

Abstract
Epithiodiketopiperazines (ETPs) are a structurally complex class of fungal natural products with potent anticancer activity. In ETPs, the diketopiperazine ring is spanned by a disulfide bond that is constrained in a high-energy eclipsed conformation. We employed computational, synthetic, and spectroscopic methods to investigate the physicochemical attributes of this atypical disulfide bond. We find that the disulfide bond is stabilized by two n→π* interactions, each with large energies (3–5 kcal/mol). The n→π* interactions in ETPs substantially decrease the disulfide reduction potential, endowing stability in physiological environments in a manner that impacts their biological activity. These data reveal a previously unappreciated means to stabilize a disulfide bond and highlight the utility of the n→π* interaction in molecular design.
Graphical Abstract

INTRODUCTION
Organisms are engaged in an incessant race to evolve strategies against selective pressures.1 Among fungi such as Chaetomium spp., the strategy to avoid predation or abate competition is manifested in the form of natural products such as epithiodiketopiperazines (ETPs).2 ETPs comprise a structurally diverse and biologically active family of fungal alkaloids characterized by a disulfide (or polysulfide) that bridges a 2,5-diketopiperazine (Figure 1A).3,4 The combination of the unique and challenging molecular architecture of ETPs3,4 and their potent biological activity2 has captured the attention of scientists across a wide range of disciplines.5–7
Figure 1.

Properties of ETPs. (A) Chemical structures of representative ETPs. (B) Depiction of the biological origin and cellular entry of an ETP. (C) Graph of the distribution of cystinyl Cβ–Sγ–Sγ′–Cβ′ dihedral angles in high-resolution (<3.0 Å) protein crystal structures (black; Table S1), dependence of the energy of the disulfide bond in dimethyl disulfide on the C–S–S–C dihedral angle (red; Table S2), and range of C–S–S–C dihedral angles in crystalline ETPs (blue; Table S3). (D) Overlap of the nS,p and πC=O* orbitals (green) in a model ETP derived from N,N-dimethylalanine. Calculations were at the M06–2X/6–311+G(d,p) level of theory.
Structure–activity relationships have revealed that the activity of ETPs is dependent upon reduction of the epidisulfide bond (Figure 1B) that spans the diketopiperazine (DKP) ring.4,5,8 Unlike those in proteins, the disulfide bonds in ETPs are locked in an eclipsed conformation (Figure 1C).9 In dimethyl disulfide, a prototypical disulfide, values for the C–S–S–C dihedral angle near 0° correspond to ~10 kcal/mol of strain energy (Figure 1C). We reasoned that a compensatory force must exist within ETPs to ameliorate the instability imposed by the eclipsed conformation.
Here, we report on the physicochemical underpinnings of the disulfide bond of ETPs. We began by considering ETP natural products and used quantum mechanical methods to search for the origin of the stability of the disulfide bond. These investigations revealed that n→π* interactions10 that arise from the overlap of the p-type lone pairs of the sulfur atoms with the π* orbitals of the amide carbonyl groups (Figure 1D) are an integral component of ETP alkaloids. We then synthesized and structurally analyzed a series of C4-substituted bisprolyl-ETPs that were designed to manipulate the energetics of the n→π* interaction, and we measured the reduction potential of these synthetic ETPs. We find that C4-substitution perturbs the n→π* interaction and correlated parameters, including the disulfide reduction potential. Our data support a role for n→π* interactions in tuning the reduction potential of ETPs and thus their biological activities.
RESULTS AND DISCUSSION
n→π* Interactions in ETPs.
To investigate the chemical forces that stabilize the strained disulfide bridge, we examined a structurally diverse catalog of natural ETPs with known crystal structures. Untethered disulfide bonds prefer a C–S–S–C dihedral angle near |θ| = 90° (Figure 1C).11 In ETPs, the value of |q| is <20° (Figure 2A), which is near an energy maximum (Figure 1C). We suspected that the strength of the two n→π* interactions in an ETP could compensate for the strain energy of its eclipsed disulfide bond and that evidence for this compensation would be apparent in the X-ray structures of the natural products. In the 1970s, pioneering crystallographic analyses of Bürgi and Dunitz revealed that the optimal angle for nucleophilic attack at a carbonyl group occurs at ~107°.12 Approach at other angles leads to less efficient orbital overlap, resulting in a smaller donation of electron density. We find that values of the S···C=O angle in ETPs are indeed close to the Bürgi–Dunitz trajectory, having a range of 119°–128° (Figure 2B).
Figure 2.
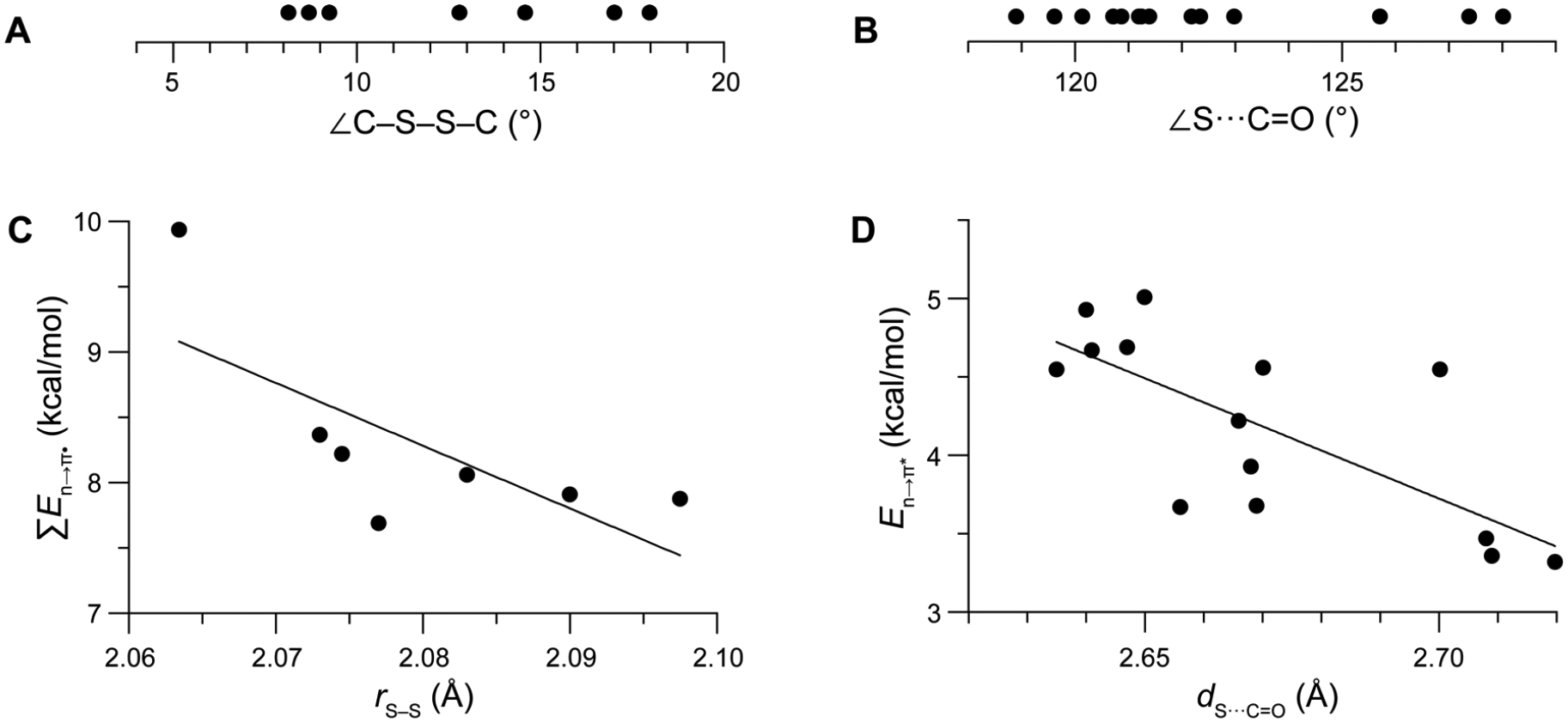
Graphs showing measured and calculated parameters of natural ETPs. (A) C–S–S–C dihedral angles. (B) Angle of the sulfur donor to the carbonyl acceptor. (C) Cumulative energy of n→π* interactions versus sulfur–sulfur bond length (R2 = 0.52). (D) Energy of an n→π* interaction versus its sulfur to carbonyl-carbon distance (R2 = 0.53). Energies on the ordinate were computed at the M06–2X/6–311+G(d,p) level of theory. Chemical structures and data are in Tables S3 and S4.
