

Abstract
Metabolomics based nuclear magnetic resonance (NMR) is widely used in disease mechanism analysis and drug discovery. One of the most important factors in NMR based metabolomics study is the accuracy of spectra bucketing which plays a critical role in data interpretation. Though various methods have been developed for automatic bucketing, the most popular approach is still the traditional rectangular bucketing method which is mainly due to the requirement of user expertise for the automatic bucketing methods. In this study, we developed a new automatic bucketing method that not only efficiently increases peak bucketing accuracy but also allows the bucketing process to be conveniently visualized and adjusted by the end-users. This method applied the line broadening (lb) factor to the average spectrum for a study set which serves as the reference spectrum, and the peak width of the reference spectrum was then set as the peak bucketing pattern. The approach to pick the bucket boundaries is simple but powerful after the line broadening factor was applied. The line broadening factors from 0 to 2 lb were tested using mouse fecal samples and the 1 lb method showed similar peak patterns and data interpretation results compared with a careful manual bucketing pattern. Besides this, the new method generated bucketing patterns could be easily visualized using the Amix software and revised by general users without excessive data science and NMR instrumentation expertise. In summary, our study showed a powerful and convenient tool in NMR peak auto bucketing with flexible visualization and adjustment ability for metabolomics studies.
Keywords: Metabolomics, Nuclear Magnetic Resonance, Bucketing, Line Broadening
Graphical Abstract
Introduction
The Nuclear Magnet Resonance (NMR) based metabolomics has been widely used in investigating disease mechanism [1, 2], drug discovery [3], nutritional science [4], plant science [5] and environmental science areas. NMR is a robust technique that can provide highly reproducible data for metabolomics which could make data collected from different time blocks comparable. To convert the NMR spectra to a data matrix for further statistical analysis or modeling, spectra bucketing is the most commonly used method which divides the spectra into smaller sizes that are usually between 0.1 to 0.5 ppm range [6, 7]. The integration of areas under the peak in the bucket size will be the peak intensity for the bucket [7], and for the whole range of a spectrum, a total number of 200 to 500 buckets are commonly used in metabolomics studies.
Though the bucketing method could be used to reduce the minor NMR peak misalignment influence due to the difference of pH, salt, and even temperature issues [6], the bucketing methods and peak alignment are two different areas for metabolomics. When peak misalignment is significant, the misalignment adjustment before bucketing is preferred for data analysis because different metabolites peaks may be easily assigned to one bucket which may lead to unreliable results. Various peak alignment methods were reported in the literature such as the correlation optimized warping [8, 9], peak alignment by beam search [10], and interval correlation shifting (icoshift) [11]. These peak alignment methods are good pre- processing methods for NMR peak bucketing when the misalignment is significant. However, when the misalignment is not significant, such as the commonly studied blood serum samples, tissue extracts, and fecal extract samples, the minor peak misalignment problem could be overcome by suitable bucketing methods. Our method was designed for metabolomics studies with minor peak misalignment problems or samples that were pre-aligned using the peak alignment methods.
The traditional NMR peak bucketing method simply divides the spectrum into hundreds of the same size of buckets which could easily split peaks or generate one bucket with various peaks. Though the traditional method may lead to inaccurate results [12], it is still the most popular method [6, 13] which is mainly due to its convenience. Various advanced methods have been developed to improve the bucketing efficiency. Gaussian binning [14] was developed with overlapping buckets possible, but the required two parameters standard deviation and step size were considered hard to tune [6]. Adaptive-intelligent binning [15] applied variable bucket sizes to analyze the spectra but is not very sensitive to lower intensity peaks as reviewed before [6]. Other techniques such as dynamic adaptive binning [16] and adaptive binning [17] were reported, and similar to the other methods mentioned above, their applications are limited by the user expertise requirement in computational methods [6, 12]. pJRES Binning Algorithm method [18] was developed recently, but focused on the noise reduction by electronics and using pJRES H-1 NMR spectra. The Optimized Bucketing Algorithm (OPA) revised the regular bucketing method using a slackness factor and local minima to find the boundary which is an easy method to reduce the probability of splitting a peak into 2 buckets. However, the performance is affected by the slackness number defined by the method. In addition, the method started with a bucket (for example 0.05 ppm) and narrow it down using the slackness factor which may potentially include more peaks into 1 bucket.
In addition to the above bucketing methods, in many cases, the NMR spectra bucketing can be done manually by overlaying the spectra and assigning the bucket range using software like AMIX 4.0 (Bruker Biospin) which allows users to assign bucket patterns manually [19, 20]. The manual bucketing method is suitable for NMR data with light misalignment or after alignment with good accuracy but is time-consuming and human errors may be involved. In this study, we designed an automatic bucketing method that could generate patterns comparable to manual bucketing, but with higher efficiency and reproducibility.
