

Abstract
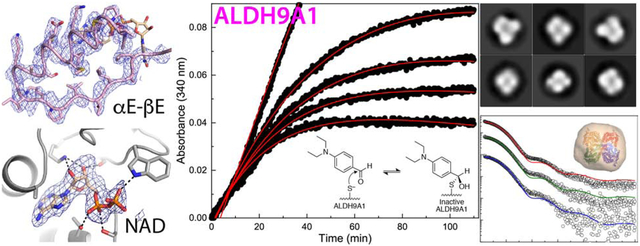
Aldehyde dehydrogenase 9A1 (ALDH9A1) is a human enzyme that catalyzes the NAD+-dependent oxidation of the carnitine precursor 4-trimethylaminobutyraldehyde to 4-N-trimethylaminobutyrate. Here we show that the broad-spectrum ALDH inhibitor diethylaminobenzaldehyde (DEAB) reversibly inhibits ALDH9A1 in a time-dependent manner. Possible mechanisms of inhibition include covalent reversible inactivation involving the thiohemiacetal intermediate and slow, tight-binding inhibition. Two crystal structures of ALDH9A1 are reported, including the first of the enzyme complexed with NAD+. One of the structures reveals the active conformation of the enzyme, in which the Rossmann dinucleotide-binding domain is fully ordered and the inter-domain linker adopts the canonical β-hairpin observed in other ALDH structures. The oligomeric structure of ALDH9A1 was investigated using analytical ultracentrifugation, small-angle X-ray scattering, and negative stain electron microscopy. These data show that ALDH9A1 forms the classic ALDH superfamily dimer-of-dimers tetramer in solution. Our results suggest that the presence of an aldehyde substrate and NAD+ promotes isomerization of the enzyme into the active conformation.
Keywords: aldehyde dehydrogenase, ALDH9A1, X-ray crystallography, small-angle X-ray scattering, analytical ultracentrifugation, negative-stain electron microscopy, time-dependent inhibition, covalent reversible inhibitor, diethylaminobenzaldehyde, DEAB
Graphical Abstract
1. Introduction
The aldehyde dehydrogenase (ALDH) structural superfamily is a large group of enzymes that catalyze the NAD+-dependent oxidation of aldehydes to carboxylic acids [1, 2]. The superfamily comprises hundreds of distinct genes, including 19 ALDHs expressed in humans. ALDHs share a common protein fold and catalytic mechanism, but subtle differences in their active sites result in different preferences for the aldehyde substrate.
ALDH9A1 is a human enzyme that catalyzes the NAD+-dependent oxidation of a variety of aldehydes including the carnitine precursor 4-trimethylaminobutyraldehyde (TMBAL, Scheme 1) [3], the GABA precursor aminobutyraldehyde [4], the dopamine metabolite 3,4-dihydroxyphenylacetaldehyde [5], and betaine aldehyde [6]. A recent comprehensive study of substrate specificity showed that ALDH9A1 has a strong (> 10-fold) preference for TMBAL [7], confirming the assertion of Vaz, et al. [3] that the major in vivo function of this enzyme is to catalyze the penultimate step of carnitine biosynthesis (E.C. 1.2.1.47), the oxidation of TMBAL to 4-N-trimethylaminobutyrate. Carnitine functions in the transport of long-chain fatty acids from the cytosol to the mitochondrial matrix for the synthesis of acyl-CoAs for β-oxidation. Because the mitochondrial membrane is impermeable to acyl-CoAs, fatty acids are conjugated to carnitine to enter mitochondria. Thus, ALDH9A1 functions indirectly in β-oxidation.
Scheme 1.
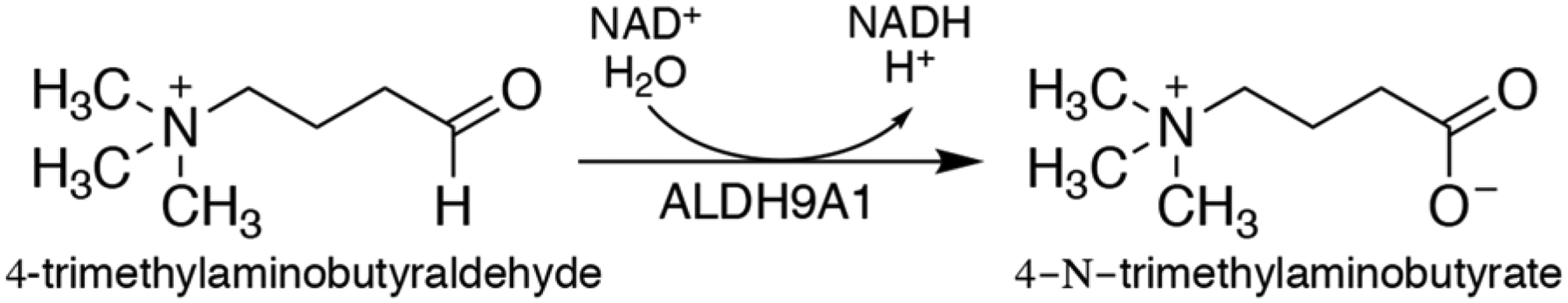
Reaction catalyzed by ALDH9A1.
Recently the first crystal structure of human ALDH9A1 was reported [7] (PDB ID: 6QAP). Although ALDH9A1 exhibits the basic ALDH superfamily fold, the structure revealed two remarkable features. First, the final α-helix and β-strand of the Rossmann dinucleotide-binding fold are disordered. Referred to as αE-βE in the closely-related betaine ALDH [8], these secondary structural elements form extensive interactions with NAD+ in other ALDHs. Further, αE forms a major part of the dimer interface in other ALDHs. The second atypical aspect of the apo ALDH9A1 structure concerns the ~25-residue inter-domain linker, which connects the catalytic domain to the oligomerization domain. In other ALDH structures, the linker folds into a long β-hairpin, wherein the strands of the β-hairpin form the floor of the aldehyde substrate tunnel, while the tip helps stabilize the NAD+ binding site. In contrast, the linker in the apo ALDH9A1 structure adopts a novel S-shaped conformation, which blocks access to the aldehyde substrate binding pocket. The apo ALDH9A1 structures appear to represent an inactive enzyme, raising the question of how αE-βE and the inter-domain linker isomerize into their active conformations.
Herein we describe a structural and biochemical study of ALDH9A1 designed to provide understanding of the catalytic function of this enzyme, in light of the atypical structural features observed previously for the apo enzyme. We demonstrate that diethylaminobenzaldehyde (DEAB) is a reversible, time-dependent inhibitor of ALDH9A1. We report two crystal structures of ALDH9A1 complexed with NAD+. In one of the structures, which was obtained from enzyme treated with DEAB and NAD+, αE-βE and the inter-domain linker are ordered and adopt the canonical secondary, tertiary, and quaternary structures observed in other ALDHs. In contrast, the other NAD+ complex structure exhibits the atypical features of the apo enzyme, showing that NAD+ binding alone is not sufficient to promote the active conformation of the enzyme. The oligomeric structure of ALDH9A1 was determined using analytical ultracentrifugation, small-angle X-ray scattering, and negative stain electron microscopy. These studies show that ALDH9A1 forms the classic ALDH superfamily dimer-of-dimers tetramer in solution. Our results provide insight into the mechanism by which ALDH9A1 is activated for catalysis.
2. Materials and methods
2.1. Expression plasmids
A synthetic gene encoding human ALDH9A1 with codons optimized for expression in Escherichia coli was purchased from Genscript. The gene was subcloned by Genscript into plasmid pET-24b(+) between NdeI/XhoI restriction sites. The expressed protein contains an N-terminal hexahistidine tag and tobacco etch virus protease cleavage site.
Removal of the His-tag from the protein expressed from the aforementioned pET-24b(+) construct was problematic, so another expression plasmid encoding ALDH9A1 fused to SUMO was created. The coding sequence of human ALDH9A1 was PCR amplified from the pET24b(+) construct, gel-purified, and cloned into pET-SUMO using BamHI and XhoI restriction sites. Positive clones were identified by colony PCR followed by DNA sequencing using T7 forward and reverse primers.
2.2. Purification of SUMO-ALDH9A1 and His-ALDH9A1
To purify SUMO-tagged ALDH9A1, the fusion protein was over-expressed in E. coli BL21 (DE3) with induction by 0.25 mM IPTG at 18°C for 20 hrs. Cells were collected by centrifugation and resuspended in buffer A (50 mM Tris pH 8.0, 300 mM NaCl, 10 mM imidazole and 5% glycerol (v/v) containing 0.1% Triton X-100. Cells were lysed by sonication and then centrifuged at 20,000 rpm in a SS-34 rotor for 30 min at 4°C to remove the remaining insoluble material. The supernatant was subjected to immobilized metal-affinity chromatography (IMAC) on a HisTrap HP column (GE Healthcare) charged with nickel ion and equilibrated with buffer A and unbound proteins were washed sequentially with ten column volumes each of buffer A with imidazole concentrations of 40, 100 and 150 mM. The His-tagged proteins were eluted with buffer A containing 200 mM imidazole. To remove the N-terminal SUMO-tag, His-tagged SUMO protease (purified separately by IMAC chromatography) was added to IMAC-purified His-SUMO-ALDH9A1 in a 1:100 ratio and subjected to dialysis for 12 – 16 h at 4°C in 25 mM Tris-HCl pH 8.0, 100 mM NaCl, 5% (v/v) glycerol and 0.5% Tris(2-arboxyethyl)phosphine (TCEP). The digested protein sample was then passed through the IMAC column, which retained the cleaved SUMO tag, SUMO protease, and uncleaved fusion protein. The pure, untagged ALDH9A1 protein was collected in the flow through and then protein quality was analyzed with SDS-PAGE. Subsequently, ALDH9A1 was concentrated using a 50 kDa MilliporeSigma Amicon Ultra centrifugal concentrator and purified further using size-exclusion chromatography (SEC) on a HiLoad 16/60 Superdex 200 column with a buffer containing 50 mM Tris-HCl pH 8.0, 600 mM NaCl, 5% (v/v) glycerol and 0.5% TCEP. The purity of the recombinant ALDH9A1 was verified by SDS-PAGE. Protein concentration was determined by absorbance using an estimated extinction coefficient calculated by ProtParam [9].
His-tagged ALDH9A1 was over-expressed from the pET-24b(+) vector in E. coli BL21 (DE3) at OD ~ 0.6 with 0.25 mM IPTG at 18°C for 18 – 21 hrs. Cells were harvested by centrifugation and resuspended in buffer A (50 mM HEPES pH 8.0, 300 mM NaCl, 10 mM imidazole, and 5% glycerol) containing 0.1% Triton X-100 and 0.4 mM PMSF. Cells were lysed by sonication and centrifuged at 16,500 rpm in a SS-34 rotor for 1 h at 4°C. The supernatant was subjected to IMAC using a HisTrap HP column (GE Healthcare) charged with nickel ion and equilibrated in buffer A. After washing the column with 100 mM imidazole, His-ALDH9A1 was eluted with 200 mM imidazole. The enzyme was concentrated with a 50 kDa MilliporeSigma Amicon Ultra centrifugal concentrator and further purified by SEC on a Superdex 200 column in the presence of 50 mM HEPES pH 8.0, 100 mM NaCl, 5% (v/v) glycerol, and 0.5 mM dithiothreitol (DTT). The purity of His-ALDH9A1 was verified with SDS-PAGE and protein concentration was determined by A280 using a NanoDrop 2000c (ThermoFisher) and confirmed via bicinchoninic acid assay (Pierce).
2.3. Kinetics of enzyme activity and inhibition
All kinetic measurements were performed using His-tag-free ALDH9A1. The steady state kinetic parameters of ALDH9A1 for the aldehyde substrate hexanal at saturating NAD+ concentration were determined. These assays monitored NADH production (340 nm) at 27°C in an Epoch 2 plate reader (BioTek, Winooski, VT, USA). The assay buffer contained 100 mM sodium pyrophosphate pH 8.0. The aldehyde substrate concentration was varied in the range of 2.5 – 150 μM, while the NAD+ concentration was fixed at 1.5 mM. The untagged enzyme was used at 500 nM. Each measurement was performed in triplicate. Initial rates were estimated by linear regression of the first five minutes of the assay. The absorbance at 340 nm was converted to NADH concentration using an extinction coefficient of 6220 M−1cm−1. Kinetic parameters were obtained by fitting the initial rate data to the Michaelis-Menten equation using Origin version 2019 (Fig. S1 of Supplementary data).
DEAB is a substrate for some ALDHs, so we first tested this possibility for ALDH9A1 using the strategy of Hurley’s group [10]. In this experiment, ALDH9A1 is combined with DEAB and NAD+, and then the absorbance at 360 nm and 300 nm are monitored. A decrease at 360 nm indicates the consumption of DEAB, while an increase at 300 nm indicates the production of the carboxylic acid product, diethylaminobenzoic acid. For these experiments, the final assay mixture (total volume of 1 mL) contained 0.5 μM ALDH9A1, 30 μM DEAB, and 1.5 mM NAD+ in a buffer containing 100 mM sodium pyrophosphate pH 8.0 and 0.1% DMSO. The reaction was initiated by the addition of enzyme. The absorbance was measured for 240 minutes using the cuvette port of a NanoDrop 2000c (ThermoFisher). Scans covering the wavelength range of 270 – 460 nm were recorded every 10 minutes.
