

Abstract
Over the past few years several methodological and data-driven advances have greatly improved our ability to robustly detect genomic signatures of selective sweeps selection in humans. New methods applied to large samples of present-day genomes provide increased power, while ancient DNA allows precise estimation of timing and tempo. However, despite these advances, we are still limited in our ability to translate these signatures into understanding about which traits were actually under selection, and why. Combining information from different populations and timescales may allow interpretation of selective sweeps. Other modes of selection have proved more difficult to detect. In particular, despite strong evidence of the polygenicity of most human traits, evidence for polygenic selection is weak, and its importance in recent human evolution remains unclear. Balancing selection and archaic introgression seem important for the maintenance of potentially adaptive immune diversity, but perhaps less so for other traits.
Keywords: Natural selection, evolution, Human Evolution
Introduction
The past few decades of human genetics research have emphasized the fundamental similarity of human populations–as demonstrated by the overwhelming support from genetic data for the recent out-of-African model of human origins, extensive gene flow between populations, and low levels of Archaic admixture. Nonetheless, the small number of differences among populations, and the even smaller number that are driven by natural selection, continue to be of great interest partly because of their potential to contribute to the explanation of how humans were able to expand occupy such a diverse range of environments. Despite this interest, and rapidly expanding datasets, there are still relatively few well-understood examples, and our overall picture of the relative importance of different modes of adaptation is limited. While recognizing that non-genetic mechanisms of adaptation such as developmental plasticity and cultural evolution are powerful forces, this review focuses on recent developments related to the detection, classification and interpretation of natural selection in the human genome.
Selective sweeps
The first generation of human genome-wide selection scans produced lists of thousands of putatively selected loci but the limited overlap, number of potential confounding factors, and lack of statistical framework to assess significance led to suspicion that these lists contained high false positive rates (1). This has led to ongoing debate about the extent of positive selection in recent human history, the contribution of “hard” and “soft” selective sweeps, of polygenic adaptation (Figure 1), and whether these features differ between populations (2–6). While these broader questions about the nature of selection remain unresolved, much recent work has focused on the identification, classification and fine-mapping of candidate loci. Many approaches (4, 7–10) combine multiple statistics and use machine learning models trained on simulated data to identify and classify sweeps. While these methods are powerful, they are still limited by our ability to simulate realistic data that incorporate effects such as background selection and heterogeneity in mutation rate.
Figure 1: Genomic signatures of adaptation.
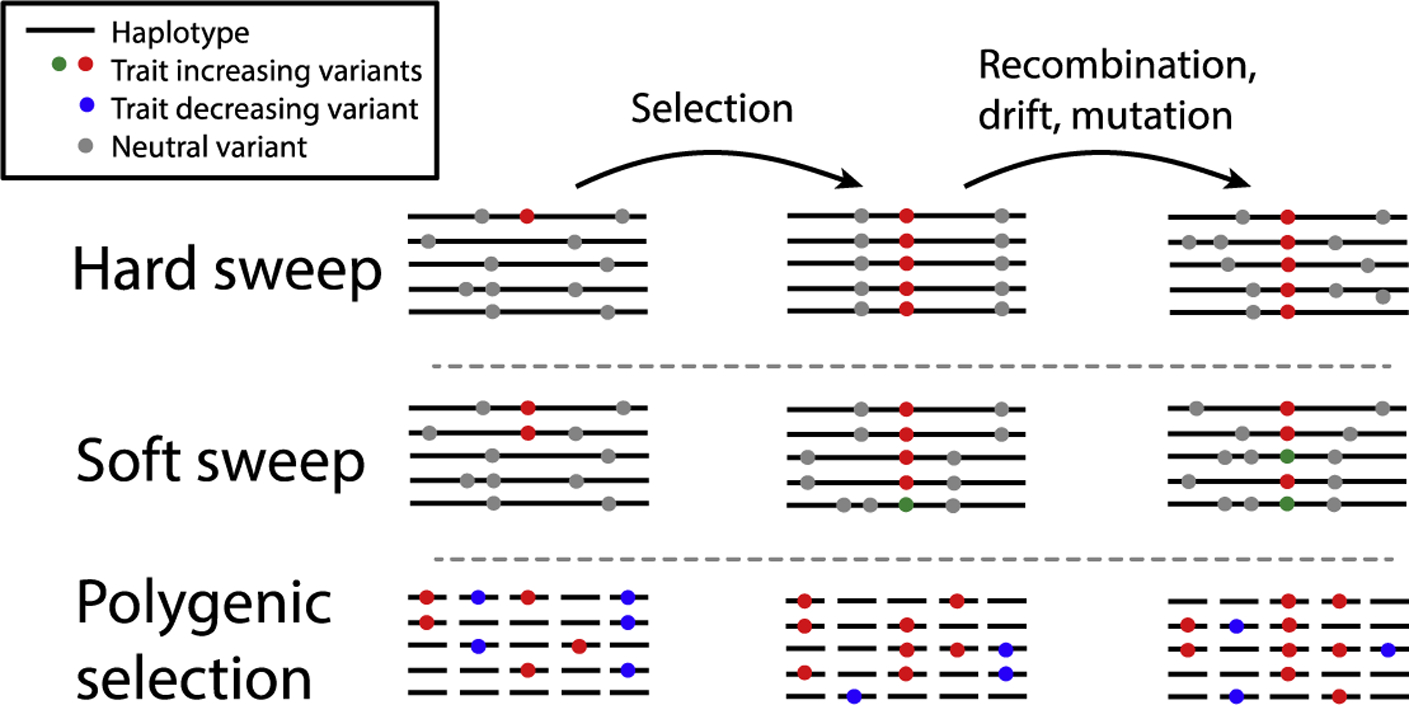
Selection on variants affecting a beneficial trait. In a hard sweep, a new (or rare) mutation (red) on a single haplotype increases rapidly in frequency. Variants on the same haplotype (grey) also increase (“hitchhike”), reducing diversity around the selected site (97). Over time, recombination, drift and mutation break down the sweep signature. In a soft sweep (98), the selected variant may already be present on multiple haplotypes (red), or there may be new selected mutations (green) as the sweep is in progress. Diversity is reduced around the sweep but not by as much as a hard sweep. In practice, this signature may be difficult to distinguish from an incomplete hard sweep. In polygenic adaptation (42), variants at many loci genome-wide change frequency; trait-increasing alleles (red) increase in frequency while trait-decreasing alleles (blue) decrease. This process is essentially a large number of weak soft sweeps, but the effects are too small to be detected at any one locus. Variants drift after selection, but mean shifts in frequency are maintained.