Next, we used quantum chemistry to investigate the chemical forces that stabilize the strained disulfide bridge of ETPs. We found that the disulfide 3p lone pairs engage intimately with the amide carbonyl groups of the diketopiperazine scaffold. We then employed second-order perturbation theory calculations within the Natural Bond Orbital theory formalism to investigate the strength of the interaction.13 We found that n→π* interactions between the sulfur 3p lone pair (n) and carbonyl π antibonding orbital (π*) have energies of 3–5 kcal/mol (Table S4). Compared to n→π* interactions studied previously in proteins (0.3–0.7 kcal/mol), these n→π* interactions are among the strongest observed to date.14
Anticipating that smaller disulfide bond lengths, rS–S, would be consistent with reduced electron–electron repulsion between the 3p orbitals of the two sulfur atoms due to donation of electron density from each nS,p into the π* orbital of a carbonyl group, we looked for evidence of n→π* interactions in the structure of natural ETPs. Specifically, we measured values of rS–S in natural ETPs, calculated values of ΣEn→π*, and found a correlation (Figure 2C). Likewise, measured values of the donor–acceptor distance, dS···C=O, decrease as values of En→π* increase (Figure 2D). Although neither of these correlations is strong, they are consistent with an n→π* interaction.
Design of ETP Model Systems.
To examine the physicochemical underpinnings of the ETP substructure in greater detail, we designed a symmetrical ETP model that reduces the complexity of the disulfide exchange equilibria while providing opportunities to examine the impact of substituents on the disulfide bond. We envisioned bisprolyl-ETP 5a (Figure 3A), and the C4-substituted derivatives 5b–e as an ideal platform for rigorous structural and physicochemical analyses.15 Substitution of pyrrolidine rings at C4 is known to influence ring puckering via a gauche effect (Figure 3B). Specifically, R-configured electron-withdrawing groups at the C-4 position in a proline residue favor C4-exo ring puckering, and S-configured groups favor C4-endo ring puckering.16 Thus, we chose to introduce flouro, hydroxy, and acetoxy groups in R or S configurations at C4 of bisprolyl-ETPs, giving rise to compounds in which the 4-substituent and disulfide bond are on the same face of the fused rings (cis) or on opposite faces (trans). Our calculations revealed that, as intended, substitution at the C4-position induces conformational changes in ETP 5 that, in turn, modulate orbital overlap and thus the energy of n→π* interactions (Figures 3C and 3D). In particular, the cis and trans configurations differ by ~1 kcal/mol.
Figure 3.

Design principles for ETP model systems. (A) C4-substituted series of symmetric bisprolyl-ETPs 5. (B) Newman projection of ETPs illustrating the gauche effect in cis and trans configurations. (C) Symmetrical n→π* interactions in model ETPs and their calculated energies. (D) Overlap of nS,p and πC=O* orbitals in bisprolyl-ETP 5a. Calculations were at the M06–2X/6–311+G(d,p) level of theory.
Synthesis of C4-Substituted Bisprolyl-ETPs.
As described above, we pursued the synthesis of the cis- and trans-C4-substituted bisprolyl-ETPs 5 (Figure 3A) to evaluate the structural parameters that modulate the reduction potential of the disulfide bond. The unsubstituted bisprolyl-ETP 5a (Figure 2, C4-H) had been used previously as a reference in cytotoxicity studies.15,17
The general approach we used to access bisprolyl-ETPs 5 is outlined in Scheme 1. We envisioned that dithiepanethione 4 could serve as an effective precursor to ETP 5 as we have demonstrated en route to related systems.4,7 We anticipated that the late-stage introduction of the trithiocarbonate to the bisprolyl-framework could provide access to both cis- and trans-dithiepanethiones 4 from a common precursor dioxasilane 3 (Scheme 1). The silyl bridge of dioxasilane 3 derived from silylation of DKP-diol 2 was anticipated to offer superior solubility in organic solvents, facilitating the sulfidation step. We expected that DKP-diol 2 could be prepared using our permanganate-mediated dihydroxylation18 of DKP 1, which itself is a cyclodipeptide derivative of the commercially available and inexpensive trans-4-hydroxy-L-proline. As described below, we found this synthetic approach to be advantageous in accessing C4-substituted derivatives through late-stage diversification.
Scheme 1.