The time-dependent inhibition of ALDH9A1 by DEAB was studied using a progress curve approach [11, 12]. Progress curve experiments were performed in a BioTek Epoch 2 plate reader in duplicate at 27°C with an assay mixture consisting of 0.5 μM ALDH9A1, 2 mM hexanal, 2.5 mM NAD+, 2 – 5.0 μM DEAB, 0.04% (v/v) DMSO, and 100 mM sodium pyrophosphate pH 8.0. The total reaction assay volume was 200 μL. The reaction was initiated by the addition of an aliquot of an enzyme- NAD+ solution and then monitored by observing the production of NADH at 340 nm. The progress curves measured at different DEAB concentrations were analyzed using global fitting to combined Equations 1a and 1b [13].
| (1a) |
| (1b) |
Equations 1a and 1b describe the inhibition model in Scheme 2B (see equations 6 and 9 of Morrison and Walsh [13]). The parameters vi and vs are the initial and steady-state velocities, t is time, ao is the initial absorbance offset parameter, and k the apparent first-order rate constant for the establishment of the equilibrium between E·I and E·I*. In Equation 1b, k5 and k6 are the forward and reverse rate constants for the isomerization of E·I to E·I*, respectively; KI is dissociation constant for the E·I complex; [I] is the inhibitor concentration; [S] is the hexanal substrate concentration; and Km is the Michaelis constant for hexanal, which was estimated to be 6.3 μM in a separate experiment performed in the absence of DEAB (Fig. S1 of Supplementary data). Global fitting was done with Origin 2019, with k5, k6, and KI considered to be global parameters (shared by all data sets), while Km, [S], and [I] were fixed at their known values. During fitting, k6 refined to a very small value (2 × 10−5 min−1) compared to k5 (0.2 min−1); however, the uncertainty of k6 was large, which prevented convergence. Therefore, k6 was fixed at 2 × 10−5 min−1 to aid convergence of the fitting algorithm.
Scheme 2.
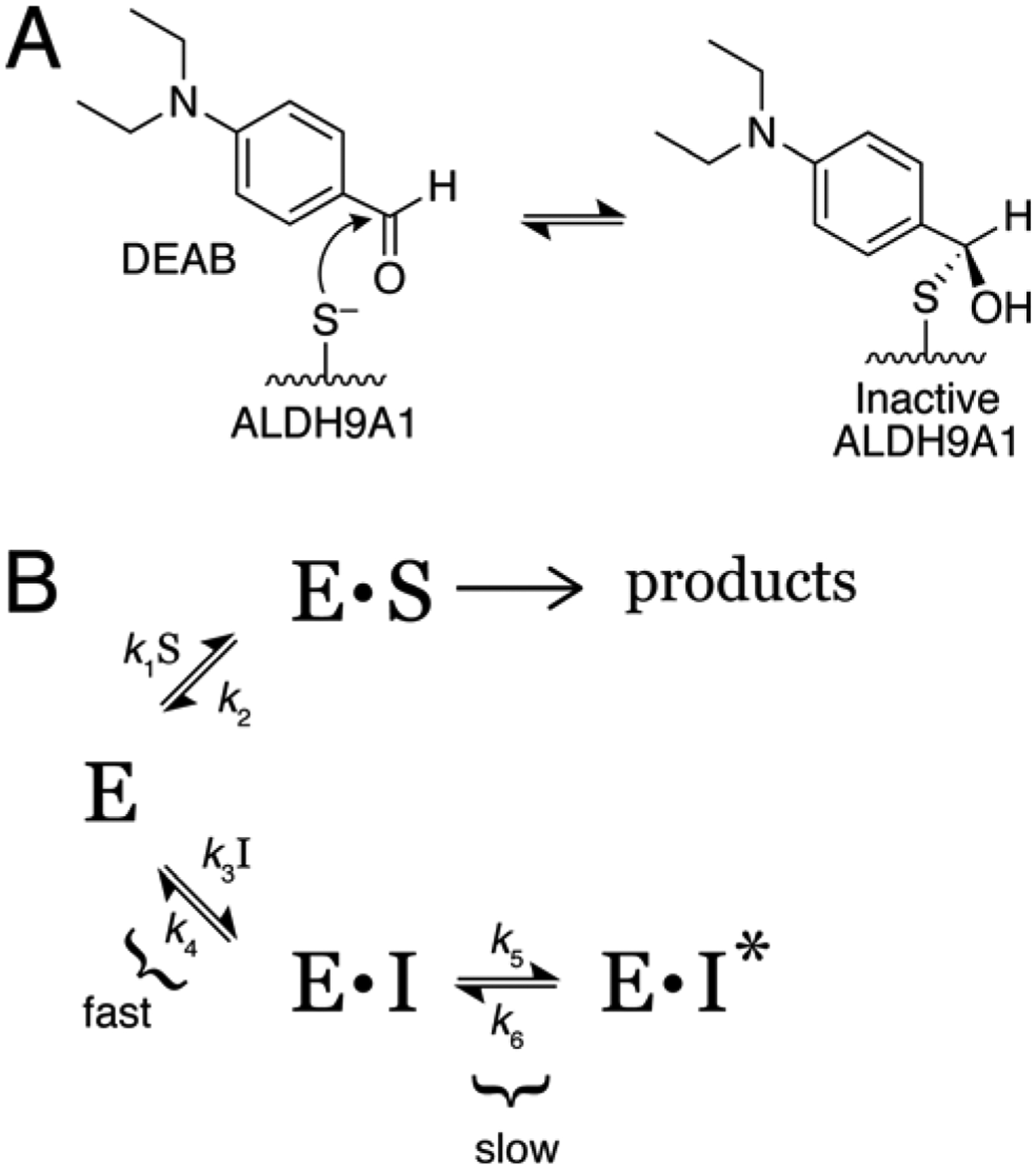
Possible mechanisms of inactivation/inhibition of ALDH9A1 by DEAB.
2.4. Crystallization of ALDH9A1-NAD+ in space group P1
Prior to crystallization, DEAB and NAD+ were added to the protein stock solution (6 mg mL−1 tag-free ALDH9A1; 50 mM Tris-HCL pH 8.0, 600 mM NaCl, 5% glycerol, and 0.5% TCEP) to final concentrations of 5 mM and 10 mM, respectively. Initial crystal screening was performed in 96-well Swissci 2 drop MRC Crystallization plates with Hampton Index at 8°C. Promising crystals were obtained in a condition containing 0.2 M NaCl, 0.1 M Bis-Tris pH 6.5, and 25% (w/v) PEG 3350. Further improvement was obtained using an additive screen approach. In this approach, the reservoir consisted of equal volumes of the aforementioned base condition (0.2 M NaCl, 0.1 M Bis-Tris pH 6.5, and 25% (w/v) PEG 3350) and an additional crystal screen condition. Improvement in the crystal quality was observed when using Hampton Index condition G8 as the additive (0.2 M ammonium acetate, 0.1 M HEPES pH 9.5, and 25% (w/v) PEG 3350). Thus, the final reservoir solution contained 0.1 M NaCl, 0.05 M Bis-Tris 6.5, 0.1 M ammonium acetate, 0.05 M HEPES pH 9.5, and 25% (w/v) PEG 3350. The crystals were cryoprotected with 0.1 M NaCl, 0.05 M Bis-Tris pH 6.5, 0.1 M ammonium acetate, 0.05 M HEPES pH 9.5, 25% (w/v) PEG 3350, 20% (v/v) hexylene glycol, 2.5 mM DEAB and 5 mM NAD+. Although the presence of DEAB in the active site was not obvious from the electron density, its inclusion in the crystallization protocol apparently facilitated growth of a new crystal form, which revealed the active conformation of the enzyme.
2.5. Crystallization of ALDH9A1-NAD+ in space group C222
Prior to crystallization, NAD+ was added to the protein stock solution (10 mg mL−1 His-tagged ALDH9A1; 50 mM HEPES pH 8.0, 100 mM NaCl, 5% (v/v) glycerol, and 0.5 mM DTT) to a final concentration of 30 mM. A crystal screen was performed in 96-well Swissci 2 drop MRC Crystallization plates at 8°C using Hampton Index with a drop ratio of 0.6/0.4 (enzyme/reservoir). Microseed matrix screening [14, 15] was used with a seed stock prepared from previously grown C222 crystals. The crystal used for data collection was harvested directly from Hampton Index condition H5 (0.1 M succinic acid pH 7.0, 15% (w/v) PEG 3350) without optimization and cryoprotected with the reservoir supplemented with 20% (v/v) hexylene glycol.
2.6. Crystal structure determination
X-ray diffraction data from a P1 crystal were collected at NECAT beamline ID-E of the Advanced Photon Source using an EIGER 16M detector in shutterless mode. The dataset used for refinement consisted of 700 frames spanning a rotation range of 140°. Diffraction data from a C222 crystal were collected at beamline 4.2.2 of the Advanced Light Source (900 frames, 180° rotation). The data from both beamlines were integrated and scaled with XDS [16], and intensities were merged and converted to amplitudes with AIMLESS [17]. The unit cell dimensions of both crystal forms are listed in Table 1. The method of Matthews [18] predicts eight chains in the P1 asymmetric unit with 49% solvent, and 2 chains in the C222 asymmetric unit with 49% solvent. Data processing statistics are listed in Table 1.
Table 1.
X-ray diffraction data collection and refinement statistics.
| Space group | P1 | C222 |
| Unit cell parameters (Å,°) | a = 82.79, b = 90.30, c = 145.24, α = 89.37, β = 84.04, γ = 73.87 | a = 158.15 , b = 163.47, c = 84.34 |
| Wavelength (Å) | 0.97918 | 1.00000 |
| Resolution (Å) | 79.09 – 2.54 (2.54 – 2.50) | 47.13 – 2.64 (2.77 – 2.64) |
| Observationsa | 177440 (9039) | 229337 (30035) |
| Unique reflectionsa | 106567 (5667) | 32497 (4262) |
| Rmerge(I)a | 0.090 (0.500) | 0.151 (1.070) |
| Rmeas(I)a | 0.127 (0.708) | 0.163 (1.156) |
| Rpim(I)a | 0.090 (0.500) | 0.061 (0.434) |
| Mean I/σa | 4.5 (1.0) | 11.6 (1.7) |
| CC1/2 | 0.991 (0.610) | 0.996 (0.803) |
| Completeness (%)a | 76.7 (82.8) | 100.0 (100.0) |
| Multiplicitya | 1.7 (1.6) | 7.1 (7.0) |
| No. of protein chains | 8 | 2 |
| No. of protein residues | 3939 | 900 |
| No. of atoms | ||
| Protein | 29077 | 6729 |
| NAD+ | 233 | 46 |
| Water | N/A | 31 |
| Rcrysta | 0.213 (0.2996) | 0.218 (0.288) |
| Rfreea,b | 0.269 (0.352) | 0.273 (0.332) |
| rmsd bonds (Å) | 0.002 | 0.002 |
| rmsd angles (°) | 0.591 | 0.511 |
| Ramachandran plotc | ||
| Favored (%) | 97.30 | 98.19 |
| Outliers (%) | 0.03 | 0.00 |
| Clashscore (PR)c | 5.73 (99) | 3.24 (100) |
| MolProbity score (PR)c | 1.96 (96) | 1.41 (100) |
| Average B (Å2) | ||
| Protein | 38.7 | 50.8 |
| NAD+ | 39.5 | 58.6 |
| Water | N/A | 30.9 |
| Coord. error (Å)d | 0.38 | 0.41 |
| PDB code | 6VR6 | 6VWF |
Values for the outer resolution shell of data are given in parenthesis.
2% test set for 6VR6; 5% test set for 6VWF.
From MolProbity. The percentile ranks (PR) for Clashscore and MolProbity score are given in parentheses.
Maximum likelihood-based coordinate error estimate from PHENIX.
Initial phases for the P1 structure were calculated using molecular replacement as implemented MOLREP [19]. The search model was derived from a protomer of cod liver betaine ALDH ([8], PDB ID: 1A4S, 70% identity to ALDH9A1). The side chains of the search model were trimmed to the C-γ atom with CHAINSAW [20]. MOLREP returned a solution consisting of eight protein chains arranged as two tetramers. We note that the molecular replacement solutions were not as clear when search models derived from the structure of apo ALDH9A1 (PDB ID: 6QAP) were used, indicating conformational differences between our structure and apo ALDH9A1.
Initial phases for the C222 structure were calculated using molecular replacement with Phaser [21]. The search model was derived from a partial structure obtained by refinement against an earlier C222 data set, which exhibited pseudo-merohedral twinning. Initial phases for the twinned data set were calculated using Phaser with a search model derived from cod liver betaine ALDH (PDB ID: 1A4S).
PHENIX [22] was used for refinement, and Coot [23] was used for model building. The B-factor model consisted of an isotropic B-factor for each non-hydrogen atoms and one TLS group per chain. Because of the modest resolution, non-crystallographic symmetry restraints were enforced during refinement of both structures. The occupancies of the NAD+ molecules in both structures were set to 1.0. The structures were validated with MolProbity [24] and the wwPDB validation service [25]. Refinement statistics are listed in Table 1.
2.7. Analytical ultracentrifugation
Sedimentation velocity experiments were performed in a Beckman XL-I analytical ultracentrifuge using an An50Ti rotor at 20°C. For this analysis, His-tagged ALDH9A1 at 3 mg mL−1 protein sample was dialyzed overnight against a buffer containing 50 mM HEPES pH 7.8, 100 mM NaCl, 5% (v/v) glycerol, and 0.5 mM DTT. A two-sector charcoal-Epon sedimentation velocity centerpiece was loaded with reference buffer and protein samples. After an equilibration period of 2 h, the sample was centrifuged at 35,000 rpm for a total of 300 radial scans spaced at 2 min intervals. Data were acquired using Rayleigh interference optics, which allowed measurement of the actual experimental protein concentration, assuming 1 mg mL−1 is equivalent to 3.33 fringes [26]. Scans 10 – 300 were analyzed and the distribution of apparent sedimentation coefficients, c(s), and distribution of apparent molecular masses, c(M), were determined using SEDFIT [27].
