

Abstract
Neurons and glia maintain central nervous system (CNS) homeostasis through diverse mechanisms of intra- and intercellular signaling. Some of these interactions include the exchange of soluble factors between cells via direct cell-to-cell contact for both short and long-distance transfer of biological materials. Transcellular transfer of mitochondria has emerged as a key example of this communication. This transcellular transfer of mitochondria are dynamically involved in the cellular and tissue response to CNS injury and play beneficial roles in recovery. This review highlights recent research addressing the cause and effect of intra- and intercellular mitochondrial transfer with a specific focus on the future of mitochondrial transplantation therapy. We believe that mitochondrial transfer plays a crucial role during bioenergetic crisis/deficit, but the quality, quantity and mode of mitochondrial transfer determines the protective capacity for the receiving cells. Mitochondrial transplantation is a new treatment paradigm and will overcome the major bottleneck of traditional approach of correcting mitochondria-related disorders.
Keywords: TRAK, Miro, Kinesin, mitochondrial transplantation, extracellular vesicles, tunneling nanotubes, mitochondrial extrusion
1. Introduction
Mitochondria are one of the major ancient double membrane systems that arose around two billion years ago (Friedman and Nunnari, 2014) from engulfment of α-proteobacterium by a precursor of the modern eukaryotic cell. Mitochondria have an inert ability to produce ATP through respiration leading them to become a driving force in evolution. Although mitochondria maintained the double membrane structure and retained the core ability to produce ATP, the overall form and composition can be drastically altered in response to the microenvironment. Apart from its traditional role as a cell powerhouse, mitochondria also possess a myriad of additional functions including calcium (Ca2+) buffering capacity (Granatiero et al., 2017; Pallafacchina et al., 2018; Szymanski et al., 2017), generation of reactive oxygen species (ROS) (Demine et al., 2019; Sena and Chandel, 2012; Singh et al., 2019; Stefanatos and Sanz, 2018), regulation of apoptosis (Bock and Tait, 2020; Galluzzi et al., 2018; Gong et al., 2019; Marchi et al., 2018), activation of endoplasmic reticulum (ER) stress response (Csordas et al., 2018; Marchi et al., 2018; Szymanski et al., 2017), as well as in the broad range sequelae of mitochondrial dysfunction. Mitochondria are also implicated in many common diseases including but not restricted to cardiovascular disease, Alzheimer’s disease (AD) and Parkinson’s disease (PD), metabolic disorders, muscular dystrophy, and in the process of normal aging (Diaz-Vegas et al., 2020).
The causes of these mitochondrial diseases are literally a collection of changes in thousands of genes that influence mitochondrial function. It has been accepted that mitochondria are comprised of over 1,000 proteins that vary within and between species in response to cellular and tissue-specific organismal needs. However, what exists in cellular mitochondria is a small, approximately 16 kbp, circular mitochondrial genome present in a vast excess of copies relative to nuclear chromosomes. Thus, mitochondrial composition is plastic in nature and is under the control of dual genomes (mitochondrial and nuclear) that add a layer of complexity to the genomic and proteomic composition of this organelle. Most of the mitochondrial proteins are encoded by nuclear genes and 13 proteins which are the core constituents of the mitochondrial electron transport chain (ETC) complexes are encoded by the mitochondrial DNA (mtDNA). Additionally, the mitochondrial genome encodes 22 transfer RNAs (tRNAs) and 2 mitochondrial ribosomal RNA (rRNA), which are essential components for the mitochondrial translational apparatus (Kopinski et al., 2019; Wallace, 2018). The nuclear-encoded mitochondrial proteins are translated on cytosolic ribosomes and actively imported and sorted into mitochondrial sub-compartments by outer and inner membrane translocase machinery. The ETC complex generates an electrochemical gradient, mitochondrial membrane potential (ΔΨm) through the coupled transfer of electrons to oxygen (O2) and the transport of protons (H+) from the matrix across the inner mitochondrial membrane (IMM) into the intermembrane space (IMS). The ΔΨm powers the terminal complex V of the ETC, the ATP synthase, which is likened to an ancient rotary turbine machine that catalyzes the synthesis of most cellular ATP. Additionally, the generated ΔΨm is linked to a critical mitochondrial function, the mitochondrial Ca2+ uptake through the mitochondrial calcium uniporter (MCU) (Baughman et al., 2011; De Stefani et al., 2011). The functional status of mitochondria depends on the ΔΨm and any perturbation of ΔΨm activates a cascade of signaling events that either activate the repair mechanism or eliminate defective mitochondria (Friedman and Nunnari, 2014).
Having established mitochondria as multivariate signal processors in nearly all eukaryotes, in recent years the diverse functionality of mitochondria has been linked to their morphological complexity. An observation made over seven decades ago revealed the plasticity of mitochondria (Porter et al., 1945), eg: fibroblast mitochondria are usually long and filamentous (1 to 10 μm in length with a constant diameter of ~700 nm), whereas, mitochondria in hepatocyte are more uniformly spheres or ovoids (Youle and van der Bliek, 2012). Additionally, mitochondria in many cell types form a complex reticulum, whereas axonal mitochondria are separated from these reticular structures and exist as discrete organelles of typically 1–3 μm in length (Chang and Reynolds, 2006). The phenotypic state of mitochondria has been linked to their capacity to produce energy, cell viability, and death (Mishra and Chan, 2014; Youle and van der Bliek, 2012). Though the link between phenotypic and functional state of mitochondria is not always observed in every context, it is true for a plethora of scenario that actively respiring mitochondria are long and filamentous, while short circular fragmented mitochondria are often associated with pathological conditions (Mishra and Chan, 2014). Thus, the pleomorphic nature of mitochondria makes it apparent that these organelles are far from static.
Mitochondria are highly dynamic organelles that constantly undergo shape change through fusion and fission to form a continuous network. Constant cycles of fission and fusion promote the intermixing and the homogenization of mitochondrial proteins, lipids, and DNA among different mitochondria within the same cell (Mishra and Chan, 2014). Also, mitochondrial dynamics allow selective elimination of mitochondria through mitophagy and mitochondrial biogenesis replenishes the mitochondrial pool, thus maintaining a healthy mitochondrial repertoire within the cell (Glancy et al., 2017). Specifically, in neurons the mitochondria are transported anterograde from the cell body to the processes of the cell, or retrograde back to cell body, enabling the removal of damaged mitochondria or replenishing healthy mitochondria (Dai et al., 2014; Suen et al., 2010; Valenci et al., 2015). Mitochondrial transport in neurons is facilitated by the microtubule-based adaptor or motor proteins trafficking system, consisting of kinesin/dynein (motor), Milton/Trak (adaptor) and Miro (mitochondrial receptor protein) (Mandal and Drerup, 2019; Schwarz, 2013).
Apart from these intracellular mitochondrial dynamics, horizontal transfer of mitochondria between mammalian cells was recently shown, thereby challenging current concepts of mitochondria and mtDNA segregation and inheritance (Torralba et al., 2016). This transcellular transfer of mitochondria promotes the incorporation of the transferred mitochondria into the endogenous network of the recipient cells, contributing to changes in the bioenergetic profile and other functional properties of the recipient cell. In addition, the intercellular transfer of mitochondria also involves the horizontal transfer of mitochondrial genes, which has important implications in physiopathology of mitochondrial dysfunction. The diverse mechanisms like formation of tunneling nanotubes, extracellular vesicles, and formation of gap junctions, naked mitochondrial ejection, or cytoplasmic fusion mediate intercellular mitochondrial transfer (Chang et al., 2019; Nakamura et al., 2020). This review focuses on both intracellular and intercellular mitochondrial movement, the contributing molecular mechanisms, molecular machinery and the impact on cell survival with a special focus on brain cells.
2. Neuronal energy demand and supply
Neuronal functioning requires high energy and a constant supply of ATP is critical for neuron growth, survival and function (Nicholls and Budd, 2000). The brain occupies only 2% of the body weight, but consumes 20% of the body’s resting energy production (Tomasi et al., 2013). Within the brain, neurons utilize 70–80% of the energy and neighboring glial cells utilize the remainder. For example, a cortical neuron in the human brain utilizes up to ~4.7 billion ATP molecules/second to power various biological processes (Zhu et al., 2012). In the brain, synapses are the primary sites of ATP consumption, where mitochondria supply ~93% of the ATP demand, with the remaining ~7% provided through glycolysis (Harris et al., 2012). Due to such a high energy demand, neurons require specialized mechanisms to maintain energy homeostasis throughout the cell, particularly in distal synapses and in axons (Saxton and Hollenbeck, 2012; Sheng, 2014). The larger size of neurons precludes the rapid diffusion of ATP from one end of the cell to the other (Hubley et al., 1996; Sun et al., 2013). This implies that the energy production must be spatially matched to local energy usage. Thus, it is necessary for the mitochondria to be located close to the sites of high energy demands, including pre- and post-synaptic domains, the axon initial segment, nodes of Ranvier and growth cones (Hollenbeck, 1996, 2005; Hollenbeck and Saxton, 2005). Axons and synapses are highly plastic and undergo spontaneous and activity-dependent remodeling, thereby changing mitochondrial distribution. Therefore, the position of mitochondria in neurons must be controlled on rapid timescales to match changes in synaptic input. Live cell imaging using mitochondrial targeted fluorescent dyes or genetically encoded mitochondrial proteins reveal that in neurons, the mitochondrial network is very dynamic, undergoing fission and fusion events with individual mitochondria exhibiting bi-directional transport, stopping, starting, persistent docking in certain regions, and changing direction (Schwarz, 2013). The mean neuronal mitochondrial transport velocities in invertebrates and vertebrates lie within the range of ~0.1–1.4 μm/s (Melkov and Abdu, 2018; Schwarz, 2013). In addition to the fast transport, a lower velocity (~50 μm/h or even slower) has been identified in developing neurons (Miller and Sheetz, 2004). In mature neurons, about 20–30% of axonal mitochondria are motile (Chen and Sheng, 2013; Kang et al., 2008); while ~15% either briefly pause or dock at synapses; and ~14% of motile mitochondria dynamically pass through presynaptic terminals. An anchored mitochondrion within the presynaptic terminals provides a stable and continuous ATP supply. Conversely, a motile axonal mitochondrion temporally supplies ATP thus, changing synaptic energy levels and synaptic activities. Mitochondria are transported between the soma and distal axon and dendritic terminals by microtubule (MT)-based motor proteins dependent transport mechanism: Kinesin-1 (KIF5) and dynein motors drive anterograde and retrograde movement, respectively (Fig. 1).
Fig. 1. Key components of the Motor-adaptor-receptor complex for intracellular mitochondrial movement.

Axonal microtubule (MT) are uniformly arranged with their plus end (+end) directed distally and minus end (−end) towards soma. (A) Plus-end directed anterograde transport (−end to +end) of mitochondria is mediated by the Kinesin motor. The N-terminal domain the motor domain with ATPase activity and binds directly the MT. The C-terminal is the cargo binding domain. Milton (or TRAK1/2) is the motor adaptor protein that links Miro (present in the outer mitochondrial membrane) to the cargo binding domain of Kinesin. (B) Minus-end directed retrograde transport (+end to −end) of mitochondria is mediated by the cytoplasmic MT-based dynein motor. Dynein contains multiple subunits including two catalytic heavy chains (DHC), several intermediate chains (DIC) and light chains (DLC). DHC is the motor domain required for dynein movement. Dynactin is a large protein complex that interacts with dynein and MT through the p150Glued subunit. Both dynein and dynactin together drive the retrograde mitochondrial movement. (C and D) Actin filament based short distance mitochondrial transport is mediated by myosin. Myosin is a two-headed motor protein with a unique globular tail domain that can interact directly with kinesin motor (C), or with DLC (D) raising the possibility of this motor to facilitate both long-range transport along MT and short-distance transport along actin filaments. (E) Myo19 was identified as the mitochondria associated myosin. Myo19 directly interacts with Miro through its C-terminal fragment of the tail region.
3. Intracellular mitochondrial transport
3.1. Motor proteins for mitochondrial movement
3.1.1. Kinesin:
Kinesin superfamily proteins (KIFs) and cytoplasmic dynein mediate long-distance transport of mitochondria and other membranous organelles or cargoes through mechanisms that depend on the polarity and organization of neuronal MTs and require ATP hydrolysis (Hirokawa et al., 2010; Martin et al., 1999). Axonal MT are uniformly arranged with their plus-end directed distally and minus end towards the soma. Of the 45 kinesin motor genes identified, the Kinesin-1 (KIF5) family drives plus-end directed anterograde transport of neuronal mitochondria (Cai et al., 2005; Gorska-Andrzejak et al., 2003; Hurd and Saxton, 1996; Pilling et al., 2006). Kinesin-1 contains two heavy chains (KHC) and two light chains (KLC). The N-terminal domain of kinesin-1 is the motor domain with ATPase activity and binds directly to the MTs and the C-terminal is the cargo-binding domain (Fig. 1A). There are three isoforms of KIF5 (KIF5A, 5B, and 5C). KIF5A is ubiquitously present; whereas, KIF5B and 5C are specific for neurons (Kanai et al., 2000). Disruption of KIF5-mitochondrial coupling impairs mitochondrial transport, thus reducing the number of mitochondria in the distal axons (Hurd and Saxton, 1996; Tanaka et al., 1998).
3.1.2. Dynein:
Cytoplasmic dynein guides MT-based retrograde transport in axons. Dynein contains multiple subunits including two catalytic heavy chains (DHC), several intermediate (DIC), light-intermediate (DLIC) and light chains (DLC). The C-terminus of the DHC is the motor domain required for the dynein movement. Dynactin is a large complex protein that binds directly to dynein and MTs through its p150Glued subunit (Waterman-Storer et al., 1995). Both dynein and dynactin associate with mitochondria and drive its retrograde transport (Fig. 1B) (Karki and Holzbaur, 1999; King and Schroer, 2000; Waterman-Storer et al., 1995). Disruption of the dynactin complex does not disrupt the binding of motor proteins to the mitochondria, but interrupts both the anterograde and retrograde transport, suggesting that dynactin is probably involved in the regulation of both KIF5 and dynein driven bi-directional mitochondrial transport (Haghnia et al., 2007).
3.1.3. Myosin:
While the MT-based motor proteins quickly transport cargoes and organelles through lengthy axons, myosin drives short distance trafficking along with actin filaments at the presynaptic terminals and growth cones (Morris and Hollenbeck, 1993). Myosins I, II, V, and VI were reported to facilitate mitochondrial transport in axons. Myosin V is a two-headed motor containing unique globular tail domain and can interact directly with either a kinesin motor or with the light-chain of dynein (Fig. 1C and 1D) (Langford, 2002), raising the possibility that the dual-motor complex may facilitate co-ordination of mitochondrial long-range transport along microtubules and short-range movement on actin filaments. For example, Myo19 was identified as a mitochondria-associated myosin and its overexpression in neuronal cell lines increased mitochondrial motility (Quintero et al., 2009). Later with the help of proximity labeling, Miro was identified as the potential binding partner of Myo19. The interaction studies showed Miro1 to bind directly to a C-terminal fragment of the Myo19 tail region. These results show Miro to coordinate microtubule and actin-based mitochondrial movement (Fig.1E) (Oeding et al., 2018)
3.2. Motor-adaptor complex for mitochondrial movement
Motor proteins recruit mitochondria by associating with their respective motor adaptor proteins and mitochondrial membrane receptors (Fig. 1). This motor/adaptor/receptor complex ensures targeted mitochondrial trafficking and precise regulation of their distribution in response to changes in neuronal activity (Brickley et al., 2005; Brickley and Stephenson, 2011; Fransson et al., 2003; Fransson et al., 2006; Glater et al., 2006; Guo et al., 2005; Stowers et al., 2002). The Drosophila protein Milton was the first identified motor adaptor protein that links mitochondrial OMM protein Miro to the KIF5 cargo binding domain (Glater et al., 2006). There are two mammalian orthologues: TRAK1 and TRAK2, sometimes referred to as GRIF1 and OIP106 (Beck et al., 2002; Brickley et al., 2005). TRAK1/2 lacks an obvious domain structure other than an extensive coiled-coil region, which includes a significant region of homology with the Huntingtin-associated protein 1 (HAP-1) (Stowers et al., 2002). The interaction of TRAK with KIF5 is direct, as TRAK contains one N-terminal KIF5 binding site and two dynein/dynactin binding sites; one at the N-terminal and the other at the C-terminus (van Spronsen et al., 2013). Thus, TRAK can mediate both KIF5 and dynein driven bi-directional transport of axonal mitochondria. Mammalian TRAK proteins are also recruited to endosomal compartments and associate with γ-aminobutyric acid (GABAA) receptors, K+ channels, and Hrs protein highlighting their role in neuronal cargo transport (Grishin et al., 2006; Kirk et al., 2006; Webber et al., 2008). Depleting TRAK1 or overexpressing a dominant negative mutant impairs axonal mitochondrial motility (Brickley and Stephenson, 2011; van Spronsen et al., 2013). Conversely, overexpression of TRAK2 robustly enhances mitochondrial motility (Chen and Sheng, 2013).
