

Abstract
Neurodegenerative, neurodevelopmental and neuropsychiatric disorders are among the greatest public health challenges, since many lack disease-modifying treatments. A major reason for this lack in effective therapies is our limited understanding of the causative molecular and cellular mechanisms. Genome-wide association studies are providing a growing catalogue of disease-associated genetic variants. The next challenge is to elucidate how these variants cause disease, and to translate this understanding into therapies. This review describes how recently developed CRISPR-based functional genomics approaches can uncover disease mechanisms and therapeutic targets in neurological diseases. The bacterial CRISPR system can be used in experimental disease models to edit genomes and to control the expression levels of genes, using CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa). These genetic perturbations can be implemented in massively parallel genetic screens to evaluate the functional consequences for human cells. CRISPR screens are particularly powerful in combination with induced pluripotent stem cell (iPSC) technology, which enables the derivation of differentiated cell types, such as neurons and glia, and brain organoids from cells obtained from patients. CRISPRi/CRISPRa-based modelling of disease-associated changes in gene expression can pinpoint causal changes. Genetic modifier screens can elucidate disease mechanisms, causal determinants of cell-type selective vulnerability, and identify therapeutic targets.
Introduction
Over the last decade, advances in next-generation sequencing have enabled the identification of genetic variants associated with the risk of many diseases, including neurodegenerative, neurodevelopmental and neuropsychiatric disorders. While the catalogue of such risk variants is growing rapidly, functional studies that uncover how these variants contribute to disease are lagging far behind1–3. A functional and mechanistic understanding will be key to translating findings from human genetics to effective therapies.
This review outlines current challenges and presents how the marriage of two recently developed technologies, CRISPR-based functional genomics and human iPSC-derived disease models, provides a new approach to systematically elucidate disease mechanisms and therapeutic targets for neurological disorders. The technologies are introduced and distinct applications with relevance to neurology are described: the elucidation of disease mechanisms using patient-derived cells and isogenic controls, the modelling of disease-associated changes in gene expression to pinpoint disease-relevant genes, the identification of causal determinants of cell-type selective vulnerability to disease, and genetic modifier screens for the identification of therapeutic targets. The review concludes with a discussion of future challenges and opportunities.
Current challenges
Despite the major advances in identifying human genetic variants linked to neurological disease, the elucidation of the mechanisms by which these variants – as well as non-genetic risk factors – cause disease remains a formidable challenge1. This section enumerates central mechanistic questions in neurological disease and describes limitations of two widely used approaches to answering those questions, namely studies in patient tissues and in mouse models.
Key mechanistic questions
The discovery of a new genetic risk variant raises a series of mechanistic questions (Fig. 1):
Figure 1: Key mechanistic questions in neurological disease.
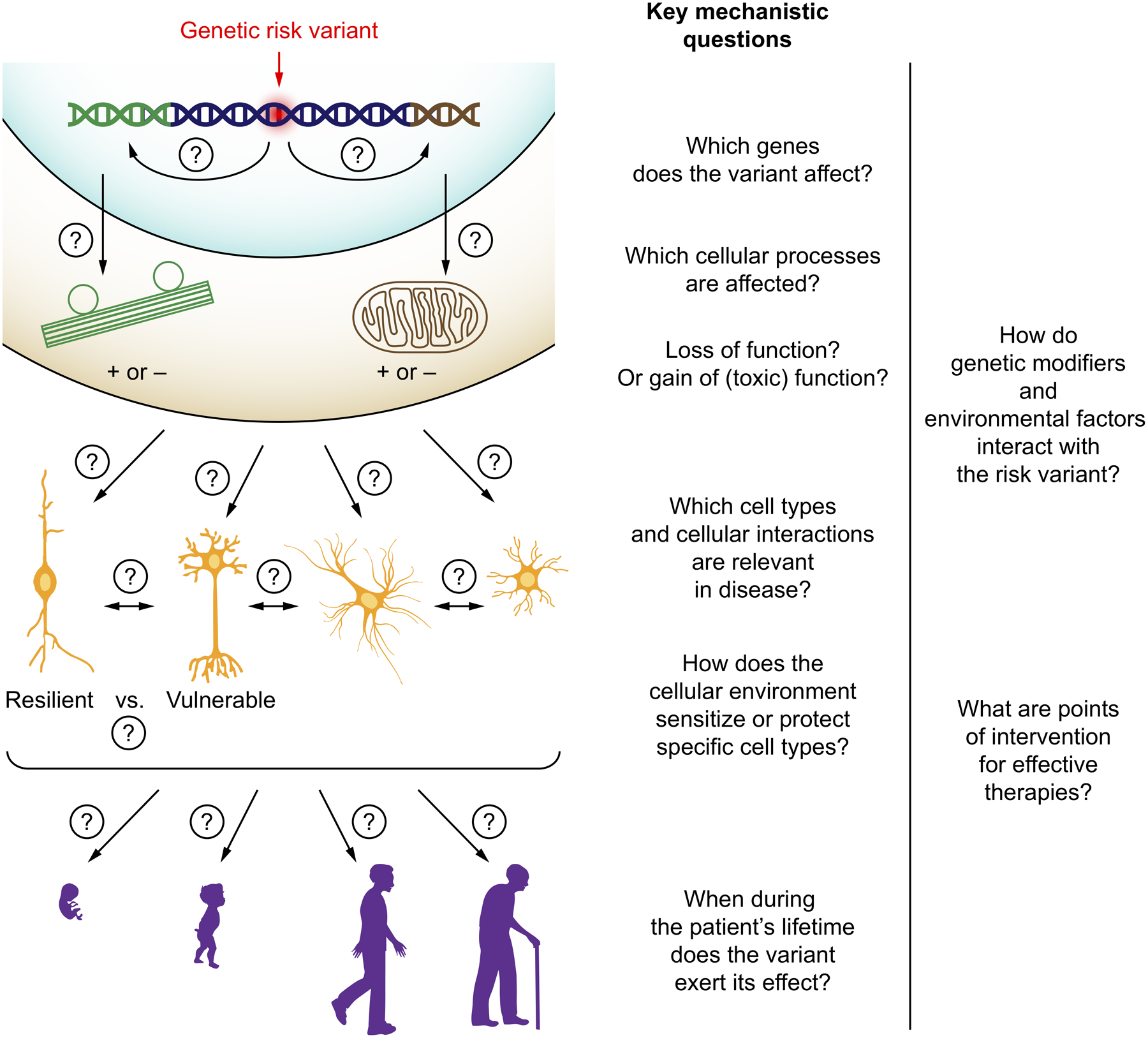
When a disease-linked genetic variant has been identified, key mechanistic questions need to be answered. See main text for details.
If the variant is not located within a coding sequence, which gene or genes does it affect?Many risk variants identified in genome-wide association studies are located outside coding regions, and are typically thought to affect the expression of one or several coding genes. However, it is challenging to pinpoint the disease-relevant genes affected by a risk variant. While the nearest coding gene is often reported when a new risk variant is identified, this approach does not necessarily name the disease-relevant gene. For example, an Alzheimer’s Disease risk locus initially attributed to the CELF1 gene was later shown to affect expression of the myeloid lineage transcription factor SPI1/PU.14. Intriguingly, variants in enhancers can exert cell-type specific effects on gene expression, as exemplified by Alzheimer’s Disease variants that affect BIN1 expression specifically in microglia, but not neurons or astrocytes5.
Which disease-relevant cellular processes are impacted by the genetic changes?For many human genes, the molecular and cellular functions of the gene product are not well understood, and many genes have multiple functions – therefore genetic changes can have pleiotropic effects, and a major challenge is to understand which of these effects are relevant for disease.
Do the changes represent a loss of function, a gain in function (possibly a gain in a novel toxic function, such as a toxic protein aggregate), or a combination of the two?The challenge of answering this question is illustrated by functional studies of the most frequent genetic cause of amyotrophic lateral sclerosis and frontotemporal dementia, a hexanucleotide repeat expansion within the C9orf72 gene. Experimental results support toxicity of RNAs containing the expanded repeat, as well as toxicity of dipeptide repeat translated from these RNAs, but also to haploinsufficiency of the C9orf72 protein as a cause of disease6. Intriguingly, these two mechanisms may synergize in disease7.
Through which cell types or cellular interactions does the variant cause disease?Risk variants can affect distinct processes in several cell types. Studies in disease models have shown that mutations in SOD1 associated with amyotrophic lateral sclerosis affect not only motor neurons, but also non-neuronal cells in ways that promote neurodegeneration8, and that the APOE ε4 variant associated with increased risk of Alzheimer’s Disease causes functional changes in neurons, astrocytes and microglia9. A major challenge is to deconvolute the contributions of different cell types and their interactions to disease.
Which factors in the cellular environment make some cell types selectively vulnerable to the effects of the genetic variant, and others resistant?The selective vulnerability of specific cell types is a hallmark of many neurological diseases and drives disease-specific symptoms. Disease-associated mutations are often found in proteins that are expressed broadly (such as tau, which is expressed in all neurons), yet specific subpopulations are selectively affected by downstream processes (such as the Von Economo neurons in frontotemporal dementia10). An understanding of the factors that make some neurons vulnerable and others comparatively resilient to disease could inform therapeutic strategies aimed at increasing resilience of vulnerable neuronal populations.
At which point in human development or aging does the variant exert its effect?This question is highly relevant for the development of effective therapies: if processes driving age-associated neurological diseases are initiated early in life, therapeutic intervention may need to start similarly early.
Through which mechanisms do genetic modifiers or environmental risk factors interact with the genetic variant?Some genetic risk factors show a striking interaction with other factors. For example, the APOE ε4 variant, which is the most common genetic risk factor for Alzheimer’s Disease, has a significantly stronger effect on disease risk in women than in men11,12, and a differential disease association in different ethnic groups13. The underlying mechanisms are not understood.
Answers to these questions have important implications for our understanding of disease mechanisms, and should clarify which therapeutic strategies are likely to be effective and disease-modifying.
Limitations of patient tissue studies
The characterization of human brain tissue has been transformed by recent advances in analytical techniques such as single-nuclear transcriptomics14, spatial transcriptomics15, multiplexed immunofluorescence16 and proteomics17. These approaches can yield a molecular description of neurological disease states at an unprecedented level of detail.
However, biopsies of the relevant tissues of the nervous system are generally not possible, and studies rely on post-mortem tissue. Due to the short-lived and non-expandable nature of human post-mortem brain tissue, studies are generally correlative, since experimental perturbations that establish causality are rarely feasible.
Similarly, advances in imaging technologies, including magnetic resonance imaging and positron emission tomography with specific molecular tracers, have enabled longitudinal tracking of disease progression in patient brains, but interventions to establish causality are only possible in the context of clinical trials.
Limitations of animal models of disease
Historically, mouse models have been widely used to investigate mechanisms of human diseases. Mice combine several advantages, including relative ease of genetic manipulation and moderate cost, the ability to study neuronal circuits and to monitor behavioral and cognitive phenotypes. They have provided important insights into neurological diseases, and can serve as pre-clinical models to evaluate new therapeutic approaches. However, mouse models for neurological disease also have several limitations.
First, there are differences between disease-relevant cell types, such as neurons and glia, between humans and mice. For example, dopaminergic neurons, which are selectively vulnerable in Parkinson’s Disease, contain substantially higher levels of dopamine in humans compared to mice, and some cellular disease processes have been proposed to depend on the high levels of dopamine found only in human dopaminergic neurons18. Even more poignantly, the type of human neuron identified as selectively vulnerable in frontotemporal dementia, the von Economo neuron, does not exist in mice10. Furthermore, mouse and human microglia, which have recently emerged as key players in Alzheimer’s Disease and other neurodegenerative diseases, show differences in particular in the expression of disease-relevant genes19, and they adopt distinctly different states in human diseased brains compared to mouse models20.
Second, there are genetic differences between humans and mice that complicate the modelling of disease variants. Coding variants that are disease-causing in humans may not cause disease in mice: the A53T mutation in alpha-synuclein is linked to Parkinson’s Disease in humans but corresponds to the wild-type residue in mice, and mouse models of A53T alpha-synuclein rely on overexpression of human proteins21. Regulation of disease-relevant genes can differ substantially between mice and humans22. Non-coding risk variants in humans often occur in non-conserved sequences in the genome, precluding direct mouse modeling of these variants, and it is challenging to determine through which coding gene or genes these variants affect disease risk3. Therefore, it is not obvious how these variants should be modelled in mice.