A more direct way to study natural selection is to use ancient DNA data to directly observe changes in allele frequency over time. Ancient DNA has revolutionized the study of demographic history and is becoming increasingly useful for the study of natural selection and phenotypic evolution (11). While even a single genome can be informative about demographic history, inference of selection requires much larger sample sizes. Ancient DNA based scans can detect strong signals of selection (12) (Figure 2A), but have limited power due to small sample sizes. However, ancient DNA can provide precise estimates of the timing of selection on particular alleles such as those associated with skin pigmentation (12, 13) and lactase persistence (14–16) (Figure 2B). It can help to resolve complex evolutionary histories, for example at the FADS locus (17–20) (Figure 2C), and help to separate the effects of selection from those of changes in ancestry. Currently, the vast majority of ancient DNA samples are from Western Eurasia and it is on this region that most ancient DNA studies of selection have focused (with exceptions (21, 22)). Large ancient DNA studies in other parts of the world should allow similar analyses.
Figure 2: Ancient DNA adds another dimension to selection scans.
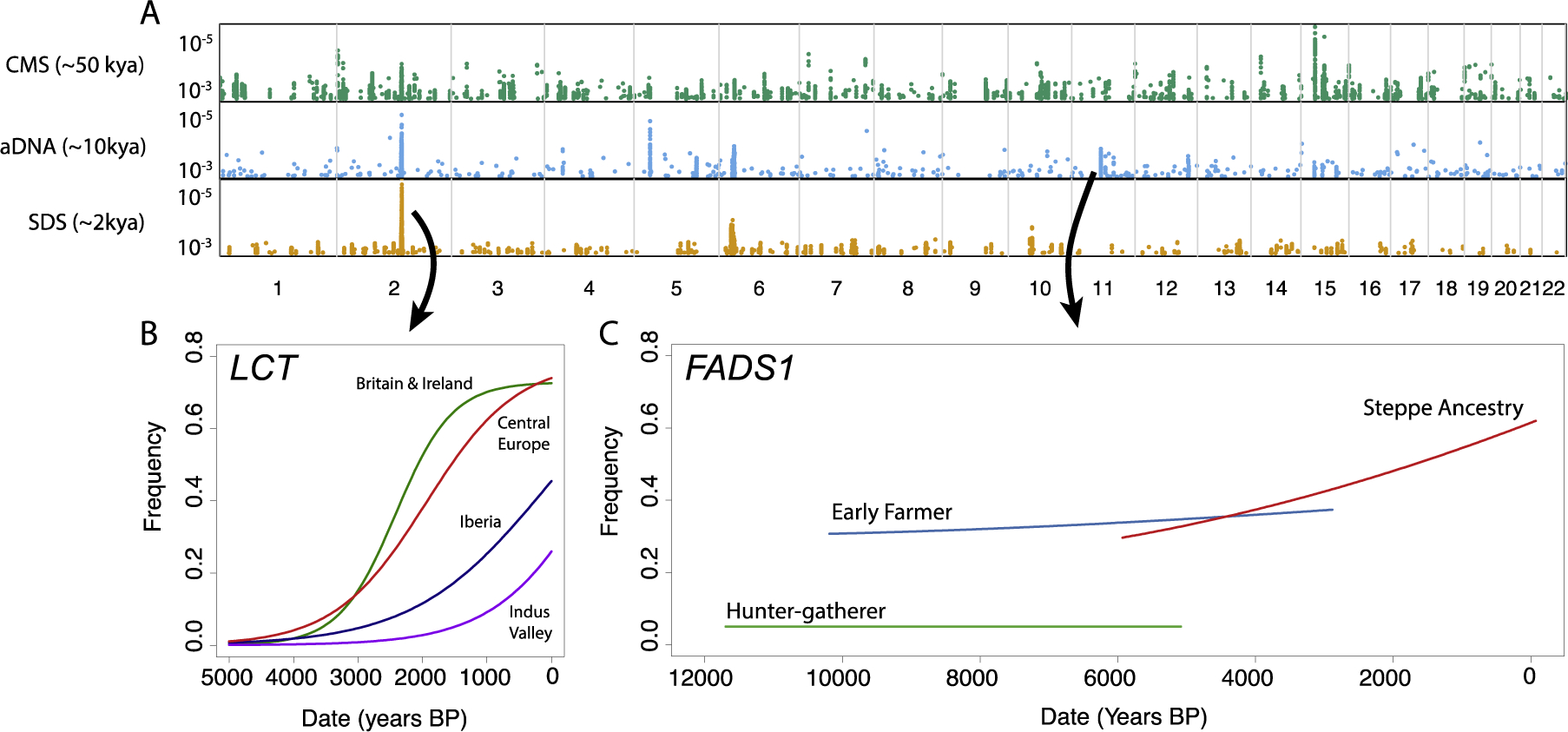
A: genome-wide selection scan signals from three different approaches (8, 12, 26) with power to detect selection over different timescales. Y-axis shows log10 quantiles for the top 0.1% of tested markers. B: stratified by geographic location, ancient DNA from 668 individuals reveals distinct trajectories of the lactase persistence allele in different parts of Europe. C: stratified by ancestry derived from the three main source populations of present-day Europe, ancient DNA reconstructs the evolution of the FADS locus (redrawn from (17)).