3.3. Motor-adaptor-receptor complex for mitochondrial movement
Miro, the mitochondrial outer membrane protein functions as the receptor of the motor/adaptor/receptor complexes by binding to the motor adaptor, Milton (or TRAK1/2) thus, recruiting KIF5 to the mitochondrial surface (Glater et al., 2006) (Fig. 1). Miro is a member of the Rho-GTPase family with two homologous domains with small GTPases that are separated by two Ca2+ binding EF-hand motifs. At the carboxyl terminus of Miro there is a trans-membrane domain that tethers it to the surface of the OMM (Fransson et al., 2006). Drosophila has a single Miro (Guo et al., 2005); whereas, mammals have two Miro orthologs, Miro-1 (RhoT1) and Miro-2 (RhoT2) and they share 60% sequence identity (Aspenstrom et al., 2007). Biochemical data originally linked Milton and Miro only to KIF5, but Drosophila phenotypes indicated that they may also be needed for dynein-mediated transport (Stowers et al., 2002).
3.4. Regulation of mitochondrial movement
3.4.1. Reversible arrest by cytosolic Ca2+
Cytosolic Ca2+ (cCa2+) is one of the best studied regulators of mitochondrial movement. Elevation of cCa2+ stops both the anterograde and the retrograde movement of mitochondria (Chang et al., 2006; Rintoul et al., 2003; Szabadkai et al., 2006). Such elevation of cCa2+ occurs during physiological stimuli including the activation of glutamate receptors in dendrites, action potential in axons, and by neuromodulators that trigger Ca2+ release from the stores (Ohno et al., 2011; Rintoul et al., 2003; Saotome et al., 2008; Yi et al., 2004). Studies have shown ~0.4 μM of cCa2+ is needed to cause 50% reduction in mitochondrial movement and complete arrest occurs at low micromolar concentrations (Saotome et al., 2008; Yi et al., 2004). The docking of mitochondria in the region of high Ca2+ maintains cCa2+ homeostasis by (i) buffering the cCa2+ and (ii) also providing ATP for the active efflux of Ca2+ from the cell. In addition, subcellular regions with inadequate ATP are likely to have elevated cCa2+ and the arrest of mitochondria in such regions will provide ATP for biological processes (Wang and Schwarz, 2009; Yang and Steele, 2000). Detailed examination of the mechanism underlying this regulation suggested that the EF hand of Miro plays a critical role in the Ca2+-mediated regulation of mitochondrial movement. Two mechanisms are proposed for the molecular mechanism downstream to Ca2+ binding: (i) Ca2+ binding to Miro triggers the dissociation of either Miro/TRAK or TRAK/KIF5 interaction, thereby uncoupling the KIF5 from the mitochondrial surface (Macaskill et al., 2009) (Fig. 2A); (ii) KIF5 switches from the active state to an inactive state by binding to the Ca2+ bound Miro through the head domain of kinesin, thus, preventing it from binding to microtubules (Wang and Schwarz, 2009) (Fig. 2B). These two proposed mechanisms are noteworthy in the context of health and disease. The first is readily reversible and will allow mitochondrial movement as soon as the cCa2+ is removed. The second explains the selective arrest of mitochondrial motility.
Fig. 2. Molecular regulation of mitochondrial movement.
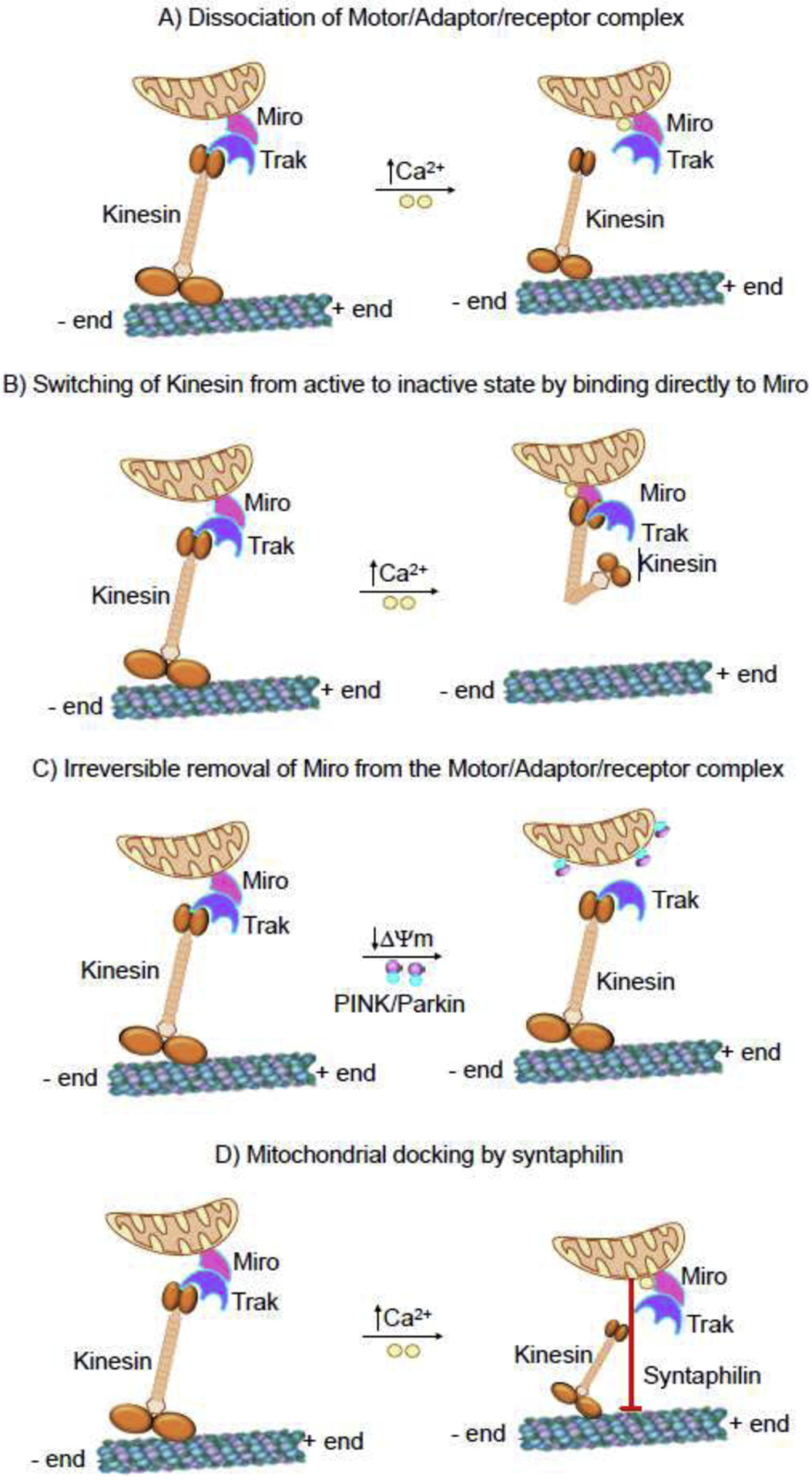
(A) Dissociation of motor/adaptor/receptor complex. Binding of Ca2+ to the Miro EF hands facilitate the dissociation of either Miro/TRAK or KIF5/TRAK interaction, thereby uncoupling the KIF5 from the mitochondrial surface. (B) Switching of kinesin from active to inactive state by directly binding to Miro. KIF5 switches from the active state to an inactive state by binding to the Ca2+ bound Miro through the head domain of kinesin, thus preventing it from binding to microtubules. (C) Irreversible removal of Miro from the complex. PINK1 binds to and phosphorylates Miro to signal Parkin. The consequence of activating the PINK/Parkin pathway is the proteasome-dependent degradation of Miro and consequent release of KIF5/TRAK complex from the mitochondrial surface. (D) Mitochondrial docking by syntaphilin. Syntaphilin (SPH) acts as a static anchor specific for axonal mitochondria. Depending on the energy requirement mitochondria binds the microtubules through the protein syntaphilin and docks at the site of high energy demand.
3.4.2. Irreversible arrest by PINK/Parkin pathway
An additional mechanism of mitochondrial arrest by PINK1 (PTEN-induced putative kinase 1)/Parkin-mediated pathway is very different from the readily reversible Ca2+-mediated mitochondrial docking. PINK1 is a protein kinase and Parkin is an E3 ubiquitin ligase, and together they initiate a pathway for the autophagic clearance of mitochondria. Physiologically, PINK1 is recruited and imported into the mitochondria and then inactivated by proteolytic cleavage. Conversely, in a damaged mitochondria with compromised mitochondrial membrane potential (ΔΨm) PINK1 accumulates on the surface of the mitochondria and recruits Parkin to the site. The potential involvement of a PINK/Parkin mechanism for the mitochondrial movement was confirmed by the observation of a biochemical association between PINK1 and Miro/Milton complex (Weihofen et al., 2009). Subsequently, Parkin was also shown to bind this complex, and this interaction was greatly increased in depolarized mitochondria (Wang et al., 2011). Binding of PINK1 phosphorylates Miro at various sites of which S156 is indispensable for the activity of Parkin. The consequence of activating the PINK/Parkin pathway is the proteasome-dependent degradation of Miro and consequent release of KIF5/Milton complex from the mitochondrial surface (Chan et al., 2011; Wang et al., 2011) (Fig. 2C). This proteolytic and irreversible mechanism of mitochondrial docking will facilitate the removal of quarantined mitochondria by mitophagy and prevent them from fusing with other healthy mitochondria.
3.5. Mitochondrial anchoring
One of the most intriguing anchoring mechanisms involves the association of mitochondria with microtubules through the protein syntaphilin (SPH) (Fig. 2D). SPH acts as a static anchor specific for axonal mitochondria (Kang et al., 2008). SPH specifically targets the outer membrane of the axonal mitochondria through its C-terminal mitochondria-targeting domain and axon-sorting sequence and arrests motile mitochondria by anchoring them to the MTs. Dynein light chain LC8 also appears to be involved with the SPH-based docking (Chen et al., 2009). Deleting SPH results in a robust increase in axonal mitochondrial motility and conversely overexpression of SPH abolishes mitochondrial motility (Kang et al., 2008). However, SPH is absent from the genome of Drosophila, although flies have the stationary pool of mitochondria like mammals in their neurons.
These observations clearly show that mitochondria are not just static organelles afloat in the cytosolic milieu, but that they are dynamic organelles that traffic, fuse and divide (Jakobs, 2006). In axons, motile mitochondria can become stationary and stationary mitochondria can be remobilized in response to changes in bioenergetic status and synaptic activity. Thus, axonal mitochondria deploy an anchoring mechanism in addition to the motor-driven transport. These anchored/docked mitochondria serve as the power plants for stable and continuous ATP supply at the synapses. Thus, SPH can serve as an attractive molecular target for investigations into mechanisms recruiting motile mitochondria into activated synapses.
4. Intercellular mitochondrial transfer
Organelle exchange represents a special form of intercellular communication that allows unidirectional or bidirectional transfer, not only of signals, small molecules, or ions but also defined intracellular structures such as mitochondria, lysosomes, endosomal vesicles and plasma membrane components (Rogers and Bhattacharya, 2013). Transport of mitochondria from one cell to another has gained importance in recent years as a significant mediator of cellular health and fitness. Mitochondrial transfer supports the exogenous replacement of damaged mitochondria, thereby rescuing mitochondrial defects (Hayakawa et al., 2016; Patananan et al., 2016). Most studies used stem cells as mitochondria donors, but some utilized immortalized or primary cells from the same or different species to serve as mitochondrial donors (Berridge et al., 2016). It was previously unknown whether mitochondrial transfer was a physiologically relevant process or was observed only in the in vitro environment. But in recent years, several reports provide evidence for transfer of mitochondria to occur in vivo and to be involved in diverse pathophysiological conditions such as tissue injury and cancer progression (Ahmad et al., 2014; Hayakawa et al., 2016; Islam et al., 2012; Moschoi et al., 2016; Tan et al., 2015). The molecular and signaling mechanism(s) by which cells containing dysfunctional mitochondria acquire functional mitochondria from other cells and how this process is regulated remains unknown. But it is hypothesized that cells likely possess mechanisms to trigger organelle exchange in response to injury signals originating from distressed recipient cells. However, the molecular cues that initiate this process remain unidentified. The first evidence of functional mitochondrial transfer was observed in intrinsic mitochondria-depleted recipient cells from mesenchymal stem cells/fibroblasts (Spees et al., 2006). Careful examination of mitochondrial and nuclear DNA polymorphisms in these rescued clones excluded cell fusion as the mechanism of mitochondrial transfer. Also, the transfer did not occur through the passive uptake of mitochondrial fragments or isolated organelles but involved an active process such as the formation of tunneling nanotubes (TNTs) or vesicular transfer of mitochondrial fragments (Spees et al., 2006). TNTs are so far described as the major cellular structure that mediates intercellular mitochondrial transfer. Other mechanisms have been proposed including, extracellular vesicles, naked mitochondria ejection, and gap junctions.
4.1. Tunneling nanotubes
TNTs are nanotubular structures produced by the outgrowth of filopodia-like cell membrane protrusions that connect with the target cell. The membrane from each cell extends to fuse together, thereby forming a tightly connected bridge that is not tethered to the substrate, but rather hovering in the extracellular space (Gerdes et al., 2007). The discovery of TNTs in 2004 emerged as a novel mechanism of cell-to-cell communication, demonstrating the ability of mammalian cells to donate or receive organelles from other cells (Rustom et al., 2004). TNTs are different from other cellular protrusions, as TNTs are straight with a small diameter (20–500nm) and a length up to 100 μm. They are dynamic structures formed de novo within minutes and display lifetimes ranging from minutes to several hours (Bukoreshtliev et al., 2009; Rustom et al., 2004). TNTs contain a skeleton mainly composed of F-actin and transport proteins that facilitate active transport of cargo and mitochondria along these structures (Ahmad et al., 2014). TNTs were first described in cultured pheochromocytoma PC12 cells (Rustom et al., 2004) and subsequent studies have shown that they connect a variety of cell types including epithelial, fibroblastic, immune, neuronal and glial cells (Abounit and Zurzolo, 2012; Gousset et al., 2009; Rustom et al., 2004; Wang and Gerdes, 2011) and an evidence confirm their existence in vivo (Chinnery et al., 2008). Studies have shown the involvement of TNTs in mitochondrial transport, the repair of cell damage, the activation of enhanced immune responses, and cell metabolic reprogramming (Bukoreshtliev et al., 2009; Gousset et al., 2009; Islam et al., 2012; Jackson et al., 2016; Sinha et al., 2016).
