

Abstract
Cellular senescence entails an irreversible growth arrest that evolved in part to prevent cancer. Paradoxically, senescent cells secrete pro-inflammatory and growth-stimulatory molecules, termed the senescence-associated secretory phenotype (SASP), which is correlated with cancer cell proliferation in culture and xenograft models. However, at what tumor stage and how senescence and the SASP act on endogenous tumor growth in vivo is unknown. To understand the role of senescence in cancer etiology, we subjected p16–3MR transgenic mice, which permit the identification and selective elimination of senescent cells in vivo, to the well-established two-step protocol of squamous cell skin carcinoma (SCC), in which tumorigenesis is initiated by a carcinogen 7,12-dimethylbenz[α]anthracene (DMBA) and then promoted by 12-O-tetradecanoyl-phorbol-13-acetate (TPA). We show that TPA promotes skin carcinogenesis by inducing senescence and a SASP. Systemic induction of senescence in non-tumor bearing p16–3MR mice using a chemotherapy followed by the two-step carcinogenesis protocol potentiated the conversion of benign papillomas to carcinomas by elevating p38 MAPK and MAPK/ERK signaling. Ablation of senescent cells reduced p38 MAPK and MAPK/ERK signaling, thereby preventing the progression of benign papillomas to carcinomas. Thus, we show for the first time that senescent cells are tumor promoters, not tumor initiators, and that they stimulate skin carcinogenesis by elevating p38 MAPK and MAPK/ERK signaling. These findings pave the way for developing novel therapeutics against senescence-fueled cancers.
Keywords: doxorubicin, senescence-associated secretory phenotype, squamous cell carcinoma, microenvironment, inflammation, transgenic mice, tumorspheres
Graphical Abstract
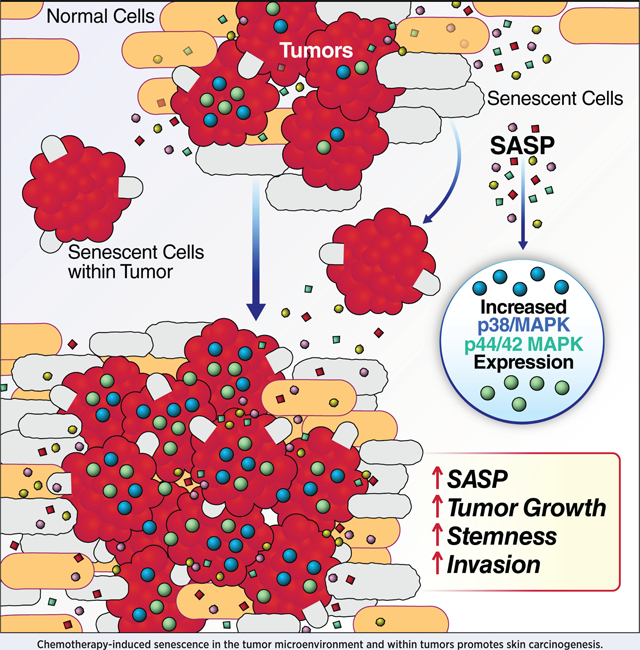
Introduction
Cellular senescence entails a permanent growth arrest wherein cells fail to proliferate but remain metabolically active. The senescence response is tumor suppressive, and is established and maintained by pathways controlled by the p53 and p16INK4a tumor suppressor proteins (1). This response is often triggered by potentially oncogenic stressors, and thus prevents stressed cells from progressing through the cell cycle and ultimately to malignancy (1). Among the stimuli that induce cellular senescence are mitochondrial dysfunction (2), telomere shortening (3), oncogene activation (4), epigenetic modifications (5), and genotoxic insults, including radiation and several anti-cancer agents (1).
Cellular senescence is paradoxical (6, 7). On the one hand, it is beneficial, optimizing certain steps in embryogenesis (8, 9), wound healing (6) and tissue reprogramming (7). On the other hand, despite protecting young organisms from cancer (1, 10), over time, senescent cells can evade the immune system and accumulate in tissues to secrete pro-inflammatory molecules, growth factors and proteases to create a pro-carcinogenic microenvironment (11). The senescent secretome, termed the senescence-associated secretory phenotype (SASP) (11), is now recognized as a significant driver of many age-related pathologies (11, 12).
Recent studies show that the selective elimination of senescent cells using drugs (termed senolytics) or transgenic mice ameliorates many age-related phenotypes and diseases (12–14). Further, we used a transgenic mouse model (p16–3MR) to show that removing chemotherapy-induced senescent cells ameliorates several deleterious side effects of these therapies (15). In p16–3MR mice, the p16INK4a (p16) promoter drives a herpes virus thymidine kinase (HSV-tk), which phosphorylates the nucleoside analog ganciclovir (GCV), converting it into a DNA chain terminator that fragments mitochondrial DNA (16), thereby inducing apoptosis selectively in p16-expressing senescent cells (6). However, how and at what stage senescent cells act on intrinsic tumor growth is not known.
A major side effect of genotoxic cancer treatments is an elevated risk of new neoplasms, unrelated to the primary cancer treated decades earlier (17). For example, childhood cancer survivors treated with radiation, which induces senescence (18), had a higher incidence of non-melanoma skin cancer years after treatment for their original cancer (17).
Squamous cell skin carcinoma (SCC) develops from epidermal squamous cells, and is the second most prevalent type of non-melanoma skin cancer (19). Unlike basal skin carcinoma, SCC has a higher rate of recurrence and metastasis (19). Indeed, cancer patients treated with the genotoxic chemotherapeutic doxorubicin (DOXO) can develop SCC years after treatment (20, 21). DOXO, an anthracycline and topoisomerase II inhibitor, induces senescence and a SASP in cultured cells and mice (15). Thus, chemotherapy-induced senescence might contribute to SCC development in vivo.
To test this idea, we employed the well-established two-step skin carcinogenesis protocol to induce SCC (22). Following this protocol, we initiated tumorigenesis in p16–3MR mouse skin with a single dose of the DNA damaging carcinogen 7,12-dimethylbenz[α]anthracene (DMBA), followed by promotion with 12-O-tetradecanoyl-phorbol-13-acetate (TPA) (22). DMBA causes H-Ras mutations, whereas TPA stimulates the growth of DMBA-initiated cells. This protocol leads to benign papillomas, which can eventually convert to carcinomas (22). We show for the first time that TPA, but not the single dose of DMBA, induces senescence and a SASP, suggesting that senescent cells might act as tumor promoters but not initiators. Multiple DMBA treatments also induce SCC (22), but the two-step protocol allowed us to determine whether senescent cells are tumor initiators or promoters. We also induced cellular senescence in p16–3MR mice using DOXO, then eliminated senescent cells with GCV followed by DMBA/TPA treatment. Senescent cells exacerbated tumor growth and the malignant conversion of benign tumors, whereas their ablation reduced tumor size and malignancy. Together, our findings provide a direct link between cellular senescence and cancer promotion in vivo, warranting the development of therapeutics against senescence-fueled cancers, including SCC.
Materials and Methods
Mice
p16–3MR mice were bred and housed at the Buck Institute for Research on Aging. All procedures were approved by the Institutional Animal Care and Use Committee. For all experiments, age-matched (3 mo old) p16–3MR homozygous female mice were randomly divided into experimental groups. Methods for senescence induction and the two-step protocol in mice are in Supplementary Materials.
Cell Lines and cell culture
Primary human neonatal keratinocytes (HEKn) (ATCC® PCS-200–010™) from the American Type Culture Collection (ATCC) (Manassas, VA, USA), were maintained in dermal basal medium, keratinocyte growth kit (ATCC) and antibiotics as per the manufacturer’s instructions. A-431 (ATCC® CRL1555™) human epidermoid carcinoma cells from ATCC, were maintained in Dulbecco’s Modified Eagle’s medium (DMEM), 10% fetal bovine serum (FBS) (Thermo Fisher Scientific, Waltham, MA) and 0.01% Penicillin/Streptomycin (Corning, NY, USA). Cells were cultured in a 20% O2, 5% CO2 humidified incubator and were authenticated by ATCC, including Short Tandem Repeat (STR) profiling. Primary HCA2 human foreskin fibroblasts were from O. Pereira-Smith (U Texas Health Science Center, San Antonio), and cultured in DMEM + FBS in a 3% O2 incubator. They were not re-authenticated. 24 h prior to lysis, cells were cultured in supplement-free media. All cells were mycoplasma-free.
Senescence induction in culture
Doxorubicin hydrochloride (DOXO) from R&D Systems (Minneapolis, MN, USA) was dissolved in dimethyl sulfoxide (DMSO). Cells were treated with DMSO or DOXO at 250 nM as described (15). We observed no cell death above the low background levels, and no reversal of senescence, as reported (23). For in vivo experiments, DOXO (2 mg/ml) was dissolved in warm endotoxin-free PBS (EMD Millipore, Hayward, CA, USA). DMBA (2.5 mg/ml) (Sigma-Aldrich, St. Louis, MO, USA) was prepared in acetone. TPA (Sigma-Aldrich) was prepared in acetone at 1.6 mM or in DMSO at 100 μM for mouse and culture experiments, respectively. For TPA-induced senescence in culture, keratinocytes were treated with TPA (1.3 nM-10 nM) for 48 h and maintained in TPA-free medium for an additional 10 days. TPA was dissolved in DMSO for cell culture experiments, and in acetone for mouse experiments, because low doses of acetone caused morphological changes in cultured human keratinocytes, whereas DMSO did not.
Bioluminescence
Mice were injected intraperitoneally (i.p.) with Xenolight RediJect Coelentarazine h (Perkin Elmer, Waltham, MA) solution and imaged using a Xenogen IVIS-200 Optical Imaging Instrument (Perkin Elmer) as described (6).
Tumorsphere assay
A-431 cells were cultured on growth factor-reduced Matrigel (Corning, NY, USA) and serum-free DMEM for 72 h to form tumorspheres (24). Tumorspheres were incubated with conditioned media (CM) from quiescent (made by a 3 d incubation in 0.2% FBS) or DOXO-induced senescent HCA2 cells. CM were generated by a 3 d incubation in serum-free media, and contained solvent, SB203580 (Selleck Chemicals, Houston, TX) or IL-1α antibody (R&D Systems). CM were replenished every 72 h. After 7 d, we assessed tumorsphere EdU incorporation.
