

Abstract
Objectives:
Apolipoprotein CIII (apoCIII) mediates the metabolism of triglyceride (TG)-rich lipoproteins. High levels of plasma apoCIII are positively correlated with the plasma TG levels and increase the cardiovascular risk. However, whether apoCIII is directly involved in the development of atherosclerosis has not been fully elucidated.
Approach and Results:
To examine the possible roles of apoCIII in lipoprotein metabolism and atherosclerosis, we generated apoCIII knockout (KO) rabbits using zinc finger nuclease technique. On a normal standard diet, apoCIII KO rabbits exhibited significantly lower plasma levels of TG than those of wild-type (WT) rabbits while total cholesterol and HDL-cholesterol levels were unchanged. Analysis of lipoproteins isolated by sequential ultracentrifugation revealed that reduced plasma TG levels in KO rabbits were accompanied by prominent reduction of VLDLs and intermediate-density lipoproteins (IDLs). In addition, KO rabbits showed faster TG clearance rate after intravenous fat load than WT rabbits. On a cholesterol-rich diet, KO rabbits exhibited constantly and significantly lower levels of plasma total cholesterol and TG than WT rabbits, which was caused by a remarkable reduction of β-VLDLs, the major atherogenic lipoproteins. β-VLDLs of KO rabbits showed higher uptake by cultured hepatocytes and were cleared faster from the circulation than β-VLDLs isolated from WT rabbits. Both aortic and coronary atherosclerosis was significantly reduced in KO rabbits compared with WT rabbits.
Conclusions:
These results indicate that apoCIII deficiency facilitates TG-rich lipoprotein catabolism and therapeutic inhibition of apoCIII expression may become a novel means not only for the treatment of hyperlipidemia but also for atherosclerosis.
Keywords: apolipoprotein CIII, TG-rich lipoproteins, hyperlipidemia, atherosclerosis, knockout rabbits
Graphical Abstract
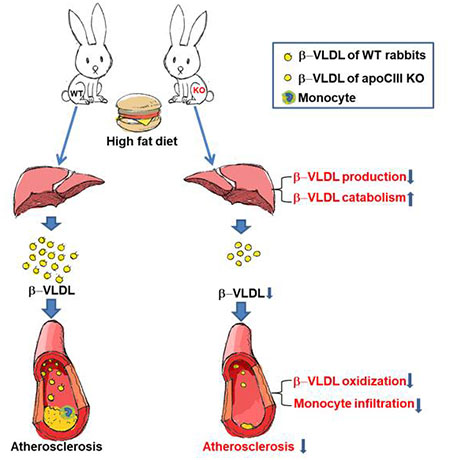
Introduction
ApolipoproteinC-III (apoCIII) is a major component of plasma chylomicrons and very low density lipoproteins (VLDL), and is a minor component of high density lipoproteins (HDL) and was first reported by Brown et al. 50 years ago1. In humans, as a glycoprotein, apoCIII contains 79 amino acids with a molecular weight of 8.8 kDa and mainly expressed in the liver and lesser in small intestine2. Normal plasma levels of apoCIII in healthy populations are at 7~12 mg/dL but can be >30 mg/dL in hypertriglyceridemic patients3, 4. It is generally believed that apoCIII is a key regulator in triglyceride-rich lipoprotein (TRL) metabolism to maintain the plasma TG homeostasis though inhibition of lipoprotein lipase activity5, 6, suppression of the hepatic uptake of TRLs7, and modulation of the assembly and secretion of VLDL particles in the liver8, 9. However, ample evidence from epidemiological, genetic and clinical studies has shown that increased plasma apoCIII is strongly associated with elevated plasma TG levels and increases the risk of ischemic heart disease10–12. Large-scale population genetic studies revealed that carriers with apoCIII “loss-of-function” mutations have lower plasma TG levels along with cardioprotective lipoprotein profiles than non-carriers13–18. In support of this, clinical studies revealed that therapeutic inhibition of apoCIII can alleviate human hypertriglyceridemia19–21. Critical roles of apoCIII in the development of hypertriglyceridemia were also demonstrated by experimental animal studies that overexpression of the human apoCIII gene in transgenic mice22–24 and rabbits25 leads to a marked hypertriglyceridemia whereas apoCIII gene deletion in mice results in hypotriglyceridemia26, 27. In spite of this widely-accepted notion, there is still uncertainty regarding whether apoCIII is also directly involved in development of atherosclerosis or whether targeting apoCIII can inhibit the progression of atherosclerosis. In fact, the role of apoCIII in atherosclerosis remains controversial28,29. Overexpression of human apoCIII in transgenic mice induces severe hypertriglyceridemia but is not atherogenic even in the setting of apoE deficiency because large-sized TRLs are considered unable to penetrate into the arterial wall to initiate the lesion formation28. Moreover, in LDL receptor knockout (KO) mice, human apoC-III overexpression exacerbated the development of atherosclerosis29 but apoCIII deficiency did not affect30. Therefore, high levels of plasma apoCIII are clearly associated with hypertriglyceridemia and high risk of ischemic heart disease, whereas it remains unclear whether apoCIII per se really plays a direct role in atherosclerosis.
To test the hypothesis that apoCIII may participate in atherosclerosis, we have generated apoCIII KO rabbits using zinc finger nuclease technology31. Rabbits have several advantages in studying human lipoprotein metabolism and atherosclerosis because they are sensitive to a cholesterol diet and develop atherosclerosis rapidly32. The major atherogenic lipoproteins in cholesterol-fed rabbits are those of cholesteryl ester-rich β-LVLDs whereas in Watanabe heritable hyperlipidemic (WHHL) rabbits, there are high levels of plasma LDLs, which is more similar to humans. Furthermore, like humans but unlike mice, rabbits have cholesteryl ester transfer protein and intestinal apoB editing, both of which are important in human lipoprotein metabolism. In addition, appropriate size of rabbits also facilitates researchers to perform multiple lipoprotein assays and turn-over studies32, 33. Our current studies revealed that genetic deletion of the apoCIII gene in KO rabbits reduces plasma levels of TG, which was basically caused by accelerated catabolism of TRLs. Importantly, we showed that apoCIII deficiency leads to the resistance of KO rabbits to a cholesterol diet-induced hyperlipidemia and inhibits atherosclerosis.
Materials and methods
The data that support the findings of this study are available from the corresponding author.
Rabbits
ApoCIII knockout (KO) rabbits were created by zinc finger nucleases (ZFN) technique as described previously31. We obtained five KO founders confirmed by PCR sequencing assays. We selected one male founder KO rabbit with an adenine insertion in the exon 3 of apoCIII gene and bred this KO with normal female rabbits to generate F1 heterozygous (apoCIII+/−) and F2 homozygous (apoCIII−/−) KO rabbits. Sex- and age-matched wild-type (WT) littermates were used as controls in the current study. All rabbits were fed with a normal standard diet (CLEA Japan Inc., Tokyo, Japan) containing 17% protein, 4% fat, and 14% crude fiber. The animals were housed in a barrier facility with a 12-h light/dark cycle at 24°C and 55% humidity. AII animal experiments were performed with the approval of the Animal Care Committee of the University of Yamanashi and Saga and conformed to the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health.
Genotyping
For genotyping detection of pups, rabbit ear (20 mg) was biopsied and digested in 700 μL lysis buffer containing 20 mM Tris-Cl (pH 8.0), 5 mM EDTA (pH 8.0), 400 mM NaCl, 1 % (w/v) SDS and 400 μg/mL proteinase K at 55℃ for overnight. Rabbit genomic DNA was extracted by phenol: chloroform: isoamyl (25:24:1) alcohol (Invitrogen, Carlsbad, CA) and precipitated by isopropanol. Concentrations of DNA were adjusted to 100ng/reaction and polymerase chain reaction (PCR) was performed with primers designed to detect specific ZFN mutation site of apoCIII (Figure 1A). The forward primer 5’-TCTGCACGCTTGGGGCTGGA-3’ was common for both WT and mutant sequences whereas the reverse primer 1 (Rmut), 5’-CTGGCCTGCTGGGCCATTTT-3’ was for detecting mutant sequence and the reverse primer 2 (R), 5’-CTGGCCTGCTGGGCCATTTG-3’ was for the WT sequence. An annealing temperature of 69°C (1 min) was used for PCR amplification. PCR products were run on a 1% agarose gel stained by ethidium bromide and visualized under a UV illuminator.
Figure 1. Generation of apoCIII knockout (KO) rabbits.
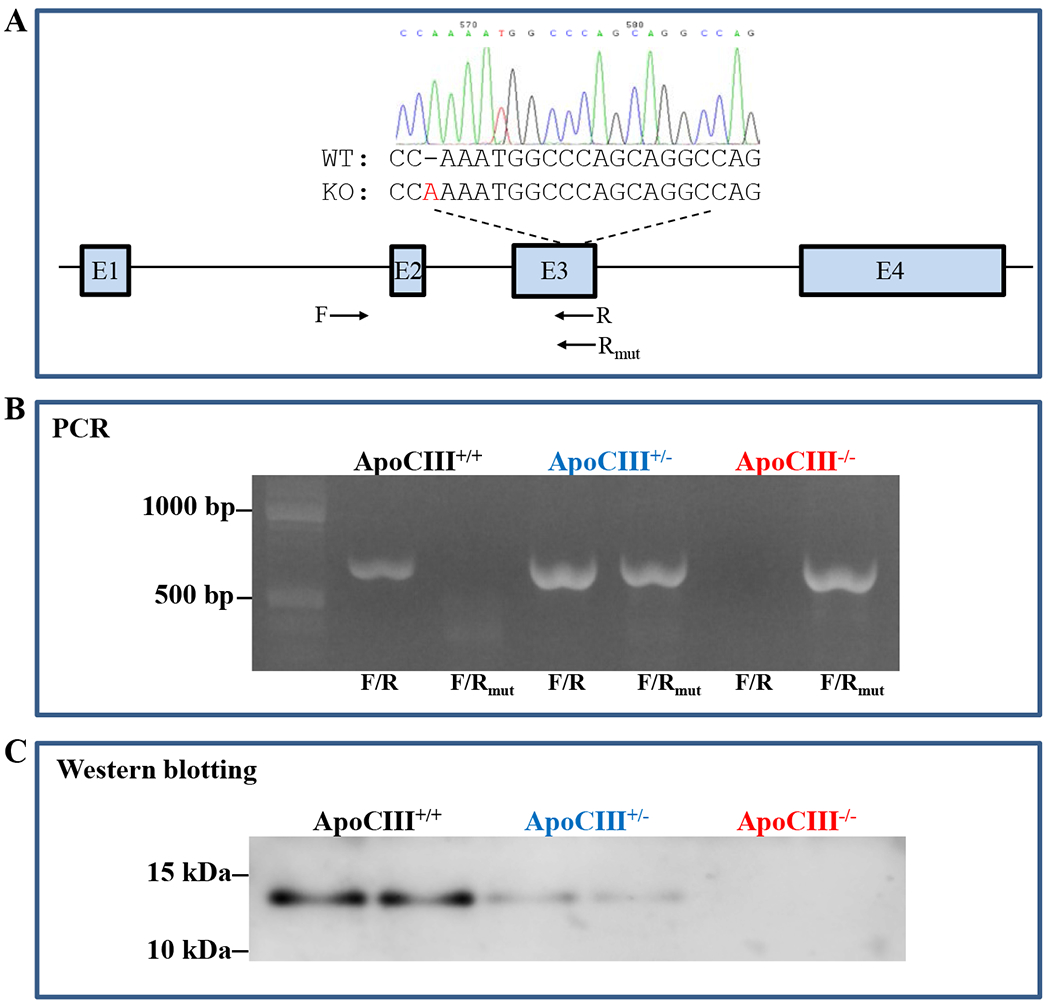
A. Schematic illustration of rabbit apoCIII gene and mutant sequences along with one forward (F) and two reverse (R and Rmut) primers for PCR analysis of KO rabbits shown below. Note: the founder KO rabbit had an adenine insertion in the apoCIII gene, which causes the frameshift.