Retrosynthesis of Designed Bisprolyl-ETPs (5)
As illustrated in Scheme 2, we targeted cis- and trans-C4-silyloxy ETPs 5c as common intermediates en route to other C4-substituted epidisulfides 5 (Figure 3A). Our synthesis commenced with the known silylation of trans-C4-hydroxy-L-proline followed by aryl boronic acid-catalyzed dehydrative-dimerization to corresponding DKP 1.19 The permanganate-mediated hydroxylation4,18 of DKP 1 using bis(pyridine)silver(I) permanganate20 in a pyridine–α,α,α-trifluorotoluene mixture (1:1) afforded DKP-diol 2 in 40% yield.21 We found that exposure of diol 2 to monosodium p-methoxybenzyl trithiocarbonate, a reagent for cis-sulfidation of DKP-diols,8b and trifluoroacetic acid in dichloromethane led to the formation of cis-and trans-dithiepanethiones 4 (d.r. 2.2:1) in 61% and 19% yield, respectively. Nonetheless, as shown in Scheme 2, we found it advantageous to utilize dioxasilane 3 as the substrate for the sulfidation reaction since it afforded the cis-and trans-dithiepanethiones 4 (d.r. 1.4:1) in 51% and 39% yield, respectively. These dithiepanethiones were readily separated and efficiently converted to the corresponding cis-and trans-C4-silyloxy epidisulfides 5c in 90% and 71% yield, respectively, upon aminolysis followed by oxidative disulfide formation with potassium triiodide.22,4
Scheme 2.

Synthesis of Substituted ETPs (5c)a
aConditions: (a) Py2AgMnO4, Pyridine, PhCF3, 23 °C, 2 h 20 min, 40%. (b) i-Pr2SiCl2, NEt3, DMAP, DMF, 0 → 23 °C, 95%. (c) sodium p-methoxybenzyl trithiocarbonate, TFA, CH2Cl2, 23 °C, 1.75 h, 51% (cis-4) + 39% (trans-4). (d) ethanolamine, acetone, 0 → 23 °C, 30 min; then KI3, pyridine, CH2Cl2, 90%. (e) ethanolamine, acetone, 0 → 23 °C, 45 min; then KI3, pyridine, CH2Cl2, 71%. Si = Si(Ph)2t-Bu.
Having secured access to epidisulfides 5c, it was necessary to determine their relative and absolute stereochemistry prior to advancing towards the water-soluble derivatives for our study (vida infra). As shown in Scheme 3, cis- and trans-C4-silyloxy ETPs 5c were desilylated upon exposure to hydrogen fluoride in pyridine–THF mixture (1:9) to give cis- and trans-C4-OH ETPs 5b in 81% and 82% yield, respectively. We were able to obtain crystals of cis- and trans-C4-OH ETPs 5b suitable for X-ray diffraction by slow evaporation from dichloromethane–methanol (10:1). We found structural information such as S–S bond length and S···C=O angle of approach observable in the solid state corelated to both the strength of n→π* interactions as well as the reduction potential of C4-substituted bisprolyl-ETPs (vida infra). Subsequent treatment of cis- and trans-C4-OH ETPs 5b with acetyl chloride and pyridine in dichloromethane led to the isolation of cis- and trans-C4-OAc ETPs 5d in 78% and 92% yield, respectively.
Scheme 3.

Synthesis of C4-Substituted ETPs 5b and 5da
aConditions: (a) HF-pyridine, pyridine, THF, 0 → 23 °C, 40 h, 81%. (b) AcCl, pyridine, CH2Cl2, 0 → 23 °C, 6 h, 78%. (c) HF-pyridine, pyridine, THF, 0 → 23 °C, 18 h, 82%. (d) AcCl, pyridine, CH2Cl2, 0 → 23 °C, 8 h, 92%. In the ORTEP representation of ETPs 5b, the thermal ellipsoids are drawn at 50% probability.
We were intrigued by the possibility of late-stage introduction of the fluoro-substituents to access cis- and trans-C4-F ETPs 5e by deoxofluorination of the available ETPs 5b–d.23 Yet, despite a wealth of precedent for stereoinvertive deoxofluorination on C4-OH-substituted proline derivatives,24 there are no reported examples of deoxofluorination in the presence of a disulfide.25 We found that exposure of cis-C4-silyloxy ETP 5c to triethylamine trihydrofluoride (10 equiv) and triethylamine (5 equiv) in dichloromethane at 23 °C for 21 h afforded cis-C4-OH ETP 5b, and that exposure of the reaction mixture to morpholinodifluorosulfinium tetrafluoroborate (XtalFluor-M, 4 equiv) at −78 °C, followed by warming, provided ent-trans-C4-F ETP 5e in 4% yield. Similarly, exposure of trans-C4-silyloxy ETP 5c to identical conditions gave ent-cis-C4-F ETP 5e in 7% yield. Alternatively, we sought to pursue a complementary approach to C4-F-ETPs 5e via the use of the corresponding C4-F DKP 6 (Scheme 4).26 Condensation of N-Boc-trans-4-F-L-proline with trans-4-F-L-proline-OMe hydrochloride26 followed by trifluoroacetic acid-promoted cyclization of the resulting dipeptide gave the desired DKP 6 in 87% yield.22 Permanganate-mediated dihydroxylation of DKP 6 was not optimal due to incomplete dihydroxylation and complications arising from the water solubility of the resulting DKP-diol.
Scheme 4.

Synthesis of cis- and trans-C4-F ETPs 5ea
aConditions: (a) NaHMDS, S8, THF, 23 °C, 2 h; NaBH4, THF, EtOH, 0 → 23 °C, 2 h; KI, I2, pyridine, DCM, 23 °C, 5 min, 19% (cis-5e) + 5% (trans-5e). In the ORTEP representation of ETPs 5e, the thermal ellipsoids are drawn at 50% probability.
Reasoning that the observed challenges were in part due to the inductive influence of the C4-F substituent,4,18a we examined the use of base-promoted electrophilic sulfidation. Exposure of DKP 6 to a solution of elemental sulfur and sodium hexamethyldisilazide (NaHMDS) in tetrahydrofuran15,27 followed by sequential reduction with sodium borohydride and oxidation with potassium triiodide gave separable mixtures of cis-C4-F and trans-C4-F epipolysulfides. The polysulfide mixtures were subjected separately to another round of reduction and oxidation to give cis- and trans-C4-F epidisulfides 5e, offering an alternative approach to access ETPs 5e for our planned study. Importantly, we obtained crystals suitable for X-ray diffraction of cis-C4-F epidisulfide 5e by recrystallization from acetone-hexanes (3:1) and of trans-C4-F epidisulfide 5e by slow evaporation of a saturated solution in acetone-hexanes (1:7), providing further opportunities for detailed structural analysis (Scheme 4).
