

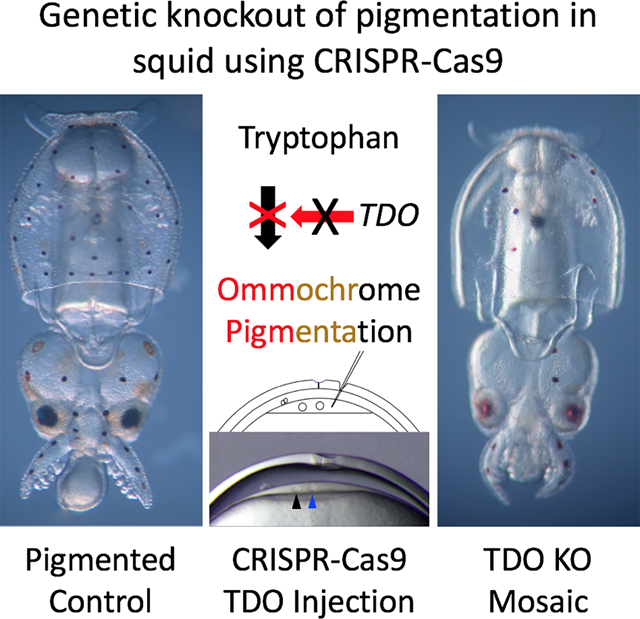
Summary
Seminal studies using squid as a model led to breakthroughs in neurobiology. The squid giant axon and synapse, for example, laid the foundation for our current understanding of the action potential (1), ionic gradients across cells (2), voltage-dependent ion channels (3), molecular motors (4–7), and synaptic transmission (8–11). Despite their anatomical advantages, the use of squid as a model receded over the past several decades as investigators turned to genetically tractable systems. Recently, however, two key advances have made it possible to develop techniques for the genetic manipulation of squid. The first is the CRISPR-Cas9 system for targeted gene disruption, a largely species-agnostic method (12, 13). The second is the sequencing of genomes for several cephalopod species (14–16). If made genetically tractable, squid and other cephalopods offer a wealth of biological novelties that could spur discovery. Within invertebrates, not only do they possess by far the largest brains, they also express the most sophisticated behaviorals (17). In this paper, we demonstrate efficient gene knockout in the squid Doryteuthis pealeii using CRISPR-Cas9. Ommochromes, the pigments found in squid retinas and chromatophores, are derivatives of tryptophan, and the first commited step in their synthesis is normally catalyzed by Tryptophan 2,3 Dioxygenase (TDO; (18–20)). Knocking out TDO in squid embryos efficiently eliminated pigmentation. By precisely timing CRISPR-Cas9 delivery during early development, the degree of pigmentation could be finely controlled. Genotyping revealed knockout efficiencies routinely greater than 90%. This study represents a critical advancement towards making squid genetically tractable.
Graphical Abstract
eTOC Blurb
Crawford et al. report the first gene knockout in a cephalopod. Using CRISPR-Cas9 in squid embryos, they target a gene encoding Tryptophan 2,3 Dioxygenase, an enzyme involved in ommochrome pigment production. Knockouts are highly efficient in G0 animals, resulting in a near complete lack of pigmentation and over 90% disruption of the tdo locus.
Results
Selection of D. pealeii as a model
D. pealeii was selected as a model because of several favorable characteristics. It is readily available, its embryos are transparent and their development has been well characterized (21). In addition, oocytes can be fertilized in vitro and the period between fertilization and the first cell division is relatively long, enabling reagents to be delivered early (22). Finally, transcriptome sequences are available (23, 24) and, although unpublished, the genome has been sequenced and was available at the onset of this study. Despite these attributes, D. pealeii requires >6 months to reach sexual maturity (25) and its life cycle has not been closed. Therefore, our goal was to determine whether we could produce G0 knockouts.
Selection of tdo for knockout
As a target for knockout, we wanted a non-essential gene with a clear phenotype during embryonic development. In cephalopods, the pigments in the eyes and chromatophores are ommochromes, a derivative of tryptophan (Figure 1A) (18–20). Ommochromes also pigment the eyes of Drosophila where their genetics and biochemistry has been studied in detail (26). In invertebrates, Tryptophan, 2,3 Dioxygenase (TDO) catalyzes the first committed step in ommochrome biosynthesis, converting Tryptophan to N-Formylkyneurenine ((27) Figure 1B). We hypothesized that its disruption would reduce or eliminate ommochrome synthesis in squid as well. To verify that TDO was an appropriate choice, we determined whether a TDO-selective inhibitor (680C91) impeded pigmentation in developing embryos. The development of D. pealeii has been divided into 30 stages and eye pigmentation appears at ~stage 25, while chromatophore pigmentation starts at stage 26 (21). We added 680C91 to developing embryos at stage 20, and it clearly blocked pigmentation in both the eyes and chromatophores, with animals developing normally otherwise (Figure 1C). These data supported our choice of TDO.
Figure 1. TDO activity is required for pigmentation in chromatophores and retinas in developing D. pealeii.
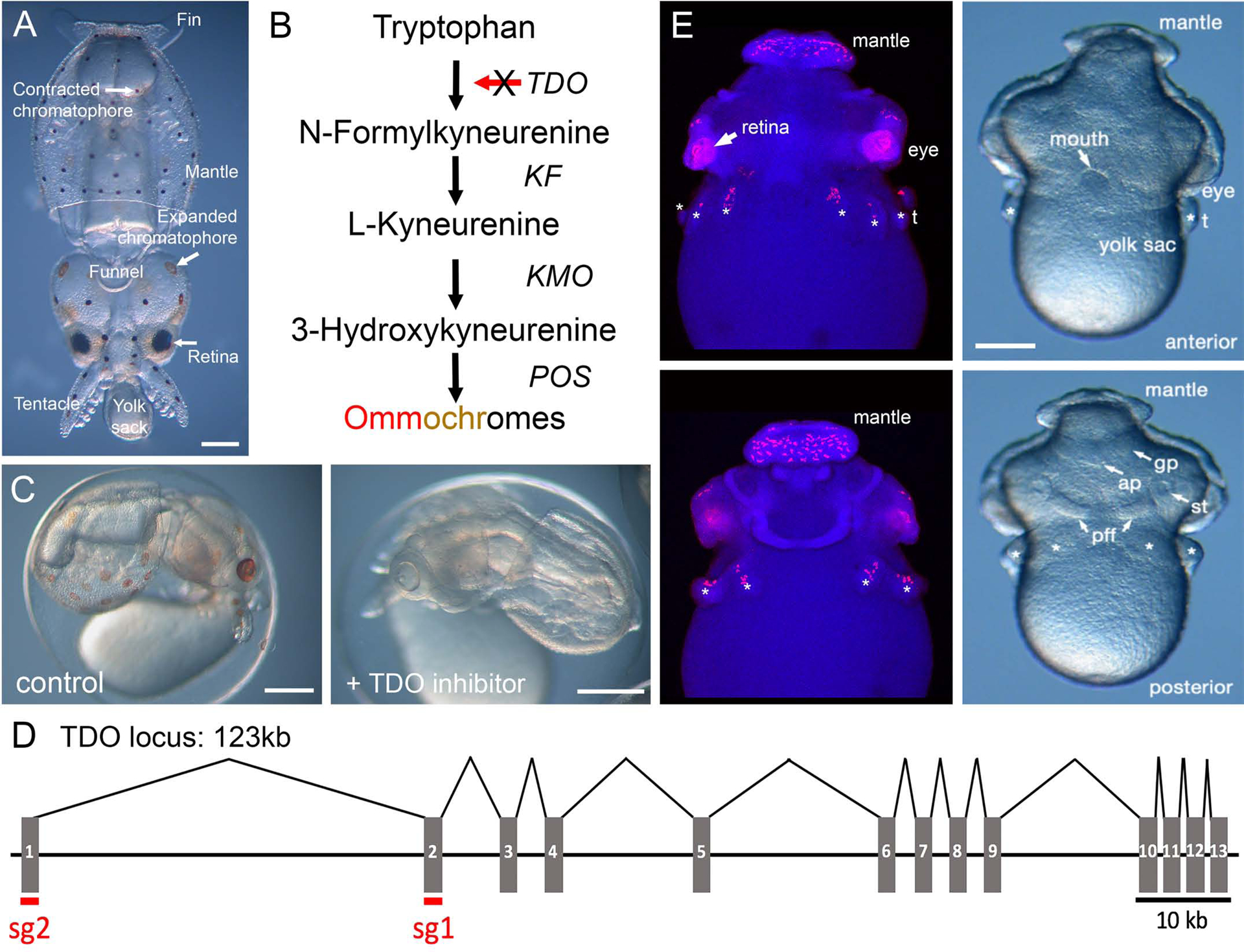
A) A recently hatched D. pealeii with relevant anatomical structures labelled (dorsal view). B) A schematic for ommochrome biosynthesis from tryptophan. KF = kyneurenine formamidase, KMO = kynurenine 3-monooxygenase, POS = phenoxazinone synthase. C) Stage 27 embryos that were treated with either DMSO (left) or 3.15 μM TDO inhibitor 680C91 (right) starting at stage 20. D) The TDO locus spans 123Kb in the D. pealeii genome, with 13 exons. Intron size is to scale. E) Fluorescent in situ hybridization for TDO in stage 22 D. pealeii embryos demonstrates expression in the developing eyes and chromatophores on the mantle, above the eyes, and on the arm primordia. Anterior (top) and posterior (bottom) views of the in situs (left) are shown along images of stage matched live embryos (right). * indicates arms, t = tentacle, pff = posterior funnel fold, ap = anal papilla, gp = gill primordia, st = statocyst. Scale bar = 250 μm. See Figure S1 for more details on TDO intentification and expression.