B. PCR detection of KO rabbits using two sets of primers designated as F/R for wild type (WT) and F/Rmut for mutant sequences. Heterozygous KO rabbits had two bands (a WT band and a mutant band) and homozygous KO rabbits only have a mutant band.
C. Western blotting showed that apoCIII KO rabbits had either reduced (hetero-) or none apoCIII (homo-) in the plasma.
Plasma lipids
Rabbits were fasted for 16 h and the blood was collected from an intermediate auricular artery and into a microtube containing 1.5 mg/ml ethylenediaminetetraacetic acid (EDTA) and 1 % (v/v) aprotinin (Sigma-Aldrich, A6279, St Louis, MO). EDTA-plasma was separated by centrifugation at 4,000 rpm for 20 min at 4°C. Total cholesterol (TC), triglycerides (TG) and HDL-cholesterol (HDL-C) concentrations were measured using Wako enzymatic assay kits (Wako Pure Chemical Industries Ltd., Osaka, Japan)34.
Analysis of lipoproteins and apolipoproteins
Plasma (2 μL) was loaded to a 1% agarose gel and lipoproteins were separated by electrophoresis (Helena Laboratories, Saitama, Japan) and neutral lipids were visualized by Fat Red 7B staining. ApoAI, apoB100, apoCIII and apoE contents in the whole plasma were determined by Western blotting as described previously34. Plasma (0.5 μL) was subjected to 4-20% sodium dodecyl sulfate poly-acrylamide gel electrophoresis (SDS-PAGE). Then, proteins were transferred to a polyvinylidene difluoride membrane and probed with goat polyclonal antibodies (Abs) against human apoB (1:1000 dilution, cat#32017, Rockland Inc., Limerick, PA), apoE (1:1000 dilution, cat#AB947, Millipore, Darmstadt, Germany), apoCIII (1:1000 dilution, cat#GWB-1F5BC8, Genway Biotec Inc., San Diego, CA) and mouse monoclonal Ab against rabbit apoAI (1:1000 dilution, cat#LS-C314186/70403, LifeSpan BioScience Inc., Seattle, WA) (see the details of Ab information in Supplement Tables). After incubation with primary Abs for 2 h at room temperature, immunocomplexed proteins were identified by reaction with a horseradish peroxidase–conjugated secondary Abs followed by chemiluminescent detection (ECL kit; GE Healthcare Dharmacon, Inc., Buckinghamshire, UK).
Plasma lipoproteins were separated by sequential density ultracentrifugation with a Beckman TLA120.2 rotor, as described previously35. Isolated individual density fractions contain different lipoproteins: the d < 1.006 g/mL fraction contains VLDL; the d = 1.006-1.02 g/mL fraction contains intermediate density lipoprotein (IDL); the d = 1.02-1.04 g/mL fraction contains large LDL; the d = 1.04-1.06 g/mL fraction contains both LDL at the β-migrating position and HDL1 at the α-migrating position; the d = 1.06-1.08 and 1.08-1.10 g/mL fractions are those of HDL2; while the d = 1.10-1.21 g/mL fraction contains HDL3. All fractions were dialyzed against phosphate-buffered saline for overnight. Lipoprotein fractions (10 μL) were loaded onto 4-20% SDS-PAGE and apolipoproteins were visualized either by Coomassie brilliant blue (CBB) staining (Wako Pure Chemical Industries) or immunoblotted with anti-apoAI, apoB, apoCIII and apoE Abs, as described above. TC ang TG contents in each fraction were measured using the Wako assay kits described above.
Intravenous fat load test
Rabbits on a either standard diet or cholesterol diet for 5 weeks were fasted overnight and subjected to an intravenous fat load test25. Rabbits received a bolus infusion of Intralipos® Injection 20% solution (Otsuka Pharmaceutical Com., Tokyo, Japan) consisting of 20% soybean oil, 1.2% lecithin, and 2.2% glycerin at 2 mL/kg BW. Blood samples were collected from an intermediate auricular artery at 0 (pre-injection), 1, 10, 20, 30, 60, 90, and 120 min after Intralipos administration. Plasma TG concentrations were measured as described above and normalized to the plasma TG levels at 1 min.
Cholesterol diet experiments
Rabbits were fed with a diet containing 0.3% cholesterol and 3% soybean oil for 16 weeks36. Rabbits were fasted overnight (16 h) before blood samples were collected from the intermediate auricular artery. Plasma levels of TC, TG, and HDL-C were measured weekly as above. For the analysis of lipoprotein profiles and apolipoproteins, plasma lipoproteins were isolated from cholesterol-fed rabbits at 8 and 15 weeks and measured as above.
Preparation of Dil-labeled β-VLDLs and clearance studies
Cholesterol-fed WT and KO rabbits were fasted overnight and bled from the auricular intermediate artery. 20~30 ml of blood was collected in a venoject® II tube (Terumo, Tokyo) and pooled from four rabbits of each group. Serum was separated by centrifugation at 3,000 rpm for 20 min at 4°C, and d<1.006 g/ml lipoproteins (β-VLDLs) were isolated by ultracentrifugation in 16x76 mm polycarbonate centrifuge tubes (Beckman Coulter, Inc., Brea, CA) with a 70.1Ti rotor (Beckman, Carlsbad, CA) at 50,000 rpm for 18 h at 4°C. Isolated lipoproteins were those of β-VLDLs as confirmed by 1% agarose gel electrophoresis stained by Fat Red 7B or probed with anti-apoB Ab (S-Figure. 1). The protein concentration was determined by a DC protein assay kit (Bio-Rad, Hercules, CA). β-VLDLs were stored in 4°C and used within three days for DiI-labeling.
β-VLDLs were labeled with fluorescence using the method described with a slight modification37, 38. Briefly, concentrated β-VLDLs were diluted in rabbit lipoprotein-deficient serum and adjusted to original serum volume (before ultracentrifugation) and then mixed with 1,1’-Dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate (DiI) (Wako Pure Chemical Industries) at a ratio of 3:1 (β-VLDLs : DiI) by mg. The mixture solution was incubated at 37°C for more than 8h. Then, the mixture solution was centrifuged at 50,000 rpm for 18 h with a 70.1Ti rotor at 4°C as above. DiI-labeled β-VLDLs were collected from the top portion of the tube and dialyzed against saline and filtrated before use.
For the measurement of DiI-labeled β-VLDL clearance rate in vivo, male WT rabbits (3 month-old) were fasted overnight, and then DiI-labeled β-VLDLs (3 mg protein/kg BW) were intravenously injected. Blood samples were collected at selected time points and collected into microtubes with EDTA. Plasma fluorescence intensity was directly measured using a fluorescent microplate reader (Molecular Devices LLC., San Jose, CA). Data are normalized to the 2 min fluorescence intensity as 100%.
Dil-labeled β-VLDL uptake assays
Two kinds of Dil-labeled β-VLDLs isolated from either KO or WT rabbits were prepared as described above and then dialyzed against 0.9% NaCl with 0.01% EDTA overnight at 4°C. Human hepatoma (HepG2) cells were purchased from ATCC (American Type Culture Collection, Manassas, VA) and grown in Dulbecco’s modified Eagle’s medium/F-12 (GIBCO-BRL, Gaithersburg, MD) supplemented with 10% fetal bovine serum (Sigma-Aldrich) and 1% antibiotic antimycotic solution (Sigma-Aldrich). The cells were maintained in a humidified incubator with 95% air and a 5% CO2 atmosphere at 37°C. For comparing β-VLDL uptake, HepG2 cells (5x105 cells/well) were seeded into 12-well plates (Falcon, Franklin, NJ) overnight and then, the medium was replaced with Dulbecco’s modified Eagle’s medium supplemented with 5% lipoprotein-deficient serum and maintained for 24h. The cells were incubated with Dil-labeled β-VLDLs (2 or 5 μg/mL) of for 2h at 37°C39. At the end, the cells were washed with PBS for 3 times and 1 ml of isopropanol was added to each well to recover the cell-associated β-VLDLs. The isopropanol extracts were then transferred to microtubes and centrifuged at 1000 rpm for 10 min. Fluorescent intensity of isopropanol-DiI samples was measured by a fluorescent microplate reader with an excitation of 520 nm and an emission of 578 nm40. All assays were performed in triplicate and repeated three times. Fluorescence intensity values were normalized to total cellular proteins. In addition to fluorescence intensity measurement, β-VLDL uptake was directly observed under fluorescent microscopy after the cells were incubated with DiI-labeled β-VLDLs (1 μg/mL) for 1h, 2h, and 4h. After that, the cells were fixed with 4% paraformaldehyde solution (Electron Microscopy Sciences, Hatfield, PA). After washing with PBS, the cell nuclei were stained with Hoechst 33342 (Dōjindo Laboratories, Kumamoto, Japan) and fluorescence signals were pictured under a fluorescence microscope (IX71; Olympus, Tokyo, Japan).
