

Abstract
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by an asymptomatic period of amyloid-β (Aβ) deposition as insoluble extracellular plaque, intracellular tau aggregation, neuronal and synaptic loss, and subsequent cognitive dysfunction and dementia. A growing public health crisis, the worldwide prevalence of AD is expected to rise from 46.8 million individuals affected in 2015 to 131.5 million in 2050. Sleep disturbances have been associated with increased future risk of AD. A bi-directional relationship is hypothesized between sleep and AD with sleep disturbances as either markers for AD pathology and/or a mechanism mediating increased risk of AD. In this review, the evidence in humans supporting this complex relationship between sleep and AD will be discussed as well as the therapeutic potential and challenges of treating sleep disturbances to prevent or delay the onset of AD.
Keywords: Sleep, Alzheimer’s disease, amyloid-beta, tau
Introduction
Alzheimer’s disease (AD) is a progressive neurodegenerative disorder characterized by an asymptomatic period of amyloid-β (Aβ) deposition as insoluble extracellular plaque, intracellular tau aggregation, neuronal and synaptic loss, and eventual cognitive dysfunction and dementia (Bateman et al., 2012; Jack et al., 2013; Vos et al., 2013). Although classically diagnosed by cognitive symptoms, AD is increasingly defined by imaging and cerebrospinal fluid (CSF) biomarkers for Aβ, tau, and neurodegeneration (Jack et al., 2016). Substantial evidence supports that amyloid deposition begins ~15–20 years before cognitive impairment (i.e., during an asymptomatic or “preclinical” stage of AD) (Price and Morris, 1999; Sperling et al., 2011). Age is the greatest risk factor for AD with the risk doubling every 5 years after the age of 65 (Jorm and Jolley, 1998). With the global population ≥60 years old expected to increase from 12.2% in 2015 to 21.2% in 2050, the prevalence of AD is also expected to rise from 46.8 million individuals affected in 2015 to 131.5 million in 2050 (Prince et al., 2015).
Sleep disturbances have been associated with future risk of both cognitive impairment and AD pathology. For example, older women self-reporting ≤5 hours of sleep/night had worse cognitive performance over the subsequent two years compared to those who slept 7 hours/night (Tworoger et al., 2006). In another study, sleep efficiency (a measure of sleep quality defined as total sleep time/time in bed) was significantly lower in cognitively normal older adults with amyloid deposition (i.e., amyloid-positive) compared to those who were amyloid-negative (Ju et al., 2013). Differences in sleep parameters, such as sleep efficiency, between asymptomatic individuals with and without AD pathology raises an important question of what came first: the sleep disturbance or AD pathology. A major challenge in the field has been determining the causal relationship between sleep and AD.
Further complicating investigations of the relationship between sleep and AD, reports of daytime sleepiness and other sleep-related symptoms also increase with normal aging (Smagula et al., 2016). Multiple measures of sleep architecture change during normal aging. In one study, sleep efficiency decreased significantly from an average of 85.7% (standard deviation (SD) 8.3) in individuals ≤54 years old to an average of 79.2% (SD 10.1) in individuals >70 years old (p<0.001) (Redline et al., 2004). In the same study, the percent of the night spent in non-rapid eye movement (NREM) sleep stage 3 or slow wave sleep (SWS) decreased significantly from 11.2% (95% confidence intervals (CI) 9.9–12.6) in men ≤54 years old to 5.5% (95% CI 4.5–6.5) in men >70 years old while women remain in the range of 14–17% across the same time period. An increased number of nighttime awakenings, increased time in NREM sleep stage 1 (N1) or drowsiness, earlier waking times, and decreased sleep spindles have also been found to change with age (Redline et al., 2004). Further, sleep disorders such as sleep-disordered breathing (e.g., obstructive sleep apnea (OSA)), insomnia, restless legs syndrome/periodic leg movement disorder (RLS/PLMD), and REM sleep behavior disorder (RBD) also produce sleep-related symptoms and increase with age (Ancoli-Israel et al., 1991a; Ancoli-Israel et al., 1991b; Asplund, 1996; Bliwise, 2005; Hoch et al., 1990; Phillips et al., 2000; Schenck et al., 1986).
An additional complication to defining the relationship between sleep and AD is that sleep-wake activity may be measured via multiple modalities, such as sleep questionnaires and logs, actigraphs, and studies based on electroencephalography (EEG) to measure electrical activity in the brain to differentiate sleep and wake states. Each of these methods have advantages and disadvantages, and studies in humans associating sleep disturbances with future risk of cognitive impairment have used each of them. Sleep questionnaires or logs may be easily deployed to large numbers of participants, but rely on subjective self-report of sleep activity and quality. More objective measures, such as actigraphy, are quantitative but rely on rest-activity rhythms as a surrogate for sleep-wake activity. Attended polysomnography is the gold standard for sleep monitoring, but may be cost-prohibitive and inconvenient for participants. EEG-based ambulatory devices are increasingly available for at-home sleep monitoring, but provide a more limited number of EEG channels than polysomnography.
Given the long period of asymptomatic preclinical AD, sleep disturbances are hypothesized to be either markers for AD pathology and/or a mechanism mediating increased risk of AD (i.e., a bi-directional relationship, Fig 1) (Brown et al., 2016a; Carroll and Macauley, 2019; Cedernaes et al., 2017; Havekes et al., 2019; Ju et al., 2014; Lucey and Bateman, 2014; Mander et al., 2016; Musiek and Holtzman, 2016; Yaffe et al., 2014). In this review, new evidence in humans will be discussed that complicates the relationship between sleep and AD but also points to exciting future directions for investigation.
Figure 1:
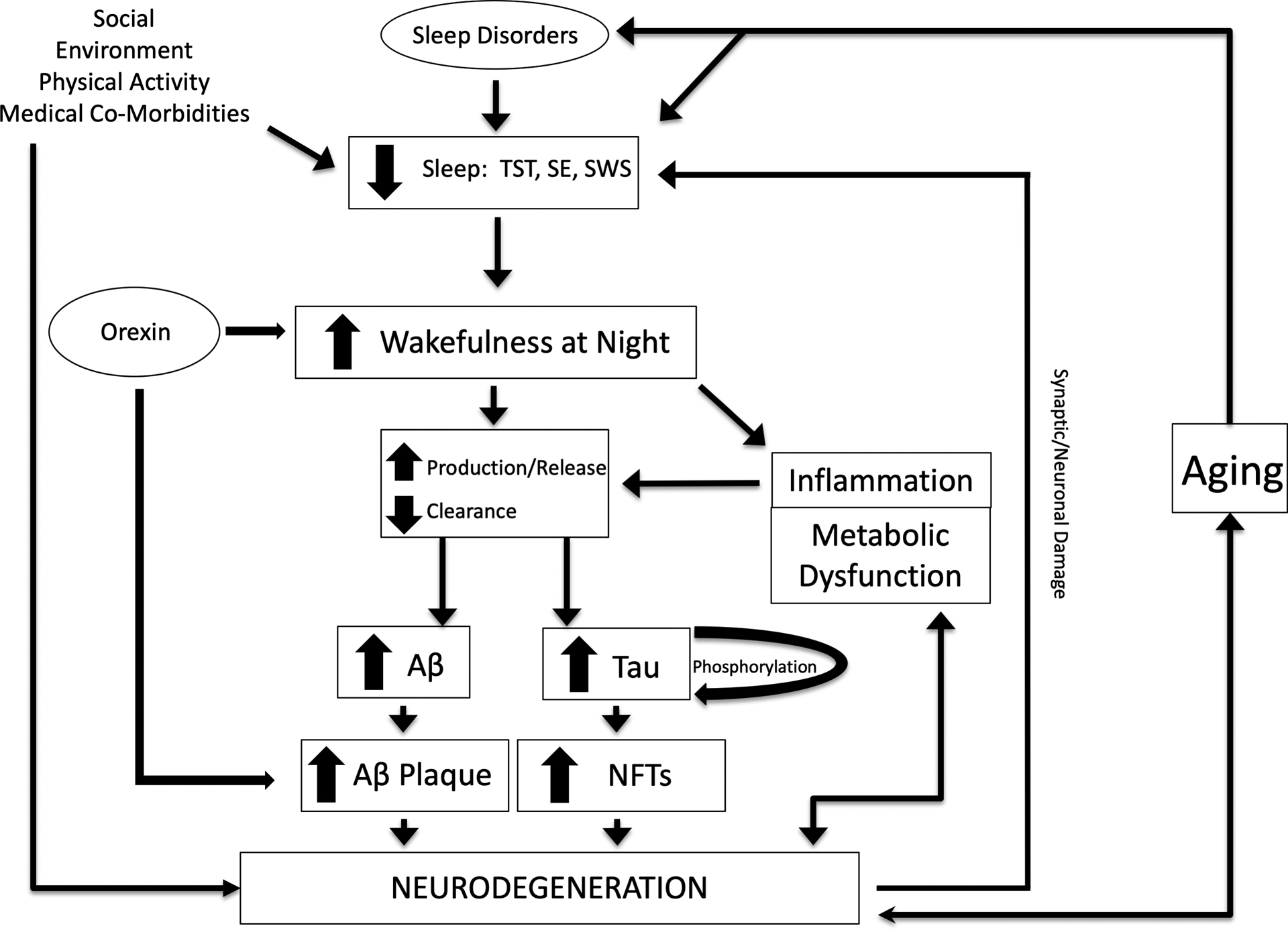
Hypothetical model of the relationship between sleep and Alzheimer’s disease. Multiple factors, including aging, sleep disorders, and environmental factors, lead to sleep disturbance and increased wakefulness at night. Decreased sleep increases the production of Aβ and release of tau, as well as decreased clearance from the CSF, promoting the formation of amyloid plaques and tau pathology. Tau phosphorylation is also altered by sleep loss. Sleep disturbance may modulate effects of inflammation and metabolic dysfunction on Aβ and tau levels as well as promote neurodegeneration. Neurodegeneration from amyloid plaques and tau pathology results in synaptic/neuronal damage that feedbacks to cause sleep disturbances. Orexin is a neuropeptide that promotes wakefulness and has been found to increase amyloid pathology.