2.8. Small-angle X-ray scattering
Shutterless SAXS data collection was performed at beamline 12.3.1 of the Advanced Light Source through the SIBYLS Mail-in High Throughput SAXS program [28]. Prior to SAXS analysis, purified His-tag-free ALDH9A1 was passed over a Superdex 200 10–30 size-exclusion chromatography column in the presence of a buffer containing 50 mM HEPES pH 8.0, 600 mM NaCl, 2% (v/v) glycerol, 1 mM DTT, and 1 mM NAD+. Samples were then supplemented with 1 mM NAD+ and dialyzed overnight at 4°C against a buffer containing 50 mM HEPES pH 8.0, 600 mM NaCl, 2% (v/v) glycerol, 1 mM DTT, and 1 mM NAD+.
SAXS data were collected on a Pilatus detector operating in shutterless mode, writing frames every 0.3 s. Buffer subtracted SAXS curves were averaged using SAXS FrameSlice. PRIMUS [29] was used to inspect the merged data and to derive SAXS parameters. The maximum particle dimension was estimated from calculations of the pair distribution function using GNOM [30] via PRIMUS. The molecular mass was estimated using the SAXSMoW [31]. Theoretical SAXS curves were calculated using FoXS [32]. DENSSWeb [33] was used for ab initio electron density determination from solution scattering data.
2.9. Negative stain electron microscopy
Preparation of negative-stained grids was carried out adapted from the previously described protocol [34]. Briefly, carbon-coated copper grids (Electron Microscopy Sciences, Hatfield, PA) were glow discharged, and a 5 μL drop of His-tagged ALDH9A1 (~0.08 mg mL−1) was applied to the grid, followed by a 2 min incubation at room temperature. Excess protein was removed by blotting with Whatman P4 filter paper. Blotting was immediately followed by two quick washes in water and a quick wash in 0.75% (w/v) uranyl formate (UF; Ted Pella, Reading, CA), blotting with filter paper in between wash steps. A final incubation, by floating the grid for 2 min on a 0.75% UF (w/v) drop, was performed before excess UF was removed from the grid with filter paper. The grid was air-dried. Protein particles were observed using a JEOL JEM 1400 transmission electron microscope (Peabody, MA) at 80 kV.
The data were analyzed with RELION-3 [35]. Contrast transfer function correction was performed using CTFFind 4.1 using a spherical aberration of 3.2 mm, amplitude contrast of 0.5, and magnified pixel size of 3.51 Å. The resolution range used for fitting was 5–30 Å. The defocus range was 5000–50000 Å with a defocus step of 500 Å and an astigmatism of 200 Å. An initial particle set was obtained using template-free auto-picking (Laplacian-of-Gaussian-based) with a minimum diameter of 50 Å, maximum diameter of 120 Å, and threshold of 3.5. This calculation generated 10373 particles. Low quality particles (e.g., random noise, overlapped protein molecules) were eliminated during several rounds of iterative 2D class averaging and selection, resulting in a set of 7430 particles that was used for the final 2D class average calculation presented here.
3. Results
3.1. Time-dependent inhibition of ALDH9A1 by DEAB
DEAB (Fig. 1A) is variously a substrate, competitive tight-binding inhibitor, or irreversible covalent inactivator of ALDHs [10, 36]. We therefore performed activity assays to understand how DEAB interacts with ALDH9A1.
Fig. 1.
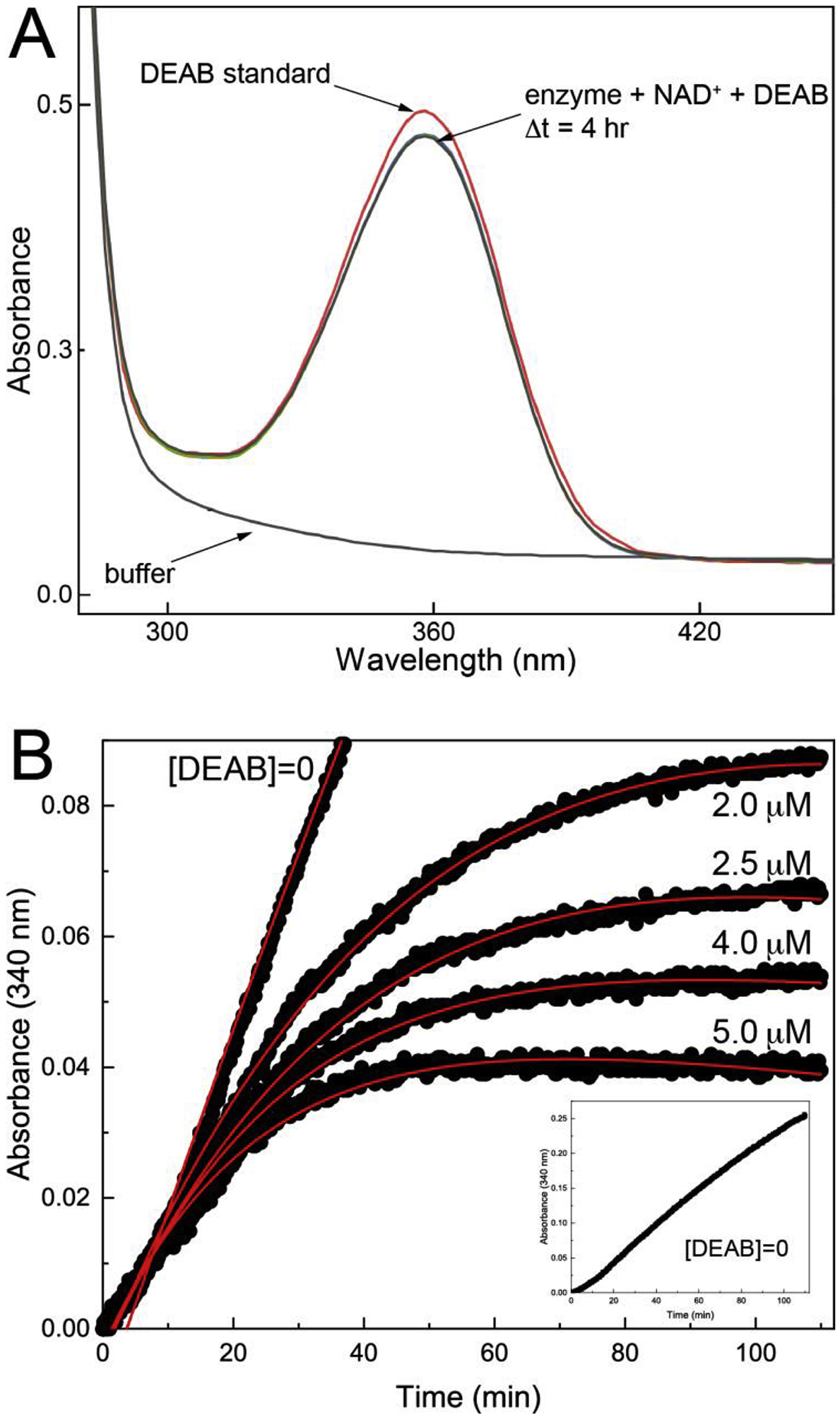
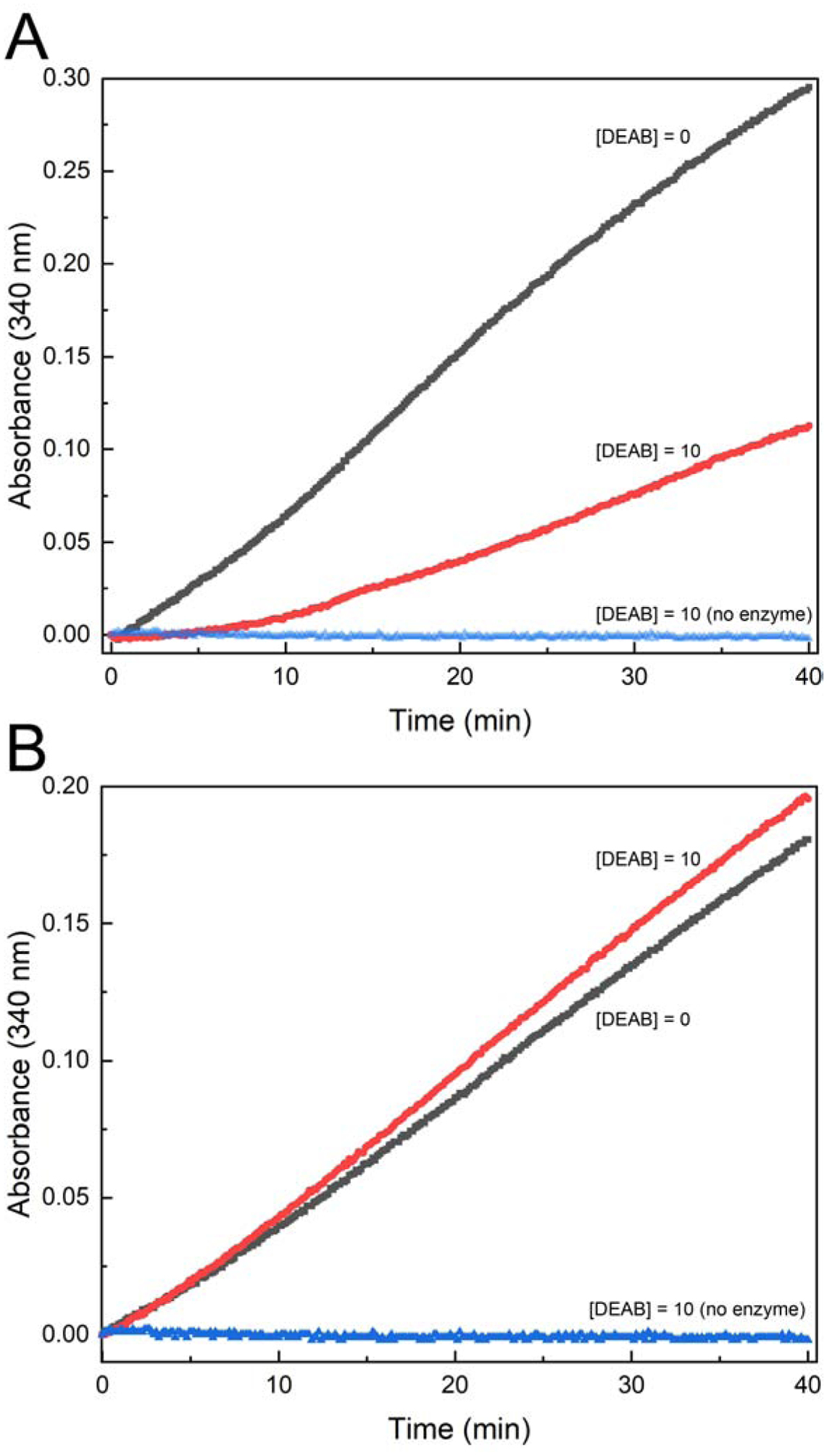
Inhibition of ALDH9A1 by DEAB. (A) Evidence that DEAB is not a substrate of ALDH9A1. ALDH9A1 (0.5 μM) was incubated with 30 μM DEAB and 1.5 mM NAD+, and spectra were collected every 10 minutes for a total of 4 hours. For reference, the red curve is the spectrum of DEAB. (B) Time-dependent inhibition of ALDH9A1 by DEAB. The black circles are experimental progress curves monitoring NADH production from ALDH9A1 in the presence of 2 mM of the aldehyde substrate hexanal, 2.5 mM NAD+, and various concentrations of DEAB. The red curves represent the global fit to combined Equations 1a and 1b. The parameters from global fitting are k5 = 0.184 ± 0.052 min−1, KI = 0.054 ± 0.018 μM. For ease of viewing, a vertical offset was applied to each data set to account for the absorbance due to the different DEAB concentrations. The inset shows the progress curve in the absence of DEAB. The unshifted progress curves with error bars are shown in Fig. S2 of Supplementary data.
We first tested whether DEAB is a substrate using the approach of Morgan et al. [10] by incubating ALDH9A1 (0.5 μM) with DEAB (30 μM) and NAD+ (1.5 mM), while monitoring the absorbance at 360 nm and 300 nm. In this assay, a decrease at 360 nm indicates consumption of DEAB, while an increase at 300 nm indicates production of the carboxylic acid product, diethylaminobenzoic acid. The spectrum of DEAB alone shows a broad peak centered at 360 nm, as expected (Fig. 1A). Spectra acquired for the mixture of ALDH9A1, DEAB, and NAD+ show little change over 4 hours (Fig. 1A). These results suggest that DEAB is not an efficient substrate for ALDH9A1.
Because we observed no significant activity with DEAB as the aldehyde substrate, we next performed time-dependent inhibition assays. A progress curve approach was used in which the enzyme (0.5 μM) was mixed with the aldehyde substrate hexanal (2 mM), NAD+ (2.5 mM), and DEAB (0 – 5.0 μM) (Fig. 1B). The initial increase in absorbance at 340 nm indicates the production of NADH associated with the catalytic turnover of hexanal to hexanoic acid. The flattening of the curves at longer time is characteristic of covalent inactivation, similar to what has been observed with ALDH2 [10] and ALDH7A1 [36]. Alternatively, time-dependent loss of activity is associated with slow, tight-binding inhibition [13, 37, 38]. A control experiment confirmed that flattening of the curves was not due to depletion of substrate or cofactor (inset, Fig. 1B). Global fitting of the data to combined Equations 1a and 1b was performed to estimate kinetic parameters for enzyme inactivation, assuming k6 ≪ k5 (see Material and Methods). These calculations yielded k5 of 0.184 ± 0.052 min−1, KI of 0.054 ± 0.018 μM, and an apparent second-order rate constant for enzyme inactivation by DEAB (k5/KI) of 57000 ± 25000 M−1s−1. This value is similar to that of ALDH2 (86000 M−1s−1) [10].