Post-heparin plasma LPL activity assays
Post-heparin plasma of rabbits fed either a standard or cholesterol diet was prepared from a blood sample taken 10 min after a bolus injection of heparin at a dose of 30 units/kg of BW. LPL and HL enzymatic activities were measured using a colorimetric method with the natural long-chain fatty acid 2-diglyceride as a substrate (Immuno-Biological Laboratories, Fujioka, Japan)41 with an auto-analyzer (BIOLIS 24i Premium, Tokyo Boeki Medisys Inc., Tokyo, Japan).
VLDL secretion studies
Hepatic VLDL secretion rate was determined using Triton WR-1339, a non-ionic detergent to inhibit VLDL clearance from the plasma as described previously42, 43. Briefly, cholesterol-fed rabbits were fasted overnight and then intravenously injected with 20% Tyloxapol (Sigma-Aldrich) solution (0.9% NaCl) at a dose of 400 mg/kg BW. Blood was collected at 0, 1, 2, 4, 6, and 8 h later after the injection. Plasma TG and VLDL-TG levels were measured as described above.
Analysis of aortic atherosclerosis
At the end of cholesterol feeding, all rabbits were euthanized by injection of an overdose of sodium pentobarbital solution. The aorta surfaces were en face stained with Sudan IV for evaluation of gross atherosclerotic lesions as described previously36. For microscopic quantification of lesion areas, the aortic arch was cut into cross segment (up to 8 for each). All specimens were embedded in paraffin and stained with hematoxylin and eosin (H&E) and elastica van Gieson (EVG). The intimal lesion size was quantitatively measured using the image analysis system WinRooF V6.4.0 (Mitani Co., Tokyo, Japan). For microscopic evaluation of the cellular components of the lesions, serial paraffin-embedded sections of the aorta were immunohistochemically stained with mAbs against macrophage (M) (RAM11), α-smooth muscle actin (HHF35), C-reactive protein (CRP), and oxidized LDL (oxLDL)44 using Histofine Simple Stain MAX-PO(M) kits (Nichirei Biosciences Inc., Tokyo, Japan). Macrophages, smooth muscle cells (SMCs), oxLDL and CRP intensity in the lesions were evaluated by quantifying the positively stained area using the image analysis system as described above. Coronary atherosclerosis was analyzed as described previously45. Rabbit hearts were fixed in 10% formalin solution and then sectioned into 5 blocks. Paraffin-embedded sections were stained with EVG and the lesions (stenosis %) of the left and right coronary arteries were quantified. Serial sections were also immunohistochemically stained for macrophages and smooth muscle cells as in aortas.
ApoB-containing lipoprotein oxidizability assays
To evaluate whether apoCIII deficiency affects the oxidizability of apoB-containing particles, we isolated four apoB-containing lipoprotein fractions [d<1.006 g/mL (β-VLDLs), d=1.006-1.02 g/mL (IDL), d=1.02-1.04 g/mL (large LDL), and d=1.04-1.06 g/mL (LDL)] from cholesterol-fed WT and apoCIII homozygous KO rabbits46. The kinetics of copper-induced lipoprotein oxidization was determined by monitoring the change of the conjugate-diene absorbance at 234 nm at 37°C with a SpectraMax 190 Absorbance Microplate Reader (Molecular Devices, Sunnyvale, CA) as described previously47. Absorbance of lag-time, maximal oxidization speed (V max), and maximal diene concentrations were calculated for the estimation of oxidization sensitivity.
Statistical analyses
All data are expressed as mean±SEM. Statistical tests performed are outlined in the figure legends and all analyses were conducted using software GraphPad Prism 7.0 (GraphPad Software, San Diego, CA). Each data set was first assessed for normality using the Shapiro-Wilk test. If data were found not to follow a normal distribution, a nonparametric test was performed as indicated. For a two-group comparison, we used either Student’s t-test or Welch’s t-test. For comparison of the lesions, we used one-way ANOVA and Dunnet post hoc test or Kruskal-Wallis test followed by Dunn post hoc test. In all cases, statistical significance was set at p<0.05.
Results
Genotyping of apoCIII KO Rabbits
As shown in Figure 1A, there was an adenine insertion in the apoCIII gene of the founder KO rabbit, which results in the frameshift and introduces early stop codons into the apoCIII gene. PCR analysis showed that WT rabbits only had a normal band whereas homozygous KO rabbits only had a mutant band and heterozygous KO rabbits had both mutant and normal bands (Figure 1B). Western blotting analysis confirmed that plasma apoCIII was reduced in heterozygous KO rabbits and undetectable in homozygous KO rabbits (Figure 1C). Examinations of KO rabbits did not reveal any abnormalities in the whole body and organs compared with WT rabbits.
Plasma lipids and lipoprotein profiles of rabbits on a standard diet
On a normal standard diet, male KO rabbits showed a significant reduction of plasma TG levels (34%↓in hetero- and 31%↓in homozygotes) compared with WT rabbits whereas there was no significant change in plasma TC and HDL-C levels (Figure 2A). TG levels of homozygous KO rabbits were not significantly reduced compared with heterozygous KO rabbits as expected, suggesting that on a normal diet, effect of apoCIII deficiency on TG metabolism was rather limited, which is different from a cholesterol diet experiment (see below). In another word, a prominent effect of apoCIII deficiency can only be seen when there are sufficient TG-rich VLDLs in the circulation as high cholesterol diet feeding. Female KO rabbits exhibited a mild reduction of TG levels (11%↓n.s.) compared with WT rabbits. In the following studies, we mainly focused on male KO rabbits unless otherwise specified. Agarose gel electrophoresis of plasma lipoproteins revealed that compared with WT rabbits, homozygous KO rabbits showed prominent reduction of pre-β-migrating lipoproteins (VLDLs) (Figure 2B). Western blotting analysis of the whole plasma showed that plasma apoB100 and apoE contents were concomitantly reduced in KO rabbits whereas apoAI along with apoAV (data not shown) contents were not changed compared to WT rabbits (Figure 2C).
Figure 2. Plasma lipids and lipoproteins of rabbits on a normal standard diet.
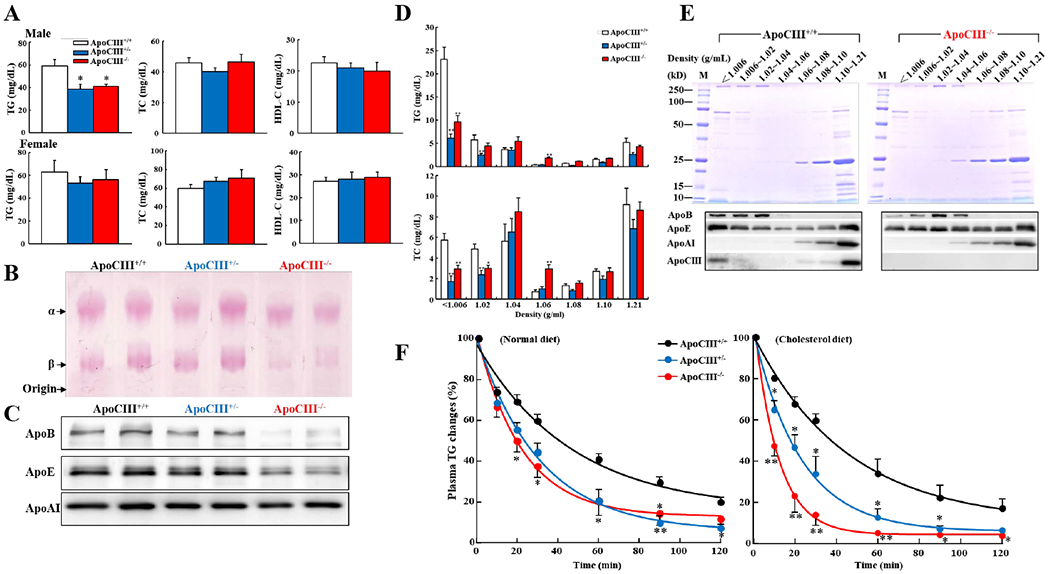
A. ApoCIII KO rabbits (male but not female) had significant lower levels of plasma TG but TC and HDL-C were not changed compared with WT rabbits. n=6-12 for each group, *p<0.05 vs WT by one-way ANOVA and Dunnet post hoc test.
B. Agarose gel electrophoresis showed that pre--migrating lipoproteins were prominently reduced in male apoCIII homozygous KO rabbits.
C. Western blotting showed apoB, apoE and apoAI contents in the whole plasma. ApoB and apoE contents in apoCIII homozygous KO rabbits were reduced. Each lane represents each individual animal sample.
D. Plasma lipoproteins were separated by sequential ultracentrifugation. TG and TC contents in each fraction were quantified (n=5-8 for each group, *p<0.05, **p<0.01 vs WT via one-way ANOVA and Dunnet post hoc test).
E. Analysis of apolipoproteins in each fraction. An equal volume (10 μL) of each fraction was resolved by electrophoresis by 4-20% SDS-PAGE. Apolipoproteins were visualized using by CBB staining (upper panel, gel) and immunobloted with anti-apoB, apoE, apoAI and apoCIII Abs (lower panel). Representative data of a single animal sample are shown.
F. Intravenous fat load test.Relative TG levels in plasma of WT and KO rabbits on either a standard (left) or cholesterol diet (right) after intravenous injection of lipid emulsion were measured and normalized to the total plasma TG. (n=4 for each group, *p<0.05, **p<0.01 vs WT by one-way ANOVA and Dunnet post hoc test).
Quantitation of TG and TC contents in each density fraction showed that both VLDLs and IDLs were significantly reduced in KO rabbits (73%↓in hetero- and 58%↓in homozygous compared with WT) (Figure 2D). In addition, fraction 4 with d=1.04-1.06 g/mL (mainly consisting of small LDLs) in KO rabbits was increased compared with WT rabbits. Analysis of lipoprotein density fractions by SDS-PAGE showed that apoCIII in WT rabbits was mainly distributed in VLDL, IDL, and HDL3. As expected, apoCIII was absent in all fractions of homozygous KO rabbits (Figure 2E). Deletion of apoCIII in KO rabbits led to two major changes in apolipoprotein compositions compared with WT rabbits. First, consistent with lipid quantitation, apoB100 along with apoE in both VLDLs and IDLs were reduced but slightly increased in small LDLs in KO rabbits (Figure 2E). Second, apoE contents were enriched in HDL2 fractions.