TST: Total sleep time; SE: sleep efficiency; SWS: slow wave sleep; Aβ: amyloid-β; NFTs: neurofibrillary tangles.
Sleep disturbance as a marker of Alzheimer’s disease risk
Neurodegenerative disorders such as AD are commonly associated with sleep disturbances (Guarnieri et al., 2012; McCurry et al., 1999; Moran et al., 2005). Evidence from multiple studies supports that sleep disturbances are a risk factor for cognitive impairment due to probable AD, however studies have shown inconsistent results regarding the nature of the relationship depending on the sleep parameter measured. For instance, individuals ≥60 years old who self-reported long sleep duration ≥11 hours/night had lower Mini-Mental State Examination (MMSE) scores compared to those who slept 7 hours/night; individuals with short sleep duration of <7 hours did not have lower cognitive function (Faubel et al., 2009). In contrast, a study of 28,670 community-dwelling older adults aged 50–85 years found that self-reported sleep durations of 3–4 hours or ≥10 hours were associated with greater odds of having memory impairment on the delayed word recall test (Xu et al., 2011). Multiple studies have replicated these results suggesting that the relationship between self-reported total sleep time and risk of memory impairment is not linear in older adults (Ding et al., 2020; Ferrie et al., 2011; Kronholm et al., 2009; Loerbroks et al., 2010; Westwood et al., 2017). Studies using self-reported sleep problems have also implicated other sleep measures such as daytime napping and excessive daytime sleepiness as predictors of cognitive decline (Keage et al., 2012).
Total sleep time measured by actigraphy, an objective measure of sleep-wake activity, in 2932 women ≥65 years old did not find a relationship with cognitive performance on the MMSE (Blackwell et al., 2006). However, in this study decreased sleep efficiency <70% correlated with cognitive impairment (MMSE <26) compared to older women with sleep efficiency ≥70%. These findings suggest that sleep quality (i.e., higher sleep efficiency or less time awake after sleep onset) rather than total sleep time is a critical factor. Studies using actigraphy have also found that increased time awake after sleep onset in cognitively normal older adults moderated the relationship between amyloid deposition and memory performance on the selective reminding test (Molano et al., 2017) as well as immediate and delayed memory (Wilckens et al., 2018). Self-report of sleep duration and other sleep parameters is subjective and may account for inconsistencies between the studies using self-reported measures and those measuring sleep-wake activity more objectively. Simultaneous measurement of objective, such as polysomnography and actigraphy, and subjective sleep parameters are needed to hone in on key measures of disturbed sleep that predict future risk of cognitive impairment. For example, a study of 25 cognitively normal and 25 mildly impaired older adults found that subjective sleep responses, like the number of nighttime awakenings and difficulty sleeping after waking, predicted SWS fragmentation in cognitively normal individuals but did not in those who were mildly impaired (Hita-Yañez et al., 2013). Studies like this will help to target the appropriate sleep measurements and populations to screen for future risk of cognitive impairment.
Further, sleep disorders such as OSA, PLMD, and insomnia have also been associated with future cognitive dysfunction (Leng et al., 2016; Osorio et al., 2011; Yaffe et al., 2011). Older women with greater than moderate or severe OSA, for example, have an increased risk of cognitive impairment over 5 years (adjusted odds ratio of 1.85) compared to those with mild or no OSA (Yaffe et al., 2011). Periodic leg movements ≥30 times per hour was associated with greater decline in cognitive function in community-dwelling older men (Leng et al., 2016) while the presence of insomnia in 346 cognitively normal older adults resulted in a 2.39 odds ratio of progressing to AD over approximately 7 years (Osorio et al., 2011).
Markers for AD pathology are also correlated with sleep disturbances in cognitively normal older adults, suggesting that preclinical AD pathology may be the cause of disrupted sleep. In cognitively normal older adults, for instance, increased amyloid deposition measured by both positron emission tomography (PET) and CSF Aβ42 levels has been associated with self-reported short sleep duration (Spira et al., 2013), self-reported excessive daytime sleepiness (Carvalho et al., 2018), longer self-reported sleep latency (Branger et al., 2016; Brown et al., 2016b), poorer self-reported sleep quality (Sprecher et al., 2015; Sprecher et al., 2017), obstructive sleep apnea (Sharma et al., 2018) and decreased sleep efficiency and increased nap frequency measured by actigraphy (Ju et al., 2013). Further, NREM slow wave activity (SWA) was found to be decreased in cognitively normal individuals with amyloid deposition (Mander et al., 2015). In another study that included both cognitively normal and mildly impaired older adults, NREM SWA also decreased with amyloid and tau pathology although the magnitude of the effect for tau pathology was greater (Lucey et al., 2019). This finding is similar to those seen in P301S mice the develop tau pathology and were found to have decreased NREM SWA (Holth et al., 2017). Finally, reduced sleep spindles and slow oscillation-spindle coupling are polysomnographic markers that appear sensitive to early tau pathology (Kam et al., 2019; Winer et al., 2019). Longitudinal studies measuring these sleep parameters in older adults with detailed cognitive assessments and AD biomarkers are needed to further define the causal nature of these relationships. Experimental or investigational studies would also help to clarify these relationships, but are challenging to perform due to night-to-night sleep variability and variability in how sleep may be measured. These problems could potentially be addressed by measuring sleep and cognition over multiple days, although this will increase participant burden and study costs.
Sleep disturbance as a promoter of Alzheimer’s pathology
Correlation of sleep-wake activity, Aβ, and tau
Aβ is a peptide produced when amyloid precursor protein (APP) is cleaved by β-secretase and γ-secretase (Selkoe, 2001). Aggregation and accumulation of Aβ is widely hypothesized to be a necessary, but not definitive, factor in AD pathogenesis (Hardy and Selkoe, 2002; Karran and De Strooper, 2016). Amyloid deposition in the brain is concentration-dependent (Meyer-Luehmann et al., 2003), therefore mechanisms that increase Aβ levels are likely to promote amyloid plaque formation. For example, over-production of Aβ in individuals with autosomal dominant AD (Potter et al., 2013) or Down syndrome (Englund et al., 2007) increase the risk of developing amyloid deposition by increasing the concentration of Aβ the brain is exposed to over time.