Major limitations of mouse models can be overcome in nonhuman primate models of neurological diseases. Nonhuman primates mirror humans much more closely in terms of their genome sequence, brain and circuit architecture than mice do, and they possess a complex behavioral and cognitive repertoire that facilitates evaluation of phenotypes relevant to neurological disease. Unlike mice, aged nonhuman primates can naturally develop symptoms reminiscent of human Alzheimer’s Disease (including cognitive and memory deficits and Aβ plaques23) and Parkinson’s Disease (including dysfunction of the nigrostriatal system and motor impairments24). An important breakthrough paving the way for genetically engineered disease models in nonhuman primates was the lentiviral delivery of a transgene that was then transmitted through the germline in marmosets25. More recently, precise genome editing techniques based on zinc finger nucleases, transcription activator-like endonucleases (TALENs) or CRISPR were successfully implemented in nonhuman primates26–28, and used to generate models of neurological and related diseases: disruption of the dystrophin gene to model Duchenne muscular dystrophy29, disruption of the MCPH1 gene to model microcephaly30, and disruption of the MECP2 gene to model Rett syndrome31.
However, experiments in animals, in particular non-human primates, are time-consuming, costly, and have important ethical considerations, limiting their scalability for the investigation of the large number of disease-associated variants that are being uncovered.
CRISPR-based gene perturbation
CRISPR technology is based on a prokaryotic antiviral defense system characterized by DNA elements termed CRISPRs (clustered regularly interspaced short palindromic repeats) and CRISPR-associated (Cas) proteins. CRISPRs are derived from bacteriophage genomes and generate RNAs that function with Cas proteins to cleave bacteriophage genomes upon infection32. The discovery, made less than 10 years ago, that a simple two-component system of a Cas protein (Streptococcus pyogenes Cas9) bound to a single guide RNA (sgRNA) can function as a programmable nuclease33 that can be deployed to cleave specific sequences in mammalian genomes34,35 has been transformative for biomedical research and the development of gene therapies.
Genome editing
The sequence of mammalian genomes can be edited by exploiting an endogenous DNA repair pathway termed homologous recombination. When a double-stranded break is introduced into genomic DNA, cells can repair the break by using a DNA molecule with high sequence similarity as a template. If an artificial template is provided while the DNA break is introduced, this pathway provides a mean to incorporate desired DNA sequences into the genome34,35 (Fig. 2a). For example, a disease-associated point mutation could be changed to the non-disease associated sequence using this strategy. Prior to the development of CRISPR technology, the targeted introduction of DNA breaks was a major challenge, since bespoke site-specific nucleases had to be engineered for each target sequence. CRISPR enables the rapid generation of a site-specific nuclease by combining the Cas9 protein (or functionally similar proteins from other microbial species) with a single guide RNA (sgRNA) that directs Cas9 to the desired target site.
Figure 2: CRISPR-based approaches to alter or perturb genes.
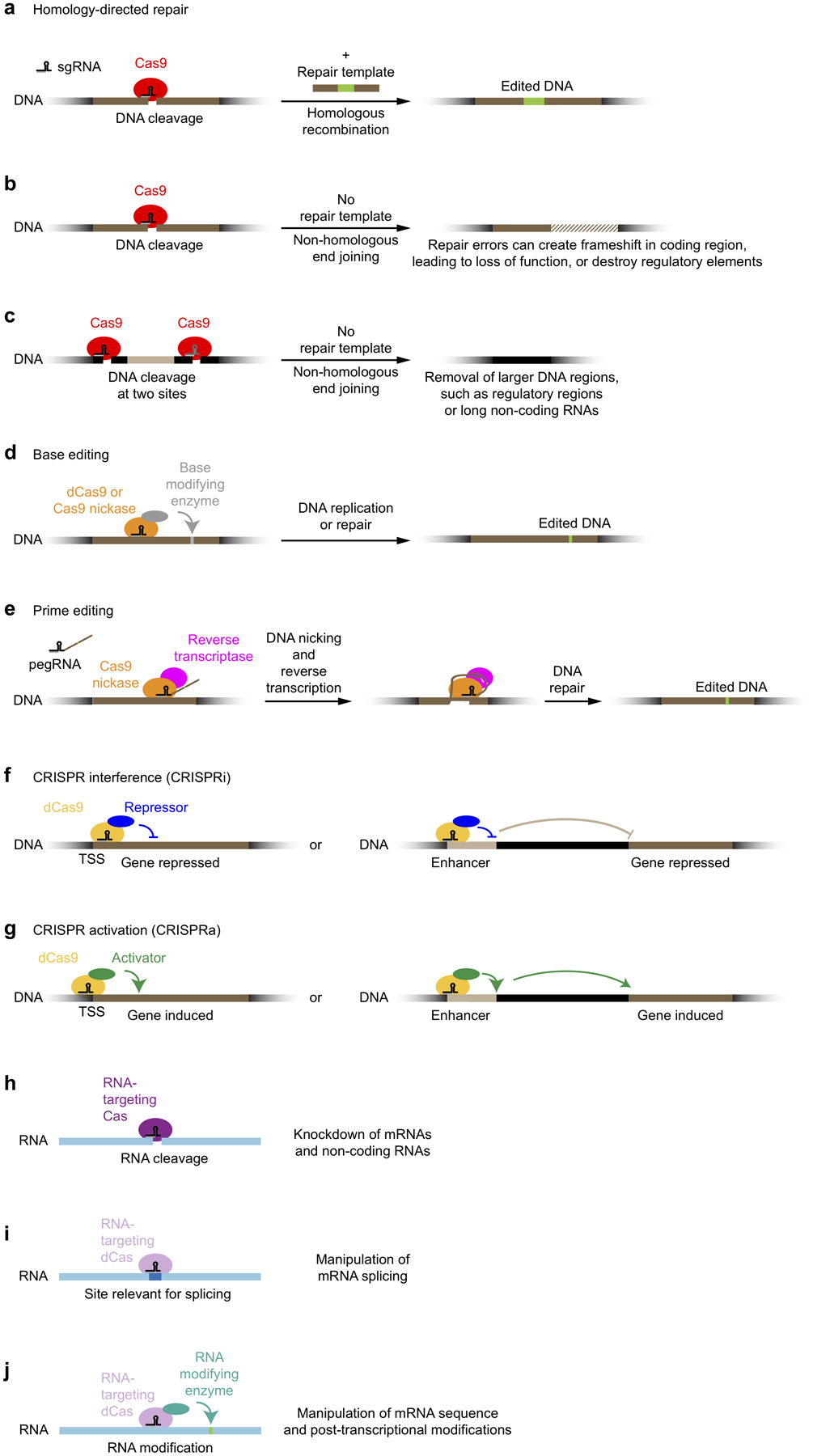
(a) Gene editing based on homology-directed repair
(b) Gene disruption based on small deletions introduced by non-homologous end-joining
(c) Removal of larger DNA regions after introduction of two DNA breaks
(d) Gene editing using base editors
(e) Prime editing
(f) CRISPR interference: Gene knockdown by transcriptional repressors recruited by dCas9
(g) CRISPR activation: Gene induction by transcriptional activators recruited by dCas9
(h) mRNA knockdown using RNA-cleaving Cas proteins
(i) Control of alternative mRNA splicing by RNA-targeting dCas proteins
(j) Post-transcriptional mRNA editing or modification by effectors recruited by RNA-targeting dCas proteins
In addition to its use as a precision tool for genome editing, CRISPR can also be deployed experimentally to knock out genes for functional studies. For this use, DNA breaks are introduced without providing a repair template, triggering repair through the non-homologous end-joining pathway. This pathway can result in perfect repair, recreating the original DNA sequence, which can be targeted again for cleavage by Cas9. However, repair errors can occur and lead to short deletions34,35. These deletions can cause frameshifts in the encoded protein, resulting in complete loss of function (Fig. 2b). Similarly, targeting of non-coding regions can reveal regulatory cis elements36 (Fig. 2b). Simultaneous delivery of two sgRNAs can result in deletion of larger DNA regions34,35, providing an additional strategy for the interrogation of regulatory elements34,35,37 or long non-coding RNAs38 (Fig. 2c).
An alternative CRISPR-based strategy for genome editing is the use of so-called base editors. Base editors recruit enzymes that chemically modify DNA bases to specific sites via a catalytically inactive version of Cas9, termed dCas9 (dead Cas9), or via a Cas9 nickase, in which one of two catalytic sites is inactivated, resulting in cleavage of only one of the DNA strands39. These base editors introduce changes that result in altered base pairing upon DNA replication or repair and thereby change the DNA sequence (Fig. 2d). Cleavage of the non-edited DNA strand by the Cas9 nickase can promote the desired repair outcome39.
A recent innovation in CRISPR-based genome editing is the so-called prime editing approach40 (Fig. 2e). A prime editor consists of Cas9 nickase fused to reverse transcriptase, and acts in complex with a prime editing guide RNA (pegRNA). The pegRNA combines an sgRNA targeting a site in the genome with a template for reverse transcriptase that contains the desired edit. During prime editing, a targeted genomic locus is nicked, the pegRNA hybridizes with the nicked DNA strand, and sequences encoded in the pegRNA are reverse-transcribed and inserted into the nicked locus by cellular repair pathways (Fig. 2e). Compared with editing approaches based on homologous recombination, prime editing has an increased rate of desired editing outcomes40.
Controlling gene expression
The CRISPR/Cas system can also be used to perturb gene function transiently, without changing the sequence of the genome. For this purpose, dCas9 is targeted to specific genomic sequences by an sgRNA; however, it does not introduce DNA breaks. Instead, it can serve as a recruitment platform for other proteins that affect gene expression.
CRISPR interference (CRISPRi).
dCas9 can be used to recruit transcriptional repressor domains (most commonly the KRAB domain, or epigenetic modifiers) to transcription start sites in the human genome to repress gene transcription (Fig. 2f). This approach is called CRISPR interference (CRISPRi)41. CRISPRi can achieve very stringent levels of knockdown (in some cases greater than 99%) of both coding and non-coding RNAs in human cells42, but it can also be used to investigate functional consequences of partial reduction in gene expression. The ability to knock down gene expression to different degrees by CRISPRi enables the investigation of the function of essential genes (knockout of which would not be compatible with live cells), and the modeling of reduced gene expression in disease states, or partial pharmacological inhibition of a cellular function. Since CRISPRi acts at the level of the transcription start site, it can selectively knockdown specific transcripts for genes with multiple transcription start sites. However, if a gene is simultaneously transcribed from multiple start sites in a cell, complete knockdown of all isoforms may not be possible with a single sgRNA. A potential source of off-target effects in CRISPRi experiments are bidirectional promoters, targeting of which can both neighboring genes43. However, CRISPRi has substantially reduced off-target effects compared with RNA interference, a previous knockdown technology41,42. CRISPRi can also be used to interrogate the function of distal regulatory elements such as enhancers44,45 (Fig 2f).
CRISPR activation (CRISPRa).
The dCas9-sgRNA complex can also serve as a recruitment platform for transcriptional activator domains to specific transcription start sites in the genome, effectively creating a highly specific custom transcription factor (Fig. 2g). This approach is called CRISPR activation (CRISPRa), and a variety of related strategies have been developed to recruit different transcriptional activation domains to a target site of interest41,46–50. Using CRISPRa, endogenous genes can be induced and overexpressed to varying degrees to evaluate functional consequences. For some genes, CRISPRi knockdown and CRISPRa knockdown will result in opposite phenotypes42. However, CRISPRi and CRISPRa frequently give distinct and complementary insights into gene function42. dCas9 fused to the histone acetyltransferase p300 enables the activation of distal regulatory elements49 (Fig. 2g).
Targeting mRNAs
Beyond its natural DNA-cleaving activity, Cas9 can be engineered to cleave RNAs51 and deployed to degrade specific mRNAs in mammalian cells52 (Fig. 2h). Furthermore, CRISPR systems from some microbial species naturally target RNA for cleavage53–57. While cleavage of mRNAs provides an alternative strategy for gene knockdown, catalytically dead versions of RNA-targeting Cas proteins can be used to manipulate mRNAs in other ways, for example to affect alternative splicing of mRNAs such as tau55, which is relevant for neurodegenerative diseases (Fig. 2i), or to control mRNA sequence and post-transcriptional modifications55 (Fig. 2j).