To examine the possible mechanism responsible for lower plasma TG and TRLs observed in apoCIII KO rabbits, we performed an intravenous fat load test and compared the clearance rate of plasma TG of KO rabbits with WT rabbits. As shown in Figure 2F, both hetero- and homozygous KO rabbits on either a standard diet or cholesterol-rich diet exhibited significantly faster TG clearance rate than WT rabbits.
Cholesterol diet experiments
To investigate the susceptibility of apoCIII KO rabbits to a cholesterol-rich diet, rabbits were fed a cholesterol-rich diet for 16 weeks. Compared with WT rabbits, apoCIII KO rabbits showed constantly and significantly “lower” hyperlipidemia: lower TC and lower TG levels throughout the experiment period but no change in HDL-C levels (Figure 3A). Analysis of lipoprotein profiles showed that lower plasma TC and TG levels present in KO rabbits were essentially owing to the reduction of all apoB-containing lipoproteins including β-VLDLs (57%↓in hetero- and 77%↓in homozygous KO), IDLs (44%↓in hetero- and 66%↓in homozygous KO), and large LDLs (49%↓in hetero- and 57%↓in homozygous KO) in which both apoB and apoE contents were reduced (Figure 3B–C).
Figure 3. Plasma lipids and lipoproteins and apolipoproteins of rabbits fed a cholesterol-rich diet.
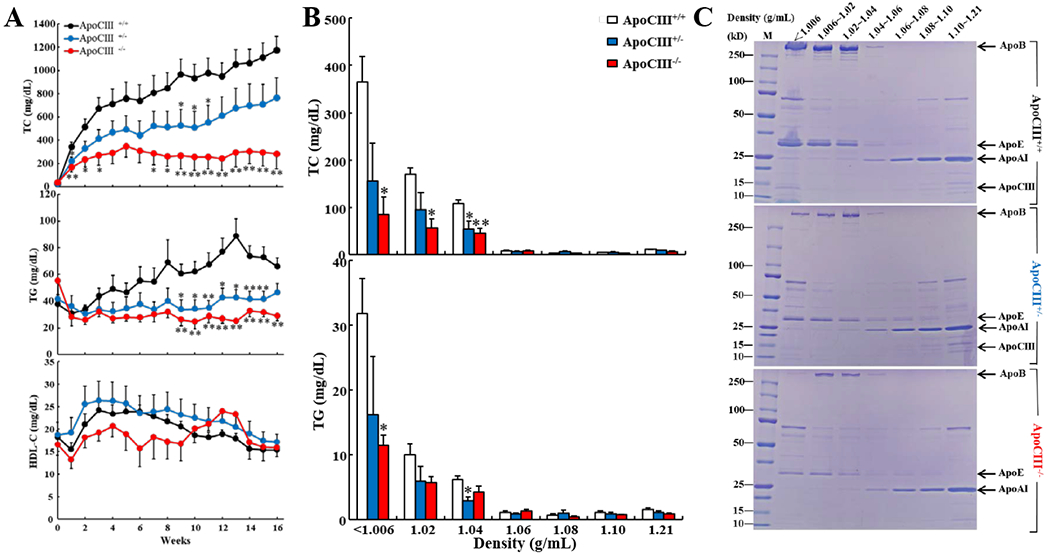
A. WT and KO rabbits were fed a cholesterol diet for 16 weeks and their plasma levels of TC, TG and HDL-C were measured (n=5-11 for each group, *p<0.05, **p<0.01 vs WT by one-way ANOVA and Dunnet post hoc test).
B. Plasma lipoproteins were separated by sequential ultracentrifugation. TC and TG contents in each fraction were quantified. (n=4-5, *p<0.05, **p<0.01 vs WT by one-way ANOVA and Dunnet post hoc test). KO rabbits had significantly lower levels of VLDL, IDL and LDL than WT rabbits.
C. Analysis of apolipoproteins in each fraction. An equal volume (10 μL) of each fraction was resolved by electrophoresis by 4-20% SDS-PAGE. Apolipoproteins were visualized after CBB staining. Representative data of a single animal sample are shown.
Taken together, these observations suggest that KO rabbits are substantially resistant to a cholesterol-diet challenge and protective against hyperlipidemia. We were prompted to investigate the possible molecular mechanisms underlying the resistance-to-cholesterol of KO rabbits. First, we isolated two kinds of β-VLDLs from either cholesterol-fed WT or homozygous KO rabbits and then labeled them with Dil-fluorescence and compared their clearance rate in WT rabbits (Figure 4A. left). In addition to absence of apoCIII, TG content in KO β-VLDLs was almost half of WT β-VLDLs (45 mg/dL in WT and 25 mg/dL in KO). When equal amounts of β-VLDLs (3 mg proteins/kg BW) were intravenously injected into WT rabbits, β-VLDLs of KO rabbits were obviously cleared faster from the circulation than those of WT rabbits (Figure 4A, right). Using the same Dil-labeled β-VLDLs, we further compared their uptake ability by cultured hepatocytes in vitro. As shown in Figure 4A–B, cultured HepG2 cells showed 40% higher uptake of KO β-VLDLs than WT β-VLDLs. Since apoCIII may mediate the catabolism of TRLs through LPL, we compared LPL and HL activities of both WT and KO rabbits fed either a standard or cholesterol diet. We found that KO rabbits exhibited relatively lower LPL and HL activities than WT rabbits (n.s.) (S-Figure 2), suggesting that the enhancement of β-VLDL catabolism in KO rabbits was independent on the LPL pathway. It was reported that apoCIII affects the assembly and secretion of VLDLs in the liver, thus we further investigated the hepatic β-VLDL secretion in vivo. Compared with WT rabbits, KO rabbits showed a trend of slower hepatic production of β-VLDLs compared with WT rabbits (n.s.)(S-Figure 3).
Figure 4. β-VLDL clearance rate in vivo and uptake by hepatocytes in vitro.
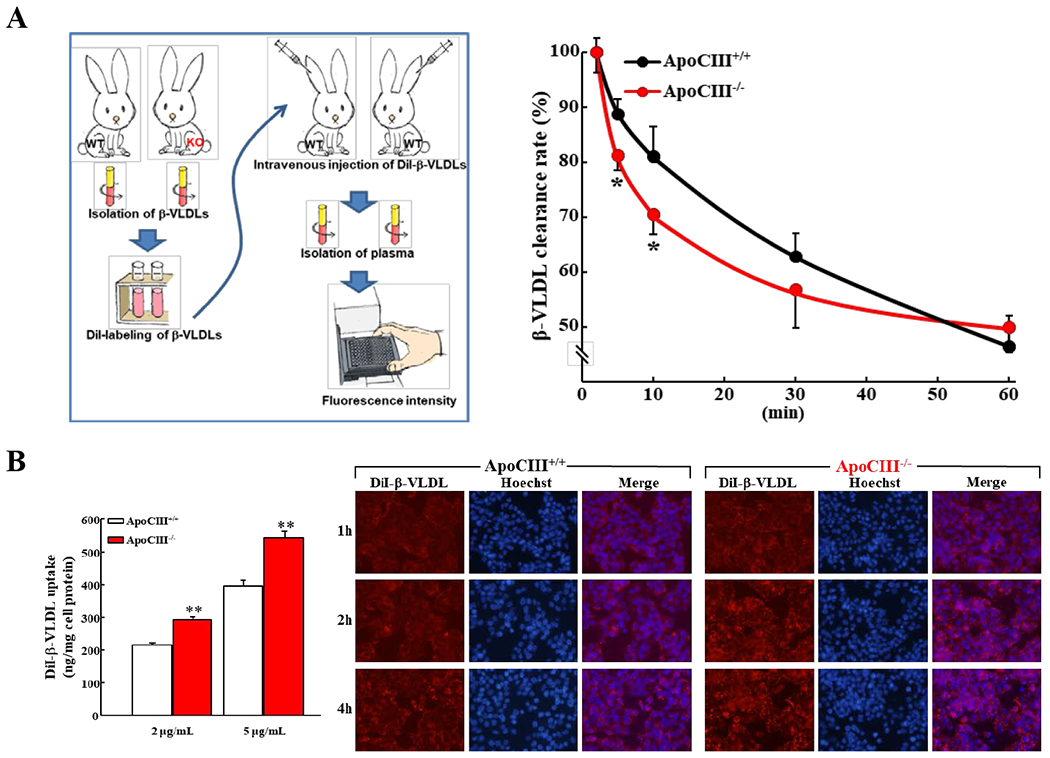
A. Plasma β-VLDLs (d<1.006 g/mL) were isolated by ultracentrifugation from either cholesterol-fed WT or apoCIII KO rabbits and then were labeled with fluorescence DiI. Dil-labeled β- VLDLs were intravenously injected into WT recipient rabbits. Blood samples were withdrawn at 2, 5, 10, 30, 60 min after Dil-labeled β-VLDLs administration (left). Plasma fluorescence intensity was measured by a fluorescent microplate reader. Data are normalized to the 2 min fluorescent intensity levels at 100% (n=3-4, *p<0.05, vs WT by Student’s t-test) (right).
B. Analysis of β- VLDL uptake assay by hepatocytes in vitro
HepG2 hepatoma cells were incubated with DiI-labeled β-VLDLs (2 g or 5 g/ml) at 37°C for 2h. The isopropanol extracts of Dil were measured by a fluorescent microplate reader. Data were normalized to total cellular protein and results are representative of three independent experiments (left)(**p<0.01 vs WT by Student’s t-test). Representative fluorescent micrographs of HepG2 hepatoma cells after incubation with DiI-labeled β-VLDLs (right). HepG2 cells were incubated with DiI-labeled β-VLDLs (1 μg/mL). Microscopic pictures were taken at 1h, 2h or 4h after incubation. Nuclei were stained with Hoechst. Representative pictures are shown.