Because complete stereochemical assignment of cis- and trans-ETPs 5b–e is critical to our analysis of the structural features impacting the reduction potential of the epidisulfide-bridge, prior to obtaining the X-ray structure for ETPs 5b and 5e, we conducted detailed nuclear magnetic resonance (NMR) studies of related derivatives (Scheme 5). We anticipated differentiating the diastereomeric pairs of cis- and trans-epidisulfides 5b and 5e by reductive methylation and selective nuclear Overhauser effect (nOe) NMR experiments relative to the C4-α-stereochemistry encoded within trans-4-hydroxy-L-proline.28,8a Accordingly, the reductive methylation of ETPs 5c and 5e using sodium borohydride and methyl iodide afforded bis(methylthioether) DKPs 7 and 8, respectively.22 The stereochemistry of the C2 methyl sulfide was secured via nOe correlations from the S-methyl to the C4-stereocenter through the C3Hα/β and C5Hα/β protons as illustrated in Scheme 5. Thestereochemical assignments of cis- and trans-sulfides 7 and 8 were consistent with the X-ray crystal structures of the corresponding cis- and trans-ETPs 5b and 5e, respectively.
Scheme 5.
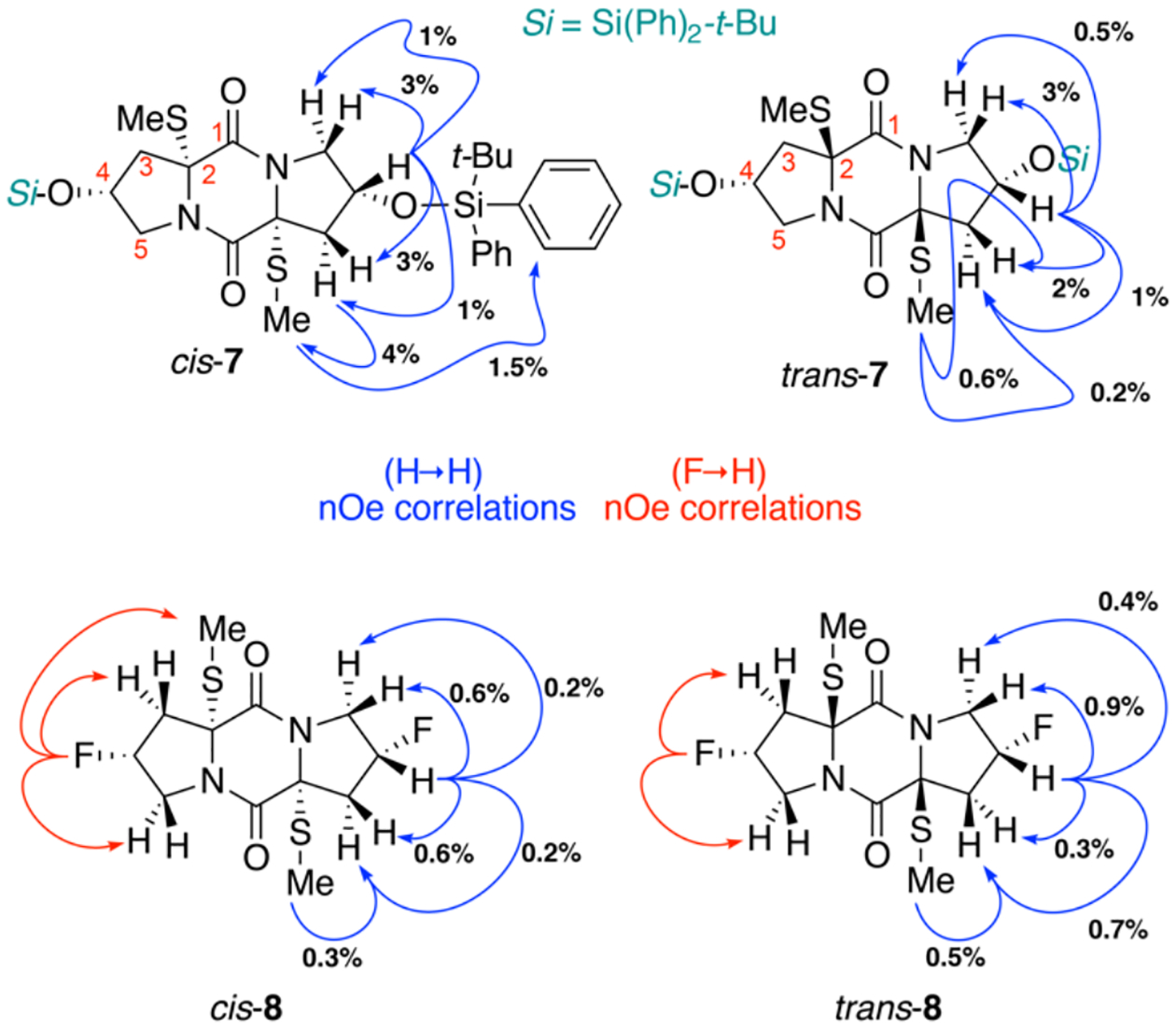
Stereochemical Assignment of Sulfides 7–8
Gauche Effect Modulates n→π* Interaction in Model ETPs.
With the model compounds in hand, we were poised to deconvolute relationships between the structural and physicochemical properties of ETPs. Consistent with our hypothesis about the role of n→π* interactions in ETPs, we found that trends in the synthetic bisprolyl-ETPs mirror those of natural ETPs (cf.: Figures 2 and 4). The most striking trend is depicted in Figure 4D, where a change in S···C=O distance of a range of 0.07 Å corresponded with a change in En→π* interaction of 2 kcal/mol.
Figure 4.

Graphs showing measured and calculated parameters of synthetic ETPs with known crystal structures. (A) C–S–S–C dihedral angles (○). (B) Angle of the sulfur donor to the carbonyl acceptor (○). (C) Cumulative energy of n→π* interactions versus sulfur–sulfur bond length (R2 = 0.82). (D) Energy of an n→π* interaction versus its sulfur to carbonyl-carbon distance (R2 = 0.90). In panels A and B, data for natural ETPs (●) are shown again for comparison. Energies on the ordinate were computed at the M06–2X/6–311+G(d,p) level of theory. Data are listed in Table S5.
Crystallographic Signature of n→π* Interactions.
Following the precedent of Bürgi and Dunitz,12 the most compelling experimental signature of an n→π* interaction has become the pyramidalization of the acceptor carbonyl group towards the electron-pair donor.29 That signature is evident in the crystal structures of both ETP natural products and model ETPs 5b and 5e (Figure 5). Moreover, the interaction is coupled with hydrogen bonding in the crystal structures of gliotoxin, sporidesmin A, and epicorazine, which can lead to aberrant pyramidalization.
Figure 5.

Graph showing two measures of the pyramidalization of carbonyl-group acceptors toward sulfur donors in natural (●) and synthetic (○) ETPs in known crystal structures. Values of Δ were determined with the CCDC program Mercury. Value of χC were determined from the ω1 and ω2 dihedral angles with the equation: χC = ω1 – ω2 + π(mod 2π).34 The origin of outlying points is indicated; red: carbonyl groups that accept hydrogen bonds in the crystal structure.