Tau is primarily an intracellular protein that modulates microtubule stability with phosphorylated tau (p-tau) reducing microtubule binding (Bramblett et al., 1993; Kellogg et al., 2018; Lindwall and Cole, 1984). P-tau promotes assembly of tau into tangles that aggregate as neurofibrillary tangles (NFTs), insoluble paired helical filaments associated with neuronal loss and cognitive symptoms (Alonso et al., 2001; Buerger et al., 2006). Kinases and phosphatases phosphorylate and dephosphorylate tau at multiple sites. For instance, different sites of tau, including serine-202 (S202) and threonine-217 (T217), are phosphorylated by a variety of kinases, such as CDK5 and GSK-3β (Liu et al., 2002; Lund et al., 2001). In AD, tau aggregation begins in the entorhinal cortex as part of normal aging and then spreads to the hippocampus and surrounding regions (Braak and Braak, 1991). Although this propagation of tau pathology is not well-understood, this transition can be detected by tau imaging changes in the inferior temporal lobe by PET scan a few years before cognitive decline (Brier et al., 2016) as well as increases in the CSF of total and phosphorylated forms of tau that mark neuronal injury beginning ~8–10 years prior to symptomatic onset (Craig-Schapiro et al., 2010; Fagan et al., 2007; Jack et al., 2010).
Soluble forms of Aβ and tau, the proteins critical to AD pathogenesis, change in CSF with sleep-wake activity (i.e., as a diurnal pattern). Longitudinal sampling of interstitial fluid (ISF) in mice via microdialysis catheters and CSF in humans via indwelling lumbar catheters found that Aβ and tau increase during wakefulness and decrease during sleep (Barthélemy et al., 2020b; Holth et al., 2019; Huang et al., 2012; Kang et al., 2009). Interestingly, in humans CSF Aβ and tau levels also linearly increase in concentration over time with serial sampling via lumbar catheter. A similar linear increase in CSF AD biomarkers with repeated lumbar punctures performed 3 days apart (Olsson et al., 2019). Although this linear rise is not completely understood, it has been associated with CSF sampling frequency and volume suggesting that sampling alters fluid dynamics in the central nervous system (Li et al., 2012; Lucey et al., 2015; Slats et al., 2012). Given AD fluid biomarker variability from changes in sleep-wake activity and CSF sampling rates, these factors need to be taken into account when designing and interpreting studies using frequent CSF sampling to measure pharmacodynamic effects of drugs or sleep interventions.
The correlation between sleep, Aβ, and tau suggested that manipulation of sleep-wake activity, such as through sleep deprivation or pharmacologically increased or enhanced sleep, would change Aβ and tau levels. In mice, sleep deprivation increased ISF Aβ concentrations and after 21 days increased amyloid deposition as insoluble plaque (Kang et al., 2009). Subsequent studies in humans showed that 1-night of sleep deprivation and selective disruption of SWS increased CSF Aβ by 10–30% (Ju et al., 2017; Lucey et al., 2018; Ooms et al., 2014). Sleep deprivation in humans or chemogenetically-induced increased wakefulness in mice increases the concentration of tau in mouse ISF, human CSF, and human plasma up to 50% (Barthélemy et al., 2020b; Benedict et al., 2020; Holth et al., 2019).
Sleep-wake activity affects the production/release of Aβ and tau
Increased production or release of Aβ and tau from neurons is one mechanism leading to this diurnal pattern. Studies in mice found that Aβ and tau are released during synaptic/neuronal activity (Cirrito et al., 2008; Cirrito et al., 2005; Kamenetz et al., 2003; Yamada et al., 2014). Other proteins released with neuronal activity in mice, such as α-synuclein (Yamada and Iwatsubo, 2018), are also increased with sleep deprivation in humans (Barthélemy et al., 2020b; Holth et al., 2019) while proteins that are not released with neuronal activity, such as glial fibrillary acidic protein and neurofilament light chain, are not (Holth et al., 2019). Additionally, serial ISF sampling for up to 168 hours via intracerebral microdialysis in humans with acute brain injury showed that the concentration of Aβ increased with improved neurological status (i.e., increased synaptic/neuronal activity (Brody et al., 2008)) and brain regions with higher levels of neuronal activity are more likely to develop amyloid deposition in both mice (Bero et al., 2011) and humans (Buckner et al., 2005; Sheline et al., 2010; Sperling et al., 2009). Neuronal activity changes with sleep-wake states. For instance, cerebral metabolic rates in humans measured by glucose utilization on 18F-fluorodeoxyglucose (FDG) PET studies are similar during wake and REM sleep, but decrease by 43.8% during N3 or slow wave sleep (Dang-Vu et al., 2010; Maquet et al., 1990). These studies support that the observed fluctuations in CSF Aβ during serial sampling are due to changes in neuronal activity, and that a likely consequence of increased neuronal activity in specific brain regions is increased amyloid deposition. Therefore, increased wakefulness during sleep periods is hypothesized to increase amyloid deposition via increased Aβ production from neuronal activity.
Sleep-mediated changes in tau concentrations are most likely due to altered release rather than production based on the findings that the half-life of tau is ~23 days in humans after translation (Sato et al., 2018) but the half-life is on the order of hours after tau is released into the brain ISF or CSF (Yanamandra et al., 2017). In comparison, the half-life of CSF Aβ is ~9 hours (Patterson et al., 2015). Further, tau peptides corresponding to truncated forms of tau from the mid-domain region (151–221 peptide) were the major tau fragments measured by mass spectrometry (MS) in the CSF of acutely sleep-deprived humans (Barthélemy et al., 2020b). No signal was detected for peptides after residue 290 including the 396–406 peptide (full length tau has 441 peptides). Further, neuronal activity releases truncated forms of tau while full length tau is released with neuronal injury (Sato et al., 2018). These findings support that increased release rather than increased production accounts for the rise in tau concentration during acute sleep deprivation.
Further evidence in humans supports that increased production of Aβ and increased release of tau are critical factors driving the changes in these proteins with sleep-wake activity. First, soluble amyloid precursor protein (APP) metabolites that form upstream from Aβ also fluctuate in human CSF with a diurnal pattern and this supports that active cleavage of APP is occurring with changes in sleep-wake activity (Dobrowolska et al., 2014). Second, stable isotope labeling kinetics (SILK) studies in cognitively normal middle-aged adults under different sleep conditions found that increased Aβ production was the necessary and critical factor affecting changes in Aβ concentration during overnight sleep deprivation (Lucey et al., 2018). SILK uses amino acids labeled with stable isotopes of carbon and nitrogen to measure in vivo production and clearance rates of proteins involved in neurodegenerative disorders including Aβ, tau, and superoxide dismutase (Bateman et al., 2006; Crisp et al., 2015; Paterson et al., 2019; Sato et al., 2018).
Sleep-wake activity affects the clearance of Aβ and tau
Decreased clearance during sleep is another mechanism hypothesized to increase soluble CSF Aβ and tau concentrations. According to this mechanism, bulk fluid flow (i.e., the “glymphatic” system) transports solutes from the ISF to the CSF (Iliff et al., 2012) which are subsequently cleared from the brain through dural lymphatics (Patel et al., 2019). During sleep, fluid flow through the glymphatic system in mice increases potentially leading to greater clearance of soluble Aβ (Xie et al., 2013) and has been implicated in increasing tau pathology in a mouse model of traumatic brain injury (Iliff et al., 2014). In mice, the glymphatic system also becomes more impaired with age (Kress et al., 2014), the greatest risk factor for AD. Clearance of Aβ from the brain is further impaired after aggregation of insoluble amyloid plaques acts as a “sink” to retain Aβ in the brain (Patterson et al., 2015); this process is not known to be affected by sleep.
Multiple studies support that the water-channel protein, aquaporin-4, mediates the glymphatic clearance mechanism. Deletion of aquaporin-4 in mice reduced ISF solute clearance, including Aβ, and resulted in the accumulation of Aβ and tau in sleep-deprived mice (Iliff et al., 2012; Zhang et al., 2020). Glymphatic clearance of brain lactate is also reduced in aquaporin-4 knock out mice (Lundgaard et al., 2017). Studies of autopsied human brains found that loss of aquaporin-4 perivascular localization, and therefore potentially reduced clearance, was associated with greater amyloid burden and increasing Braak stage (Zeppenfeld et al., 2017). Additionally, variations in the human aquaporin-4 gene modulate both the progression of cognitive decline in AD (Burfeind et al., 2017) and the relationship between sleep and amyloid deposition (Rainey-Smith et al., 2018).