Analysis of aortic atherosclerosis
We compared the aortic atherosclerosis area after staining with Sudan IV. Gross lesion area of the whole aorta of KO rabbits was significantly smaller (52%↓hetero- and 69%↓in homozygous) than WT rabbits: there was 67%↓ in aortic arch, 75%↓ in thoracic aorta, and 65%↓ in abdominal aorta of the lesion areas in homozygous KO rabbits (Figure 5A).
Figure 5. -Quantification of aortic atherosclerotic lesions.
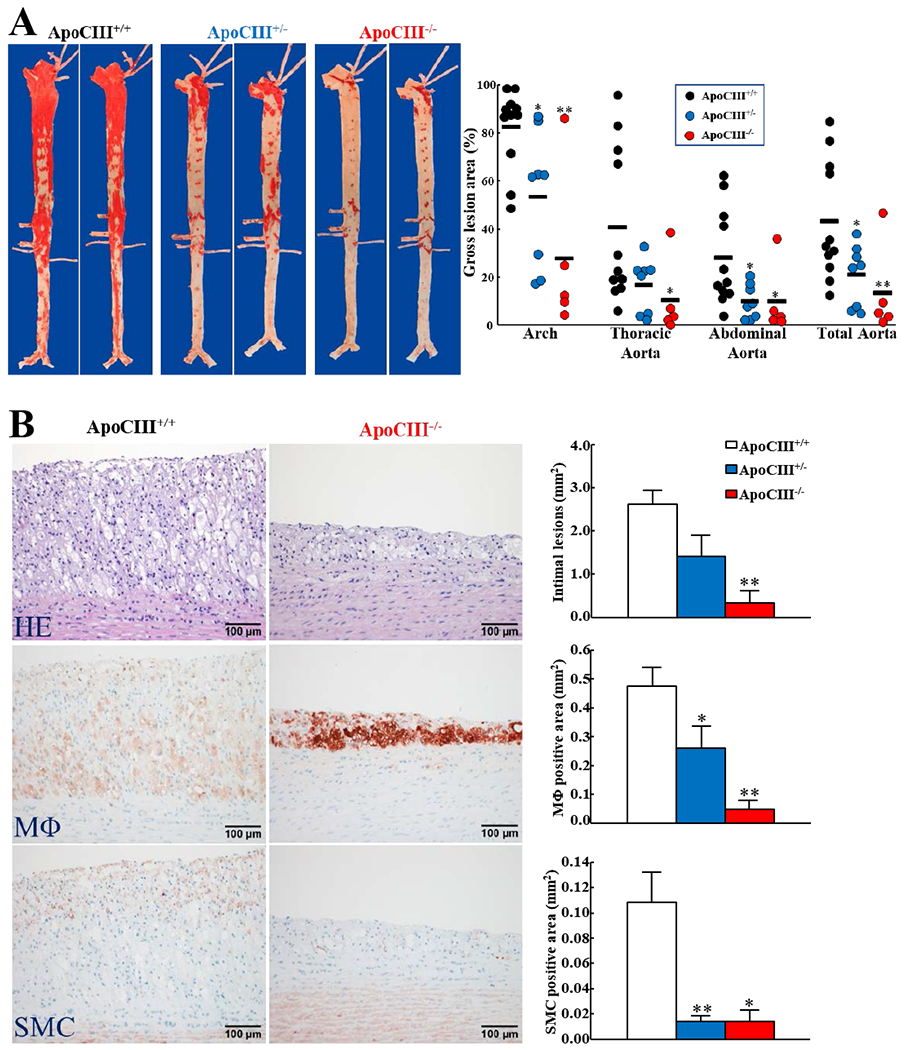
A. WT and KO rabbits were fed a cholesterol diet for 16 weeks and representative pictures of aortas stained with Sudan IV are shown (left). The lesion area (defined by the sudanophilic area) was quantified using an image analysis system (right). Each dot represents each individual animal. (*p<0.05, **p<0.01 vs WT by Kruskal-Wallis test followed by Dunn post hoc test).
B. Serial paraffin sections of aortic arch were stained with hematoxylin-eosin (HE) or immunohistochemically stained with mAbs against macrophages (MΦ) or α-smooth muscle actin for smooth muscle cells (SMCs). Representative micrographs are shown (left). Microscopic lesion areas were quantified using elastica van Gieson (EVG)-stained sections and positively-stained areas of MΦs and SMCs were measured with an image analysis system (right). (n=5-11 for each group, *p<0.05, **p<0.01 vs WT by Kruskal-Wallis test followed by Dunn post hoc test).
Histological analysis showed that the lesions of aortic atherosclerosis in cholesterol-fed rabbits were composed of macrophages and smooth muscle cells intermingled with extracellular matrix (Figure 5B, left). The microscopic lesion areas of the aortic arch were significantly reduced: 46%↓in hetero- and 87%↓in homozygous KO rabbits compared with WT rabbits (Figure 5B, right). Immunohistochemical staining showed that the lesions of KO rabbits were characterized by decreased number of macrophages and smooth muscle cells: 45%↓and 90%↓of lesional macrophages and 87%↓and 87%↓of lesional smooth muscle cells in hetero- and homozygous KO rabbits compared with WT rabbits (Figure 5B, right).
Coronary atherosclerosis
In addition to aortic lesions, coronary atherosclerosis was also significantly reduced in KO rabbits. Stenosis rate was reduced by 31% and 75% in the left and 29% and 77% in the right coronary arteries of hetero- and homozyougs KO rabbits compared with WT rabbits (Figure 6). Similar to aortic lesions, the coronary lesions of KO rabbits contained significantly less macrophages and smooth muscle cells (S-Figure 4).
Figure 6. Quantification of coronary atherosclerotic lesions.
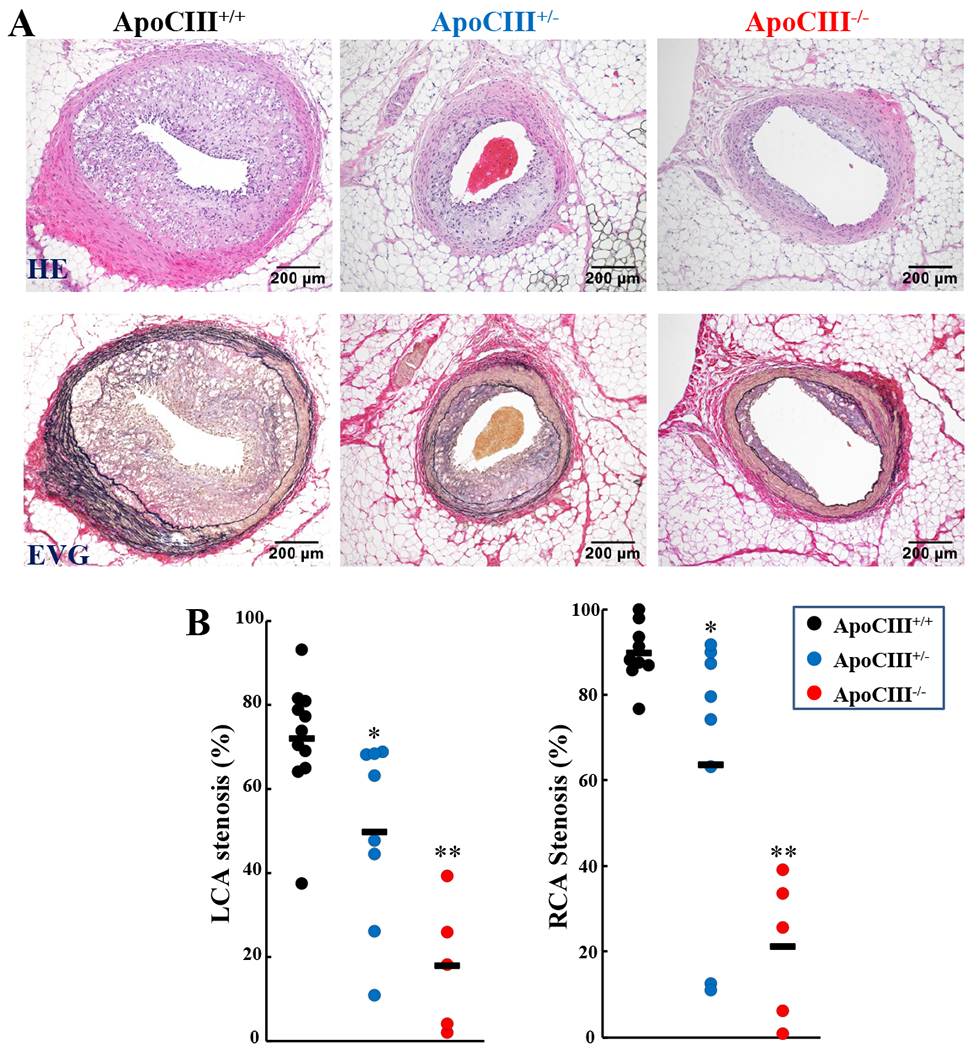
Representative micrographs of coronary atherosclerotic lesions from each group are shown (A). The specimens are stained with either hematoxylin-eosin (HE) or elastica van Gieson (EVG). The lesion size was measured and expressed as stenosis% (B). Each dot represents each individual animal. (*p<0.05, **p<0.01 vs WT by Kruskal-Wallis test followed by Dunn post hoc test).
β-VLDLs of apoCIII KO rabbits exhibited resistance to copper-induced oxidization
We also examined whether deletion of apoCIII in apoB-containing lipoproteins would affect their oxidizability. Among the four apoB-containing particles, β-VLDLs, the major atherogenic lipoproteins in cholesterol-fed rabbits, were the most sensitive to copper-induced oxidization. Compared with β-VLDLs of WT rabbits, oxidizability of β-VLDLs (but not other fractions) of KO rabbits was significantly diminished (Figure 7A–B). We also investigated the lesional oxLDL depositions and found that oxLDL staining along with CRP, an inflammatory marker, in KO rabbits was weaker than that in WT rabbits (Figure 7C). Based on these observations along with others, we outlined postulated mechanisms for apoCIII mediated-TRL metabolism and apoCIII deficiency-induced inhibition of hypercholesterolemia and atherosclerosis in Figure 8.
Figure 7. Analysis of apoB-containing particle oxidizability.