Disulfide Photophysics Enables Electrochemical Measurements.
The C–S–S–C dihedral angle (θ) correlates with the UV–vis absorption maximum of the disulfide.11 For example, oxidized lipoic acid (E°′ = −288 mV30) has C–S–S–C dihedral angle near θ = 45° and an absorption maximum of 330 nm (Figure S1),11 which is distinct from those of ETPs (Table S6), though there is some spectral overlap (Figures S2 and S3). Using this assay, we were able to detect the concentration of lipoic acid in an ETP ⇄ lipoic acid equilibrium to a concentration as low as 1 μM.
We found that the reduction potentials of synthetic ETPs range from −221 to −267 mV (Table 1). Thus, each synthetic ETP would be reduced nearly completely upon cytosolic entry. The stability afforded by two n→π* interactions decreases the ETP E°′ values substantially (Table 1), enabling the disulfide bond to remain intact in a wide range of physiological environments, including the endoplasmic reticulum (which has E°′ ≈ −210 mV33,32c). In the cytosol, however, the high ratio of reduced glutathione to oxidized glutathione leads to a reduction potential of E°′ ≈ −320 mV.32 Hence, the measured reduction potentials indicate that the n→π* interactions in ETPs are responsible for a balance between extracellular stability and intracellular activity. Without its n→π* interactions, the disulfide bond in an ETP would be much less stable than, for example, that in dithietane (C2H4S2; E°′ = −239 mV30), which has a disulfide bond within a 4-membered ring. Conversely, if the n→π* interactions were too strong, intracellular reduction to the active bisthiol form would not occur.
Table 1.
Reduction Potentials (E°′) of Synthetic ETPs
| ETP | E°′ (mV)a | E°′−n→π* (mV)b |
|---|---|---|
| C4-H (5a) | −254 ± 5 | — |
| cis-C4-OH (cis-5b) | −244 ± 7 | −57 ± 7 |
| trans-C4-OH (trans-5b) | −242 ± 6 | −69 ± 6 |
| cis-C4-OAc (cis-5d) | −230 ± 4 | — |
| trans-C4-OAc (trans-5d) | −267 ± 3 | — |
| cis-C4-F (cis-5e) | −264 ± 3 | −121 ± 3 |
| trans-C4-F (trans-5e) | −221 ± 5 | −93 ± 5 |
Values (± SD) of E°′ were derived from the thiol and disulfide concentrations of a solution equilibrated with reduced and oxidized lipoic acid.31
Values of E°′−n→π* were calculated at 25 °C from the measured values of E°′ and the calculated values of ΣEn→π* for synthetic ETPs with known crystal structures (Figure 4C).
The n→π* interactions within ETPs make their reduction potential responsive to the environment. Specifically, n→π* interactions to a carbonyl group are stronger in protic environments because a hydrogen bond polarizes the carbonyl group, making it a superior acceptor of an n→π* interaction.35 Thus, we anticipate that the disulfide bond in an ETP will be more vulnerable to reduction in a hydrophobic or otherwise desolvated environment, such as the ligand-binding site of a protein or the active site of an enzyme.
Energetic Basis for ETP Electrochemical Equilibria.
Finally, we sought to understand the reduction potentials of ETPs in light of the strain in their disulfide bonds. We found a correlation between two energies: the reduction potential of the disulfide bond and its UV–vis absorption maximum (Figure 6). Specifically, the reduction potential of an ETP is larger (which is indicative of being more easily reduced to the bisthiol) when the absorption maximum is larger (which correlates with a more eclipsed C–S–S–C dihedral angle11). To our knowledge, this is the first example of a correlation between these two manifestations of energy: E°′ ∝ λmax. This correlation is consistent with the intrinsic stability (Figure 1C) and photophysics11 of disulfide bonds.
Figure 6.

Correlations between the absorption maxima and standard reduction potential of the disulfide bond in synthetic ETPs 5e. (A) Graph showing the absorption maximum and reduction potential. Values of λmax were measured experimentally (black; R2 = 0.53) or calculated with the M06–2X (blue; R2 = 0.62) or PBE0 (red; R2 = 0.62) DFT functionals. Data are listed in Table S6. (B) Natural transition orbitals for the S0→S1 transition of cis-C4-F ETP 5e (top) and trans-C4-F ETP 5e (bottom).
To investigate the electronic basis of the relationship between E°′ and λmax, we computed the natural transition orbitals (NTOs) of synthetic model ETPs 5. At such eclipsed C–S–S–C dihedral angles, the ground state (S0) is composed primarily of p-type lone pair density (Figure 6B).11 Promotion of electrons to the first excited state (S1) populates an NTO with antibonding character, consistent with the observed correlation of E°′ and λ max (Figure 6A).
CONCLUSIONS
We discovered an important aspect of ETP natural products: strong n→π* interactions in which electron density is donated from the sulfur atoms of the disulfide bond into the carbonyl groups of the diketopiperazine. Two strong n→π* interactions in each ETP nearly completely compensate for the ~10 kcal/mol of instability imposed by the eclipsed conformation of the disulfide bond. This discovery could elucidate structure–activity relationships of ETPs and inform the design of new ETPs with desirable properties.
Supplementary Material
ACKNOWLEDGMENT
We thank Dr. Charlene Tsay and Dr. Peter Müller (Department of Chemistry, Massachusetts Institute of Technology) for assistance with single crystal X-ray diffraction. M.M. and C.R.O. thank Dr. Brandon M. Nelson for helpful discussions related to DKP-dioxasilanes.
Funding Sources
K.A.D. acknowledges the Natural Sciences and Engineering Research Council of Canada (NSERC) for a PGS-D3 scholarship. This work was supported by Grants R01 GM089732 and R01 GM044783 (NIH), and made use of the Extreme Science and Engineering Discovery Environment (XSEDE), which is supported by Grant ACI-1548562 (NSF).
Footnotes
ASSOCIATED CONTENT
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jacs.xxxxxxx.
| ETP cis-5b | CCDC 1965015 |
| ETP trans-5b | CCDC 1965016 |
| ETP cis-5e | CCDC 1965017 |
| ETP trans-5e | CCDC 1965018 |
The authors declare no competing financial interests.