CRISPR screens in mammalian cells
The fact that CRISPR-based tools can be programmed by a short sgRNA to target genes of interest has enabled the scaling of CRISPR-based gene perturbation to genome-wide screens. Such screens can query a large set of genes (such as all ~20,000 protein-coding human genes) for their contribution to a cellular function of interest. This section will provide an overview of different modes of screening and highlight examples of recent CRISPR screens with relevance to neurological disease.
Pooled genetic screens
The most scalable strategy for CRISPR-based genetic screens is pooled screening. This enabled the rapid development of screening platforms based on different CRISPR-based tools, including CRISPR knockout58–61, CRISPRi42 and CRISPRa42,50. Pooled screens take advantage of two technologies: first, the synthesis of complex pools of oligonucleotides, which are used to generate pooled sgRNA expression libraries, and second, the monitoring of those libraries in cells using next-generation sequencing. Pooled screens can be used to evaluate a range of cellular phenotypes, as delineated below.
Screens based on survival/proliferation and sensitivity to insults.
Conceptually, the simplest type of pooled screen is one based on cellular survival or proliferation (Fig. 3a). In this paradigm, sgRNA libraries are introduced into cultured cells, and samples of the cells are collected at different time points. Quantification of the frequencies of cells expressing specific sgRNAs reveals genes perturbation of which affects rates of proliferation or survival. An important application is the systematic determination of genes essential for specific cancer cell lines.
Figure 3: Types of genetic screens in human cells.
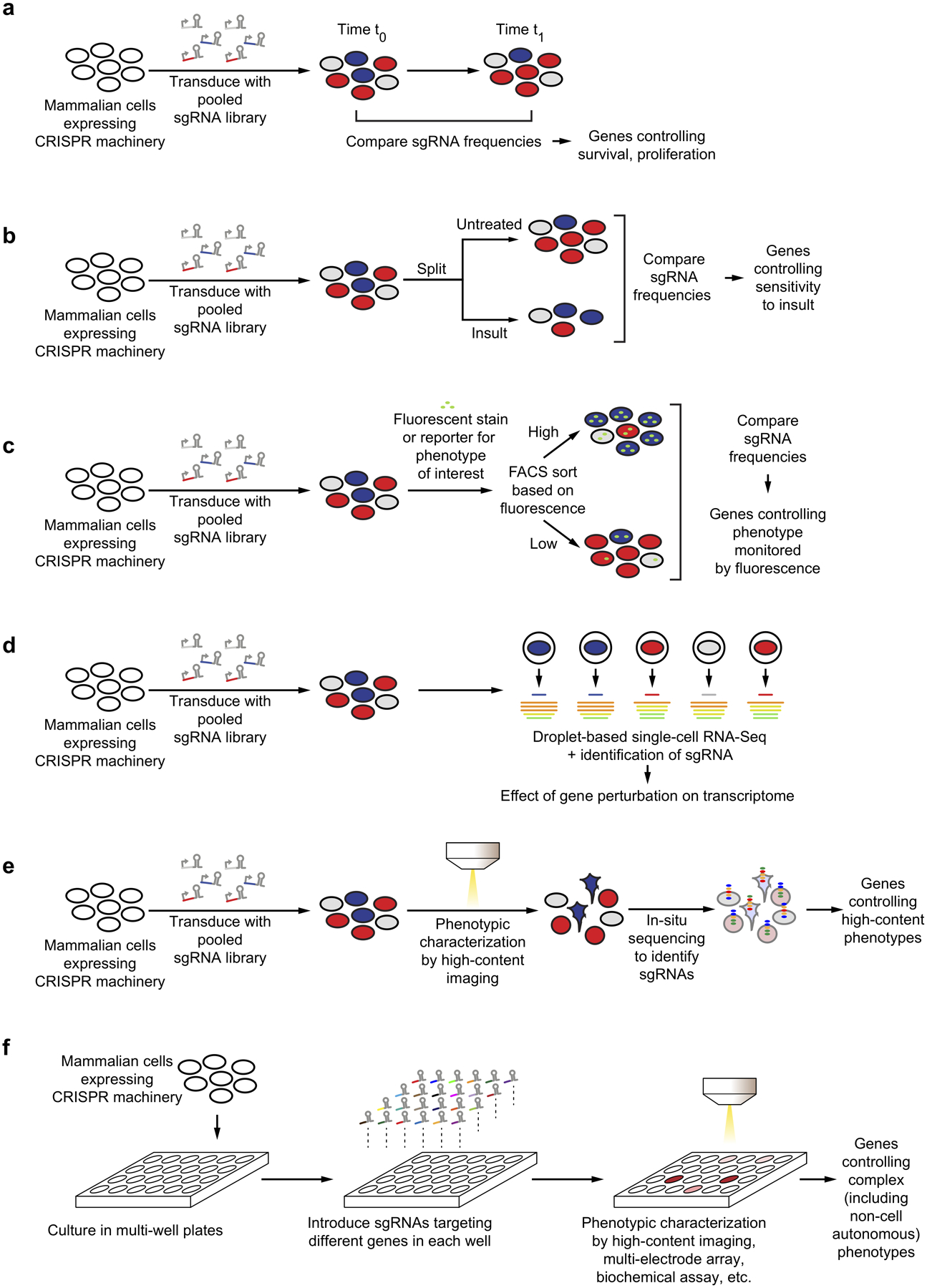
(a) Pooled screen for genes controlling cellular proliferation / survival.
(b) Pooled screen for genes controlling the sensitivity to insults, such as stressors, toxins, drugs.
(c) Pooled FACS-based screen for genes controlling cellular phenotypes monitored by fluorescent reporters or probes.
(d) Pooled screen coupled to single-cell RNA sequencing, to identify consequences of gene perturbation for high-dimensional transcriptomic phenotypes.
(e) Pooled optical screen for genes affecting cellular phenotypes monitored by microscopy.
(f) Arrayed screen for complex phenotypes, such as non-cell autonomous phenotypes or neuronal activity.
Survival-based screens can be carried out in parallel under standard culture conditions and in the presence of a toxic insult (Fig 3b). Such a screen design will reveal genes controlling sensitivity and resistance to the insult, and there are numerous applications to medically relevant questions. The insult can be a cytotoxic compound, such as an anti-cancer drug, for which genetic modifier screens can reveal the mechanism of action, potential biomarkers to stratify patients, and possible mechanisms of drug resistance62. The insult can be a stress condition, with the goal of identifying cellular pathways mediating stress sensitivity or resistance. The insult can also be the expression of a disease-linked gene that affects cellular survival or a toxic agent linked to disease, such as a pathogen or toxic protein species, in order to uncover the mechanisms underlying toxicity, genetic modifiers, host factors or potential therapeutic targets. An example with relevance to neurodegenerative disease is a recent screen for genes modulating the sensitivity of human cells to toxic dipeptide repeats63, which have been proposed to contribute to disease in ALS and frontotemporal dementia patients with repeat expansions in C9orf72. This screen pointed to nucleocytosolic transport as an important target for dipeptide repeat toxicity, but also uncovered a role for endoplasmic reticulum (ER) stress and showed that knockdown of the ER-localized thioredoxin TMX2 was protective for cells exposed to dipeptide repeats63.
Screens based on cellular functions.
Many cellular functions of interest cannot be captured by survival-based screens. However, these functions can still be interrogated in a pooled screen if they can be coupled to physical separation of cells. Most commonly, this is achieved by fluorescent activated cell sorting (FACS). FACS can separate cells based on fluorescent signals, which in turn can report on the level of proteins of interest (which are detected by fluorescently labeled antibodies), cellular activities monitored by fluorescent probes (which can be small molecules or genetically encoded reporters), or the uptake of fluorescently labeled molecules. In such screens, pools of cells expressing different sgRNAs are separated by FACS based on the biological readout of interest, and next-generation sequencing determines the frequencies of cells expressing each sgRNA in the separated populations to reveal genes controlling the process of interest (Fig. 3c). In a variation of this approach, magnetic activated cell sorting (MACS) can be used to separate cells64.
A number of recent studies have applied such screening strategies to uncover genes controlling processes relevant to neurological diseases. Several recent screens addressed mechanisms underlying the prion-like spreading of tau aggregation, which has been proposed to drive disease progression in Alzheimer’s Disease and primary tauopathies. The uptake of tau into cells was found to be mediated by specific heparan sulfate proteoglycan species65,66. After uptake, tau seeds need to escape the endolysosomal pathway to template aggregation of cytosolic tau. The ESCRT complex was found to control integrity of the endolysosomal compartment, and thereby escape of tau to the cytosol67. Cellular levels of the Parkinson’s Disease-linked protein alpha-synuclein were found to be modulated by a large number of genes, including doublecortin-like kinase 168, knockdown of which also mitigated a-synuclein toxicity69. Expression of the protein parkin, defects in which are associated with Parkinson’s Disease, was found to be negatively regulated by the transcription factor THAP1170. The RNA helicase DDX3X was found to control repeat-associated non-AUG (RAN) translation of C9orf72 hexanucleotide repeat expansions71. A screen for regulators of phagocytosis uncovered a role for the Alzheimer’s Disease risk gene TM2D3 in modulating uptake of Aβ aggregates64.
Screens based on gene expression.
While screens based on survival or FACS monitor low-dimensional phenotypes, pooled CRISPR screens can also be coupled to single-cell RNA sequencing to provide high-dimensional readouts of the effects of gene perturbation72–75 (Fig. 3d). Such screens provide rich information on cellular processes such as differentiation72, immune responses72,73,75, stress responses74 and compensatory responses76 and enable high-throughput mapping of regulatory elements77.
Pooled optical screens.
Complex morphological phenotypes that cannot be detected by FACS, but are apparent by fluorescence microscopy, can be evaluated in pooled optical screens78,79. In such screens, sgRNA libraries are introduced into cells which are then fixed, stained and characterized by microscopy. Subsequently, the sgRNA expressed in each cell is identified by in-situ sequencing (Fig. 3e).
Arrayed genetic screens
Despite the broad utility of pooled screens, there are cellular phenotypes that cannot be interrogated using pooled strategies. The most obvious category of such phenotypes are non-cell autonomous phenotypes, in which the relevant phenotype readout is not physically coupled to the cell in which a gene is perturbed. Examples with relevance to neurological disease include the secretion of factors from cells (such as cytokines, neuropeptides or Aβ peptides), the excitation of target cells by neurons (and, by extension, the electrophysiological activity of a group of neurons), or interactions between neurons and glia. Furthermore, phenotypes related to cellular morphology can in theory be evaluated in pooled optical screens; in practice, however, it is difficult to segment microscopic images of cells with complex morphologies, such as neurons and glia, which is a prerequisite for assigning all parts of a cell to the correct sgRNA.
Phenotypes that are not compatible with pooled screens can be investigated in arrayed screens, for which sgRNAs targeting different genes are delivered separately to cells in different wells of a multi-well plate (Fig. 3f). A multitude of readouts can be applied to cells in this format, which mirrors small-molecule screens used for drug discovery. These include high-content imaging of fixed and stained cells, longitudinal live-cell imaging, measurements of electrophysiological activity by calcium or voltage imaging or multi-electrode arrays, quantification of secreted factors and other biochemical assays.
The most significant disadvantage of arrayed screening approaches is that they require the generation and delivery of arrayed sgRNA libraries and automated platforms for high-throughput measurement of the phenotypes of interest. While small-scale arrayed screens are commonly used as secondary screens to characterize top hits from a genome-wide primary screen, most academic labs do not have the resources to conduct large-scale arrayed screens. (An arrayed screen targeting all human protein-coding genes each with a single sgRNA would require handling, tissue culture, and analysis of approximately 200 96-well plates. A more robust experimental design would include at least two independent sgRNAs per gene in separate wells, and at least two technical replicates for each well, resulting in approximately 800 96-well plates.)