Though the transport of mitochondria through TNTs is defined (Fig. 3A), the directionality of mitochondrial transfer and other indispensable factors essential to initiate and promote TNT formation are not fully understood. A study identified the involvement of S100A4, an extracellular protein and its receptor RAGE (Receptor for Advanced Glycation End Product) in guiding TNTs growth direction. During stress, hippocampal neurons and astrocytes initiated the formation of TNT’s after p53-mediated activation of caspase 3. Activated caspase 3 cleaves S100A4 creating a gradient of low levels of S100A4 in initiating cells (injured cells) towards a higher concentration in astrocyte target cells (Sun et al., 2012). The results from this study reveal that damaged cells (neurons) transfer cellular contents to astrocytes to spread the danger signals to initiate mitochondrial transfer. But no information regarding mitochondrial participation was provided by this study. These studies on TNT formation and mitochondrial transfer raise intriguing questions (i) on the degree of cellular damage required to initiate intercellular mitochondrial transfer and (ii) the mechanism(s) by which healthy cells detects the degree of metabolic shutdown of the stressed cells and makes a judgment to restore the functionality of the stressed cells, instead of permitting apoptosis. This line of investigation is of great interest in deciding the fate of terminally differentiated cells, including adult neurons and cardiomyocytes.
Fig. 3. Mode of intercellular mitochondrial transfer.
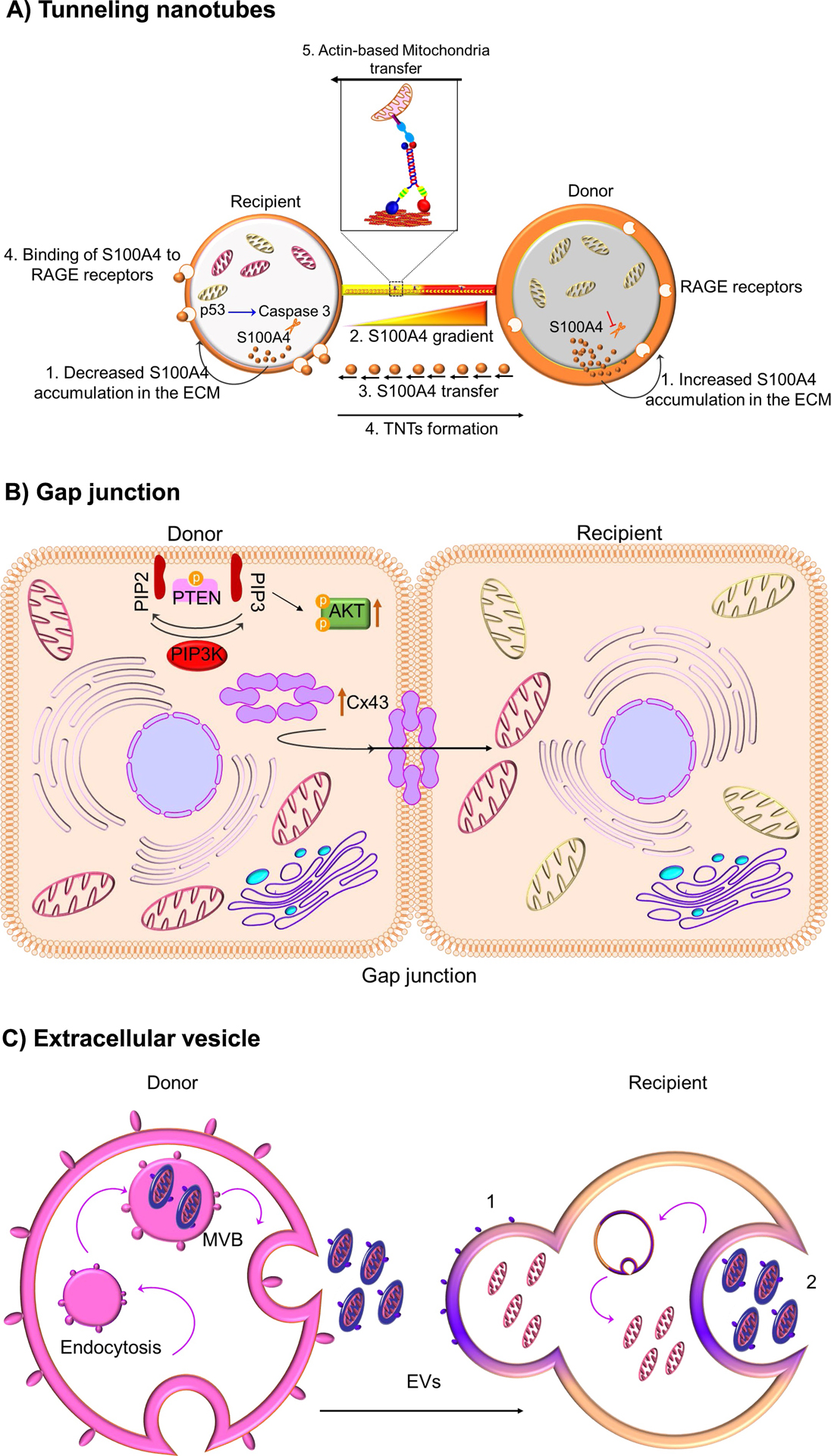
(A) Tunneling nanotubes.P53-mediated activation of caspase 3 cleaves S100A4, creating a gradient of low levels of S100A4 in initiating or recipient cells (injured cells) towards a higher concentration in target or donor cells (healthy cells). It is suggested that RAGE in recipient cells act as the putative receptor for the high concentration of S100A4 in donor cells and this co-ordination of S100A4 and RAGE guides TNT direction. Mitochondria transfer through nanotunnels is believed to be facilitated by Actin/Miro-based transport machinery. (B) Gap junction. Connexin, specifically connexin 43 oligomerizes to form a channel at the gap junction which facilitates the transfer of mitochondria. Mechanistically, it was shown that ROS-induced oxidative stress regulates the opening of connexin channels in a system mediated by phosphoinositide 3-kinase (PI3K). (C) Extracellular vesicle. The multivesicular bodies (MVB) release extracellular vesicles (EVs) from the donor cell. These released EVs can fuse with recipient cells (1) or engulfed (2) to release the EVs’ content
Spees et al. described the transfer of mitochondria from mesenchymal stem cells (MSCs) to respiration-deficient cancer cells through TNTs (Spees et al., 2006), but the direction of transfer or whether the MSCs received any signal from cancer cells to facilitate mitochondrial transfer was not described. Koyanagi et al., showed the transfer of mitochondria exclusively through TNTs from human endothelial progenitor cells (EPCs) to neonatal undifferentiated cardiomyocytes in a process intending to sustain their maturation (Koyanagi et al., 2005). Yang et al., while using microfluidic channels to track TNT formation and exchange of biological materials, observed MSCs forming TNTs and transferring mitochondria to cardiomyocytes, as opposed to fibroblasts (Yang et al., 2016). But why MSCs have a greater propensity to form TNTs compared to other cells remain unanswered. Similarly, MSC improve the survival of endothelial cells and cardiomyocytes after ischemic-reperfusion injury (in an in vitro model system deprived of glucose and oxygen and then reoxygenated) by transferring its mitochondria independent of cellular transdifferentiation or the release of paracrine factors (Han et al., 2016; Liu et al., 2014b). Mitochondrial transfer from MSCs to lung epithelium attenuated cigarette-smoke induced lung damage (Li et al., 2014), while transfer between MSC and innate immune cells via TNTs enhanced the capacity of alveolar macrophages to engulf invading bacteria in a pneumonia model (Jackson et al., 2016). A follow-up study reported that Miro1 regulated the efficiency of intercellular movement of mitochondria. Overexpression of Miro1 in MSC increased the transfer of mitochondria from stem cells to stressed alveolar epithelial cells via intercellular TNTs, thus attenuating epithelial cell apoptosis, and reducing inflammatory cell infiltration, collagen deposition, and mucus hypersecretion in the lungs (Ahmad et al., 2014). Cancer cells also acquire mitochondria from the surrounding healthy cells to optimize and repair damaged metabolic machinery and promote survival. One such critical donor of healthy mitochondria to cancer cells are stromal cells. Cancer and stromal cells communicate through TNTs and mitochondrial transfer via TNTs is shown to enhance the chemoresistance to doxorubicin and survival (Pasquier et al., 2013). In addition, mitochondrial transfer from bone marrow mesenchymal cells to acute myelogenous leukemia cells in vivo confers chemoresistance and survival (Moschoi et al., 2016).
4.2. Gap Junctions
Gap junctions have evolved into specialized cell to cell structures between eukaryotic cells and allow direct metabolic and electrical communication between almost all cell types. Generally, gap junction channels allow the passive diffusion of molecules of up to 1,000 Daltons and they transport nutrients, metabolites, second messengers, cations, anions and mitochondria (Evans and Martin, 2002; Islam et al., 2012; Mistry et al., 2019; Otsu et al., 2009; Yao et al., 2018). Nearly 20 connexin genes and 3 pannexin genes contribute to gap junction proteins in mice and humans. Connexin oligomerizes and forms channel-like structures or gap junctions between cells. Live optical imaging showed gap-junction-mediated intercellular mitochondrial transfer between bone marrow derived stromal cells (BMSC) and alveolar-epithelial cells in a lipopolysaccharide (LPS)-injured mouse model. BMSC infused to the trachea of LPS-treated mice attached to the alveolar epithelial cells by a mechanism involving connexin, particularly connexin 43. BMSC established Cx43-containing gap junctional channels (GJCs) with the alveolar epithelia releasing mitochondria-containing microvesicles that were engulfed by the epithelial cells (Fig. 3B). The presence of BMSC-derived mitochondria in the epithelia was confirmed microscopically (Islam et al., 2012). Mitochondrial acquisition by the alveolar cells restored ATP concentration and increased the secretion of pulmonary surfactant. Additionally, silencing of Rieske protein (RISP), a subunit of the mitochondrial complex III in BMSCs reduced their capacity to restore ATP levels in alveolar cells, preventing protection from LPS-injury. This implicated mitochondrial transfer as one of the plausible mechanisms of MSC-mediated therapy in models of acute lung injury and other inflammatory diseases. Recent study demonstrated that overexpression of Cx43 in iPSC derived MSCs facilitated mitochondrial transfer between iPSC-MSCs and epithelial cells in both in vitro and in vivo model of asthma inflammation (Yao et al., 2018). Cx43-based intercellular gap junctional communication also occurred in co-culture of MSCs and endothelial cells (Otsu et al., 2009). Recently, it was shown that the transfer of mitochondria from BMSCs to hematopoietic stem cells (HSCs) is an early physiological event in the mammalian response to acute bacterial infection (Mistry et al., 2019) . Mechanistically, it was observed that ROS-induced oxidative stress regulates the opening of connexin channels in a system mediated by phosphoinositide 3-kinase (PI3K) activation, which allowed the transfer of mitochondria from BMSCs to HSCs (Mistry et al., 2019).
4.3. Extracellular Vesicles
Every cell type releases a heterogenous population of vesicles to the extracellular environment. These vesicles range from 40 to 1000 nm and are referred to as extracellular vesicles (EVs). EVs can be divided into exosomes (30 to 100 nm in diameter), microvesicles (MVs) (100 nm to 1 μm diameter), and apoptotic bodies (1 to 2 μm) depending on their origin and molecular composition (Zappulli et al., 2016). Apoptotic bodies have been less studied due to their rapid elimination by phagocytic cells (Pitt et al., 2016). Exosomes and microvesicles were initially described as vesicles meant to remove archaic proteins in immune cells and reticulocytes (Paolicelli et al., 2019). More recently, they have been shown to originate from almost all cell types and are reported to mediate cell-to-cell communication in a growing number of physiological and pathological situations (Paolicelli et al., 2019). Growing evidence indicates that EVs containing lipids, proteins, RNAs and mitochondria represent an efficient way to transfer functional cargoes from one cell to another. In this line, the discovery of EVs have brought a new format of interactions to an already multifaceted communication network and a new mechanism of signal transmission (Hervera et al., 2018; Valadi et al., 2007).
Mitochondrial components have been detected in EVs (Fig. 3C), although the mechanisms by which mitochondrial proteins or mtDNA are loaded in EVs are still unknown. Smaller EVs like exosomes can transport mostly small RNAs (Valadi et al., 2007), but genomic and mtDNA have also been detected (Guescini et al., 2010; Sansone et al., 2017). Larger EVs such as MVs can contain entire mitochondria (Berridge and Neuzil, 2017). MVs carrying mitochondria can be secreted by different cell types, as observed in astrocytes, neurons and mesenchymal stem cells (Davis et al., 2014; Guescini et al., 2010; Hayakawa et al., 2016; Sinha et al., 2016) and received by epithelial cells, immune cells, astrocytes, and neurons (Torralba et al., 2016). The process of mitochondrial transfer does not always protect damaged cells but also functions to recycle organelles in other cells by a transcellular degradation process (Davis et al., 2014; Hayakawa et al., 2016). Additionally, MSC-derived EVs containing mitochondria have been shown to modulate macrophage function by enhancing their respiration and phagocytic activity (Morrison et al., 2017). In a recent study, mitochondria was observed in EVs derived from the pro-inflammatory HLA-DR+ subsets of airway myeloid derived regulatory cells (MDRCs), which are known regulators of T cell responses in asthma. Importantly, these exosomally transferred mitochondria co-localize with the mitochondrial network and generate reactive oxygen species within recipient T cells (Morrison et al., 2017).
Apart from mitochondria being expelled in EVs, mitochondria possess the capacity to produce their own vesicles in order to transport mitochondrial proteins and lipids to other organelles within the cell (Bozi et al., 2016; Bragoszewski et al., 2017; Braschi et al., 2010; Cadete et al., 2016; McLelland et al., 2014; Neuspiel et al., 2008; Soubannier et al., 2012a; Soubannier et al., 2012b; Sugiura et al., 2014). These mitochondria-derived vesicles are an alternative means of quality control alongside the action of mitochondrial proteases, ubiquitin-mediated proteosomal degradation, and mitophagy (Bragoszewski et al., 2017). As one of mitochondria’s first line of defense, MDVs expel damaged proteins in order to prevent complete mitophagy. These MDVs are formed by selective incorporation of protein cargoes which can include outer/inner membrane and matrix components (McLelland et al., 2014; Neuspiel et al., 2008; Soubannier et al., 2012a; Soubannier et al., 2012b; Sugiura et al., 2014). The mechanisms that dictate cargo selection are still unclear, however proteins are known to be selected based on their target destination and the nature of mitochondrial stress. MDVs destined for lysosomal degradation contain protein kinase PINK1 and cytosolic ubiquitin E3 ligase Parkin (McLelland et al., 2014; Soubannier et al., 2012a; Soubannier et al., 2012b), and those following the peroxisome pathway carry the retromer complex and MAPL protein (Braschi et al., 2010; Neuspiel et al., 2008).