The reversibility of inhibition was assessed by monitoring the return of enzyme activity upon dilution. ALDH9A1 (5 μM) was incubated with 10 μM DEAB and 1 mM NAD+ at 4°C for 60 minutes, and then assayed for activity by diluting 10-fold into assay buffer containing 0.5 mM hexanal and 1 mM NAD+. The progress curve for the DEAB-treated sample shows a ~5-minute lag in NADH production, followed by an increase in activity (Fig. 2A). The steady-state velocity of the DEAB-treated sample was ~70% compared to a control that was treated identically but lacked DEAB. These results show that inhibition of ALDH9A1 by DEAB is reversible.
Fig. 2.
Inhibition of ALDH9A1 by DEAB is reversible. (A) Progress curves from a dilution assay. ALDH9A1 (5 μM) was incubated with 10 μM DEAB and 1 mM NAD+ at 4°C for 60 minutes, and then assayed for activity by diluting 10-fold into assay buffer containing 0.5 mM hexanal and 1 mM NAD+. (B) Residual activity after removing excess DEAB. ALDH9A1 (5 μM) was incubated with 10 μM DEAB and 1 mM NAD+ for 60 minutes, and then the excess DEAB was removed by spin ultrafiltration using Amicon Ultra-0.5 mL centrifugal filter units. The activity was then measured in an assay containing 0.5 mM hexanal and 1 mM NAD+. Prior to the enzyme assays, the enzyme concentration was measured and used to adjust the progress curves.
The reversibility of inhibition was confirmed by measuring the return of enzyme activity after removal of DEAB. ALDH9A1 (5 μM) was incubated with 10 μM DEAB and 1 mM NAD+ for 60 minutes, and then the excess DEAB was removed by spin ultrafiltration. The activity was then measured in an assay containing 0.5 mM hexanal and 1 mM NAD+. The progress curves for the DEAB-treated and control samples are very similar (Fig. 2B). Note the progress curve for the DEAB-treated sample does not exhibit a lag. These results are consistent with a covalent reversible mechanism of inhibition.
3.2. The fold of ALDH9A1 complexed with NAD+
Two structures of ALDH9A1 complexed with NAD+ were determined. A 2.5 Å resolution structure with space group P1 was obtained using enzyme that had been incubated with both NAD+ and DEAB (Table 1). Also, a 2.64 Å structure in space group C222 was determined using enzyme incubated with NAD+, without DEAB. We note these crystal forms are different from those used for structure determination of apo ALDH9A1 [7].
As expected, ALDH9A1 complexed with NAD+ exhibits the 3-domain architecture of apo ALDH9A1, as well as other ALDHs (Fig. 3A). The NAD+-binding domain features a 5-stranded Rossmann-dinucleotide binding fold at its core. The catalytic domain has an α/β fold and contains the catalytic cysteine residue (Cys288). The oligomerization domain adopts the expected β- substructure protruding from the NAD+-binding domain.
Fig. 3.
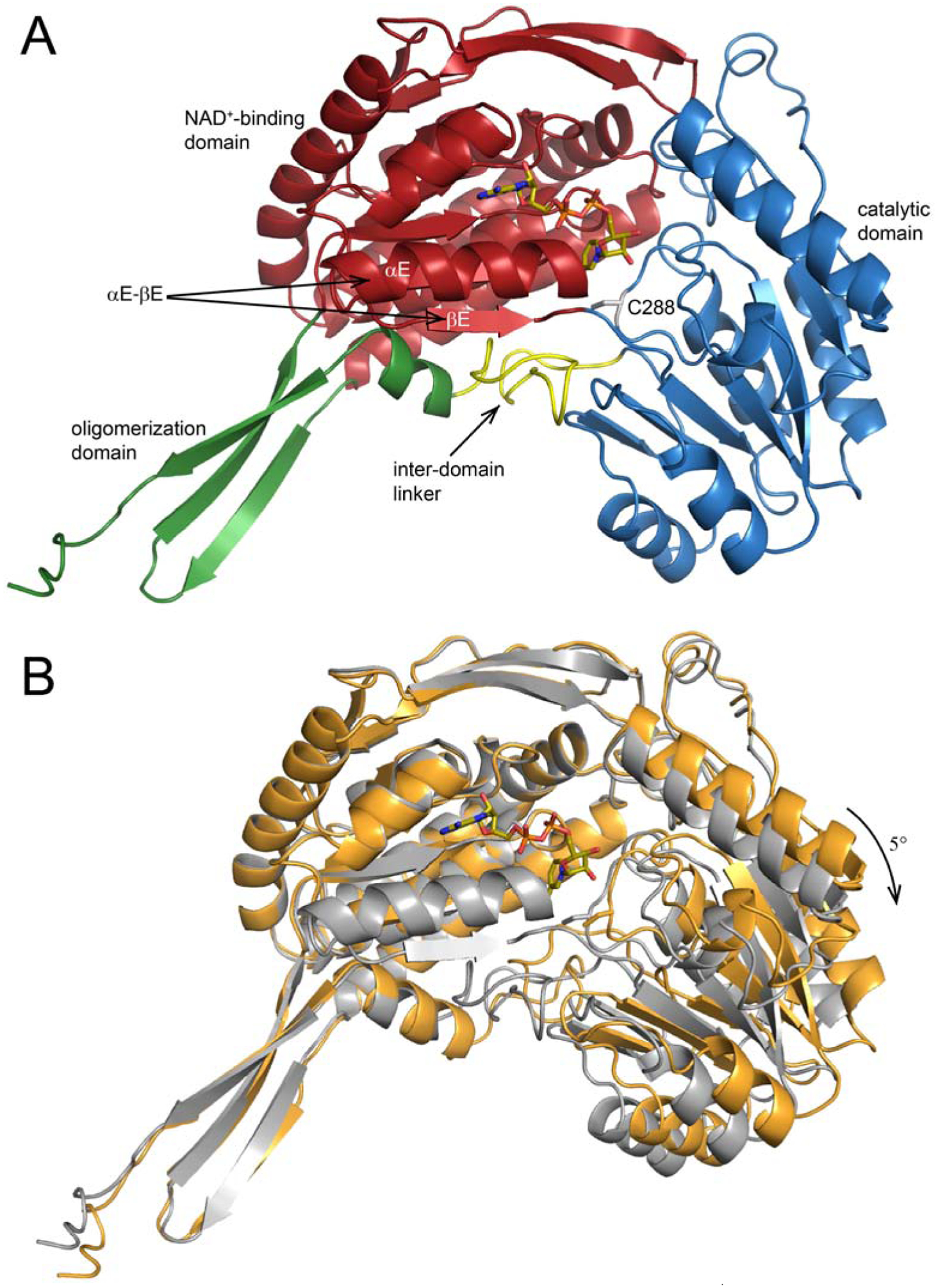
The crystal structure of P1 ALDH9A1 complexed with NAD+. (A) One protomer (chain A) of ALDH9A1 complexed with NAD+ in space group P1. The NAD+-binding, catalytic, and oligomerization domains have different colors. Arrows indicate αE-βE of the Rossmann fold domain and the β-hairpin inter-domain linker (yellow). NAD+ is shown in sticks. (B) Superposition of NAD+-bound (gray, PDB ID: 6VR6) and apo ALDH9A1 (gold, PDB ID: 6QAP). The superposition calculation was based on the NAD+-binding domains to accentuate the difference in the orientations of the catalytic domains. The arrow shows the direction of domain closure associated with NAD+ binding.
The structures of ALDH9A1-NAD+ were compared to each other and with the contents of the PDB using PDBeFold [39] to identify the closest structural neighbors. Table 2 lists the pairwise RMSD values between the chains of the various structures used in the analysis. Comparison of our P1 and C222 NAD+ structures yields RMSD values of 1.0 – 1.3 Å. This range is 3–5 times larger than the chain-chain RMSDs within either structure, suggesting that the two structures represent distinct conformations. This idea was supported by comparing our structures to the PDB. We found that the P1 ALDH9A1-NAD+ structure is most similar to betaine ALDH from cod liver (RMSD = 0.6 – 0.7), whereas the C222 ALDH9A1-NAD+ structure most resembles apo ALDH9A1 (RMSD = 0.4 – 0.6 Å).
Table 2.
Pairwise RMSDs between ALDH structures.a
| ALDH9A1-NAD+ (P1) | ALDH9A1-NAD+ (C222) | ALDH9A1 apo (6QAP) | Betaine ALDH apo (1A4S) | Betaine ALDH-NAD+ (1BPW) | |
|---|---|---|---|---|---|
| ALDH9A1-NAD+ (P1) | 0.00 – 0.24 | 1.01 – 1.26 | 1.35 – 1.51 | 0.65 – 0.69 | 0.62 – 0.65 |
| ALDH9A1-NAD+ (C222) | 0.00 – 0.28 | 0.44 – 0.58 | 1.27 – 1.36 | 1.21 – 1.30 | |
| ALDH9A1 apo (6QAP) | 0.00 – 0.24 | 1.46 – 1.56 | 1.41 – 1.51 | ||
| Betaine ALDH apo (1A4S) | 0.00 – 0.09 | 0.31 – 0.32 | |||
| Betaine ALDH-NAD+ (1BPW) | 0.00 – 0.00 |
Calculated with PDBeFold. The range was calculated from all pairwise chain-chain comparisons.
The RMSD data suggest that the current ALDH9A1 structures can be classified into two groups, one corresponding to the P1 NAD+ complex structure, and the other consisting of the C222 NAD+ complex structure and the apo structures. Additional analysis shows that the two groups differ by a domain rotation. For example, when the NAD+-binding domains of the P1 NAD+ complex and apo ALDH9A1 are superimposed, the catalytic domains differ by an apparent rigid body rotation of ~5° (Fig. 3B). Apparently, interactions of the enzyme with DEAB and NAD+ caused the NAD+-binding and catalytic domains to rotate closer together.
3.3. Conformation and interactions of NAD+ bound to ALDH9A1
Electron density for NAD+ was observed in all eight chains of the P1 asymmetric unit. In chain A, electron density was present for the entire cofactor (Fig. 4A), whereas in the other chains only the ADP portion could be modeled with confidence (Fig. S3 of Supplementary data). Similarly, only the AMP fragment could be modeled in the C222 structure (Fig. S4 of Supplementary data). We note that weak density for the nicotinamide riboside of NAD+ is common in ALDHs [40–44]. Electron density extending from catalytic Cys288 was present in all eight chains of the P1 structure, possibly indicating covalent modification by DEAB; however, we were not able to model DEAB covalently bound to Cys288 satisfactorily into these features. The lack of convincing density for the covalent adduct is perhaps consistent with the apparent reversibility of inactivation (Fig. 2).
Fig. 4.
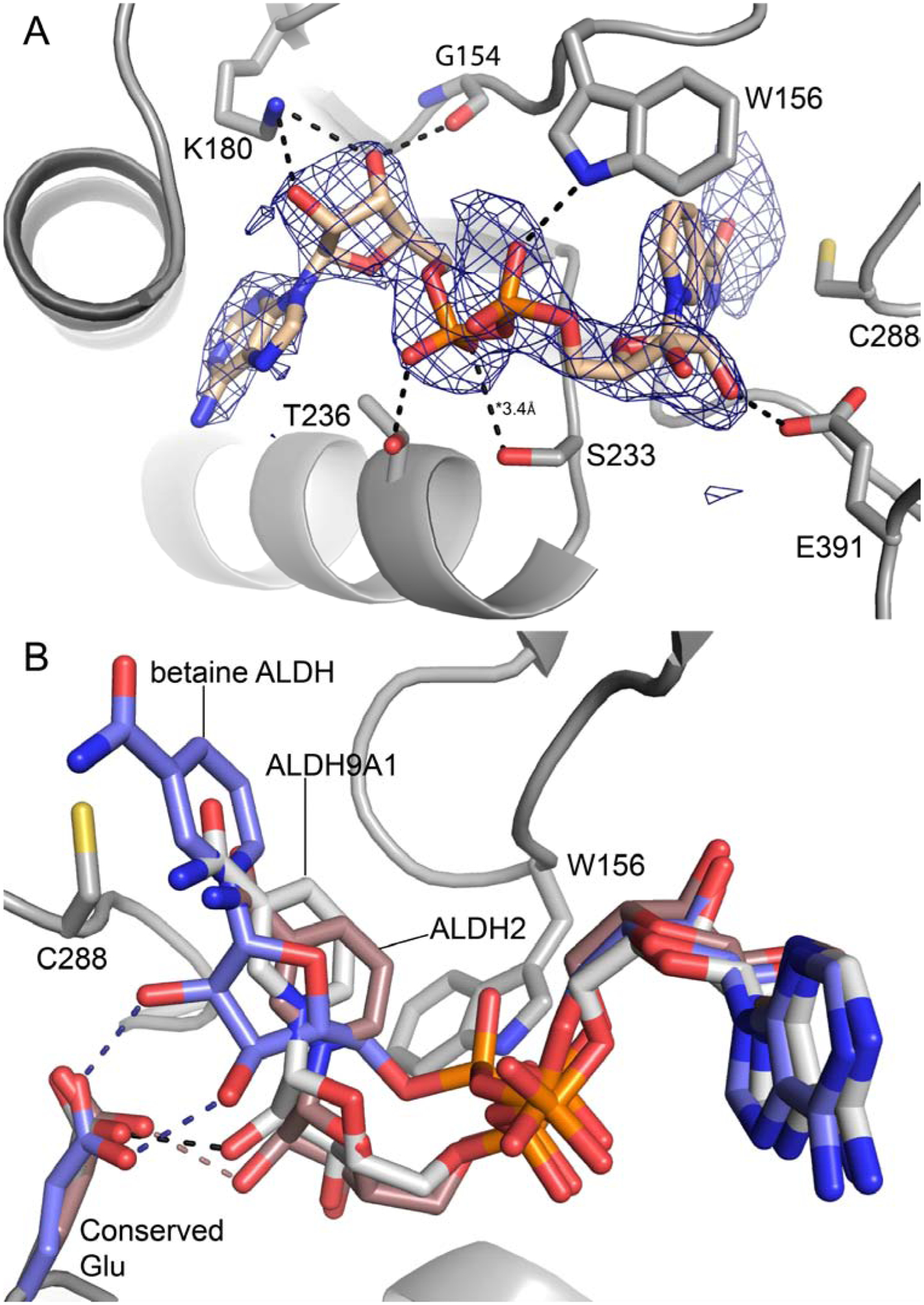
Conformation of NAD+ bound to chain A of the P1 ALDH9A1 (PDB ID: 6VR6). (A) Electron density and interactions for NAD+. The cage represents a polder omit map (3.5σ). The dashed lines indicate interactions less than 3.2 Å, except where noted. (B) Superposition of the cofactors of P1 ALDH9A1 (gray), betaine ALDH (blue, PDB ID: 1BPW), and ALDH2 (brown, PDB ID: 1O02). The betaine ALDH and ALDH2 structures show the “hydride transfer” and “hydrolysis” conformations, respectively. Hydrogen bonds with the conserved glutamate residue are colored as follows: black, ALDH9A1; blue, betaine ALDH; brown, ALDH2.