Sleep-wake activity affects tau phosphorylation
Interestingly, overnight sleep deprivation affects phosphorylation of each tau form differently (i.e., it is site-specific) in CSF collected from acute sleep-deprived cognitively normal middle-aged adults. For example, phosphorylated threonine-181 (pT181) increased similarly to unphosphorylated threonine-181 (T181) and the pT181/T181 ratio did not change with sleep deprivation. In contrast, the ratio of phosphorylated S202 (pS202) to S202 (pS202/S202) declined in sleep-deprived participants compared to controls while the ratio of tau phosphorylated at T217 (pT217) to T217 (pT217/T217) increased 15–20% during sleep deprivation (Barthélemy et al., 2020b). Recent work from the Dominantly Inherited Alzheimer Network found that in individuals with dominantly inherited AD pT217 increases approximately 21 years prior to their estimated age of symptom onset and at approximately the time amyloid deposition begins (Barthélemy et al., 2020a). pT181 begins to increase a few years later at approximately 19 years prior to estimated age of symptom onset. These findings suggest that sleep deprivation increases phosphorylated tau forms that are seen in the very earliest stages of AD pathogenesis.
The mechanism for how sleep deprivation alters p-tau is unknown but possible explanations have been proposed (Barthélemy et al., 2020b). Sleep deprivation may alter physiologic processes that modulate site-specific phosphorylation of tau and lead to tau hyperphosphorylation. Different sites of tau, such as serine-202 (S202) and threonine-217 (T217), are phosphorylated by multiple kinases, including CDK5 and GSK-3β (Liu et al., 2002). Site-specific differences in tau phosphorylation, including increased T217, as well as kinase and phosphatase activity were observed in the hippocampus of fasting mice (Li et al., 2006; Planel et al., 2001). Also in mice, sleep loss increased phosphorylation of the brain proteome, including kinases such as microtubule affinity regulating kinase 2 (MARK2) (Wang et al., 2018). MARK2 is activated by phosphorylation (Kosuga et al., 2005) and in turn phosphorylates tau and inhibits tau-microtubule interactions (Augustinack et al., 2002). Additional studies in animal models show that protein phosphorylation is altered by changes in synaptic activity such as during sleep (Bruüning et al., 2019; Chen et al., 2019). Activation or deactivation of kinases and phosphatases during sleep-wake activity may lead to tau hyperphosphorylation and account for the observed differences in truncated p-tau forms. Alternatively, release of p-tau from neurons may be dependent on the specific site of phosphorylation. If so, then decreased CSF p-tau forms may result from increased intracellular aggregation, such as NFTs, similar to CSF Aβ42 concentrations in the presence of amyloid plaques.
Tau spreading and other potential mechanisms
A recent study found that prolonged sleep deprivation in mice promoted spreading of tau pathology in the locus coeruleus and may represent a possible mechanism for how sleep disturbance promotes AD pathogenesis (Holth et al., 2019). Trans-synaptic transmission of tau protein, similar to the spread of prions in Creutzfeldt-Jakob disease, is a hypothesized mechanism to explain tau propagation since neurons with tau pathology are anatomically connected (Frost and Diamond, 2010; Wu et al., 2016). Several lines of evidence support a prion-like transmission of tau with soluble tau spreading through the interstitial fluid and seeding new aggregates (DeVos et al., 2018; Mudher et al., 2017). Tau is released extracellularly during neuronal activity both in vivo and in cultured cells (Sato et al., 2018; Yamada et al., 2014). In both in vitro and in vivo studies, exogenous tau aggregates are imported into neurons and act as “seeds” to induce the aggregation of other tau proteins. Injection of tau aggregates as “seeds” induces the spreading of tau pathology from the injection site to synaptically connected brain regions. For instance, transgenic mice that express human tau only in the entorhinal cortex were found to have tau aggregates composed of human tau and endogenous mouse tau in brain regions downstream in the synaptic circuit such as the dentate gyrus, CA fields of the hippocampus, and cingulate cortex (Wang et al., 2017). Since no expression of human tau was detected in these regions, human tau in these areas should derive from the entorhinal cortex.
Other potential mechanisms for sleep deprivation to increase Aβ is by increased stress, disrupted circadian rhythms, or increased inflammation. Both acute stress (Kang et al., 2007) and disrupted circadian clock function (Kress et al., 2018) have been found to increase ISF Aβ and amyloid deposition in mice. A recent study measured CSF and plasma cortisol rhythms in sleep-deprived participants compared to non-sleep-deprived individuals (Blattner et al., 2020). Cortisol is both a marker of stress, such as from motion sickness (Eversmann et al., 1978) or delirium (Pearson et al., 2010), and has an endogenous circadian rhythm (Weitzman et al., 1971). No significant group differences were found, strongly suggesting that increased stress or disrupted endogenous circadian rhythms as measured by cortisol do not account for the rise in CSF Aβ concentrations under sleep deprivation conditions.
Finally, sleep disturbances affect inflammation and metabolism that are also risk factors for Alzheimer’s disease (Carroll and Macauley, 2019; Irwin and Vitiello, 2019). Individuals with poor sleep quality are at increased risk of developing type 2 diabetes (Kawakami et al., 2004) and individuals with type 2 diabetes who have untreated OSA were found to have worse glucose control (Aronsohn et al., 2010). Further, studies in mice showed that hyperglycemia modulates Aβ concentrations and neuronal activity (Macauley et al., 2015) connecting back to mechanisms we have discussed for how sleep disturbances may increase AD risk. For inflammation, sleep disturbances and long sleep duration, but not short sleep duration, were associated with higher levels of C-reactive protein and interleukin-6 in humans (Irwin et al., 2016). In chronically sleep restricted rats, levels of inflammatory factors such as interleukin-1β, tumor necrosis factor-α, and nitric oxide were increased and positively correlated amyloid deposition in the brain (Liu et al., 2020), suggesting that sleep loss increased inflammation and that this increase was further increased in the presence of AD pathology. Although further investigations are needed, inflammation and metabolic dysfunction are both highly promising and biologically plausible potential mechanisms linking sleep and AD risk.
Sleep as a modifiable risk factor for Alzheimer’s disease
A key question is whether increased or enhanced sleep will decrease Aβ and tau concentrations, and potentially decrease the risk of developing AD. Since neuronal activity is lowest during SWS and disrupting SWS increased CSF Aβ, enhanced SWS is a proposed target to lower CSF Aβ concentrations and potentially prevent or delay AD. Unfortunately, acute treatment for 1-night with sodium oxybate, a GABA-B receptor agonist known to increase SWS, did not lower CSF Aβ concentrations compared to controls (Lucey et al., 2018) suggesting that drugs working through this mechanism will not be effective treatments to lower CSF Aβ and/or that this effect is dependent on different neurotransmitter networks. Additional therapies that increase SWS, such as acoustic stimulation, are promising but their effect on CSF Aβ have not been tested in humans (Grimaldi et al., 2020).
OSA is a common and treatable sleep disorder where frequent respiratory events occur during sleep and lead to sleep disturbance. OSA is also a risk factor for AD and has recently been shown to modify CSF AD biomarkers. A study of 20 middle-aged patients with untreated OSA found that CSF Aβ42/40 was negatively correlated with the number of respiratory events per hour of sleep (Liguori et al., 2019). Treatment of OSA with continuous positive airway pressure (CPAP) therapy in 18 participants for 1–4 months showed that the greater the reduction in sleep-related respiratory events after treatment with CPAP, the greater the reduction in CSF Aβ and tau concentrations from their pre-treatment baselines (Ju et al., 2019). Although further investigation is needed, these studies strongly suggest that treating OSA has the potential to reduce AD risk.
Another potential target for intervention to decrease CSF Aβ and potentially other AD biomarkers to prevent/delay AD is the orexin system. Orexin-A and orexin-B (also known as hypocretin-1 and hypocretin-2) are wake-promoting neuropeptides of 33 and 28 amino acids encoded by a common precursor polypeptide, prepro-orexin (Tsujino and Sakurai, 2009). Neurons producing orexin are exclusively localized to the perifornical area and the lateral and posterior hypothalamic area and project to the brainstem nuclei, amygdala, hippocampus, and cerebral cortex (Date et al., 1999; Elias et al., 1998; Nambu et al., 1999; Peyron et al., 1998). Orexins bind to two G protein-coupled receptors, orexin receptor 1 (OXR1) and orexin receptor 2 (OXR2) (Tsujino and Sakurai, 2009). The orexin system regulates sleep-wake activity, feeding behavior, energy homeostasis, and the reward system (Tsujino and Sakurai, 2009). Orexin deficiency causes narcolepsy, a sleep disorder resulting in excessive daytime sleepiness, sleep paralysis, sleep-related hallucinations, and cataplexy (Kryger MH, 2005).