In NMR processing, the windows function is a process that multiplies the time domain signal by an exponentially decaying function. The exponential multiply (EM) is commonly applied in NMR data as a pretreatment to reduce noise and increase the signal-noise ratio [21, 22] which is called line broadening (lb). The EM method applies multiplication of the free induction decay (FID) with exponential of line broadening factors (exp(−lb × t) [23]. In many software, for example, Bruker Topspin, the lb values can be easily changed. Though the general value for metabolomics data is around 0.3 [24], if an average of all spectra in a study was applied as a reference spectrum, the reference spectrum will not be smooth. This is mainly due to the misalignment problem (Figure 1A) and is also why the peak width range cannot be applied directly as the bucket range. Therefore, in this study, we applied the line broadening factor to smooth the average spectrum first, which will make the local minimum a great choice as the boundary of the buckets. When the spectra misalignment was not significant, the human manually selected bucket is usually more accurate for metabolomics studies but is time-consuming and requires instrumentation and data interpretation expertise. Hence a careful manual bucketing was adapted as a reference to test the performance of our new bucketing method. The method was tested using a mouse fecal study which has commonly overserved minor misalignment in fecal samples and regular sample size for mouse studies. In addition, the automatic bucketing pattern can be easily plotted in AMIX 4.0 for users which will provide visible results for general users to analyze and revise.
Figure 1.
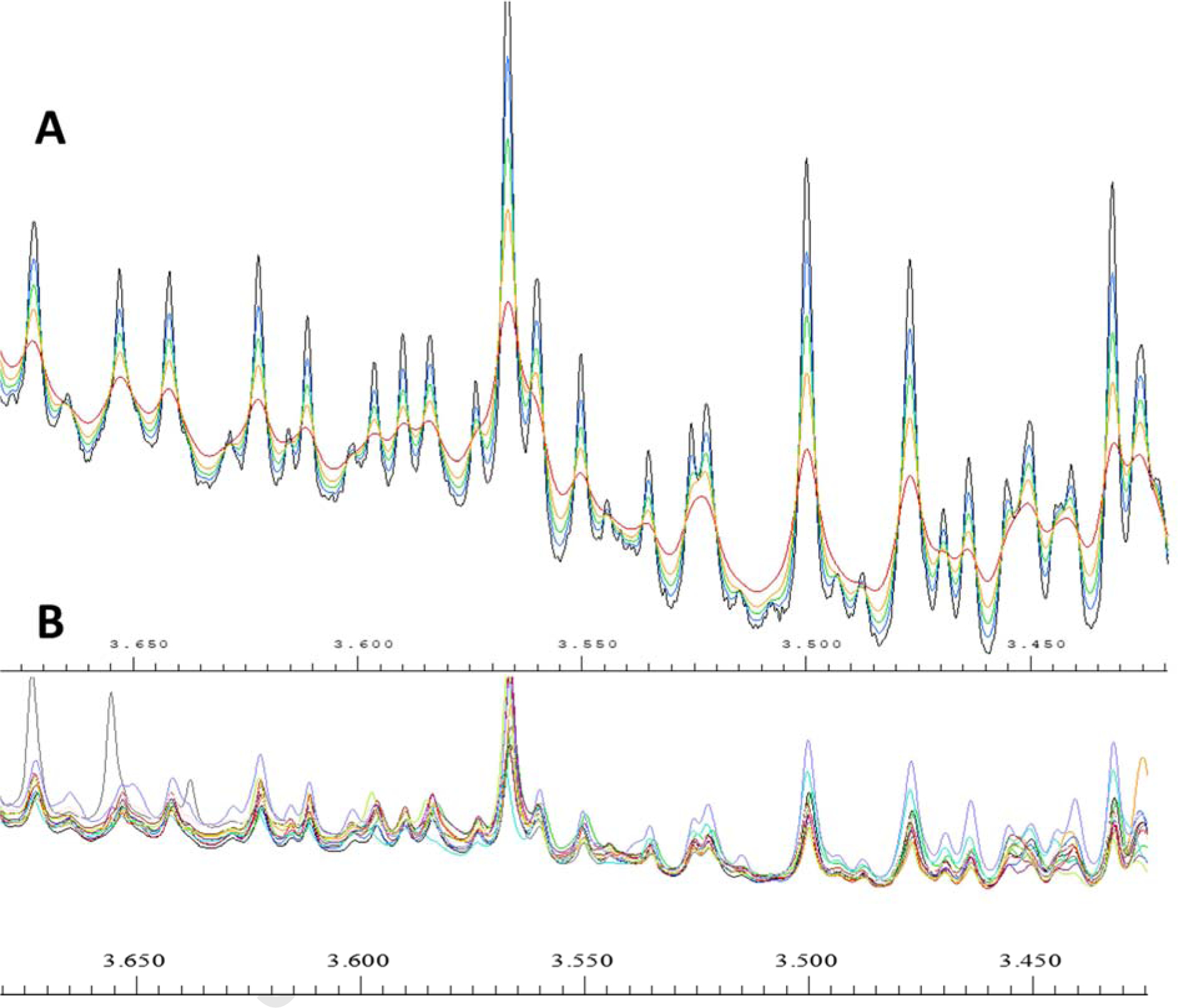
A, The line broadening factor to the reference peak. The trough order from bottom to top (or crest from top to bottom) is no line broadening, 0.3lb, 0.6lb, 1lb, and 2lb representatively. B, The overlay of all the 12 spectra in the study in the same range of Figure 1A. All the spectra were normalized to the total intensity of the total spectrum range using Amix 4.0.
