its interactions with other dystrophin-glycoprotein complex (DGC)3, 4. The dystrophin protein is highly expressed in skeletal and cardiac muscles.
Studies of DMD prevalence suggest an average age at diagnosis in United States is from 5 to 9 years of age, affecting approximately 1 per 5,000 boys5. DMD has typically resulted in severe muscle wasting and loss of ambulation in their early teens, subsequently lead to respiratory and cardiac dysfunction and death between 20 and 40 years of age1. Activation of innate inflammatory response has been well characterized in the pathology of muscular dystrophy due to the sarcolemmal instability after the disruption of DGC complex with the release of cytoplasmic contents and increased inflammatory immune cells infiltration including neutrophil and monocytes to the damage site and muscle tissue6–8. In severe muscle damage, the muscle tissue fails to repair where the regenerative capacity of myofibers is compromised. Excessive inflammation has been reported to be associated with the muscle tissue loss and fibrosis by increased production of proinflammatory cytokines9. Clinically, the overexpression of the proinflammatory cytokines such as tumor necrosis factor alpha (TNF-α), transforming growth factor beta (TGF-β), interleukin-1 (IL-1), and interleukin-6 (IL-6) has been reported in the muscle biopsies and serum samples from DMD patients, indicating that the inflammation may play an important role in pathogenesis of DMD10–13.
In recent years, increasing attention has been focused on the sterile inflammatory response that is activated by a large multimeric protein complex, known as inflammasome. Inflammasomes belong to the innate immune system and are composed of a sensor protein, an adaptor protein and a zymogen, procaspase-1. Several inflammasome complexes have been described including NLRP3 (nucleotide-binding domain leucine-rich repeat (NLR) and pyrin domain containing receptor 3), NLRP1, NLRC4, AIM2 (absent in melanoma 2). To date, the most described inflammasome is NLRP3 inflammasome, which consists of NLRP3, PYCARD (PYRIN-PAAD-DAPIN domain [PYD] and caspase recruitment domain [CARD] containing) adaptor, frequently referred to as apoptosis-associated speck-like protein (ASC), and pro-caspase-114, 15. Inflammasome is activated in the response to tissue damage or infection, which in turn activates procaspase-1 into active caspase-1. The active caspase-1 proteolytically activates cytokines interleukin‑1β (IL‑1β) and interleukin‑18 (IL‑18) and eventually induces an inflammatory form of cell death called pyroptosis15–17. Several studies have suggested a role of NLRP3 inflammasome in the pathogenesis of pulmonary hypertension, type 2 diabetes, liver damage, obesity-associated glomerular injury and muscular dystrophy18–21. Rawat et al (2010) has previously shown that the inflammasomes components and pro-inflammatory cytokine such as ASC, pro-caspase-1 and IL‑1β were upregulated in both dystrophin-deficient human (DMD) and mdx mouse skeletal muscle, indicating dystrophin-deficient induced activation of inflammasomes pathway. However, the contribution of the inflammasomes in the pathophysiology of DMD has not been well defined. This study aims to explore the effect of ASC deficiency on the pathology of muscular dystrophy.
2. Materials and methods
2.1. Mice.
All animal studies were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of the Ohio State University. The mdx (C57BL/10ScSn-Dmdmdx/J) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). ASC deficient mice22 (a gift from Dr. Sutterwala) and mdx mice were intercrossed to generate mutant and control mice. Genotyping of ASC and mdx mice was performed by tail DNA PCR as previously described22, 23. All mice were maintained at The Ohio State University Laboratory Animal Resources in accordance with animal use guidelines. All animal studies were authorized by the Animal Care, Use, and Review Committee of The Ohio State University.
2.2. Serum cytokine detection by ELISA.
The blood samples were collected from 8-week-old male WT, mdx and mdx/ASC−/− mice before sacrificed. The blood was allowed to clot for 15 min to 30 min and centrifuged at 8000 rpm for 15 min in room temperature. The supernatant was collected as serum and stored at −80 °C for the cytokine detection assay. Cytokine levels were detected using MSD (Meso Scale Discovery, Rockville, MD) 10 panels V-PLEX mouse ELISA kit, following the manufacturer’s protocol. The signal was collected with MESO QUICKPLEX SQ 120 and the data were analyzed using MSD Workbench 4.0 software.
2.3. Western blotting.
Quadriceps muscles were harvested from 8-week-old male WT, mdx and mdx/ASC−/− mice. The tissues were homogenized with KINEMATICA Polytron™ PT 1300D Homogenizers (Bohemia, NY) and lysed with cold RIPA buffer (150 mM Sodium Chloride, 1% Triton X100, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris-HCl, 5 mM EDTA, pH 7.4) supplemented with protease inhibitors. The lysates were then centrifuged at 20,000 g for 20 min and supernatant was collected for Western blotting. The protein concentration of the supernatant was determined using the Lowry assay (Bio-Rad Laboratories, Hercules, CA, USA). Equal amount of extracted protein samples was separated by SDS-PAGE (BioRad, 4–20%) and transferred onto PVDF membranes (0.45 μm). The blots were blocked for non-specific binding with 5% milk in Tris-buffered saline containing 0.1 % Tween 20 (TBS, pH 7.4) for 1 hour at room temperature with gentle shaking. After rinsing in TBS-T, the blots were incubated at 4°C overnight with primary antibodies against ASC (AL177, AG-25B-0006-C100, Adipogen International, San Diego, CA), caspase-1 (sc-514, Santa Cruz Biotechnology, Dallas, TX), P2X7 (ab109246, Abcam, Cambridge, MA), IL-18 (5180R; Biovision, Milpitas, California), IL‑1β (ab205924, Abcam) and dystrophin (E2660, Spring Bioscience, Pleasanton, CA). The blots were washed in TBS-T and incubated with respective secondary antibodies conjugated to horseradish peroxidise for 2 hours at room temperature. The blots were developed using ECL Western blotting substrate (Pierce Biotechnology, Rockford, IL) and images were captured using the ChemiDoc system (Bio-Rad Laboratories). The densitometric analysis was performed using Image Lab™ software, version 5.2.1 (Bio-Rad Laboratories). The respective protein expression levels were normalized to the housekeeping protein β-actin.