4.4. Mitochondrial extrusion
Mitochondrial extrusion is another possible mechanism for mitochondria to transfer from one cell to another. Extrusion allows the release of mitochondria or mitochondrial components from cells under specific conditions during which mitochondria become unfit to remain in cells. Retaining damaged mitochondria can produce large quantities of ROS (Lyamzaev et al., 2008) and under such circumstances, cells tend to dispose of their mitochondria into the intercellular space. During TNFα-induced cell death, cytoplasmic vacuoles engulf mitochondria, which subsequently fuse with the plasma membrane, releasing free mitochondria to the extracellular medium (Maeda and Fadeel, 2014). Likewise, LPS stimulation promotes the fusion of autophagolysosomes with the plasma membrane and releases mitochondrial components into extracellular space (Unuma et al., 2015). Neutrophils can extrude mitochondrial components including specifically oxidized mitochondrial nucleoids (Caielli et al., 2016). Under high concentrations of ROS, HeLa cells can extrude fragments of mitochondria through selective elimination or mitoptosis (Lyamzaev et al., 2008). Basophils release mtDNA in an ROS-dependent manner to create extracellular DNA traps to fight bacteria (Morshed et al., 2014). Likewise, eosinophils rapidly release mtDNA in the extracellular space to bind to and kill infectious bacteria (Yousefi et al., 2008). Mitochondrial extrusion not only occurs in vitro but also in vivo. A handful of studies have shown the release of naked or encapsulated mitochondria into the extracellular milieu. Nakajima et al., in 2008 used a mouse model to confirm the release of naked mitochondria into the intercellular space after an anti-Fas antibody injection. In response to this treatment, cytoplasmic vacuoles engulfed fragmented mitochondria and extrude them from apoptotic hepatocytes (Nakajima et al., 2008). Likewise, activated platelets released respiration-competent mitochondria, both as free organelles or mitochondrial encapsulated within microparticles (Boudreau et al., 2014). A number artificial techniques that is independent of biological interaction between the isolated organelle and the recipient cells were developed for the transfer of naked mitochondria. Examples include mitochondrial transformation (Kesner et al., 2016), magnetomitotransfer (Macheiner et al., 2016), MitoCeption (Caicedo et al., 2015), and mitochondrial invasion (Popkov et al., 2017). In 2016, transfer of naked mitochondria into somatic mammalian cells using photothermal nanoblade was devised (Wu et al., 2016). The use of photothermal nanoblade bypasses endocytosis and cell fusion and showed to rescue the pyrimidine auxotroph phenotype and respiration of ρ0 cells that lack mtDNA.
5. Mitochondrial transfer in the central nervous system
Astrocytes have broad and diverse roles in the central nervous system (CNS), and are involved in the regulation of neurodevelopment, neurotransmission, metabolism and blood flow (Attwell et al., 2010; Iadecola and Nedergaard, 2007; Khakh and Sofroniew, 2015). Under normal conditions, astrocytes protect neurons against oxidative stress and excitotoxicity and provide neurotrophic support (Ouyang et al., 2013; Wang and Cynader, 2001). By contrast, astrocytes under stressful conditions may withdraw this protection or release deleterious factors such as inflammatory cytokines that damage neurons (Khakh et al., 2017; Liddelow et al., 2017; Trias et al., 2018; Yun et al., 2018). Recent proof-of-concept studies demonstrate that the neuroprotective functions of astrocytes may include transfer of mitochondria to damaged neurons (Hayakawa et al., 2016; Huang et al., 2019; Lippert and Borlongan, 2019; Voloboueva et al., 2007). Not just astrocytes transfer mitochondria, but recent emerging data suggest the entire nervous system to benefit from cell to cell transfer of mitochondria for different purposes and under different physiological conditions. For example, (i) the transfer of mitochondria to astrocytes from neurons allowed cells to breakdown nonfunctional mitochondria (transmitophagy) (Davis et al., 2014); (ii) the transfer of healthy mitochondria from astrocytes to stressed neurons promoted neuroprotection and neurorepair (Hayakawa et al., 2016; Huang et al., 2019; Lippert and Borlongan, 2019); (iii) the transfer of functional mitochondria from endothelial progenitor cells (EPCs) to brain endothelial cells improved cell viability and barrier function (Hayakawa et al., 2018a; Hayakawa et al., 2018b); (iv) transfer of hematopoietic stem and progenitor cells’ (HSPCs) mitochondria to neurons restored mitochondrial function along with frataxin expression in a mouse model of Friedreich’s ataxia (Rocca et al., 2017).
5.1. Transmitophagy in the CNS
It is generally accepted that healthy cells degrade their own mitochondria. But recently, recycling of these organelles by other cells in a process called transcellular degradation of mitochondria or transmitophagy has been reported. The transmitophagy process is mediated by the cellular evulsions containing mitochondria from neurons, in which these structures are taken up by astrocytes and then recycled (Davis et al., 2014). The reason for transmitophagy is not known, but it has been hypothesized that focal axon damage stimulates the process. Another theory posits that transporting the damaged mitochondria back to the neuronal soma is energy demanding and thus the CNS makes use of astrocytes to perform the quality control process of removing the unfunctional mitochondria (Davis et al., 2014). It was observed that the retinal ganglion cell axons of wild type (WT) mice shed mitochondria at the optic nerve head (ONH). Based on the frequency of axonal protrusions and evulsions, within the ONH of adult WT mice, it was observed that most axons contain at least one such structure filled with mitochondria at various stages of degeneration. Further, considering the number of mitochondria per protrusion/evulsion, ~1 million retinal ganglion cell mitochondria were estimated to be degraded at the ONH of mice at any one time. But this study left several questions unanswered: (i) it is unknown whether the axonal mitochondria removed at the ONH are only resident mitochondria within axons at this location or whether other axonal mitochondria fated to undergo degradation are actively transported to the ONH for disposal; (ii) why axonal mitochondria are degraded selectively at the ONH. But morphologically similar structures filled with degrading mitochondria were also found in the cortex at a 10-fold lower density, suggesting transmitophagy of axonal mitochondria by astrocytes is a widespread phenomenon in the CNS (Davis et al., 2014).
Additionally, astrocytes were shown to phagocytose whole synapses elsewhere in the visual pathway (Chung et al., 2013). Because synapses usually contain mitochondria, it is quite possible that the molecular machinery used by astrocytes in the phagocytosis of synapses may also be used in the phagocytosis of axonal mitochondrial evulsions in the ONH and elsewhere. Although this description of a transmitophagy process is new for the nervous system, transcellular degradation of mitochondria is also seen upon fertilization. Because mitochondrial genome is believed to be maternally inherited, immediately after fertilization sperm-derived paternal mitochondria trigger localized autophagy and active lysosomal degradation in the oocyte cytoplasm (Sato and Sato, 2011). Similarly, autophagy-assisted phagocytosis is also involved in the turnover of photoreceptor outer segments by retinal pigment epithelial cells (Kim et al., 2013), and thus may be a common mechanism used in the nervous system.
5.2. Transfer of healthy mitochondria to stressed neurons
Mitochondria comprise the intracellular cores for energetics and viability, but under some conditions mitochondria might also be released into the extracellular space and taken up by recipient cells (Falchi et al., 2013). The ability to exchange mitochondria represents a tangible mode of cell-to-cell signaling in the CNS. Considering the new concept of intercellular organelle transfer, in 2016, Hayakawa et al., (Hayakawa et al., 2016) showed CD38 signaling mediated release of functional mitochondria from activated astrocytes. The released mitochondria were then taken up by damaged cortical neurons where ATP levels were restored after oxygen-glucose deprivation/reperfusion (OGD/R) injury. Additionally, treatment with extracellular mitochondria containing particles, released from cultured astrocytes in a mouse model of focal cerebral ischemia provided neuroprotection (Hayakawa et al., 2016). Thus, in vitro astrocyte-to-neuron mitochondrial delivery and in vivo astrocyte-derived mitochondrial transfer promoted neuronal survival, plasticity, as well as improved energetic capacity and neurological functioning. Suppression of CD38 signaling by short interfering RNA reduced extracellular mitochondria transfer and worsened neurological outcomes. These findings suggest a new mechanism of neuro-glial crosstalk that may contribute to endogenous neuroprotective and neurorecovery mechanisms after stroke (Hayakawa et al., 2016).
If astrocyte-derived mitochondria can be shown to be inside damaged neurons, it is possible that bioenergetic support to the damaged neuron could be provided. Mitochondria could serve as a direct therapeutic agent in numerous metabolic degenerative diseases. By simply increasing the number of functional mitochondria with intact motility in injured neurons may provide better regenerative capacity both in the peripheral and central nervous systems. However, mechanistic explanations of how these mitochondria are released into the cytoplasm or used to provide bioenergetic and metabolic support for damaged neurons needs to be addressed. One possibility is that the mitochondria are independent from receptor-mediated or protein coupled signaling to induce their effects. Cytokines, miRNA, transcription factors and other cell effectors usually require the activation of specific signaling pathways to induce a response in the target cell (Godlewski et al., 2015). We can speculate that the exogenous mitochondria once inside the cell can begin functioning and possibly fuse with resident mitochondria to build a pool of intact mitochondria to sustain bioenergetic demands during recovery. It is possible that transfer of replication competent functional mitochondria into recipient cells with damaged mitochondria in vivo requires membrane tunneling nanotube connections or intercellular bridges with cytoskeletal structures to direct movement, as observed in cell culture systems (Rustom et al., 2004). On the other hand, transfer of mitochondria via discrete membrane-bound particles is likely to result in transcellular mitochondrial degradation or recycling.
6. Mitochondrial transplantation: a new paradigm of therapeutic strategy
Without doubt, mitochondria are the master organelle of cellular energetics, fueling multiple process like proliferation, migration, differentiation and stress resistance. Thus, the concept of “mitochondrial medicine”, medical intervention targeting mitochondria, launches a new line of biomedical exploration. So far, more than 400 clinical trials for mitochondrial-targeted medical intervention including completed trials have been registered at ClinicalTrials.gov. Mitochondrial therapy aims to restore mitochondrial functions, however there is currently no treatment to cure mitochondria-related diseases caused by bioenergetic crisis or by accumulation of mitochondrial ROS. Therefore, a new paradigm of mitochondrial therapy based on either direct or systemic delivery of mitochondria from autologous sources was established (Birsa et al., 2013; Gollihue et al., 2018a). Such supplementation of exogenous healthy mitochondria to replace damaged mitochondria improved cellular bioenergetics, reversed excessive ROS production, and restored mitochondrial function (Gollihue and Rabchevsky, 2017; Liu et al., 2014a). In fact, growing body of evidence demonstrate promising outcome for mitochondrial transplantation strategies under many experimental conditions such as cardiac ischemia, ischemia reperfusion of liver, and neurodegenerative models (Nakamura et al., 2020).
Notably in a pioneering clinical study, mitochondrial transplantation was performed on five pediatric patients dependent upon extracorporeal membrane oxygenation (ECMO) support due to myocardial dysfunction related to ischemia and reperfusion (IR) (Emani et al., 2017). Autologous mitochondria isolated from the abdominal muscles were injected intracardially and all patients showed marked improvement in their myocardial function. Additionally, in a recent safety and efficacy study by the same group of researchers reported that intracorononary injection of mitochondria resulted in rapid uptake and specific biodistribution of exogenous mitochondria throughout the healthy swine heart. They also showed the mitochondrial administration to be safe with no effect on coronary patency. Moreover, they demonstrated enhanced myocardial function following IR injury (Shin et al., 2019).
However, a view point article raised several concerns regarding their findings (Bertero et al., 2018) questioning: (i) how mitochondria could survive the extracellular environment with high Ca2+, (ii) whether the mitochondria remain viable and efficiently produce ATP, and (iii) did the mitochondria pass through cell membranes to produce sufficient ATP to boost cardiac contractile function. In direct response, the authors (McCully et al., 2020) affirmed (i) the presence of cell-free mitochondria in the blood containing physiological Ca2+ and Na+ concentrations (Al Amir Dache et al., 2020), (ii) several studies have demonstrated functional integration of exogenous mitochondria into recipient cells in media containing 1.8 mM Ca2+, representing physiological concentration (Cowan et al., 2017; Kesner et al., 2016; Pacak et al., 2015), (iii) mitochondria transplanted by either direct or intracoronary injection increased total tissue ATP content, oxygen uptake, contractile function, and upregulated proteins associated with mitochondrial function (Masuzawa et al., 2013; Shin et al., 2019), and (iv) transplanted mitochondria produced no systemic inflammatory responses (Emani et al., 2017; Masuzawa et al., 2013; Ramirez-Barbieri et al., 2019; Shin et al., 2019). As the first successful clinical trial for mitochondrial transplantation and based on similar cell death pathways as cardiac IR injury, it is logical to foster the emerging concept of mitochondrial transplantation after CNS injury to support oxidative phosphorylation in recipient cells (Gollihue et al., 2018b; Hayakawa et al., 2018a; Hayakawa et al., 2016).
6.1. Therapeutic use of exogenous mitochondria for CNS injury or disease.
6.1.1. Parkinson’s disease.
Mitochondrial dysfunction aggravates the progression of PD, manifested by increased oxidative stress dysregulated bioenergetic homeostasis, and reduced viability of affected SN dopaminergic neurons. Though mitochondria targeting antioxidants proved to have great potential and promising outcomes in animal models and preclinical tests (Jin et al., 2014; Park et al., 2018), existing agents have limited effect in preventing neuronal deterioration. In this regard, instead of targeting a specific aspect of mitochondrial function, supplementing exogenous mitochondria to damaged regions may potentially be an innovative therapeutic strategy. Chang et al. demonstrated that cell-penetrating peptide-based (Pep-1) mitochondrial delivery in 6-hydroxydopamine (OHDA)-treated PC12 cells rescued mitochondrial respiratory function, improved cell viability and promoted neurite growth in the presence of nerve growth factor (Chang et al., 2016). Additionally, xenogenic/allogenic injection of mitochondria into medical forebrain bundle (MFB) of 6-OHDA-unilaterally infused PD rats enhanced the survival of dopaminergic neurons and effectively sustained mitochondrial functions by restoring normal levels of mitochondrial complex I–IV and relieving mitochondrial oxidative stress in vivo (Chang et al., 2016). In another study by Shi et al., MPP (1-methyl-4-phenyl-pyridinium)-treated SH-SY5Y neuroblastoma neuron-like cells incubated with intact mitochondria increased cell viability in a dose-dependent manner (Shi et al., 2017). ATP production, mitochondrial complex I activity and cell survival were rescued after mitochondrial supplementation, while levels of ROS significantly decreased. Also, the initial report by Shi et al., showed that systemic intravenous mitochondrial administration to the MPTP (1-methyl-4-phenyl-1,2,3,6-tertrahydropyridine)-induced PD mouse model prevented disease progression (Shi et al., 2017). In vivo distribution of intravenously-injected mitochondria was found in multiple organs, including brain within 2h of injection.
6.1.2. Ischemic stroke.
Current intervention for stroke is very limited due to narrow therapeutic time window after the occurrence of ischemic stroke. After ischemic stroke, lack of glucose and oxygen supply disturbs ATP synthesis in mitochondria, results in energy imbalances, dysregulation of cellular homeostasis and finally neuronal cell death. Therefore targeting mitochondria is a promising approach for neuroprotection after stroke (Russo et al., 2018). In light of intercellular mitochondrial transfer in stroke, exogenously injected mitochondria was observed to be distributed in neuron, astrocytes, and microglia in the peri-infarct area at 4 weeks after transient focal ischemia. Concomitantly, mitochondrial transplantation significantly improved motor function and decreased the infarct area, and TUNEL-positive cells (Huang et al., 2016). More recently, mitochondria allografted from skeletal muscle decreased the infarct volume, and attenuated neurological deficits, cellular oxidative stress, and apoptosis at 28 days after stroke in a rat model of focal ischemia (Zhang et al., 2019).
Studies implicate that stem cells may have the ability to transfer mitochondria into injured cells through tunneling nanotubes, microvesicles, gap junctions, cell fusion or direct uptake (Heyck et al., 2019; Liu et al., 2018; Paliwal et al., 2018) and stem cell mediated mitochondrial transfer may be a key mechanism for neuroprotection and neurorepair. It has been reported that mitochondria are transferred from mesenchymal multipotent stromal cells to co-cultured neurons (Babenko et al., 2015). In this context, studies showed that intravenous injection of mesenchymal multipotent stromal cells to middle cerebral artery occlusion (MCAO) rats reduced the infarction area and improved post-stroke neurological indexes. Treatment of primed stem cells which had been previously co-cultured with neurons caused a more pronounced beneficial outcome in rats after stroke (Babenko et al., 2015). Very recently, Liu and colleagues demonstrated the transfer of mitochondria from transplanted mesenchymal stem cells (MSCs) to damaged brain endothelial cells after transient focal cerebral ischemia in rats (Liu et al., 2019).