The UK Biobank (23) and UK10 (24) projects have enabled particularly deep investigation of recent selection in the British population (25–27). Recently, similar scans have become possible in the Japanese population thanks to BioBank Japan and other cohorts (28, 29). These scans reveal a qualitatively similar landscape of selection between Britain and Japan. Both populations show evidence of selection on dietary, immune and anthropometric phenotypes but carry relatively few strong sweeps. Curiously, in both cases, the strongest signals of selection are at loci associated with a specific agricultural product; ability to consume milk in Britain (the LCT locus), and inability to consume alcohol in Japan (the ADH locus). On the other hand, the Japanese population does not show the very strong recent signals of selection for pigmentation-associated variation that the British population does (29). More broadly, these data provide the opportunity to assess the extent of parallel adaptation at the level of individual genes, pathways, phenotypes or classes of phenotype in the two populations. It is widely believed that the development of agriculture was one of the strongest forces in recent human evolution and demography, and it represents one of the few repeated experiments in human evolution. The demographic transitions associated with the introduction of agriculture in both the UK and Japan were very similar (30, 31) and the phenotypic associations available through UK Biobank and BioBank Japan provide an excellent opportunity to test whether the adaptive responses were also similar.
Although lacking the phenotypic information from large biobanks, genetic data from diverse cohorts from Africa (32–35), East Asia (36, 37) and other parts of the world are enabling a broader assessment of human adaptation. These studies confirm that the immune system is a frequent, perhaps the most frequent, target of positive selection and help to identify the genetic basis of putative local adaptations such as short stature in African rainforest hunter-gatherers (33, 34, 38–41).
Polygenic adaptation
Genome-wide association studies (GWAS) indicate that many human traits are highly polygenic-controlled by a large number of variants-many of which have extremely small marginal effects. This observation, coupled with the relatively limited number of selective sweeps, suggested that polygenic adaptation might be an important force in human evolution (42). In this model, complex traits evolve as the result of small shifts in frequency of large number of variants. These shifts are too small to produce classical signals of selection but can be identified in aggregate. Over the past decade, several different studies supported this expectation (12, 26, 43–47). Many of these focused on differential selection for height across Europe, although other traits were also implicated. Recently, with the release and analysis of the UK Biobank dataset, it became clear that the signals of selection of height had been overestimated (48, 49). Specifically, the GWAS on which previous analyses had relied had not fully corrected for the effect of population stratification, leading to overestimation of the effect of selection. If the results for height in Europe–apparently the clearest example of polygenic selection-cannot be trusted, how can we trust evidence for other traits, which surely suffer from similar problems? What about evidence of selection in non-European ancestry populations, which is likely further biased by non-transferability of GWAS effect size estimates (50)?
So, in 2020, the question of the contribution of polygenic adaptation to human evolution is largely back to where it was in 2010. In many cases, patterns of phenotypic variation are highly suggestive of local adaptation (51–53). Many traits are highly polygenic and it seems that we should expect polygenic adaptation to be common. On the other hand, the empirical evidence is relatively weak and polygenic selection tests are highly sensitive to artefacts. Some authors have argued that using GWAS effect sizes estimated in an outgroup population avoids bias associated with population stratification (54, 55). Others have attempted to use effect sizes re-estimated within sibling pairs (56), which should be more resistant to stratification. However, neither of these approaches is totally satisfactory. In any case, even if there is some residual signal of selection, the fact that polygenic selection tests turned out to be unexpectedly vulnerable to population stratification probably warrants additional caution before we accept such claims. Given that we expect polygenic selection to be common, why is it so hard to find?
One possibility is that polygenic adaptation is, in fact, relatively rare. Despite hundreds or thousands of loci with nonzero effects on a trait, widespread pleiotropy might mean that adaptation is driven by shifts in the frequency of a relatively small proportion of loci. That is, adaptation on polygenic traits may be more oligogenic than polygenic (57, 58). Alternatively, polygenic adaptation could occur without leaving a clearly detectable signature of consistent frequency shifts because effects vary in time due to allelic heterogeneity or interactions (either genetic or environmental), or because selection pressures or effect sizes are fluctuating (58) (Figure 3). Finally, recent theoretical work shows that the details of the response to selection on a polygenic trait are sensitive to the details of the genetic architecture (59–61) so might leave more complex genomic signatures than commonly assumed.