2.4. Immunofluorescence staining and histology.
Frozen muscle sections of 8 μm were prepared from quadriceps of 8-week-old male WT, mdx and mdx/ASC−/− mice. Immunofluorescence staining was performed as described previously24. In brief, the slides were fixed with 4% paraformaldehyde for 15 minutes at room temperature and washed with PBS before blocked with 5% BSA for 1 hour. The slides were incubated with primary antibodies against eMyHC (F1.652, Developmental Studies Hybridoma Bank, University of Iowa, IA) and laminin α2 (4H8/2, Alexis, San Diego, CA) at 4 °C overnight. After that, the slides were washed with PBS and incubated with secondary antibodies (Alexa Fluor 488 goat anti-rat IgG, Invitrogen, Carlsbad, CA or Alexa Fluor 594 goat anti-mouse IgG, Invitrogen) for 1h at room temperature. The slides were sealed with VECTASHIELD Antifade Mounting Medium with DAPI (vector laboratory, Burlingame, CA). Embryonic-MyHC-positive muscle fibers were counted and expressed as percentage of total number of positive fibers over total number of fibers in 10 fields. All images were taken under a Nikon Ti-E fluorescence microscope, magnification, X200 (Nikon, Melville, NY).
2.5. Histology.
For histological examinations, frozen muscle sections of 8 μm were prepared from each group and were fixed in 4% paraformaldehyde for 15 minutes at room temperature. The slides were then proceeded to the standard protocol of hematoxylin and eosin (H&E), and Masson-trichrome staining to show myofiber morphology and assess collagen content. All images were taken under a Nikon Ti-E fluorescence microscope, magnification, X200 (Nikon, Melville, NY).
2.6. Serum Creatine Kinase Levels.
Creatine kinase (CK) levels were analyzed using the Creatine Kinase-SL assay kit (Sekisui Diagnostics, Lexington, MA) according to manufacturers’ instructions. Briefly, 6 μL of diluted serum sample was added accordingly to the 96-well UV microplate and following by 300 μL of CK reagent. The plate was incubated at 30°C for 5 minutes and read in Flexstation 3 microplate reader (Molecular Devices, Sunnyvale, CA) for 2 min at 30 sec intervals.
2.7. Central nucleation counting
Frozen muscle sections were stained with anti-laminin α2 antibody to delineate muscle fibers, and VECTASHIELD® Mounting Medium with DAPI was used for nuclear staining. All fibers, except those in direct contact with fascia, were analyzed for the location of their nuclei. For each sample group, the number of fibers with centrally localized nuclei relative to the total number of fibers was recorded. For each individual mouse, about 2500–3500 fibers were counted.
2.8. Gene expression of IL-18 and IL-1β.
Total RNA was extracted using RNeasy Mini Kit (Qiagen, USA) according to manufacturer’s instruction. Then 5μg RNA was reverse transcribed using RevertAid RT Reverse Transcription Kit (Thermo Scientific, USA). Quantitative PCR was carried out using the Radiant™ SYBR Green qPCR kits (Alkali Scientific, Pompano Beach, FL). The IL-18 forward and reverse primer sequences were 5′-AAAGTGCCAGTGAACCC-3′ and 5′-TTTGATGTAAGTTAGTGAGAGTGA-3′ respectively. The IL-1β forward and reverse primer sequences were 5′-CACAGCAGCACATCAACAAG-3′ and 5′-GTGCTCATGTCCTCATCCTG-3′ respectively. Each assay was run on a LightCycler® 480 System (Roche Diagnostics Corporation, IN, USA) in triplicate and the results are calculated using the relative standard curve method. All gene expressions were normalized against the respective housekeeping gene GAPDH.
2.9. Statistics.
The data were expressed as mean ± SEM and analyzed with GraphPad Prism 5.0 software (San Diego, USA). Statistical significance was determined using one-way ANOVA followed by Bonferroni post hoc-tests or unpaired 2-tailed Student’s t test whenever appropriate. A P value of less than 0.05 is regarded as significant.
3. Results
3.1. Serum inflammatory cytokine profile in mdx mice
When cells die by necrosis they stimulate a robust acute inflammatory response in vivo25. Repeated muscle injury in the absence of dystrophin occurs, which persistently activates the immune system and causes muscle inflammation. We first examined the inflammatory profile in mdx mice by measuring inflammatory cytokines in the serum samples of 8-week-old mdx mice. The pro-inflammatory cytokines such as TNF-α, IL-6, KC/GRO and IL-10 were significantly increased in mdx mice compared to WT (Figure 1). The serum levels of IL-1β appeared to be increased in mdx mice, although the difference did not reach statistical significance.
Figure 1.