6.1.3. Traumatic brain injury and spinal cord injury.
Traumatic brain injury (TBI) and spinal cord injury (SCI) have been some of the most important medical issues worldwide given the lack of effective treatment. The initial study investigating the feasibility of mitochondrial transplantation in SCI reported that supplementation of a pool of healthy mitochondria into L1/L2 contusion SCI rat model acutely sustained cellular bioenergetics in the injured spinal cord and improved locomotor activity, whereas, long-term effects on neuroprotection and tissue sparing were not observed (Gollihue et al., 2018a). However, transplanted mitochondria were found to distributed and localized within various cell types including microglia/brain macrophages, endothelial cells, pericytes, astrocytes, and oligodendrocytes and no evidence of uptake in neurons (Gollihue et al., 2018a). In a TBI rat model, supplement of freshly isolated mitochondria derived from rat cortical neurons into injured hippocampal neurons promoted neurite re-growth and restored membrane potential of the injured neurons (Chien et al., 2018).
6.1.4. Schizophrenia.
Effectiveness of mitochondrial transplantation for schizophrenia has recently been described. Bilateral injection of freshly isolated mitochondria into the intra-prefrontal cortex in SZ model rats, prevented attention-deficit characterized cognitive impairment in SZ along with an improvement in mitochondrial membrane potential (Robicsek et al., 2018). However, fundamental mechanisms of how transplanted mitochondria influence brain function in a SZ rat model remain to be fully understood.
6.2. Clinical application of mitochondrial transplantation techniques.
Previous proposals for treating mitochondrial dysfunction have been targeting specific mitochondrial residents and fusion and fission regulators (El-Hattab et al., 2017; Wang et al., 2016). The outcome of these approaches has not been satisfactory and the emerging line of approach is to supplement freshly isolated mitochondria, ie., mitochondrial transplantation to injury sites. A number of in vivo studies documented feasible approaches of mitochondrial transplantation, including microinjection directly into affected sites in SCI, stroke and PD models (Chang et al., 2016; Gollihue et al., 2018a; Kaza et al., 2017; Masuzawa et al., 2013), and intravenous administration in PD and fatty liver models (Fu et al., 2017; Shi et al., 2017) (Table 1). Nonetheless, it is conceivable that clinical translation of mitochondrial transplantation would face great challenge. The effectiveness of mitochondrial therapy is expected to be variable among patients due to the heterogeneity of pathogenesis and efficiency of mitochondrial internalization into affected tissues. Also, the therapeutic outcome of mitochondrial transplantation largely depends upon the isolation protocol, numbers and quality of isolated mitochondria, the proper route of organelle delivery and tissue-specific differential uptake. The major pros and cons that has to be addressed before the implementation of mitochondrial transplantation in clinical settings are:
Table 1:
Exogenous mitochondria transplantation in CNS disease and injury
| No | Disease/injury model | Source of mitochondria | Recipient cells | Mode/Route of delivery | Outcome | Reference |
|---|---|---|---|---|---|---|
| 1 | In vitro ischemia | Mesenchymal multipotent stromal cells | Cortical neurons and astrocytes | Co-culture | Improved cell viability | (Babenko et al., 2015) |
| 2 | Traumatic brain injury (TBI mouse) | Cortical neurons | Hippocampal neurons | Add in medium | Enhanced neuroregeneration | (Chien et al., 2018) |
| 3 | Transient focal cerebral ischemia (mouse) | Mouse cortical astrocytes | Peri-infarct cortex | Direction injection or autologous secretion | Promoted adjacent neuronal survival and plasticity after injury transfer | (Hayakawa et al., 2016) |
| 4 | Parkinson’s disease (PD rats) | PC12: human osteosarcoma cybrids | Brain neurons | Intracerebroventricular | Improved motor function and attenuated the deterioration of dopaminergic neurons | (Chang et al., 2016) |
| 5 | Parkinson’s disease (PD mouse) | HepG2 | Multiple tissues including brain | Intravenous injection | Improved behavior test, increased ETC activity, ATP, decreased ROS, apoptosis and necrosis | (Shi et al., 2017) |
| 6 | Spinal cord injury L1/L2 contusion (SCI rats) | PC12/rat skeletal muscle (allograft) | Brain macrophages, pericytes, endothelial cells, glia | Microinjection at mediolateral grey matter | Maintenance of acute mitochondrial bioenergetics, and enhanced behavioral recovery | (Gollihue et al., 2018a) |
| 7 | Schizophrenia (SZ rats) | Human lymphocyte/rat brain (heterograft/allograft) | Prefrontal cortex | Direct injection | Prevented dissipation of mitochondrial membrane potential and attentional deficit | (Robicsek et al., 2018) |
| 8 | Ischemic Reperfusion brain injury (IR rats) | BHK cells | Neuron, astrocytes, microglia in peri-infarct area of the ischemic hemisphere | Intrafemoral artery injection | Improve motor function, decrease infarct area and cell death | (Huang et al., 2016) |
| 9 | Ischemic Reperfusion brain injury (IR rats) | Pectoralis major Muscle (autologous) | Neurons around the ischemic penumbra | Intracerebroventricular | Decrease infarct volume, neurological deficits, cellular oxidative stress, apoptosis, and gliosis, promote neurogenesis | (Zhang et al., 2019) |
| 10 | Ischemic Reperfusion brain injury (IR rats) | Mesenchymal stromal cells | Per-infarct area | Intra-arterial | Improve mitochondrial function in peri- infarct area and functional recovery | (Liu et al., 2019) |
6.2.1. Source and quality of mitochondrial isolation.
Functional homogeneity of isolated mitochondria is critical for achieving therapeutic efficiency. By comparing the mitochondria enriched fractions from various tissues for mitochondrial number, purity, and membrane potential, it was identified that muscle-derived mitochondria may be a good candidate for isolation and transplantation. In protein adjusted mitochondrial suspension, muscle had the highest mitochondrial number and the MitoTracker/JC1 values (Zhang et al., 2019). Future studies are warranted to define mitochondrial dosages with a common unit ie., number of mitochondria or protein concentration. Additionally, MitoTracker dependent mitochondrial labeling has been commonly used to assess mitochondrial quality and number. But membrane-potential dependent labeling with MitoTracker dyes may cause background signals and non-specific detections when used at higher concentrations. So, mitochondrial labeling with fluorescent proteins can be an alternative approach. Molecular probes that drive expression of fluorescent proteins in cell-specific mitochondria may be useful to quantify the mitochondrial dose to be used for transplantation.
6.2.2. Storage of isolated mitochondria.
Mitochondrial storage is one of the key aspect of clinical translation. It is known that the function of isolated mitochondria could be influenced by freeze-thaw cycles and also by storage temperature (Araki, 1977). Thus, freshly isolated mitochondria are the best option for mitochondrial transplantation (Roushandeh et al., 2019). The isolation and preparation of autogenic mitochondria is rapid and purified mitochondria are viable within 30 min, a time frame within the clinical time-frame for use in surgery (Kaza et al., 2017; McCully et al., 2016). But is it impossible to store mitochondria? Back in 2006, it was shown that ~50% of normal respiratory function was maintained in brain-derived mitochondria that was cryopreserved in 10% (v/v) dimethylsulfoxide at −80°C (Nukala et al., 2006). Another study showed trehalose-frozen mitochondria to preserve mitochondrial ultrastructure along with retaining ability to produce ATP and to import proteins (Yamaguchi et al., 2007). If we can freeze and thaw isolated mitochondria without functional disruption, mitochondrial transplantation therapy will be potentially applicable at the patient bedside. Further studies are warranted to investigate this idea and assess the feasibility of storing mitochondria until transplantation.
6.2.3. Cellular uptake of transplanted mitochondria.
We do not yet understand how transplanted mitochondria maintain oxygen consumption rate (OCR), rescue impaired mitochondrial function, attenuate oxidative damage, and sustain neuronal survival. It is also not known whether mitochondria must be internalized into cells in order to improve the disease condition or whether they can function as extracellular mitochondrial vesicles/particles (Hayakawa et al., 2018a). If mitochondria are maintained extracellular, it is unlikely that mitochondria can survive in high extracellular Ca2+ milieu. In that case, to maintain the mitochondrial integrity, Acetyl-l-carnitine (ALC)/ N-acetylcysteine amide (NACA) can be co-administered to maintain both host and grafted mitochondria. But if mitochondria are internalized, delivering mitochondria within specialized hydrogels that control mitochondrial release or understanding the mechanisms by which mitochondria enter cells will be intriguing to determine optimal therapeutic time windows for mitochondrial transplantation after injury.
More importantly, in CNS injury, in addition to maintenance of cell survival, regenerative outcome characterized by neurite re-growth, de novo synaptogenesis and the restoration of neuronal activity should be inclusively evaluated. Therefore, increased understanding of the mitochondrial quality, dose, mechanisms underlying mitochondrial delivery and cellular uptake will facilitate the translation of mitochondrial transplantation in clinic.
6.3. Clinical trials on mitochondrial transplantation.
The burgeoning of mitochondrial therapy opened a new era of reversing mitochondria function in human diseases. Thus far registered clinical trials for treating diseases based on mitochondrial delivery technique have been launched. Till date there is only one completed clinical trial which aimed to treat infertility by autologous mitochondrial injection into oocytes (NCT #02586298). An ongoing trial trues to demonstrate the feasibility of mitochondrial transplantation, using autologous mitochondrial injection (NCT #02851758), for rehabilitating myocardial ischemia/reperfusion injury and is currently recruiting participants. The usefulness of mitochondria within translational medicine can not only be exemplified by artificial mitochondrial transplantation therapy, but can also be evidenced in other areas such as the use of the mitochondria or the mtDNA as a biomarker of the severity of the disease. In a study, the quantity and activity of extracellular mitochondria in the cerebrospinal fluid (CSF) was measured by the fluorescent probe JC1, which showed to be an effective prognostic factor after subarachnoid hemorrhage (SAH) in mice and patients (Chou et al., 2017). By measuring the red and green fluorescence it was determined that SAH patients with better prognosis at 3 months had more active mitochondria (red/green) in the CSF. Additionally, the use of MitoTracker Red CMXRos defined the origin of the mitochondrial flow showing that patients with the best prognoses had mitochondria of astrocytic origin when compared with those of microglial, endothelial or platelet origin (Chou et al., 2017).
7. Concluding Remarks
Considering the complex impact of mitochondrial motility within and between cells, it is important to emphasize that mitochondria are highly dynamic organelles especially in neurons. They travel the length of the axons from the cell body to supply energy at distance processes and travel back to the cell body. Motile mitochondria target to the place of increased energy demand, provide energy and then re-mobilize. This pattern of mitochondrial motility (anterograde and retrograde) was considered the classical mechanism of mitochondrial movement. But recently, transcellular transfer of mitochondria has been identified as a new mechanism for mitochondrial movement. In this case, mitochondria are transferred to supply energy to the recipient cells or to undergo transmitophagy. The type of mitochondrial transfer, aimed to rescue the recipient cellular functions needs to be considered with caution, since whether transferred mitochondria remain functional remains a point of contention. Thus, while donor cells can transfer and establish functional mitochondria into recipient cells with damaged mitochondrial DNA, deliberation will continue over the mode of delivery, the cellular source, and the mechanism(s) of transfer and cellular internalization of these mitochondria (Berridge et al., 2016). Since mitochondria originated from bacteria, the mitochondrial DNA and their components are highly immunogenic and care must be taken to determine the nature and reactivity of the mitochondria that is being used for mitochondrial transfer/transplant. Depending on the nature of mitochondria, inter- and intracellular signals vary, resulting in either survival or death of the recipient cells. Because of the prominent role of mitochondria in both neuronal physiology and pathology, priority should be to determine whether the transcellular transfer of mitochondria contributes to health or disease in various research and clinical settings.
Highlights.
This review highlights recent advances and provided comprehensive summary on the pathophysiological implications of inter and intracellular mitochondrial transfer.
Different regulatory mechanisms and the molecular machinery involved in intracellular mitochondrial movement are discussed.
Several promising results on artificial mitochondrial supplementation therapy are reviewed.
A new paradigm for mitochondrial therapy based on organelle delivery as a strategy to treat neurological disorders is described.