2. Methodology
2.1. Sample preparation
A group of mouse fecal extracts was applied as examples to demonstrate the bucketing method. The 12 fecal samples including a control group and a case group, and 6 samples a group is regular for the mouse-[25, 26] and cell line- [19, 27] based metabolomics studies. The fecal samples were extracted using water with previously established methods [28]. A phosphate buffer of D2O was added to the extracted samples, and the final samples contained 10% of D2O with 0.1 M phosphate buffer (pH = 7.4) and 0.5mM trimethylsilylpropanoic acid (TSP). The samples were transferred to 5 mm NMR tubes after being centrifuged for further NMR acquisition.
2.2. NMR acquisition and data pretreatment
A Bruker Ascend 400 MHz high-resolution NMR with a sampleXpress autosampler was applied in this study and all the experiments were carried out using ICON-NMR software (Bruker Biospin) and controlled by ICON-NMR. All the experiments were carried out after 3D shimming and 1D shimming. A 1D NOESY experiment with water suppression (noesygppr1d) was carried out with 32k increments, 64 transients which took around 7 min. All the spectra were carefully phased and calibrated to TSP in Bruker Topspin 4.06 (Bruker Biospin).
2.3. Bucketing methods
The bucketing methods include 4 steps and were mostly carried out using Bruker Amix 4.0 and Topspin 4.06 with a home written Matlab script for bucketing boundary discovery.
First, an average of all the spectra was calculated in Amix 4.0, and a line broadening factor was applied to the average spectrum in Topspin 4.06 to smooth the spectrum. One example of the line shape of the average spectrum after different line broadening factor is listed in Figure 1.
Second, the automatic bucketing method was applied using the trough of each peak. The process was carried out using a simple algorithm. 1) The differences between all the neighboring peaks were calculated to get an array. 2) The products of all the neighboring values of the step 1 array were also calculated and all the crests and troughs have negative values because they are the positions where the values change from increasing to decreasing or the opposite way. Briefly, from left to right, a crest has increasing values on the left and decreasing values on the right, so it has positive values in the array of step 2) on the left and negative values on the right. A trough has negative values of step 2) array on the left and positive values on the right. 3) Once we got all the crest and trough values by step 2), we can get the trough positions if the position has negative values in array 2 and their left values in step 1 is positive. All the trough positions were converted into chemical shift values (ppm).
Third, the trough positions were filtered by removing the bucket size smaller than 0.005 ppm and larger than 0.3 ppm, since no real NMR peak would be in those ranges.
At last, the bucket table was imported to AMIX 4.0 as a pattern file, and large peaks could be split manually when necessary, but not required. No manual adjustment was needed for the demonstration data.
Detailed instruction with a simple example is in the supporting materials.
2.4. Data interpretation
All the NMR bucketing processing was carried out in Amix 4.0 (Bruker BioSpin) and exported to Excel (Microsoft) for further data analysis. The Pearson correlation relationship, student t-test (two tails) and coefficient of variation (CV) were calculated in Excel (Microsoft). The box plots were plotted in Matlab (R 2018, Mathworks) and the principal component analysis (PCA) was carried out in PLS-toolbox (Eigenvector Research).
3. Results
3.1. The line broadening factor for fecal samples
Line broadening is commonly used in NMR based metabolomics studies, a value of 0.3 lb was commonly applied to the NMR spectrum for metabolomics studies (Figure S1). When the values become larger, the line became broader and it is more effective to remove some small peaks (Figure 1A). When we use line broadening factor on the reference peaks, the extremely small buckets could be combined to their neighbor large buckets which help to reduce the peak splitting problem (one peak split into two buckets). However, when the line width was too broad, for example, 2lb in this case, it increases the probability of including two peaks in one bucket (3.525 ppm in Figure 1A red line).
In the fecal sample study, even when each spectrum was phased carefully and 0.3lb line broadening factors were applied before data analysis, the direct average was not a good choice for a reference spectrum because a large number of small peaks were created by averaging the spectra (Figure 1A blue line). The small peaks which look like noise were mainly produced from the spectra misalignment, although the misalignment was minor.