Systematic genetic interaction screens
Genes typically do not function in isolation, but in combination with other genes. Examples are genes encoding nodes in a signaling pathway, enzymes in a metabolic pathway, or subunits of a multi-component protein complex. While the simplest implementation of the screening strategies described above evaluates gene function by perturbing one gene at a time, it is possible to perturb combinations of genes in a systematic manner to quantify genetic interactions and discover functional relationships between gene products. This approach was pioneered in mammalian cells using RNA interference80,81, but CRISPR-based platforms have enabled the generation of substantially larger genetic interaction maps82, including CRISPRa-based gain-of-function maps83 and maps that combine gain- and loss-of-function perturbations to map directional pathways84.
Genetic interactions are of great interest to neurological disease, since genetic modifiers can yield insights into disease mechanisms and potential therapeutic strategies. Genetic modifiers for neurodegeneration have been identified successfully through human genetics, such as RAB10 for APOE65 in Alzheimer’s Disease (variants in the RAB10 locus are linked to resilience in individuals carrying the high-risk APOE ε4 allele85), and TMEM106B for GRN66, 67 in frontotemporal dementia (SNPs affecting TMEM106B expression are protective in carriers of disease-associated GRN mutations86,87). However, this approach is likely to be currently underpowered to identify many relevant genetic interactions. CRISPR modifier screens in cells with genetic risk variants and systematic genetic interaction maps have the potential to identify additional genetic interactions, the disease relevance of which will then have to be validated. A recent example for a genetic interaction screen with relevance to neurological disease is a screen for genes loss of which was synthetic lethal with C9orf72 loss88. The strongest synthetic lethal gene was FIS1, which encodes a protein that mediating mitochondrial fission and mitophagy, and follow-up experiments also pointed to a role in regulating inflammatory signaling88.
Screens in iPSC-derived disease models
The majority of CRISPR screens published to date, including those investigating processes relevant to neurological disease, were conducted in cancer cell lines. Even though such screens have been able to uncover genes that were then validated in relevant cell types such as neurons63,65,68, they have obvious limitations for the study of neurological disease. First, most neurological diseases are characterized by changes in the function of specific cell types of the nervous system, and such functions can only be appropriately studied in the relevant cell types. Second, even if aspects of the disease process can be modeled in other cell types, genetic screens may fail to detect disease-relevant genes if those genes are not expressed in the cell line used for the screen, or if the genes have cell-type specific roles. Third, cellular survival is a relevant phenotype for neurodegenerative diseases. However, survival of cancer cell lines is controlled very differently than survival of untransformed cells, and essential genes in cancer cells differ from those in stem cells or neurons76. These differences limit the usefulness of cancer cells to study cell death in neurodegeneration. As a case in point, there was little correlation between genes found to control sensitivity to toxic dipeptide repeats in cancer cells versus primary neurons63. Fourth, a key feature and important topic of research for several neurological diseases is the selective vulnerability of specific cell types. Screens aimed at uncovering the factors controlling selective vulnerability (and, by extension, potential therapeutic targets to reduce vulnerability) will have to be implemented in relevant cell types.
Primary human cells of the nervous system are difficult to obtain. Neurons in particular are post-mitotic and therefore non-expandable. An attractive, recently established approach is the derivation of relevant cell types from human iPSCs. This section introduces iPSC technology and discusses iPSC-derived disease models and their use for CRISPR screening.
iPSC technology
Primary cell types readily obtained from human adult donors, such as skin fibroblasts or peripheral blood cells, can be reprogrammed to become induced pluripotent stem cells (iPSCs) by the transient expression of four transcription factors, such as the so-called “Yamanaka factors”89,90 (Fig. 4a). More recently, iPSCs were successfully obtain from post-mortem human brain tissue, specifically dura mater fibroblasts91. Such induced pluripotent stem cells (iPSCs) can be expanded and differentiated into a variety of cell types, including neuronal subtypes and glia, by protocols that mimic environmental developmental cues, or express specific transcription factors (Fig. 4a) Fibroblasts can also be directly reprogrammed into differentiated cell types such as neurons92.
Figure 4: Induced pluripotent stem cell (iPSC) approaches to study neurological disease.
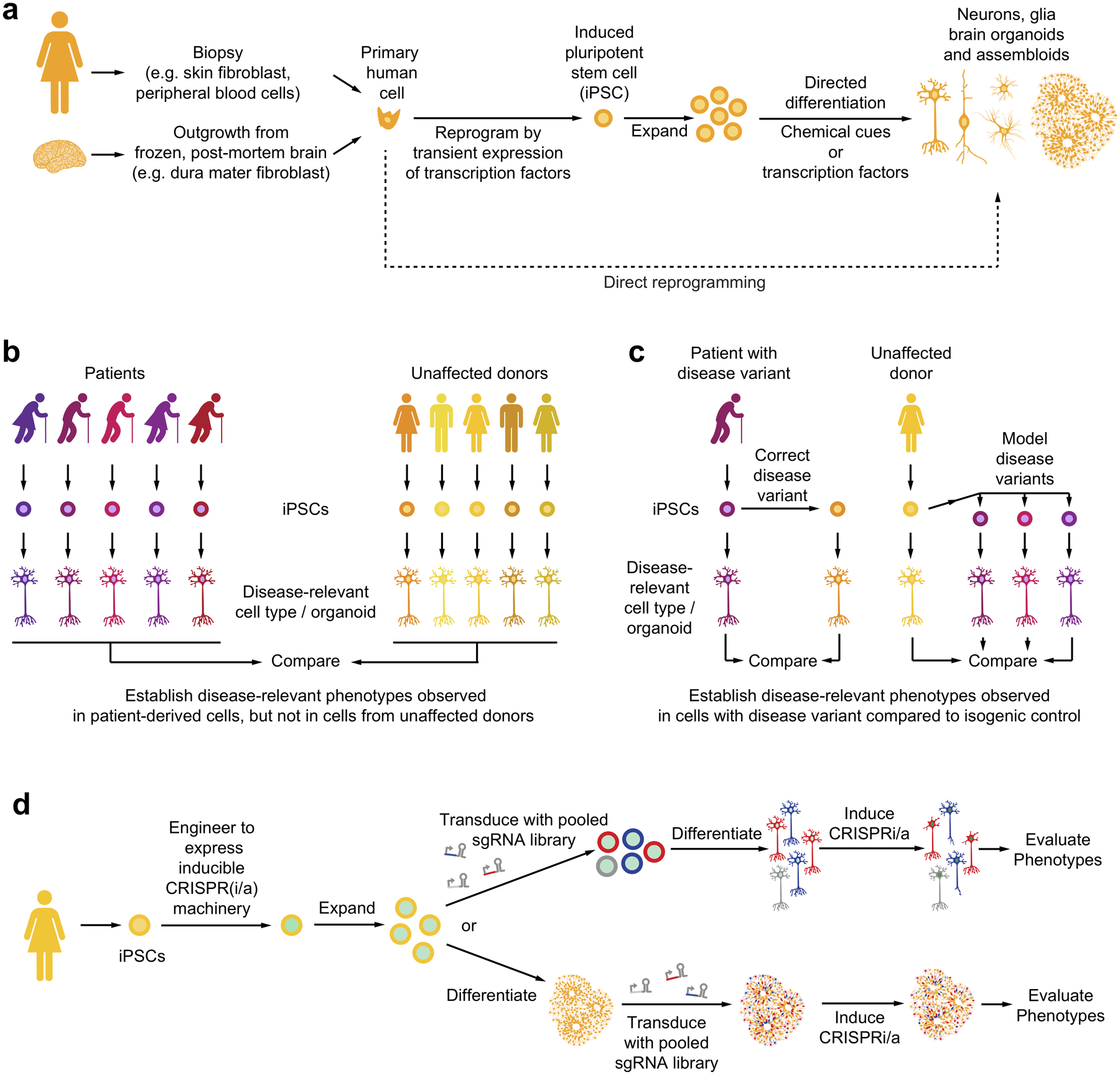
(a) Primary human cells can be reprogrammed to iPSCs and differentiated into neurons, glia and brain organoids.
(b) Panels of iPSC-derived cells from patients and unaffected donors can reveal disease-relevant cellular phenotypes specifically in patient-derived cells.
(c) CRISPR-based correction of genetic disease variants in patient-derived iPSCs or modeling of genetic disease variants in iPSCs from unaffected donors enables evaluation of phenotypes caused by disease variants in cells with isogenic backgrounds.
(d) Strategies for CRISPR-based genetic screens in iPSC-derived cell types or organoids.
Human pluripotent stem cells can also be differentiated into three-dimensional cultures recapitulating features of the human cortex93, termed brain organoids (Fig. 4a). Furthermore, iPSC-derived cell types from different lineages can be combined into defined three-dimensional cultures, termed assembloids9,94–98. For example, microglia can be introduced into brain organoids to investigate their interaction with other brain cell types9,94,96,98.
An important motivation for iPSC technology is the development of cell-based therapies in regenerative medicine. However, iPSC-derived cells also provide powerful disease models for biomedical research, as outlined below.
iPSC-derived cellular disease models
iPSCs can be derived from healthy donors as well as from patients with familial or sporadic diseases. Intriguingly, cells derived from patients with a variety of neurological diseases have been found to recapitulate aspects of the disease99 (Fig. 4b). For example, several cellular and molecular features of Alzheimer’s Disease have been recapitulated in iPSC-derived cell types. Neurons derived from familial as well as sporadic Alzheimer’s patient cells show enlarged early endosomes100, phosphorylated tau100, and increased production of Aβ peptides100 and oligomers101. Similar phenotypes are observed in neurons carrying the APOE ε4 risk allele9,102. APOE ε4 also affects properties of iPSC-derived astrocytes and microglia, notably reducing their ability to clear Aβ9,102. Neurodevelopmental disorders can be modeled in patient iPSC-derived brain organoids103,104.
A challenge in establishing robust cellular phenotypes that distinguish iPSC-derived cells from patients versus healthy donors is variability between donors, compounded by variability introduced during iPSC generation. Genome-editing technology has enabled a strategy to overcome these sources of variability: the generation of isogenic iPSC lines105, in which a disease-associated genetic variant is corrected in a patient line or introduced into a healthy donor line (Fig. 4c). Several studies related to neurological disease successfully reverted a disease-associated cellular phenotype in patient-derived cells by correcting the disease-associated mutation, for example in the MAPT gene106,107. Vice versa, CRISPR-based genome editing can be used to model panels of disease-associated genetic variants on the background of an parent line derived from an unaffected donor108. These two complementary strategies can test whether a genetic variant is necessary or sufficient, respectively, for disease-relevant cellular phenotypes. Importantly, linkage disequilibrium can make it difficult to assign causality to a specific genetic variant based on natural genetic variation in the human population, and the use of isogenic lines can overcome this problem.
A further technical challenge is variability introduced during differentiation, which can necessitate the screening of large numbers of cells in order to detect phenotypic differences with statistical significance. A strategy that can substantially reduce variability in differentiation is the use of protocols in which differentiation is driven by the inducible expression of transcription factors – in particular when the expression cassette for the inducible transcription factors is integrated in a defined genomic safe-harbor locus in iPSCs109.
CRISPR screens in iPSC-derived cells
Recently, we implemented large-scale CRISPR-based genetic screens for the first time in a human iPSC-derived differentiated cell type, namely neurons76. To this aim, CRISPRi machinery was expressed from a safe-harbor locus in iPSCs and sgRNA libraries were introduced via lentiviral infection. iPSCs were then differentiated into neurons, and screened for different phenotypes, including survival, single-cell transcriptomics, and morphology (Fig 4d). To control the time point of genetic perturbation, the expression of CRISPR machinery can be induced at later time points during differentiation76, or sgRNAs can be introduced post-differentiation (Fig. 4d). Several findings from these first screens in iPSC-derived neurons supported the importance of conducting CRISPR screens in relevant cell types.
First, the set of genes controlling survival of neurons were distinct from those controlling cancer cell or iPSC survival. CRISPR screening recovered a previously known key regulator of neuronal cell death, MAP3K12/DLK76.
Second, housekeeping genes that were essential for survival of both iPSCs and neurons may play different roles in the two cell types, based on vastly different transcriptomic consequences of their knockdown76. A prominent example was the ubiquitin-activating E1 enzyme UBA176. Intriguingly, UBA1 is widely expressed in the human body, but loss-of-function mutations are linked to neurodegenerative diseases110.