Figure 3: Limits to polygenic adaption. A:
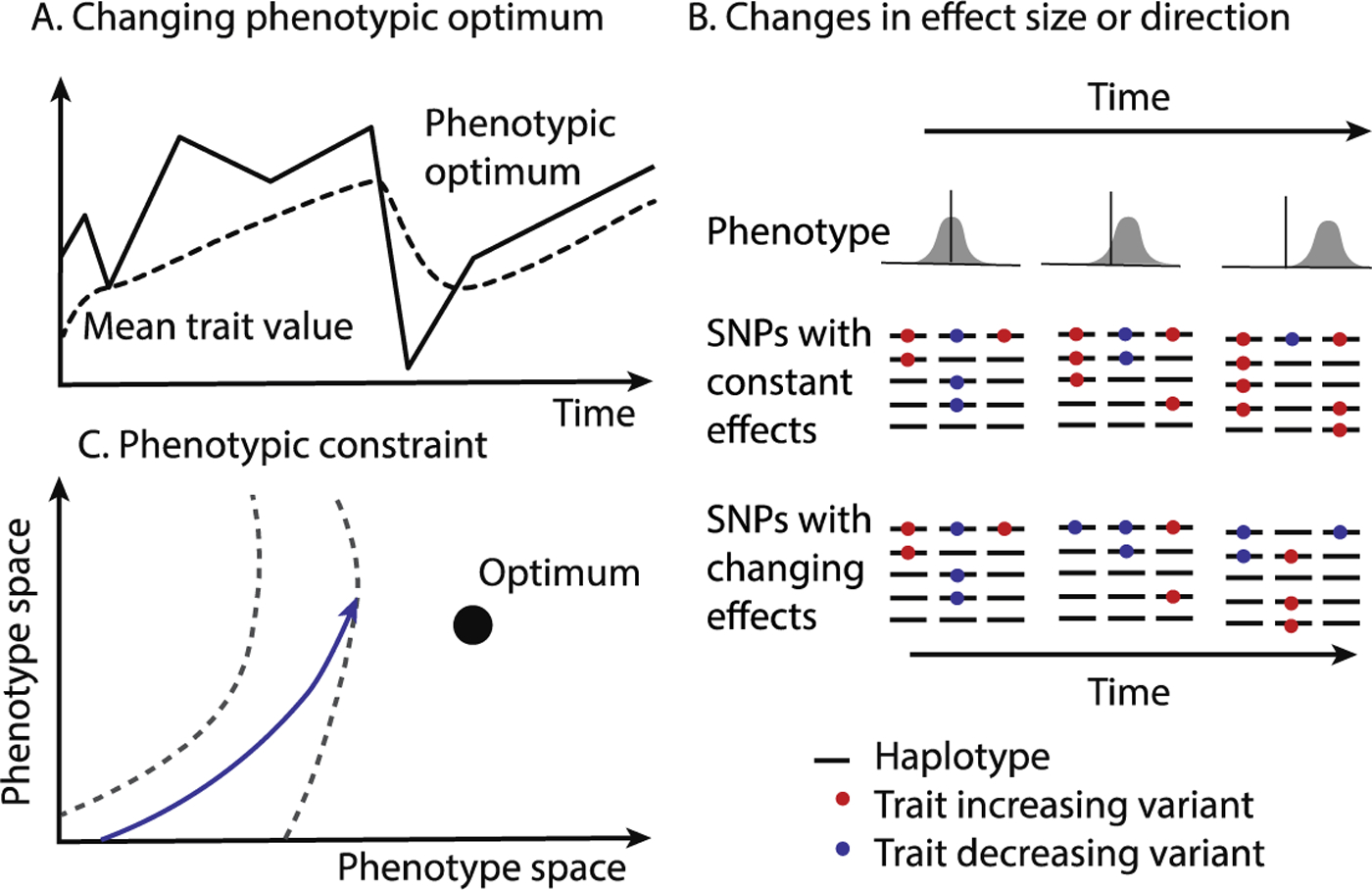
If the phenotypic optimum changes over time, polygenic adaptation will not leave a consistent signal of frequency shift. B: Similarly, if effect sizes or direction changes over time due to allelic heterogeneity, or interactions, then polygenic adaption will occur, but will not leave a consistent pattern of frequency shifts. This cartoon shows a population of five haplotypes with three trait-associated SNPs over three time periods, with selection for an increased phenotype. If SNP effects are constant, then trait-increasing SNPs consistently increase in frequency and trait-decreasing SNPs decrease in frequency. On the other hand, if effects change over time, then this signal would be obscured over the long term, even though polygenic adaptation is still occurring. C: Finally, polygenic adaptation may be fundamentally limited by pleiotropy, which constrains the range of possible phenotypes that can be reached (between the dashed lines), or the set of variants that can respond to selection.
Adaptive archaic introgression
A long-standing hypothesis is that Neanderthals (and Denisovans), who lived in Eurasia for hundreds of thousands of years, carried adaptations to that environment that would have been beneficial to modern humans on their arrival. While, broadly speaking, archaic ancestry was deleterious and selected against in modern humans (62–65), this idea has gained some empirical support from the observation that some specific variants were positively selected (66). Perhaps the best example is of a Denisovan haplotype at EPAS1 that is associated with altitude adaptation in present-day Tibetans (67). However, even this case is not so simple. The functional variant tagged by the Denisovan haplotype is unclear and not necessarily of Denisovan origin. Even if the functional variant was present in Denisovans, the haplotype was carried by Denisovans living at low altitude (Denisova cave is only 700m above sea level), so did not necessarily represent an altitude adaptation. It may have just been part of Denisovan physiology that happened to later become adaptive in modern humans in the high-altitude environment.