Western blotting, cell proliferation assays, immunostaining and SA-β-gal staining
Western blotting was performed as described (6, 15). Primary and secondary antibodies are listed in Supplementary Table 1. Cell proliferation was assessed using a Z1 Coulter Particle Counter (Beckman Coulter, Fullerton, CA, USA) and EdU incorporation using the Molecular Probes Click iT EdU Alexa Fluor 488 HCS Assay kit (Thermo-Fisher Scientific) as per the manufacturer’s instructions. Immunostaining was performed as described (15). SA-β-gal staining was assessed using the Biovision Kit (Milpitas, CA, USA) as per the supplier’s instructions. Images were quantified from 3 independent fields from 3 biological replicates using Image J software and the Image J cell counter tool.
qRT-PCR analysis
RNA was isolated from cultured cells using the Bioline Isolate II RNA Mini Kit (Taunton, MA, USA). RNA was isolated from homogenized tissues using TRIzol reagent (Thermo-Fisher Scientific) with the Direct-zol™ RNA MiniPrep kit (Genesee Scientific, San Diego, CA, USA) as recommended by the supplier. cDNA synthesis and qRT-pCR were performed as described (15). Primers are listed in Supplementary Tables 2 and 3.
Immunohistochemistry and SA-β-gal staining of skin tissues
Human SCC and normal skin tissue arrays (SK208) were from US Biomax (Rockville, MD). Mouse skin was fixed in 10% buffered formalin (Thermo-Fisher Scientific) for 7 d, transferred to 70% ethanol for 24 h and paraffin-embedded. Tissues were cut into 7 μm sections, deparaffinized, rehydrated, washed with PBS and stained with Hematoxylin and Eosin (H&E) (6). For immunostaining, tissue sections were rinsed in PBS followed by antigen retrieval using citrate buffer (Cell Signaling Technology) and the Cell Signaling Technology protocol. Skin sections were then stained overnight with antibodies from Cell Signaling, listed in Supplementary Table 1, per the manufacturer’s instructions.
SA-β-gal staining was performed as described (6). Images were obtained using an Olympus BX20 microscope; 3 independent fields were quantified using Image J software and the Image J cell counter tool. For vimentin quantification, 3 different fields were analyzed using the Color Deconvolution plug-in in Image J. The plugin includes a built-in vector that separates the image into three color channels. The brown channel was set to include only vimentin positive staining and then quantified for percent area.
Statistical analysis
Statistics were assessed using GraphPad Software (San Diego, CA). For time dependent tumor growth, statistical significance was determined by two-way ANOVA. Tukey’s test for multiple comparisons was used for all post-analyses. For in vivo experiments with multiple comparisons, one-way ANOVA and Sidak’s test for multiple comparisons were used. For pairwise comparisons, data were analyzed using the unpaired two-tailed Student t test. Differences between means were considered significant when values were *p<0.05 or lower; ns denotes non-significant. Data are presented as mean values ±SEM or ± SD for in vivo and cell culture experiments, respectively. All culture experiments were replicated at least 3 times.
Results
TPA induces senescence and a SASP in human keratinocytes and mouse skin
Previous studies detected senescent cells in the dermal and stromal layers of DMBA/TPA-induced papillomas and within papillomas themselves (25, 26), implicating a role for senescent cells in SCC development. Because these studies did not reveal when or how senescent cells contributed to tumor growth, we asked whether DMBA or TPA induced senescence. We treated human keratinocytes, the SCC cells of origin (22), with a single dose of increasing concentrations of DMBA (2–8 μM) or DMSO (control). The cells proliferated for 10 d after treatment, suggesting that DMBA did not induce senescence (Figure S1A).
To determine whether DMBA induces senescence in vivo, we treated dorsal skin of p16–3MR mice with a single dose of DMBA (0.25 mg/ml) or acetone (control) and analyzed whole skin and the epidermis for p16 mRNA levels 1 mo later. We followed the standard two-step skin carcinogenesis protocol of a single dose of DMBA for initiation because multiple DMBA treatments would not allow us to determine whether senescent cells act as tumor initiators or promoters. We also tested skin for senescence-associated beta-galactosidase (SA-β-gal) (27) activity and cell proliferation. A single-dose of DMBA compared to acetone yielded no differences in p16 mRNA levels, SA-β-gal activity or cell proliferation (assessed by immunostaining for Ki-67). Thus, a single dose of DMBA alone does not induce senescence in cultured keratinocytes or skin (Figure S1B–F).
We then treated human keratinocytes with DMSO or various concentrations of TPA (1.3–10 nM) and assessed senescence 12 d later. TPA inhibited cell proliferation in a dose dependent manner, with greatest inhibition at 5 and 10 nM (Figure S1G). TPA (10 nM) also induced SA-β-gal activity (27) in >80% of cells (Figure 1A–B), along with the inhibition of cell proliferation (Figure 1C). Compared to DMSO-treatment, TPA reduced Lamin B1 (LMNB1) mRNA levels (28) and elevated mRNA levels of the senescence markers p16 and p21, and the SASP factors interleukin-6 (IL-6), matrix metalloproteinases (MMPs) MMP3 and MMP9, and chemokine C-X-C motif 10 (CXCL10) (15) (Figure 1D). Further, TPA (5 and 10 nM), but not DMSO, decreased Lamin B1 (Lmnb1) and increased p16 protein levels (Figure 1E).
Figure 1: TPA induces senescence and a SASP in human keratinocytes and mouse skin.
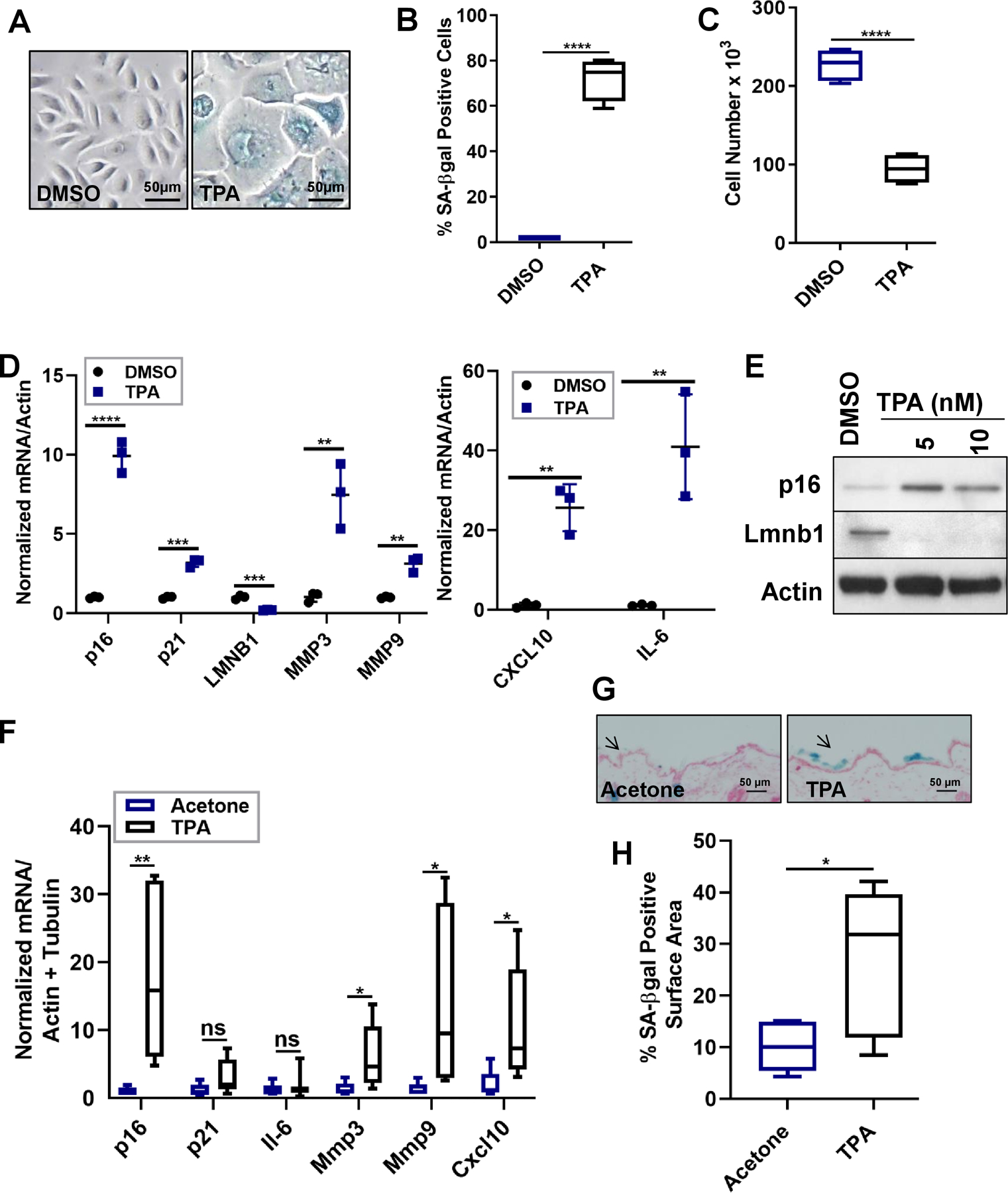
(A-C) Human keratinocytes were treated with DMSO (control) or TPA (10 nM) for 48 h. 12 d later, the cells were stained for SA-ß-gal (A-B) and assessed for cell proliferation (C). N=3 independent experiments. (D) Total RNA was isolated from human keratinocytes and analyzed for p16, p21 and SASP mRNAs normalized to actin. N=3 independent experiments, presented as means ±SD; **p<0.01, ***p<0.001, ****p<0.0001. (E) Protein lysates from human keratinocytes treated with DMSO or TPA (5 nM or 10 nM) were evaluated for Lmnb1 and p16 levels by immunoblotting. (F) Total RNA was isolated from mouse skin topically treated with either acetone or TPA for 2 mos. One month after the last treatments, the skin was analyzed for the indicated mRNAs, normalized to actin and tubulin. N=5 for the acetone group, N=7 for the TPA group. Shown are means ±SEM; *p<0.05, **p<0.01 (Student t-test). (G-H) Frozen tissue sections of acetone- or TPA-treated skin were stained for SA-ß-gal (blue) and nuclei (red). (G) Representative images are shown (4x magnification), and arrows denote SA-ß-gal positive areas. (H) Percent of SA-ß-gal positive surface area of skin compared to unstained skin. N=5 for acetone group, N=7 for TPA group. Shown are means ±SEM; *p<0.05 (Student t-test).