With the increase of the line broadening, the noise-like peaks were decreasing and the main peaks showed up, which indicated that the peak width range after smoothing could be applied as the boundary of a bucket. The 0.3 lb showed a better peak smoothing but still contained a large number of small peaks which are noise in most cases. For example, the small peaks around 3.515 ppm in Figure 1A cannot be removed using 0.3lb. Therefore, 0.6 lb was used as the minimum value for the following bucketing studies. The 2 lb method showed high noise suppression, however, many small peaks were combined in the process. For example, the doublet peak at 3.525 ppm became a singlet, so values larger than 2 lb were not investigated in this study. The line broadening process can overcome the problem caused by minor misalignment such as the peaks at 3.645 ppm by using a broader bucket size than the average peak width. The line broadening method is suitable for studies with minor misalignment such as fecal, tissue, and cell line studies. When the misalignment problem was significant, NMR peak alignment methods such as icoshift need to be applied before this method [11].
3.2. The line broadening influence to the bucket numbers and sizes
Fecal samples are commonly used in metabolomics and the water extracts of fecal samples usually have small misalignment, but not significant. A representative overlay of NMR spectra is listed in Figure 2, and the range selected showed slight misalignment which is normal in fecal metabolomics studies. The traditional rectangular bucketing method has a bucket size of 0.03 ppm (the most popular bucket size in literature) which will very likely split one peak into two different buckets and also could include several peaks into one bucket. This may not highly affect the chemometric models like PCA, which has noise suppression function but will highly affect statistical significance analysis, such as a t-test because one bucket may have several peaks from more than one metabolite which makes the t-test for one metabolite inaccurate.
Figure 2.
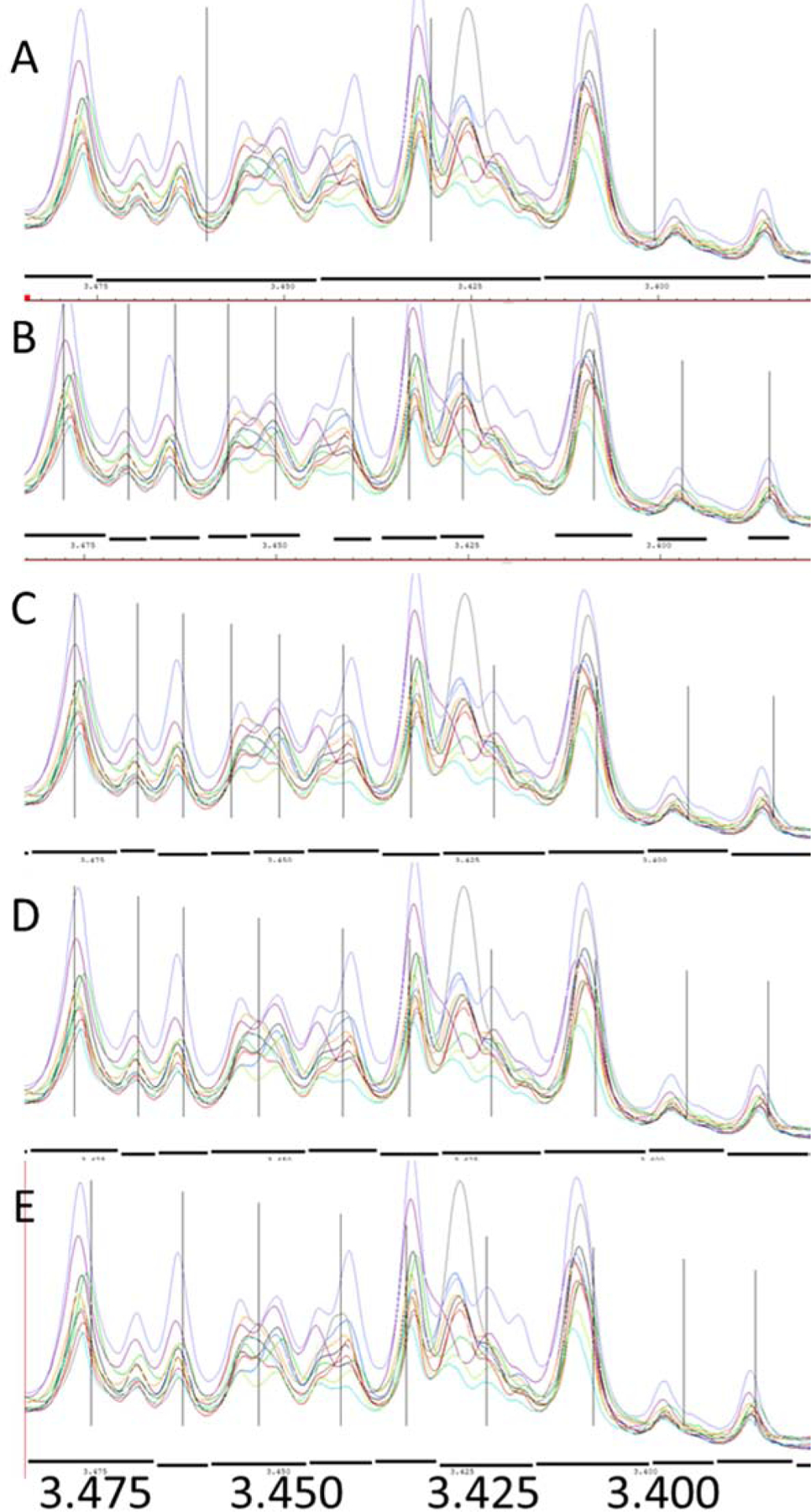
An example of the difference between the traditional rectangular bucketing and various line broadening was applied in a series of fecal extract NMR spectra. The black dashes under the spectra are the bucket sizes and the vertical lines are the center of the buckets. A. the traditional rectangular bucketing method with a bucket size of 0.03 ppm (typical in literature, traditional method reference); B. no line broadening factor on the average spectrum (no lb reference), C, Line broadening factor is 0.6; D, Line broadening factor is 1; E. Line broadening factor is 2. The bucket pattern was generated by Amix 4.0.