Four apoB-containing lipoprotein fractions (β-VLDLs, IDLs, large LDLs, and LDLs) were isolated from cholesterol-fed WT and KO rabbits and analyzed for copper-induced lipoprotein oxidation potential by monitoring the dynamic changes of the conjugate-diene absorbance at 234 nm. Representative dynamic changes are shown in (A), and lag-time, maximal oxidation speed (V max), and maximal diene concentrations are shown in (B). (n=3-4 for each group, *p<0.05, *p<0.01 vs WT by Student’s t-test).
C. Immunohistochemical analysis of oxidized LDL depositions in the lesions of aortas.
Serial sections of aortic arch were stained with HE or mAbs against oxidized LDL and macrophage (MΦ) and representative micrographs are shown (C). OxLDL-positive areas are largely co-localized with macrophages. The positive area of ox-LDL was quantified (D). (n=5 for each group, **p<0.01 vs WT by Student’s t-test).
Figure 8. Postulated mechanisms for apoCIII involvement in TG-rich lipoprotein metabolism and cholesterol diet-induced hypercholesterolemia and atherosclerosis in KO rabbits.

On a normal diet, apoCIII deficiency enhances the hepatic catabolism of TG-rich lipoproteins thus lowers the plasma TG in KO rabbits (left panel). On a cholesterol diet, normal rabbits develop massive hypercholesterolemia due to elevation of plasma β-VLDLs and atherosclerosis (right panel). ApoCIII deficiency may inhibit the development of atherosclerosis though two possible mechanisms: (1) lipid-lowering effects due to increased hepatic uptake and catabolism along with reduced secretion of β-VLDLs, and (2) direct roles on the arterial walls including suppression of β-VLDL oxidization/deposition and possible inhibition of inflammatory reaction.
Discussion
It has been proposed that apoCIII confers an atherogenic effect, but direct evidence in vivo is lacking. In the current study, we tested the hypothesis whether apoCIII plays a direct role in the development of atherosclerosis using apoCIII KO rabbits. We found that apoCIII deficiency not only enhances the hepatic catabolism of TRLs (pre-β-migrating VLDLs) but also protects against a cholesterol diet-induced hypercholesterolemia (β-migrating VLDLs rich in cholesteryl esters) atherosclerosis in KO rabbits (Figure 8).
ApoCIII deficiency in KO rabbits led to a dramatic reduction of both aortic (69% reduction over WT rabbits) and coronary (75% reduction) atherosclerosis and the lesions were associated with decreased macrophage infiltration (90% reduction) and smooth muscle cell proliferation (87% reduction) in aortic atherosclerosis. Atheroprotective effects of apoCIII deficiency shown in KO rabbits appear to be involved in multiple mechanisms (Figure 8). First, apoCIII KO rabbits had lower hyperlipidemia with reduced levels of plasma atherogenic lipoproteins, β-VLDLs, than WT rabbits. This lipid-lowering effect of apoCIII deficiency was possibly attributed to the enhancement of the hepatic catabolism of β-VLDLs. This notion was supported by the demonstration that β-VLDLs isolated from KO rabbits were cleared faster from the circulation than those of WT rabbits. ApoCIII was reported to impair the clearance of apoB-containing lipoproteins by interfering with their binding to hepatic lipoprotein receptors48. Apparently, β-VLDLs without apoCIII enhance their binding to and uptake by hepatocytes as shown in our study, presumably through the apoE-mediated LDL family receptors49. Although it has been generally believed that high doses of apoCIII inhibits LPL activity thereby affecting TRL hydrolysis in vitro10, 50 , some studies suggest that this mechanism does not seem to occur in vivo49, 51–53. In the current study, apoCIII deficiency in KO rabbits fed either a standard diet or a cholesterol diet showed relatively lower LPL and HL activity than WT rabbits, which eliminates the possibility that apoCIII deficiency can enhance LPL activity or LPL-mediated pathway plays the major role in the enhancement of TRL catabolism in KO rabbits although it is still unknown whether apoCIII deficiency affects LPL expression. Second, the lower levels of β-VLDLs in KO rabbits may be partially owing to reduced secretion of β-VLDLs in the liver because intracellular apoCIII promotes assembly and secretion of VLDLs in the hepatocytes54. In the current study, we found that apoCIII KO rabbits showed a lower tendency of β-VLDL secretion in the liver even though not significant. Taken together, these observations strongly suggest that apoCIII deficiency reduces plasma atherogenic lipoproteins in the circulation.
In addition to the lipid-lowering effect, atheroprotective effects of apoCIII deficiency may be pertaining to apoCIII participation in lipoprotein oxidization or apoCIII direct functions on the arterial wall thereby prohibiting the initiation and progression of atherosclerosis in KO rabbits. For example, apoCIII-deficient β-VLDLs are less susceptible to copper-induced oxidization and the lesions of KO rabbits contained less oxLDL depositions with less CRP than WT, which may attenuate monocyte adhesion to endothelial cells and infiltration into the intima as proposed by others55, 56. It has been reported that in patients with hypertriglyceridemia and type 2 diabetes mellitus, apoCIII-enriched LDLs exhibited an altered lipid compositions, increased binding to proteoglycans and lipoprotein retention, and increased LDL susceptibility to hydrolysis and aggregation by sphingomyelinase57, 58. If these events are indeed operative in vivo, it can be conceived that inflammatory reaction in the arterial wall was presumably diminished in apoCIII KO rabbits. In contrast to KO rabbits, previous studies using apoCIII KO26, 27 or transgenic mice22, 23, 28 failed to demonstrate the relationship between apoCIII and atherogenicity. In the setting of LDL receptor deletion, apoCIII deficiency does not affect atherosclerosis30 although overexpression of human apoCIII increases atherosclerosis29. It is likely that this discrepancy between apoCIII KO mice and KO rabbits regarding apoCIII atherogenecity may be attributed to intrinsic metabolic differences between two species32. For example, mice do not have CETP and have both intestinal and hepatic apoB editing activity so both internally derived chylomicrons and hepatically derived VLDLs and LDLs contain apoB48. It has been known that apoB48-containing particles are catabolized faster than those apoB100 containing particles59. Furthermore, mouse apoCIII molecule has only one glycosylated isoform24 whereas both human and rabbit apoCIII have three types of glycosylated isoform: non-glycosylated isoform (apoCIII0) and glycosylated isoform with either one or two moles of sialic acid (apoCIII1 and apoCIII2 respectively)60, 61, although it is still unknown how these apoCIII isoforms are actually involved in development of atherosclerosis. Nevertheless, our current studies not only support the notion that lowering apoCIII can improve plasma TRL metabolism19, 62, 63 but also shed a light on the apoCIII functional roles in the development of atherosclerosis. Recently, therapeutic inhibition of apoCIII using antisense oligonucleotide (ASO) such as volanesorsen and AKCEA-APOCIII-LRx have been under clinical trials for the treatment of hypertriglyceridemia16, 19–21, 64. The next urgent question is “can we also use apoCIII inhibition as a new therapeutic strategy to treat atherosclerosis?”. Obviously, this supposition is awaiting vigorous studies to prove in future.
One of the limitations of the current study was gender differences between male and female KO rabbits because the plasma lipid phenotypes described above were less obvious in female apoCIII KO rabbits compared with male KO counterparts. The mechanisms underlying the gender effect on apoCIII functions require extensive investigations in future. Nevertheless, our preliminary study showed that female apoCIII homozygous KO rabbits as male KO counterparts also exhibited faster catabolism of TRLs and resistance to cholesterol diet-induced hypercholesterolemia and atherosclerosis (S-Figure 5).
In conclusion, our results demonstrated that apoCIII deficiency in KO rabbits reduces plasma TG levels through enhancement of TRL catabolism. KO rabbits showed resistance to a cholesterol diet-induced hyperlipidemia and atherosclerosis. These data support the concept that therapeutic inhibition of apoCIII is beneficial to patients with hypertriglyceridemia. It is worth testing whether apoCIII inhibition therapeutics can treat atherosclerosis in future.
Supplementary Material
Highlights.
We generated apoCIII knockout (KO) rabbits using zinc finger nuclease technique.
ApoCIII deficiency reduces plasma triglycerides in apoCIII KO rabbits on a normal standard diet due to reduction of VLDLs.
On a cholesterol-rich diet, KO rabbits protects against hypercholesterolemia and atherosclerosis.
ApoCIII deficiency enhances β-VLDL catabolism in the liver.
Acknowledgements
We thank Mrs. Kato, Y. for her technical assistance in making pathological specimens and Mrs. Suzuki, R. (Admission Center of Yamanashi University) for preparing the graphic work.
Sources of funding
This work was supported in part by the Research grant from Ono Medical Foundation, JSPS KAKENHI (JP17K08783 and JP15H04718), the National Natural Science Foundation of China (No. 81941001 and 81770457), the Natural Science Foundation of Shaanxi Province (2017JZ028), JSPS-CAS under the Japan-China Research Cooperative Program, and NIH grant (R01HL117491 and R01HL129778).