Substantial evidence supports a role for the orexin system in the development of amyloid deposition. In humans, patients with narcolepsy (i.e., with orexin deficiency) have reduced CSF Aβ, tau, p-tau, and amyloid deposition on amyloid PET compared to age- and sex-matched controls (Gabelle et al., 2019; Jennum et al., 2017). Further, knocking out the orexin gene in amyloid precursor protein (APP) transgenic mice led to increased sleep time and a marked decrease in amyloid pathology in the brain while over-expression of orexin in the hippocampus did not (Roh et al., 2014). In contrast, increasing wakefulness by rescue of orexin neurons in APP/PS1 mice lacking orexin increased the amount of Aβ pathology in the brain. Additional studies in APP transgenic mice that develop amyloid deposition found that treatment with a dual orexin receptor antagonist, almorexant, decreased soluble Aβ concentrations while intra-cerebroventricular administration of orexin increased them (Kang et al., 2009). Further, prolonged treatment with almorexant for 8 weeks decreased amyloid deposition; this effect was recently replicated in mice with suvorexant, a dual orexin receptor antagonist approved by the Food and Drug Administration for the treatment of insomnia (Zhou et al., 2020). Although the effect of a dual orexin receptor antagonist on soluble CSF Aβ and tau or amyloid deposition in the brain has not been tested in humans, these findings strongly suggest that blocking orexin will modulate amyloid pathology in the brain.
Future Directions
Extensive evidence implicates sleep disturbances as both a marker for AD pathology and future risk of developing AD, and suggests that improving disturbances in sleep-wake activity could prevent/delay the onset of AD. A major unanswered issue is if an intervention to improve sleep will decrease CSF Aβ, tau, and p-tau over the long-term and ultimately slow/halt AD pathogenesis. To address this issue, it is critical to answer several questions.
First, when do sleep disturbances begin in AD relative to the development of pathology and clinical symptoms? For instance, do sleep disturbances precede or follow the development of amyloid deposition? The Aβ diurnal oscillation, particularly Aβ42, attenuates in the presence of amyloid pathology in mice (Roh et al., 2012) and humans with autosomal dominant AD (Roh et al., 2012) and sporadic AD (Lucey et al., 2017). This is presumably due to Aβ42 aggregating as insoluble plaque rather than clearing to the CSF and suggests that decreasing Aβ concentrations by treating sleep disturbances may not alter the trajectory of amyloid deposition once an individual is amyloid-positive (i.e., sleep therapy would need to be used as primary prevention of AD). Furthermore, the effect of improved sleep on tau or p-tau in the presence of amyloid deposition is not known. If tau and/or p-tau were decreased with improved sleep in amyloid-positive individuals, then treating sleep disturbances in this population may reduce progression to symptomatic AD (i.e., sleep therapy could be used as secondary prevention of AD). Although current evidence supports that sleep disturbances begin during preclinical AD, longitudinal studies are needed to establish these temporal relationships.
Second, what sleep disturbances need to be treated? Establishing what sleep disturbance(s) and the age range when it is critical to measure as a marker for AD or to target for intervention is a major issue for the field to address. As discussed above, sleep disorders (e.g. insomnia, obstructive sleep apnea), sleep symptoms (e.g. daytime sleepiness), and sleep parameters (e.g. sleep efficiency, NREM SWA) have all been associated with AD. Sleep complaints, such as insomnia and daytime sleepiness, are common in older adults and are potentially due to numerous causes. In addition to primary sleep disorders such as obstructive sleep apnea and insomnia, sleep disturbances in the elderly are common, multifactorial, and may be due normal aging, medical comorbidities, polypharmacy, psychosocial and cognitive factors, or a combination (Fragoso and Gill, 2007; Pack et al., 2006). For instance, self-reported health and social factors were recently found to increase the likelihood of older adults reporting short sleep duration (Scarlett et al., 2020). Future investigations will likely be needed to evaluate specific sleep disturbances and their relationship with AD.
Third, does a sleep intervention decrease CSF Aβ or tau sufficiently to alter the trajectory of AD pathogenesis? Studies in mice revealed that pharmacological reduction of CSF Aβ by 20–25% decreased amyloid plaque formation and growth (Yan et al., 2009). In humans, the APP mutation A673T decreases Aβ approximately 40% in vitro and is protective against AD (Jonsson et al., 2012). To be effective, sleep interventions will most likely need to decrease CSF Aβ a similar amount.
Fourth, what is the mechanism(s) mediating the relationship between sleep and AD? Is it increased production/release of Aβ and tau, altered tau phosphorylation, increased tau spreading between neurons, or an alternative possibility such as increased inflammation, metabolic dysfunction, or synaptic damage? Future studies need to test multiple fluid biomarkers beyond just Aβ and tau for AD, including inflammatory markers, markers of synaptic function, and markers of metabolism to explore these potential mechanisms.
If sleep is ultimately found to be a reliable marker for AD risk or a potential target for intervention, then effective delivery of sleep therapies will be essential to prevent/delay the onset of AD throughout the population. Following successful early phase I and II studies translating findings from animal models to humans, subsequent research will need to focus on phase III clinical trials and eventually translation to patients and clinical practice. Unfortunately, implementation research is under-developed in sleep medicine. Improving adherence to treatments for sleep-disordered breathing, for example, has been identified as a high priority need for future research (Parthasarathy et al., 2016). There may be additional challenges implementing sleep measurement devices and sleep interventions in older adults that require investigation. Given the time required for these studies, implementation research in sleep medicine needs to increase to speed the translation of potential new sleep therapies for AD to patients.
Highlights:
Sleep disturbances are associated with increased risk of cognitive impairment and Alzheimer’s disease pathology.
Alzheimer’s disease pathology may lead to sleep disturbances.
Modifying sleep-wake activity alters soluble cerebrospinal fluid Aβ and tau.
Funding:
This work was supported by the National Institutes of Health (K76 AG054863).
Footnotes
Declarations of Interest: No relevant conflicts of interest. Dr. Lucey consults for Merck.