To determine whether TPA induces senescence in skin, we topically treated dorsal skin with either acetone or TPA twice a week for 2 mos. We chose a 2 mo treatment because in the skin carcinogenesis protocol tumors develop 8 wks after TPA treatment, suggesting that senescence might occur earlier. To exclude reported inflammatory effects of TPA, the skin was analyzed for senescence 1 mo after the last treatment. TPA induced senescence in the skin as demonstrated by elevated levels of p16 and SASP factor (Mmp3, Mmp9, and Cxcl10) mRNAs (Figure 1F). Unlike the increased p21 and IL-6 mRNA levels in response to TPA in human keratinocytes, there was no increase in p21 and Il-6 mRNA levels in mouse skin, suggesting that p21 and Il-6 are not major factors in TPA-induced senescence in this tissue (Figure 1F). TPA-induced senescence was confirmed by the presence of SA-β-gal-positive cells in TPA- but not acetone-treated skin (Figure 1G–H). These data indicate that TPA induces senescence and a SASP in human keratinocytes and mouse skin.
TPA-induced senescence (pre-treatment) stimulates tumor growth
To explore the consequences of TPA-induced senescence, we asked whether TPA pre-treatment facilitates tumor growth in the skin. We first induced senescence with TPA in p16–3MR mouse skin as described above. A week after the last treatment, we injected the mice with PBS or GCV to remove p16-positive senescent cells. We then implemented the two-step skin carcinogenesis protocol. To ensure the elimination of senescent cells, PBS or GCV was injected for 10 d every 3 wks until the end of the study (Figure 2A). Tumor growth was monitored for 24 wks after DMBA initiation and 8 wks after the last TPA treatment.
Figure 2: TPA-induced senescence (pre-treatment) stimulates skin tumor growth.
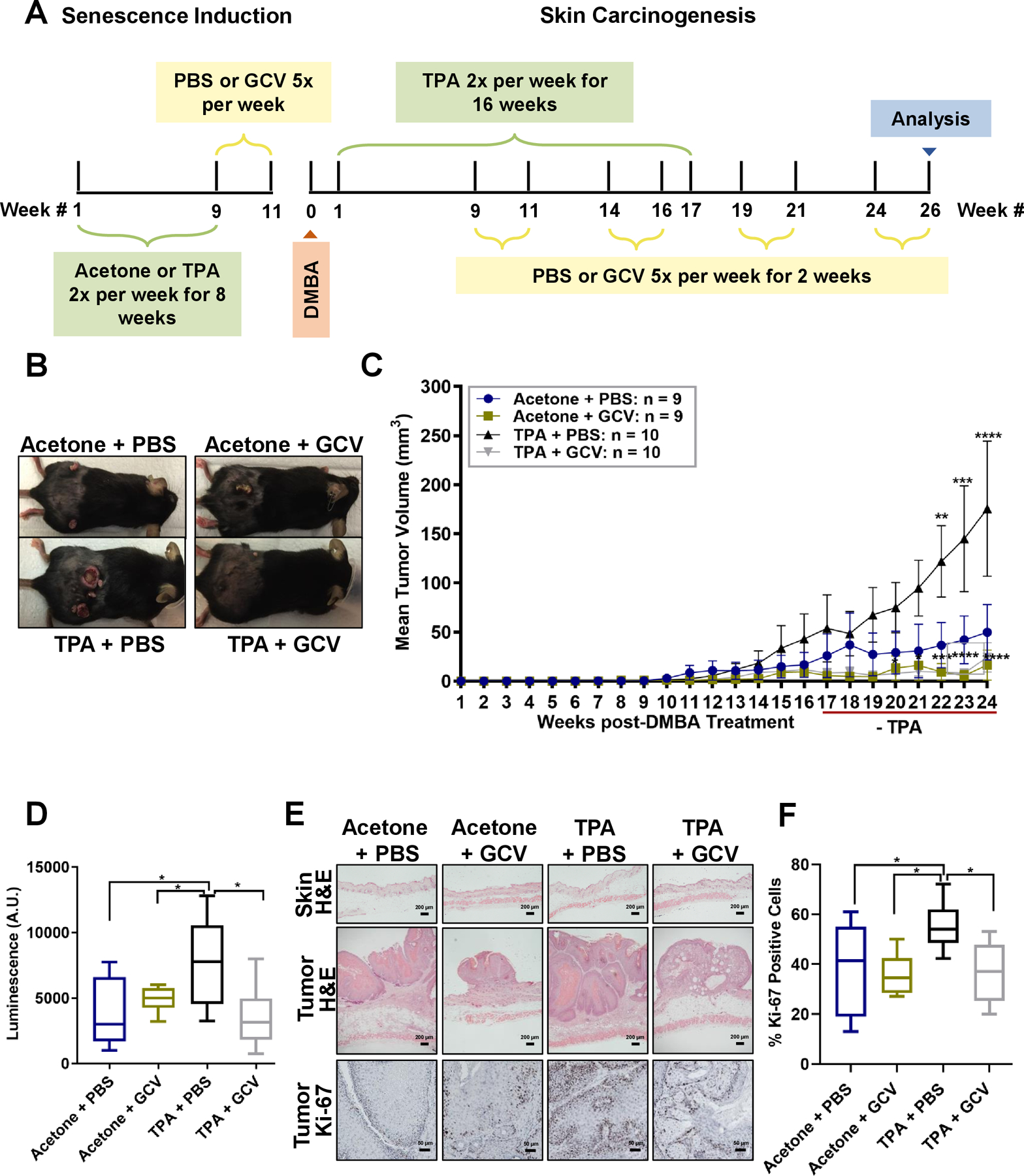
(A) Schematic of treated p16–3MR mice. (B) Representative images of tumor- bearing mice, 24 wks after DMBA treatment. (C) Tumors of Acetone + PBS, Acetone + GCV, TPA + PBS, TPA + GCV groups were monitored once a wk; mean tumor volume over 24 wks is shown. Shown are means ±SEM; **p<0.01, ***p<0.001, ****p<0.0001 (two-way ANOVA, Tukey’s test for multiple comparisons was used post-analyses). (D) Luminescence of tumor-bearing skin in arbitrary units (A.U.) units 24 wks after DMBA treatment. N = 8–10 per group. Shown are means ±SEM; *p<0.05 (one-way ANOVA, Sidak’s multiple comparisons test was used post-analyses). (E) Representative images of H&E staining of skin (top panels) and tumors (middle panels) (200 μm and 50 μm respectively), and Ki-67 staining of tumors (bottom panels) (50 μm). (F) Percentage of Ki-67-positive cells in tumors from the 4 treatment groups. Shown are means ±SEM; *p<0.05 (one-way ANOVA, Sidak’s multiple comparisons test was used post-analyses).
Starting 22 wks after DMBA and 5 wks after the last TPA treatment, TPA + PBS treated mice developed significantly larger tumors than Acetone + PBS, Acetone + GCV and TPA + GCV treated mice (Figure 2B–C). Ultimately, the mean tumor volume of TPA + PBS treated mice was 175.5 mm3, while mean tumor volume of Acetone + PBS treated mice was 49.76 mm3. Importantly, removing senescent cells reduced tumor size in mice treated with Acetone + GCV or TPA + GCV (mean volumes of 16.2 mm3 and 24.2 mm3, respectively; Figure 2C). Notably, GCV markedly reduced tumor volume in mice pre-treated with TPA, suggesting that the senescent milieu in these mice promoted tumor growth.
To assess senescent cells in mouse skin, we used the luciferase reporter in the p16–3MR transgene (6) and measured luminescence in mice given Acetone + PBS, Acetone + GCV, TPA + PBS or TPA + GCV 24 wks after DMBA treatment. Luminescence was significantly elevated in mice treated with TPA + PBS compared to treatment with Acetone + PBS or Acetone + GCV. Eliminating senescent cells decreased luminescence in TPA + GCV treated mice, but there was no difference in luminescence between the Acetone + PBS and Acetone + GCV groups (Figure 2D). Further, H&E staining of tumors and untreated skin revealed that TPA-induced senescence generates benign papillomas that were larger than tumors in mice treated with Acetone + PBS, Acetone + GCV and TPA + GCV (Figure 2E).
To determine whether the increased tumor size in TPA + PBS-treated mice was due to increased proliferation, we immunostained tumors for the proliferation marker Ki-67. The larger tumor size correlated with increased Ki-67 levels in the tumors. Importantly, eliminating senescent cells reduced Ki-67 protein levels (Figure 2E–F). Thus, TPA promotes tumor growth in the skin by inducing senescence and eliminating senescent cells reduces tumor size.
Doxorubicin induces senescence and a SASP in human keratinocytes and mouse skin
The long-term effects of DOXO in cancer patients can be deleterious, often leading to secondary cancers including SCC (20, 21, 29). To understand how DOXO-induced senescence might contribute to SCC, we treated human keratinocytes with a single dose of DOXO (250 nM) or DMSO (vehicle) for 24 h and assessed senescence 10 d later. DOXO induced senescence as evidenced by SA-β-gal activity (27) in >80 % of cells (Figure 3A–B), and a significant decrease in cell proliferation (Figure 3C). DOXO, but not vehicle, also elevated mRNA levels of p16 and p21 and the SASP factors IL-6, MMP3, MMP9 and CXCL10 (11), and decreased LMNB1 mRNA levels (28) (Figure 3D). We confirmed elevated p16 and reduced LMNB1 expression at the protein levels by western analyses (Figure 3E).