A single TDO gene was annotated in the D. pealeii genome based on similarity to homologs from other species. The gene is large and highly fragmented, consisting of 13 exons spanning over 120 KB (Figure 1D). A phylogenetic comparison of this sequence with diverse TDOs, and indolamine-2,3-dioxygenases (a different enzyme that catalyzes the same reaction), supported its identity as TDO (Figure S1). We next analyzed the expression profile of D. pealeii TDO. In situ hybridization of stage 22 embryos showed punctae on the arms and mantle, consistent with chromatophore expression (Figure 1E). Expression was robust within the eyes as well. Comparative expression calculated from RNAseq data yielded a similar picture for adult specimens, with highest expression in the retina and the chromatophore layer of the skin (Figure S1B). Taken together, these data suggested that disrupting tdo would lead to a loss of pigmentation in the chromatophores and eyes.
Microinjection in D. pealeii embryos
To knockout tdo, we adopted a standard approach using CRISPR gRNAs and Cas9 nuclease. Single gRNAs were designed to exons 1 and 2 of tdo (Figure 1D) and synthesized chemically with protecting groups. These were mixed with recombinant Streptococcus pyogenes Cas9 protein to form ribonuclear complexes. Our intention was to inject these components into early stage embryos, however there were several obstacles to overcome (Figure 2). First, the chorion surrounding D. pealeii embryos is thick and tough, resisting even beveled, quartz injection needles. To overcome this issue, we made partial cuts through the chorion above the site of injection using “micro-scissors” fashioned from #5 forceps (see methods; Figure 2 B&C). This was a delicate process: if the cuts were too large, the embryo and yolk would extrude through them, arresting development. For stability, embryos were nested blastodisc side up in agarose and beveled quartz needles were passed through the cuts. A second challenge became apparent following the preliminary injections of dyes. Unlike zebrafish embryos, which form their blastodisc via microfilament-guided streams directing cytoplasm up through the yolk cell (28), squid embryos form their blastodiscs via microtubule driven cortical streaming (29, 30). Therefore, injections had to be made directly into the blastodisc, a relatively shallow target with a depth of 20–40μm. Figure 2D shows a small bolus of DiI (1,1’-dioctadecyl-3,3,3’3’-tetramethylindocarbocyanine perchlorate) injected through a cut in the chorion. This panel shows an embryo injected after 4th cleavage to demonstrate the precision of our injections. Figure 2E summarizes the features relevant to injections. After injections, embryos were cultured until hatching and monitored for pigmentation.
Figure 2. Microinjection method for squid embryos.
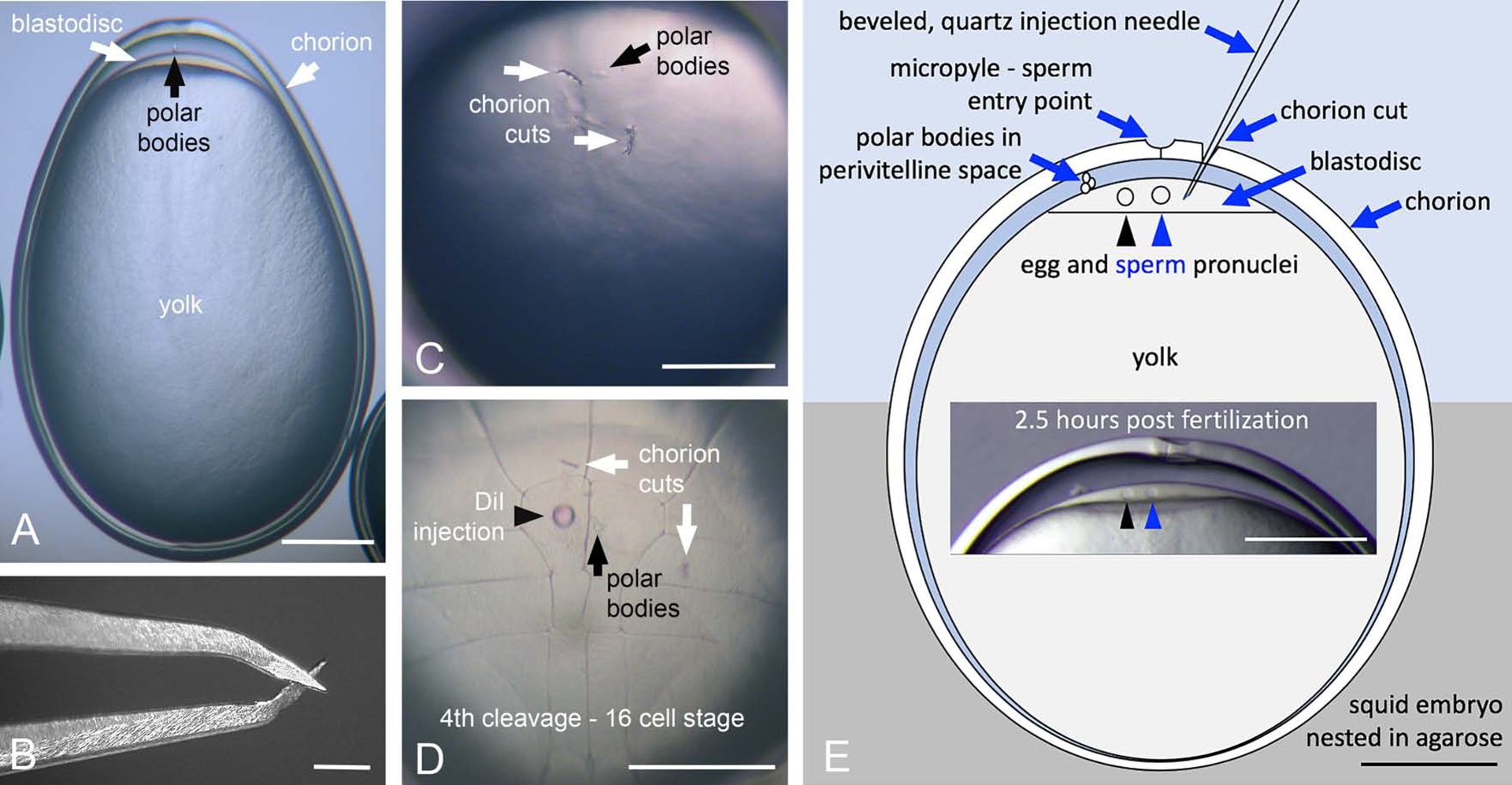
A) Side view whole embryo. The tough outer chorion (white arrow) is visible along with the blastodisc (at the animal pole, white arrow) and yolk in this telolecithal embryo. The polar bodies (black arrow) are also visible. B) Close up view of micro-scissors used to clip small cuts in the chorion. C) Animal pole view showing the chorion cuts (white arrows) and polar bodies on the surface of the zygote. D) Reference view, DiI injected embryo (black arrowhead). This embryo was injected at the 8-cell stage and has progressed through 4th cleavage. The chorion cuts (white arrows) and polar bodies (black arrow) are shown. The polar bodies reside near or in the anterior mid-line. E) Diagram of an embryo nested in agarose in which a beveled quartz injection needle has been passed through the chorion cut and into the blastodisc layer. The egg (black arrowhead) and sperm (blue arrowhead) pronuclei are represented in this panel. Inset, depicts the relative position of the polar bodies in an embryo ~2.5 hpf. Scale bars = 250 μm.