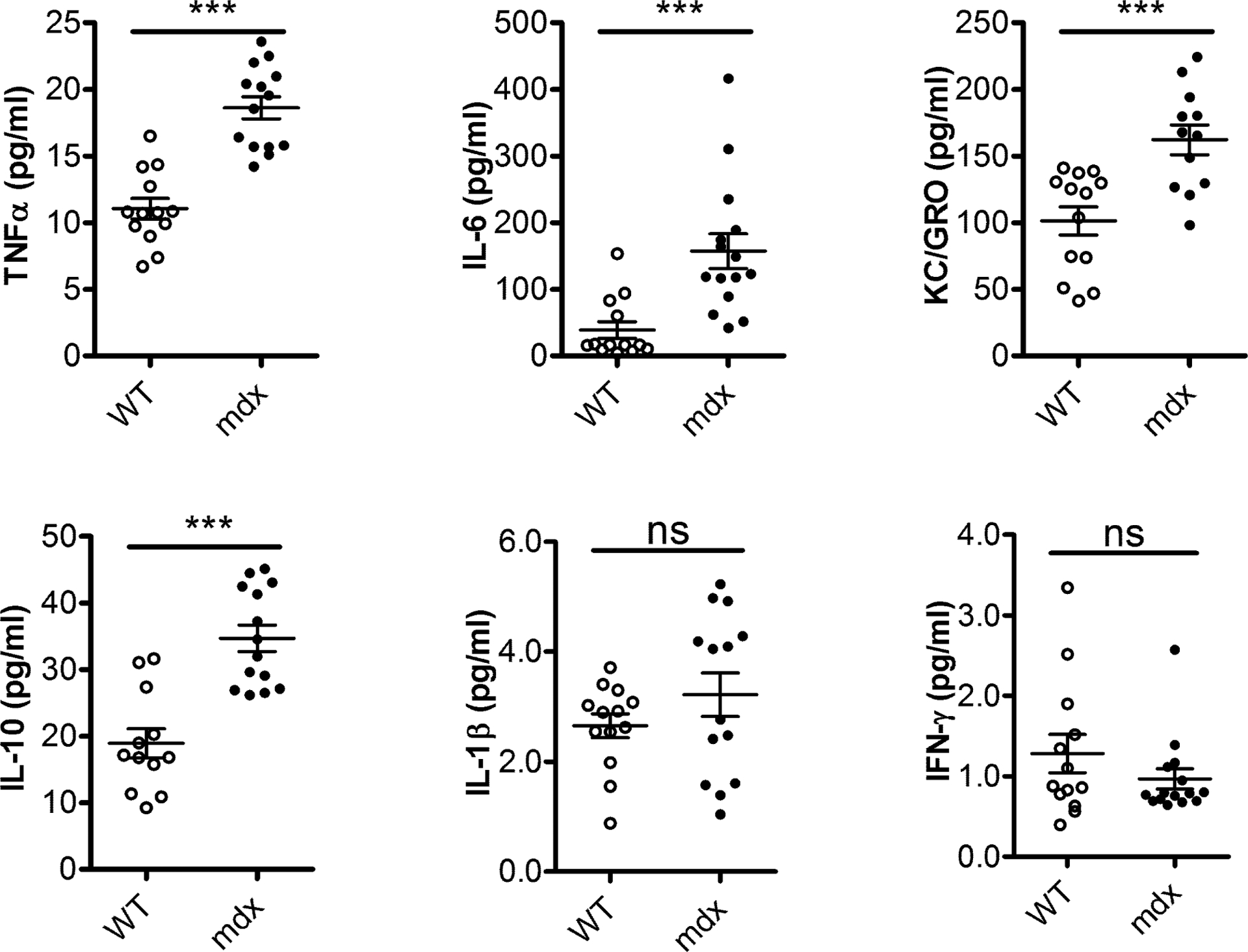
Upregulation of pro-inflammatory cytokines in mdx mice. The serum cytokines profile was measured by ELISA in WT and mdx mice. The pro-inflammatory cytokines, TNF-α, IL-6, KC/GRO and IL-10 were increased in 8-week-old mdx mice compared to WT. Results are shown as mean ± SEM, n = 12–14, ***P < 0.001 compared with WT.
3.2. Up-regulation of inflammasome proteins in mdx muscles
A number of cellular contents such as uric acid, HMGB1, SAP130, IL-1α, IL-33, DNA, S100 proteins, heat shock proteins and others, are released upon necrotic cell injury, which are believed to contribute to death-induced inflammation. The NLRP3 inflammasome, part of innate immune system, has been known to participate in the sterile inflammatory response, which may further exacerbate muscle pathology in muscular dystrophy. Next, our Western blotting results demonstrated a significant increase of P2X7 receptor, an ATP-gated ion channel in mdx mice relative to WT control. As expected, the biomarkers for activation of inflammasomes protein complex were found upregulated including caspase-1, ASC and IL-18, in muscle lysates from mdx mice (Figure 2). Our data also confirmed the signature protein expression of dystrophin was absent in mdx mice (Figure 2A).
Figure 2.