Figure 3: DOXO induces senescence and a SASP in human keratinocytes and mouse skin.
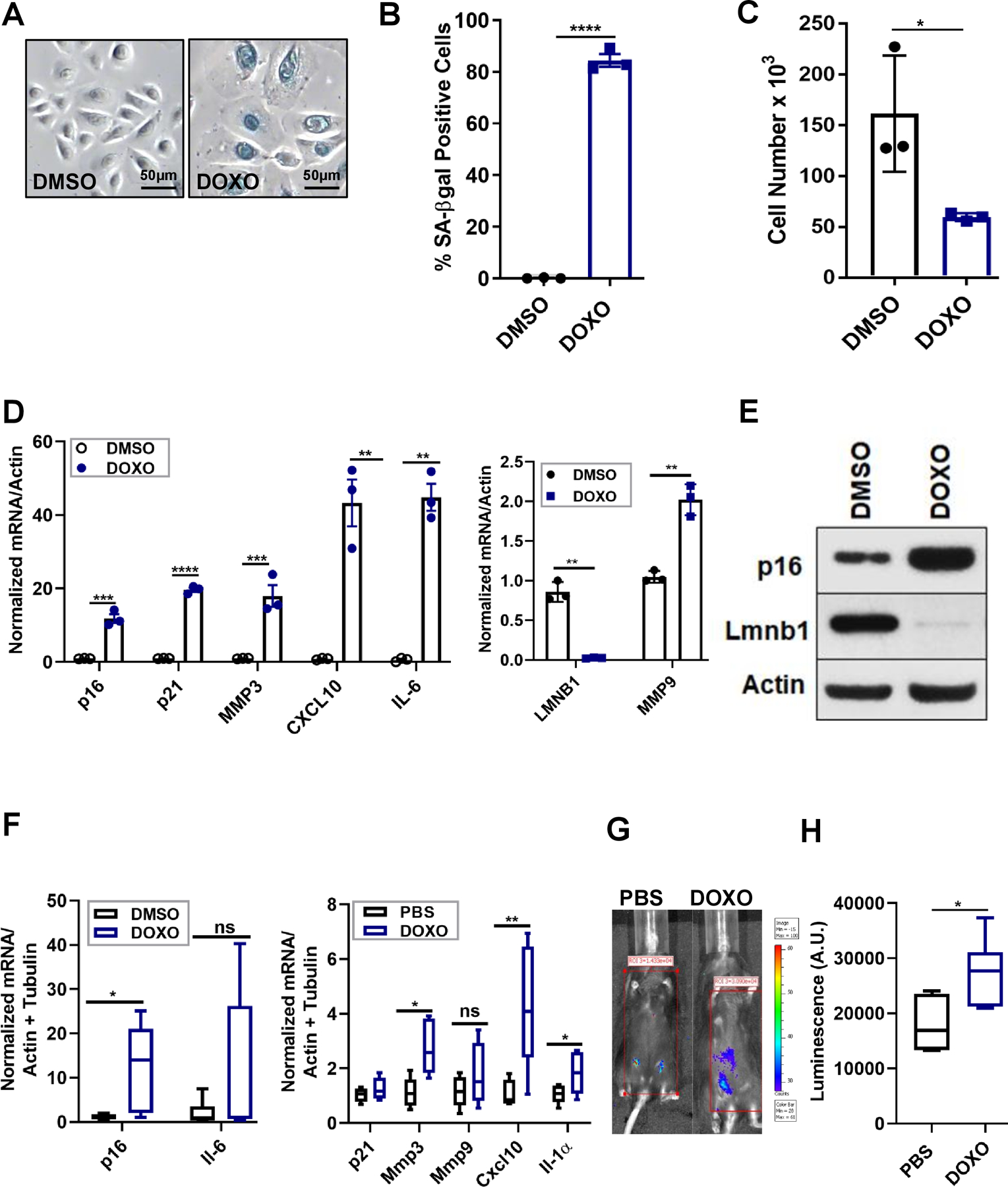
(A-C) Human keratinocytes were treated with DMSO or DOXO (250 nM) for 24 h. 10 d later, the cells were stained for SA-ß-gal (A-B) and assessed for cell proliferation (C). N=3 independent experiments. Shown are means ±SD, *p<0.5, ***p<0.001, (Student t test). (D) Total RNA was isolated from the indicated cells and analyzed for p16, p21 and SASP mRNAs, normalized to actin. N=3 independent experiments. Shown are means ±SD; **p<0.01, ***p<0.001. (E) Protein lysates from human keratinocytes were evaluated for p16 and LMNB1 by immunoblotting. (F) Total RNA was isolated from the skin of PBS- or DOXO-treated mice and analyzed for the indicated mRNAs, normalized to actin and tubulin. N=6 per each treatment group. Shown are means ±SEM; *p<0.05, **p<0.01 (Student t-test). (G-H) Representative image (G) and quantification in arbitrary units (A.U.) of whole body luminescence of p16–3MR mice one mo after PBS or DOXO treatments. N=6 per each treatment group. Shown are means ±SEM; *p<0.05 (Student t test).
To determine whether DOXO induced senescence in the skin in vivo, we injected p16–3MR mice with PBS or DOXO (i.p., 12 mg/kg), a dose known to induce senescence in mice (15). One mo later, we assess the presence of senescent cells. Systemic DOXO but not PBS significantly increased mRNA levels of p16 and the SASP factors Mmp3, Cxcl10 and Il-1α, but not Mmp9, Il6 or p21. Unlike TPA-induced senescence, Mmp9 mRNA levels did not increase upon systemic DOXO treatment. Further, p21 and Il-6 mRNA levels did not change upon DOXO treatment, consistent with our findings in TPA-treated skin (Figure 3F). Notably, whole body luminescence increased significantly 1 mo after DOXO, but not PBS, treatment, confirming widespread senescence induced by DOXO (Figure 3G–H). Importantly, senescent cells persisted in mouse skin 1 mo after DOXO exposure, creating an environment permissive for tumor growth.
DOXO-induced senescence fuels skin tumor growth
To determine whether DOXO-induced senescence drives SCC, we injected p16–3MR mice with DOXO (12 mg/kg) with a second injection (5 mg/kg) 3 d later (to ensure the persistence of senescent cells over a 6 mo period), followed by the skin carcinogenesis protocol (Figure 4A). A DOXO dose of 17 mg/kg is not toxic to these mice (15). The presence of senescent cells in DOXO-treated mice was confirmed by whole body luminescence, and by p16 mRNA. As expected, GCV significantly reduced whole body luminescence and p16 mRNA levels in the skin (Figure 4B–C). Thus, DOXO-induced senescent cells were cleared prior to DMBA treatment.
Figure 4: DOXO-induced senescence fuels skin tumor growth.
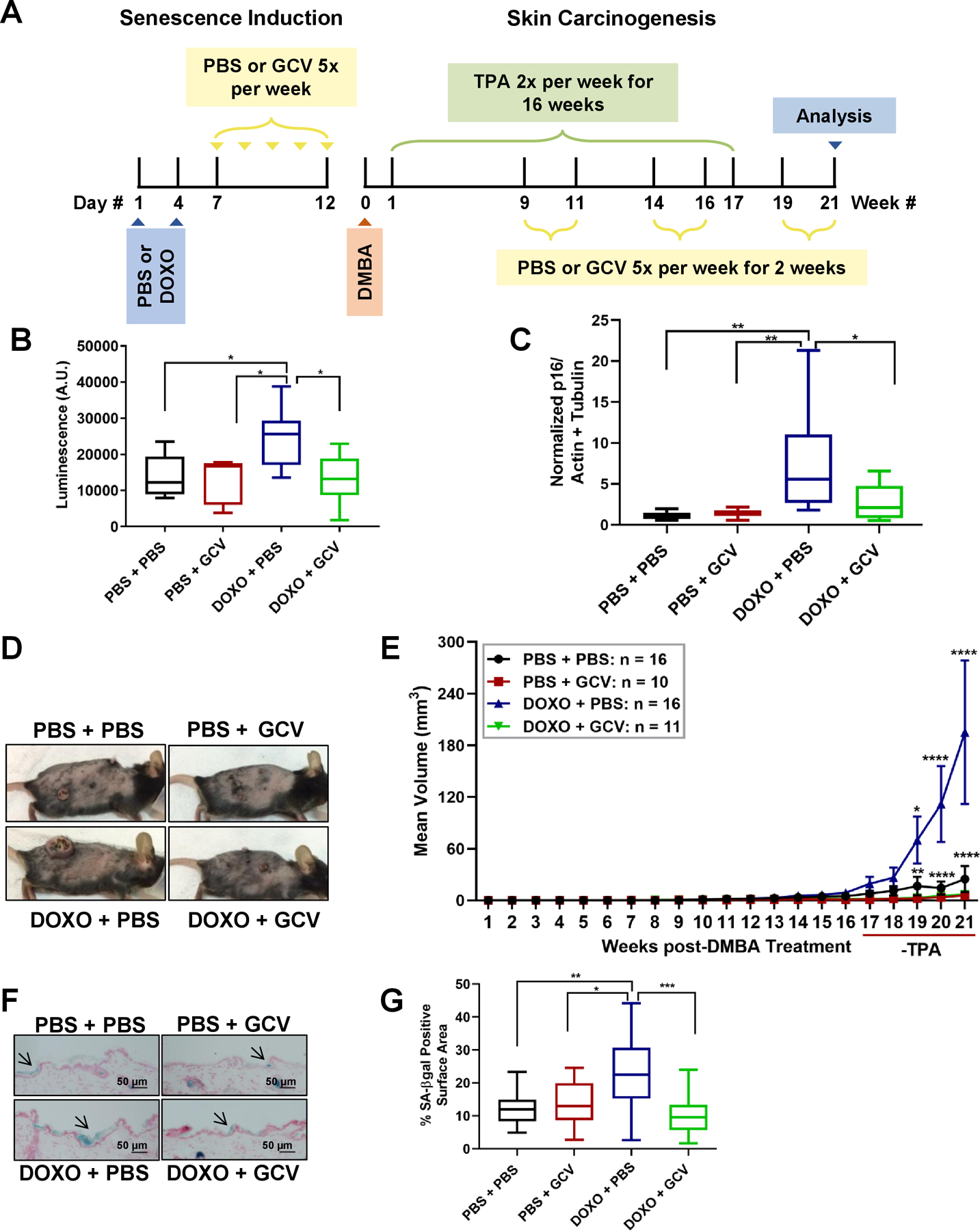
(A) A schematic of the PBS/DOXO and skin carcinogenesis regimens of p16–3MR mice. TPA treatment lasted 16 wks. (B) Quantification of whole body luminescence of p16–3MR mice 10 d after PBS/DOXO treatments and one wk after DMBA initiation. Quantification is in arbitrary units (A.U.). Shown are means ±SEM; *p<0.05 (one-way ANOVA, Sidak’s multiple comparisons test was used post-analyses). (C) Total RNA was isolated from the skin of the 4 treatments groups and analyzed for p16 normalized to actin and tubulin (PBS + PBS: n=10, PBS + GCV: n=8, DOXO + PBS: n=9, DOXO + GCV: n=8). Shown are means ±SEM; *p<0.05, **p<0.01 (one-way ANOVA, Sidak’s multiple comparisons test was used post-analyses). (D) Representative images of tumor-bearing mice 21 wks after DMBA treatment and 5 wks after TPA treatment in PBS + PBS, PBS + GCV, DOXO + PBS, DOXO + GCV groups. (E) The mean tumor volume of the same mice is shown over 21 wks. Shown are means ±SEM; *p<0.05, **p<0.01, ****p<0.0001 (two way ANOVA, Tukey’s test for multiple comparisons was used post-analyses). (F-G) Frozen skin sections of (PBS + PBS: n=10, PBS + GCV: n=10, DOXO + PBS: n=14, DOXO + GCV: n=14) treatment groups were stained for SA-ß-gal (blue) and nuclei (red). (F) Representative images are shown (50 μm), and arrows denote SA-ß-gal positive areas. (G) Percent of SA-ß-gal positive surface area of skin compared to non-stained skin. Shown are means ±SEM; *p<0.5, **p<0.01, ***p<0. 001 (one-way ANOVA, Sidak’s multiple comparisons test was used post-analyses).