While there is some evidence that classically adaptive traits such as skin pigmentation experienced a contribution from adaptive archaic introgression, by far the strongest evidence for an important role in recent evolution involves the immune system. Specific targets include the toll-like receptor genes TLR1, 6 and 10 (68, 69), the oligoadenylate synthetases OAS2 and 3 (70–72), and the interferon pathway (69, 73). However, recent analyses suggest that the effects go beyond these individual loci and apply more broadly over large classes of immune-associated genes (74, 75), largely through regulatory effects (76–78). In some cases, these archaic alleles may have provided protection against pathogens, or classes of pathogen, transmitted directly from archaic to modern humans (75, 79). However, in other cases, for example the TLR cluster, positive selection on the archaic allele occurred tens of thousands of years after introgression (69). Like EPAS1, much of the adaptive archaic admixture may have contributed to the reservoir of potentially adaptive standing variation, rather than being of immediate advantage, although this remains to be systematically tested.
Balancing selection
One reason why the immune system, in particular, might retain a large reservoir of potential archaic targets of positive selection is that a relatively large proportion of immune-associated variation is under balancing selection. Recently a number of tests specifically designed to detect balancing selection within (80, 81) and between species (82–84) have been developed. These tests broadly confirm enrichment of balancing selection at immune-associated genes, but also highlight a number of intriguing potential new signals. These include loci associated with reproductive biology or behavior, including the CADM2 and ESR1 loci (81, 83, 85, 86). Signatures of balancing selection can be generated by a number of different evolutionary processes-frequency dependent selection, overdominance, and fluctuating selection, for example-and the relative contribution of these processes remains unclear. A key question for future work is to determine whether it is possible, perhaps using ancient DNA, to distinguish the effects of these processes both at specific loci, and more generally across the genome.
Can we do better than just-so stories?
New datasets and statistical approaches have made the detection of genomic signatures of selection much easier. While there is still relatively little overlap between methods, the ability to combine statistics and directly replicate signals with ancient DNA has allowed us to robustly identify parts of the genome under selection. What is less clear, in almost every case, is why those parts of the genome were selected. As has recently been observed (87), even for the clearest and best-known examples of selective sweeps, many of which involve alleles with pleiotropic effects, we almost never know which phenotype is actually under selection. Even when we can make a good guess at the phenotype, we almost never know the mechanism by which it affects fitness. How might we do better?
One approach, as already described, is to borrow information across loci and look for enrichment of selection signals in pathways, or sets of loci associated with a particular trait. But even if we can identify such a trait, it is hard to know whether we should assume that was the specific trait under selection. In parallel, we can try to borrow information across populations (88). Correlating shared environment with shared and parallel adaptation across populations can provide clues to the underlying drivers of that selection. A particularly powerful way to do this is to investigate the few repeated experiments in human evolution. The introduction of agriculture, already discussed, is the most striking example, but others include migration to extreme latitudes or altitudes, urbanization, or response to particular classes of pathogen. Key here is the incorporation of external information, for example archaeological data about subsistence, lifestyle and diet. Finally, we can use the precise temporal information provided by ancient DNA to directly test hypotheses about drivers of selection and we should be ruthless about rejecting or reformulating hypotheses that are contradicted by direct evidence.
Simulations provide an important tool for developing intuition about different demographic and selective scenarios, for evaluating the performance of different methods and for training machine learning models. This has been made much easier by recent developments in population genetic tools including 1) msprime, which enables fast coalescent simulations (89) 2) stdpopsim, a library of standard demographic models (90) and, in particular 3) SLiM, which allows the forward simulations needed to model complex selection (91). Development and maintenance of these tools is critical to enabling future selection studies.
Finally, while most work has focused on ancient selection, very large datasets provide the opportunity to investigate very recent selection-over the past few or even current generations (92–94). Some authors have worried that relaxation of selection due to modern medical care will lead to an overall decrease in fitness (95, 96). On the other hand, much selection happens in utero and large effective population sizes might be expected to increase the efficacy of both negative and positive selection. Modern environmental conditions might promote selection for traits that were previously neutral or deleterious. Despite much speculation about these sorts of effects, there has been relatively little analysis, leaving many opportunities for studies of human adaptation over the past 40, rather than 40,000 years.
Acknowledgments
I.M. was supported by a Research Fellowship from the Alfred P. Sloan Foundation (FG-2018-10647), a New Investigator Research Grant from the Charles E. Kaufman Foundation (KA2018-98559), and NIGMS award number (R35GM133708). The content is solely the responsibility of the author and does not necessarily represent the official views of the National Institutes of Health or other funders.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Akey JM (2009) Constructing genomic maps of positive selection in humans: where do we go from here? Genome Res 19(5):711–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hernandez RD, et al. (2011) Classic selective sweeps were rare in recent human evolution. Science 331(6019):920–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Enard D, Messer PW, & Petrov DA (2014) Genome-wide signals of positive selection in human evolution. Genome Res 24(6):885–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schrider DR & Kern AD (2017) Soft Sweeps Are the Dominant Mode of Adaptation in the Human Genome. Mol Biol Evol 34(8):1863–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Harris RB, Sackman A, & Jensen JD (2018) On the unfounded enthusiasm for soft selective sweeps II: Examining recent evidence from humans, flies, and viruses. PLoS Genet 14(12):e1007859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Laval G, Patin E, Boutillier P, & Quintana-Murci L (2019) A genome-wide Approximate Bayesian Computation approach suggests only limited numbers of soft sweeps in humans over the last 100,000 years. bioRxiv 10.1101/2019.12.22.886234. [DOI] [Google Scholar]
- 7.Sheehan S & Song YS (2016) Deep Learning for Population Genetic Inference. PLoS Comput Biol 12(3):e1004845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grossman SR, et al. (2013) Identifying recent adaptations in large-scale genomic data. Cell 152(4):703–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pybus M, et al. (2015) Hierarchical boosting: a machine-learning framework to detect and classify hard selective sweeps in human populations. Bioinformatics 31(24):3946–3952. [DOI] [PubMed] [Google Scholar]
- 10.Sugden LA, et al. (2018) Localization of adaptive variants in human genomes using averaged one-dependence estimation. Nat Commun 9(1):703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Marciniak S & Perry GH (2017) Harnessing ancient genomes to study the history of human adaptation. Nat Rev Genet 18(11):659–674. [DOI] [PubMed] [Google Scholar]