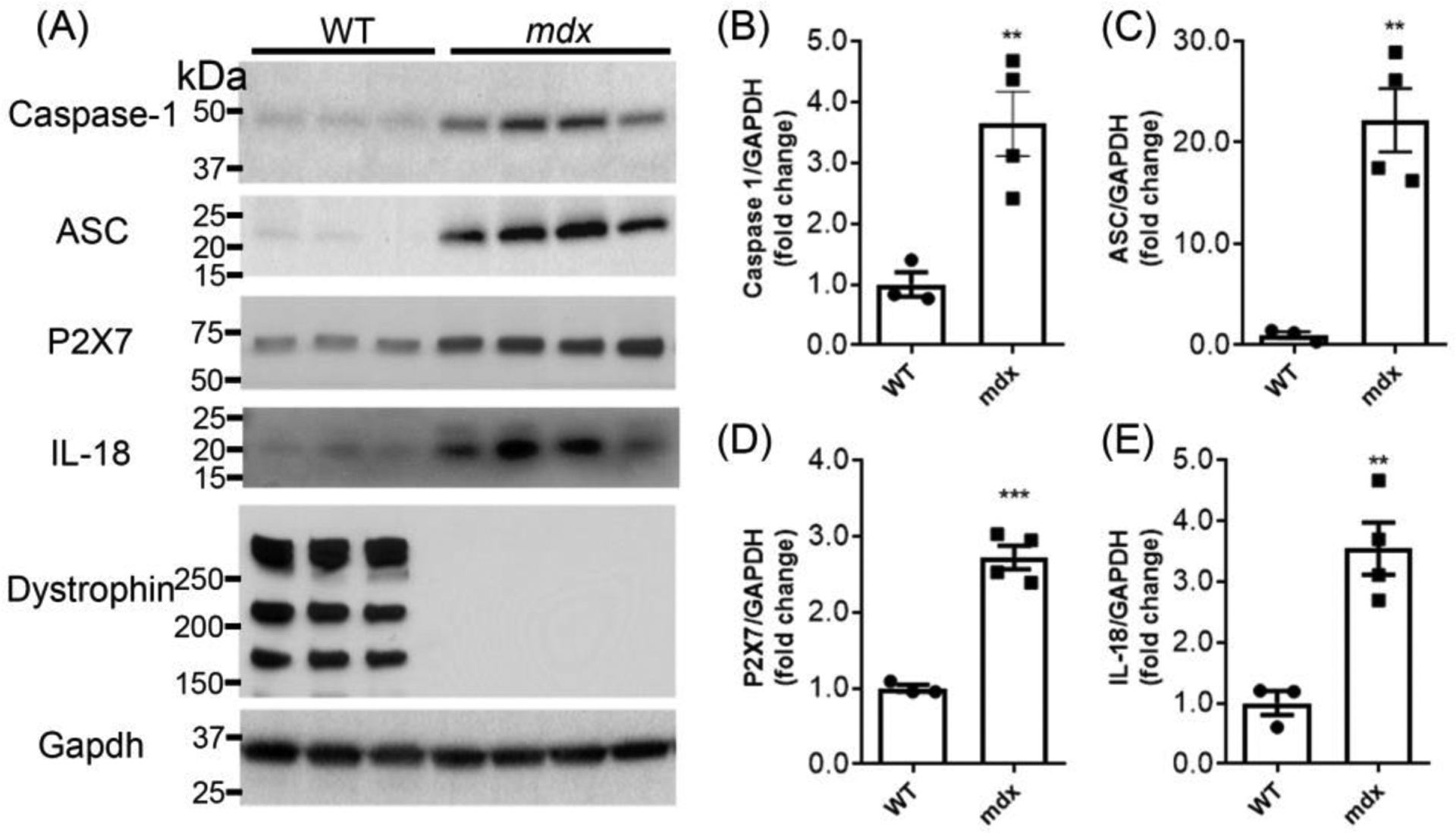
Inflammasome signaling proteins upregulated in mdx mice. (A) The Western blotting shows upregulation of P2X7, caspase-1, ASC and IL-18 and absence of dystrophin protein in muscle lysates from mdx mice. (B-E) Graphs show quantitation analysis of Western blots. **P < 0.01, ***P < 0.001 compared with WT.
3.3. Effect of ASC deficiency on muscle pathology in mdx mice
To study if blockade of the NLRP3 inflammasome ameliorates muscular dystrophy pathology, we generated a double knockout (DKO) mouse strain with deficiency in both dystrophin and ASC, an inflammasome adaptor protein. The mice genotyped by tail DNA PCR were verified by Western blot analysis. Disruption of dystrophin and ASC expression was confirmed in the muscle lysates of the mdx/ASC DKO mice (Figure 3).
Figure 3.
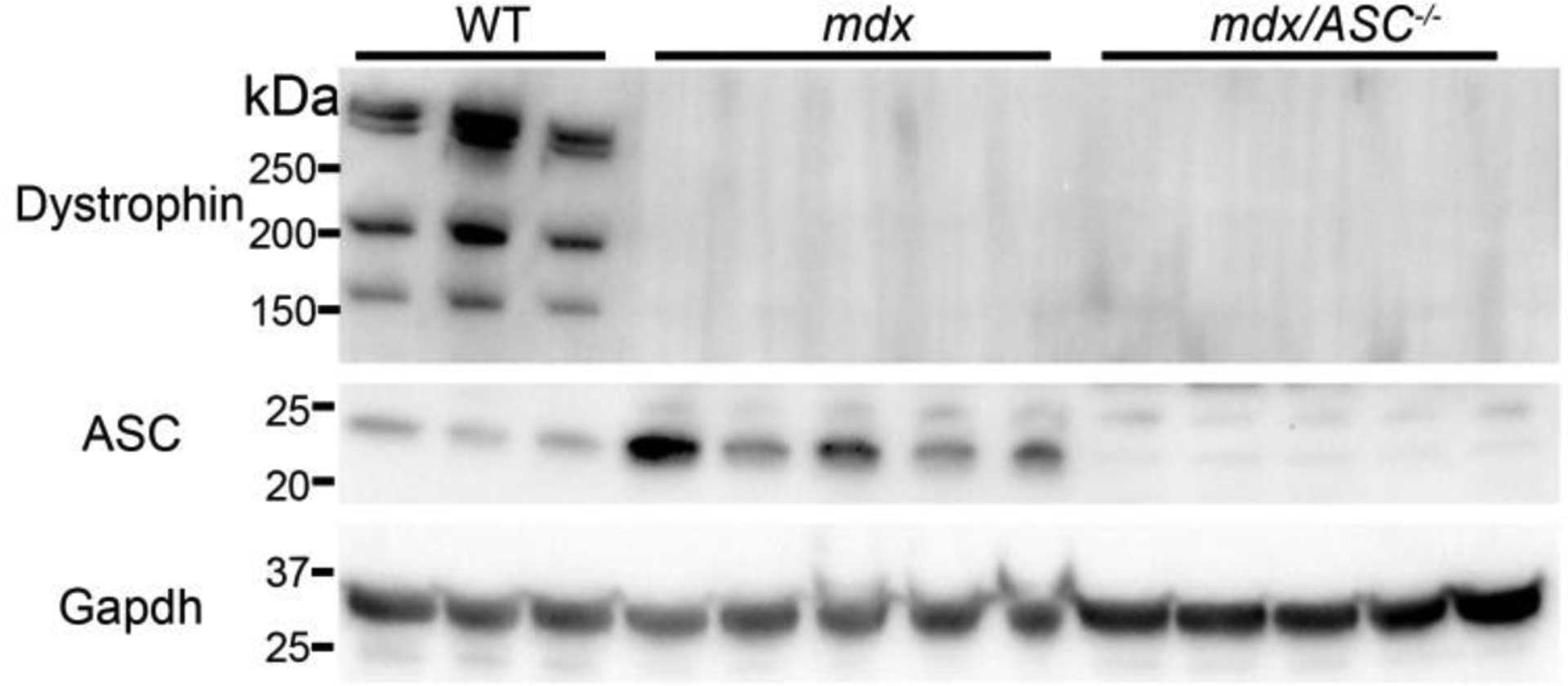
Western blot analysis of muscle lysates from WT, mdx and mdx/ASC DKO mice. Western blotting shows upregulation of ASC and absence of dystrophin protein in muscle lysates from mdx mice and absence of both dystrophin and ASC protein in mdx/ASC DKO mice.