For no line broadening method (Figure 2 B) which acted as the no line broadening reference, the bucket size is close to the peak width of the overlay peaks. Since the method deleted small buckets (buckets smaller than 0.003 ppm), distances between two buckets were significant (for example, between the right two buckets). The biggest problem is that this method produced too many tiny buckets, and in many cases, one peak could be divided into many small peaks (<0.003pm) and could be completely deleted by the program due to the small bucket filtration (for example, peaks around 3.42 ppm). For the whole screen, 5499 buckets were discovered but only 257 of them (around 5%) were considered as valid (Table 1) due to the bucket size. Hence, line broadening is important to overcome the low-efficiency problem and to produce more accurate results. When the line broadening from 0.6 to 2 lb was applied, small buckets were significantly reduced. With the increase of the line broadening, the possibility of cutting off peaks to two buckets was decreased. The total valid buckets were significantly increased with the line broadening factor, and the 1 lb and 2 lb methods have obtained between 78% and 97% valid buckets (Table 1), representatively. While the bucket size is more reasonable with a large line broadening numbers (like 2 lb), it also increased the chance of combining peaks into one bucket (Figure 2E and Table S1). Therefore, a series of tests were applied further to investigate the lb value influence on the fecal samples in metabolomics studies.
Table 1.
The total buckets generated by different bucketing methods.
| Buckets identified (potentially) | Total valid buckets * | Total buckets picked | Valid percentage** | |
|---|---|---|---|---|
| No lb | 87 | 257 | 5499 | 0.047 |
| 0.6 lb | 103 | 793 | 1766 | 0.449 |
| 1 lb | 117 | 800 | 1027 | 0.779 |
| 2 lb | 107 | 494 | 510 | 0.969 |
| Manual | 129 | - | - |
Total valid buckets are buckets larger than 0.005 ppm and smaller than 0.3 ppm.
Valid percentage was calculated by the total valid buckets divided by the total buckets picked.
3.3. The metabolites data analysis compared with a manual bucketing method
In order to investigate the influence of different methods to peak intensities, a PCA study was applied to the same data generated by all methods. The metabolites peaks obtained by different methods varied, hence only the metabolite peaks picked by all methods were used in this study. According to the PCA score plot, all the methods showed high in-group consistency, but the different groups showed a clear difference (Figure 3). The PCA score plot showed the principal information of all the peak intensity information in a study, so the clear separation in the PCA score plot (Figure 3) indicated that distinct differences in peak intensities could be observed when different line broadening factors applied. The first principal component (PC1) represented more variances than the second principal component (PC2) with 43.43% vs 23.86%, so PC1 is relatively more important than PC2. Though the 1 lb and 2lb showed a similar distance to the manual separation, the 1 lb method is closer with the manual bucketing method in the PC1 direction, so it has a higher similarity compared to the manual bucketing method than the 2 lb method. The No lb and 0.6 lb showed a distinct difference in the score plot and the loading plot suggested that high ppm range contributes more in the separation (Figure S2) which may be due to the relatively low signal noise in the high chemical shift range (Figure S1).
Figure 3.