To maximize the potential knockout efficiency, we performed the majority of our injections before the first cleavage. Embryos were fertilized in vitro. It took approximately 3.5 hours post fertilization (hpf) to reach the onset of the first cell division, defining our window for injections. As D. pealeii oocytes are transparent, we could monitor the events leading to the first cell division to time injections precisely. Following fertilization, the zygote membrane pulls away from the chorion surface and the blastodisc begins to form (29, 30). The egg nucleus resides within the thin layer of cortical cytoplasm eccentric to the micropyle in the unfertilized egg and has yet to complete meiosis. With fertilization, meiosis is completed before egg pronuclear migration and fusion. At room temperature (20–21°C), the first polar body forms 20 minutes post fertilization, while the second polar body is released 1.5 hpf. Egg pronuclear migration toward the sperm nucleus, located beneath the micropyle, begins shortly thereafter and terminates in nuclear contact 3 hpf. The realtive proximity of the pronuclei is an excellent indicator of the time post-fertilization (Figures 2E and 4 “I” panels).
Figure 4. Efficient tdo gene disruption using CRISPR-Cas9.
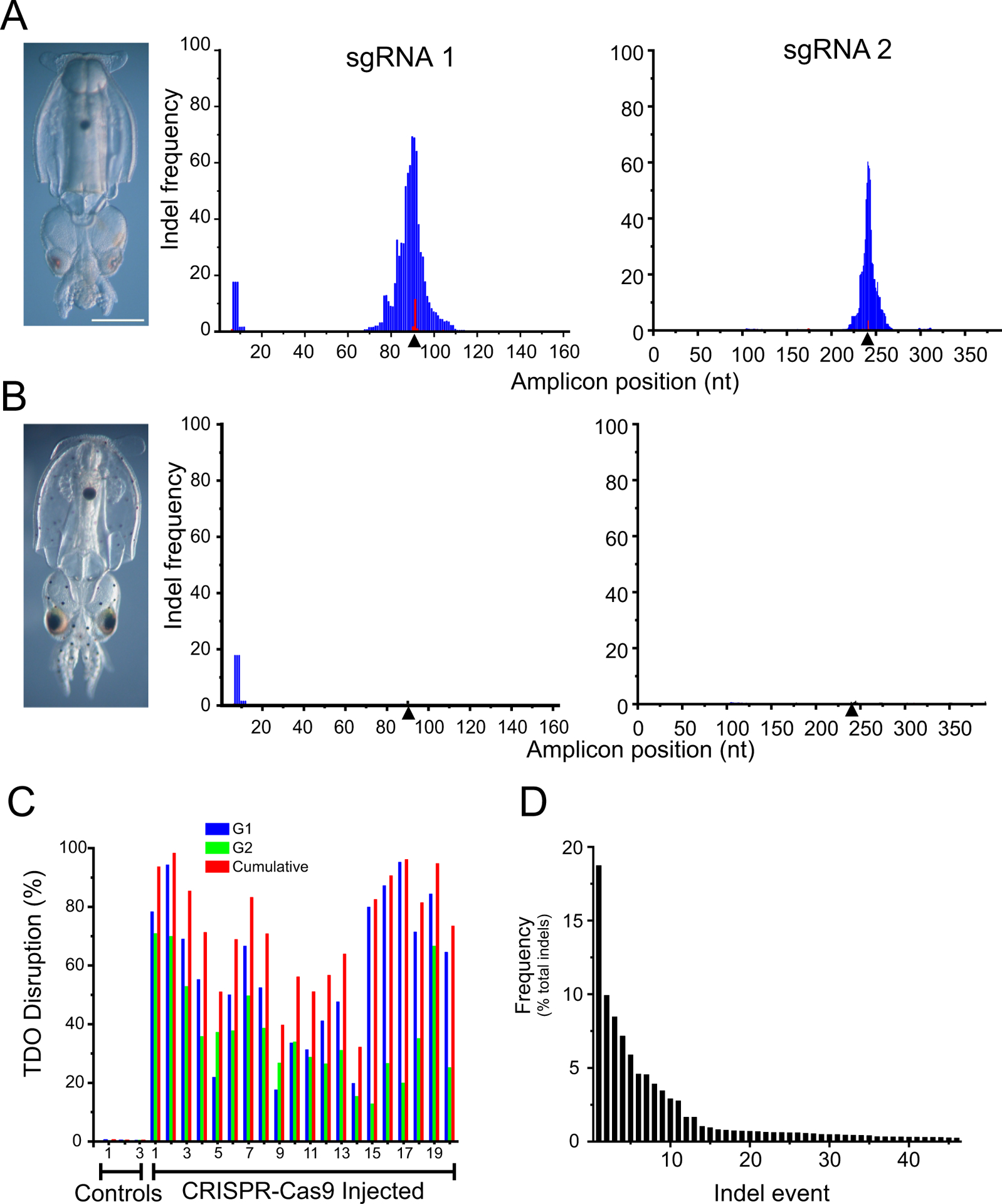
Hatchlings are shown for an individual that was injected with Cas9 and two sgRNAs targeting tdo (A) vs a control embryo (B). Pictures reveal a loss of pigmentation in the chromatophores of the experimental animal. Genomic DNA was extracted from these animals and amplicons surrounding the target sites for sgRNA 1 and sgRNA2 were generated by PCR and then sequenced with MiSeq. Histograms show the frequency that each position in the amplicon is deleted (blue) or contains an insertion (red). Triangles indicated the Cas9 cut site as specified by the CRISPR sgRNA. C) In similar experiments on 3 control animals and 20 CRISPR-Cas9 injected animals, the percent tdo gene disruption was determined for each sgRNA alone and together. D) A representative histogram of the frequency of specific indels (46 in total) are presented for a CRISPR-Cas9 injected individual. See Figure S2 for more details on indel characterizations.
Knockout phenotypes
The injection of two sgRNAs and Cas9 resulted in a reduction of chromatophore and eye pigmentation, however the extent depended on the precise time of injection (Figure 3). Panel 3A shows an example of a control embryo at hatching and one that was injected 2 hpf (30 minutes post 2nd polar body formation). For embryos injected at this stage, pigmentation is completely absent from the chromatophores and is minimal in the eyes which are light red (n=9). The large black spot near the posterior of the mantle is the ink sack. Ink pigment contains melanins, not ommochromes, and is unaffected by TDO disruption. Panel 3B shows more examples of embryos injected during early pronuclear migration (2.5 hpf). Most, but not all, of the chromatophores are missing pigmentation and the eyes are light red (n= 14, 8 white, 4 nearly white, 2 with darker eyes and a few pigmented chromatophores). In panel 3C, injections were after nuclear contact (3 hpf) but before first cleavage (3.5 hpf). These produced mosaic patterns of chromatophore pigmentation (n=21, all with mosaic pigmentation) and a range of eye pigmentation from pink to dark brown, often in the same embryo. Injection into a single blastomere following first cleavage (3.75 hpf) often produced embryos where chromatophores were missing over half of the mantle (Panels 3D&E). Thus, stronger phenotypes were produced with earlier injections; however, the blastodisc is narrower at earlier stages and a more difficult target. In addition, embryo survival was poorer at earlier times of injection (~40–50% loss for embryos injected between 1.5 – 2.0 hpf). These data showed that tdo sgRNAs produced appropriate and consistent reductions of pigmentation.