Acknowledgements
This work was supported by NIH R01 MH107340, NIH CNAC pilot grant, and NIH K99/R00 4R00HL138268-03.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of Interest
No potential conflict of interest was reported by the authors
References
- Abounit S, Zurzolo C, 2012. Wiring through tunneling nanotubes--from electrical signals to organelle transfer. J Cell Sci 125, 1089–1098. [DOI] [PubMed] [Google Scholar]
- Ahmad T, Mukherjee S, Pattnaik B, Kumar M, Singh S, Kumar M, Rehman R, Tiwari BK, Jha KA, Barhanpurkar AP, Wani MR, Roy SS, Mabalirajan U, Ghosh B, Agrawal A, 2014. Miro1 regulates intercellular mitochondrial transport & enhances mesenchymal stem cell rescue efficacy. EMBO J 33, 994–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al Amir Dache Z, Otandault A, Tanos R, Pastor B, Meddeb R, Sanchez C, Arena G, Lasorsa L, Bennett A, Grange T, El Messaoudi S, Mazard T, Prevostel C, Thierry AR, 2020. Blood contains circulating cell-free respiratory competent mitochondria. FASEB J 34, 3616–3630. [DOI] [PubMed] [Google Scholar]
- Araki T, 1977. Freezing injury in mitochondrial membranes. II. Degradation of phospholipid in rabbit liver mitochondria during freezing and storage at low temperatures. Cryobiology 14, 151–159. [DOI] [PubMed] [Google Scholar]
- Aspenstrom P, Ruusala A, Pacholsky D, 2007. Taking Rho GTPases to the next level: the cellular functions of atypical Rho GTPases. Exp Cell Res 313, 3673–3679. [DOI] [PubMed] [Google Scholar]
- Attwell D, Buchan AM, Charpak S, Lauritzen M, Macvicar BA, Newman EA, 2010. Glial and neuronal control of brain blood flow. Nature 468, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babenko VA, Silachev DN, Zorova LD, Pevzner IB, Khutornenko AA, Plotnikov EY, Sukhikh GT, Zorov DB, 2015. Improving the Post-Stroke Therapeutic Potency of Mesenchymal Multipotent Stromal Cells by Cocultivation With Cortical Neurons: The Role of Crosstalk Between Cells. Stem Cells Transl Med 4, 1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK, 2011. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476, 341–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck M, Brickley K, Wilkinson HL, Sharma S, Smith M, Chazot PL, Pollard S, Stephenson FA, 2002. Identification, molecular cloning, and characterization of a novel GABAA receptor-associated protein, GRIF-1. J Biol Chem 277, 30079–30090. [DOI] [PubMed] [Google Scholar]
- Berridge MV, Neuzil J, 2017. The mobility of mitochondria: Intercellular trafficking in health and disease. Clin Exp Pharmacol Physiol 44 Suppl 1, 15–20. [DOI] [PubMed] [Google Scholar]
- Berridge MV, Schneider RT, McConnell MJ, 2016. Mitochondrial Transfer from Astrocytes to Neurons following Ischemic Insult: Guilt by Association? Cell Metab 24, 376–378. [DOI] [PubMed] [Google Scholar]
- Bertero E, Maack C, O’Rourke B, 2018. Mitochondrial transplantation in humans: “magical” cure or cause for concern? J Clin Invest 128, 5191–5194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsa N, Norkett R, Higgs N, Lopez-Domenech G, Kittler JT, 2013. Mitochondrial trafficking in neurons and the role of the Miro family of GTPase proteins. Biochem Soc Trans 41, 1525–1531. [DOI] [PubMed] [Google Scholar]
- Bock FJ, Tait SWG, 2020. Mitochondria as multifaceted regulators of cell death. Nat Rev Mol Cell Biol 21, 85–100. [DOI] [PubMed] [Google Scholar]
- Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, Pare A, Rousseau M, Naika GS, Levesque T, Laflamme C, Marcoux G, Lambeau G, Farndale RW, Pouliot M, Hamzeh-Cognasse H, Cognasse F, Garraud O, Nigrovic PA, Guderley H, Lacroix S, Thibault L, Semple JW, Gelb MH, Boilard E, 2014. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase A2 to promote inflammation. Blood 124, 2173–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bozi LH, Bechara LR, Dos Santos AF, Campos JC, 2016. Mitochondrial-derived vesicles: a new player in cardiac mitochondrial quality control. J Physiol 594, 6077–6078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bragoszewski P, Turek M, Chacinska A, 2017. Control of mitochondrial biogenesis and function by the ubiquitin-proteasome system. Open Biol 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braschi E, Goyon V, Zunino R, Mohanty A, Xu L, McBride HM, 2010. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr Biol 20, 1310–1315. [DOI] [PubMed] [Google Scholar]
- Brickley K, Smith MJ, Beck M, Stephenson FA, 2005. GRIF-1 and OIP106, members of a novel gene family of coiled-coil domain proteins: association in vivo and in vitro with kinesin. J Biol Chem 280, 14723–14732. [DOI] [PubMed] [Google Scholar]
- Brickley K, Stephenson FA, 2011. Trafficking kinesin protein (TRAK)-mediated transport of mitochondria in axons of hippocampal neurons. J Biol Chem 286, 18079–18092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukoreshtliev NV, Wang X, Hodneland E, Gurke S, Barroso JF, Gerdes HH, 2009. Selective block of tunneling nanotube (TNT) formation inhibits intercellular organelle transfer between PC12 cells. FEBS Lett 583, 1481–1488. [DOI] [PubMed] [Google Scholar]
- Cadete VJ, Deschenes S, Cuillerier A, Brisebois F, Sugiura A, Vincent A, Turnbull D, Picard M, McBride HM, Burelle Y, 2016. Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J Physiol 594, 5343–5362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Gerwin C, Sheng ZH, 2005. Syntabulin-mediated anterograde transport of mitochondria along neuronal processes. J Cell Biol 170, 959–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caicedo A, Fritz V, Brondello JM, Ayala M, Dennemont I, Abdellaoui N, de Fraipont F, Moisan A, Prouteau CA, Boukhaddaoui H, Jorgensen C, Vignais ML, 2015. MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci Rep 5, 9073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caielli S, Athale S, Domic B, Murat E, Chandra M, Banchereau R, Baisch J, Phelps K, Clayton S, Gong M, Wright T, Punaro M, Palucka K, Guiducci C, Banchereau J, Pascual V, 2016. Oxidized mitochondrial nucleoids released by neutrophils drive type I interferon production in human lupus. J Exp Med 213, 697–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC, 2011. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20, 1726–1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang CY, Liang MZ, Chen L, 2019. Current progress of mitochondrial transplantation that promotes neuronal regeneration. Transl Neurodegener 8, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, Honick AS, Reynolds IJ, 2006. Mitochondrial trafficking to synapses in cultured primary cortical neurons. J Neurosci 26, 7035–7045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang DT, Reynolds IJ, 2006. Differences in mitochondrial movement and morphology in young and mature primary cortical neurons in culture. Neuroscience 141, 727–736. [DOI] [PubMed] [Google Scholar]
- Chang JC, Wu SL, Liu KH, Chen YH, Chuang CS, Cheng FC, Su HL, Wei YH, Kuo SJ, Liu CS, 2016. Allogeneic/xenogeneic transplantation of peptide-labeled mitochondria in Parkinson’s disease: restoration of mitochondria functions and attenuation of 6-hydroxydopamine-induced neurotoxicity. Transl Res 170, 40–56 e43. [DOI] [PubMed] [Google Scholar]
- Chen Y, Sheng ZH, 2013. Kinesin-1-syntaphilin coupling mediates activity-dependent regulation of axonal mitochondrial transport. J Cell Biol 202, 351–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen YM, Gerwin C, Sheng ZH, 2009. Dynein light chain LC8 regulates syntaphilin-mediated mitochondrial docking in axons. J Neurosci 29, 9429–9438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chien L, Liang MZ, Chang CY, Wang C, Chen L, 2018. Mitochondrial therapy promotes regeneration of injured hippocampal neurons. Biochim Biophys Acta Mol Basis Dis 1864, 3001–3012. [DOI] [PubMed] [Google Scholar]
- Chinnery HR, Pearlman E, McMenamin PG, 2008. Cutting edge: Membrane nanotubes in vivo: a feature of MHC class II+ cells in the mouse cornea. J Immunol 180, 5779–5783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou SH, Lan J, Esposito E, Ning M, Balaj L, Ji X, Lo EH, Hayakawa K, 2017. Extracellular Mitochondria in Cerebrospinal Fluid and Neurological Recovery After Subarachnoid Hemorrhage. Stroke 48, 2231–2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WS, Clarke LE, Wang GX, Stafford BK, Sher A, Chakraborty C, Joung J, Foo LC, Thompson A, Chen C, Smith SJ, Barres BA, 2013. Astrocytes mediate synapse elimination through MEGF10 and MERTK pathways. Nature 504, 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan DB, Yao R, Thedsanamoorthy JK, Zurakowski D, Del Nido PJ, McCully JD, 2017. Transit and integration of extracellular mitochondria in human heart cells. Sci Rep 7, 17450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Csordas G, Weaver D, Hajnoczky G, 2018. Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol 28, 523–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai Y, Zheng K, Clark J, Swerdlow RH, Pulst SM, Sutton JP, Shinobu LA, Simon DK, 2014. Rapamycin drives selection against a pathogenic heteroplasmic mitochondrial DNA mutation. Hum Mol Genet 23, 637–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CH, Kim KY, Bushong EA, Mills EA, Boassa D, Shih T, Kinebuchi M, Phan S, Zhou Y, Bihlmeyer NA, Nguyen JV, Jin Y, Ellisman MH, Marsh-Armstrong N, 2014. Transcellular degradation of axonal mitochondria. Proc Natl Acad Sci U S A 111, 9633–9638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R, 2011. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 476, 336–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demine S, Renard P, Arnould T, 2019. Mitochondrial Uncoupling: A Key Controller of Biological Processes in Physiology and Diseases. Cells 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Vegas A, Sanchez-Aguilera P, Krycer JR, Morales PE, Monsalves-Alvarez M, Cifuentes M, Rothermel BA, Lavandero S, 2020. Is mitochondrial dysfunction a common root of noncommunicable chronic diseases? Endocr Rev. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Hattab AW, Zarante AM, Almannai M, Scaglia F, 2017. Therapies for mitochondrial diseases and current clinical trials. Mol Genet Metab 122, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emani SM, Piekarski BL, Harrild D, Del Nido PJ, McCully JD, 2017. Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J Thorac Cardiovasc Surg 154, 286–289. [DOI] [PubMed] [Google Scholar]
- Evans WH, Martin PE, 2002. Gap junctions: structure and function (Review). Mol Membr Biol 19, 121–136. [DOI] [PubMed] [Google Scholar]
- Falchi AM, Sogos V, Saba F, Piras M, Congiu T, Piludu M, 2013. Astrocytes shed large membrane vesicles that contain mitochondria, lipid droplets and ATP. Histochem Cell Biol 139, 221–231. [DOI] [PubMed] [Google Scholar]
- Fransson A, Ruusala A, Aspenstrom P, 2003. Atypical Rho GTPases have roles in mitochondrial homeostasis and apoptosis. J Biol Chem 278, 6495–6502. [DOI] [PubMed] [Google Scholar]
- Fransson S, Ruusala A, Aspenstrom P, 2006. The atypical Rho GTPases Miro-1 and Miro-2 have essential roles in mitochondrial trafficking. Biochem Biophys Res Commun 344, 500–510. [DOI] [PubMed] [Google Scholar]
- Friedman JR, Nunnari J, 2014. Mitochondrial form and function. Nature 505, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu A, Shi X, Zhang H, Fu B, 2017. Mitotherapy for Fatty Liver by Intravenous Administration of Exogenous Mitochondria in Male Mice. Front Pharmacol 8, 241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, Alnemri ES, Altucci L, Amelio I, Andrews DW, Annicchiarico-Petruzzelli M, Antonov AV, Arama E, Baehrecke EH, Barlev NA, Bazan NG, Bernassola F, Bertrand MJM, Bianchi K, Blagosklonny MV, Blomgren K, Borner C, Boya P, Brenner C, Campanella M, Candi E, Carmona-Gutierrez D, Cecconi F, Chan FK, Chandel NS, Cheng EH, Chipuk JE, Cidlowski JA, Ciechanover A, Cohen GM, Conrad M, Cubillos-Ruiz JR, Czabotar PE, D’Angiolella V, Dawson TM, Dawson VL, De Laurenzi V, De Maria R, Debatin KM, DeBerardinis RJ, Deshmukh M, Di Daniele N, Di Virgilio F, Dixit VM, Dixon SJ, Duckett CS, Dynlacht BD, El-Deiry WS, Elrod JW, Fimia GM, Fulda S, Garcia-Saez AJ, Garg AD, Garrido C, Gavathiotis E, Golstein P, Gottlieb E, Green DR, Greene LA, Gronemeyer H, Gross A, Hajnoczky G, Hardwick JM, Harris IS, Hengartner MO, Hetz C, Ichijo H, Jaattela M, Joseph B, Jost PJ, Juin PP, Kaiser WJ, Karin M, Kaufmann T, Kepp O, Kimchi A, Kitsis RN, Klionsky DJ, Knight RA, Kumar S, Lee SW, Lemasters JJ, Levine B, Linkermann A, Lipton SA, Lockshin RA, Lopez-Otin C, Lowe SW, Luedde T, Lugli E, MacFarlane M, Madeo F, Malewicz M, Malorni W, Manic G, Marine JC, Martin SJ, Martinou JC, Medema JP, Mehlen P, Meier P, Melino S, Miao EA, Molkentin JD, Moll UM, Munoz-Pinedo C, Nagata S, Nunez G, Oberst A, Oren M, Overholtzer M, Pagano M, Panaretakis T, Pasparakis M, Penninger JM, Pereira DM, Pervaiz S, Peter ME, Piacentini M, Pinton P, Prehn JHM, Puthalakath H, Rabinovich GA, Rehm M, Rizzuto R, Rodrigues CMP, Rubinsztein DC, Rudel T, Ryan KM, Sayan E, Scorrano L, Shao F, Shi Y, Silke J, Simon HU, Sistigu A, Stockwell BR, Strasser A, Szabadkai G, Tait SWG, Tang D, Tavernarakis N, Thorburn A, Tsujimoto Y, Turk B, Vanden Berghe T, Vandenabeele P, Vander Heiden MG, Villunger A, Virgin HW, Vousden KH, Vucic D, Wagner EF, Walczak H, Wallach D, Wang Y, Wells JA, Wood W, Yuan J, Zakeri Z, Zhivotovsky B, Zitvogel L, Melino G, Kroemer G, 2018. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 25, 486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerdes HH, Bukoreshtliev NV, Barroso JF, 2007. Tunneling nanotubes: a new route for the exchange of components between animal cells. FEBS Lett 581, 2194–2201. [DOI] [PubMed] [Google Scholar]
- Glancy B, Hartnell LM, Combs CA, Femnou A, Sun J, Murphy E, Subramaniam S, Balaban RS, 2017. Power Grid Protection of the Muscle Mitochondrial Reticulum. Cell Rep 19, 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glater EE, Megeath LJ, Stowers RS, Schwarz TL, 2006. Axonal transport of mitochondria requires milton to recruit kinesin heavy chain and is light chain independent. J Cell Biol 173, 545–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godlewski J, Krichevsky AM, Johnson MD, Chiocca EA, Bronisz A, 2015. Belonging to a network--microRNAs, extracellular vesicles, and the glioblastoma microenvironment. Neuro Oncol 17, 652–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollihue JL, Patel SP, Eldahan KC, Cox DH, Donahue RR, Taylor BK, Sullivan PG, Rabchevsky AG, 2018a. Effects of Mitochondrial Transplantation on Bioenergetics, Cellular Incorporation, and Functional Recovery after Spinal Cord Injury. J Neurotrauma 35, 1800–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollihue JL, Patel SP, Rabchevsky AG, 2018b. Mitochondrial transplantation strategies as potential therapeutics for central nervous system trauma. Neural Regen Res 13, 194–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollihue JL, Rabchevsky AG, 2017. Prospects for therapeutic mitochondrial transplantation. Mitochondrion 35, 70–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong YN, Crawford JC, Heckmann BL, Green DR, 2019. To the edge of cell death and back. FEBS J 286, 430–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorska-Andrzejak J, Stowers RS, Borycz J, Kostyleva R, Schwarz TL, Meinertzhagen IA, 2003. Mitochondria are redistributed in Drosophila photoreceptors lacking milton, a kinesin-associated protein. J Comp Neurol 463, 372–388. [DOI] [PubMed] [Google Scholar]
- Gousset K, Schiff E, Langevin C, Marijanovic Z, Caputo A, Browman DT, Chenouard N, de Chaumont F, Martino A, Enninga J, Olivo-Marin JC, Mannel D, Zurzolo C, 2009. Prions hijack tunnelling nanotubes for intercellular spread. Nat Cell Biol 11, 328–336. [DOI] [PubMed] [Google Scholar]
- Granatiero V, De Stefani D, Rizzuto R, 2017. Mitochondrial Calcium Handling in Physiology and Disease. Adv Exp Med Biol 982, 25–47. [DOI] [PubMed] [Google Scholar]
- Grishin A, Li H, Levitan ES, Zaks-Makhina E, 2006. Identification of gamma-aminobutyric acid receptor-interacting factor 1 (TRAK2) as a trafficking factor for the K+ channel Kir2.1. J Biol Chem 281, 30104–30111. [DOI] [PubMed] [Google Scholar]