Repeated muscle degeneration and regeneration is a histopathological hallmark of muscular dystrophy, as indicated by the presence of muscle necrosis and central nucleated muscle fibers (CNF). The quadriceps and gastrocnemius muscles were assessed in H&E staining. As shown in the top panel of Figure 4A, both the mdx and mdx/ASC DKO muscles exhibited various size of muscle fibers, presence of internal nuclei, and increased fibrotic area. To quantify some of the pathological features, we stained the muscle sections with laminin α2 (to demarcate the muscle fibers) and 4’,6-diamidino-2-phenylindole (DAPI, to label nuclei) (Figure 4A, bottom). Both mdx and mdx/ASC DKO muscles had significantly increased CNFs as compared to WT controls, but there was no significant difference between mdx and mdx/ASC DKO muscles (Figure 4B). Due to the presence of smaller, regenerating muscle fibers in dystrophic muscles, the average cross-sectional area (CSA) of muscle fibers were significantly decreased in both mdx and mdx/ASC DKO muscles (Figure 4C). Again, we observed no difference in CSA between mdx and mdx/ASC DKO muscles (Figure 4C). Moreover, the fiber size distribution was similarly altered in mdx and mdx/ASC DKO muscles as compared to WT controls (Figure 4D). Consistently, we observed that the serum creatine kinase (CK) levels were similarly increased in both mdx and mdx/ASC DKO mice (Figure 4E).