Figure 3. Timing of CRISPR-Cas9 injection affects pigmentation.
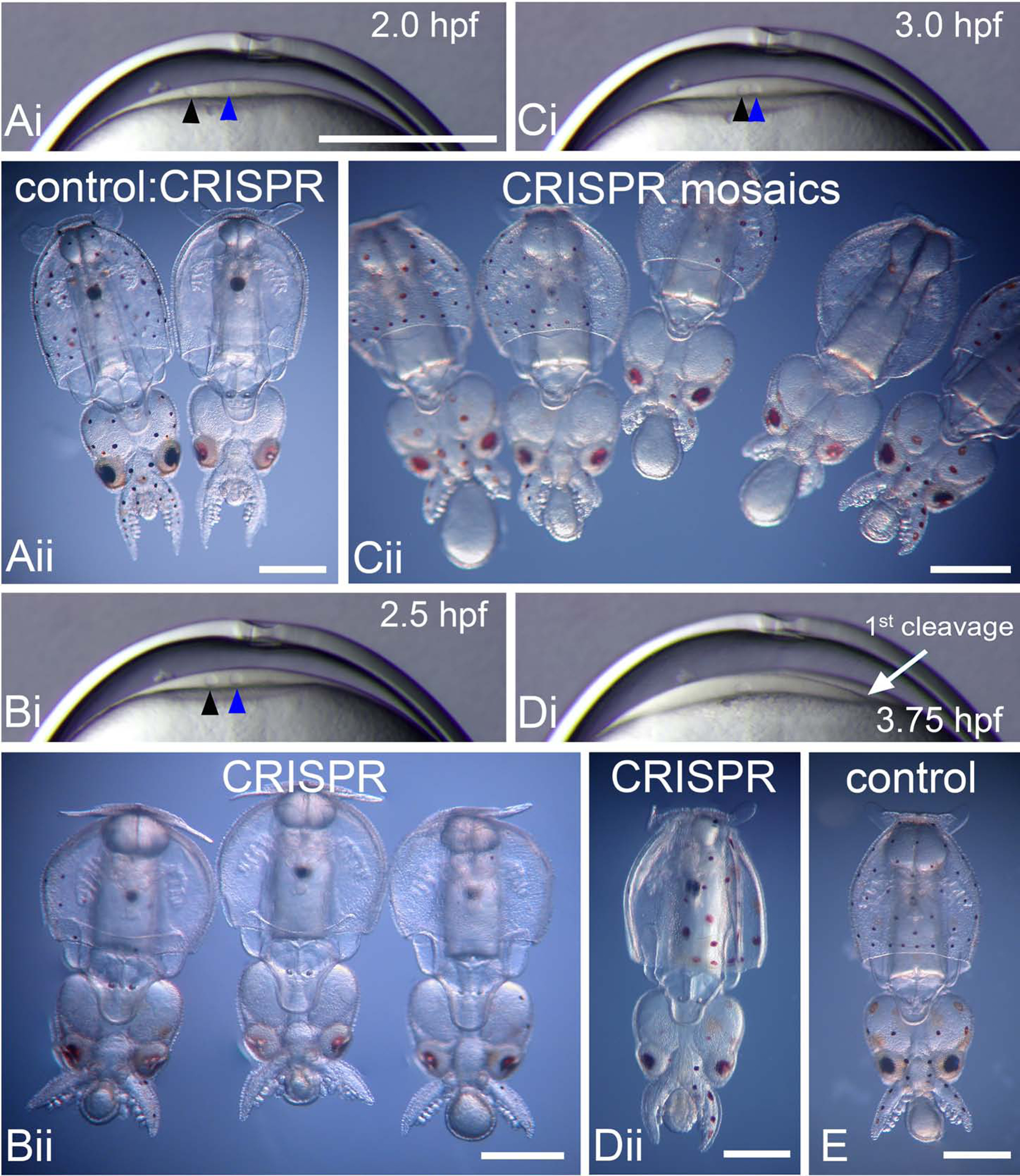
Ai) Embryo blastodisc 2.0 hpf; Bi) 2.5 hpf; Ci) 3.0 hpf; and Di) 3.75 hpf. The blastodisc (a thin lens of cytoplasm) increases in thickness throughout this series of images. The egg pronucleus (black arrowhead) approaches the sperm pronucleus (blue arrowhead) in Ai, Bi, and Ci. Di) One of the nuclei of first cleavage, along with the meroblastic cleavage furrow (white arrow) are visible in this panel. Aii) The embryo on the left is a control hatchling; note the black and reddish brown chromatophores evenly placed across its mantle, head and tentacles. In contrast, the embryo on the right was injected with 2 CRISPR sgRNAs αTDO and Cas9 at 2 hpf and has very few pigmented chromatophores, in addition to light pink to light red eyes. Close inspection reveals that there are 2 diminutive pigmented cells positioned medial to each eye. Bii) CRISPR-Cas9 αTDO embryos injected at 2.5 hpf. These embryos are missing nearly all their pigment and exhibit a range of eye color from faint pink to darker red. Cii). These embryos were injected post nuclear contact, 3.0 hpf and exhibit a range of mosaic patterns, from embryos missing some pigmented chromatophores to others missing large patches or regions of pigmented chromatophores. A range of eye pigmentation from deep brown to light red is also typical in this group. Dii) CRISPR-Cas9 TDO embryo injected post first cleavage, ~3.75 hpf, into one cell only. This embryo is missing pigmentation on half its body, the side that would form from the injected cell. E. Control embryo with normal chromatophore pigmentation and patterning. All views are ventral. Ai, Bi, Ci, and Di are all at the same magnification, scale bar in Ai = 250μm. Aii, Bii, Cii, Dii, and E, Scale bar = 500 μm.
Knockout genotypes
To corroborate the phenotypes produced by the sgRNA injections, we examined genetic disruptions of tdo. At hatching, DNA was extracted from whole embryos and amplicons bracketing the targets for sgRNA1 and sgRNA2 were amplified by PCR. Individual amplicons were sequenced using MiSeq, yielding on average ~ 50,000 paired-end reads that covered the entire amplicon. Figure 4A shows an example of a hatchling that was injected before first cleavage with both sgRNAs. Most pigmentation in the chromatophores and the eyes is missing. For amplicons covering the targets of each sgRNA, we then calculated the frequency that each base was either missing (blue) or had an insertion at that position (red). For each amplicon, there is a peak at the position specified by the CRISPR gRNA for Cas9 cleavage (see triangle). Deletions were more common than insertions and the shape of the plot indicates that multiple deletion events occured.
Assuming that indels at each guide are independent, we estimate that the total tdo disruption in this individual was ~90%. Figure 4B shows the same analysis for a control embryo where no indels are present at the specified sites (for sgRNA1 there is a small peak of deletions at ñt10 present in all control and the experimental samples that was due to a PCR error over highly repetitive intronic sequence). We performed a similar analysis on 20 CRISPR-Cas9 injected individuals and 3 controls (Figure 4C). The cumulative disruption of tdo ranged from ~30–95%. sgRNA1 performed marginally better than sgRNA2, but they both were effective. Figure 4D presents a breakdown of the individual indel events from a representative specimen. In this case 46 indels were identified, the most frequent representing less than 20% of the total. Other specimens had a large range of events (from 8–78) and in few cases did a single event occur at a frequency of more than 20% (Figure S2A&B). Across samples, there was a large range of indel sizes: for sgRNA 1, 95% of the deletions were less than 43 bp and 95% of the insertions were less than 8 nt (median deletion = 9nt, median insertion = 2 bp) and for sgRNA 2, 95% of the deletions were less than 25bp and 95% of the insertions were less than 7bp (median deletion = 7 nt, median insertion = 3 bp; Figure S2 A&B). These data indicate that CRISPR-Cas9 injections produced multiple indels at the targeted locations.