Throughout the skin carcinogenesis protocol, tumor incidence (Figure S2A) and number (Figure S2B) did not significantly change across treatment groups (PBS + PBS, PBS + GCV, DOXO + PBS and DOXO + GCV). However, 21 wks after DMBA initiation, tumors in the DOXO-treated mice were an average size of 183 mm3 compared to 29 mm3 in the PBS-treated group. Mean tumor volumes of the PBS + GCV and DOXO + GCV-treated groups were 7.1 mm3 and 5.4 mm3, respectively, indicating that removal of senescent cells suppressed tumor growth (Figure 4D–E). Further, 21 wks after DMBA initiation, DOXO-injected mice weighed significantly less than PBS-treated mice, while GCV prevented this weight loss (Figure S2C). To confirm that DOXO-induced senescent cells persist independent of the skin carcinogenesis treatment regimen, skin that was not treated with DMBA/TPA from the 4 groups was stained for SA-β-gal activity at the end of the study. DOXO-treated mice had a higher percentage of skin area positive for SA-β-gal compared to PBS-treated mice, and this percentage declined upon elimination of senescent cells (Figure 4F–G).
These data support the hypothesis that senescent cells can drive skin carcinogenesis, and that genotoxic drugs such as DOXO can accelerate tumor growth by inducing senescence in vivo.
Eliminating DOXO-induced senescent cells prevents malignant progression
To identify the reason behind the increased tumor size in DOXO- vs PBS-treated mice, we characterized skin tumors from PBS + PBS, PBS + GCV, DOXO + PBS and DOXO + GCV-treated mice. For each mouse and treatment group, we compared untreated skin (side without DMBA and TPA treatment) with DMBA- and TPA-treated skin. H&E staining revealed that ~31% (5/16) of DOXO-exposed mice developed carcinomas, while the other groups developed only benign papillomas (Figure 5A). Consistent with tumor size, Ki-67 positive cells were more abundant in tumors from DOXO-treated mice compared to other treatment groups (Figure 5B). Further, vimentin, an epithelial-mesenchymal transition marker (11, 30), was elevated at the epithelial invasive front, underlying connective tissue and the tumor itself, and vascularization was more prominent as evidenced by increased levels of CD-31-positive vessels in DOXO- compared to PBS-treated tumors. Vimentin and CD-31 levels were significantly reduced by GCV (Figure 5C–D). Together, the results suggest that DOXO-induced senescence potentiates the conversion of benign skin papillomas to carcinomas.
Figure 5: Eliminating DOXO-induced senescent cells prevents malignant tumor development.
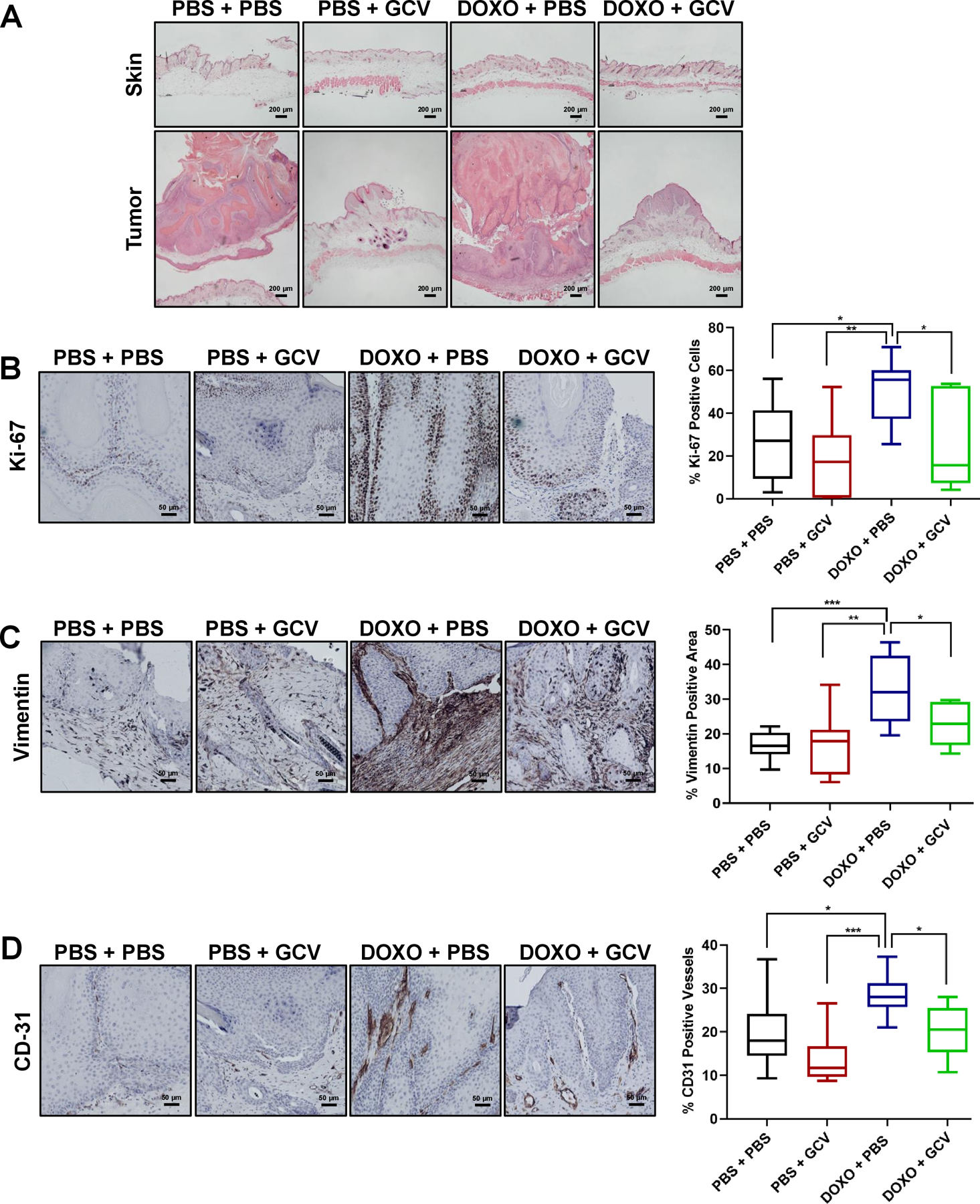
Representative images of H&E staining of the skin (top panels) and tumors (bottom panels) in PBS + PBS, PBS + GCV, DOXO + PBS, DOXO + GCV treated-groups (200 μm). (B-D) In the same 4 groups (PBS + PBS: n =9, PBS + GCV: n=7, DOXO + PBS: n=11, DOXO + GCV: n=7), representative IHC images for Ki-67 (B), vimentin (C) and CD-31 (D) (50 μm) with the corresponding quantification of percent positive cells in the right panels. Shown are means ±SEM; *p<0.05, **p<0.01, ***p<0. 001 (one-way ANOVA, Sidak’s multiple comparisons test was used post-analyses).