NAD+ binds in the expected site at the C-termini of the β-strands of the Rossmann fold. NAD+ forms several electrostatic interactions with the protein (Fig. 4A). The adenine ribose hydrogen bonds with Lys180. The pyrophosphate interacts with Trp156, Ser233, and Thr236. The nicotinamide ribose of the one complete NAD+ forms a hydrogen bond with Glu391, a residue identically conserved in the ALDH superfamily [45].
The conformation of the complete NAD+ (chain A of P1 structure) is unusual in that the nicotinamide riboside group is shifted by 3 – 4 Å out of the active site compared to the normal (a.k.a. hydride transfer) conformation (Fig. 4B). For example, a structure of betaine ALDH shows the hydride transfer conformation (Fig. 4B). The retracted pose in our structure is similar to the hydrolysis conformation described by the Hurley group in their studies of ALDH2 [40] (Fig. 4B). A key difference between the hydride transfer and hydrolysis conformations is that the former makes two hydrogen bonds to a conserved glutamate residue, whereas the latter makes only one (Fig. 4B). Note the NAD+ in our structure exhibits this feature of the hydrolysis conformation.
3.4. Conformations of the αE-βE region of the Rossmann fold and the inter-domain linker
The αE-βE region of the Rossmann fold (residues 232 – 258) exhibits a well-defined conformation in the P1 NAD+ complex, in contrast to the C222 NAD+-complex and apo enzyme structures in which these residues are disordered. The electron density is strong and continuous for the main chain, and most of the side chains also have clear electron density indicating their conformations (Fig. 5A). Residues 234 – 247 form an α-helix, and residues 251 – 254 form a β-strand (Fig. 5A); these are the final two secondary structural elements of the Rossmann dinucleotide binding fold. Comparison to betaine ALDH shows that the αE-βE region of P1 ALDH9A1-NAD+ adopts the canonical secondary and tertiary structure of the ALDH superfamily (Fig. 5B). Furthermore, the αE helices of the domain-swapped dimer interact across a molecular two-fold axis, as in other ALDHs (Fig. 5C).
Fig. 5.
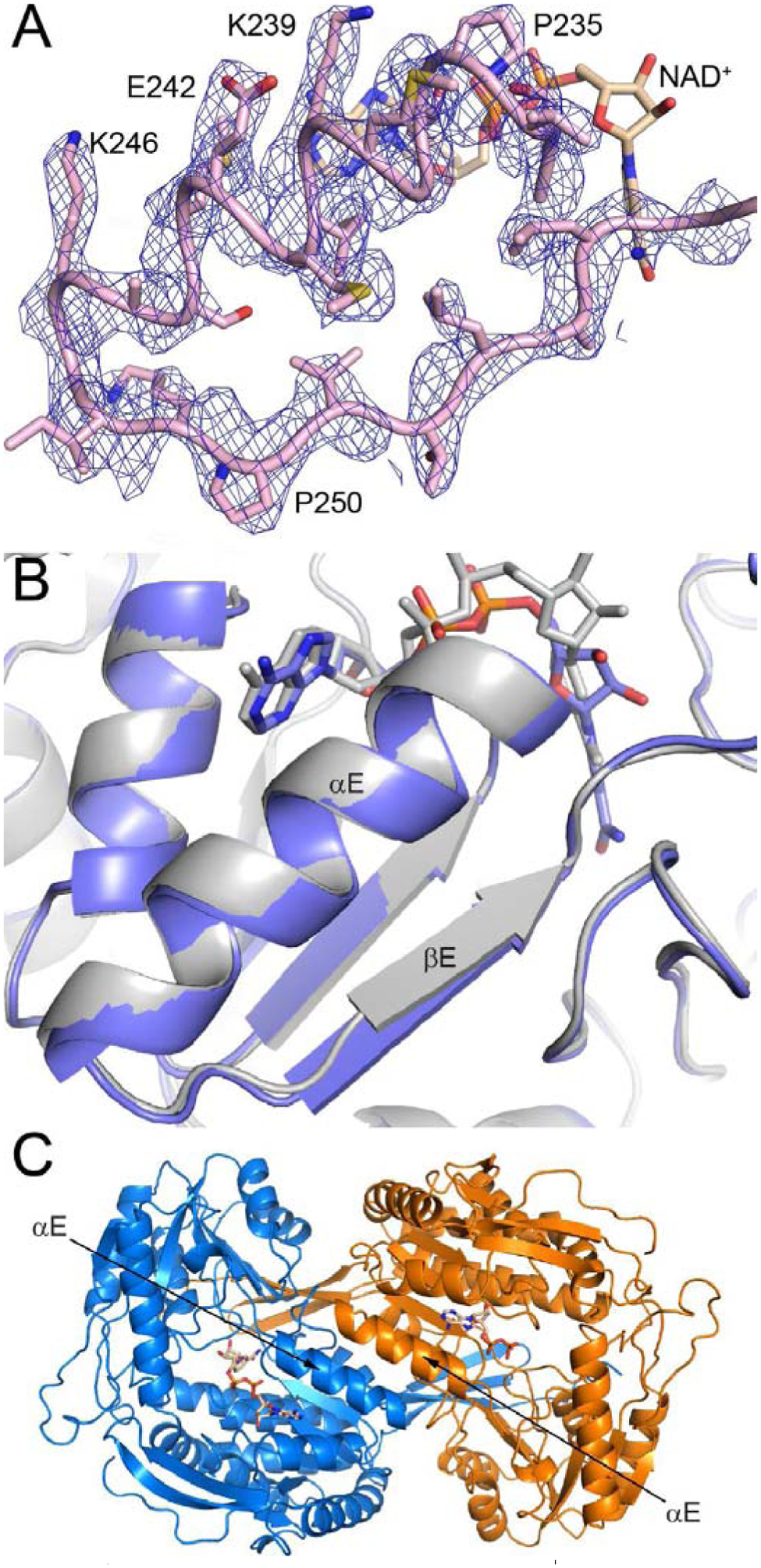
Conformation of αE-βE in the P1 ALDH9A1-NAD+ structure. (A) Electron density for αE-βE. The cage represents a refined 2Fo-Fc map (1σ). (B) Comparison of the αE-βE regions of P1 ALDH9A1-NAD+ (gray, PDB ID: 6VR6) and betaine ALDH (blue, PDB: 1BPW). (C) The domain-swapped dimer of ALDH9A1, viewed down the 2-fold axis that passes between the αE helixes of adjacent protomers. The two protomers have different colors.
The electron density maps also clearly defined the conformation of the inter-domain linker in the P1 NAD+ complex (residues 451 – 470) (Fig. 6A). This section of the polypeptide chain connects the catalytic and oligomerization domains, and forms part of the active site. The linker in our P1 structure adopts the β-hairpin structure typical of the ALDH superfamily, as shown by the comparison with betaine ALDH (Fig. 6B). Several interactions within the β-hairpin stabilize its conformation, including three main-chain hydrogen bonds and charged-hydrogen bonds between Arg467 and the carbonyls of Gly466 and Gly459. These interactions are also present in betaine ALDH. The canonical β-hairpin is very different from the S-shaped conformation in apo ALDH9A1 (Fig. 6C). We note that the electron density for the inter-domain linker in the C222 NAD+ complex is weak and discontinuous, but nevertheless consistent with the S-conformation.
Fig. 6.
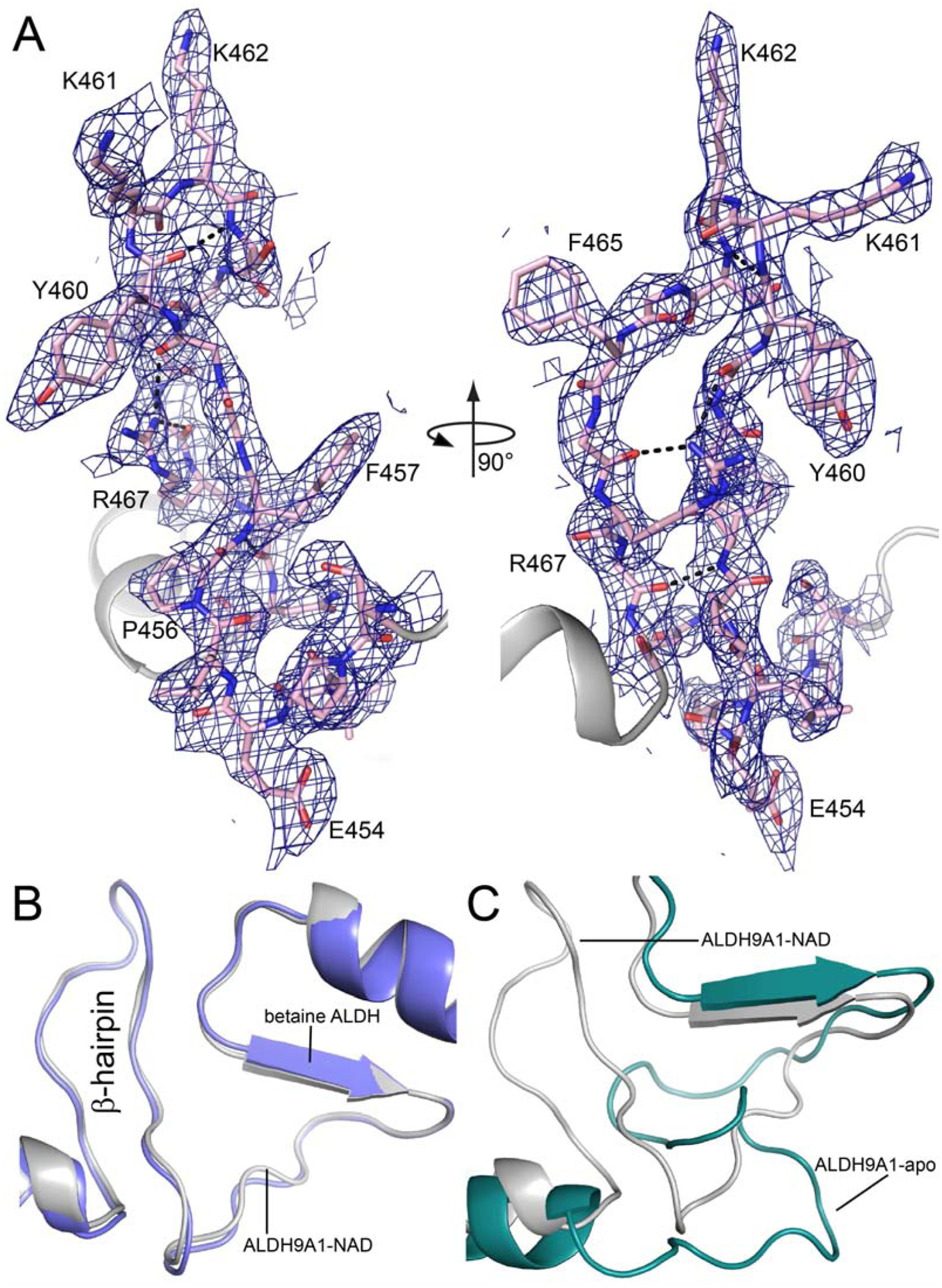
Conformation of the inter-domain linker in the P1 ALDH9A1-NAD+ structure. (A) Two views of electron density for the linker (refined 2Fo-Fc map at 1σ). (B) Comparison of the inter-domain linkers of P1 ALDH9A1-NAD+ (gray, PDB ID: 6VR6) and betaine ALDH (blue, PDB: 1BPW). (C) Comparison of the inter-domain linkers of P1 ALDH9A1-NAD+ (gray, PDB ID: 6VR6) and apo ALDH9A1 (green, PDB ID: 6QAP).