Figure 4.
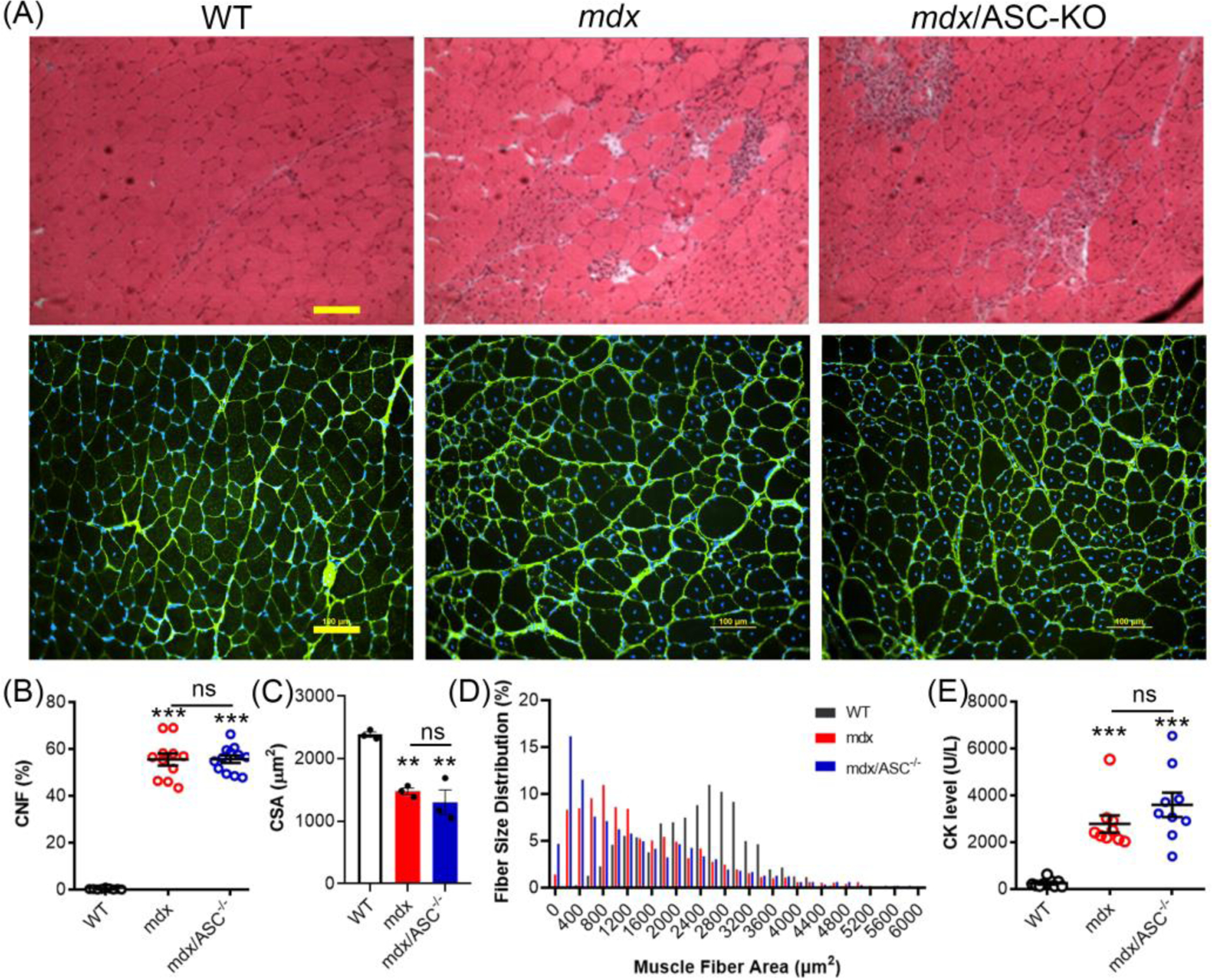
ASC disruption had minimal impact on the dystrophic pathology of mdx mice. (A) H&E (top panel) and laminin α2/DAPI (bottom panel) stained histological sections of quadriceps from WT, mdx and mdx/ASC−/− mice. (B) Percentage of central nucleated fibers (CNF) in muscle sections of WT, mdx and mdx/ASC−/− mice. (C) Average cross-sectional area (CSA) of muscle fibers in WT, mdx and mdx/ASC−/− mice. (D) Muscle fiber size distribution of WT, mdx and mdx/ASC−/− mice. (E) Serum creatine kinase (CK) levels of WT, mdx and mdx/ASC−/− mice. ***P < 0.001 compared with WT; ns, not statistically significant.
To further investigate if ASC disruption has any impact on muscle regeneration in mdx mice, embryonic myosin heavy chain (eMyHC), a marker of newly regenerated muscle fibers, was assessed. Both mdx and mdx/ASC DKO muscles exhibited increased number of eMyHC-positive fibers compared to WT (Figure 5A & B). However, no statistically significant changes were observed between mdx/ASC DKO and mdx.
Figure 5.
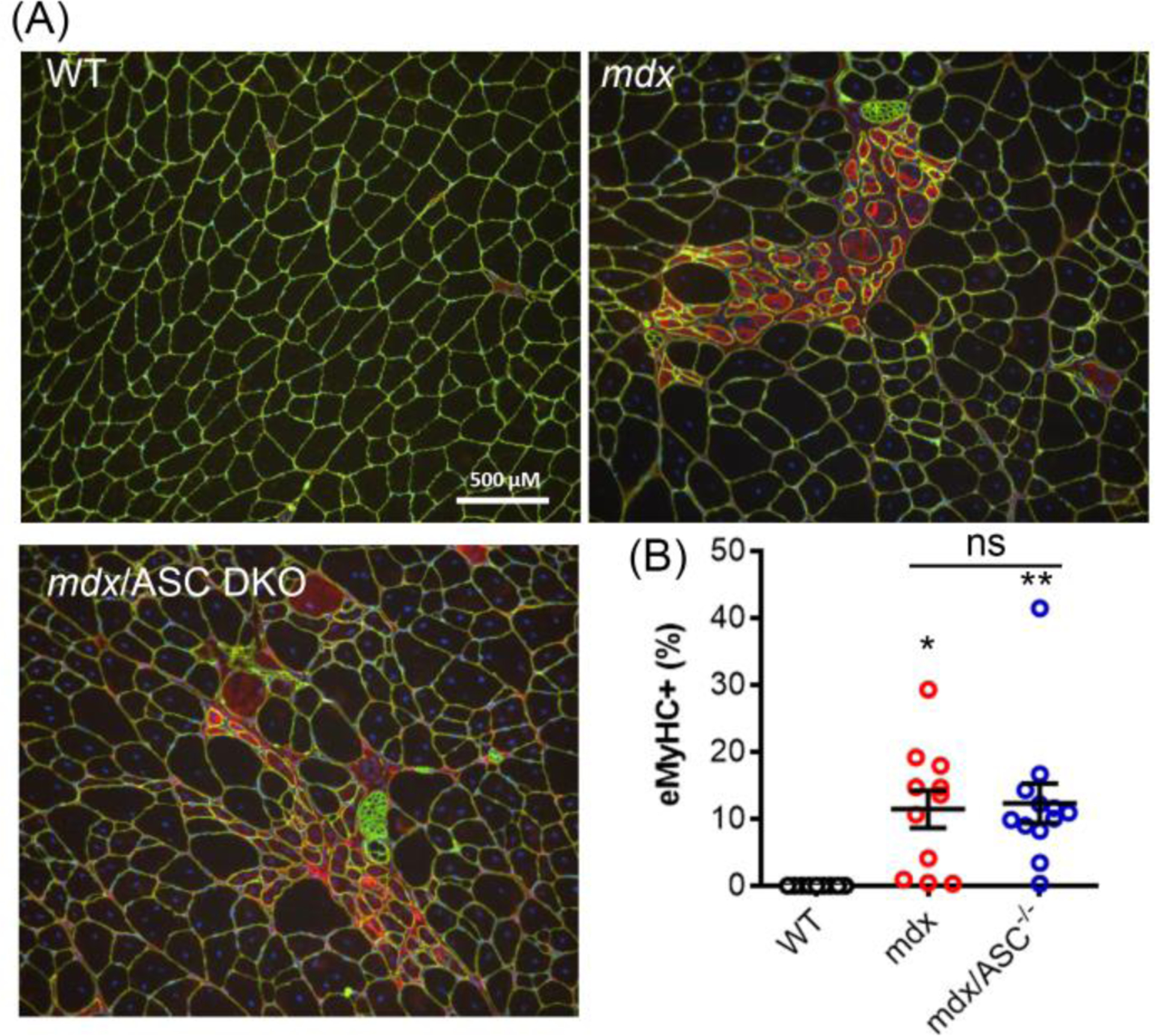
Muscle regeneration was similar in mdx and mdx/ASC DKO mice. Representative immunofluorescence images (A) and quantitation (B) of eMyHC (red) from quadriceps of different mouse groups. Muscle fibers were co-stained with laminin 2α (green). *P < 0.05, **P < 0.01, compared with WT; ns, not statistically significant.