- 12.Mathieson I, et al. (2015) Genome-wide patterns of selection in 230 ancient Eurasians. Nature 528(7583):499–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wilde S, et al. (2014) Direct evidence for positive selection of skin, hair, and eye pigmentation in Europeans during the last 5,000 y. Proceedings of the National Academy of Sciences 111(13):4832–4837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burger J, Kirchner M, Bramanti B, Haak W, & Thomas MG (2007) Absence of the lactase-persistence-associated allele in early Neolithic Europeans. Proc Natl Acad Sci U S A 104(10):3736–3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olalde I, et al. (2017) The Beaker Phenomenon And The Genomic Transformation Of Northwest Europe. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Olalde I, et al. (2019) The genomic history of the Iberian Peninsula over the past 8000 years. Science 363(6432):1230–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mathieson S & Mathieson I (2018) FADS1 and the Timing of Human Adaptation to Agriculture. Mol Biol Evol 35(12):2957–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ye K, Gao F, Wang D, Bar-Yosef O, & Keinan A (2017) Dietary adaptation of FADS genes in Europe varied across time and geography. Nat Ecol Evol 1:167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Buckley MT, et al. (2017) Selection in Europeans on Fatty Acid Desaturases Associated with Dietary Changes. Mol Biol Evol 34(6):1307–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mathieson I (2020) Limited evidence for selection at the FADS locus in Native American populations. Mol Biol Evol:msaa064. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper, along with references 17–19, uses ancient DNA to describe the history of the FADS locus, which experienced multiple episodes of selection throughout human evolution.
- 21.Jeong C, et al. (2016) Long-term genetic stability and a high-altitude East Asian origin for the peoples of the high valleys of the Himalayan arc. Proceedings of the National Academy of Sciences 113(27):7485–7490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Skoglund P, et al. (2017) Reconstructing Prehistoric African Population Structure. Cell 171(1):59–71 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bycroft C, et al. (2018) The UK Biobank resource with deep phenotyping and genomic data. Nature 562(7726):203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Consortium UK (2015) The UK10K project identifies rare variants in health and disease. Nature 526(7571):82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Galinsky KJ, Loh PR, Mallick S, Patterson NJ, & Price AL (2016) Population Structure of UK Biobank and Ancient Eurasians Reveals Adaptation at Genes Influencing Blood Pressure. Am J Hum Genet 99(5):1130–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Field Y, et al. (2016) Detection of human adaptation during the past 2000 years. Science 354(6313):760–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Palamara PF, Terhorst J, Song YS, & Price AL (2018) High-throughput inference of pairwise coalescence times identifies signals of selection and enriched disease heritability. Nat Genet 50(9):1311–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]; An efficient algorithm allows selection scans to be extended to samples that are an order of magnitude larger than previously
- 28.Okada Y, et al. (2018) Deep whole-genome sequencing reveals recent selection signatures linked to evolution and disease risk of Japanese. Nat Commun 9(1):1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yasumizu Y, et al. (2020) Genome-wide natural selection signatures are linked to genetic risk of modern phenotypes in the Japanese population. Mol Biol Evol. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper, along with reference 23, use sequence and biobank data to perform comprehensive selection scans in the Japanese population.
- 30.Brace S, et al. (2019) Ancient genomes indicate population replacement in Early Neolithic Britain. Nat Ecol Evol 3(5):765–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kanzawa-Kiriyama H, et al. (2017) A partial nuclear genome of the Jomons who lived 3000 years ago in Fukushima, Japan. J Hum Genet 62(2):213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Patin E, et al. (2017) Dispersals and genetic adaptation of Bantu-speaking populations in Africa and North America. Science 356(6337):543–546. [DOI] [PubMed] [Google Scholar]
- 33.Fan S, et al. (2019) African evolutionary history inferred from whole genome sequence data of 44 indigenous African populations. Genome Biol 20(1):82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lopez M, et al. (2019) Genomic Evidence for Local Adaptation of Hunter-Gatherers to the African Rainforest. Curr Biol 29(17):2926–2935 e2924. [DOI] [PubMed] [Google Scholar]
- 35.Owers KA, et al. (2017) Adaptation to infectious disease exposure in indigenous Southern African populations. Proc Biol Sci 284(1852). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chiang CWK, Mangul S, Robles C, & Sankararaman S (2018) A Comprehensive Map of Genetic Variation in the World’s Largest Ethnic Group-Han Chinese. Mol Biol Evol 35(11):2736–2750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu X, et al. (2017) Characterising private and shared signatures of positive selection in 37 Asian populations. Eur J Hum Genet 25(4):499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jarvis JP, et al. (2012) Patterns of ancestry, signatures of natural selection, and genetic association with stature in Western African pygmies. PLoS Genet 8(4):e1002641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lachance J, et al. (2012) Evolutionary history and adaptation from high-coverage whole-genome sequences of diverse African hunter-gatherers. Cell 150(3):457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Perry GH, et al. (2014) Adaptive, convergent origins of the pygmy phenotype in African rainforest hunter-gatherers. Proc Natl Acad Sci U S A 111(35):E3596–3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bergey CM, et al. (2018) Polygenic adaptation and convergent evolution on growth and cardiac genetic pathways in African and Asian rainforest hunter-gatherers. Proc Natl Acad Sci U S A 115(48):E11256–E11263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pritchard JK, Pickrell JK, & Coop G (2010) The genetics of human adaptation: hard sweeps, soft sweeps, and polygenic adaptation. Curr Biol 20(4):R208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Turchin MC, et al. (2012) Evidence of widespread selection on standing variation in Europe at height-associated SNPs. Nat Genet 44(9):1015–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Berg JJ & Coop G (2014) A population genetic signal of polygenic adaptation. PLoS Genet 10(8):e1004412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Robinson MR, et al. (2015) Population genetic differentiation of height and body mass index across Europe. Nat Genet 47(11):1357–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guo J, et al. (2018) Global genetic differentiation of complex traits shaped by natural selection in humans. Nat Commun 9(1):1865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Racimo F, Berg JJ, & Pickrell JK (2017) Detecting polygenic adaptation in admixture graphs. bioRxiv. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Berg JJ, et al. (2019) Reduced signal for polygenic adaptation of height in UK Biobank. Elife 8:e39725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sohail M, et al. (2019) Polygenic adaptation on height is overestimated due to uncorrected stratification in genome-wide association studies. Elife 8:e39702. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper, along with reference 48 showed that previously reported signals of polygenic adaptation on height in Europe were overestimated due to population stratification in GWAS results.