The SASP drives skin carcinogenesis
The senescent fibroblast SASP is known to enhance tumor cell proliferation and invasion in culture (11, 31). Upstream SASP regulators have been identified, including p38MAPK and IL-1α (32, 33). We therefore assessed p38 phosphorylation (Thr180/Tyr182) (p-p38) by western blotting. DOXO-induced senescence elevated p38 (Thr180/Tyr182) (Figure S3A). To determine whether this increase facilitates cancer proliferation, we used human A-431 skin carcinoma 3D-tumorspheres. We incubated the tumorspheres for 3 d in serum-free media, then incubated with conditioned media (CM) from non-senescent or DOXO-induced senescent HCA2 skin fibroblasts treated or not with SB203580 (SB) (10 μM), a pan p38MAPK inhibitor. Prior to treating the tumorspheres, we verified fibroblast phenotypes by SA-β-gal staining, DNA damage (γ-H2AX) foci and p16 mRNA levels (Figure S3B–D). Treatment of skin fibroblasts with SB reduced DOXO-induced SASP factors (Figure S3E). Importantly, tumorspheres exposed to CM from senescent, but not non-senescent, fibroblasts incorporated more EdU, indicative of increased proliferation, which was reduced by p38MAPK inhibition (Figure 6A–B). Since IL-1α mRNA expression also increased in mouse skin in response to DOXO, we inhibited IL-1α signaling in DOXO-treated skin fibroblasts using an IL-1α neutralizing antibody; the antibody reduced the DOXO-induced SASP (Figure S3F). IL-1α inhibition also reduced tumorsphere proliferation (EdU incorporation; Figure 6C–D). Finally, p38MAPK expression was elevated in tumors from DOXO-treated mice and this elevation declined upon removal of senescent cells with GCV (Figure 6A). These findings show that DOXO-induced senescence promotes SCC progression by elevating p38MAPK and IL-1α signaling, two upstream mediators of the SASP, in the skin microenvironment.
Figure 6: The SASP drives skin carcinogenesis.
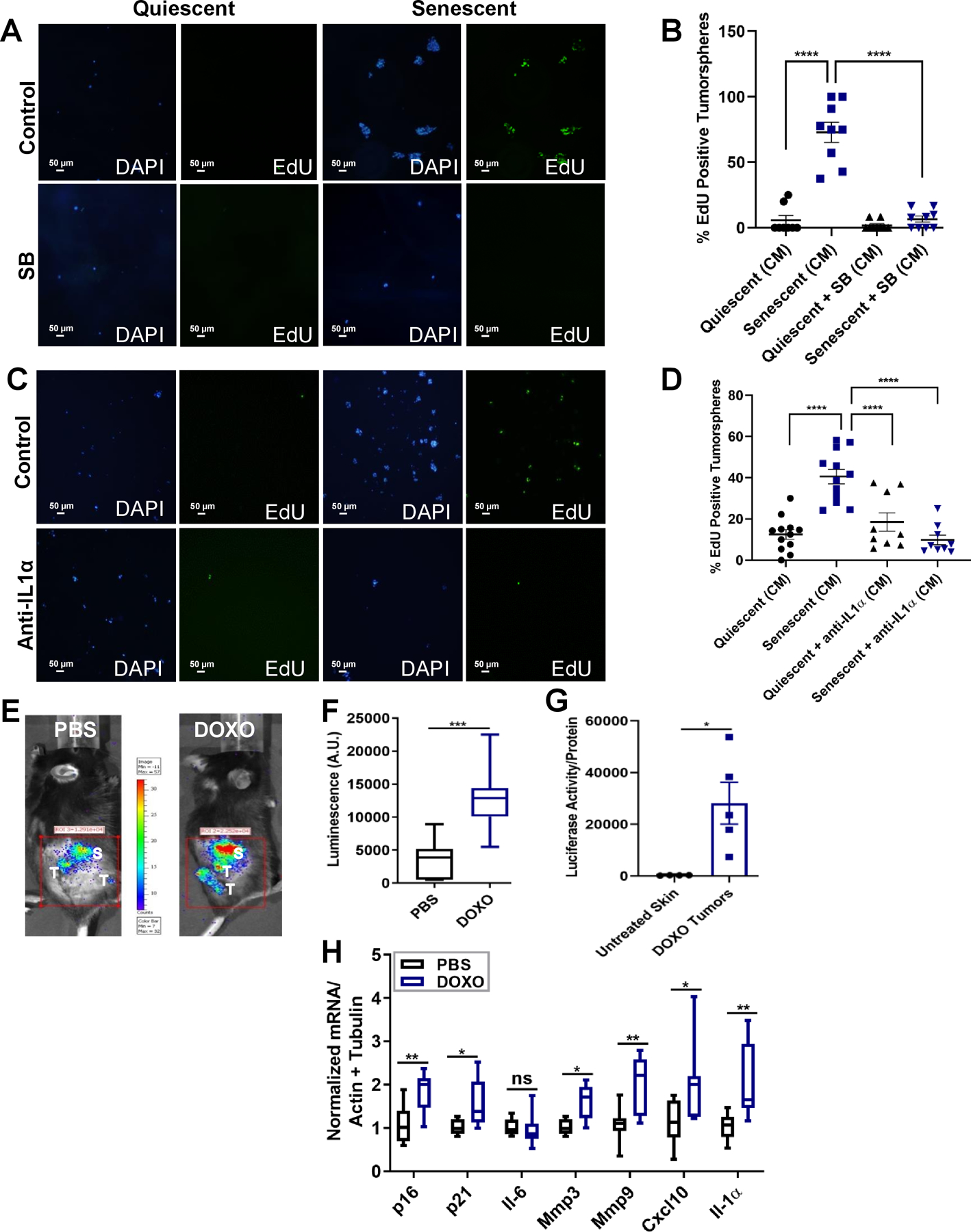
(A-D) Representative images and quantification of tumorspheres of A-431 cells embedded in reduced growth factor Matrigel incubated for 7 d with conditioned media from non-senescent (quiescent) or senescent HCA2 fibroblasts with or without SB203580 (A-B) or anti-IL1α (C-D). (A and C) Tumorspheres are shown stained with DAPI (blue) and EdU (green). (B and D) Quantification of percentage EdU-positive cells. N=3 independent experiments. Shown are mean ±SEM; **p<0.01, ****p<0.0001 (one-way ANOVA, Sidak’s multiple comparisons test was used post-analyses). (E) Representative tumor and adjacent skin luminescence images of PBS- or DOXO-treated mice, 21 wks after DMBA treatment and 5 wks after the last TPA treatment. Luminescent skin and tumors are labeled on the images as (S) and (T), respectively. (F) Luminescence quantification of tumors and adjacent skin shown in (E) in arbitrary units (A.U.). N= 8 per each treatment group. Shown are mean ±SEM; ***p<0.001 (Student t test). (G) Luciferase activity of untreated skin compared to DOXO-tumor, protein lysates were normalized to total protein content. N= 4–5 for skin and DOXO tumors respectively. Shown are mean ±SEM; *p<0.05 (Student t test). (H) Total RNA was isolated from skin adjacent to tumors from mice in (E-F) and analyzed for the indicated mRNAs, normalized to actin and tubulin. N= 8 per treatment group. Shown are mean ±SEM; *p<0.5, **p<0.001 (Student t test).
To test these findings in mice, we asked whether senescence and SASP markers were present in skin adjacent to tumors and/or within the tumors themselves. Senescent cells adjacent to tumors would indicate that senescence in the microenvironment can fuel cancer growth, whereas senescent cells within tumors would suggest that tumor-associated senescence can exacerbate tumor growth.
To verify the chronic presence of senescent cells throughout the body months after DOXO treatment, we assessed whole body luminescence in PBS- or DOXO-treated mice 21 wks after DMBA initiation and 5 wks after TPA removal. Luminescence was higher in DOXO-treated, compared to PBS-treated, mice, suggesting that senescent cells persist weeks after DOXO exposure (Figure S3G–H). To evaluate senescent cells adjacent to tumors, we imaged DMBA/TPA-treated skin of PBS or DOXO-injected mice 21 wks after DMBA treatment. Luminescence was apparent in skin adjacent to and within tumors (Figure 6E–F). Further, DOXO-injected mice had significantly higher luminescence compared to PBS-treated mice (Figure 6E–F), and luminescence was higher in DOXO-treated tumors compared to untreated skin, indicating the presence of senescent cells within the tumors (Figure 6G). Moreover, mRNA levels of p16, p21, and the SASP factors Mmp9, Mmp3, Cxcl-10 and Il-1α were higher in skin adjacent to tumors in DOXO-treated compared to PBS-treated mice (Figure 6H). This elevation was accompanied by increased mRNA levels of the skin stem cell genes Cd34, Lgr6 and Lrig1 (26) (Figure S3I). These findings suggest that DOXO-induced senescence and the SASP promote skin carcinogenesis by enhancing stemness in the skin microenvironment.
A subset of macrophages was reported to express p16 and SA-β-gal in mouse tissues (34), and neutrophils, important orchestrators of inflammation, secrete pro-inflammatory molecules similar to the SASP (35). Thus, neutrophils and macrophages might contribute to the microenvironment that promotes cancer development (35, 36). To test the possibility that macrophages and/or neutrophils, not senescent cells, drive SCC, we assessed mRNA levels of the macrophage marker Cd68 (37) and neutrophil markers elastase (Elane) (38) and myeloperoxidase (Mpo) (39) in the skin of PBS- or DOXO-treated mice. There were no differences in these markers between the two groups (Figure S4A). To confirm these findings, we stained tissue sections from PBS- and DOXO-treated mice for the macrophage marker F4/80 (37) and the neutrophil marker Ly6G (40). Consistent with the mRNA data, we observed no difference in the levels of F4/80 and Ly6G in PBS- and DOXO-treated mice (Figure S4B–C). These findings suggest that DOXO induces senescence and a SASP in the skin, independent of macrophages and neutrophils.
To confirm that senescent cells were present within skin tumors, we compared luciferase activity in untreated skin and DOXO-induced tumors in p16–3MR mice. Luminescence was evident only in tumors, not in untreated skin, further indicating that p16-positive senescent cells are present in the tumors (Figure 6G). Together, these findings suggest that DOXO-induced senescence and the SASP occur throughout the body, but also both in skin adjacent to tumors and within tumors. Additionally, they suggest that the SASP within tumors may facilitate tumor growth in addition to the SASP in the skin microenvironment. Thus, senescent cells in tumors might reinforce tumor growth fueled by the senescent microenvironment.
p38MAPK and p44/42 ERK/MAPK signaling increase in tumors from DOXO-treated mice
p38MAPK and ERK/MAPK (p44/42) signaling are known to promote the SASP and tumor progression, respectively (32, 41). The expression of p-p38 (Thr180/Tyr182) MAPK in skin tumors in vivo was elevated in DOXO-treated, compared to PBS-treated, mice. Moreover, this elevation declined upon removing senescent cells with GCV (Figure 7A). Likewise, p-p44/42 MAPK increased in tumors from DOXO-treated compared to PBS-treated mice and this increase declined upon elimination of senescent cells (Figure 7B).