Discussion
This study demonstrates that squid genes can be efficiently disrupted using the CRISPR-Cas9 system. We routinely disrupted tdo at efficiencies >90 %, resulting in an almost complete lack of pigmentation. Given that both sgRNAs worked well, we expect similar efficiencies at other targets. Similarly efficient G0 knockouts have proven to be useful research tools for other organisms, including butterflies, amphipods and even zebrafish (31–35). We expect that they will be similarly useful with squid. D. pealeii is not culturable in captivity at this point. We routinely raise embryos through hatching, but mortality is high thereafter. Accordingly, this species will not be used to establish genetic lines, but it still offers great utility, particularly for developmental studies. Life-cycle culture is possible for other squid species, including Euprymna scolopes, a model with a published genome that is commonly used to investigate bacterial-animal symbioses (16, 36, 37). We expect that our methods, with modifications, will be transferable.
Interestingly, injecting before the first cleavage produced high knockout efficiencies; however, it also produced large numbers of indels, and single events did not dominate (Figure 4D and Figure S2). The average number of distinct indels was 38 ± 13 (SD) per specimen and the average frequency of the most frequent event was 15 ± 8% (SD). In no case did we observe an event at a frequency greater than 30%. This indicates that indels were not formed before the first cleavage, and that they were being created continuously across subsequent cell divisions. While delivering CRISPR-Cas9 prior to the first cleavage ensures that it is present in subsequent blastomeres, it is unclear why the precise timing of reagent delivery before the first division is important. Considering that Cas9 protein was injected and its activity was still delayed, the injection of Cas9 mRNA would probably be less effective.
Our methods for gene knockout should be readily adopted by other research groups. Loliginid squid are available worldwide and our methods do not require specialized equipment. When the D. pealeii genome is released, CRISPR sgRNAs can be designed to avoid off-target edits. The ability to knockout genes in squid will enable us to ask new questions. Some examples include: how does the cephalopod brain encode complex behaviors in comparison to the vertebrate brain? What is the mechanistic basis of high-level mRNA recoding in cephalopods and how is it deployed to respond to the environment (23, 24, 38)? How is camouflage produced structurally and controlled by the brain? And what controls development of the unique cephalopod body plan? This study provides a way forward to investigate these questions, and many others.
Star Methods
Resource availability
Lead contact: Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Joshua Rosenthal (jrosenthal@mbl.edu).
Materials availability:
This study did not generate unique reagents.
Data and code availability
The scripts (Cumulative.py, indels.py, Event_frequency.py) generated during this study are available at Github (https://github.com/pipedq/CRISPR_G0_Genotyping). The sequence for the D. pealeii TDO locus is available at Genbank (MT648678). RNAseq reads are deposited as Bioproject PRJNA641326. Reads from the amplicon sequencing are available on request.
Experimental Model and Subject Details
Adult specimens of D. pealeii were obtained from the Vineyard Sound by otter trawl between July and October 2019 by the Marine Resources Center of the Marine Biological Laboratories, Woods Hole, MA. Embryos were obtained via in vitro fertilization and maintained at 18 °C in well aerated 0.22μM filtered seawater (FSW).
Statement on the ethical treatment of animals
Although the use of cephalopods for research is not currently regulated in the USA, the Marine Biological Laboratory has implemented strict internal policies to ensure their ethical and humane treatment. All specimens of Doryteuthis pealeii used in this study conformed to the Marine Biological Laboratory’s “Policy for the use of cephalopods for research and teaching.”
METHOD DETAILS
Identifying the TDO gene in D. pealeii
The human TDO and IDO protein sequence was used as bait to search the proteomes of D. pealeii, Octopus bimaculoides, Euprymna scolopes, Architeuthis dux, Crassostrea gigas, Capitella teleta, Drosophila melanogaster, Tribolium castaneum, Mus musculus, and Homo sapiens with BLASTP. The identified sequences were aligned using MUSCLE (39), and an approximately-maximum likelihood tree was built with FastTree2 (40) and illustrated with FigTree (http://tree.bio.ed.ac.uk/software/figtree). The TDO sequence was mapped using BLAST onto the D. pealeii genome assembly (in preparation, provided by C Albertin, a member of the genome project) to determine the structure of the locus. The sequence of the tdo locus is deposited in Genbank (MT648678).
Specimen collection and oocyte preparation
Oocytes and spermatophores were removed by dissection from adult D. pealeii and fertilized in vitro at room temperature as previously described (22). Upon sacrifice and dissection, mature oocytes were collected from the oviduct of the gravid female and placed into well aerated FSW in a medium sized (4.5 inches) glass fingerbowl. Eggs were washed 2x with FSW by gentle swirling and decanting. Next, several spermatophores (5–6) were collected from the distal tip of the penis of the male, placed into a small (2 inch) glass Syracuse dish half filled with aerated FSW, and compressed to induce the release of sperm. Concentrated drops of eggs, collected with a fire polished large bore glass Pasteur pipette are next added to the Syracuse dish with the FSW sperm mixture. After 20 minutes, the egg/sperm mixture is washed back into FSW in a medium finger bowl and washed several times with FSW to remove excess sperm. Following fertilization, embryos were cultured in 60-mm plastic Petri dishes lined with 0.2% agarose (Type II-A, Sigma), and filled with FSW supplemented with 1:100 dilution of Penicillin-Streptomycin 100x (Gibco, 15-140-122) at 18°C. Agarose was prepared by microwaving in FSW until melted, allowed to cool and then poured to line (non-plasma treated) Petri dishes. Dishes were stored at 4°C in closed plastic containers until needed. Freshly fertilized embryos 10–20 embryos were plated per dish in well aerated FSW-PS.
Oocyte injection
After second polar body formation (1.5 hpf), embryos laying naturally on their sides were held gently in place against the agarose cushion with a standard pair of watchmaker’s forceps. Using the modified “scissors” made from forceps (see below), a partial clip was made to the apex of the chorion to create two small cuts (~8–10μm long cuts) angled away from each other. It is important that these cuts are partial, and the two blades of the scissors do not fully close. When done properly, each blade should make a small incision in the chorion. Partial cuts heal after injection allowing the chorion to elevate normally. If the cut is too great and the blades brought too close together, a large “V” shaped incision results and the gel-like embryo/yolk extrudes through the small opening, destroying the embryo.
Once multiple embryos are clipped, they are positioned blastodisc side up within the soft agarose base using a polished Pasteur pipette formed into the shape of a “hockey stick” (~1mm in diameter) over a flame. In this position, the micropyle, chorion cuts, polar bodies (marking the outer membrane surface of the zygote) and deeper yolk are easily visible. Embryos were injected by passing a beveled quartz needle (2–3 μm tip with a 15° angled bevel) through one of the chorion cuts and up to but not through the zygotic membrane. Using diffracted light, it is possible to see the outer membrane of the embryo begin to indent from the pressure of the needle and with that, a gentle tap to the long end of the micromanipulator control knob will “pop” the needle tip into the blastodisc, but not into the yolk. Driving the needle into the blastodisc with the micromanipulator can easily result in overshooting the blastodisc and wounding the yolk. Significant trauma to the yolk layer results in death.