- Guescini M, Genedani S, Stocchi V, Agnati LF, 2010. Astrocytes and Glioblastoma cells release exosomes carrying mtDNA. J Neural Transm (Vienna) 117, 1–4. [DOI] [PubMed] [Google Scholar]
- Guo X, Macleod GT, Wellington A, Hu F, Panchumarthi S, Schoenfield M, Marin L, Charlton MP, Atwood HL, Zinsmaier KE, 2005. The GTPase dMiro is required for axonal transport of mitochondria to Drosophila synapses. Neuron 47, 379–393. [DOI] [PubMed] [Google Scholar]
- Haghnia M, Cavalli V, Shah SB, Schimmelpfeng K, Brusch R, Yang G, Herrera C, Pilling A, Goldstein LS, 2007. Dynactin is required for coordinated bidirectional motility, but not for dynein membrane attachment. Mol Biol Cell 18, 2081–2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han H, Hu J, Yan Q, Zhu J, Zhu Z, Chen Y, Sun J, Zhang R, 2016. Bone marrow-derived mesenchymal stem cells rescue injured H9c2 cells via transferring intact mitochondria through tunneling nanotubes in an in vitro simulated ischemia/reperfusion model. Mol Med Rep 13, 1517–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris JJ, Jolivet R, Attwell D, 2012. Synaptic energy use and supply. Neuron 75, 762–777. [DOI] [PubMed] [Google Scholar]
- Hayakawa K, Bruzzese M, Chou SH, Ning M, Ji X, Lo EH, 2018a. Extracellular Mitochondria for Therapy and Diagnosis in Acute Central Nervous System Injury. JAMA Neurol 75, 119–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Chan SJ, Mandeville ET, Park JH, Bruzzese M, Montaner J, Arai K, Rosell A, Lo EH, 2018b. Protective Effects of Endothelial Progenitor Cell-Derived Extracellular Mitochondria in Brain Endothelium. Stem Cells 36, 1404–1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayakawa K, Esposito E, Wang X, Terasaki Y, Liu Y, Xing C, Ji X, Lo EH, 2016. Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hervera A, De Virgiliis F, Palmisano I, Zhou L, Tantardini E, Kong G, Hutson T, Danzi MC, Perry RB, Santos CXC, Kapustin AN, Fleck RA, Del Rio JA, Carroll T, Lemmon V, Bixby JL, Shah AM, Fainzilber M, Di Giovanni S, 2018. Reactive oxygen species regulate axonal regeneration through the release of exosomal NADPH oxidase 2 complexes into injured axons. Nat Cell Biol 20, 307–319. [DOI] [PubMed] [Google Scholar]
- Heyck M, Bonsack B, Zhang H, Sadanandan N, Cozene B, Kingsbury C, Lee JY, Borlongan CV, 2019. Highlight article: The brain and eye: Treating cerebral and retinal ischemia through mitochondrial transfer. Exp Biol Med (Maywood) 244, 1485–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Niwa S, Tanaka Y, 2010. Molecular motors in neurons: transport mechanisms and roles in brain function, development, and disease. Neuron 68, 610–638. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, 1996. The pattern and mechanism of mitochondrial transport in axons. Front Biosci 1, d91–102. [DOI] [PubMed] [Google Scholar]
- Hollenbeck PJ, 2005. Mitochondria and neurotransmission: evacuating the synapse. Neuron 47, 331–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ, Saxton WM, 2005. The axonal transport of mitochondria. J Cell Sci 118, 5411–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Nakamura Y, Lo EH, Hayakawa K, 2019. Astrocyte Signaling in the Neurovascular Unit After Central Nervous System Injury. Int J Mol Sci 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang PJ, Kuo CC, Lee HC, Shen CI, Cheng FC, Wu SF, Chang JC, Pan HC, Lin SZ, Liu CS, Su HL, 2016. Transferring Xenogenic Mitochondria Provides Neural Protection Against Ischemic Stress in Ischemic Rat Brains. Cell Transplant 25, 913–927. [DOI] [PubMed] [Google Scholar]
- Hubley MJ, Locke BR, Moerland TS, 1996. The effects of temperature, pH, and magnesium on the diffusion coefficient of ATP in solutions of physiological ionic strength. Biochim Biophys Acta 1291, 115–121. [DOI] [PubMed] [Google Scholar]
- Hurd DD, Saxton WM, 1996. Kinesin mutations cause motor neuron disease phenotypes by disrupting fast axonal transport in Drosophila. Genetics 144, 1075–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iadecola C, Nedergaard M, 2007. Glial regulation of the cerebral microvasculature. Nat Neurosci 10, 1369–1376. [DOI] [PubMed] [Google Scholar]
- Islam MN, Das SR, Emin MT, Wei M, Sun L, Westphalen K, Rowlands DJ, Quadri SK, Bhattacharya S, Bhattacharya J, 2012. Mitochondrial transfer from bone-marrow-derived stromal cells to pulmonary alveoli protects against acute lung injury. Nat Med 18, 759–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MV, Morrison TJ, Doherty DF, McAuley DF, Matthay MA, Kissenpfennig A, O’Kane CM, Krasnodembskaya AD, 2016. Mitochondrial Transfer via Tunneling Nanotubes is an Important Mechanism by Which Mesenchymal Stem Cells Enhance Macrophage Phagocytosis in the In Vitro and In Vivo Models of ARDS. Stem Cells 34, 2210–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakobs S, 2006. High resolution imaging of live mitochondria. Biochim Biophys Acta 1763, 561–575. [DOI] [PubMed] [Google Scholar]
- Jin H, Kanthasamy A, Ghosh A, Anantharam V, Kalyanaraman B, Kanthasamy AG, 2014. Mitochondria-targeted antioxidants for treatment of Parkinson’s disease: preclinical and clinical outcomes. Biochim Biophys Acta 1842, 1282–1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanai Y, Okada Y, Tanaka Y, Harada A, Terada S, Hirokawa N, 2000. KIF5C, a novel neuronal kinesin enriched in motor neurons. J Neurosci 20, 6374–6384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang JS, Tian JH, Pan PY, Zald P, Li C, Deng C, Sheng ZH, 2008. Docking of axonal mitochondria by syntaphilin controls their mobility and affects short-term facilitation. Cell 132, 137–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki S, Holzbaur EL, 1999. Cytoplasmic dynein and dynactin in cell division and intracellular transport. Curr Opin Cell Biol 11, 45–53. [DOI] [PubMed] [Google Scholar]
- Kaza AK, Wamala I, Friehs I, Kuebler JD, Rathod RH, Berra I, Ericsson M, Yao R, Thedsanamoorthy JK, Zurakowski D, Levitsky S, Del Nido PJ, Cowan DB, McCully JD, 2017. Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J Thorac Cardiovasc Surg 153, 934–943. [DOI] [PubMed] [Google Scholar]
- Kesner EE, Saada-Reich A, Lorberboum-Galski H, 2016. Characteristics of Mitochondrial Transformation into Human Cells. Sci Rep 6, 26057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Beaumont V, Cachope R, Munoz-Sanjuan I, Goldman SA, Grantyn R, 2017. Unravelling and Exploiting Astrocyte Dysfunction in Huntington’s Disease. Trends Neurosci 40, 422–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakh BS, Sofroniew MV, 2015. Diversity of astrocyte functions and phenotypes in neural circuits. Nat Neurosci 18, 942–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Zhao H, Martinez J, Doggett TA, Kolesnikov AV, Tang PH, Ablonczy Z, Chan CC, Zhou Z, Green DR, Ferguson TA, 2013. Noncanonical autophagy promotes the visual cycle. Cell 154, 365–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King SJ, Schroer TA, 2000. Dynactin increases the processivity of the cytoplasmic dynein motor. Nat Cell Biol 2, 20–24. [DOI] [PubMed] [Google Scholar]
- Kirk E, Chin LS, Li L, 2006. GRIF1 binds Hrs and is a new regulator of endosomal trafficking. J Cell Sci 119, 4689–4701. [DOI] [PubMed] [Google Scholar]
- Kopinski PK, Janssen KA, Schaefer PM, Trefely S, Perry CE, Potluri P, Tintos-Hernandez JA, Singh LN, Karch KR, Campbell SL, Doan MT, Jiang H, Nissim I, Nakamaru-Ogiso E, Wellen KE, Snyder NW, Garcia BA, Wallace DC, 2019. Regulation of nuclear epigenome by mitochondrial DNA heteroplasmy. Proc Natl Acad Sci U S A 116, 16028–16035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyanagi M, Brandes RP, Haendeler J, Zeiher AM, Dimmeler S, 2005. Cell-to-cell connection of endothelial progenitor cells with cardiac myocytes by nanotubes: a novel mechanism for cell fate changes? Circ Res 96, 1039–1041. [DOI] [PubMed] [Google Scholar]
- Langford GM, 2002. Myosin-V, a versatile motor for short-range vesicle transport. Traffic 3, 859–865. [DOI] [PubMed] [Google Scholar]
- Li X, Zhang Y, Yeung SC, Liang Y, Liang X, Ding Y, Ip MS, Tse HF, Mak JC, Lian Q, 2014. Mitochondrial transfer of induced pluripotent stem cell-derived mesenchymal stem cells to airway epithelial cells attenuates cigarette smoke-induced damage. Am J Respir Cell Mol Biol 51, 455–465. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, Wilton DK, Frouin A, Napier BA, Panicker N, Kumar M, Buckwalter MS, Rowitch DH, Dawson VL, Dawson TM, Stevens B, Barres BA, 2017. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lippert T, Borlongan CV, 2019. Prophylactic treatment of hyperbaric oxygen treatment mitigates inflammatory response via mitochondria transfer. CNS Neurosci Ther 25, 815–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu CS, Chang JC, Kuo SJ, Liu KH, Lin TT, Cheng WL, Chuang SF, 2014a. Delivering healthy mitochondria for the therapy of mitochondrial diseases and beyond. Int J Biochem Cell Biol 53, 141–146. [DOI] [PubMed] [Google Scholar]
- Liu F, Lu J, Manaenko A, Tang J, Hu Q, 2018. Mitochondria in Ischemic Stroke: New Insight and Implications. Aging Dis 9, 924–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Guo L, Zhou Z, Pan M, Yan C, 2019. Mesenchymal stem cells transfer mitochondria into cerebral microvasculature and promote recovery from ischemic stroke. Microvasc Res 123, 74–80. [DOI] [PubMed] [Google Scholar]
- Liu K, Ji K, Guo L, Wu W, Lu H, Shan P, Yan C, 2014b. Mesenchymal stem cells rescue injured endothelial cells in an in vitro ischemia-reperfusion model via tunneling nanotube like structure-mediated mitochondrial transfer. Microvasc Res 92, 10–18. [DOI] [PubMed] [Google Scholar]
- Lyamzaev KG, Nepryakhina OK, Saprunova VB, Bakeeva LE, Pletjushkina OY, Chernyak BV, Skulachev VP, 2008. Novel mechanism of elimination of malfunctioning mitochondria (mitoptosis): formation of mitoptotic bodies and extrusion of mitochondrial material from the cell. Biochim Biophys Acta 1777, 817–825. [DOI] [PubMed] [Google Scholar]
- Macaskill AF, Rinholm JE, Twelvetrees AE, Arancibia-Carcamo IL, Muir J, Fransson A, Aspenstrom P, Attwell D, Kittler JT, 2009. Miro1 is a calcium sensor for glutamate receptor-dependent localization of mitochondria at synapses. Neuron 61, 541–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macheiner T, Fengler VH, Agreiter M, Eisenberg T, Madeo F, Kolb D, Huppertz B, Ackbar R, Sargsyan K, 2016. Magnetomitotransfer: An efficient way for direct mitochondria transfer into cultured human cells. Sci Rep 6, 35571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maeda A, Fadeel B, 2014. Mitochondria released by cells undergoing TNF-alpha-induced necroptosis act as danger signals. Cell Death Dis 5, e1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandal A, Drerup CM, 2019. Axonal Transport and Mitochondrial Function in Neurons. Front Cell Neurosci 13, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchi S, Patergnani S, Missiroli S, Morciano G, Rimessi A, Wieckowski MR, Giorgi C, Pinton P, 2018. Mitochondrial and endoplasmic reticulum calcium homeostasis and cell death. Cell Calcium 69, 62–72. [DOI] [PubMed] [Google Scholar]
- Martin M, Iyadurai SJ, Gassman A, Gindhart JG Jr., Hays TS, Saxton WM, 1999. Cytoplasmic dynein, the dynactin complex, and kinesin are interdependent and essential for fast axonal transport. Mol Biol Cell 10, 3717–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuzawa A, Black KM, Pacak CA, Ericsson M, Barnett RJ, Drumm C, Seth P, Bloch DB, Levitsky S, Cowan DB, McCully JD, 2013. Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 304, H966–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCully JD, Emani SM, Del Nido PJ, 2020. Letter by McCully et al Regarding Article, “Mitochondria Do Not Survive Calcium Overload”. Circ Res 126, e56–e57. [DOI] [PubMed] [Google Scholar]
- McCully JD, Levitsky S, Del Nido PJ, Cowan DB, 2016. Mitochondrial transplantation for therapeutic use. Clin Transl Med 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA, 2014. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 33, 282–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melkov A, Abdu U, 2018. Regulation of long-distance transport of mitochondria along microtubules. Cell Mol Life Sci 75, 163–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KE, Sheetz MP, 2004. Axonal mitochondrial transport and potential are correlated. J Cell Sci 117, 2791–2804. [DOI] [PubMed] [Google Scholar]
- Mishra P, Chan DC, 2014. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol 15, 634–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mistry JJ, Marlein CR, Moore JA, Hellmich C, Wojtowicz EE, Smith JGW, Macaulay I, Sun Y, Morfakis A, Patterson A, Horton RH, Divekar D, Morris CJ, Haestier A, Di Palma F, Beraza N, Bowles KM, Rushworth SA, 2019. ROS-mediated PI3K activation drives mitochondrial transfer from stromal cells to hematopoietic stem cells in response to infection. Proc Natl Acad Sci U S A 116, 24610–24619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris RL, Hollenbeck PJ, 1993. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci 104 ( Pt 3), 917–927. [DOI] [PubMed] [Google Scholar]
- Morrison TJ, Jackson MV, Cunningham EK, Kissenpfennig A, McAuley DF, O’Kane CM, Krasnodembskaya AD, 2017. Mesenchymal Stromal Cells Modulate Macrophages in Clinically Relevant Lung Injury Models by Extracellular Vesicle Mitochondrial Transfer. Am J Respir Crit Care Med 196, 1275–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morshed M, Hlushchuk R, Simon D, Walls AF, Obata-Ninomiya K, Karasuyama H, Djonov V, Eggel A, Kaufmann T, Simon HU, Yousefi S, 2014. NADPH oxidase-independent formation of extracellular DNA traps by basophils. J Immunol 192, 5314–5323. [DOI] [PubMed] [Google Scholar]
- Moschoi R, Imbert V, Nebout M, Chiche J, Mary D, Prebet T, Saland E, Castellano R, Pouyet L, Collette Y, Vey N, Chabannon C, Recher C, Sarry JE, Alcor D, Peyron JF, Griessinger E, 2016. Protective mitochondrial transfer from bone marrow stromal cells to acute myeloid leukemic cells during chemotherapy. Blood 128, 253–264. [DOI] [PubMed] [Google Scholar]
- Nakajima A, Kurihara H, Yagita H, Okumura K, Nakano H, 2008. Mitochondrial Extrusion through the cytoplasmic vacuoles during cell death. J Biol Chem 283, 24128–24135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura Y, Park JH, Hayakawa K, 2020. Therapeutic use of extracellular mitochondria in CNS injury and disease. Exp Neurol 324, 113114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, McBride HM, 2008. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol 18, 102–108. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL, 2000. Mitochondria and neuronal survival. Physiol Rev 80, 315–360. [DOI] [PubMed] [Google Scholar]
- Nukala VN, Singh IN, Davis LM, Sullivan PG, 2006. Cryopreservation of brain mitochondria: a novel methodology for functional studies. J Neurosci Methods 152, 48–54. [DOI] [PubMed] [Google Scholar]
- Oeding SJ, Majstrowicz K, Hu XP, Schwarz V, Freitag A, Honnert U, Nikolaus P, Bahler M, 2018. Identification of Miro1 and Miro2 as mitochondrial receptors for myosin XIX. J Cell Sci 131. [DOI] [PubMed] [Google Scholar]
- Ohno N, Kidd GJ, Mahad D, Kiryu-Seo S, Avishai A, Komuro H, Trapp BD, 2011. Myelination and axonal electrical activity modulate the distribution and motility of mitochondria at CNS nodes of Ranvier. J Neurosci 31, 7249–7258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otsu K, Das S, Houser SD, Quadri SK, Bhattacharya S, Bhattacharya J, 2009. Concentration-dependent inhibition of angiogenesis by mesenchymal stem cells. Blood 113, 4197–4205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang YB, Xu L, Lu Y, Sun X, Yue S, Xiong XX, Giffard RG, 2013. Astrocyte-enriched miR-29a targets PUMA and reduces neuronal vulnerability to forebrain ischemia. Glia 61, 1784–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacak CA, Preble JM, Kondo H, Seibel P, Levitsky S, Del Nido PJ, Cowan DB, McCully JD, 2015. Actin-dependent mitochondrial internalization in cardiomyocytes: evidence for rescue of mitochondrial function. Biol Open 4, 622–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paliwal S, Chaudhuri R, Agrawal A, Mohanty S, 2018. Regenerative abilities of mesenchymal stem cells through mitochondrial transfer. J Biomed Sci 25, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pallafacchina G, Zanin S, Rizzuto R, 2018. Recent advances in the molecular mechanism of mitochondrial calcium uptake. F1000Res 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paolicelli RC, Bergamini G, Rajendran L, 2019. Cell-to-cell Communication by Extracellular Vesicles: Focus on Microglia. Neuroscience 405, 148–157. [DOI] [PubMed] [Google Scholar]