The ordering of αE-βE and the remodeling of the inter-domain linker result in the formation of new tertiary and quaternary structural interactions (Fig. 7). For example, βE makes three main chain hydrogen bonds with the inter-domain linker to form a two-stranded anti-parallel β-sheet structure (Fig. 7A). And the tip of the inter-domain linker forms several hydrogen bonds with the catalytic domain. The αE helices of adjacent protomers pack together to form a new dimer interface (Fig. 7B). This interface involves the nonpolar face of αE, which includes Met238, Met241, Ala245, and Ile248. Also, Phe465 of the linker intercalates between the βE strands of two adjacent protomers, forming intermolecular nonpolar contacts with Ile248 and Pro250 (Fig. 7B). In summary, the isomerization of the αE-βE region and the inter-domain linker into their canonical conformations results in the formation of many noncovalent interactions that stabilize the active form of ALDH9A1.
Fig. 7.
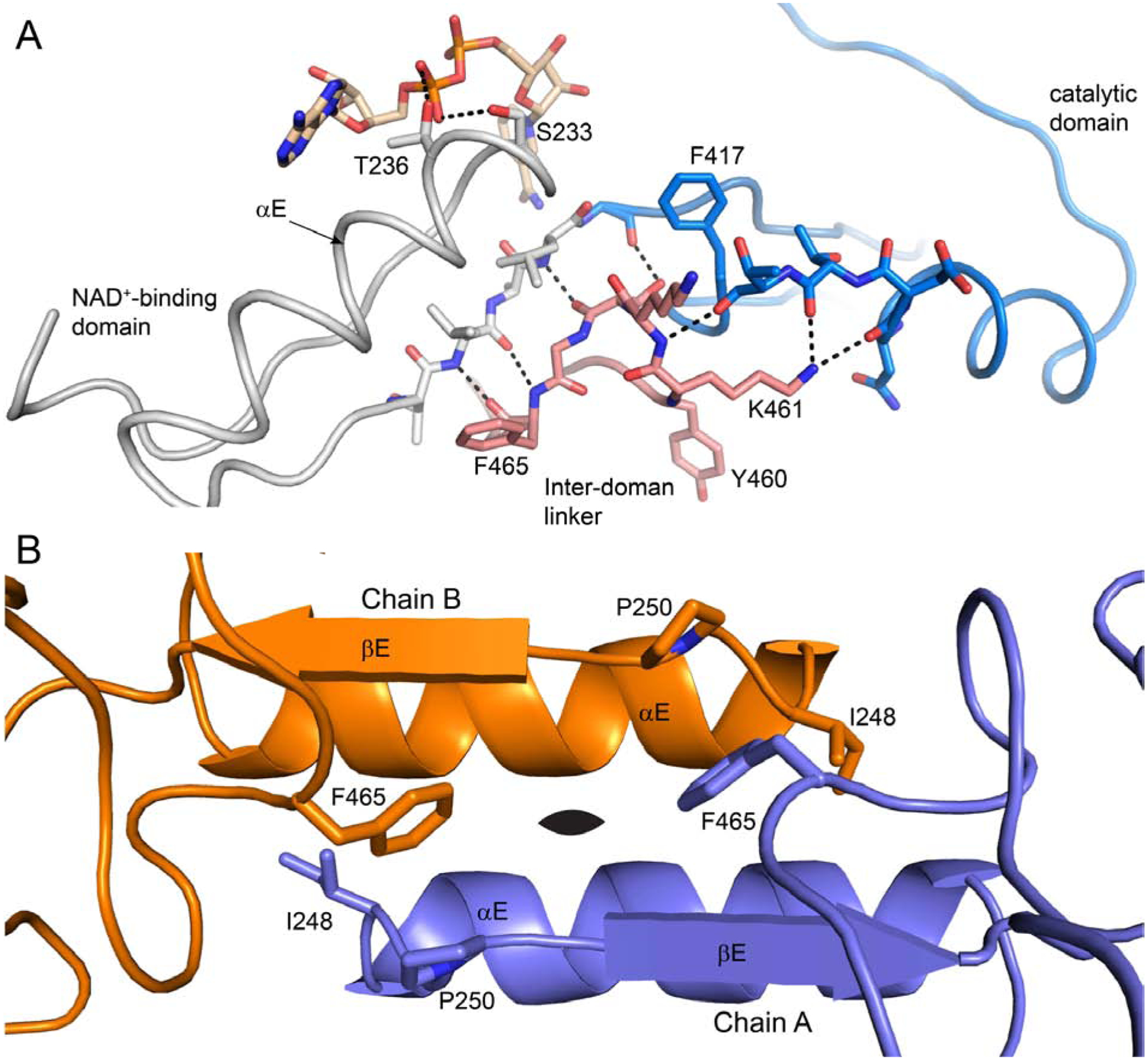
Interactions involving the αE-βE region and the inter-domain linker in the P1 ALDH9A1 structure. (A) Interaction of the inter-domain linker (pink) with αE-βE (gray) and the catalytic domain (blue). (B) The dimer interface formed by the αE-βE regions of adjacent protomers. The black oval denotes the 2-fold axis of the dimer.
3.5. Oligomeric structure of ALDH9A1
Sedimentation velocity analytical ultracentrifugation was used to determine the oligomeric state of ALDH9A1 in solution. The distribution of apparent sedimentation coefficients, c(s), of apo (NAD+-free) His-ALDH9A1 at 3 mg mL−1 revealed a single major peak at 6.8 S (Fig. 8). Likewise, the distribution of molecular masses, c(M), revealed a single major peak corresponding to a molecular mass of 223 kDa, which is within 1% of the molecular mass of the His-ALDH9A1 tetramer (theoretical molecular mass: 224.5 kDa). Thus, sedimentation velocity suggests apo His-ALDH9A1 is tetrameric in solution at 3 mg mL−1. This result is consistent with the observation of the classic dimer-of-dimers ALDH tetramer in both of our structures, as well as the crystal forms reported by Koncitikova et al. [7].
Fig. 8.

Sedimentation velocity analysis of apo ALDH9A1. The graph shows the distribution of molecular masses, c(M), (black curve), and the distribution of sedimentation coefficients, c(s) (red curve).
The in-solution quaternary structure of ALDH9A1 was determined using SAXS. Static SAXS data were collected at three nominal protein concentrations in the range of 1 – 5 mg mL−1, in the presence of 1 mM NAD+ (Fig. 9A) The average Rg from Guinier analysis of 37.9 ± 0.3 Å (Table 3) is in agreement with the SAXS Rg of 38 Å for the ALDH7A1 tetramer [43, 46] and the Rg of 35.5 Å calculated from the ALDH9A1 crystallographic tetramer. For each concentration, the theoretical SAXS curve generated from the crystallographic tetramer provided a reasonably good fit to the experimental data (Fig. 9A). Imposition of a multi-body fit to simulate a mixture of dimer and tetramer did not statistically improve the fits to the experimental data. Finally, the ab initio shape reconstruction shows good agreement with the tetramer (Fig. 9B). Overall, these data suggest ALDH9A1 is primarily tetrameric under the conditions used, consistent with the sedimentation velocity data.
Fig. 9.
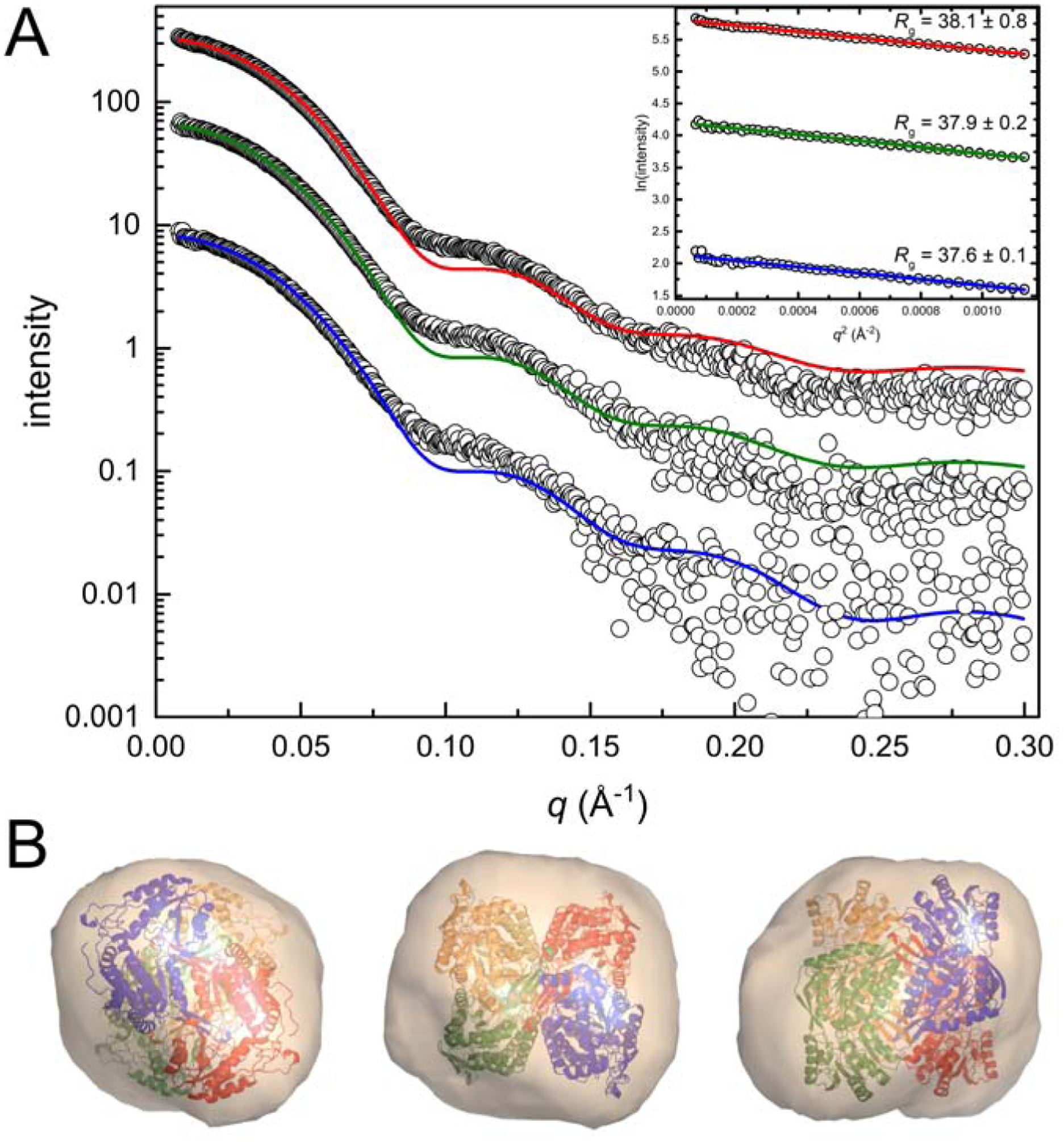
SAXS analysis of ALDH9A1 in the presence of 1 mM NAD+. (A) SAXS experimental data (open circles) and the corresponding FoXS single-body fit to the ALDH9A1 crystallographic tetramer (blue, green, and red curves) at three nominal protein concentrations. (B) Three views of the electron density ab initio shape reconstruction generated using DENSSWeb. The envelope is contoured to 1.0σ with the crystallographic tetramer of ALDH9A1 fit into the map using the “fit in map” utility of Chimera [66, 67].
Table 3.
ALDH9A1 solution structural parameters from SAXS
| Concentration (mg mL−1) | 1.25 | 2.5 | 5 |
| Guinier analysis | |||
| Points used | 1–47 | 1–48 | 1–48 |
| qRg range | 0.31–1.28 | 0.31–1.30 | 0.30–1.29 |
| Rg (Å) | 38.1 ± 0.8 | 37.9 ± 0.2 | 37.6 ± 0.1 |
| I(0) | 8.5 ± 0.1 | 16.68 ± 0.04 | 33.39 ± 0.06 |
| P(r) analysis | |||
| Dmax (Å) | 109 | 108 | 105 |
| Rg (Å) | 37.0 ± 0.5 | 37.3 ± 0.9 | 37.0 ± 0.6 |
| Porod volume (Å3) | 252,000 | 237,000 | 238,000 |
| Mass from SAXSMoW (kDa) | 203 | 182 | 167 |
| FoXS χ2 | 0.24 | 1.0 | 2.1 |
| SASBDB Code | SASDHV5 | SASDHW5 | SASDHX5 |
The quaternary structure of apo ALDH9A1 in solution was confirmed with negative stain electron microscopy (Fig. 10). The micrographs exhibited good contrast and particle separation (Fig. 10A). The 2D class averages calculated from 7430 particles are shown in Fig. 10B, and for comparison, views of the ALDH9A1 tetramer and dimer are shown in Fig. 10C. Most of the 2D classes appear to resemble the tetramer viewed down one of its three mutually orthogonal 2-fold axes (classes 1, 4, 6–10). In these classes, which account for ~80% of the particles, the four subunits of the tetramer and a two-fold symmetry axis perpendicular to the page are apparent. Classes 3 and 5 (16% of the particles) appear to represent the tetramer rotated by 45° around a 2-fold axis. Class 2 may represent a dimer viewed down its 2-fold axis. Note the dimer represents only 4% of the particles. Thus, apo ALDH9A1 is predominantly tetrameric under the conditions used for negative stain EM. In summary, the conclusion from multiple biophysical measurements - X-ray crystallography, sedimentation velocity, SAXS, and electron microscopy - is that ALDH9A1 exists in solution primarily as the classic ALDH superfamily dimer-of-dimers tetramer under the conditions used.
Fig. 10.

Negative-stain EM analysis of apo ALDH9A1. (A) Representative micrograph. (B) 2D class averages calculated from 7430 particles. (C) Surface representations of the ALDH9A1 tetramer viewed in various orientations relative to a reference orientation, and a dimer viewed down its 2-fold axis.