4. Discussion
In this study, we found that the inflammasome proteins were up-regulated in the skeletal muscle of mdx mice, which also displayed an altered inflammation cytokine profile in the serum. However, genetic disruption of the inflammasome adaptor ASC in the mdx mice did not significantly improve the muscle pathology. Our results cast doubt on the feasibility of targeting inflammasome as a therapeutic strategy for DMD.
During sterile tissue damage, the immune system, particularly the inflammasome, has been implicated in many pathological conditions including DMD. Rawat et al. (2010) showed an increased expression of ASC and capspase-120 in muscle biopsies of DMD patients. In consistent with these previous observations, we also found that the expression of P2X7, ASC, caspase-1 an IL-1β were significantly increased. These findings suggest a potential role of inflammasome activation in the pathogenesis of muscular dystrophy. Indeed, a previous study showed that genetic ablation of NLRP3 appeared to reduce the muscle pathology, inflammation and oxidative stress in mdx mice26. Ghrelin, a circulating hormone was found to suppress muscle inflammation and ameliorate disease phenotype in mdx mice via inhibition of NLRP3 inflammasome27. In addition, several other studies have shown that pharmacological blockade of P2X7 receptors reduced muscle inflammation and improve muscle pathology in mdx mice28–30.
Given the promising previous observations mentioned above, it is surprising that genetic disruption of the inflammasome adaptor protein ASC had little impact on the muscle pathology in dystrophic mice as shown by our present study. As a central player of the NLRP3 inflammasome, ASC is a bipartite molecule that contains both an N-terminal PYD and a C-terminal CARD, allowing it to bridge the sensor NLRP3 and the effector pro-caspase-1[32]. The deletion or silencing of ASC has been reported to reduce hypoxia-induced pulmonary hypertension and high fat diet-induced podocyte injury18, 21. It is unclear what exactly underlies the discrepancy between our study and others, however, numerous studies have shown that NLRP3 may act via an inflammasome-independent mechanism, which may or may not require ASC. For example, hypoxia induced significant increase of NLRP3, independent of ASC, caspase-1 and IL-1β, which mediated mitochondrial regulation in renal injury31. NLRP3-deficient and ASC-deficient mice show difference in survival and early renal dysfunction following renal ischemic injury32. NLRP3 deficiency or inhibition via β-hydroxybutyrate precursor 1,3-butanediol protected the mice from nephrocalcinosis-related chronic kidney disease (CKD), but IL-1 inhibitor had no such protective effect, suggesting that NLRP3 has an IL-1-independent function in CKD33. During the NLRP3-mediated Epithelial-Mesenchymal Transition process induced by TNF-α or TGF-β1, the cleaved caspase-1 and ASC speck were not detected in mesenchymal-like colon cancer cells, indicating that NLRP3 functions in an inflammasome-independent way34. Moreover, NLRP3 was found to be required for resistance to pneumococcal pneumonia, whereas caspase-1 and caspase-11 were dispensable in S. pneumoniae-infected mice35. Thus, it is possible that the ASC-independent role of NLRP3 is involved in muscle pathology of mdx mice. Even more complicated, the sterile inflammatory responses are often only partially reduced and in some cases seem largely independent of the inflammasome components as demonstrated by the use of NLRP3-deficient animals36. Other cellular processes such as autophagy may also be involved in tissue inflammation37. Therefore it is possible that the mechanisms operating in vivo are more complicated and may involve redundant pathways. Disruption of one aspect of the immune system may be well compensated by the presence of the other redundant pathways.
5. Conclusions
This study shows that ASC knockout has minimal impact on muscle pathology of mdx mice. This finding suggests that targeting ASC may not be sufficient for alleviating muscle pathology in DMD. Further investigations are needed to better understand the inflammasome-dependent and independent functions of ASC and NLRP3 in muscular dystrophy.