References
- Alonso A. d. C., et al. , 2001. Hyperphosphorylation induces self-assembly of tau into tangles of paired helical filaments/straight filaments. Proc Natl Acad Sci USA. 98, 6923–6928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ancoli-Israel S, et al. , 1991a. Periodic limb movements in sleep in community-dwelling elderly. Sleep. 14, 496–500. [DOI] [PubMed] [Google Scholar]
- Ancoli-Israel S, et al. , 1991b. Sleep-disordered breathing in community-dwelling elderly. Sleep. 14, 486–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aronsohn RS, et al. , 2010. Impact of untreated obstructive sleep apnea on glucose control in type 2 diabetes. Am J Respir Crit Care Med. 181, 507–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asplund R, 1996. Daytime sleepiness and napping amongst the elderly in relation to somatic health and medical treatment. J Intern Med. 239, 261–7. [DOI] [PubMed] [Google Scholar]
- Augustinack JC, et al. , 2002. Specific tau phosphorylation sites correlate with severity of neuronal cytopathology in Alzheimer’s disease. Acta Neuropathol. 103, 26–35. [DOI] [PubMed] [Google Scholar]
- Barthélemy NR, et al. , 2020a. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nat Med. 26, 398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barthélemy NR, et al. , 2020b. Sleep deprivation affects tau phosphorylation in human cerebrospinal fluid. Ann Neurol. 87, 700–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, et al. , 2006. Human amyloid-β synthesis and clearance rates as measured in cerebrospinal fluid in vivo. Nat Med. 12, 856–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman RJ, et al. , 2012. Clinical and biomarker changes in dominantly inherited Alzheimer’s disease. N Engl J Med. 367, 795–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedict C, et al. , 2020. Effects of acute sleep loss on diurnal plasma dynamics of CNS health biomarkers in young men. Neurology. 94, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bero AW, et al. , 2011. Neuronal activity regulates the regional vulnerability to amyloid-β deposition. Nat Neurosci. 14, 750–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackwell T, et al. , 2006. Poor sleep is associated with impaired cognitive function in older women: the study of osteoporotic fractures. J Gerontol A Biol Sci Med Sci. 61, 405–410. [DOI] [PubMed] [Google Scholar]
- Blattner MS, et al. , 2020. Increased CSF Aβ during sleep deprivation in healthy middle-aged adults is not due to stress or circadian disruption. J Alzheimers Dis. 75, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliwise DL, Normal Aging In: Kryger MH, et al. , (Eds.), Principles and Practice of Sleep Medicine. Elsevier, Philadelphia, PA, 2005, pp. 24–38. [Google Scholar]
- Braak H, Braak E, 1991. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Bramblett GT, et al. , 1993. Abnormal tau phosphorylation at Ser396 in Alzheimer’s disease recapitulates development and contributes to reduced microtubule binding. Neuron. 10, 1089–1099. [DOI] [PubMed] [Google Scholar]
- Branger P, et al. , 2016. Relationships between sleep quality and brain volume, metabolism, and amyloid deposition in late adulthood. Neurobiol Aging. 41, 107–114. [DOI] [PubMed] [Google Scholar]
- Brier MR, et al. , 2016. Tau and Aβ imaging, CSF measures, and cognition in Alzheimer’s disease. Sci Transl Med. 8, 338ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, et al. , 2008. Amyloid-β dynamics correlate with neurological status in the injured human brain. Science. 321, 1221–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BM, et al. , 2016a. Exploring the bi-directional relationship between sleep and beta-amyloid. Curr Opin Psychiatry. 29, 397–401. [DOI] [PubMed] [Google Scholar]
- Brown BM, et al. , 2016b. The relationship between sleep quality and brain amyloid burden. Sleep. 39, 1063–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruüning F, et al. , 2019. Sleep-wake cycles drive daily dynamics of synaptic phosphorylation. Science. 366. [DOI] [PubMed] [Google Scholar]
- Buckner RL, et al. , 2005. Molecular, structural, and functional characterization of Alzheimer’s disease: evidence for a relationship between default activity, amyloid, and memory. J Neurosci. 25, 7709–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buerger K, et al. , 2006. CSF phosphorylated tau protein correlates with neocortical neurofibrillary pathology in Alzheimer’s disease. Brain. 129, 3035–3041. [DOI] [PubMed] [Google Scholar]
- Burfeind KG, et al. , 2017. The effects of noncoding aquaporin-4 single-nucleotide polymorphisms on cognition and functional progression of Alzheimer’s disease. Alzheimers Dement (N Y). 3, 348–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll CM, Macauley SL, 2019. The interaction between sleep and metabolism in Alzheimer’s disease: cause or consequence of disease? Front Aging Neurosci. 11, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho DZ, et al. , 2018. Association of excessive daytime sleepiness with longitudinal β-amyloid accumulation in elderly persons without dementia. JAMA Neurol. 75, 672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cedernaes J, et al. , 2017. Candidate mechanisms underlying the association between sleep-wake disruptions and Alzheimer’s disease. Sleep Med Rev. 31, 102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, et al. , 2019. Changes of protein phosphorylation are associated with synaptic functions during the early stage of Alzheimer’s disease. ACS Chem Neurosci. 10, 3986–3996. [DOI] [PubMed] [Google Scholar]
- Cirrito JR, et al. , 2008. Endocytosis is required for synaptic activity-dependent release of Amyloid-beta in vivo. Neuron. 58, 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cirrito JR, et al. , 2005. Synaptic activity regulates interstitial fluid amyloid-β levels in vivo. Neuron. 48, 913–922. [DOI] [PubMed] [Google Scholar]
- Craig-Schapiro R, et al. , 2010. YKL-40: a novel prognostic fluid biomarker for preclinical Alzheimer’s disease. Biol Psychiatry. 68, 903–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crisp MJ, et al. , 2015. In vivo kinetic approach reveals slow SOD1 turnover in the CNS. J Clin Invest. 125, 2772–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang-Vu TT, et al. , 2010. Functional neuroimaging insights into the physiology of human sleep. Sleep. 33, 1589–1603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Date Y, et al. , 1999. Orexins, orexigenic hypothalamic peptides, interact with autonomic, neuroendocrine and neuroregulatory systems. Proc Natl Acad Sci USA. 96, 748–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVos SL, et al. , 2018. Synaptic tau seeding precedes tau pathology in human Alzheimer’s disease brain. Front Neurosci. 12, 267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding G, et al. , 2020. Both short and long sleep durations are associated with cognitive impairment among community-dwelling Chinese older adults. Medicine (Baltimore). 99, e19667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobrowolska JA, et al. , 2014. Diurnal patterns of soluble amyloid precursor protein metabolites in the human central nervous system. PLoS ONE. 9, e89998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias CF, et al. , 1998. Chemically defined projections linking the mediobasal hypothalamus and the lateral hypothalamic area. J Comp Neurol. 402, 442–459. [PubMed] [Google Scholar]
- Englund H, et al. , 2007. Increase in β-amyloid levels in cerebrospinal fluid of children with Down syndrome. Dement Geriatr Cogn Disord. 24, 369–374. [DOI] [PubMed] [Google Scholar]
- Eversmann T, et al. , 1978. Increased secretion of growth hormone, prolactin, antidiuretic hormone, and cortisol induced by the stress of motion sickness. Aviat Space Environ Med. 49, 53–57. [PubMed] [Google Scholar]
- Fagan AM, et al. , 2007. Cerebrospinal fluid tau/beta-amyloid(42) ratio as a prediction of cognitive decline in nondemented older adults. Arch Neurol. 64, 343–349. [DOI] [PubMed] [Google Scholar]
- Faubel R, et al. , 2009. Usual sleep duration and cognitive function in older adults in Spain. J Sleep Res. 18, 427–435. [DOI] [PubMed] [Google Scholar]
- Ferrie JE, et al. , 2011. Change in sleep duration and cognitive function: findings from the Whitehall II study. Sleep. 34, 565–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fragoso CAV, Gill TM, 2007. Sleep complaints in community-living older persons: a multifactorial geriatric syndrome. J Am Geriatr Soc. 55, 1853–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost B, Diamond MI, 2010. Prion-like mechanisms in neurodegenerative diseases. Nat Rev Neurosci. 11, 155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabelle A, et al. , 2019. Reduced brain amyloid burden in elderly patients with narcolepsy type 1. Ann Neurol. 85, 74–83. [DOI] [PubMed] [Google Scholar]
- Grimaldi D, et al. , 2020. Neurostimulation techniques to enhance sleep and improve cognition in aging. Neurobiol Dis. 141, 104865. [DOI] [PubMed] [Google Scholar]
- Guarnieri B, et al. , 2012. Prevalence of sleep disturbances in mild cognitive impairment and dementing disorders: a multicenter Italian clinical cross-sectional study on 431 patients. Dement Geriatr Cogn Disord. 33, 50–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy J, Selkoe DJ, 2002. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 297, 353–356. [DOI] [PubMed] [Google Scholar]
- Havekes R, et al. , 2019. Alzheimer’s disease pathogenesis: the role of disturbed sleep in attenuated brain plasticity and neurodegenerative processes. Cell Signal. 64, 109420. [DOI] [PubMed] [Google Scholar]