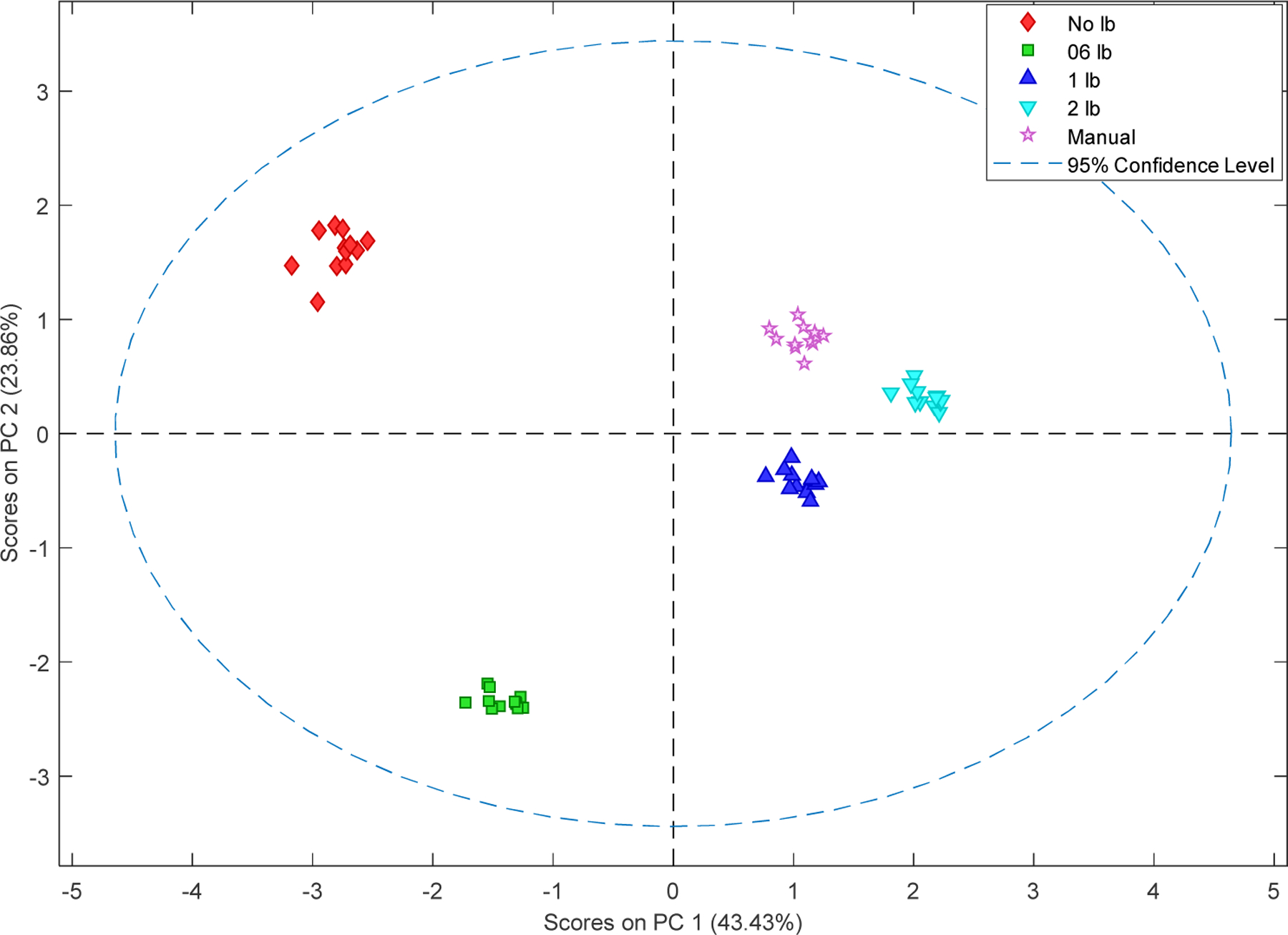
PCA of metabolites with the chemical shift from all the 5 methods. All five methods showed good reproducibility. Both No lb and 0.6 lb showed a large difference with the manual, 1 lb and 2 1lb methods.
Pearson correlation relationship is an indicator to compare the similarity of two groups of data. Considering the same data were analyzed by different methods, one can expect high R values between different line broadening methods versus the manual bucketing indicated high similarity with the manual bucketing method. Since the manual bucketing is the best bucketing pattern with the author’s best ability, higher R values indicated higher bucketing quality.
To compare the performance of every metabolite in each method, the Pearson correlation relationship R values were calculated for each peak between each line broadening value and the manual bucketing method. The R values of the representative peaks for each metabolite were listed in Table 2. High R values were observed from each method. The 1 lb method showed the highest R-value which is 0.9866, and 93% metabolites have R values higher than 0.97 (Table 2). The 0.6 lb showed the highest percentage (59%) of R values higher than 0.99, which is higher than 1 lb, which might be because of the smaller bucket size. The smaller bucket size may be more accurate for peaks with negligible misalignment.
Table 2.
The Pearson correlation relationship of metabolites between each line broadening method and the manual bucketing for each metabolite. Only one representative peak was selected for each metabolite if the metabolite has multiple peaks.