Acknowledgements
This work was supported by US National Institutes of Health grants R01-HL116546 and R01-AR064241 to R.H.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
None
References
- 1.D’Amario D et al. A current approach to heart failure in Duchenne muscular dystrophy. Heart (2017). [DOI] [PubMed] [Google Scholar]
- 2.Nowak KJ & Davies KE Duchenne muscular dystrophy and dystrophin: pathogenesis and opportunities for treatment. EMBO reports 5, 872–876 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gao QQ & McNally EM The Dystrophin Complex: Structure, Function, and Implications for Therapy. Comprehensive Physiology 5, 1223–1239 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Allen DG, Whitehead NP & Froehner SC Absence of Dystrophin Disrupts Skeletal Muscle Signaling: Roles of Ca2+, Reactive Oxygen Species, and Nitric Oxide in the Development of Muscular Dystrophy. Physiological reviews 96, 253–305 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Romitti PA et al. Prevalence of Duchenne and Becker muscular dystrophies in the United States. Pediatrics 135, 513–521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Falzarano MS, Scotton C, Passarelli C & Ferlini A Duchenne Muscular Dystrophy: From Diagnosis to Therapy. Molecules 20, 18168–18184 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tidball JG & Villalta SA Regulatory interactions between muscle and the immune system during muscle regeneration. American journal of physiology. Regulatory, integrative and comparative physiology 298, R1173–1187 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rosenberg AS et al. Immune-mediated pathology in Duchenne muscular dystrophy. Science translational medicine 7, 299rv294 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Costamagna D, Costelli P, Sampaolesi M & Penna F Role of Inflammation in Muscle Homeostasis and Myogenesis. Mediators of inflammation 2015, 805172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.De Paepe B & De Bleecker JL Cytokines and chemokines as regulators of skeletal muscle inflammation: presenting the case of Duchenne muscular dystrophy. Mediators of inflammation 2013, 540370 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Saito K et al. A sensitive assay of tumor necrosis factor alpha in sera from Duchenne muscular dystrophy patients. Clinical chemistry 46, 1703–1704 (2000). [PubMed] [Google Scholar]
- 12.Wright CR et al. A Reduction in Selenoprotein S Amplifies the Inflammatory Profile of Fast-Twitch Skeletal Muscle in the mdx Dystrophic Mouse. Mediators of inflammation 2017, 7043429 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cruz-Guzman Odel R, Rodriguez-Cruz M & Escobar Cedillo RE Systemic Inflammation in Duchenne Muscular Dystrophy: Association with Muscle Function and Nutritional Status. BioMed research international 2015, 891972 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.He Y, Hara H & Nunez G Mechanism and Regulation of NLRP3 Inflammasome Activation. Trends in biochemical sciences 41, 1012–1021 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vanaja SK, Rathinam VA & Fitzgerald KA Mechanisms of inflammasome activation: recent advances and novel insights. Trends in cell biology 25, 308–315 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Latz E, Xiao TS & Stutz A Activation and regulation of the inflammasomes. Nature reviews. Immunology 13, 397–411 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Guo H, Callaway JB & Ting JP Inflammasomes: mechanism of action, role in disease, and therapeutics. Nature medicine 21, 677–687 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cero FT et al. Absence of the inflammasome adaptor ASC reduces hypoxia-induced pulmonary hypertension in mice. American journal of physiology. Lung cellular and molecular physiology 309, L378–387 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Masters SL et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nature immunology 11, 897–904 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rawat R et al. Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. The American journal of pathology 176, 2891–2900 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Boini KM et al. Activation of inflammasomes in podocyte injury of mice on the high fat diet: Effects of ASC gene deletion and silencing. Biochimica et biophysica acta 1843, 836–845 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sutterwala FS et al. Critical role for NALP3/CIAS1/Cryopyrin in innate and adaptive immunity through its regulation of caspase-1. Immunity 24, 317–327 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Shin JH, Hakim CH, Zhang K & Duan D Genotyping mdx, mdx3cv, and mdx4cv mice by primer competition polymerase chain reaction. Muscle Nerve 43, 283–286 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu L et al. CRISPR-mediated Genome Editing Restores Dystrophin Expression and Function in mdx Mice. Molecular therapy : the journal of the American Society of Gene Therapy 24, 564–569 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen CJ et al. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nature medicine 13, 851–856 (2007). [DOI] [PubMed] [Google Scholar]
- 26.Boursereau R, Abou-Samra M, Lecompte S, Noel L & Brichard SM Downregulation of the NLRP3 inflammasome by adiponectin rescues Duchenne muscular dystrophy. BMC Biol 16, 33 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang L et al. Ghrelin improves muscle function in dystrophin-deficient mdx mice by inhibiting NLRP3 inflammasome activation. Life Sci 232, 116654 (2019). [DOI] [PubMed] [Google Scholar]
- 28.Al-Khalidi R et al. Zidovudine ameliorates pathology in the mouse model of Duchenne muscular dystrophy via P2RX7 purinoceptor antagonism. Acta Neuropathol Commun 6, 27 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gazzerro E et al. Enhancement of Muscle T Regulatory Cells and Improvement of Muscular Dystrophic Process in mdx Mice by Blockade of Extracellular ATP/P2X Axis. Am J Pathol 185, 3349–3360 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Sinadinos A et al. P2RX7 purinoceptor: a therapeutic target for ameliorating the symptoms of duchenne muscular dystrophy. PLoS Med 12, e1001888 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SM et al. Inflammasome-Independent Role of NLRP3 Mediates Mitochondrial Regulation in Renal Injury. Front Immunol 9, 2563 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Iyer SS et al. Necrotic cells trigger a sterile inflammatory response through the Nlrp3 inflammasome. P Natl Acad Sci USA 106, 20388–20393 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anders HJ et al. The macrophage phenotype and inflammasome component NLRP3 contributes to nephrocalcinosis-related chronic kidney disease independent from IL-1-mediated tissue injury. Kidney Int 93, 656–669 (2018). [DOI] [PubMed] [Google Scholar]
- 34.Wang H et al. Inflammasome-independent NLRP3 is required for epithelial-mesenchymal transition in colon cancer cells. Exp Cell Res 342, 184–192 (2016). [DOI] [PubMed] [Google Scholar]
- 35.Fang RD et al. ASC and NLRP3 maintain innate immune homeostasis in the airway through an inflammasome-independent mechanism. Mucosal Immunol 12, 1092–1103 (2019). [DOI] [PubMed] [Google Scholar]
- 36.Rock KL, Latz E, Ontiveros F & Kono H The sterile inflammatory response. Annual review of immunology 28, 321–342 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Saitoh T et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1beta production. Nature 456, 264–268 (2008). [DOI] [PubMed] [Google Scholar]