Nonstandard Abbreviations and Acronyms
- Apo
apolipoprotein
- BW
body weight
- CETP
cholesteryl ester transfer protein
- CRP
C-reactive protein
- Dil
1,1’-Dioctadecyl-3,3,3’,3’-tetramethylindocarbocyanine perchlorate
- EDTA
ethylenediaminetetraacetic acid
- EVG
elastica van Gieson
- HDL
high-density lipoprotein
- HDL-C
high-density lipoprotein-cholesterol
- HE
hematoxylin and eosin
- HL
hepatic lipase
- IDL
intermediate-density lipoprotein
- KO
knockout
- LDL
low-density lipoprotein
- LPL
lipoprotein lipase
- oxLDL
oxidized low-density lipoprotein
- PCR
polymerase chain reaction
- MO
macrophage
- SDS-PAGE
sodium dodecyl sulfate poly-acrylamide gel electrophoresis
- SMC
smooth muscle cell
- TC
total cholesterol
- TG
triglycerides
- TRLs
triglyceride-rich lipoproteins
- VLDL
very low-density lipoprotein
- WT
wild-type
- ZFN
zinc finger nuclease
Footnotes
Disclosure
None
References
- 1.Brown WV, Levy RI, Fredrickson DS. Studies of the proteins in human plasma very low density lipoproteins. J. Biol. Chem 1969;244:5687–5694 [PubMed] [Google Scholar]
- 2.Brewer HB Jr., Shulman R, Herbert P, Ronan R, Wehrly K. The complete amino acid sequence of alanine apolipoprotein (apoc-3), and apolipoprotein from human plasma very low density lipoproteins. J. Biol. Chem 1974;249:4975–4984 [PubMed] [Google Scholar]
- 3.Huff MW, Breckenridge WC, Strong WL, Wolfe BM. Metabolism of apolipoproteins C-II, C-III, and B in hypertriglyceridemic men. Changes after heparin-induced lipolysis. Arteriosclerosis. 1988;8:471–479 [DOI] [PubMed] [Google Scholar]
- 4.Cohn JS, Patterson BW, Uffelman KD, Davignon J, Steiner G. Rate of production of plasma and very-low-density lipoprotein (vldl) apolipoprotein C-III is strongly related to the concentration and level of production of vldl triglyceride in male subjects with different body weights and levels of insulin sensitivity. J. Clin. Endocrinol. Metab 2004;89:3949–3955 [DOI] [PubMed] [Google Scholar]
- 5.Brown WV, Baginsky ML. Inhibition of lipoprotein-lipase by an apoprotein of human very low-density lipoprotein. Biochem. Biophys. Res. Commun 1972;46:375-& [DOI] [PubMed] [Google Scholar]
- 6.Ginsberg HN, Le NA, Goldberg IJ, Gibson JC, Rubinstein A, Wang-Iverson P, Norum R, Brown WV. Apolipoprotein b metabolism in subjects with deficiency of apolipoproteinsC-III and ai. Evidence that apolipoprotein C-III inhibits catabolism of triglyceride-rich lipoproteins by lipoprotein lipase in vivo. J. Clin. Invest 1986;78:1287–1295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Windler E, Havel RJ. Inhibitory effects of C-apolipoproteins from rats and humans on the uptake of triglyceride-rich lipoproteins and their remnants by the perfused rat-liver. J. Lipid Res 1985;26:556–565 [PubMed] [Google Scholar]
- 8.Sundaram M, Zhong S, Bou Khalil M, Links PH, Zhao Y, Iqbal J, Hussain MM, Parks RJ, Wang Y, Yao Z. Expression of apolipoprotein C-III in mca-rh7777 cells enhances vldl assembly and secretion under lipid-rich conditions. J. Lipid Res 2010;51:150–161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Qin W, Sundaram M, Wang YW, Zhou H, Zhong SM, Chang CC, Manhas S, Yao EF, Parks RJ, McFie PJ, Stone SJ, Jiang ZHG, Wang CR, Figeys D, Jia WP, et al. Missense mutation in apoc3 within the c-terminal lipid binding domain of human apoC-III results in impaired assembly and secretion of triacylglycerol-rich very low density lipoproteins evidence that apoC-III plays a major role in the formation of lipid precursors within the microsomal lumen. J. Biol. Chem 2011;286:27769–27780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ramms B, Gordts P. Apolipoprotein C-III in triglyceride-rich lipoprotein metabolism. Curr. Opin. Lipidol 2018;29:171–179 [DOI] [PubMed] [Google Scholar]
- 11.Norata GD, Tsimikas S, Pirillo A, Catapano AL. Apolipoprotein C-III: From pathophysiology to pharmacology. Trends Pharmacol. Sci 2015;36:675–687 [DOI] [PubMed] [Google Scholar]
- 12.Huff MW, Hegele RA. Apolipoprotein C-III: Going back to the future for a lipid drug target. Circ. Res 2013;112:1405–1408 [DOI] [PubMed] [Google Scholar]
- 13.Jorgensen AB, Frikke-Schmidt R, Nordestgaard BG, Tybjaerg-Hansen A. Loss-of-function mutations in apoc3 and risk of ischemic vascular disease. N. Engl. J. Med 2014;371:32–41 [DOI] [PubMed] [Google Scholar]
- 14.Pollin TI, Damcott CM, Shen H, Ott SH, Shelton J, Horenstein RB, Post W, McLenithan JC, Bielak LF, Peyser PA, Mitchell BD, Miller M, O’Connell JR, Shuldiner AR. A null mutation in human apoc3 confers a favorable plasma lipid profile and apparent cardioprotection. Science. 2008;322:1702–1705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tg, Hdl Working Group of the Exome Sequencing Project NHL, Blood I, Crosby J, Peloso GM, Auer PL, Crosslin DR, Stitziel NO, Lange LA, Lu Y, Tang ZZ, Zhang H, Hindy G, Masca N, Stirrups, et al. Loss-of-function mutations in apoc3, triglycerides, and coronary disease. N. Engl. J. Med 2014;371:22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Khetarpal SA, Zeng X, Millar JS, Vitali C, Somasundara AVH, Zanoni P, Landro JA, Barucci N, Zavadoski WJ, Sun Z, de Haard H, Toth IV, Peloso GM, Natarajan P, Cuchel M, et al. A human apoc3 missense variant and monoclonal antibody accelerate apoC-III clearance and lower triglyceride-rich lipoprotein levels. Nat. Med 2017;23:1086–1094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crosby J, Peloso GM, Auer PL, Crosslin DR, Stitziel NO, Lange LA, Lu Y, Tang ZZ, Zhang H, Hindy G, Masca N, Stirrups K, Kanoni S, Do R, Jun G, et al. Loss-of-function mutations in apoc3, triglycerides, and coronary disease. N. Engl. J. Med 2014;371:22–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Saleheen D, Natarajan P, Armean IM, Zhao W, Rasheed A, Khetarpal SA, Won HH, Karczewski KJ, O’Donnell-Luria AH, Samocha KE, Weisburd B, Gupta N, Zaidi M, Samuel M, Imran A, et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature. 2017;544:235–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Graham MJ, Lee RG, Bell TA 3rd, Fu W, Mullick AE, Alexander VJ, Singleton W, Viney N, Geary R, Su J, Baker BF, Burkey J, Crooke ST, Crooke RM. Antisense oligonucleotide inhibition of apolipoprotein C-III reduces plasma triglycerides in rodents, nonhuman primates, and humans. Circ. Res 2013;112:1479–1490 [DOI] [PubMed] [Google Scholar]
- 20.Witztum JL, Gaudet D, Freedman SD, Alexander VJ, Digenio A, Williams KR, Yang Q, Hughes SG, Geary RS, Arca M, Stroes ESG, Bergeron J, Soran H, Civeira F, Hemphill L, et al. Volanesorsen and triglyceride levels in familial chylomicronemia syndrome. N. Engl. J. Med 2019;381:531–542 [DOI] [PubMed] [Google Scholar]
- 21.Alexander VJ, Xia S, Hurh E, Hughes SG, O’Dea L, Geary RS, Witztum JL, Tsimikas S. N-acetyl galactosamine-conjugated antisense drug to apoc3 mrna, triglycerides and atherogenic lipoprotein levels. Eur. Heart J 2019;40:2785–2796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ito Y, Azrolan N, O’Connell A, Walsh A, Breslow JL. Hypertriglyceridemia as a result of human apoC-III gene expression in transgenic mice. Science. 1990;249:790–793 [DOI] [PubMed] [Google Scholar]
- 23.de Silva HV, Lauer SJ, Wang J, Simonet WS, Weisgraber KH, Mahley RW, Taylor JM. Overexpression of human apolipoprotein C-III in transgenic mice results in an accumulation of apolipoprotein b48 remnants that is corrected by excess apolipoprotein e. J. Biol. Chem 1994;269:2324–2335 [PubMed] [Google Scholar]
- 24.Aalto-Setala K, Fisher EA, Chen X, Chajek-Shaul T, Hayek T, Zechner R, Walsh A, Ramakrishnan R, Ginsberg HN, Breslow JL. Mechanism of hypertriglyceridemia in human apolipoprotein (apo) ciii transgenic mice. Diminished very low density lipoprotein fractional catabolic rate associated with increased apoC-III and reduced apo E on the particles. J. Clin. Invest 1992;90:1889–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ding Y, Wang Y, Zhu H, Fan J, Yu L, Liu G, Liu E. Hypertriglyceridemia and delayed clearance of fat load in transgenic rabbits expressing human apolipoprotein CIII. Transgenic Res. 2011;20:867–875 [DOI] [PubMed] [Google Scholar]
- 26.Maeda N, Li H, Lee D, Oliver P, Quarfordt SH, Osada J. Targeted disruption of the apolipoprotein C-III gene in mice results in hypotriglyceridemia and protection from postprandial hypertriglyceridemia. J. Biol. Chem 1994;269:23610–23616 [PubMed] [Google Scholar]
- 27.Jong MC, Rensen PC, Dahlmans VE, van der Boom H, van Berkel TJ, Havekes LM. Apolipoprotein C-III deficiency accelerates triglyceride hydrolysis by lipoprotein lipase in wild-type and apoe knockout mice. J. Lipid Res 2001;42:1578–1585 [PubMed] [Google Scholar]
- 28.Ebara T, Ramakrishnan R, Steiner G, Shachter NS. Chylomicronemia due to apolipoproteinC-III overexpression in apolipoprotein e-null mice. Apolipoprotein ciii-induced hypertriglyceridemia is not mediated by effects on apolipoprotein e. J. Clin. Invest 1997;99:2672–2681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masucci-Magoulas L, Goldberg IJ, Bisgaier CL, Serajuddin H, Francone OL, Breslow JL, Tall AR. A mouse model with features of familial combined hyperlipidemia. Science. 1997;275:391–394 [DOI] [PubMed] [Google Scholar]
- 30.Li H, Han Y, Qi R, Wang Y, Zhang X, Yu M, Tang Y, Wang M, Shu YN, Huang W, Liu X, Rodrigues B, Han M, Liu G. Aggravated restenosis and atherogenesis in apociii transgenic mice but lack of protection in apociii knockouts: The effect of authentic triglyceride-rich lipoproteins with and without apoC-III. Cardiovasc. Res 2015;107:579–589 [DOI] [PubMed] [Google Scholar]