Third, an arrayed longitudinal imaging screens uncovered consequences of gene knockdown for neuron-specific morphological features, such as neurite length and branching patterns76, which could not have been evaluated in other cell types.
Several groups are actively developing CRISPR-based screening platforms in other iPSC-derived cell types. On a technical level, different modes of CRISPR perturbation will have specific advantages and disadvantages for applications in iPSC-derived cell types. CRISPR knockout strategies may result in stronger phenotypes and do not require ongoing expression of the CRISPR machinery. However, CRISPR-induced DNA breaks are particularly toxic to iPSCs111, reducing cell survival and potentially confounding the observed phenotypes. CRISPRi-based gene knockdown may result in weaker phenotypes than full gene knockout, however it is non-toxic in iPSCs and iPSC-derived cells76, inducible76 and reversible42, thereby enabling time-resolved analysis of gene function76. Partial knockdown of genes achieved by CRISPRi makes it possible to functionally characterize essential genes76. CRISPRa evaluates gain-of-function phenotypes and can provide complementary insight to CRISPR knockout or CRISPRi-based loss-of-function screens42. On a biological level, it will be highly instructive to directly compare the results of parallel screens in different cell types in order to elucidate cell type-specific roles of human genes in health and disease.
Applications to neurological disease
The recent development of CRISPR screens in human iPSC-derived cells has paved the way for applications to neurological disease. This section provides a roadmap and describes how four major disease-relevant questions could be addressed by such screens.
Mechanisms of disease variants
Genetic variants associated with familial disease or with disease risk in sporadic patients are being identified at a rapid pace. However, the mechanism by which these variants contribute to disease is often unknown or controversial. A mechanistic understanding of disease-linked variants is an important pre-requisite for the development of new therapeutic strategies.
CRISPR-based modifier screens in isogenic cell lines have the potential to provide insights into the mechanism through which a disease variant affects relevant cellular functions (Fig. 5). Ideally, such screens should be implemented in isogenic cell lines.
Figure 5: Elucidating disease mechanisms and therapeutic targets through modifier screens in patient-derived cells and isogenic controls.
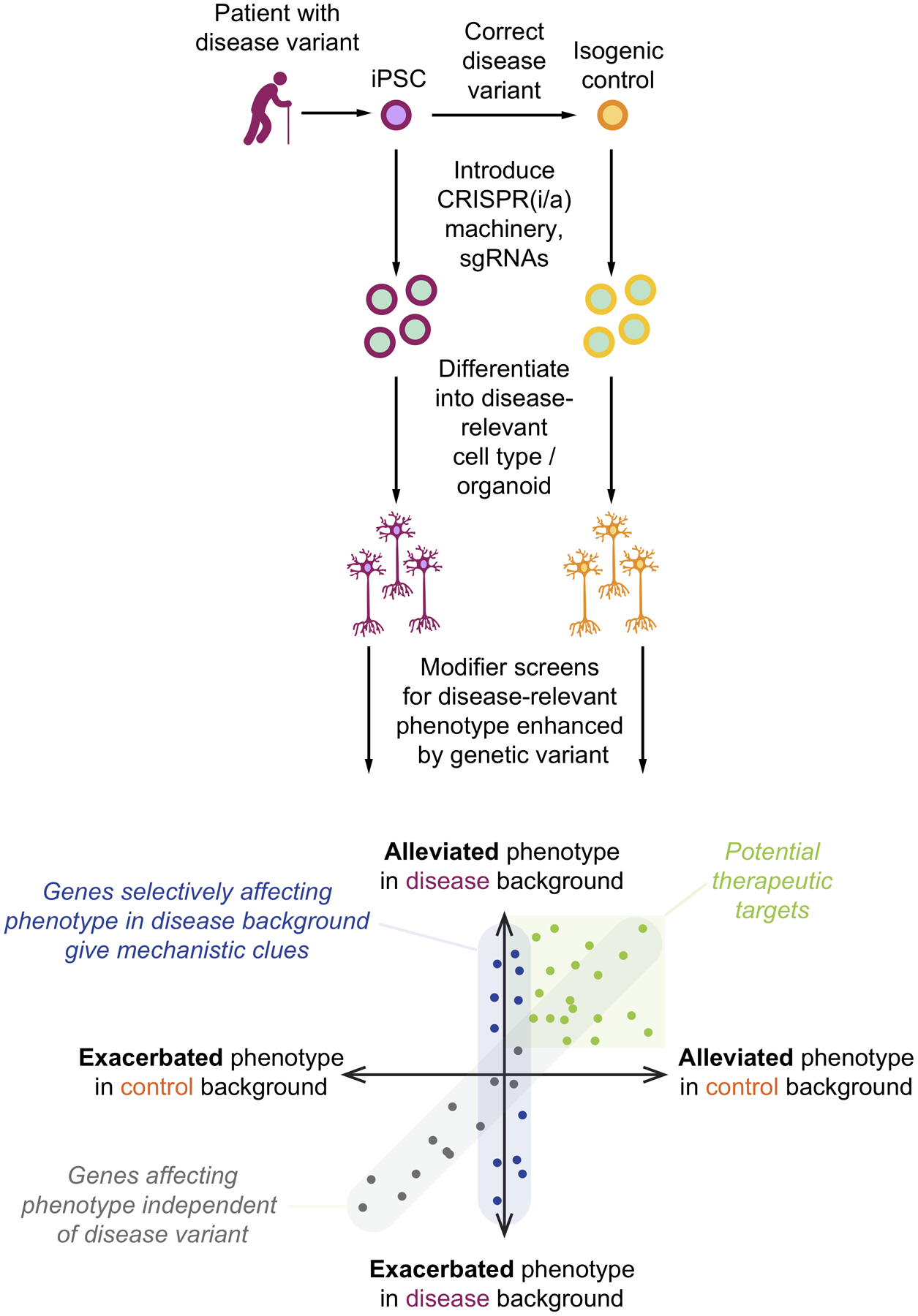
Strategy for parallel modifier screens in cells with a disease-associated variant and isogenic control cells for a disease-relevant phenotype observed more strongly in the disease background. Comparison of hits from the two screens can reveal three important classes of genes: Modifiers of the phenotype in both the disease and control background (grey dots), modifiers that alleviate the phenotype and are therefore potential therapeutic targets (green dots), and modifiers of the phenotype in disease background, but control cells (blue dots). This latter category can be thought of genetically interacting with the disease variant, and give clues about the mechanism by which the disease variant causes the phenotype.
The first challenge is to establish a disease-relevant phenotype observed in a disease-relevant cell type derived from patient iPSCs, but not in isogenic controls. Ideally, the phenotype should closely mirror a phenotype observed in patients – such as changes in neuronal or glial functions, survival, morphology or gene expression, or the aggregation of specific proteins.
The second challenge is to design a screen based on the phenotype of interest. Cell-autonomous phenotypes that can be monitored in survival- or FACS-based screens should be prioritized due to the scalability of such screens, which enables genome-wide screens. Screens based on single-cell transcriptomics yield rich phenotypes but are currently expensive to implement. Non-cell autonomous screens, or screens for complex functional or morphological phenotypes typically require arrayed screening approaches. Arrayed and single-cell transcriptomic screens will typically require the selection of a focused gene set for perturbation.
Next, parallel genetic modifier screens for the phenotype of interest are conducted in the isogenic cells (Fig. 5). Conceptually, the screen design is similar to screens for sensitivity to an insult (Fig. 2b) – except that the insult is the disease-associated genetic variant. In modifier screens for sensitivity to toxic drugs, knockdown of genes that are the direct target of the drug, or are relevant for the cellular function targeted by the drug, synergizes with drug treatment62. Identification of such drug-gene interactions has been a highly successful strategy for the identification of drug mechanisms of action62. Similarly, genetic modifiers of the phenotype caused by a genetic variant represent gene-gene interactions (in the case of survival screens, synthetic lethal genes), which can reveal the mechanism of action of the genetic variant (Fig. 5).
The final challenge is to validate the generality of findings from large scale genetic screens. Hit genes should be tested in larger panels of patient-derived cells, including, where available, different disease-associated mutations in the gene of interest. Ideally, the mechanistic hypothesis should lead to predictions that can be directly tested in patient tissue.
Discovery of therapeutic targets
An important motivation for the development of cell-based disease models is their use for drug discovery in arrayed phenotypic small-molecule screens. An alternative to pharmacological screening is genetic screening, which seeks to identify genetic perturbations that ameliorate disease-relevant phenotypes and thereby point to potential therapeutic targets (Fig. 5). Genetic screens can be an attractive alternative for two reasons:
First, while small-molecule screens need to be implemented in an arrayed format and therefore require automation infrastructure, genetic screens for some phenotypes can be implemented as pooled screens, making them scalable to genome-wide screens without requiring automation.
Second, while small-molecule screens yield compounds with beneficial effects, it is often a major challenge to identify their molecular targets and mechanism of action. By contrast, genetic screens directly identify relevant targets and mechanisms, for which effective compounds must then be developed. Importantly, genetic screens are also a strategy for the identification of the mechanisms of actions of novel compounds from phenotypic screens62.
To design a genetic screen for the discovery of therapeutic targets for neurological disease, the same challenges must be addressed as for the identification of disease mechanisms, described in the previous subsection. In fact, a well-designed modifier screen can simultaneously reveal disease mechanisms and therapeutic targets (Fig. 5).
Causative changes in gene expression
CRISPRi and CRISPRa enable the systematic control of the expression levels of specific genes in human cells. Therefore, they are ideal tools to dissect changes in gene expression that causally contribute to disease states. This strategy can be applied to answer several questions relevant to neurological disease.
First, GWAS studies frequently uncover disease-associated genetic variants outside protein-coding regions. Such variants can affect the expression of other genes, thus acting as expression quantitative trait loci (eQTLs). eQTLs can affect the expression of nearby, but also distant genes, and their effect can be cell-type and cell-state specific. Therefore, it is not trivial to determine the genes through which a disease-associated eQTL affects disease processes. Even if the set of genes affected in their expression by a genetic variant is known, it is not clear which of these candidate genes is relevant for disease. CRISPRi/a enables the manipulation of expression levels of candidate genes individually and in specific combinations in order to evaluate the consequences for cellular function (Fig. 6a). This strategy was recently implemented to analyze the effects of schizophrenia-associated variants on the function of iPSC-derived neurons112. While this study focused on variants thought to affect only the expression of one protein-coding gene each, future large-scale screens could investigate eQTLs that affect the expression of many genes, in order to uncover convergent phenotypes that point to those changes in gene expression that are likely causative in disease, and the cell types they act in (Fig. 6a).
Figure 6: Modeling disease-associated changes in gene expression to pinpoint causal changes.