| Metabolites | ppm | No lb | 06 lb | 1 lb | 2 lb |
|---|---|---|---|---|---|
| Acetate | 1.92 | 0.9943 | 0.9983 | 0.9981 | 0.9981 |
| Alanine | 1.49 | 0.9971 | 0.9876 | 0.9852 | 0.9818 |
| Arabinose | 4.53 | 0.829 | 0.9814 | 0.9781 | 0.9398 |
| Aspartate | 2.71 | 0.9559 | 0.9973 | 0.9963 | 0.9957 |
| Butyrate | 1.61 | 0.9717 | 0.9917 | 0.9933 | 0.9898 |
| Cholate | 0.93 | 0.9956 | 0.9966 | 0.9962 | 0.9972 |
| Choline | 3.2 | 0.9762 | 0.9971 | 0.9973 | 0.9971 |
| Ethanol | 1.19 | 0.9959 | 0.9983 | 0.9981 | 0.9968 |
| Fumarate | 6.52 | 0.988 | 0.9996 | 0.9997 | 0.9645 |
| Glucose | 3.25 | 0.8149 | 0.9075 | 0.9968 | 0.9959 |
| Glutamate | 2.06 | 0.9917 | 0.9915 | 0.9897 | 0.992 |
| Glutamine | 2.45 | 0.9291 | 0.9842 | 0.9789 | 0.9254 |
| Glycine | 3.57 | 0.9923 | 0.9871 | 0.9877 | 0.9368 |
| Histidine may | 3.17 | 0.9103 | 0.9659 | 0.9317 | 0.9664 |
| Hypoxanthine | 8.2 | 0.6633 | 0.7512 | 0.9828 | 0.99 |
| Isoleucine | 1.02 | 0.9947 | 0.9932 | 0.9974 | 0.9922 |
| Lactate | 1.34 | 0.9838 | 0.995 | 0.9943 | 0.994 |
| Leucine | 1.76 | 0.991 | 0.9954 | 0.9969 | 0.9966 |
| Phenylalanine | 7.39 | 0.9675 | 0.9902 | 0.9872 | 0.9879 |
| Propionate | 1.06 | 0.9919 | 0.9908 | 0.9825 | 0.9792 |
| Sarcosine | 2.74 | 0.9751 | 0.9664 | 0.9326 | 0.9398 |
| Succinate | 2.41 | 0.9954 | 0.992 | 0.9887 | 0.9201 |
| Taurine | 3.28 | 0.9888 | 0.9917 | 0.9812 | 0.9872 |
| Threonine | 4.26 | 0.8773 | 0.9623 | 0.9762 | 0.9704 |
| Trimethylamine | 2.9 | 0.9718 | 0.8749 | 0.9893 | 0.9896 |
| Tyrosine | 6.91 | 0.9852 | 0.593 | 0.9922 | 0.9891 |
| Uracil | 5.82 | 0.9187 | 0.9931 | 0.9968 | 0.956 |
| Valine | 1.05 | 0.9876 | 0.9841 | 0.988 | 0.9929 |
| Xylose | 5.21 | 0.9958 | 0.9988 | 0.9975 | 0.9973 |
| Average | 0.9528 | 0.9606 | 0.9866 | 0.9779 | |
| Percentage (>0.99) | 0.38 | 0.59 | 0.48 | 0.41 | |
| Percentage (>0.98) | 0.55 | 0.76 | 0.83 | 0.66 | |
| Percentage (>0.97) | 0.69 | 0.76 | 0.93 | 0.72 |
Statistical significance analysis such as t-tests is usually applied in metabolomics studies, and for one metabolite, the p-value represents the confidence of the intensity difference between two groups [20]. The smaller the p values, the higher the probability that the values in the two groups are different. Table 3 showed p values for metabolites ranked by the manual method from low to high for peaks with p values lower than 0.1. As a result, the 1 lb method showed the closest average p values with the manual method and the No lb method produced the highest difference.
Table 3.
The p values of student t-test for selected peaks between the case and control groups. Metabolites with multiple peaks were reserved to study the method influence to the t-test p values. The p values were ordered from smallest to largest based on the manual method.
| ppm | No lb | 06 lb | 1 lb | 2 lb | Manual | |
|---|---|---|---|---|---|---|
| Cholate | 0.93 | 1.82E-04 | 6.78E-05 | 3.13E-05 | 3.64E-05 | 6.19E-05 |
| Leucine | 1.71 | 3.65E-02 | 2.20E-05 | 2.34E-03 | 8.11E-05 | 2.26E-03 |
| Leucine | 0.95 | 1.16E-03 | 4.41E-03 | 6.97E-03 | 6.19E-03 | 2.67E-03 |
| Isoleucine | 0.94 | 5.57E-03 | 1.31E-02 | 1.04E-02 | 5.80E-03 | 4.08E-03 |
| Butyrate | 1.61 | 7.42E-03 | 2.74E-03 | 2.71E-03 | 2.15E-03 | 5.87E-03 |
| Leucine | 1.76 | 2.35E-02 | 7.31E-03 | 9.01E-03 | 8.30E-03 | 9.82E-03 |
| Leucine | 1.74 | 2.68E-02 | 1.01E-02 | 1.05E-02 | 9.01E-03 | 1.08E-02 |
| Leucine | 1.76 | 1.75E-02 | 9.04E-03 | 1.14E-02 | 1.02E-02 | 1.21E-02 |
| Saccharopine | 3.08 | 1.23E-02 | 1.27E-02 | 1.19E-02 | 1.11E-02 | 1.26E-02 |
| Trimethylamine | 2.93 | 4.83E-01 | 3.79E-03 | 9.08E-03 | 1.07E-02 | 1.35E-02 |
| Trimethylamine | 2.90 | 3.35E-02 | 1.92E-02 | 2.28E-02 | 2.14E-02 | 1.61E-02 |
| Valine | 0.98 | 4.66E-02 | 3.17E-02 | 3.09E-02 | 3.03E-02 | 3.70E-02 |
| Isoleucine | 3.68 | 1.80E-01 | 1.61E-02 | 1.42E-02 | 1.77E-02 | 4.20E-02 |
| Leucine | 0.98 | 4.74E-02 | 9.60E-02 | 7.15E-02 | 7.25E-02 | 5.04E-02 |
| Ethanol | 3.65 | 5.67E-02 | 3.48E-02 | 3.54E-02 | 3.83E-02 | 5.23E-02 |
| Lactate | 4.13 | 2.02E-01 | 1.88E-02 | 2.52E-02 | 1.59E-02 | 5.42E-02 |
| Glutamate | 2.02 | 8.75E-02 | 6.82E-01 | 1.94E-01 | 4.65E-01 | 5.87E-02 |
| Threonine | 4.26 | 5.05E-01 | 8.05E-02 | 5.44E-02 | 1.77E-02 | 7.35E-02 |
| Lactate | 1.34 | 1.72E-01 | 5.37E-02 | 5.70E-02 | 5.55E-02 | 8.01E-02 |
| Average | 1.02E-01 | 5.77E-02 | 3.05E-02 | 4.20E-02 | 2.83E-02 |
4. Discussion
4.1. The line broadening methods influence to a t-test