4. Discussion
The new structures described here provide insight into the mechanism by which ALDH9A1 is activated for catalysis. The inactive conformation first observed in the apo enzyme is characterized by disorder of αE-βE and a novel S-shaped inter-domain linker [7]. Because these atypical features are also present in our C222 NAD+-complex structure, it appears that NAD+-binding is not sufficient to activate the enzyme. Instead, we found that treatment of ALDH9A1 with DEAB and the cofactor NAD+ apparently induced αE-βE and the inter-domain linker to assume their canonical conformations, as observed in the P1 structure. We conclude that the presence of an aldehyde substrate and NAD+ promotes isomerization of the enzyme into the active conformation.
The disorder-order transitions discussed here are not unique to ALDH9A1. Disorder in the interdomain linker has also been observed in ALDH1A2 (PDB ID: 1BI9) [47], ALDH4A1 (PDB IDs: 4OE5/6) [42], and ALDH7A1 (PDB IDs: 4ZVX/Y) [43]. In ALDH1A2, ordering of the linker is thought to be related to aldehyde binding, similar to ALDH9A1 [48]. In ALDH4A1 and ALDH7A1, no clear connection between ligand binding and disorder-order transitions has been established. Disorder in αE-βE is rarer. The best example is a naturally-occurring disease variant of ALDH2 (ALDH2*2) in which Glu487 is mutated to Lys (PDB ID: 1ZUM) [49]. Glu487 in wild-type ALDH2 forms intersubunit ion pairs with two arginine residues, one located on βE (Arg264), and another in the interdomain linker (Arg475). The introduction of Lys487 obviously disrupts these stabilizing interactions, resulting in substantial disorder in both αE-βE and the interdomain linker. ALDH9A1 does not have an analogous ionic interaction network, due to the substitution of a glutamine for Glu487, and perhaps more importantly, a proline in place of Arg264. The absence of these interactions may explain the propensity for disorder in ALDH9A1. On the other hand, the close ALDH9A1 homolog, betaine ALDH, also lacks the cross-subunit ion pairs, yet it exhibits canonical αE-βE and interdomain linker conformations, even in the absence of bound ligands (PDB ID: 1A4S). Although the mechanisms of structural ordering in ALDHs remain to be determined, the observation of large swaths of disorder in the active sites of multiple ALDHs suggests disorder-order transitions may be an inherent feature of the ALDH superfamily fold and important for catalytic function.
The NAD+ in our P1 structure unexpectedly has a retracted pose (Fig. 4). Two conformations of the cofactor bound to ALDHs have been described: “hydride transfer” and “hydrolysis”. These designations refer to steps in the catalytic mechanism. NAD+ tends to bind in the hydride transfer conformation, which positions the hydride acceptor of NAD+ (C4 atom) ~3 Å from the S-atom of the catalytic cysteine, ready to accept a hydride ion from the thiohemiacetal intermediate. Following hydride transfer and formation of the acyl-enzyme intermediate, the newly formed NADH retracts from the active site into the hydrolysis conformation. This movement allows room for a water molecule to enter the active site for hydrolysis of the acyl-enzyme intermediate. NAD+ in crystal structures typically adopts the hydride transfer pose; however, NAD+ has also been observed in the hydrolysis pose in ALDH1 (PDB ID: 1BXS) [50] and ALDH2 (PDB ID: 1O00) [40]. Also, mutation of the conserved glutamate that hydrogen bonds to the nicotinamide ribose in ALDH7A1 causes NAD+ to switch to a hydrolysis-like pose (PDB ID: 6O4L) [51]. NADH bound to ALDH2 adopts the mechanistically-expected hydrolysis pose (PDB ID: 1O02) [40]. On the other hand, a structure of ALDH7A1 shows NADH in the hydride transfer pose (PDB ID: 2J6L) [52]. Altogether, the crystal structures of ALDHs provide evidence of the flexibility of the bound cofactor, which is essential for catalysis. Why NAD+ in our structure is retracted into a hydrolysis-like conformation is unclear. It is possible that covalent modification of the catalytic cysteine by DEAB, which is suggested by the electron density but could not be modeled with certainty, prevents NAD+ from moving into the hydride transfer conformation.
We showed that ALDH9A1 forms the classic ALDH superfamily dimer-of-dimers tetramer in solution. This is not surprising considering that the closest sequence homologs of ALDH9A1 in the PDB also form the tetramer (at least in crystallo), including betaine ALDHs (40 – 70% identical [8, 53]) and ALDH2 (43% identical).
ALDH9A1 may be added to the list of ALDHs that are inhibited by DEAB. This information is pertinent to studies that use the ALDEFLUOR flow cytometry assay to detect cancer stem cells based on ALDH activity (ALDH-bright cells) [54–58]. DEAB is used in the control arm of the ALDEFLUOR assay, and any ALDHs that are reversibly inhibited or covalently inactivated by DEAB will contribute to a cell being labeled as ALDH-bright and thus classified as a cancer stem cell. DEAB was once thought to be specific for ALDH1A1, which led researchers to assume that the ALDEFLUOR assay was specific for the detection of ALDH1A1. Recent work has proven this assumption incorrect, as DEAB inhibits or inactivates several ALDHs including ALDH1A1, ALDH1A3, ALDH1B1, ALDH5A1, ALDH1A2, ALDH2, and ALDH7A1 [10, 36, 59].
Hurley’s group proposed a general mechanism for the inhibition of ALDHs by DEAB. They proposed that DEAB functions as a substrate through the hydride transfer step, but that the acyl-enzyme intermediate is protected against hydrolysis by resonance stabilization [10]. The extent of resonance stabilization, and therefore the potency of inhibition, depends on the noncovalent interactions between the acyl-enzyme and the active site, which varies among ALDH isozymes. For example, the active sites of ALDH1A2, ALDH2 and ALDH7A1 apparently provide exceptional stabilization, since DEAB is a covalent irreversible inhibitor of these enzymes [10, 36]. For ALDH1A1, ALDH1A3, ALDH1B1, and ALDH5A1, DEAB inhibits by virtue of being a slow substrate; the active sites of these enzymes apparently provide less efficient stabilization of the acyl-enzyme than in ALDH1A2, ALDH2 and ALDH7A1.
We found that the inhibition of ALDH9A1 by DEAB is time-dependent, consistent with covalent inactivation. In contrast to ALDH2 and ALDH7A1, the inhibition was diminished by jump-dilution and abrogated by removal of excess DEAB, consistent with a covalent, reversible mechanism. The irreversible inactivation of ALDH2 and ALDH7A1 by DEAB in the presence of NAD+ is due to formation of the acyl-enzyme [10, 36]. In principle, covalent reversible inhibition of an ALDH can arise by slow hydrolysis of the acyl-enzyme intermediate to generate active enzyme and diethylaminobenzoic acid. This is not likely to be the case for ALDH9A1, given the rapid return of enzyme activity upon removal of DEAB coupled with the fact that we find no evidence that DEAB serves as a substrate for this enzyme (see Fig. 1A). A more plausible mechanism is formation of a reversible thiohemiacetal covalent intermediate (Scheme 2A) that fails to progress through the hydride transfer step of the normal catalytic mechanism that would generate the acyl-enzyme intermediate. The covalent, reversible inhibition due to thiohemiacetal formation with an active site cysteine residue proposed here finds a direct mechanistic analogy in the inhibition cysteine proteases by aldehydes [60–62] and is mechanistically related to the inhibition of viral proteases by ketoamide compounds that form a reversible, covalent tetrahedral species with the catalytic serine [63–65]. This mechanism (Scheme 2A) could also explain the covalent, reversible inhibition of ALDH1A2 by DEAB in the absence of NAD+ [10].
The progress curves in Figure 1B and the reversibility of inhibition are also consistent with slow, tight-binding inhibition (Scheme 2B) [13]. In this mechanism, inhibitor binding could involve the rapid formation of an initial collision complex (E·I) that subsequently undergoes a slow isomerization reaction to E·I* (with or without formation of the thiohemiacetal covalent intermediate). Presumably the initial encounter would involve the apo, inactive enzyme, followed by an isomerization step resulting in an enzyme conformation similar to the P1 structure described here. Isomerization between these two conformations likely would be quite slow (seconds to minutes), as it involves very large structural changes, including folding of the αE-βE region and reorganization of the interdomain linker.
Supplementary Material
Highlights.
Diethylaminobenzaldehyde is a time-dependent, reversible inhibitor of ALDH9A1
Two crystal structures of ALDH9A1 complexed with NAD+
First crystal structure of the active conformation of ALDH9A1
ALDH9A1 forms the classic ALDH superfamily dimer-of-dimers tetramer in solution
Acknowledgements
We thank K. Burnett for collecting the SAXS data through the SIBYLS mail-in program, J. Schuermann for help with data collection at beamline 24-ID-E, J. Nix for help with data collection at beamline 4.2.2, and T. White for help with EM data collection. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16M detector on 24-ID-E beam line is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. Part of this work was conducted at the Advanced Light Source (ALS), a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the Department of Energy, Office of Basic Energy Sciences, through the Integrated Diffraction Analysis Technologies (IDAT) program, supported by DOE Office of Biological and Environmental Research. Additional support comes from the National Institute of Health project ALS-ENABLE (P30 GM124169) and a High-End Instrumentation Grant S10OD018483.
Funding
Research reported in this publication was supported by the NIGMS of the National Institutes of Health under award number R01GM093123 (to J.J.T.), and an administrative supplement to NIH grant R01GM065546 (to J.J.T., Collaborative Supplements for Cryo-Electron Microscopy Technology Transfer). I.A.Q. was supported by an ICMR International Fellowship from the Indian Council of Medical Research, India.
Abbreviations:
- ALDH
aldehyde dehydrogenase
- ALDH9A1
aldehyde dehydrogenase 9A1
- DEAB
diethylaminobenzaldehyde
- IMAC
immobilized metal-affinity chromatography
- SAXS
small-angle X-ray scattering
- SEC
size-exclusion chromatography
- TCEP
tris(2-carboxyethyl)phosphine
- TMBAL
4-trimethylaminobutyraldehyde
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Databases: Coordinates and structure factor amplitudes have been deposited in the Protein Data Bank under accession codes 6VR6 and 6VWF. SAXS data sets have been deposited in the Small-Angle Scattering Biological Data Bank under the following accession codes: SASDHV5, SASDHW5, and SASDHX5.
Conflict of Interest: The authors declare no competing financial interest.