REFERENCES
- (1).(a) Darwin C On the Origin of Species by Means of Natural Selection, or Preservation of Favoured Races in the Struggle for Life. John Murray: London, UK, 1859. [Google Scholar]; (b) Mayr E The Growth of Biological Thought: Diversity, Evolution, and Inheritance. Harvard University Press: Cambridge, MA, 1982. [Google Scholar]
- (2).(a) Keller NP Fungal secondary metabolism: Regulation, function and drug discovery. Nat. Rev. Microbiol 2019, 17, 167–180. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Künzler M How fungi defend themselves against microbial competitors and animal predators. PLoS Pathog. 2019, 14, e1007184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).(a) Hino T; Nakagawa M Chemistry and reactions of cyclic tautomers of tryptamines and tryptophans In The Alkaloids: Chemistry and Pharmacology, Brossi A, Ed. Academic Press: New York, NY, 1989; Vol. 34, pp 1–75. [Google Scholar]; (b) Waring P; Beaver J Gliotoxin and related epipolythiodioxopiperazines. Gen. Pharmacol 1996, 27, 1311–1316. [DOI] [PubMed] [Google Scholar]; (c) Gardiner DM; Waring P; Howlett BJ The epipolythiodioxopiperazine (ETP) class of fungal toxins: Distribution, mode of action, functions, and biosynthesis. Microbiology 2005, 151, 1021–1032. [DOI] [PubMed] [Google Scholar]; (d) Kim J; Movassaghi M Biogenetically inspired syntheses of alkaloid natural products. Chem. Soc. Rev 2009, 38, 3035–3050. [DOI] [PubMed] [Google Scholar]; (e) Jiang C-S; Guo Y-W Epipolythiodioxopiperazines from fungi: Chemistry and bioactivities. Mini Rev. Med. Chem 2011, 11, 728–745. [DOI] [PubMed] [Google Scholar]; (f) Welch TR; Williams RM Epidithiodioxopiperazines: Occurence, synthesis, and biogenesis. Nat. Prod. Rep 2014, 31, 1376–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Kim J; Movassaghi M Biogenetically-inspired total synthesis of epidithioketopiperazines and related alkaloids Acc. Chem. Res 2015, 48, 1159–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).(a) Vigushin DM; Mirsaidi N; Brooke G; Sum C; Pace P; Inman L; Moody CJ; Coombes RC Gliotoxin is a dual Inhibitor of farnesyltransferase and geranylgeranyltransferase I with antitumor activity against breast cancer in vivo. Med. Oncol 2004, 21, 21–30. [DOI] [PubMed] [Google Scholar]; (b) Zheng CJ; Kim CJ; Bae KS; Kim Y; Kim WG Bionectins A-C, epidithiodioxopiperazines with anti-MRSA activity from Bionectra byssicola F120. J. Nat. Prod 2006, 69, 1816–1819. [DOI] [PubMed] [Google Scholar]; (c) Isham CR; Tibodeau JD; Jin W; Xu R; Timm M; Bible KC Chaetocin: A promising new antimyeloma agent with in vitro and in vivo activity mediated via imposition of oxidative stress. Blood 2007, 109, 2579–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Cook KM; Hilton ST; Mecinovic J; Motherwell W; Figg WD; Schofield CJ Epidithiodiketopiperazines block the interaction between hypoxia-inducible-factor-1α (HIF-1α) and p300 by a zinc ejection mechanism. J. Biol. Chem 2009, 284, 26831–26838. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Liu F; Liu Q; Yang D; Bollag WB; Robertson K; Wu P; Liu L Verticillin A overcomes apoptosis resistance in human colon carcinoma through DNA methylation-dependent upregulation of BNIP3. Cancer Res. 2011, 71, 6807–6816. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Dubey R; Levin MD; Szabo LZ; Laszlo CF; Kushal S; Singh JB; Oh P; Schnitzer JE; Olenyuk BZ Suppression of tumor growth by designed dimeric epidithiodiketopiperazine targeting hypoxia-inducible transcription factor complex J. Am. Chem. Soc 2013, 135, 4537–4549. [DOI] [PubMed] [Google Scholar]; (g) Saleh AA; Jones GW; Tinley FC; Delaney SF; Alabbadi SH; Fenlon K; Doyle S; Owens RA Systems impact of zinc chelation by the epipolythiodioxopiperazine dithiol gliotoxin in Aspergillus fumigatus: A new direction in natural product functionality. Metallomics 2018, 10, 854–866. [DOI] [PubMed] [Google Scholar]; (h) Dewangan J; Srivastava S; Mishra S; Pandey PK; Kivakar A; Rath SK Chetomin induces apoptosis in human triple-negative breast cancer cells by promoting calcium overload and mitochondrial dysfunction. Biochem. Biophys. Res. Commun 2018, 495, 1915–1921. [DOI] [PubMed] [Google Scholar]; (i) Asquith CRM; Sil BC; Laitinen T; Tizzaard GJ; Coles SJ; Poso A; Hofmann-Ehmann R; Hilton ST Novel epidithiodiketopiperazines as anti-viral zinc ejectors of the feline immunodeficiency virus (FIV) nucleocapsinprotein as a model for HIV infection. Bioorg. Med. Chem. Lett 2019, 27, 4174–4184. [DOI] [PubMed] [Google Scholar]
- (6).For representative syntheses of ETPs, see:; (a) Fukuyama T; Nakatsu S-I; Kishi Y Total synthesis of gliotoxin, dehydrogliotoxin, and hyalodendrin. Tetrahedron 1981, 37, 2045–2078. [Google Scholar]; (b) Overman LE; Sato T Construction of epdidithiodioxopiperazines by directed oxidation of hydroxyproline derived dioxopiperazines. Org. Lett 2007, 9, 5267–5270. [DOI] [PubMed] [Google Scholar]; (c) Iwasa E; Hamashima Y; Fujishiro S; Higuchi E; Ito A; Yoshida M; Sodeoka M Total synthesis of (+)-chaetocin and its analogues: Their histone methyltransferase G9a inhibitory activity. J. Am. Chem. Soc 2010, 132, 4078–4079. [DOI] [PubMed] [Google Scholar]; (d) Codelli JA; Puchlopek AL; Reisman SE Enantioselective total synthesis of (−)-acetylaranotin, a dihydrooxepine epidithiodiketopiperazine. J. Am. Chem. Soc 2012, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Takeuchi R; Shimokawa J; Fukuyama T Development of a route to chiral epidithiodioxopiperazine moieties and application to the asymmetric synthesis of (+)-hyalodendrin. Chem. Sci 2014, 5, 2003–2006. [Google Scholar]; (f) Baumann M; Dieskau AP; Loertscher B,M; Walton MC; Nam S; Xie J; Horne D; Overman LE Tricyclic analogues of epidithiodioxopiperazine alkaloids with promising in vitro and in vivo antitumor activity Chem. Sci 2015, 6, 4451–4457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).For representative syntheses of ETPs from our laboratory, see:; (a) Kim J; Ashenhurst JA; Movassaghi M Total synthesis of (+)-11,11′-dideoxyverticillin A. Science 2009, 324, 238–241. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kim J; Movassaghi M General approach to epipolythiodiketopiperazine alkaloids: Total synthesis of (+)-chaetocins A and C and (+)-12,12′-dideoxychetracin A. J. Am. Chem. Soc 2010, 132, 14376–14378. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Coste A; Kim J; Adams TC; Movassaghi M Concise total synthesis of (+)-bionectins A and C. Chem. Sci 2013, 4, 3191–3197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Boyer N; Morrison KC; Kim J; Hergenrother PJ; Movassaghi M Synthesis and anticancer activity of epipolythiodiketopiperazine alkaloids. Chem. Sci 2013, 4, 1311–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Olsson CR; Payette JN; Cheah JH; Movassaghi M Synthesis of potent cytotoxic epidithiodiketopiperazines designed for derivatization. J. Org. Chem 2020, 85, 4648–4662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).(a) Borthwick AD 2,5-Diketopiperazines: Synthesis, reactions, medicinal chemistry, and bioactive natural products. Chem. Rev 2012, 112, 3641–3716. [DOI] [PubMed] [Google Scholar]; (b) Zong L; Bartolami E; Abegg D; Adibekian A; Sakai N; Matile S Epidithiodiketopiperazines: Strain-promoted thiol-mediated cellular uptake at the highest tension. ACS Cent. Sci 2017, 3, 449–453. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chuard N; Poblador-Bahamonde AI; Zong L; Bartolami E; Hildebrandt J; Weigand W; Sakai N; Matile S Diselenolane-mediated cellular uptake. Chem. Sci 2018, 9, 1860–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Kilgore HR; Raines RT n→π* Interactions modulate the properties of cysteine residues and disulfide bonds in proteins. J. Am. Chem. Soc 2018, 140, 17606–17611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kilgore HR; Raines RT Disulfide chromophores arise from stereoelectronic effects. J. Phys. Chem. B 2020, 124, 3931–3935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).(a) Bürgi HB; Dunitz JD; Shefter E Geometric reaction coordinates. II. Nucleophilic addition to a carbonyl group. J. Am. Chem. Soc 1973, 95, 5065–5067. [Google Scholar]; (b) Bürgi HB; Dunitz JD; Shefter E Chemical reaction paths. IV. Aspects of O···C=O interactions in crystals. Acta Crystallogr., Sect. B: Struct. Crystallogr Cryst. Chem 1974, 30, 1517–1527. [Google Scholar]; (c) Bürgi HB; Dunitz JD; Lehn JM; Wipff G Stereochemistry of reaction paths at carbonyl centres. Tetrahedron 1974, 30, 1563–1572. [Google Scholar]
- (13).(a) Weinhold F; Landis CR Discovering Chemistry with Natural Bond Orbitals. John Wiley & Sons: Hoboken, NJ, 2012. [Google Scholar]; (b) Weinhold F Natural bond orbital analysis: A critical overview of relationships to alternative bonding perspectives. J. Comput. Chem 2012, 33, 2363–2379. [DOI] [PubMed] [Google Scholar]; (c) Glendening ED; Badenhoop JK; Reed AE; Carpenter JE; Bohmann JA; Morales CM; Landis CR; Weinhold F NBO 6.0, Theoretical Chemistry Institute, University of Wisconsin–Madison: Madison, WI, 2013. [Google Scholar]
- (14).Newberry RW; Raines RT The n→π* interaction. Acc. Chem. Res 2017, 50, 1838–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).For a prior synthesis of 5a, see:; Öhler E; Poisel H; Tataruch F; Schmidt U Syntheseversuche in der Reihe der 3.6 Epidithio-2.5-dioxo-piperazin-Antibiotika Gliotoxin, Sporidesmin, Aranotin und Chaetocin, IV. Synthese des Epidithio-L-prolyl-Lprolinanhydrids. Chem. Ber 1972, 105, 635–641. [DOI] [PubMed] [Google Scholar]
- (16).(a) Shoulders MD; Satyshur KA; Forest KT; Raines RT Stereoelectronic and steric effects in side chains preorganize a protein main chain. Proc. Natl. Acad. Sci. USA 2010, 107, 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pandey AK; Naduthambi D; Thomas KM; Zondlo NJ Proline editing: A general and practical approach to the synthesis of functionally and structurally diverse peptides. Analysis of steric versus stereoelectronic effects of 4-substituted prolines on conformation within peptides. J. Am. Chem. Soc 2013, 135, 4333–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Bernando PH; Brasch N; Chai CLL; Waring P Novel redox mechanism for the glutathione-dependent reversible uptake of a fungal toxin in cells. J. Biol. Chem 2003, 187, 46549–46555. [DOI] [PubMed] [Google Scholar]
- (18).(a) Bischoff AJ; Nelson BM; Niemeyer ZL; Sigman MS; Movassaghi M Quantitative modeling of bis(pyridine)silver(I) permanganate oxidation of hydantoin derivatives: Guidelines for predicting the site of oxidiation in complex substrates. J. Am. Chem. Soc 2017, 139, 15539–15547. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Haines BE; Nelson BM; Grandner JM; Kim J; Houk KN; Movassaghi M; Musaev DG Mechanism of permanganate promoted dihydroxylation of complex diketopiperazines: Critical roles of counter cation and ion-pairing J. Am. Chem. Soc 2018, 140, 13375–13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).(a) Ishihara K; Ohara S; Yamamoto H 3,4,5-Trifluorobenzeneboronic acid as an extremely active amidiation catalyst. J. Org. Chem 1996, 61, 4196. [DOI] [PubMed] [Google Scholar]; (b) Delaney JP; Brozinski HL; Henderson LC Synergistic effects with a C2-symmetric organocatalyst: The potential formation of a chiral catalytic pocket. Org. Biomol. Chem 2013, 11, 2951–2960. [DOI] [PubMed] [Google Scholar]
- (20).Firouzabadi H; Vessal B; Naderi M Bispyridinesilver permanganate [Ag(C5H5N)2]MnO4: An efficient oxidizing reagent for organic substrates. Tetrahedron Lett. 1982, 23, 1847–1850. [Google Scholar]
- (21). This solvent combination offered optimal substrate solubility and oxidation, as DKP 1 is insoluble in α,α,α-trifluorotoluene alone as solvent. The oxidation of DKP 1 with bis(pyridine)silver(I) permanganate in dichloromethane gave diol 2 in 8% yield with 46% recovered starting material, whereas the same oxidation in pyridine gave diol 2 in 12% yield with 63% recovered starting material.