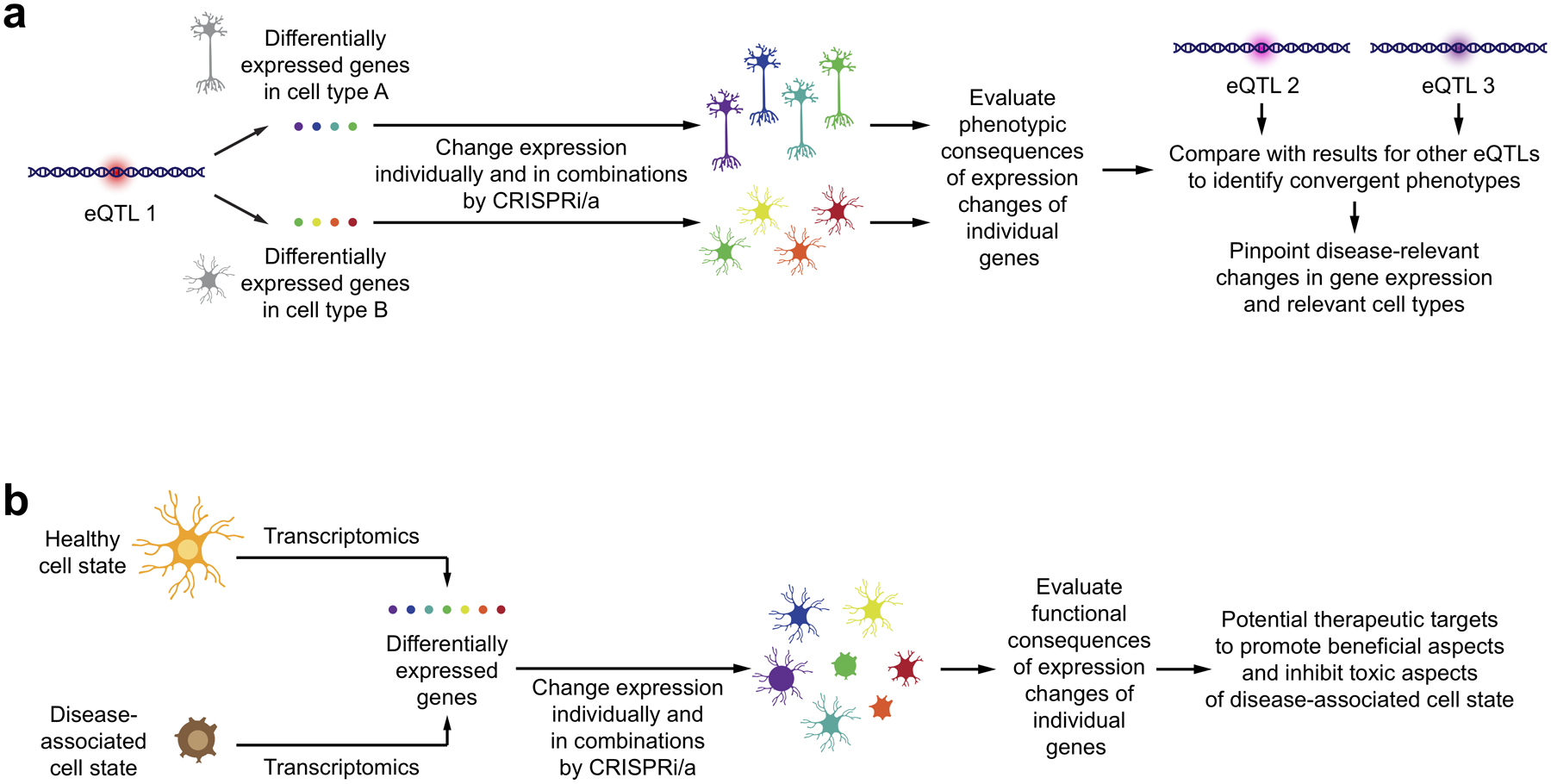
(a) Genetic variants constituting an expression quantitative trait locus (eQTL) can affect expression of different sets of genes in different cell types. To identify which of these changes are likely disease-causing, the expression of genes can be controlled individually or in combination in different cell types by CRISPRi/a. The resulting phenotypes can be characterized and compared to those observed for expression changes associated with other eQTLs linked to the same disease. Convergent phenotypes for genes affected by different eQTLs can point those expression changes likely to be causal, and to the cell types in which they likely act.
(b) Disease-associated states of cell types involve expression changes in multiple genes, some of which could be beneficial, while others could be detrimental in the context of disease. Controlling the expression of these genes in the relevant cell type individually and in combination using CRISPRi/a, functional consequences can be linked to specific genes, uncovering potential therapeutic strategies to enhance beneficial aspects and inhibit detrimental aspects of the disease-associated cell state.
Second, mouse models and single-cell and single-nuclear RNA sequencing of patient tissues have revealed disease-associated cell states, in particular of microglia20,113 and astrocytes114. Importantly, the phenotypes measured in CRISPRi/a screens with single-cell RNA sequencing readouts are transcriptomics signatures, which can be directly compared to transcriptomics signatures from patient tissues. These disease-associated states are characterized by expression changes in numerous genes, but it is generally not clear which of these changes have either beneficial or detrimental effects in disease. CRISPRi/a screens enable the targeted modeling of gene expression changes individually or in combination, in order to evaluate the consequences on cellular function in the relevant cell type (Fig. 6b). A detailed understanding of the genes expression changes of which are detrimental would prioritize these genes as potential therapeutic targets.
Cell type-selective vulnerability
A hallmark of many neurological disorders, in particular neurodegenerative diseases, is the selective vulnerability of specific cell types. For example, motor neurons are selectively vulnerable in amyotrophic lateral sclerosis (ALS). Intriguingly, the expression of ALS risk genes is not restricted to motor neurons, indicating that specific factors in the cellular environment of motor neurons make them more vulnerable to ALS. Conversely, mutations in alpha-synuclein can cause Parkinson’s Disease, where dopaminergic neurons in the substantia nigra are selectively vulnerable, but motor neurons are less vulnerable, even though alpha-synuclein is widely expressed in neurons. Hence, selective vulnerabilities of specific neuronal subtypes are disease-specific. An understanding of the cellular factors controlling selective vulnerability is highly desirable, since it would both illuminate mechanisms driving disease and uncover potential therapeutic strategies that could vulnerable neurons into resilient neurons.
The in-depth molecular characterization of neuronal subtypes in the human brain115 provides a detailed description of factors that are differentially expressed between vulnerable and resilient neurons. However, most differentially expressed factors are likely to reflect the different functional specializations of neurons, and it is not clear which of these factors are causal determinants of vulnerability to specific diseases.
As described in the previous subsection, CRISPRi/a-based control of the expression levels of individual genes in relevant iPSC-derived cell types makes it possible to enable the functional impact of expression changes. Thus, genes that are differentially expressed between vulnerable and resilient neuronal subtypes can be tested for effects on disease-relevant cellular phenotypes (Fig. 7). While it is likely that not all determinants of selective vulnerability are cell autonomous or amenable to modeling in iPSC-derived cells, recent studies highlight the promise of iPSC models of selective vulnerability. iPSC-derived GABAergic neurons were selectively vulnerable to expression of the e4 variant of APOE associated with Alzheimer’s Disease102. iPSC-derived dopaminergic neurons recapitulate molecular processes characteristic of Parkinson’s Disease, dependent on dopamine levels18.
Figure 7: Uncovering causal determinants of cell-type selective vulnerability.
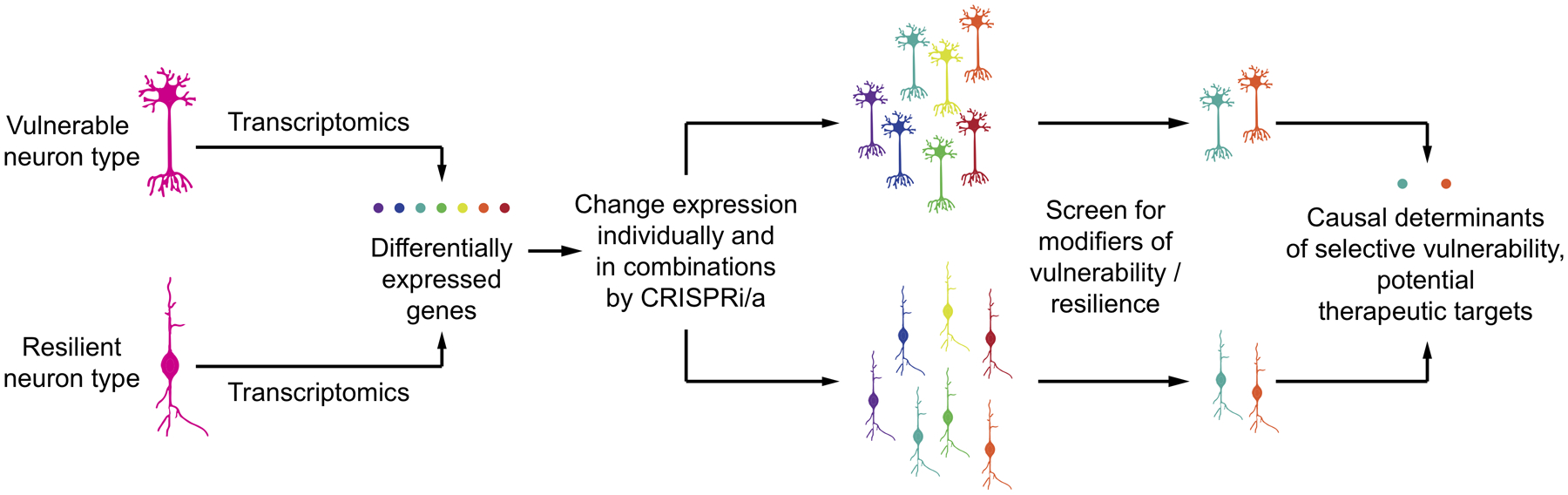
Transcriptomics approaches catalogue differentially expressed genes between vulnerable and resilient types of neurons. CRISPRi/a can be used to change the expression levels of these genes in iPSC-derived models of the vulnerable and resilient neuronal types to pinpoint those genes that are causal determinants of selective vulnerability. Such genes are potential therapeutic targets to turn vulnerable neurons into resilient neurons.
In conclusion, CRISPRi/a-based screens in iPSC-derived cells that systematically alter the expression levels of genes differentially expressed between vulnerable and resilient cell types have the potential to reveal causal determinants of selective vulnerability, which are potential therapeutic targets116.
Conclusions and future directions
Recent advances in CRISPR-based genetic screening and iPSC-based disease modeling, and especially the integration of these technologies have enormous potential for the discovery of mechanisms and therapeutic targets for neurologic disorders. CRISPR-based functional genomics enable the specific perturbation of genes of interests, thereby making it feasible to establish causality.
Several challenges remain. Some are technical in nature, such as the silencing of transgenes during iPSC differentiation for some cell types. A number of important challenges exist with respect to the faithful recapitulation of disease processes in iPSC-derived models.
First, we currently lack procedures to generate all disease-relevant cell types from iPSCs. The development of new differentiation protocols for cell types relevant to neurological disease is an active area of research117.
Second, iPSC-derived neurons resemble young neurons, whereas many neurological disorders affect older neurons, and aging is the major risk factor for neurodegenerative diseases. Several strategies are being investigated to model aging in iPSC-derived cells118,119. Neurons directly reprogrammed from fibroblasts maintain some features reflecting the age of the donor and mirroring some properties of aged neurons120,121; however they are not readily obtained in quantities required for large-scale screens.
Third, most or all neurological disorders involve interactions between different cell types in a tissue context. iPSC-derived brain organoids and assembloids have great promise for the study of such interactions. Ongoing efforts aim to improve the recapitulation of brain tissue architecture, including important features such as vasculature.
Fourth, neurological diseases manifest as dysfunction of the nervous system, and involve processes at the level of neuronal circuits. Further technology development is needed to recapitulate relevant aspects of brain circuit function in iPSC-derived models, and to replicate defects characteristic of neurological diseases.
New innovations in the rapidly evolving field of CRISPR technology are likely to open up exciting new possibilities for screens in iPSC-based disease models. For example, the recently developed prime editing technology40 could enable large-scale screens in which disease-associated genetic variants are introduced in the genome of iPSCs and phenotypic consequences are evaluated in relevant cell types.
Beyond its use as research tool, CRISPR technology has the potential to be used therapeutically in neurological disease. In particular, it would enable the editing of disease-associated variants in relevant tissues. Clinical trials are currently underway for CRISPR-based therapies targeting cells that are more easily accessible, such as blood cells and photoreceptor cells. Effective delivery to the central nervous system will be a major challenge that needs to be overcome to pave the way for CRISPR-based therapy of neurological disease. In the meantime, research applications of CRISPR technology, as described in this review, have the potential to identify therapeutic strategies that can be implemented using traditional small-molecule therapeutics.
Key Points.
CRISPR technology enables editing of the human genome sequence, making it possible to model or correct disease-associated genetic variants.
CRISPR interference (CRISPRi) and CRISPR activation (CRISPRa) enable changes in the expression levels of genes in human cells.
Perturbation of large numbers of genes in CRISPR-based genetic screens facilitates the identification of genes relevant for specific cellular functions.
Induced pluripotent stem cells (iPSCs) can be derived from patients and differentiated into neurons, glia, and brain organoids to generate models of neurological disease.
CRISPR screens in iPSC-derived disease models can elucidate disease mechanisms and potential therapeutic targets.