References
- [1].Sophos NA, Vasiliou V, Aldehyde dehydrogenase gene superfamily: the 2002 update, Chem. Biol. Interact 143–144 (2003) 5–22. [DOI] [PubMed] [Google Scholar]
- [2].Koppaka V, Thompson DC, Chen Y, Ellermann M, Nicolaou KC, Juvonen RO, Petersen D, Deitrich RA, Hurley TD, Vasiliou V, Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application, Pharmacol. Rev 64(3) (2012) 520–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Vaz FM, Fouchier SW, Ofman R, Sommer M, Wanders RJ, Molecular and biochemical characterization of rat gamma-trimethylaminobutyraldehyde dehydrogenase and evidence for the involvement of human aldehyde dehydrogenase 9 in carnitine biosynthesis, J Biol Chem 275(10) (2000) 7390–4. [DOI] [PubMed] [Google Scholar]
- [4].Kurys G, Ambroziak W, Pietruszko R, Human aldehyde dehydrogenase. Purification and characterization of a third isozyme with low Km for gamma-aminobutyraldehyde, J Biol Chem 264(8) (1989) 4715–21. [PubMed] [Google Scholar]
- [5].Ambroziak W, Pietruszko R, Human aldehyde dehydrogenase. Activity with aldehyde metabolites of monoamines, diamines, and polyamines, J Biol Chem 266(20) (1991) 13011–8. [PubMed] [Google Scholar]
- [6].Chern MK, Pietruszko R, Human aldehyde dehydrogenase E3 isozyme is a betaine aldehyde dehydrogenase, Biochemical and biophysical research communications 213(2) (1995) 561–8. [DOI] [PubMed] [Google Scholar]
- [7].Koncitikova R, Vigouroux A, Kopecna M, Sebela M, Morera S, Kopecny D, Kinetic and structural analysis of human ALDH9A1, Bioscience reports 39(4) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Johansson K, El-Ahmad M, Ramaswamy S, Hjelmqvist L, Jornvall H, Eklund H, Structure of betaine aldehyde dehydrogenase at 2.1 A resolution, Protein Sci 7(10) (1998) 2106–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A, Protein identification and analysis tools on ExPASy server, Humana Press, Totowa, NJ, 2005. [Google Scholar]
- [10].Morgan CA, Parajuli B, Buchman CD, Dria K, Hurley TD, N,N-diethylaminobenzaldehyde (DEAB) as a substrate and mechanism-based inhibitor for human ALDH isoenzymes, Chem Biol Interact 234 (2015) 18–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Juncosa JI, Takaya K, Le HV, Moschitto MJ, Weerawarna PM, Mascarenhas R, Liu D, Dewey SL, Silverman RB, Design and Mechanism of (S)-3-Amino-4-(difluoromethylenyl)cyclopent-1-ene-1-carboxylic Acid, a Highly Potent gamma-Aminobutyric Acid Aminotransferase Inactivator for the Treatment of Addiction, J Am Chem Soc 140(6) (2018) 2151–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Salminen KA, Leppanen J, Venalainen JI, Pasanen M, Auriola S, Juvonen RO, Raunio H, Simple, direct, and informative method for the assessment of CYP2C19 enzyme inactivation kinetics, Drug Metab Dispos 39(3) (2011) 412–8. [DOI] [PubMed] [Google Scholar]
- [13].Morrison JF, Walsh CT, The behavior and significance of slow-binding enzyme inhibitors, Adv Enzymol Relat Areas Mol Biol 61 (1988) 201–301. [DOI] [PubMed] [Google Scholar]
- [14].D’Arcy A, Bergfors T, Cowan-Jacob SW, Marsh M, Microseed matrix screening for optimization in protein crystallization: what have we learned?, Acta Crystallogr F Struct Biol Commun 70(Pt 9) (2014) 1117–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Till M, Robson A, Byrne MJ, Nair AV, Kolek SA, Shaw Stewart PD, Race PR, Improving the success rate of protein crystallization by random microseed matrix screening, J Vis Exp (78) (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kabsch W, XDS, Acta Crystallogr D Biol Crystallogr 66(Pt 2) (2010) 125–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Evans PR, Murshudov GN, How good are my data and what is the resolution?, Acta Crystallogr. D Biol. Crystallogr 69(Pt 7) (2013) 1204–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Matthews BW, Solvent content of protein crystals, J. Mol. Biol 33 (1968) 491–497. [DOI] [PubMed] [Google Scholar]
- [19].Vagin A, Teplyakov A, MOLREP: an automated program for molecular replacement, J. Appl. Crystallogr 30 (1997) 1022–1025. [Google Scholar]
- [20].Stein N, CHAINSAW: a program for mutating pdb files used as templates in molecular replacement, J. Appl. Crystallogr 41(3) (2008) 641–643. [Google Scholar]
- [21].McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, Phaser crystallographic software, J. Appl. Crystallogr 40(Pt 4) (2007) 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Afonine PV, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH, Adams PD, Towards automated crystallographic structure refinement with phenix.refine, Acta Crystallogr. D Biol. Crystallogr 68(Pt 4) (2012) 352–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Emsley P, Lohkamp B, Scott WG, Cowtan K, Features and development of Coot, Acta Crystallogr D Biol Crystallogr 66(Pt 4) (2010) 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr. D Biol. Crystallogr D66(Pt 1) (2010) 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Gore S, Sanz Garcia E, Hendrickx PMS, Gutmanas A, Westbrook JD, Yang H, Feng Z, Baskaran K, Berrisford JM, Hudson BP, Ikegawa Y, Kobayashi N, Lawson CL, Mading S, Mak L, Mukhopadhyay A, Oldfield TJ, Patwardhan A, Peisach E, Sahni G, Sekharan MR, Sen S, Shao C, Smart OS, Ulrich EL, Yamashita R, Quesada M, Young JY, Nakamura H, Markley JL, Berman HM, Burley SK, Velankar S, Kleywegt GJ, Validation of Structures in the Protein Data Bank, Structure 25(12) (2017) 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Cole JL, Lary JW, T PM, Laue TM , Analytical ultracentrifugation: sedimentation velocity and sedimentation equilibrium, Methods Cell Biol 84 (2008) 143–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Schuck P, Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling, Biophys. J 78(3) (2000) 1606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dyer KN, Hammel M, Rambo RP, Tsutakawa SE, Rodic I, Classen S, Tainer JA, Hura GL, High-throughput SAXS for the characterization of biomolecules in solution: a practical approach, Methods Mol Biol 1091 (2014) 245–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Konarev PV, Volkov VV, Sokolova AV, Koch MHJ, Svergun DI, IUCr PRIMUS: a Windows PC-based system for small-angle scattering data analysis, Journal of applied crystallography 36(5) (2003) 1277–1282. [Google Scholar]
- [30].Svergun DI, IUCr, Determination of the regularization parameter in indirect-transform methods using perceptual criteria, Journal of applied crystallography 25(4) (1992) 495–503. [Google Scholar]
- [31].Piiadov V, Ares de Araujo E, Oliveira Neto M, Craievich AF, Polikarpov I, SAXSMoW 2.0: Online calculator of the molecular weight of proteins in dilute solution from experimental SAXS data measured on a relative scale, Protein Sci 28(2) (2019) 454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Schneidman-Duhovny D, Hammel M, Tainer JA, Sali A, FoXS, FoXSDock and MultiFoXS: Single-state and multi-state structural modeling of proteins and their complexes based on SAXS profiles., Nucleic Acids Res 44(W1) (2016) W424–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Grant TD, Ab initio electron density determination directly from solution scattering data, Nat Methods 15(3) (2018) 191–193. [DOI] [PubMed] [Google Scholar]
- [34].Rames M, Yu Y, Ren G, Optimized negative staining: a high-throughput protocol for examining small and asymmetric protein structure by electron microscopy, J Vis Exp (90) (2014) e51087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zivanov J, Nakane T, Forsberg BO, Kimanius D, Hagen WJ, Lindahl E, Scheres SH, New tools for automated high-resolution cryo-EM structure determination in RELION-3, Elife 7 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Luo M, Gates KS, Henzl MT, Tanner JJ, Diethylaminobenzaldehyde is a covalent, irreversible inactivator of ALDH7A1, ACS Chem. Biol 10(3) (2015) 693–7. [DOI] [PubMed] [Google Scholar]
- [37].Pandhare J, Dash C, Rao M, Deshpande V, Slow tight binding inhibition of proteinase K by a proteinaceous inhibitor: conformational alterations responsible for conferring irreversibility to the enzyme-inhibitor complex, J Biol Chem 278(49) (2003) 48735–44. [DOI] [PubMed] [Google Scholar]
- [38].Dash C, Vathipadiekal V, George SP, Rao M, Slow-tight binding inhibition of xylanase by an aspartic protease inhibitor: kinetic parameters and conformational changes that determine the affinity and selectivity of the bifunctional nature of the inhibitor, J Biol Chem 277(20) (2002) 17978–86. [DOI] [PubMed] [Google Scholar]
- [39].Krissinel E, Henrick K, Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions, Acta Crystallogr. D Biol. Crystallogr 60(Pt 12 Pt 1) (2004) 2256–68. [DOI] [PubMed] [Google Scholar]
- [40].Perez-Miller SJ, Hurley TD, Coenzyme isomerization is integral to catalysis in aldehyde dehydrogenase, Biochemistry 42(23) (2003) 7100–9. [DOI] [PubMed] [Google Scholar]
- [41].Srivastava D, Singh RK, Moxley MA, Henzl MT, Becker DF, Tanner JJ, The three-dimensional structural basis of type II hyperprolinemia, J Mol Biol 420(3) (2012) 176–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Pemberton TA, Srivastava D, Sanyal N, Henzl MT, Becker DF, Tanner JJ, Structural Studies of Yeast Delta(1)-Pyrroline-5-carboxylate Dehydrogenase (ALDH4A1): Active Site Flexibility and Oligomeric State, Biochemistry 53(8) (2014) 1350–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Luo M, Tanner JJ, Structural basis of substrate recognition by aldehyde dehydrogenase 7A1, Biochemistry 54(35) (2015) 5513–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Korasick DA, Koncitikova R, Kopecna M, Hajkova E, Vigouroux A, Morera S, Becker DF, Sebela M, Tanner JJ, Kopecny D, Structural and Biochemical Characterization of Aldehyde Dehydrogenase 12, the Last Enzyme of Proline Catabolism in Plants, J Mol Biol 431(3) (2019) 576–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Perozich J, Nicholas H, Wang BC, Lindahl R, Hempel J, Relationships within the aldehyde dehydrogenase extended family, Protein Sci 8(1) (1999) 137–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Korasick DA, White TA, Chakravarthy S, Tanner JJ, NAD(+) promotes assembly of the active tetramer of aldehyde dehydrogenase 7A1, FEBS Lett 592(19) (2018) 3229–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Lamb AL, Newcomer ME, The structure of retinal dehydrogenase type II at 2.7 A resolution: implications for retinal specificity, Biochemistry 38(19) (1999) 6003–11. [DOI] [PubMed] [Google Scholar]
- [48].Bordelon T, Montegudo SK, Pakhomova S, Oldham ML, Newcomer ME, A disorder to order transition accompanies catalysis in retinaldehyde dehydrogenase type II, J. Biol. Chem 279(41) (2004) 43085–91. [DOI] [PubMed] [Google Scholar]
- [49].Larson HN, Weiner H, Hurley TD, Disruption of the coenzyme binding site and dimer interface revealed in the crystal structure of mitochondrial aldehyde dehydrogenase “Asian” variant, J. Biol. Chem 280(34) (2005) 30550–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Moore SA, Baker HM, Blythe TJ, Kitson KE, Kitson TM, Baker EN, Sheep liver cytosolic aldehyde dehydrogenase: the structure reveals the basis for the retinal specificity of class 1 aldehyde dehydrogenases, Structure 6(12) (1998) 1541–51. [DOI] [PubMed] [Google Scholar]
- [51].Laciak AR, Korasick DA, Gates KS, Tanner JJ, Structural analysis of pathogenic mutations targeting Glu427 of ALDH7A1, the hot spot residue of pyridoxine-dependent epilepsy, Journal of inherited metabolic disease (2019) Epub 2019/10/28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Brocker C, Lassen N, Estey T, Pappa A, Cantore M, Orlova VV, Chavakis T, Kavanagh KL, Oppermann U, Vasiliou V, Aldehyde dehydrogenase 7A1 (ALDH7A1) is a novel enzyme involved in cellular defense against hyperosmotic stress, J. Biol. Chem 285(24) (2010) 18452–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Gonzalez-Segura L, Rudino-Pinera E, Munoz-Clares RA, Horjales E, The crystal structure of a ternary complex of betaine aldehyde dehydrogenase from Pseudomonas aeruginosa Provides new insight into the reaction mechanism and shows a novel binding mode of the 2’-phosphate of NADP+ and a novel cation binding site, J Mol Biol 385(2) (2009) 542–57. [DOI] [PubMed] [Google Scholar]
- [54].Storms RW, Trujillo AP, Springer JB, Shah L, Colvin OM, Ludeman SM, Smith C, Isolation of primitive human hematopoietic progenitors on the basis of aldehyde dehydrogenase activity, Proc Natl Acad Sci U S A 96(16) (1999) 9118–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhou L, Sheng D, Wang D, Ma W, Deng Q, Deng L, Liu S, Identification of cancer-type specific expression patterns for active aldehyde dehydrogenase (ALDH) isoforms in ALDEFLUOR assay, Cell Biol Toxicol 35(2) (2019) 161–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Mele L, Liccardo D, Tirino V, Evaluation and Isolation of Cancer Stem Cells Using ALDH Activity Assay, Methods Mol Biol 1692 (2018) 43–48. [DOI] [PubMed] [Google Scholar]
- [57].Marcato P, Dean CA, Giacomantonio CA, Lee PW, Aldehyde dehydrogenase: its role as a cancer stem cell marker comes down to the specific isoform, Cell Cycle 10(9) (2011) 1378–84. [DOI] [PubMed] [Google Scholar]
- [58].Pors K, Moreb JS, Aldehyde dehydrogenases in cancer: an opportunity for biomarker and drug development?, Drug Discov Today 19(12) (2014) 1953–63. [DOI] [PubMed] [Google Scholar]
- [59].Moreb JS, Ucar D, Han S, Amory JK, Goldstein AS, Ostmark B, Chang LJ, The enzymatic activity of human aldehyde dehydrogenases 1A2 and 2 (ALDH1A2 and ALDH2) is detected by Aldefluor, inhibited by diethylaminobenzaldehyde and has significant effects on cell proliferation and drug resistance, Chem. Biol. Interact 195(1) (2012) 52–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Lewis CA Jr., Wolfenden R, Thiohemiacetal formation by inhibitory aldehydes at the active site of papain, Biochemistry 16(22) (1977) 4890–5. [DOI] [PubMed] [Google Scholar]
- [61].Otto HH, Schirmeister T, Cysteine Proteases and Their Inhibitors, Chemical reviews 97(1) (1997) 133–172. [DOI] [PubMed] [Google Scholar]
- [62].Dufour E, Storer AC, Menard R, Peptide aldehydes and nitriles as transition state analog inhibitors of cysteine proteases, Biochemistry 34(28) (1995) 9136–43. [DOI] [PubMed] [Google Scholar]
- [63].Perni RB, Almquist SJ, Byrn RA, Chandorkar G, Chaturvedi PR, Courtney LF, Decker CJ, Dinehart K, Gates CA, Harbeson SL, Heiser A, Kalkeri G, Kolaczkowski E, Lin K, Luong YP, Rao BG, Taylor WP, Thomson JA, Tung RD, Wei Y, Kwong AD, Lin C, Preclinical profile of VX-950, a potent, selective, and orally bioavailable inhibitor of hepatitis C virus NS3–4A serine protease, Antimicrob Agents Chemother 50(3) (2006) 899–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Malcolm BA, Liu R, Lahser F, Agrawal S, Belanger B, Butkiewicz N, Chase R, Gheyas F, Hart A, Hesk D, Ingravallo P, Jiang C, Kong R, Lu J, Pichardo J, Prongay A, Skelton A, Tong X, Venkatraman S, Xia E, Girijavallabhan V, Njoroge FG, SCH 503034, a mechanism-based inhibitor of hepatitis C virus NS3 protease, suppresses polyprotein maturation and enhances the antiviral activity of alpha interferon in replicon cells, Antimicrob Agents Chemother 50(3) (2006) 1013–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Romano KP, Ali A, Aydin C, Soumana D, Ozen A, Deveau LM, Silver C, Cao H, Newton A, Petropoulos CJ, Huang W, Schiffer CA, The molecular basis of drug resistance against hepatitis C virus NS3/4A protease inhibitors, PLoS Pathog 8(7) (2012) e1002832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE, UCSF Chimera--a visualization system for exploratory research and analysis, J Comput Chem 25(13) (2004) 1605–12. [DOI] [PubMed] [Google Scholar]
- [67].Goddard TD, Huang CC, Ferrin TE, Visualizing density maps with UCSF Chimera, J Struct Biol 157(1) (2007) 281–7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.