Figure 7: Phospho-p38 and phospho-p44/42 MAPK signaling is increased in tumors from DOXO-treated mice.
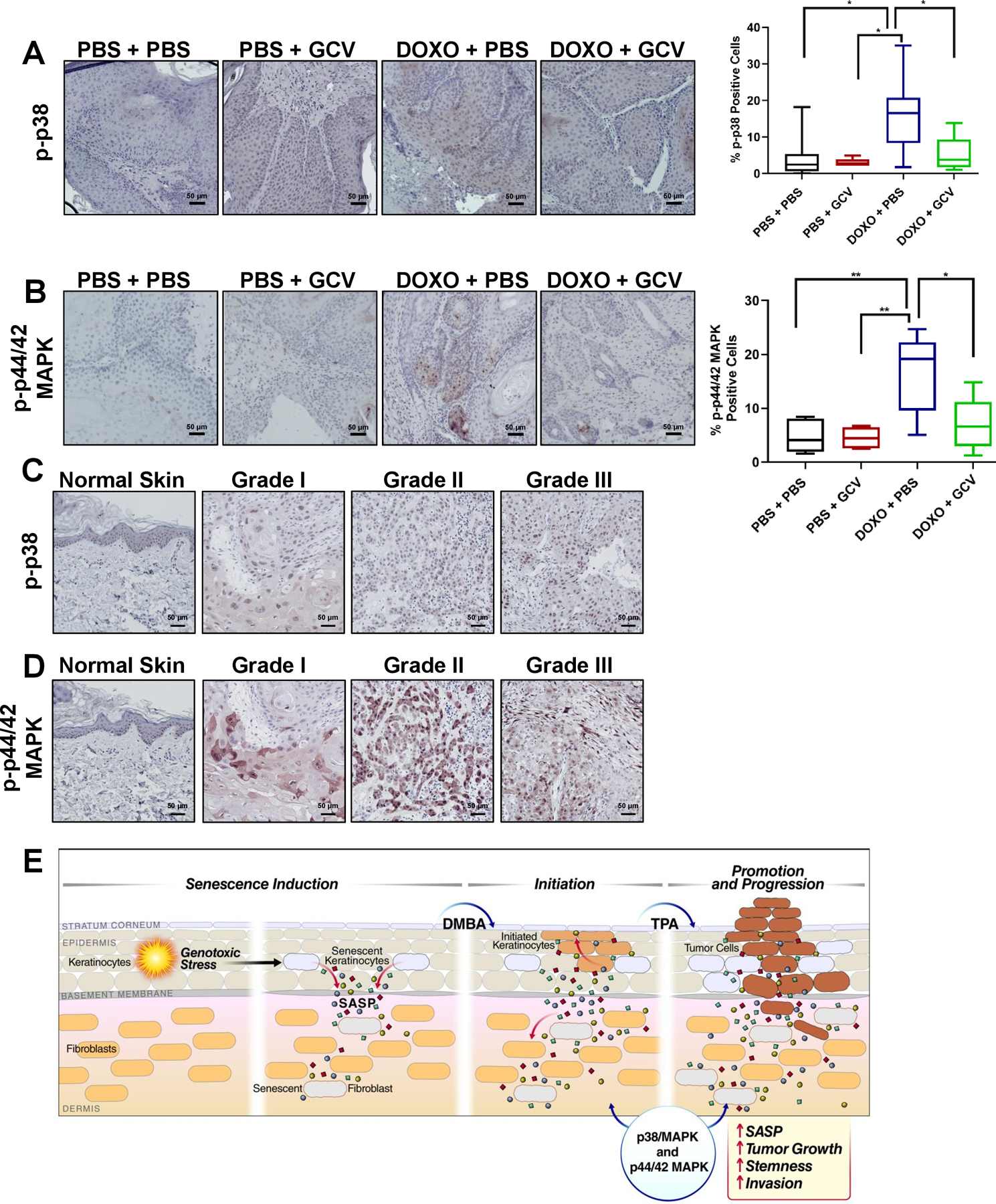
(A-B) Representative IHC images of tumors from PBS + PBS: n =9, PBS + GCV: n=7, DOXO + PBS: n=11, DOXO + GCV: n=7, stained with p-p38 (A) or p-p44/42 (B) with the corresponding quantification of percent positive cells in the right panels. Shown are means ±SEM; *p<0.05, **p<0.01, (one-way ANOVA, Sidak’s multiple comparisons test was used post-analyses). (C-D) Representative IHC images of normal human skin and hSCC lesions (grades I-III) stained with p-p38 (C) and p-p44/42 (D). (E) Working model showing that DOXO induces senescence and a SASP in keratinocytes and fibroblasts, creating a microenvironment permissive for tumor growth and invasion. Once tumors are formed, senescent cells within the tumors also reinforce tumor growth by elevating p-p38 and p-p44/42 signaling.
To determine the translational significance of p38 and p44/42 MAPK activation, we compared protein expression of p-p38 (Thr180/Tyr182) and p-p44/42 in commercially available human (h) SCC and normal skin tissue arrays. p38 and p44/42 MAPK phosphorylation was elevated in 77% of hSCC lesions (grades I-III) compared to normal skin, supporting our finding that these pathways might contribute to malignancy after induction of senescence (Figure 7C–D). Thus, DOXO-induced senescence and the SASP might contribute to p38 and p44/42 MAPK signaling within skin tumors and in the microenvironment surrounding the tumors, reinforcing tumor growth by increasing stemness and converting benign papillomas to carcinomas (Figure 7E).
Discussion
SCC incidence rises after genotoxic cancer treatments (20, 21). The mechanism behind this increase, decades after chemotherapy, remains elusive. Using two senescence inducers (TPA and DOXO) and p16–3MR mice, we show that cellular senescence drives skin carcinogenesis. TPA is an established tumor promoter that can stimulate cell proliferation (22). We show that TPA also promotes carcinogenesis by inducing p16-dependent senescence, accompanied by a SASP. Strikingly, removal of TPA-induced senescent cells in p16–3MR mice reduced tumor growth. Acute TPA treatment can also lead to the secretion of inflammatory chemokines, cytokines, proteases and growth factors that stimulate tumor growth, analogous to the SASP. However, these effects of TPA can be reversed, depending on the number of applications and treatment duration (42). We show that once TPA is removed, tumor growth accelerates, suggesting that senescent cells, not the inflammation caused by TPA, drive tumor growth.
Chemotherapeutic agents, such as DOXO, induce cellular senescence and a SASP (15). Therefore, chemotherapy-induced senescence might contribute to SCC development. Indeed, treating mice with DOXO resulted in a persistent senescent microenvironment in the skin, which was conducive to tumor growth. After DOXO and DMBA/TPA treatments, senescent cells were present throughout the body, including the skin adjacent to tumors and within the tumors. Thus, not only are DOXO- induced senescent cells present throughout the body, they are also present adjacent to and within the tumors, exacerbating tumor growth. Since DOXO was administered systemically, it is difficult to discern to what extent senescent cells in organs other than skin, or SASP factors in the circulation, contribute to skin cancer growth. For example, we previously showed that DOXO induces senescence and a SASP in the liver, concomitant with an elevation in the SASP factors Il-6 and Cxcl1 in sera (15). It is therefore plausible that senescent cells throughout the body might also contribute to skin tumor progression.
One deleterious side effect of chemotherapy in cancer patients is weight loss (43). We show here that removal of senescent cells periodically for 21 wks prevents the weight loss caused by DOXO. Thus, removing senescent cells might improve cancer patients’ overall well-being.
The senescence fueled skin tumor growth in our study is primarily p16-dependent. TPA and DOXO treatments alone elevated p16, but not p21, mRNA levels 1 mo after treatment. However, DOXO, DMBA and TPA treatments together increased p21 mRNA in skin adjacent to tumors. These data suggest that a higher senescence burden, or different quality of senescent cells, might occur later during skin carcinogenesis. Additionally, not all SASP factors increased in the skin after DOXO or TPA treatment alone. For instance, TPA-induced senescence elevated expression of the SASP factors Mmp3, Mmp9 and Cxcl10, whereas DOXO-induced senescence elevated Mmp3, Il-1α and Cxcl10, but not Mmp9, mRNA levels.
Cxcl-10, which acts through its receptor CXCR3, is highly expressed in several cancers (44), and is associated with metastasis and poor survival (45). MMPs degrade the extracellular matrix and elevated levels of MMPs often mark aggressive forms of cancer (46, 47). For example, elevated MMP9 in stromal cells of the skin enhances SCC development (47), and IL1α promotes the growth of several cancers (48). Collectively, these studies support our data that the SASP, including Cxcl10 and MMPs, might promote senescence-induced skin carcinogenesis.
The chronic presence of senescent cells has been linked to tumor cell proliferation. For example, we showed that senescent fibroblasts promote premalignant epithelial cell growth in culture, and caused premalignant cells to form tumors in immune-compromised mice (49). Additionally, our recent study revealed that DOXO treatment enhanced lung metastases of Polyoma Virus middle T antigen-expressing breast cancer cells transplanted into the mammary fat pad, and that systemic elimination of senescent cells reduced these metastases (15). Although these studies show an association between senescent cells and carcinogenesis, they used exogenously administered cancer cell lines in vivo. In our study, we monitored skin tumor growth throughout the initiation, promotion and progression stages of endogenous tumor development. Thus, our study more closely recapitulates human tumor pathogenesis, identifying senescence as a potential culprit behind tumor promotion and the conversion of benign papillomas to carcinomas.
Cancer recurrence and the development of second neoplasms in cancer survivors, decades after chemotherapy, is an emerging public health concern (17, 50). Chemotherapy-induced senescence might contribute to cancer recurrence and second cancer development. Our study suggests a direct link between the presence of DOXO-induced senescent cells and tumor promotion in a model of SCC. Our findings may help develop senolytics aimed at eliminating senescence-fueled skin cancer.
Supplementary Material
Significance.