The microinjection set-up consisted of a Xenoworks Digital Pressure Injector (Sutter Instruments, Novato CA), mounted on a Discovery V8 steroscope (Zeiss) using an MMO-202ND manual 3-axis manipulator (Narishige). Quartz micropipettes were pulled on a P-2000G Pipette Puller (Sutter Instruments) and beveled using a BV-10 Micropipetted Beveller (Sutter Instruments). The program used to pull the pipettes consisted of one line with the following settings: heat 750, filament 4, velocity 60, delay 140, pull 175. After pulling, they were bevelled for 30 seconds at a 20° angle. Settings on the injector were Pressure 74, Width 0.21, and Positive Pressure Flow 10. Using these settings, we estimated that 0.221 pL were injected per oocyte by measuring the volume under these settings injected into a drop of oil.
CRISPR sgRNAs and Cas9 protein
Chemically modified CRISPR sgRNAs were synthesized by Synthego (Menlo Park, CA) as was recombinant 2X NLS Cas9 protein (Cas9 2NLS Nuclease). The gene-specific sequence for sgRNA1 was CAUCCAAUCAGUGCCGAAGC and for sgRNA2 was UGGCAGCUGAGGUUCGUGUU. The solution that was injected into oocytes consisted of 34 μM sgRNA (17 μM each), 7μM Cas9 protein and 1.7X PBS. Given the volume that was injected, we estimate that 3.76 amol of each sgRNA and 1.54 amol of Cas9 protein were injected per oocyte.
Post-injection care of embryos
Following injections, embryos were observed daily and moved using clean, large bore polished glass pipettes to fresh culture dishes with MFSW-PS every two days. Some embryos were cultured within their chorions while others were mechanically dechorionated using fine forceps during early organogenesis (after 5 or 6 dpf) to facilitate photography. Dechorionated and chorionated embryos were co-cultured in the same dishes. Dechorionated embryos were not distinguishable developmentally from their siblings. Embryos were cultured for at least 18 days or until control embryos began to hatch from their chorions. For imaging, embryos were anesthetized in 6% Ethanol in FSW and imaged with a Nikon CoolPix 995 camera mounted on a Zeiss Stemi 2000-C Trinocular scope. Embryos were staged according to (21).
Forceps-scissor fabrication
Using a new set of forceps (Inox; #5), the final ~1 mm of each tip is bent 15 to 30 degrees using the fine pliers of a forceps repair kit. The tips should both bend inward. The tips should engage as miniature scissors. A high magnification image of the scissors used in this study are presented in Figure 2.
TDO Inhibitor Treatment
Embryos were treated with 3.15 μM TDO inhibitor 680C91 (Sigma-Aldrich) in MFSW-PS from early organogenesis stage 20 (21), through late organogenesis (stage 27). Dilutions were prepared from a 42 mM stock solution dissolved in dimethyl sulfoxide (DMSO, 10 mg/ml). Dishes and treatment solutions were refreshed every other day. Control embryos were cultured in the presence of DMSO without the inhibitor.
In situ hybridizations
In situ hybridization was carried out as in Shigeno et al. with several modifications (41). A 751 bp sequence of the D. pealeii TDO gene (nt 417–1167) was amplified by PCR (using primers GAGCAATCGCGTCAAGTACA and GGCGTTGTCTTCAGGGTAGA) and cloned into pGEM T-Easy. Digoxigenin-labeled nucleotides (Roche 11277073910) were incorporated into antisense riboprobes generated with SP6 reverse transcriptase (New England Biolabs M0207L) according to the manufacturer’s instructions. Embryos were anesthetized in 2% ethanol in FSW and fixed overnight at 4°C in 4% paraformaldehyde (Electron Microscopy Sciences 15714) in filtered sea water. Fixed embryos were washed for five minutes two times and for 30 minutes once in DEPC-PBS (phosphate buffered saline treated with diethyl pyrocarbonate) and stored in hybridization solution (50% formamide, 5xSSC, 1% SDS, 250g yeast RNA, and 0.1g heparin sulfate per 500mL) at −20°C until use. Tissue was prehybridized for at least 1 hour at 72°C and incubated overnight with 30μL antisense RNA probe reaction at 72°C. Tissue was washed once quickly, four times for 30 minutes, and once for 1h in preheated Solution × (50% formamide, 1% SDS, 2xSSC). Embryos were then washed three times for 15 minutes in TBST (25mL 1M Tris-HCl, 8g NaCl, 0.2g KCl per 1L with 1% Tween) and blocked overnight at 4°C in 10% Roche blocking buffer in TBST. Riboprobes were detected with an anti-DIG antibody coupled to horseradish peroxidase (Sigma 11207733910) diluted to 1:250 in 10% Roche blocking buffer in TBST and incubated at room temperature for 2 hours. Following antibody incubation, embryos were washed three times for 15 minutes and three times for 30 minutes with TBST, followed by two 5 minute washes in TNT (0.1M Tris HCl pH 7.5, 0.15M NaCl, and 0.05%. Tween). Embryos were washed in 50μL amplification diluent and incubated for 1 hour in the dark with Cy5 tyramide (Perkin Elmer) diluted 1:50 in amplification diluent. Embryos were washed three times for 15 minutes in TBST, and twice in Solution × preheated to 72°C and stored in TBST until imaging. Embryos were mounted in Fluoromount-G with Dapi (Southern Biotech) and imaged on an LSM-710 confocal microscope (Zeiss).
TDO expression in the transcriptome
RNAseq reads generated from the D. pealeii genome project (Bioproject PRJNA641326) were mapped onto the genome with Star 2.7.0 (42). Transcripts per million (TPM) for selected tissues were plotted using Rstudio.
Genotyping CRISPR-Cas9 embryos
In order to genotype embryos, genomic DNA was isolated from whole individuals according to a method originally used for Ciona, which uses 25 uL of a Ciona DNA Extraction buffer (made of 1% Triton X, 100 mM NaCl, 20 mM Tris-HCL pH 7.8 and 1mM EDTA), 25 uL H20 and 2.5 uL Proteinase K to digest the ground up embryo, all incubated at 55 °C for 2 hours (42). Samples then underwent an extraction with phenol: chloroform (1:1) followed by a further purification using the Monarch DNA kit (NEB). Nested PCR amplifications surrounding sgRNA-targeted sequences in TDO were the performed. For sgRNA1 we used primer pair GCCTCAAAACAACCCATATTATTGAGG + GAGTTGTAGCGCATCTGAGCAC followed by primer pair TAAATACTTGTGTTCATAGGGTACAC + GGTAAACCCGCTCTGAGTTATTTCCC which resulted in a 215 bp amplicon. For sgRNA2 we used primer pair GCGTGCTATTCTGCATTAGCAC + CGTTAAACCAGTTCTGCCCTCAAG followed by primer pair CCCTAACCATAACCTTAACGTCTC + GCATTCTGTACGATGACACTAAGC which resulted in a 390 bp amplicon. In some cases, we added partial Illumina tags to the nested reactions (Forward ACACTCTTTCCCTACACGACGCTCTTCCGATCT and reverse GACTGGAGTTCAGACGTGTGCTCTTCCGATCT) but in other cases they were added by Genewiz during library preparation. PCR products were then gel extracted and quantified prior to sequencing on the MiSeq platform (paired end 250 nt) using the Amplicon EZ service at Genewiz (South Plainfield, NJ).
Reads were aligned to the amplicon reference sequence using Bowtie2 with the local configuration (43). Because we were expecting misalignment to the reference sequence due to indels, we reduced the gap open and extend penalty from 5 and 3 respectively (default setting) to 3 and 1. Indel analysis was performed by processing the aligned reads using the mpileup function in the SAMtools package filtering out base-calls with Phred Quality (Q) <40 (44). A summary of alignment and filtering results are shown in Table S1. From the mpileup files that were generated, the Indel.py script was used to determine the percentage of deletions per position (number of reads that were missing a position over the total number of reads covering the position). The percentage of insertions per position were calculated in the same way, and both percentages were used to graph the InDel histograms presented in Fig 4&B. To calculate the total disruption per animal, the script Cumulative.py examined a region spanning −1nt to +1nt from the expected Cas9 cut site designated by the CRISPR sgRNA (total number of reads that contain a deletion and/or insertion event and divided by the total number of reads). Finally, to identify specific indels, the Event_frequency.py script (GitHub) was used to determine the number of reads at a position that have a distinct deletion (e.g. −1A, −2AG etc…) or insertion (e.g. +1A, +1G etc…) event and analysis was limited a region spanning −5nt to +5nt from the expected Cas9 cut site designated by the CRISPR sgRNA. Events that occurred at a frequency higher than 0.1% were used for analysis.