- Park JS, Davis RL, Sue CM, 2018. Mitochondrial Dysfunction in Parkinson’s Disease: New Mechanistic Insights and Therapeutic Perspectives. Curr Neurol Neurosci Rep 18, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquier J, Guerrouahen BS, Al Thawadi H, Ghiabi P, Maleki M, Abu-Kaoud N, Jacob A, Mirshahi M, Galas L, Rafii S, Le Foll F, Rafii A, 2013. Preferential transfer of mitochondria from endothelial to cancer cells through tunneling nanotubes modulates chemoresistance. J Transl Med 11, 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patananan AN, Wu TH, Chiou PY, Teitell MA, 2016. Modifying the Mitochondrial Genome. Cell Metab 23, 785–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilling AD, Horiuchi D, Lively CM, Saxton WM, 2006. Kinesin-1 and Dynein are the primary motors for fast transport of mitochondria in Drosophila motor axons. Mol Biol Cell 17, 2057–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitt JM, Kroemer G, Zitvogel L, 2016. Extracellular vesicles: masters of intercellular communication and potential clinical interventions. J Clin Invest 126, 1139–1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popkov VA, Plotnikov EY, Silachev DN, Zorova LD, Pevzner IB, Jankauskas SS, Zorov SD, Andrianova NV, Babenko VA, Zorov DB, 2017. Bacterial Therapy and Mitochondrial Therapy. Biochemistry (Mosc) 82, 1549–1556. [DOI] [PubMed] [Google Scholar]
- Porter KR, Claude A, Fullam EF, 1945. A Study of Tissue Culture Cells by Electron Microscopy : Methods and Preliminary Observations. J Exp Med 81, 233–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintero OA, DiVito MM, Adikes RC, Kortan MB, Case LB, Lier AJ, Panaretos NS, Slater SQ, Rengarajan M, Feliu M, Cheney RE, 2009. Human Myo19 is a novel myosin that associates with mitochondria. Curr Biol 19, 2008–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez-Barbieri G, Moskowitzova K, Shin B, Blitzer D, Orfany A, Guariento A, Iken K, Friehs I, Zurakowski D, Del Nido PJ, McCully JD, 2019. Alloreactivity and allorecognition of syngeneic and allogeneic mitochondria. Mitochondrion 46, 103–115. [DOI] [PubMed] [Google Scholar]
- Rintoul GL, Filiano AJ, Brocard JB, Kress GJ, Reynolds IJ, 2003. Glutamate decreases mitochondrial size and movement in primary forebrain neurons. J Neurosci 23, 7881–7888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robicsek O, Ene HM, Karry R, Ytzhaki O, Asor E, McPhie D, Cohen BM, Ben-Yehuda R, Weiner I, Ben-Shachar D, 2018. Isolated Mitochondria Transfer Improves Neuronal Differentiation of Schizophrenia-Derived Induced Pluripotent Stem Cells and Rescues Deficits in a Rat Model of the Disorder. Schizophr Bull 44, 432–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocca CJ, Goodman SM, Dulin JN, Haquang JH, Gertsman I, Blondelle J, Smith JLM, Heyser CJ, Cherqui S, 2017. Transplantation of wild-type mouse hematopoietic stem and progenitor cells ameliorates deficits in a mouse model of Friedreich’s ataxia. Sci Transl Med 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers RS, Bhattacharya J, 2013. When cells become organelle donors. Physiology (Bethesda) 28, 414–422. [DOI] [PubMed] [Google Scholar]
- Roushandeh AM, Kuwahara Y, Roudkenar MH, 2019. Mitochondrial transplantation as a potential and novel master key for treatment of various incurable diseases. Cytotechnology 71, 647–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russo E, Nguyen H, Lippert T, Tuazon J, Borlongan CV, Napoli E, 2018. Mitochondrial targeting as a novel therapy for stroke. Brain Circ 4, 84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustom A, Saffrich R, Markovic I, Walther P, Gerdes HH, 2004. Nanotubular highways for intercellular organelle transport. Science 303, 1007–1010. [DOI] [PubMed] [Google Scholar]
- Sansone P, Savini C, Kurelac I, Chang Q, Amato LB, Strillacci A, Stepanova A, Iommarini L, Mastroleo C, Daly L, Galkin A, Thakur BK, Soplop N, Uryu K, Hoshino A, Norton L, Bonafe M, Cricca M, Gasparre G, Lyden D, Bromberg J, 2017. Packaging and transfer of mitochondrial DNA via exosomes regulate escape from dormancy in hormonal therapy-resistant breast cancer. Proc Natl Acad Sci U S A 114, E9066–E9075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saotome M, Safiulina D, Szabadkai G, Das S, Fransson A, Aspenstrom P, Rizzuto R, Hajnoczky G, 2008. Bidirectional Ca2+-dependent control of mitochondrial dynamics by the Miro GTPase. Proc Natl Acad Sci U S A 105, 20728–20733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M, Sato K, 2011. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 334, 1141–1144. [DOI] [PubMed] [Google Scholar]
- Saxton WM, Hollenbeck PJ, 2012. The axonal transport of mitochondria. J Cell Sci 125, 2095–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwarz TL, 2013. Mitochondrial trafficking in neurons. Cold Spring Harb Perspect Biol 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Chandel NS, 2012. Physiological roles of mitochondrial reactive oxygen species. Mol Cell 48, 158–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng ZH, 2014. Mitochondrial trafficking and anchoring in neurons: New insight and implications. J Cell Biol 204, 1087–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Zhao M, Fu C, Fu A, 2017. Intravenous administration of mitochondria for treating experimental Parkinson’s disease. Mitochondrion 34, 91–100. [DOI] [PubMed] [Google Scholar]
- Shin B, Saeed MY, Esch JJ, Guariento A, Blitzer D, Moskowitzova K, Ramirez-Barbieri G, Orfany A, Thedsanamoorthy JK, Cowan DB, Inkster JA, Snay ER, Staffa SJ, Packard AB, Zurakowski D, Del Nido PJ, McCully JD, 2019. A Novel Biological Strategy for Myocardial Protection by Intracoronary Delivery of Mitochondria: Safety and Efficacy. JACC Basic Transl Sci 4, 871–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A, Kukreti R, Saso L, Kukreti S, 2019. Oxidative Stress: A Key Modulator in Neurodegenerative Diseases. Molecules 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha P, Islam MN, Bhattacharya S, Bhattacharya J, 2016. Intercellular mitochondrial transfer: bioenergetic crosstalk between cells. Curr Opin Genet Dev 38, 97–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM, 2012a. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22, 135–141. [DOI] [PubMed] [Google Scholar]
- Soubannier V, Rippstein P, Kaufman BA, Shoubridge EA, McBride HM, 2012b. Reconstitution of mitochondria derived vesicle formation demonstrates selective enrichment of oxidized cargo. PLoS One 7, e52830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spees JL, Olson SD, Whitney MJ, Prockop DJ, 2006. Mitochondrial transfer between cells can rescue aerobic respiration. Proc Natl Acad Sci U S A 103, 1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stefanatos R, Sanz A, 2018. The role of mitochondrial ROS in the aging brain. FEBS Lett 592, 743–758. [DOI] [PubMed] [Google Scholar]
- Stowers RS, Megeath LJ, Gorska-Andrzejak J, Meinertzhagen IA, Schwarz TL, 2002. Axonal transport of mitochondria to synapses depends on milton, a novel Drosophila protein. Neuron 36, 1063–1077. [DOI] [PubMed] [Google Scholar]
- Suen DF, Narendra DP, Tanaka A, Manfredi G, Youle RJ, 2010. Parkin overexpression selects against a deleterious mtDNA mutation in heteroplasmic cybrid cells. Proc Natl Acad Sci U S A 107, 11835–11840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiura A, McLelland GL, Fon EA, McBride HM, 2014. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J 33, 2142–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Qiao H, Pan PY, Chen Y, Sheng ZH, 2013. Motile axonal mitochondria contribute to the variability of presynaptic strength. Cell Rep 4, 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, Wang Y, Zhang J, Tu J, Wang XJ, Su XD, Wang L, Zhang Y, 2012. Tunneling-nanotube direction determination in neurons and astrocytes. Cell Death Dis 3, e438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabadkai G, Simoni AM, Bianchi K, De Stefani D, Leo S, Wieckowski MR, Rizzuto R, 2006. Mitochondrial dynamics and Ca2+ signaling. Biochim Biophys Acta 1763, 442–449. [DOI] [PubMed] [Google Scholar]
- Szymanski J, Janikiewicz J, Michalska B, Patalas-Krawczyk P, Perrone M, Ziolkowski W, Duszynski J, Pinton P, Dobrzyn A, Wieckowski MR, 2017. Interaction of Mitochondria with the Endoplasmic Reticulum and Plasma Membrane in Calcium Homeostasis, Lipid Trafficking and Mitochondrial Structure. Int J Mol Sci 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan AS, Baty JW, Dong LF, Bezawork-Geleta A, Endaya B, Goodwin J, Bajzikova M, Kovarova J, Peterka M, Yan B, Pesdar EA, Sobol M, Filimonenko A, Stuart S, Vondrusova M, Kluckova K, Sachaphibulkij K, Rohlena J, Hozak P, Truksa J, Eccles D, Haupt LM, Griffiths LR, Neuzil J, Berridge MV, 2015. Mitochondrial genome acquisition restores respiratory function and tumorigenic potential of cancer cells without mitochondrial DNA. Cell Metab 21, 81–94. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Kanai Y, Okada Y, Nonaka S, Takeda S, Harada A, Hirokawa N, 1998. Targeted disruption of mouse conventional kinesin heavy chain, kif5B, results in abnormal perinuclear clustering of mitochondria. Cell 93, 1147–1158. [DOI] [PubMed] [Google Scholar]
- Tomasi D, Wang GJ, Volkow ND, 2013. Energetic cost of brain functional connectivity. Proc Natl Acad Sci U S A 110, 13642–13647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torralba D, Baixauli F, Sanchez-Madrid F, 2016. Mitochondria Know No Boundaries: Mechanisms and Functions of Intercellular Mitochondrial Transfer. Front Cell Dev Biol 4, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trias E, Barbeito L, Yamanaka K, 2018. Phenotypic heterogeneity of astrocytes in motor neuron disease. Clin Exp Neuroimmunol 9, 225–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unuma K, Aki T, Funakoshi T, Hashimoto K, Uemura K, 2015. Extrusion of mitochondrial contents from lipopolysaccharide-stimulated cells: Involvement of autophagy. Autophagy 11, 1520–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO, 2007. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 9, 654–659. [DOI] [PubMed] [Google Scholar]
- Valenci I, Yonai L, Bar-Yaacov D, Mishmar D, Ben-Zvi A, 2015. Parkin modulates heteroplasmy of truncated mtDNA in Caenorhabditis elegans. Mitochondrion 20, 64–70. [DOI] [PubMed] [Google Scholar]
- van Spronsen M, Mikhaylova M, Lipka J, Schlager MA, van den Heuvel DJ, Kuijpers M, Wulf PS, Keijzer N, Demmers J, Kapitein LC, Jaarsma D, Gerritsen HC, Akhmanova A, Hoogenraad CC, 2013. TRAK/Milton motor-adaptor proteins steer mitochondrial trafficking to axons and dendrites. Neuron 77, 485–502. [DOI] [PubMed] [Google Scholar]
- Voloboueva LA, Suh SW, Swanson RA, Giffard RG, 2007. Inhibition of mitochondrial function in astrocytes: implications for neuroprotection. J Neurochem 102, 1383–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace DC, 2018. Mitochondrial genetic medicine. Nat Genet 50, 1642–1649. [DOI] [PubMed] [Google Scholar]
- Wang W, Karamanlidis G, Tian R, 2016. Novel targets for mitochondrial medicine. Sci Transl Med 8, 326rv323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Gerdes HH, 2011. Long-distance electrical coupling via tunneling nanotubes. Biochim Biophys Acta 1818, 2082–2086. [DOI] [PubMed] [Google Scholar]
- Wang X, Schwarz TL, 2009. The mechanism of Ca2+ -dependent regulation of kinesin-mediated mitochondrial motility. Cell 136, 163–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL, 2011. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147, 893–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XF, Cynader MS, 2001. Pyruvate released by astrocytes protects neurons from copper-catalyzed cysteine neurotoxicity. J Neurosci 21, 3322–3331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman-Storer CM, Karki S, Holzbaur EL, 1995. The p150Glued component of the dynactin complex binds to both microtubules and the actin-related protein centractin (Arp-1). Proc Natl Acad Sci U S A 92, 1634–1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webber E, Li L, Chin LS, 2008. Hypertonia-associated protein Trak1 is a novel regulator of endosome-to-lysosome trafficking. J Mol Biol 382, 638–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ, 2009. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry 48, 2045–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu TH, Sagullo E, Case D, Zheng X, Li Y, Hong JS, TeSlaa T, Patananan AN, McCaffery JM, Niazi K, Braas D, Koehler CM, Graeber TG, Chiou PY, Teitell MA, 2016. Mitochondrial Transfer by Photothermal Nanoblade Restores Metabolite Profile in Mammalian Cells. Cell Metab 23, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi R, Andreyev A, Murphy AN, Perkins GA, Ellisman MH, Newmeyer DD, 2007. Mitochondria frozen with trehalose retain a number of biological functions and preserve outer membrane integrity. Cell Death Differ 14, 616–624. [DOI] [PubMed] [Google Scholar]
- Yang H, Borg TK, Ma Z, Xu M, Wetzel G, Saraf LV, Markwald R, Runyan RB, Gao BZ, 2016. Biochip-based study of unidirectional mitochondrial transfer from stem cells to myocytes via tunneling nanotubes. Biofabrication 8, 015012. [DOI] [PubMed] [Google Scholar]
- Yang Z, Steele DS, 2000. Effects of cytosolic ATP on spontaneous and triggered Ca2+-induced Ca2+ release in permeabilised rat ventricular myocytes. J Physiol 523 Pt 1, 29–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Fan XL, Jiang D, Zhang Y, Li X, Xu ZB, Fang SB, Chiu S, Tse HF, Lian Q, Fu QL, 2018. Connexin 43-Mediated Mitochondrial Transfer of iPSC-MSCs Alleviates Asthma Inflammation. Stem Cell Reports 11, 1120–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi M, Weaver D, Hajnoczky G, 2004. Control of mitochondrial motility and distribution by the calcium signal: a homeostatic circuit. J Cell Biol 167, 661–672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM, 2012. Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousefi S, Gold JA, Andina N, Lee JJ, Kelly AM, Kozlowski E, Schmid I, Straumann A, Reichenbach J, Gleich GJ, Simon HU, 2008. Catapult-like release of mitochondrial DNA by eosinophils contributes to antibacterial defense. Nat Med 14, 949–953. [DOI] [PubMed] [Google Scholar]
- Yun SP, Kam TI, Panicker N, Kim S, Oh Y, Park JS, Kwon SH, Park YJ, Karuppagounder SS, Park H, Kim S, Oh N, Kim NA, Lee S, Brahmachari S, Mao X, Lee JH, Kumar M, An D, Kang SU, Lee Y, Lee KC, Na DH, Kim D, Lee SH, Roschke VV, Liddelow SA, Mari Z, Barres BA, Dawson VL, Lee S, Dawson TM, Ko HS, 2018. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med 24, 931–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappulli V, Friis KP, Fitzpatrick Z, Maguire CA, Breakefield XO, 2016. Extracellular vesicles and intercellular communication within the nervous system. J Clin Invest 126, 1198–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Ma Z, Yan C, Pu K, Wu M, Bai J, Li Y, Wang Q, 2019. Muscle-derived autologous mitochondrial transplantation: A novel strategy for treating cerebral ischemic injury. Behav Brain Res 356, 322–331. [DOI] [PubMed] [Google Scholar]
- Zhu XH, Qiao H, Du F, Xiong Q, Liu X, Zhang X, Ugurbil K, Chen W, 2012. Quantitative imaging of energy expenditure in human brain. Neuroimage 60, 2107–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]