Acknowledgements
I thank members of the Kampmann lab and Alex Pollen for discussions, and referees for their insightful comments. Research on neurodegenerative and neuropsychiatric diseases in the Kampmann lab is supported by a Ben Barres Early Career Acceleration Award from the Chan Zuckerberg Initiative, the Tau Consortium, an Allen Distinguished Investigator Award (Paul G. Allen Family Foundation), a Chan-Zuckerberg Biohub Investigator Award, an NIH Director’s New Innovator Award (NIH/NIGMS DP2 GM119139), NIH/NIA grants (R01 AG062359 and R56 AG057528), and the NINDS Tau Center Without Walls (NIH/NINDS U54 NS100717), and the UCSF/UC Berkeley Innovative Genomics Institute (IGI).
Glossary
- Genome-wide association studies (GWAS).
Studies aiming to identify genetic variants associated with disease risk or other traits in human populations.
- Pleiotropic
Affecting several traits.
- Single-nucleus transcriptomics
Sequencing-based quantification of mRNA molecules in individual nuclei isolated from tissues to characterize gene expression programmes at single-cell resolution.
- Spatial transcriptomics
Methods detecting abundance and localization of multiple RNA molecules of interest in the tissue context, based on hybridization or in situ sequencing approaches.
- Multiplexed immunofluorescence
Methods detecting abundance and localization of multiple proteins and protein states of interest in the tissue context, based on parallel and sequential probing with antibodies.
- Synthetic lethal
Two genetic variants or gene perturbations that are lethal to cells or organisms in combination but not individually.
- Linkage disequilibrium
Co-occurrence of genetic variants at different loci within individuals in a population at frequencies different from those expected by chance.
- Safe-harbour locus
A site in the genome considered suitable for the integration of transgenes, because it enables stable expression of the transgene without disrupting the function of endogenous genes.
Footnotes
Competing Interests
M.K. has filed a patent application related to CRISPRi and CRISPRa screening (PCT/US15/ 40449) and serves on the Scientific Advisory Board of Engine Biosciences.
References
- 1.Gallagher MD & Chen-Plotkin AS The Post-GWAS Era: From Association to Function. American journal of human genetics 102, 717–730, doi: 10.1016/j.ajhg.2018.04.002 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Claussnitzer M et al. A brief history of human disease genetics. Nature 577, 179–189, doi: 10.1038/s41586-019-1879-7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pimenova AA, Raj T & Goate AM Untangling Genetic Risk for Alzheimer’s Disease. Biol Psychiatry 83, 300–310, doi: 10.1016/j.biopsych.2017.05.014 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Huang KL et al. A common haplotype lowers PU.1 expression in myeloid cells and delays onset of Alzheimer’s disease. Nature neuroscience 20, 1052–1061, doi: 10.1038/nn.4587 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nott A et al. Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139, doi: 10.1126/science.aay0793 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Balendra R & Isaacs AM C9orf72-mediated ALS and FTD: multiple pathways to disease. Nat Rev Neurol 14, 544–558, doi: 10.1038/s41582-018-0047-2 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boivin M et al. Reduced autophagy upon C9ORF72 loss synergizes with dipeptide repeat protein toxicity in G4C2 repeat expansion disorders. The EMBO journal, e100574, doi: 10.15252/embj.2018100574 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yamanaka K et al. Mutant SOD1 in cell types other than motor neurons and oligodendrocytes accelerates onset of disease in ALS mice. Proceedings of the National Academy of Sciences of the United States of America 105, 7594–7599, doi: 10.1073/pnas.0802556105 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin YT et al. APOE4 Causes Widespread Molecular and Cellular Alterations Associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron 98, 1294, doi: 10.1016/j.neuron.2018.06.011 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Seeley WW et al. Early frontotemporal dementia targets neurons unique to apes and humans. Ann Neurol 60, 660–667, doi: 10.1002/ana.21055 (2006). [DOI] [PubMed] [Google Scholar]
- 11.Payami H et al. Alzheimer’s disease, apolipoprotein E4, and gender. JAMA 271, 1316–1317 (1994). [PubMed] [Google Scholar]
- 12.Poirier J et al. Apolipoprotein E polymorphism and Alzheimer’s disease. Lancet 342, 697–699, doi: 10.1016/0140-6736(93)91705-q (1993). [DOI] [PubMed] [Google Scholar]
- 13.Tang MX et al. Relative risk of Alzheimer disease and age-at-onset distributions, based on APOE genotypes among elderly African Americans, Caucasians, and Hispanics in New York City. American journal of human genetics 58, 574–584 (1996). [PMC free article] [PubMed] [Google Scholar]
- 14.Lake BB et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590, doi: 10.1126/science.aaf1204 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.La Manno G et al. Molecular Diversity of Midbrain Development in Mouse, Human, and Stem Cells. Cell 167, 566–580 e519, doi: 10.1016/j.cell.2016.09.027 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Uhlen M et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419, doi: 10.1126/science.1260419 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Hosp F & Mann M A Primer on Concepts and Applications of Proteomics in Neuroscience. Neuron 96, 558–571, doi: 10.1016/j.neuron.2017.09.025 (2017). [DOI] [PubMed] [Google Scholar]
- 18.Burbulla LF et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 357, 1255–1261, doi: 10.1126/science.aam9080 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gosselin D et al. An environment-dependent transcriptional network specifies human microglia identity. Science 356, doi: 10.1126/science.aal3222 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Friedman BA et al. Diverse Brain Myeloid Expression Profiles Reveal Distinct Microglial Activation States and Aspects of Alzheimer’s Disease Not Evident in Mouse Models. Cell Rep 22, 832–847, doi: 10.1016/j.celrep.2017.12.066 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Giasson BI et al. Neuronal alpha-synucleinopathy with severe movement disorder in mice expressing A53T human alpha-synuclein. Neuron 34, 521–533, doi: 10.1016/s0896-6273(02)00682-7 (2002). [DOI] [PubMed] [Google Scholar]
- 22.Maloney B, Ge YW, Alley GM & Lahiri DK Important differences between human and mouse APOE gene promoters: limitation of mouse APOE model in studying Alzheimer’s disease. J Neurochem 103, 1237–1257, doi: 10.1111/j.1471-4159.2007.04831.x (2007). [DOI] [PubMed] [Google Scholar]
- 23.Price DL et al. Aged non-human primates: an animal model of age-associated neurodegenerative disease. Brain Pathol 1, 287–296, doi: 10.1111/j.1750-3639.1991.tb00672.x (1991). [DOI] [PubMed] [Google Scholar]
- 24.Emborg ME et al. Age-related declines in nigral neuronal function correlate with motor impairments in rhesus monkeys. J Comp Neurol 401, 253–265 (1998). [PubMed] [Google Scholar]
- 25.Sasaki E et al. Generation of transgenic non-human primates with germline transmission. Nature 459, 523–527, doi: 10.1038/nature08090 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Niu Y et al. Generation of gene-modified cynomolgus monkey via Cas9/RNA-mediated gene targeting in one-cell embryos. Cell 156, 836–843, doi: 10.1016/j.cell.2014.01.027 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Liu H et al. TALEN-mediated gene mutagenesis in rhesus and cynomolgus monkeys. Cell Stem Cell 14, 323–328, doi: 10.1016/j.stem.2014.01.018 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato K et al. Generation of a Nonhuman Primate Model of Severe Combined Immunodeficiency Using Highly Efficient Genome Editing. Cell Stem Cell 19, 127–138, doi: 10.1016/j.stem.2016.06.003 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Chen Y et al. Functional disruption of the dystrophin gene in rhesus monkey using CRISPR/Cas9. Hum Mol Genet 24, 3764–3774, doi: 10.1093/hmg/ddv120 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ke Q et al. TALEN-based generation of a cynomolgus monkey disease model for human microcephaly. Cell Res 26, 1048–1061, doi: 10.1038/cr.2016.93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen Y et al. Modeling Rett Syndrome Using TALEN-Edited MECP2 Mutant Cynomolgus Monkeys. Cell 169, 945–955 e910, doi: 10.1016/j.cell.2017.04.035 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hale CR et al. RNA-guided RNA cleavage by a CRISPR RNA-Cas protein complex. Cell 139, 945–956, doi: 10.1016/j.cell.2009.07.040 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jinek M et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821, doi: 10.1126/science.1225829 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cong L et al. Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823, doi: 10.1126/science.1231143 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mali P et al. RNA-guided human genome engineering via Cas9. Science 339, 823–826, doi: 10.1126/science.1232033 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Canver MC et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197, doi: 10.1038/nature15521 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Groschel S et al. A single oncogenic enhancer rearrangement causes concomitant EVI1 and GATA2 deregulation in leukemia. Cell 157, 369–381, doi: 10.1016/j.cell.2014.02.019 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Han J et al. Efficient in vivo deletion of a large imprinted lncRNA by CRISPR/Cas9. RNA Biol 11, 829–835, doi: 10.4161/rna.29624 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424, doi: 10.1038/nature17946 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anzalone AV et al. Search-and-replace genome editing without double-strand breaks or donor DNA. Nature 576, 149–157, doi: 10.1038/s41586-019-1711-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilbert LA et al. CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451, doi: 10.1016/j.cell.2013.06.044 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gilbert LA et al. Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661, doi: 10.1016/j.cell.2014.09.029 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Rosenbluh J et al. Complementary information derived from CRISPR Cas9 mediated gene deletion and suppression. Nat Commun 8, 15403, doi: 10.1038/ncomms15403 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kearns NA et al. Functional annotation of native enhancers with a Cas9-histone demethylase fusion. Nat Methods 12, 401–403, doi: 10.1038/nmeth.3325 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thakore PI et al. Highly specific epigenome editing by CRISPR-Cas9 repressors for silencing of distal regulatory elements. Nat Methods 12, 1143–1149, doi: 10.1038/nmeth.3630 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Maeder ML et al. CRISPR RNA-guided activation of endogenous human genes. Nat Methods 10, 977–979, doi: 10.1038/nmeth.2598 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Perez-Pinera P et al. RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods 10, 973–976, doi: 10.1038/nmeth.2600 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chavez A et al. Highly efficient Cas9-mediated transcriptional programming. Nat Methods 12, 326–328, doi: 10.1038/nmeth.3312 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hilton IB et al. Epigenome editing by a CRISPR-Cas9-based acetyltransferase activates genes from promoters and enhancers. Nat Biotechnol 33, 510–517, doi: 10.1038/nbt.3199 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Konermann S et al. Genome-scale transcriptional activation by an engineered CRISPR-Cas9 complex. Nature 517, 583–588, doi: 10.1038/nature14136 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.O’Connell MR et al. Programmable RNA recognition and cleavage by CRISPR/Cas9. Nature 516, 263–266, doi: 10.1038/nature13769 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Batra R et al. Elimination of Toxic Microsatellite Repeat Expansion RNA by RNA-Targeting Cas9. Cell 170, 899–912 e810, doi: 10.1016/j.cell.2017.07.010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Abudayyeh OO et al. C2c2 is a single-component programmable RNA-guided RNA-targeting CRISPR effector. Science 353, aaf5573, doi: 10.1126/science.aaf5573 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.East-Seletsky A et al. Two distinct RNase activities of CRISPR-C2c2 enable guide-RNA processing and RNA detection. Nature 538, 270–273, doi: 10.1038/nature19802 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Konermann S et al. Transcriptome Engineering with RNA-Targeting Type VI-D CRISPR Effectors. Cell 173, 665–676 e614, doi: 10.1016/j.cell.2018.02.033 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yan WX et al. Cas13d Is a Compact RNA-Targeting Type VI CRISPR Effector Positively Modulated by a WYL-Domain-Containing Accessory Protein. Molecular cell 70, 327–339 e325, doi: 10.1016/j.molcel.2018.02.028 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rousseau BA, Hou Z, Gramelspacher MJ & Zhang Y Programmable RNA Cleavage and Recognition by a Natural CRISPR-Cas9 System from Neisseria meningitidis. Molecular cell 69, 906–914 e904, doi: 10.1016/j.molcel.2018.01.025 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Zhou Y et al. High-throughput screening of a CRISPR/Cas9 library for functional genomics in human cells. Nature 509, 487–491, doi: 10.1038/nature13166 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C & Yusa K Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol 32, 267–273, doi: 10.1038/nbt.2800 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Shalem O et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science 343, 84–87, doi: 10.1126/science.1247005 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wang T, Wei JJ, Sabatini DM & Lander ES Genetic screens in human cells using the CRISPR-Cas9 system. Science 343, 80–84, doi: 10.1126/science.1246981 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Kampmann M Elucidating drug targets and mechanisms of action by genetic screens in mammalian cells. Chemical communications, doi: 10.1039/c7cc02349a (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kramer NJ et al. CRISPR-Cas9 screens in human cells and primary neurons identify modifiers of C9ORF72 dipeptide-repeat-protein toxicity. Nature genetics 50, 603–612, doi: 10.1038/s41588-018-0070-7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Haney MS et al. Identification of phagocytosis regulators using magnetic genome-wide CRISPR screens. Nature genetics 50, 1716–1727, doi: 10.1038/s41588-018-0254-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Rauch JN et al. Tau Internalization is Regulated by 6-O Sulfation on Heparan Sulfate Proteoglycans (HSPGs). Sci Rep 8, 6382, doi: 10.1038/s41598-018-24904-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stopschinski BE et al. Specific glycosaminoglycan chain length and sulfation patterns are required for cell uptake of tau versus alpha-synuclein and beta-amyloid aggregates. The Journal of biological chemistry 293, 10826–10840, doi: 10.1074/jbc.RA117.000378 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Chen JJ et al. Compromised function of the ESCRT pathway promotes endolysosomal escape of tau seeds and propagation of tau aggregation. The Journal of biological chemistry, doi: 10.1074/jbc.RA119.009432 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Rousseaux MWC et al. A Druggable Genome Screen Identifies Modifiers of alpha-Synuclein Levels via a Tiered Cross-Species Validation Approach. J Neurosci 38, 9286–9301, doi: 10.1523/JNEUROSCI.0254-18.2018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Vazquez-Velez GE et al. Doublecortin-like Kinase 1 Regulates alpha-Synuclein Levels and Toxicity. J Neurosci 40, 459–477, doi: 10.1523/JNEUROSCI.1076-19.2019 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Potting C et al. Genome-wide CRISPR screen for PARKIN regulators reveals transcriptional repression as a determinant of mitophagy. Proceedings of the National Academy of Sciences of the United States of America 115, E180–E189, doi: 10.1073/pnas.1711023115 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Cheng W et al. CRISPR-Cas9 Screens Identify the RNA Helicase DDX3X as a Repressor of C9ORF72 (GGGGCC)n Repeat-Associated Non-AUG Translation. Neuron, doi: 10.1016/j.neuron.2019.09.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jaitin DA et al. Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 167, 1883–1896 e1815, doi: 10.1016/j.cell.2016.11.039 (2016). [DOI] [PubMed] [Google Scholar]
- 73.Dixit A et al. Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866 e1817, doi: 10.1016/j.cell.2016.11.038 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Adamson B et al. A Multiplexed Single-Cell CRISPR Screening Platform Enables Systematic Dissection of the Unfolded Protein Response. Cell 167, 1867–1882 e1821, doi: 10.1016/j.cell.2016.11.048 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Datlinger P et al. Pooled CRISPR screening with single-cell transcriptome readout. Nat Methods 14, 297–301, doi: 10.1038/nmeth.4177 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tian R et al. CRISPR Interference-Based Platform for Multimodal Genetic Screens in Human iPSC-Derived Neurons. Neuron 104, 239–255 e212, doi: 10.1016/j.neuron.2019.07.014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Gasperini M et al. A Genome-wide Framework for Mapping Gene Regulation via Cellular Genetic Screens. Cell 176, 377–390 e319, doi: 10.1016/j.cell.2018.11.029 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Feldman D et al. Optical Pooled Screens in Human Cells. Cell 179, 787–799 e717, doi: 10.1016/j.cell.2019.09.016 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wang C, Lu T, Emanuel G, Babcock HP & Zhuang X Imaging-based pooled CRISPR screening reveals regulators of lncRNA localization. Proceedings of the National Academy of Sciences of the United States of America 116, 10842–10851, doi: 10.1073/pnas.1903808116 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bassik MC et al. A systematic mammalian genetic interaction map reveals pathways underlying ricin susceptibility. Cell 152, 909–922, doi: 10.1016/j.cell.2013.01.030 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kampmann M, Bassik MC & Weissman JS Integrated platform for genome-wide screening and construction of high-density genetic interaction maps in mammalian cells. Proceedings of the National Academy of Sciences of the United States of America 110, E2317–2326, doi: 10.1073/pnas.1307002110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Horlbeck MA et al. Mapping the Genetic Landscape of Human Cells. Cell 174, 953–967 e922, doi: 10.1016/j.cell.2018.06.010 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Norman TM et al. Exploring genetic interaction manifolds constructed from rich single-cell phenotypes. Science 365, 786–793, doi: 10.1126/science.aax4438 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Boettcher M et al. Dual gene activation and knockout screen reveals directional dependencies in genetic networks. Nat Biotechnol 36, 170–178, doi: 10.1038/nbt.4062 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ridge PG et al. Linkage, whole genome sequence, and biological data implicate variants in RAB10 in Alzheimer’s disease resilience. Genome Med 9, 100, doi: 10.1186/s13073-017-0486-1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Finch N et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology 76, 467–474, doi: 10.1212/WNL.0b013e31820a0e3b (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Cruchaga C et al. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol 68, 581–586, doi: 10.1001/archneurol.2010.350 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Chai N et al. Genome-wide synthetic lethal CRISPR screen identifies FIS1 as a genetic interactor of ALS-linked C9ORF72. Brain Res 1728, 146601, doi: 10.1016/j.brainres.2019.146601 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Takahashi K & Yamanaka S Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676, doi: 10.1016/j.cell.2006.07.024 (2006). [DOI] [PubMed] [Google Scholar]
- 90.Takahashi K et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell 131, 861–872, doi: 10.1016/j.cell.2007.11.019 (2007). [DOI] [PubMed] [Google Scholar]
- 91.Sproul AA et al. Generation of iPSC lines from archived non-cryoprotected biobanked dura mater. Acta Neuropathol Commun 2, 4, doi: 10.1186/2051-5960-2-4 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vierbuchen T et al. Direct conversion of fibroblasts to functional neurons by defined factors. Nature 463, 1035–1041, doi: 10.1038/nature08797 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Eiraku M et al. Self-organized formation of polarized cortical tissues from ESCs and its active manipulation by extrinsic signals. Cell Stem Cell 3, 519–532, doi: 10.1016/j.stem.2008.09.002 (2008). [DOI] [PubMed] [Google Scholar]
- 94.Pasca SP Assembling human brain organoids. Science 363, 126–127, doi: 10.1126/science.aau5729 (2019). [DOI] [PubMed] [Google Scholar]
- 95.Schwartz MP et al. Human pluripotent stem cell-derived neural constructs for predicting neural toxicity. Proceedings of the National Academy of Sciences of the United States of America 112, 12516–12521, doi: 10.1073/pnas.1516645112 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Brownjohn PW et al. Functional Studies of Missense TREM2 Mutations in Human Stem Cell-Derived Microglia. Stem cell reports 10, 1294–1307, doi: 10.1016/j.stemcr.2018.03.003 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Park J et al. A 3D human triculture system modeling neurodegeneration and neuroinflammation in Alzheimer’s disease. Nature neuroscience 21, 941–951, doi: 10.1038/s41593-018-0175-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Abud EM et al. iPSC-Derived Human Microglia-like Cells to Study Neurological Diseases. Neuron 94, 278–293 e279, doi: 10.1016/j.neuron.2017.03.042 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Han SS, Williams LA & Eggan KC Constructing and deconstructing stem cell models of neurological disease. Neuron 70, 626–644, doi: 10.1016/j.neuron.2011.05.003 (2011). [DOI] [PubMed] [Google Scholar]
- 100.Israel MA et al. Probing sporadic and familial Alzheimer’s disease using induced pluripotent stem cells. Nature 482, 216–220, doi: 10.1038/nature10821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kondo T et al. Modeling Alzheimer’s disease with iPSCs reveals stress phenotypes associated with intracellular Abeta and differential drug responsiveness. Cell Stem Cell 12, 487–496, doi: 10.1016/j.stem.2013.01.009 (2013). [DOI] [PubMed] [Google Scholar]
- 102.Wang C et al. Gain of toxic apolipoprotein E4 effects in human iPSC-derived neurons is ameliorated by a small-molecule structure corrector. Nature medicine, doi: 10.1038/s41591-018-0004-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bershteyn M et al. Human iPSC-Derived Cerebral Organoids Model Cellular Features of Lissencephaly and Reveal Prolonged Mitosis of Outer Radial Glia. Cell Stem Cell 20, 435–449 e434, doi: 10.1016/j.stem.2016.12.007 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lancaster MA et al. Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379, doi: 10.1038/nature12517 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Soldner F et al. Generation of isogenic pluripotent stem cells differing exclusively at two early onset Parkinson point mutations. Cell 146, 318–331, doi: 10.1016/j.cell.2011.06.019 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Fong H et al. Genetic correction of tauopathy phenotypes in neurons derived from human induced pluripotent stem cells. Stem cell reports 1, 226–234, doi: 10.1016/j.stemcr.2013.08.001 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jiang S et al. Integrative system biology analyses of CRISPR-edited iPSC-derived neurons and human brains reveal deficiencies of presynaptic signaling in FTLD and PSP. Transl Psychiatry 8, 265, doi: 10.1038/s41398-018-0319-z (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Kwart D et al. A Large Panel of Isogenic APP and PSEN1 Mutant Human iPSC Neurons Reveals Shared Endosomal Abnormalities Mediated by APP beta-CTFs, Not Abeta. Neuron, doi: 10.1016/j.neuron.2019.07.010 (2019). [DOI] [PubMed] [Google Scholar]
- 109.Wang C et al. Scalable Production of iPSC-Derived Human Neurons to Identify Tau-Lowering Compounds by High-Content Screening. Stem cell reports 9, 1221–1233, doi: 10.1016/j.stemcr.2017.08.019 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Groen EJN & Gillingwater TH UBA1: At the Crossroads of Ubiquitin Homeostasis and Neurodegeneration. Trends in molecular medicine 21, 622–632, doi: 10.1016/j.molmed.2015.08.003 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ihry RJ et al. p53 inhibits CRISPR-Cas9 engineering in human pluripotent stem cells. Nature medicine 24, 939–946, doi: 10.1038/s41591-018-0050-6 (2018). [DOI] [PubMed] [Google Scholar]
- 112.Schrode N et al. Synergistic effects of common schizophrenia risk variants. Nature genetics, doi: 10.1038/s41588-019-0497-5 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Deczkowska A et al. Disease-Associated Microglia: A Universal Immune Sensor of Neurodegeneration. Cell 173, 1073–1081, doi: 10.1016/j.cell.2018.05.003 (2018). [DOI] [PubMed] [Google Scholar]
- 114.Liddelow SA et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487, doi: 10.1038/nature21029 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hodge RD et al. Conserved cell types with divergent features in human versus mouse cortex. Nature 573, 61–68, doi: 10.1038/s41586-019-1506-7 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Kampmann M A CRISPR Approach to Neurodegenerative Diseases. Trends in molecular medicine 23, 483–485, doi: 10.1016/j.molmed.2017.04.003 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Tsunemoto R et al. Diverse reprogramming codes for neuronal identity. Nature 557, 375–380, doi: 10.1038/s41586-018-0103-5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Miller JD et al. Human iPSC-based modeling of late-onset disease via progerin-induced aging. Cell Stem Cell 13, 691–705, doi: 10.1016/j.stem.2013.11.006 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Vera E, Bosco N & Studer L Generating Late-Onset Human iPSC-Based Disease Models by Inducing Neuronal Age-Related Phenotypes through Telomerase Manipulation. Cell Rep 17, 1184–1192, doi: 10.1016/j.celrep.2016.09.062 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mertens J et al. Directly Reprogrammed Human Neurons Retain Aging-Associated Transcriptomic Signatures and Reveal Age-Related Nucleocytoplasmic Defects. Cell Stem Cell 17, 705–718, doi: 10.1016/j.stem.2015.09.001 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kim Y et al. Mitochondrial Aging Defects Emerge in Directly Reprogrammed Human Neurons due to Their Metabolic Profile. Cell Rep 23, 2550–2558, doi: 10.1016/j.celrep.2018.04.105 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]