Quantification and statistical analyses
To quantify the overall frequency of specific deletions or insertions for all animals, the script Cumulative_Event_frecuency.py was used. The scripts takes in indel frequency data of all animals and create a histogram that records how frequent the deletion or insertion of × number of bases in all reads is in all reads in all the animals for each CRISPR sgRNA. The script also encodes for equations needed for obtaining the parameters of descriptive statistics (mean, median, quartiles, e.g) used to characterize the frequency distribution.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-Digoxigenin-POD Fab fragment | Sigma-Aldrich | 11207733910 |
| Bacterial and Virus Strains | ||
| N/A | ||
| Biological Samples | ||
| N/A | ||
| Chemicals, Peptides, and Recombinant Proteins | ||
| Agarose, Type II-A, Medium EEO | Sigma-Aldrich | A9918 |
| Penicillin-Streptomycin 100x | Gibco | 15-140-122 |
| TDO Inhibitor 680C91 | Sigma-Aldrich | SML0287 |
| Dimethysulfoxide (DMSO) | Sigma-Aldrich | D8418 |
| Diethyl pyrocarbonate | Sigma-Aldrich | D5758 |
| Paraformaldehyde | Electron microscopy sciences | 15714 |
| 2X NLS Cas9 protein | Synthego | |
| Formamide | Sigma | 47670–2.5L-F |
| DIG wash and block set (Roche blocking buffer) | Sigma | 11585762001 |
| RNA from yeast | Sigma | 10109223001 |
| SP6 RNA polymerase | Roche | M0207L |
| DIG-labeling nucleotide mix | Roche | 11277073910 |
| Fluoromount-G | Southern biotech | 0100–20 |
| Ethanol | Pharmco | 111000200 |
| Critical Commercial Assays | ||
| pGEM t Easy vector | Promega | A1360 |
| Cy5 TSA plus kit | Perkin Elmer | NEL752001KT |
| Monarch DNA Kit | NEB | T3010L |
| Monarch Gel Extraction kit | NEB | T2010L |
| Deposited Data | ||
| TDO locus of Doryteuthis pealeii | GenBank | MT648678 |
| Doryteuthis pealeii RNAseq data | Bioproject | PRJNA641326 |
| Experimental Models: Cell Lines | ||
| N/A | ||
| Experimental Models: Organisms/Strains | ||
| Adult Doryteuthis pealeii | Wild caught in the Vineyard Sound near Woods Hole | |
| Oligonucleotides | ||
| In situ PCR primer 1 | IDT | GAGCAATCGCGTCAAGTACA |
| In situ PCR primer 2 | IDT | GGCGTTGTCTTCAGGGTAGA |
| sgRNA 1 PCR forward primer nested reaction 1 | IDT | GCCTCAAAACAACCCATATTATTGAGG |
| sgRNA 1 PCR reverse primer nested reaction 1 | IDT | GAGTTGTAGCGCATCTGAGCAC |
| sgRNA 1 PCR forward primer nested reaction 2 | IDT | TAAATACTTGTGTTCATAGGGTACAC |
| sgRNA 1 PCR reverse primer nested reaction 2 | IDT | GGTAAACCCGCTCTGAGTTATTTCC |
| sgRNA 2 PCR forward primer nested reaction 1 | IDT | GCGTGCTATTCTGCATTAGCAC |
| sgRNA 2 PCR reverse primer nested reaction 1 | IDT | CGTTAAACCAGTTCTGCCCTCAAG |
| sgRNA 2 PCR forward primer nested reaction 2 | IDT | CCCTAACCATAACCTTAACGTCTC |
| sgRNA 2 PCR reverse primer nested reaction 1 | IDT | GCATTCTGTACGATGACACTAAGC |
| CRISPR sgRNAs | ||
| sgRNA1 | Synthego | CAUCCAAUCAGUGCCGAAGC |
| sgRNA2 | Synthego | UGGCAGCUGAGGUUCGUGUU |
| Software and Algorithms | ||
| MUSCLE | (39) | |
| Fig tree Software | http://tree.bio.ed.ac.uk/software/figtree | |
| Fast tree 2 software | (40) | http://www.microbesonline.org/fasttree/ |
| Star v. 2.7.0 | (42) | |
| Bowtie2 | (44) | |
| SAMtools | Genome Research Limited | http://www.htslib.org/ |
| Indel.py | This study | https://github.com/pipedq/CRISPR_G0_Genotyping |
| Cumulative.py | This study | https://github.com/pipedq/CRISPR_G0_Genotyping |
| Event_frequency.py | This study | https://github.com/pipedq/CRISPR_G0_Genotyping |
| Other | ||
| Quartz glass capillaries for making injection needles | Sutter Instruments | QF100-70-10 |
Highlights.
The first gene knockout in a cephalopod
Tryptophan 2,3 Dioxygenase is required for pigmentation in Doryteuthis pealeii
CRISPR-Cas9 is highly efficient in squid embryos
Acknowledgements
This work was supported by NSF IOS 1827509, NSF IOS 1664767, NSF DBI 1723141, The Binational Science Foundation award 2013094, The Grass Foundation for the Doryteuthis Genome Project, The MBL Whitman Fellowship Program and Faculty Development Grant support from St. Mary’s College of Maryland for K. Crawford, The Hibbitt Family for their support of C. Albertin, the office of the NIH Director 1DP5OD023111-01 and the John Harvard Distinguished Science Fellowship for their support of K. Koenig, and Charles and Patricia Robertson and The Owens Family Foundation for their support of J. Rosenthal. We thank Emily Garcia and Cheyenne Rodriguez who assisted with TDO probe generation and in situ hybridization. We thank the Cephalopod Initiative at the Marine Biological Laboratory for providing animals. We also thank Dr. Daniel Rokhsar and the D. pealeii genome project for providing the gene sequence for TDO and for access to RNAseq expression data. Finally, we thank Antonio Giraldez of Yale University for designing and fabricating the critical micro-scissors used in this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare no competing interests
References
- 1.Hodgkin AL, and Huxley AF (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol 117, 500–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Weer P, and Geduldig D (1973). Electrogenic sodium pump in squid giant axon. Science 179, 1326–1328. [DOI] [PubMed] [Google Scholar]
- 3.Armstrong CM, and Bezanilla F (1973). Currents related to movement of the gating particles of the sodium channels. Nature 242, 459–461. [DOI] [PubMed] [Google Scholar]
- 4.Vale RD, Schnapp BJ, Mitchison T, Steuer E, Reese TS, and Sheetz MP (1985). Different axoplasmic proteins generate movement in opposite directions along microtubules in vitro. Cell 43, 623–632. [DOI] [PubMed] [Google Scholar]
- 5.Brady ST, Lasek RJ, and Allen RD (1982). Fast axonal transport in extruded axoplasm from squid giant axon. Science 218, 1129–1131. [DOI] [PubMed] [Google Scholar]
- 6.Vale RD, Reese TS, and Sheetz MP (1985). Identification of a novel force-generating protein, kinesin, involved in microtubule-based motility. Cell 42, 39–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vale RD, Schnapp BJ, Reese TS, and Sheetz MP (1985). Organelle, bead, and microtubule translocations promoted by soluble factors from the squid giant axon. Cell 40, 559–569. [DOI] [PubMed] [Google Scholar]
- 8.Bloedel J, Gage PW, Llinás R, and Quastel DMJ (1966). Transmitter release at the squid giant synapse in the presence of tetrodotoxin. Nature 212, 49–50. [DOI] [PubMed] [Google Scholar]