- (22). See the Supporting Information for further details.
- (23).L’Hereux A; Beauliue F; Bennett C; Bill DR; Clayton S; LaFlamme F; Mirmehrabi M; Tdayon S; Tovell D; Couturier M Aminodifluorosulfinium salts: Selective fluorination reagents with enhanced thermal stability and ease of handling. J. Org. Chem 2010, 75, 3401–3411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Newberry RW; Raines RT 4-Fluoroprolines: Conformational analysis and effects on the stability and folding of peptides and proteins. Top. Heterocycl. Chem 2017, 48, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Thiocarbonyl derivatives are converted to gem-difluorides using Deoxo-Fluor, see:; Lal GS; Lobach E; Evans A Fluorination of thiocarbonyl compounds with bis(2-methoxythyl)aminosulfur trifluoride (Deoxo-Fluor Reagent): A facile synthesis of gem-difluorides J. Org. Chem 2000, 65, 4830–4832. [DOI] [PubMed] [Google Scholar]
- (26). The direct boronic acid catalyzed dimerization approach used to prepare diketopiperazine diketopiperazine 1 was not optimal for use with trans-C4-fluoro-L-proline due to poor solubility.
- (27).Nicolaou KC; Giguère D; Totokotsòpoulos S; Sun Y-P A practical sulfenylation of 2,5-diketopiperazines. Angew. Chem., Int. Ed 2012, 51, 728–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Boyer N; Movassaghi M Concise total synthesis of (+)-gliocladins B and C. Chem. Sci 2012, 3, 1798–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).(a) Choudhary A; Gandla D; Krow GR; Raines RT Nature of amide carbonyl–carbonyl interactions in proteins. J. Am. Chem. Soc 2009, 131, 7244–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Choudhary A; Raines RT Signature of n→π* interactions in α-helices. Protein Sci. 2011, 20, 1077–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Choudhary A; Kamer KJ; Raines RT An n→π* interaction in aspirin: Implications for structure and reactivity. J. Org. Chem 2011, 76, 7933–7937. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Kamer KJ; Choudhary A; Raines RT Intimate interactions with carbonyl groups: Dipole–dipole or n→π*? J. Org. Chem 2013, 78, 2099–2103. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Newberry RW; Raines RT n→π* Interactions in poly(lactic acid) suggest a role in protein folding. Chem. Commun 2013, 49, 7699–7701. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Newberry RW; VanVeller B; Guzei IA; Raines RT n→π* Interactions of amides and thioamides: Implications for protein stability. J. Am. Chem. Soc 2013, 135, 7843–7846. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Choudhary A; Fry CG; Kamer KJ; Raines RT An n→π* interaction reduces the electrophilicity of the acceptor carbonyl group. Chem. Commun 2013, 49, 8166–8168. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Guzei IA; Choudhary A; Raines RT Pyramidalization of a carbonyl C atom in (2S)-N-(selenoacetyl)proline methyl ester. Acta Crystallogr., Sect. E: Struct. Rep. Online 2013, 69, o805–806. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Newberry RW; Bartlett GJ; VanVeller B; Woolfson DN; Raines RT Signatures of n→π* interactions in proteins. Protein Sci. 2014, 23, 284–288. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Newberry RW; Raines RT A key n→π* interaction in N acyl homoserine lactones. ACS Chem. Biol 2014, 9, 880–883. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Choudhary A; Newberry RW; Raines RT n→π* Interactions engender chirality in carbonyl groups. Org. Lett 2014, 16, 3421–3423. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Wilhelm P; Lewandowski B; Trapp N; Wennemers H A crystal structure of an oligoproline PPII-helix, at last. J. Am. Chem. Soc 2014, 136, 15829–15832. [DOI] [PubMed] [Google Scholar]; (m) Newberry RW; Raines RT Crystal structure of N-(3-oxobutanoyl)-L-homoserine lactone. Acta Crystallogr., Sect. E: Struct. Rep. Online 2016, 72, 136–139. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Rahim A; Sahariah B; Sarma BK N,N′ Di(acylamino)-2,5-diketopiperazines: Strategic incorporation of reciprocal n→π* Interactions in a druglike scaffold. Org. Lett 2018, 20, 5743–5746. [DOI] [PubMed] [Google Scholar]
- (30).Lees WJ; Whitesides GM Equilibrium constants for thiol–disulfide interchange reactions: A coherent, corrected set. J. Org. Chem 1993, 58, 642–647. [Google Scholar]
- (31). Because of systematic spectral overlap, these values are upper limits. The actual values of E°′ could be <30 mV lower, that is, more negative.
- (32).(a) Ostergaard H; Tachibana C; Winther JR Monitoring disulfide bond formation in the eukaryotic cytosol. J. Cell Biol 2004, 166, 337–345. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Morgan B; Ezerina D; Amoako TN; Riemer J; Seedorf M; Dick TP Multiple glutathione disulfide removal pathways mediate cytosolic redox homeostasis. Nat. Chem. Biol 2013, 9, 119–125. [DOI] [PubMed] [Google Scholar]; (c) Schwarzländer M; Dick TP; Meyer AJ; Morgan B Dissecting redox biology using fluorescent protein sensors. Antioxid. Redox Signal 2016, 24, 680–712. [DOI] [PubMed] [Google Scholar]
- (33).Birk J; Meyer M; Aller I; Hansen HG; Odermatt A; Dick TP; Meyer AJ; Appenzeller-Herzog C Endoplasmic reticulum: Reduced and oxidized glutathione revisited. J. Cell Sci 2013, 126, 1604–1617. [DOI] [PubMed] [Google Scholar]
- (34).Winkler FK; Dunitz JD The non-planar amide group. J. Mol. Biol 1971, 59, 169–182. [DOI] [PubMed] [Google Scholar]
- (35).(a) Shoulders MD; Kotch FW; Choudhary A; Guzei IA; Raines RT The aberrance of the 4S diastereomer of 4-hydroxyproline. J. Am. Chem. Soc 2010, 132, 6651–6653. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Erdmann RS; Wennemers H Importance of ring puckering versus interstrand hydrogen bonds for the conformational stability of collagen. Angew. Chem., Int. Ed 2011, 50, 6835–6838. [DOI] [PubMed] [Google Scholar]; (c) Erdmann RS; Wennemers H Effect of sterically demanding substituents on the conformational stability of the collagen triple helix. J. Am. Chem. Soc 2012, 134, 17117–17124. [DOI] [PubMed] [Google Scholar]; (d) Siebler C; Erdmann RS; Wennemers H Switchable proline derivatives: Tuning the conformational stability of the collagen triple helix by pH changes. Angew. Chem., Int. Ed 2014, 53, 10340–10344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.