- 50.Martin AR, et al. (2019) Clinical use of current polygenic risk scores may exacerbate health disparities. Nat Genet 51(4):584–591. [DOI] [PMC free article] [PubMed] [Google Scholar]; An empirical demostration that polygenic risk scores (PRS) do not transfer across populations, and therefore that even significant differences in PRS cannot be interpreted as evidence of selection.
- 51.Jablonski NG & Chaplin G (2010) Colloquium paper: human skin pigmentation as an adaptation to UV radiation. Proc Natl Acad Sci U S A 107 Suppl 2:8962–8968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zaidi AA, et al. (2017) Investigating the case of human nose shape and climate adaptation. PLoS Genet 13(3):e1006616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Savell KR, Auerbach BM, & Roseman CC (2016) Constraint, natural selection, and the evolution of human body form. Proc Natl Acad Sci U S A 113(34):9492–9497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chen M, et al. (2019) Evidence of polygenic adaptation at height-associated loci in mainland Europeans and Sardinians. bioRxiv doi: 10.1101/776377. [DOI] [Google Scholar]
- 55.Tucci S, et al. (2018) Evolutionary history and adaptation of a human pygmy population of Flores Island, Indonesia. Science 361(6401):511–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cox SL, Ruff CB, Maier RM, & Mathieson I (2019) Genetic contributions to variation in human stature in prehistoric Europe. Proc Natl Acad Sci U S A 116(43):21484–21492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bell G (2009) The oligogenic view of adaptation. Cold Spring Harb Symp Quant Biol 74:139–144. [DOI] [PubMed] [Google Scholar]
- 58.Bell G (2010) Fluctuating selection: the perpetual renewal of adaptation in variable environments. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 365(1537):87–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hayward LK & Sella G (2019) Polygenic adaptation after a sudden change in environment. biorXiv doi: 10.1101/792952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hollinger I, Pennings PS, & Hermisson J (2019) Polygenic adaptation: From sweeps to subtle frequency shifts. PLoS Genet 15(3):e1008035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Thornton KR (2019) Polygenic Adaptation to an Environmental Shift: Temporal Dynamics of Variation Under Gaussian Stabilizing Selection and Additive Effects on a Single Trait. Genetics 213(4):1513–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Petr M, Paabo S, Kelso J, & Vernot B (2019) Limits of long-term selection against Neandertal introgression. Proc Natl Acad Sci U S A 116(5):1639–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]; Selection against Neanderthal variation did not cause a long slow decline in Neanderthal ancestry in Europeans. Rather, the decline can be explained by gene flow with African popualtions.
- 63.Harris K & Nielsen R (2016) The Genetic Cost of Neanderthal Introgression. Genetics 203(2):881–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Juric I, Aeschbacher S, & Coop G (2016) The Strength of Selection against Neanderthal Introgression. PLoS Genet 12(11):e1006340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sankararaman S, et al. (2014) The genomic landscape of Neanderthal ancestry in present-day humans. Nature 507(7492):354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Racimo F, Sankararaman S, Nielsen R, & Huerta-Sanchez E (2015) Evidence for archaic adaptive introgression in humans. Nat Rev Genet 16(6):359–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Huerta-Sanchez E, et al. (2014) Altitude adaptation in Tibetans caused by introgression of Denisovan-like DNA. Nature 512(7513):194–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dannemann M, Andres AM, & Kelso J (2016) Introgression of Neandertal- and Denisovan-like Haplotypes Contributes to Adaptive Variation in Human Toll-like Receptors. Am J Hum Genet 98(1):22–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Deschamps M, et al. (2016) Genomic Signatures of Selective Pressures and Introgression from Archaic Hominins at Human Innate Immunity Genes. Am J Hum Genet 98(1):5–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sams AJ, et al. (2016) Adaptively introgressed Neandertal haplotype at the OAS locus functionally impacts innate immune responses in humans. Genome Biol 17(1):246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mendez FL, Watkins JC, & Hammer MF (2012) Global genetic variation at OAS1 provides evidence of archaic admixture in Melanesian populations. Mol Biol Evol 29(6):1513–1520. [DOI] [PubMed] [Google Scholar]
- 72.Mendez FL, Watkins JC, & Hammer MF (2012) A haplotype at STAT2 Introgressed from neanderthals and serves as a candidate of positive selection in Papua New Guinea. Am J Hum Genet 91(2):265–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jagoda E, et al. (2018) Disentangling Immediate Adaptive Introgression from Selection on Standing Introgressed Variation in Humans. Mol Biol Evol 35(3):623–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gouy A & Excoffier L (2020) Polygenic patterns of adaptive introgression in modern humans are mainly shaped by response to pathogens. Mol Biol Evol. [DOI] [PubMed] [Google Scholar]
- 75.Enard D & Petrov DA (2018) Evidence that RNA Viruses Drove Adaptive Introgression between Neanderthals and Modern Humans. Cell 175(2):360–371 e313. [DOI] [PMC free article] [PubMed] [Google Scholar]; Argues that viral interacting proteins (VIPs) show strong signatures of adaptive introgression from Neanderthals and that response to viruses was therefore a major target of selection.