- 9.Bullock TH, and Hagiwara S (1957). Intracellular recording from the giant synapse of the squid. J. Gen. Physiol 40, 565–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hagiwara S, and Tasaki I (1958). A study on the mechanism of impulse transmission across the giant synapse of the squid. J. Physiol 143, 114–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Katz B, and Miledi R (1967). A study of synaptic transmission in the absence of nerve impulses. J. Physiol 192, 407–436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, and Charpentier E (2012). A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang H, Yang H, Shivalila CS, Dawlaty MM, Cheng AW, Zhang F, and Jaenisch R (2013). One-step generation of mice carrying mutations in multiple genes by CRISPR/cas-mediated genome engineering. Cell 153, 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Albertin CB, Simakov O, Mitros T, Wang ZY, Pungor JR, Edsinger-Gonzales E, Brenner S, Ragsdale CW, and Rokhsar DS (2015). The octopus genome and the evolution of cephalopod neural and morphological novelties. Nature 524, 220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.da Fonseca RR, Couto A, Machado AM, Brejova B, Albertin CB, Silva F, Gardner P, Baril T, Hayward A, Campos A, et al. (2020). A draft genome sequence of the elusive giant squid, Architeuthis dux. Gigascience 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belcaid M, Casaburi G, McAnulty SJ, Schmidbaur H, Suria AM, Moriano-Gutierrez S, Sabrina Pankey M, Oakley TH, Kremer N, Koch EJ, et al. (2019). Symbiotic organs shaped by distinct modes of genome evolution in cephalopods. Proc. Natl. Acad. Sci. U. S. A 116, 3030–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanlon RT, and Messenger JB (2018). Cephalopod Behaviour (Cambridge University Press; ) [Google Scholar]
- 18.Williams TL, DiBona CW, Dinneen SR, Labadie SFJ, Chu F, and Deravi LF (2016). Contributions of Phenoxazone-Based Pigments to the Structure and Function of Nanostructured Granules in Squid Chromatophores. Langmuir 32, 3754–9. [DOI] [PubMed] [Google Scholar]
- 19.Schwinck I (1953). Über den Nachweis eines Redox-Pigmentes (Ommochrom) in der Haut von Sepia officinalis. Naturwissenschaften 40, 365. [Google Scholar]
- 20.Aubourg SP, Torres-Arreola W, Trigo M, and Ezquerra-Brauer JM (2016). Partial characterization of jumbo squid skin pigment extract and its antioxidant potential in a marine oil system. Eur. J. Lipid Sci. Technol 118, 1293–1304. [Google Scholar]
- 21.Arnold JM (1965). Normal embryonic stages of the squid,Loligo pealeii (Leseur). Biol. Bull 128, 24–32. [Google Scholar]
- 22.Crawford K (2002). Culture method for in vitro fertilization to hatching of the squid, Loligo pealeii. In Biological Bulletin (Marine Biological Laboratory), pp. 216–217. [DOI] [PubMed] [Google Scholar]
- 23.Liscovitch-Brauer N, Alon S, Porath HT, Elstein B, Unger R, Ziv T, Admon A, Levanon EY, Rosenthal JJC, and Eisenberg E (2017). Trade-off between Transcriptome Plasticity and Genome Evolution in Cephalopods. Cell 169, 191–202.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Alon S, Garrett SC, Levanon EY, Olson S, Graveley BR, Rosenthal JJC, and Eisenberg E (2015). The majority of transcripts in the squid nervous system are extensively recoded by A-to-I RNA editing. Elife 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Macy W (2001). Seasonal maturity and size at age of Loligo pealeii in waters of southern New England. ICES J. Mar. Sci 58, 852–864. [Google Scholar]
- 26.Yamamoto M, Howells AJ, and Ryall RL (1976). The ommochrome biosynthetic pathway in Drosophila melanogaster: The head particulate phenoxazinone synthase and the developmental onset of xanthommatin synthesis. Biochem. Genet 14, 1077–1090. [DOI] [PubMed] [Google Scholar]
- 27.Figon F, and Casas J (2019). Ommochromes in invertebrates: biochemistry and cell biology. Biol. Rev 94, 156–183. Available at: http://doi.wiley.com/10.1111/brv.12441 [Accessed April 9, 2020]. [DOI] [PubMed] [Google Scholar]
- 28.Leung CF, Webb SE, and Miller AL (2000). On the mechanism of ooplasmic segregation in single-cell zebrafish embryos. Dev. Growth Differ 42, 29–40. [DOI] [PubMed] [Google Scholar]
- 29.Crawford K (2000). The role of microtubules during blastodisc formation of the squid, loligo pealei. In Biological Bulletin (Marine Biological Laboratory), pp. 207–208. [DOI] [PubMed] [Google Scholar]
- 30.Crawford K (2001). Ooplasm segregation in the squid embryo, Loligo pealeii. In Biological Bulletin (Marine Biological Laboratory), pp. 251–252. [DOI] [PubMed] [Google Scholar]
- 31.Li X, Fan D, Zhang W, Liu G, Zhang L, Zhao L, Fang X, Chen L, Dong Y, Chen Y, et al. (2015). Outbred genome sequencing and CRISPR/Cas9 gene editing in butterflies. Nat. Commun 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang L, and Reed RD (2016). Genome editing in butterflies reveals that spalt promotes and Distal-less represses eyespot colour patterns. Nat. Commun 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Banerjee T, and Monteiro A (2018). CRISPR-Cas9 Mediated Genome Editing in Bicyclus anynana Butterflies. Methods Protoc. 1, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wu RS, Lam II, Clay H, Duong DN, Deo RC, and Coughlin SR (2018). A Rapid Method for Directed Gene Knockout for Screening in G0 Zebrafish. Dev. Cell 46, 112–125.e4. [DOI] [PubMed] [Google Scholar]
- 35.Martin A, Serano JM, Jarvis E, Bruce HS, Wang J, Ray S, Barker CA, O’Connell LC, and Patel NH (2016). CRISPR/Cas9 Mutagenesis Reveals Versatile Roles of Hox Genes in Crustacean Limb Specification and Evolution. Curr. Biol 26, 14–26. [DOI] [PubMed] [Google Scholar]
- 36.Hanlon RT, Claes MF, Ashcraft SE, and Dunlap PV (1997). Laboratory culture of the sepiolid squid Euprymna scolopes: A model system for bacteria-animal symbiosis. Biol. Bull 192, 364–374. [DOI] [PubMed] [Google Scholar]
- 37.McFall-Ngai MJ (2014). The Importance of Microbes in Animal Development: Lessons from the Squid-Vibrio Symbiosis. Annu. Rev. Microbiol 68, 177–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Garrett S, and Rosenthal JJC (2012). RNA editing underlies temperature adaptation in K+ channels from polar octopuses. Science 335, 848–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Edgar RC (2004) MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Price MN Dehal PS, Arkin AP (2010). FastTree 2 - Approximately maximum-likelihood trees for large alignments. PLoS One 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shigeno S, Parnaik R, Albertin CB, Ragsdale CW (2015). Evidence for a cordal, not ganglionic, pattern of cephalopod brain neurogenesis. Zool. Lett 1, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C Jha S, Batut P, Chaisson M, Gringeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Veeman MT, Chiba S, Smith WC (2011). Ciona genetics. Methods Mol. Biol 770, 401–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Langmead B, Salzberg SL (2012) Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, & 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The scripts (Cumulative.py, indels.py, Event_frequency.py) generated during this study are available at Github (https://github.com/pipedq/CRISPR_G0_Genotyping). The sequence for the D. pealeii TDO locus is available at Genbank (MT648678). RNAseq reads are deposited as Bioproject PRJNA641326. Reads from the amplicon sequencing are available on request.