- Hita-Yañez E, et al. , 2013. Polysomnographic and subjective sleep markers of mild cognitive impairment. Sleep. 36, 1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoch CC, et al. , 1990. Comparison of sleep-disordered breathing among healthy elderly in the seventh, eighth, and ninth decades of life. Sleep. 13, 502–11. [DOI] [PubMed] [Google Scholar]
- Holth JK, et al. , 2019. The sleep-wake cycle regulates extracellular tau in mice and humans. Science. 363, 880–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holth JK, et al. , 2017. Altered sleep and EEG power in the P301S tau transgenic mouse model. Ann Clin Transl Neurol. 4, 180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, et al. , 2012. Effects of age and amyloid deposition on Aβ dynamics in the human central nervous system. Arch Neurol. 69, 51–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, et al. , 2014. Impairment of glymphatic pathway function promotes tau pathology after traumatic brain injury. J Neurosci. 34, 16180–16193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliff JJ, et al. , 2012. A paravascular pathway facilitates CSF flow through the brain parenchyma and the clearance of interstitial solutes, including Amyloid β. Sci Transl Med. 4, 147ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin MR, et al. , 2016. Sleep Disturbance, Sleep Duration, and Inflammation: A Systematic Review and Meta-Analysis of Cohort Studies and Experimental Sleep Deprivation. Biol Psychiatry. 80, 40–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin MR, Vitiello MV, 2019. Implications of sleep disturbance and inflammation for Alzheimer’s disease dementia. Lancet Neurol. 18, 296–306. [DOI] [PubMed] [Google Scholar]
- Jack CR, et al. , 2016. A/T/N: An unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 87, 539–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, et al. , 2013. Tracking pathophysiological processes in Alzheimer’s disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 12, 207–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack CR, et al. , 2010. Brain beta-amyloid measures and magnetic resonance imaging atrophy both predict time-to-progression from mild cognitive impairment to Alzheimer’s disease. Brain. 133, 3336–3348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jennum PJ, et al. , 2017. Cerebrospinal Fluid Biomarkers of Neurodegeneration Are Decreased or Normal in Narcolepsy. Sleep. 40. [DOI] [PubMed] [Google Scholar]
- Jonsson T, et al. , 2012. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 488, 96–99. [DOI] [PubMed] [Google Scholar]
- Jorm A, Jolley D, 1998. The incidence of dementia: a meta-analysis. Neurology. 51, 728–733. [DOI] [PubMed] [Google Scholar]
- Ju Y-E, et al. , 2014. Sleep and Alzheimer disease pathology-a bidirectional relationship. Nat Rev Neurol. 10, 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Y-E, et al. , 2013. Sleep quality and preclinical Alzheimer’s disease. JAMA Neurol. 70, 587–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Y-ES, et al. , 2017. Slow wave sleep disruption increases cerebrospinal fluid amyloid-β levels. Brain. 140, 2104–2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Y-ES, et al. , 2019. Obstructive sleep apnea treatment, slow wave activity, and amyloid-β. Ann Neurol. 85, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kam K, et al. , 2019. Sleep oscillation-specific associations with Alzheimer’s disease CSF biomarkers: novel roles for sleep spindles and tau. Mol Neurodegener. 14, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamenetz F, et al. , 2003. APP processing and synaptic function. Neuron. 37, 925–937. [DOI] [PubMed] [Google Scholar]
- Kang J-E, et al. , 2007. Acute stress increases interstitial fluid amyloid-β via corticotropin-releasing factor and neuronal activity. Proc Natl Acad Sci USA. 104, 10673–10678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang J-E, et al. , 2009. Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science. 326, 1005–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran E, De Strooper B, 2016. The amyloid cascade hypothesis: are we poised for success or failure? J Neurochem. 139 Suppl 2, 237–252. [DOI] [PubMed] [Google Scholar]
- Kawakami N, et al. , 2004. Sleep disturbance and onset of type 2 diabetes. Diabetes Care. 27, 282–3. [DOI] [PubMed] [Google Scholar]
- Keage HAD, et al. , 2012. What sleep characteristics predict cognitive decline in the elderly? Sleep Med. 13, 886–892. [DOI] [PubMed] [Google Scholar]
- Kellogg EH, et al. , 2018. Near-atomic model of microtubule-tau interactions. Science. 360, 1242–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosuga S, et al. , 2005. GSK-3β directly phosphorylates and activates MARK2/PAR-1. J Biol Chem. 280, 42715–42722. [DOI] [PubMed] [Google Scholar]
- Kress BT, et al. , 2014. Impairment of paravascular clearance pathways in the aging brain. Ann Neurol. 76, 845–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress GJ, et al. , 2018. Regulation of amyloid-β dynamics and pathology by the circadian clock. J Exp Med. 215, 1059–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kronholm E, et al. , 2009. Self-reported sleep duration and cognitive functioning in the general population. J Sleep Res. 18, 436–446. [DOI] [PubMed] [Google Scholar]
- Kryger MH, R. T, Dement WC, 2005. Principles and Practice of Sleep Medicine. Elsevier Saunders, Philadelphia, PA. [Google Scholar]
- Leng Y, et al. , 2016. Periodic limb movements in sleep are associated with greater cognitive decline in older men without dementia. Sleep. 39, 1807–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, et al. , 2006. Concurrent alterations of O-GlcNAcylation and phosphorylation of tau in mouse brains during fasting. Eur J Neurosci. 23, 2078–2086. [DOI] [PubMed] [Google Scholar]
- Liguori C, et al. , 2019. Obstructive sleep apnea may induce orexinergic system and cerebral β-amyloid metabolism dysregulation: is it a further proof for Alzheimer’s disease risk? Sleep Med. 56, 171–176. [DOI] [PubMed] [Google Scholar]
- Lindwall G, Cole RD, 1984. Phosphorylation affects the ability of tau protein to promote microtubule assembly. J Biol Chem. 259, 5301–5305. [PubMed] [Google Scholar]
- Liu F, et al. , 2002. Involvement of aberrant glycosylation in phosphorylation of tau by cdk5 and GSK-3β. FEBS Lett. 530, 209–214. [DOI] [PubMed] [Google Scholar]
- Liu P, et al. , 2020. Activation of Inflammation is Associated with Amyloid-β Accumulation Induced by Chronic Sleep Restriction in Rats. J Alzheimers Dis. 74, 759–773. [DOI] [PubMed] [Google Scholar]
- Loerbroks A, et al. , 2010. Nocturnal sleep duration and cognitive impairment in a population-based study of older adults. Int J Geriatr Psychiatry. 25, 100–109. [DOI] [PubMed] [Google Scholar]
- Lucey BP, Bateman RJ, 2014. Amyloid-β diurnal pattern: possible role of sleep in Alzheimer’s disease pathogenesis. Neurobiol Aging. 35, S29–S34. [DOI] [PubMed] [Google Scholar]
- Lucey BP, et al. , 2018. Effect of sleep on overnight CSF amyloid-β kinetics. Ann Neurol. 83, 197–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucey BP, et al. , 2017. Associations between β-amyloid kinetics and the β-amyloid diurnal pattern in the central nervous system. JAMA Neurol. 74, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucey BP, et al. , 2019. Reduced non-rapid eye movement sleep is associated with tau pathology in early Alzheimer’s disease. Sci Transl Med. 11, eaau6550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund ET, et al. , 2001. Characterization of the in vitro phosphorylation of human tau by tau protein kinase II (cdk5/p20) using mass spectrometry. J Neurochem. 76, 1221–1232. [DOI] [PubMed] [Google Scholar]
- Lundgaard I, et al. , 2017. Glymphatic clearance controls state-dependent changes in brain lactate concentration. J Cereb Blood Flow Metab. 37, 2112–2124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley SL, et al. , 2015. Hyperglycemia modulates extracellular amyloid-β concentrations and neuronal activity in vivo. J Clin Invest. 125, 2463–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mander BA, et al. , 2015. β-amyloid deposition in the human brain disrupts NREM slow wave sleep and associated hippocampus-dependent long-term memory. Nat Neurosci. 18, 1051–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mander BA, et al. , 2016. Sleep: a novel mechanistic pathway, biomarker, and treatment target in the pathology of Alzheimer’s disease? Trends Neurosci. 39, 552–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maquet P, et al. , 1990. Cerebral glucose utilization during sleep-wake cycle in man determined by positron emission tomography and [18F]2-fluoro-2-deoxy-D-glucose method. Brain Res. 513, 136–143. [DOI] [PubMed] [Google Scholar]
- McCurry SM, et al. , 1999. Characteristics of sleep disturbance in community-dwelling Alzheimer’s disease patients. J Geriatr Psychiatry Neurol. 12, 53–59. [DOI] [PubMed] [Google Scholar]
- Meyer-Luehmann M, et al. , 2003. Extracellular amyloid formation and associated pathology in neural grafts. Nat Neurosci. 6, 370–377. [DOI] [PubMed] [Google Scholar]
- Molano JR, et al. , 2017. The interaction of sleep and amyloid deposition on cognitive performance. J Sleep Res. 26, 288–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran M, et al. , 2005. Sleep disturbance in mild to moderate Alzheimer’s disease. Sleep Med. 6, 347–352. [DOI] [PubMed] [Google Scholar]
- Mudher A, et al. , 2017. What is the evidence that tau pathology spreads through prion-like propagation? Acta Neuropathol Commun. 5, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musiek ES, Holtzman DM, 2016. Mechanisms linking circadian clocks, sleep, and neurodegeneration. Science. 354, 1004–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nambu T, et al. , 1999. Distribution of orexin neurons in the adult rat brain. Brain Res. 827, 243–60. [DOI] [PubMed] [Google Scholar]