Findings identify chemotherapy-induced senescence as a culprit behind tumor promotion, suggesting that elimination of senescent cells after chemotherapy may reduce occurrence of second cancers decades later.
Acknowledgments
This work was supported by NIH grants F32 AG044946 to FA and AG09909 and AG017242 to JC, and funds from the Hillblom Foundation. We thank Dr. Aseem Lal for histological analysis of the skin and tumors.
Footnotes
Conflict of Interests
JC is a co-founder of Unity Biotechnology and MD owns equity in Unity Biotechnology. All other authors declare no competing financial interests.
References
- 1.Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011;192:547–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wiley CD, Velarde MC, Lecot P, Liu S, Sarnoski EA, Freund A et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab 2016;23:303–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Harley CB, Futcher AB, Greider CW. Telomeres shorten during aging of human fibroblasts. Nature 1990;345:458–60. [DOI] [PubMed] [Google Scholar]
- 4.Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997;88:593–602. [DOI] [PubMed] [Google Scholar]
- 5.Zhang R, Adams PD. Heterochromatin and its relationship to cell senescence and cancer therapy. Cell Cycle 2007;6:784–9. [DOI] [PubMed] [Google Scholar]
- 6.Demaria M, Ohtani N, Youssef SA, Rodier F, Toussaint W, Mitchell JR et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Dev Cell 2014;31:722–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mosteiro L, Pantoja C, Alcazar N, Marion RM, Chondronasiou D, Rovira M et al. Tissue damage and senescence provide critical signals for cellular reprogramming in vivo. Science 2016;354:aaf4445. [DOI] [PubMed] [Google Scholar]
- 8.Storer M, Mas A, Robert-Moreno A, Pecoraro M, Ortells MC, Di Giacomo V et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 2013;155:1119–30. [DOI] [PubMed] [Google Scholar]
- 9.Munoz-Espin D, Canamero M, Maraver A, Gomez-Lopez G, Contreras J, Murillo-Cuesta S et al. Programmed cell senescence during mammalian embryonic development. Cell 2013;155:1104–18. [DOI] [PubMed] [Google Scholar]
- 10.Lecot P, Alimirah F, Desprez PY, Campisi J, Wiley C. Context-dependent effects of cellular senescence in cancer development. Br J Cancer 2016;114:1180–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Coppe JP, Patil CK, Rodier F, Sun Y, Munoz DP, Goldstein J et al. Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 2008;6:2853–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 2016;530:184–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bussian TJ, Aziz A, Meyer CF, Swenson BL, van Deursen JM, Baker DJ. Clearance of senescent glial cells prevents tau-dependent pathology and cognitive decline. Nature 2018;562:578–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, Atkinson EJ et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 2017;8:14532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Demaria M, O’Leary MN, Chang J, Shao L, Liu S, Alimirah F et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov 2017;7:165–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Laberge RM, Adler D, DeMaria M, Mechtouf N, Teachenor R, Cardin GB et al. Mitochondrial DNA damage induces apoptosis in senescent cells. Cell Death Dis 2013;4:e727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Turcotte LM, Neglia JP, Reulen RC, Ronckers CM, van Leeuwen FE, Morton LM et al. Risk, Risk Factors, and Surveillance of Subsequent Malignant Neoplasms in Survivors of Childhood Cancer: A Review. J Clin Oncol 2018;36:2145–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Le ON, Rodier F, Fontaine F, Coppe JP, Campisi J, DeGregori J et al. Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging Cell 2010;9:398–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Que SKT, Zwald FO, Schmults CD. Cutaneous squamous cell carcinoma: Incidence, risk factors, diagnosis, and staging. J Am Acad Dermatol 2018;78:237–47. [DOI] [PubMed] [Google Scholar]
- 20.Pezzoli M, Bona Galvagno M, Bongioannini G. Oral squamous cell carcinoma in a patient treated with long-term pegylated liposomal doxorubicin for recurrent ovarian cancer. BMJ Case Rep 2015;2015:bcr2014204056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lauvin R, Miglianico L, Hellegouarc’h R. Skin cancer occurring 10 years after the extravasation of doxorubicin. N Engl J Med 1995;332:754. [DOI] [PubMed] [Google Scholar]
- 22.Abel EL, Angel JM, Kiguchi K, DiGiovanni J. Multi-stage chemical carcinogenesis in mouse skin: fundamentals and applications. Nat Protoc 2009;4:1350–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Beausejour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003;22:4212–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shaw FL, Harrison H, Spence K, Ablett MP, Simoes BM, Farnie G et al. A detailed mammosphere assay protocol for the quantification of breast stem cell activity. J Mammary Gland Biol Neoplasia 2012;17:111–7. [DOI] [PubMed] [Google Scholar]
- 25.Collado M, Gil J, Efeyan A, Guerra C, Schuhmacher AJ, Barradas M et al. Tumour biology: senescence in premalignant tumours. Nature 2005;436:642. [DOI] [PubMed] [Google Scholar]
- 26.Ritschka B, Storer M, Mas A, Heinzmann F, Ortells MC, Morton JP et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev 2017;31:172–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci U S A 1995;92:9363–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Freund A, Laberge RM, Demaria M, Campisi J. Lamin B1 loss is a senescence-associated biomarker. Mol Biol Cell 2012;23:2066–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cannon TL, Lai DW, Hirsch D, Delacure M, Downey A, Kerr AR et al. Squamous cell carcinoma of the oral cavity in nonsmoking women: a new and unusual complication of chemotherapy for recurrent ovarian cancer. Oncologist 2012;17:1541–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Satelli A, Li S. Vimentin in cancer and its potential as a molecular target for cancer therapy. Cell Mol Life Sci 2011;68:3033–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rodier F, Coppe JP, Patil CK, Hoeijmakers WA, Munoz DP, Raza SR et al. Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion. Nat Cell Biol 2009;11:973–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Freund A, Patil CK, Campisi J. p38MAPK is a novel DNA damage response-independent regulator of the senescence-associated secretory phenotype. EMBO J 2011;30:1536–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Orjalo AV, Bhaumik D, Gengler BK, Scott GK, Campisi J. Cell surface-bound IL-1alpha is an upstream regulator of the senescence-associated IL-6/IL-8 cytokine network. Proc Natl Acad Sci U S A 2009;106:17031–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hall BM, Balan V, Gleiberman AS, Strom E, Krasnov P, Virtuoso LP et al. Aging of mice is associated with p16(Ink4a)- and beta-galactosidase-positive macrophage accumulation that can be induced in young mice by senescent cells. Aging (Albany NY) 2016;8:1294–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rosales C. Neutrophil: A Cell with Many Roles in Inflammation or Several Cell Types. Front Physiol 2018;9:113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jackaman C, Tomay F, Duong L, Abdol Razak NB, Pixley FJ, Metharom P et al. Aging and cancer: The role of macrophages and neutrophils. Ageing Res Rev 2017;36:105–16. [DOI] [PubMed] [Google Scholar]
- 37.Xu M, Pirtskhalava T, Farr JN, Weigand BM, Palmer AK, Weivoda MM et al. Senolytics improve physical function and increase lifespan in old age. Nat Med 2018;24:1246–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lammers AM, van de Kerkhof PC, Schalwijk J, Mier PD. Elastase, a marker for neutrophils in skin infiltrates. Br J Dermatol 1986;115:181–6. [DOI] [PubMed] [Google Scholar]
- 39.Lau D, Mollnau H, Eiserich JP, Freeman BA, Daiber A, Gehling UM et al. Myeloperoxidase mediates neutrophil activation by association with CD11b/CD18 integrins. Proc Natl Acad Sci U S A 2005;102:431–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.John DS, Aschenbach J, Kruger B, Sendler M, Weiss FU, Mayerle J et al. Deficiency of cathepsin C ameliorates severity of acute pancreatitis by reduction of neutrophil elastase activation and cleavage of E-cadherin. J Biol Chem 2019;294:697–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Sullivan RJ, Infante JR, Janku F, Wong DJL, Sosman JA, Keedy V et al. First-in-Class ERK1/2 Inhibitor Ulixertinib (BVD-523) in Patients with MAPK Mutant Advanced Solid Tumors: Results of a Phase I Dose-Escalation and Expansion Study. Cancer Discov 2018;8:184–95. [DOI] [PubMed] [Google Scholar]
- 42.Rundhaug JE, Fischer SM. Molecular mechanisms of mouse skin tumor promotion. Cancers (Basel) 2010;2:436–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takayoshi K, Uchino K, Nakano M, Ikejiri K, Baba E. Weight Loss During Initial Chemotherapy Predicts Survival in Patients With Advanced Gastric Cancer. Nutr Cancer 2017;69:408–15. [DOI] [PubMed] [Google Scholar]
- 44.Liu M, Guo S, Stiles JK. The emerging role of CXCL10 in cancer (Review). Oncol Lett 2011;2:583–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wightman SC, Uppal A, Pitroda SP, Ganai S, Burnette B, Stack M et al. Oncogenic CXCL10 signalling drives metastasis development and poor clinical outcome. Br J Cancer 2015;113:327–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kerkela E, Saarialho-Kere U. Matrix metalloproteinases in tumor progression: focus on basal and squamous cell skin cancer. Exp Dermatol 2003;12:109–25. [DOI] [PubMed] [Google Scholar]
- 47.Coussens LM, Tinkle CL, Hanahan D, Werb Z. MMP-9 supplied by bone marrow-derived cells contributes to skin carcinogenesis. Cell 2000;103:481–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lewis AM, Varghese S, Xu H, Alexander HR. Interleukin-1 and cancer progression: the emerging role of interleukin-1 receptor antagonist as a novel therapeutic agent in cancer treatment. J Transl Med 2006;4:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Krtolica A, Parrinello S, Lockett S, Desprez PY, Campisi J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: a link between cancer and aging. Proc Natl Acad Sci U S A 2001;98:12072–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guida JL, Ahles TA, Belsky D, Campisi J, Cohen HJ, DeGregori J et al. Measuring Aging and Identifying Aging Phenotypes in Cancer Survivors. J Natl Cancer Inst 2019;111:1245–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.