- 31.Yang D, Zhang J, Xu J, Zhu T, Fan Y, Fan J, Chen YE. Production of apolipoprotein C-III knockout rabbits using zinc finger nucleases. J Vis Exp 2013:doi: 10.3791/50957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fan J, Kitajima S, Watanabe T, Xu J, Zhang J, Liu E, Chen YE. Rabbit models for the study of human atherosclerosis: From pathophysiological mechanisms to translational medicine. Pharmacol. Ther 2015;146:104–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fan J, Chen Y, Yan H, Niimi M, Wang Y, Liang J. Principles and applications of rabbit models for atherosclerosis research. J Atheroscler Thromb. 2018;25:213–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang C, Nishijima K, Kitajima S, Niimi M, Yan H, Chen Y, Ning B, Matsuhisa F, Liu E, Zhang J, Chen YE, Fan J. Increased hepatic expression of endothelial lipase inhibits cholesterol diet-induced hypercholesterolemia and atherosclerosis in transgenic rabbits. Arterioscler. Thromb. Vasc. Biol 2017;37:1282–1289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Fan J, Unoki H, Kojima N, Sun H, Shimoyamada H, Deng H, Okazaki M, Shikama H, Yamada N, Watanabe T. Overexpression of lipoprotein lipase in transgenic rabbits inhibits diet-induced hypercholesterolemia and atherosclerosis. J. Biol. Chem 2001;276:40071–40079 [DOI] [PubMed] [Google Scholar]
- 36.Koike T, Kitajima S, Yu Y, Nishijima K, Zhang J, Ozaki Y, Morimoto M, Watanabe T, Bhakdi S, Asada Y, Chen YE, Fan J. Human c-reactive protein does not promote atherosclerosis in transgenic rabbits. Circulation. 2009;120:2088–2094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yan H, Niimi M, Wang C, Chen Y, Zhou H, Matsuhisa F, Nishijima K, Kitajima K, Zhang B, Yokomichi H, Nakanjima K, Murakami M, Zhang J, Chen YE, Fan J. Endothelial lipase exerts its anti-atherogenic effect through increased catabolism of β-vldls. J Atheroscler Thromb. 2020;in press epub ahead of print [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pitas RE, Innerarity TL, Weinstein JN, Mahley RW. Acetoacetylated lipoproteins used to distinguish fibroblasts from macrophages in vitro by fluorescence microscopy. Arteriosclerosis. 1981;1:177–185 [DOI] [PubMed] [Google Scholar]
- 39.Ji ZS, Lauer SJ, Fazio S, Bensadoun A, Taylor JM, Mahley RW. Enhanced binding and uptake of remnant lipoproteins by hepatic lipase-secreting hepatoma cells in culture. J. Biol. Chem 1994;269:13429–13436 [PubMed] [Google Scholar]
- 40.Stephan ZF, Yurachek EC. Rapid fluorometric assay of LDL receptor activity by DiI-labeled ldl. J. Lipid Res 1993;34:325–330 [PubMed] [Google Scholar]
- 41.Nakajima K, Machida T, Imamura S, Kawase D, Miyashita K, Fukamachi I, Maeda M, Muraba Y, Koga T, Kobayashi J, Kimura T, Murakami M. An automated method for measuring lipoprotein lipase and hepatic triglyceride lipase activities in post-heparin plasma. Clin. Chim. Acta 2018;487:54–59 [DOI] [PubMed] [Google Scholar]
- 42.Hornick CA, Kita T, Hamilton RL, Kane JP, Havel RJ. Secretion of lipoproteins from the liver of normal and watanabe heritable hyperlipidemic rabbits. Proc. Natl. Acad. Sci. U. S. A 1983;80:6096–6100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Koike T, Kitajima S, Yu Y, Li Y, Nishijima K, Liu E, Sun H, Waqar AB, Shibata N, Inoue T, Wang Y, Zhang B, Kobayashi J, Morimoto M, Saku K, et al. Expression of human apoaii in transgenic rabbits leads to dyslipidemia: A new model for combined hyperlipidemia. Arterioscler. Thromb. Vasc. Biol 2009;29:2047–2053 [DOI] [PubMed] [Google Scholar]
- 44.Ning B, Chen Y, Waqar AB, Yan H, Shiomi M, Zhang J, Chen YE, Wang Y, Itabe H, Liang J, Fan J. Hypertension enhances advanced atherosclerosis and induces cardiac death in watanabe heritable hyperlipidemic rabbits. Am J Pathol. 2018;188:2936–2947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li S, Wang YN, Niimi M, Ning B, Chen Y, Kang D, Wang Z, Yu Q, Waqar AB, Liu E, Zhang J, Shiomi M, Chen YE, Fan J. Angiotensin II destabilizes coronary plaques in Watanabe heritable hyperlipidemic rabbits. Arterioscler. Thromb. Vasc. Biol 2016;36:810–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Fan J, Ji ZS, Huang Y, de Silva H, Sanan D, Mahley RW, Innerarity TL, Taylor JM. Increased expression of apolipoprotein e in transgenic rabbits results in reduced levels of very low density lipoproteins and an accumulation of low density lipoproteins in plasma. J. Clin. Invest 1998;101:2151–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Ichikawa T, Kitajima S, Liang J, Koike T, Wang X, Sun H, Okazaki M, Morimoto M, Shikama H, Watanabe T, Yamada N, Fan J. Overexpression of lipoprotein lipase in transgenic rabbits leads to increased small dense LDL in plasma and promotes atherosclerosis. Lab. Invest 2004;84:715–726 [DOI] [PubMed] [Google Scholar]
- 48.Clavey V, Lestavel-Delattre S, Copin C, Bard JM, Fruchart JC. Modulation of lipoprotein b binding to the ldl receptor by exogenous lipids and apolipoproteins CI, CII, CIII, and E. Arterioscler. Thromb. Vasc. Biol 1995;15:963–971 [DOI] [PubMed] [Google Scholar]
- 49.Gordts PL, Nock R, Son NH, Ramms B, Lew I, Gonzales JC, Thacker BE, Basu D, Lee RG, Mullick AE, Graham MJ, Goldberg IJ, Crooke RM, Witztum JL, Esko JD. ApoC-III inhibits clearance of triglyceride-rich lipoproteins through ldl family receptors. J. Clin. Invest 2016;126:2855–2866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wang CS, McConathy WJ, Kloer HU, Alaupovic P. Modulation of lipoprotein lipase activity by apolipoproteins. Effect of apolipoprotein C-III. J. Clin. Invest 1985;75:384–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kockx M, Kritharides L. Triglyceride-rich lipoproteins. Cardiol. Clin 2018;36:265–275 [DOI] [PubMed] [Google Scholar]
- 52.Mendivil CO, Zheng C, Furtado J, Lel J, Sacks FM. Metabolism of very-low-density lipoprotein and low-density lipoprotein containing apolipoprotein C-III and not other small apolipoproteins. Arterioscler. Thromb. Vasc. Biol 2010;30:239–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zheng C, Khoo C, Furtado J, Sacks FM. Apolipoprotein C-III and the metabolic basis for hypertriglyceridemia and the dense low-density lipoprotein phenotype. Circulation. 2010;121:1722–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yao Z Human apolipoprotein C-III - a new intrahepatic protein factor promoting assembly and secretion of very low density lipoproteins. Cardiovascular & hematological disorders drug targets. 2012;12:133–140 [DOI] [PubMed] [Google Scholar]
- 55.Kawakami A, Aikawa M, Alcaide P, Luscinskas FW, Libby P, Sacks FM. ApolipoproteinC-III induces expression of vascular cell adhesion molecule-1 in vascular endothelial cells and increases adhesion of monocytic cells. Circulation. 2006;114:681–687 [DOI] [PubMed] [Google Scholar]
- 56.Kawakami A, Aikawa M, Libby P, Alcaide P, Luscinskas FW, Sacks FM. ApolipoproteinC-III in apolipoprotein b lipoproteins enhances the adhesion of human monocytic cells to endothelial cells. Circulation. 2006;113:691–700 [DOI] [PubMed] [Google Scholar]
- 57.Fogelstrand P, Boren J. Retention of atherogenic lipoproteins in the artery wall and its role in atherogenesis. Nutrition, metabolism, and cardiovascular diseases : NMCD. 2012;22:1–7 [DOI] [PubMed] [Google Scholar]
- 58.Hiukka A, Stahlman M, Pettersson C, Levin M, Adiels M, Teneberg S, Leinonen ES, Hulten LM, Wiklund O, Oresic M, Olofsson SO, Taskinen MR, Ekroos K, Boren J. ApoC-III-enriched ldl in type 2 diabetes displays altered lipid composition, increased susceptibility for sphingomyelinase, and increased binding to biglycan. Diabetes. 2009;58:2018–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Li XH, Catalina F, Grundy SM, Patel S. Method to measure apolipoprotein b-48 and b-100 secretion rates in an individual mouse: Evidence for a very rapid turnover of VLDL and preferential removal of b-48-relative to b-100-containing lipoproteins. J. Lipid Res 1996;37:210–220 [PubMed] [Google Scholar]
- 60.Vaith P, Assmann G, Uhlenbruck G. Characterization of the oligosaccharide side chain of apolipoprotein C-III from human plasma very low density lipoproteins. Biochim. Biophys. Acta 1978;541:234–240 [DOI] [PubMed] [Google Scholar]
- 61.Zhang LH, Kotite L, Havel RJ. Identification, characterization, cloning, and expression of apolipoprotein c-iv, a novel sialoglycoprotein of rabbit plasma lipoproteins. J. Biol. Chem 1996;271:1776–1783 [DOI] [PubMed] [Google Scholar]
- 62.Gaudet D, Alexander VJ, Baker BF, Brisson D, Tremblay K, Singleton W, Geary RS, Hughes SG, Viney NJ, Graham MJ, Crooke RM, Witztum JL, Brunzell JD, Kastelein JJ. Antisense inhibition of apolipoprotein C-III in patients with hypertriglyceridemia. N. Engl. J. Med 2015;373:438–447 [DOI] [PubMed] [Google Scholar]
- 63.Gaudet D, Brisson D, Tremblay K, Alexander VJ, Singleton W, Hughes SG, Geary RS, Baker BF, Graham MJ, Crooke RM, Witztum JL. Targeting apoc3 in the familial chylomicronemia syndrome. N. Engl. J. Med 2014;371:2200–2206 [DOI] [PubMed] [Google Scholar]
- 64.Staels B, Vu-Dac N, Kosykh VA, Saladin R, Fruchart JC, Dallongeville J, Auwerx J. Fibrates downregulate apolipoprotein C-III expression independent of induction of peroxisomal acyl coenzyme a oxidase. A potential mechanism for the hypolipidemic action of fibrates. J. Clin. Invest 1995;95:705–712 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.