- Olsson M, et al. , 2019. Repeated lumbar punctures within 3 days may affect CSF biomarker levels. Fluids Barriers CNS. 16, 37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ooms S, et al. , 2014. Effect of 1 night of total sleep deprivation on cerebrospinal fluid β-amyloid 42 in healthy middle-aged men: a randomized clinical trial. JAMA Neurol. 71, 971–977. [DOI] [PubMed] [Google Scholar]
- Osorio RS, et al. , 2011. Greater risk of Alzheimer’s disease in older adults with insomnia. J Am Geriatr Soc. 59, 559–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pack AI, et al. , 2006. Risk factors for excessive sleepiness in older adults. Ann Neurol. 59, 893–904. [DOI] [PubMed] [Google Scholar]
- Parthasarathy S, et al. , 2016. Implementation of Sleep and Circadian Science: Recommendations from the Sleep Research Society and National Institutes of Health Workshop. Sleep. 39, 2061–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TK, et al. , 2019. Dural lymphatics regulate clearance of extracellular tau from the CNS. Mol Neurodegener. 14, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson RW, et al. , 2019. SILK studies — capturing the turnover of proteins linked to neurodegenerative diseases. Nat Rev Neurol. 15, 419–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson BW, et al. , 2015. Age and amyloid effects on human CNS amyloid-beta kinetics. Ann Neurol. 78, 439–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson A, et al. , 2010. Cerebrospinal fluid cortisol levels are higher in patients with delirium versus controls. BMC Res Notes. 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peyron C, et al. , 1998. Neurons containing hypocretin (orexin) project to multiple neuronal systems. J Neurosci. 18, 9996–10015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips B, et al. , 2000. Epidemiology of restless legs symptoms in adults. Arch Intern Med. 160, 2137–41. [DOI] [PubMed] [Google Scholar]
- Planel E, et al. , 2001. Inhibition of protein phosphatase 2A overrides tau protein kinase I/glycogen synthase kinase 3 and cyclin-dependent kinase 5 inhibition and results in tau hyperphosphorylation in the hippocampus of starved mouse. J Biol Chem. 276, 34298–34306. [DOI] [PubMed] [Google Scholar]
- Potter R, et al. , 2013. Increased in vivo amyloid-β42 production, exchange, and loss in presenilin mutation carriers Sci Transl Med. 5, 189ra77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price JL, Morris JC, 1999. Tangles and plaques in nondemented aging and “preclinical” Alzheimer’s disease. Ann Neurol. 45, 358–368. [DOI] [PubMed] [Google Scholar]
- Prince M, et al. , World Alzheimer Report 2015: The Global Impact of Dementia: An Analysis of Prevalence, Incidence, Cost and Trends. London, 2015. [Google Scholar]
- Rainey-Smith SR, et al. , 2018. Genetic variation in Aquaporin-4 moderates the relationship between sleep and brain Aβ-amyloid burden. Transl Psychiatry. 8, 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redline S, et al. , 2004. The effects of age, sex, ethnicity, and sleep-disordered breathing on sleep architecture. Arch Intern Med. 164, 406–418. [DOI] [PubMed] [Google Scholar]
- Roh JH, et al. , 2014. Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer’s disease. J Exp Med. 211, 2487–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roh JH, et al. , 2012. Disruption of the sleep-wake cycle and diurnal fluctuation of amyloid-β in mice with Alzheimer’s disease pathology. Sci Transl Med. 4, 150ra122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato C, et al. , 2018. Tau kinetics in neurons and the human central nervous system. Neuron. 97, 1284–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarlett S, et al. , 2020. Objective sleep duration in older adults: results from The Irish Longitudinal Study on Ageing. J Am Geriatr Soc. 68, 120–128. [DOI] [PubMed] [Google Scholar]
- Schenck CH, et al. , 1986. Chronic behavioral disorders of human REM sleep: a new category of parasomnia. Sleep. 9, 293–308. [DOI] [PubMed] [Google Scholar]
- Selkoe DJ, 2001. Alzheimer’s disease: genes, proteins, and therapy. Physiol Rev. 81, 741–66. [DOI] [PubMed] [Google Scholar]
- Sharma RA, et al. , 2018. Obstructive Sleep Apnea Severity Affects Amyloid Burden in Cognitively Normal Elderly. A Longitudinal Study. Am J Respir Crit Care Med. 197, 933–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheline YI, et al. , 2010. Amyloid plaques disrupt resting state default mode network connectivity in cognitively normal elderly. Biol Psychiatry. 67, 584–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smagula SF, et al. , 2016. Risk factors for sleep disturbances in older adults: evidence from prospective studies. Sleep Med Rev. 25, 21–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, et al. , 2011. Toward defining the preclinical stages of Alzheimer’s disease: Recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimer’s & Dementia. 7, 280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperling RA, et al. , 2009. Amyloid deposition is associated with impaired default network function in older persons without dementia. Neuron. 63, 178–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spira AP, et al. , 2013. Self-reported sleep and β-amyloid deposition in community-dwelling older adults. JAMA Neurol. 70, 1537–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprecher KE, et al. , 2015. Amyloid burden is associated with self-reported sleep in nondemented late middle-aged adults. Neurobiol Aging. 36, 2568–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprecher KE, et al. , 2017. Poor sleep is associated with CSF biomarkers of amyloid pathology in cognitively normal adults. Neurology. 89, 445–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsujino N, Sakurai T, 2009. Orexin/Hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol Rev. 61, 162–176. [DOI] [PubMed] [Google Scholar]
- Tworoger SS, et al. , 2006. The association of self-reported sleep duration, difficulty sleeping, and snoring with cognitive function in older women. Alzheimer Dis Assoc Disord. 20, 41–48. [DOI] [PubMed] [Google Scholar]
- Vos SJ, et al. , 2013. Preclinical Alzheimer’s disease and its outcome: a longitudinal cohort study. Lancet Neurol. 12, 957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, et al. , 2017. The release and trans-synaptic transmission of tau via exosomes. Mol Neurodegener. 12, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, et al. , 2018. Quantitative phosphoproteomic analysis of the molecular substrates of sleep need. Nature. 558, 435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weitzman ED, et al. , 1971. Twenty-four hour pattern of the episodic secretion of cortisol in normal subjects. J Clin Endocrinol Metab. 33, 14–22. [DOI] [PubMed] [Google Scholar]
- Westwood AJ, et al. , 2017. Prolonged sleep duration as a marker of early neurodegeneration predicting incident dementia. Neurology. 88, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilckens KA, et al. , 2018. Sleep moderates the relationship between amyloid beta and memory recall. Neurobiol Aging. 71, 142–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winer JR, et al. , 2019. Sleep as a potential biomarker of tau and β-amyloid burden in the human brain. J Neurosci. 39, 6315–6324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JW, et al. , 2016. Neuronal activity enhances tau propagation and tau pathology in vivo. Nat Neurosci. 19, 1085–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie L, et al. , 2013. Sleep drives metabolite clearance from the adult brain. Science. 342, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, et al. , 2011. Short or long sleep duration is associated with memory impairment in older Chinese: the Guangzhou biobank cohort study. Sleep. 34, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe K, et al. , 2014. Connections between sleep and cognition in older adults. Lancet Neurol. 13, 1017–1028. [DOI] [PubMed] [Google Scholar]
- Yaffe K, et al. , 2011. Sleep-disordered breathing, hypoxia, and risk of mild cognitive impairment and dementia in older women. JAMA. 306, 613–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, et al. , 2014. Neuronal activity regulates extracellular tau in vivo. J Exp Med. 211, 387–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada K, Iwatsubo T, 2018. Extracellular α-synuclein levels are regulated by neuronal activity. Mol Neurodegener. 13, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan P, et al. , 2009. Characterizing the appearance and growth of amyloid plaques in APP/PS1 mice. J Neurosci. 29, 10706–10714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanamandra K, et al. , 2017. Anti-tau antibody administration increases plasma tau in transgenic mice and patients with tauopathy. Sci Transl Med. 9, eaal2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeppenfeld DM, et al. , 2017. Association of Perivascular Localization of Aquaporin-4 With Cognition and Alzheimer Disease in Aging Brains. JAMA Neurol. 74, 91–99. [DOI] [PubMed] [Google Scholar]
- Zhang R, et al. , 2020. Aquaporin 4 deletion exacerbates brain impairments in a mouse model of chronic sleep disruption. CNS Neurosci Ther. 26, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou F, et al. , 2020. Suvorexant ameliorates cognitive impairments and pathology in APP/PS1 transgenic mice. Neurobiol Aging. 91, 66–75. [DOI] [PubMed] [Google Scholar]