- 76.Nedelec Y, et al. (2016) Genetic Ancestry and Natural Selection Drive Population Differences in Immune Responses to Pathogens. Cell 167(3):657–669 e621. [DOI] [PubMed] [Google Scholar]
- 77.Quach H, et al. (2016) Genetic Adaptation and Neandertal Admixture Shaped the Immune System of Human Populations. Cell 167(3):643–656 e617. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper, and reference 76, combine genetic and experimental evidence to identify selection on pathogen response across popualtions.
- 78.Silvert M, Quintana-Murci L, & Rotival M (2019) Impact and Evolutionary Determinants of Neanderthal Introgression on Transcriptional and Post-Transcriptional Regulation. Am J Hum Genet 104(6):1241–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Houldcroft CJ & Underdown SJ (2016) Neanderthal genomics suggests a pleistocene time frame for the first epidemiologic transition. Am J Phys Anthropol 160(3):379–388. [DOI] [PubMed] [Google Scholar]
- 80.Bitarello BD, et al. (2018) Signatures of Long-Term Balancing Selection in Human Genomes. Genome Biol Evol 10(3):939–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Siewert KM & Voight BF (2017) Detecting Long-Term Balancing Selection Using Allele Frequency Correlation. Mol Biol Evol 34(11):2996–3005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cheng X & DeGiorgio M (2019) Detection of Shared Balancing Selection in the Absence of Trans-Species Polymorphism. Mol Biol Evol 36(1):177–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.DeGiorgio M, Lohmueller KE, & Nielsen R (2014) A model-based approach for identifying signatures of ancient balancing selection in genetic data. PLoS Genet 10(8):e1004561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gao Z, Przeworski M, & Sella G (2015) Footprints of ancient-balanced polymorphisms in genetic variation data from closely related species. Evolution 69(2):431–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Boutwell B, et al. (2017) Replication and characterization of CADM2 and MSRA genes on human behavior. Heliyon 3(7):e00349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Day FR, et al. (2016) Physical and neurobehavioral determinants of reproductive onset and success. Nat Genet 48(6):617–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Szpak M, Xue Y, Ayub Q, & Tyler-Smith C (2019) How well do we understand the basis of classic selective sweeps in humans? FEBS Lett 593(13):1431–1448. [DOI] [PubMed] [Google Scholar]; Makes the important point that even many of the strongest signals of selection in humans are poorly understood in terms of the underlying selective pressures.
- 88.Johnson KE & Voight BF (2018) Patterns of shared signatures of recent positive selection across human populations. Nat Ecol Evol 2(4):713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Kelleher J, Etheridge AM, & McVean G (2016) Efficient Coalescent Simulation and Genealogical Analysis for Large Sample Sizes. PLoS Comput Biol 12(5):e1004842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Adrion JR, et al. (2019) A community-maintained standard library of population genetic models. bioRxiv: 10.1101/2019.1112.1120.885129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Haller BC & Messer PW (2019) SLiM 3: Forward Genetic Simulations Beyond the Wright-Fisher Model. Mol Biol Evol 36(3):632–637. [DOI] [PMC free article] [PubMed] [Google Scholar]; The software SLiM greatly expands the possibilities to model complex selection, allowing fast forward simulation of almost arbitrarily complex models.
- 92.Mostafavi H, et al. (2017) Identifying genetic variants that affect viability in large cohorts. PLoS Biol 15(9):e2002458. [DOI] [PMC free article] [PubMed] [Google Scholar]; Change in allele frequency with age in a large cohort allows the identification of variants that affect viability. Perhaps surprisingly, few such variants are found.
- 93.Kong A, et al. (2017) Selection against variants in the genome associated with educational attainment. Proc Natl Acad Sci U S A 114(5):E727–E732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Beauchamp JP (2016) Genetic evidence for natural selection in humans in the contemporary United States. Proc Natl Acad Sci U S A 113(28):7774–7779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lynch M (2016) Mutation and Human Exceptionalism: Our Future Genetic Load. Genetics 202(3):869–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kondrashov AS (2017) Crumbling Genome: The Impact of Deleterious Mutations on Humans (Wiley-Blackwell, Hoboken: ). [Google Scholar]
- 97.Smith JM & Haigh J (1974) The hitch-hiking effect of a favourable gene. Genet Res 23(1):23–35. [PubMed] [Google Scholar]
- 98.Hermisson J & Pennings PS (2005) Soft sweeps: molecular population genetics of adaptation from standing genetic variation. Genetics 169(4):2335–2352. [DOI] [PMC free article] [PubMed] [Google Scholar]