In metabolomics area, chemometric methods such as PCA is commonly used for comparing groups. PCA describes the general data distribution and it has noise suppression function. So, the bucketing method may not have significant influences on the PCA results. In this study, most of the bucketing methods, except the no line broadening method, showed similar score plots with the traditional rectangular bucketing method (Figure S3). However, the accuracy of peak intensities would highly affect the t-test results since it requires high peak intensity accuracy. P values are usually one of the most important tests to distinguish the difference between groups for scientists from various areas [20]. As we described before, the traditional bucketing methods have common problems in peak combination which could easily combine two metabolite peaks into one bucket, so the traditional rectangular method has a natural disadvantage compared with the new method, and will not be investigated in this study. The manual bucketing method which was carefully designed to avoid bucket combination and split was used as a reference for this research purpose. As a result, both the 1 lb and 2 lb methods showed promising results compared to the manual method, which indicates that they provided accurate bucketing patterns. Since p values from a t-test are probabilities, the differences between the 1 lb, 2 lb, and manual methods usually cannot change the results of a t-test in a study unless they are close to a cut off value, for example, 0.05. Metabolomics is an area to study the simultaneous changes of metabolites in the response of various factors [29], hence metabolic pathway analysis may be considered to avoid false-positive problems when p values are close to the acceptable cut-off (for example, 0.05 in many cases).
4.2. The metabolites peak intensity
For the NMR bucketing method, peak intensity consistency in one set of samples is usually critical in metabolomics studies. This is because if a method could pick peaks from every sample equally, the comparison between control and case study groups will be accurately analyzed, and the statistical methods such as a t-test will also be more accurate. To investigate the peak intensity reliability, calculations based on the comparison of each peak with the manual bucketing method was designed. First, every peak obtained by each line broadening method was normalized to its corresponding peak obtained by the manual method. For example, the lactate peak intensity at 1.34 ppm for sample #1 obtained by the 1 lb method will be divided by the lactate peak intensity at 1.34 ppm for sample #1 obtained by the manual bucketing method. Therefore, a total of 12 samples with normalized numbers were calculated for lactate (1.34 ppm) and their values are theoretically very close to each other. Second, the coefficient of variation (CV) of the 12 normalized values of lactate (1.34 ppm) will be calculated to evaluate the similarity of the 12 numbers. In analytical chemistry, a CV smaller than 10% is usually considered as very good reproducibility [30, 31], and lower than 5% was considered as high robustness [32]. While low CV numbers were observed in most cases, it is clear to see the CVs tend to have the lowest mean around 0.6 and 1 lb methods, and the 1 lb method has all CV values lower than 10%, and even lower than 5%, except for the outliers (Figure 4). This indicates that the 1 lb method showed great reproducibility around all the samples for all peaks in comparison with the manual method. Considering the p values study in 4.1 and the R values study in 3.3, we can observe that the optimized value for the line broadening factor is around 1 lb in this study, and the peak combination problem caused by the broader bucket size may result in problems for peak intensity accuracy. The results are consistent with the raw visualized results in Figure 2, which showed a clear peak combination in the 2 lb bucket plot. Since every study is different, the best line broadening factor for each study may vary, the 1 lb to 2 lb methods are suggested for studies with minor overlap. In addition, the increase of line broadening factor (1 lb in this study) to the reference spectrum can increase the performance of peak border detection. As a comparison, the increase of line broadening factor to each spectrum has limited affection to the traditional rectangular bucketing method (Figure S4). Since bucket visualization could be easily obtained using Amix software, a visual check for representative areas is recommended in every study.
Figure 4.
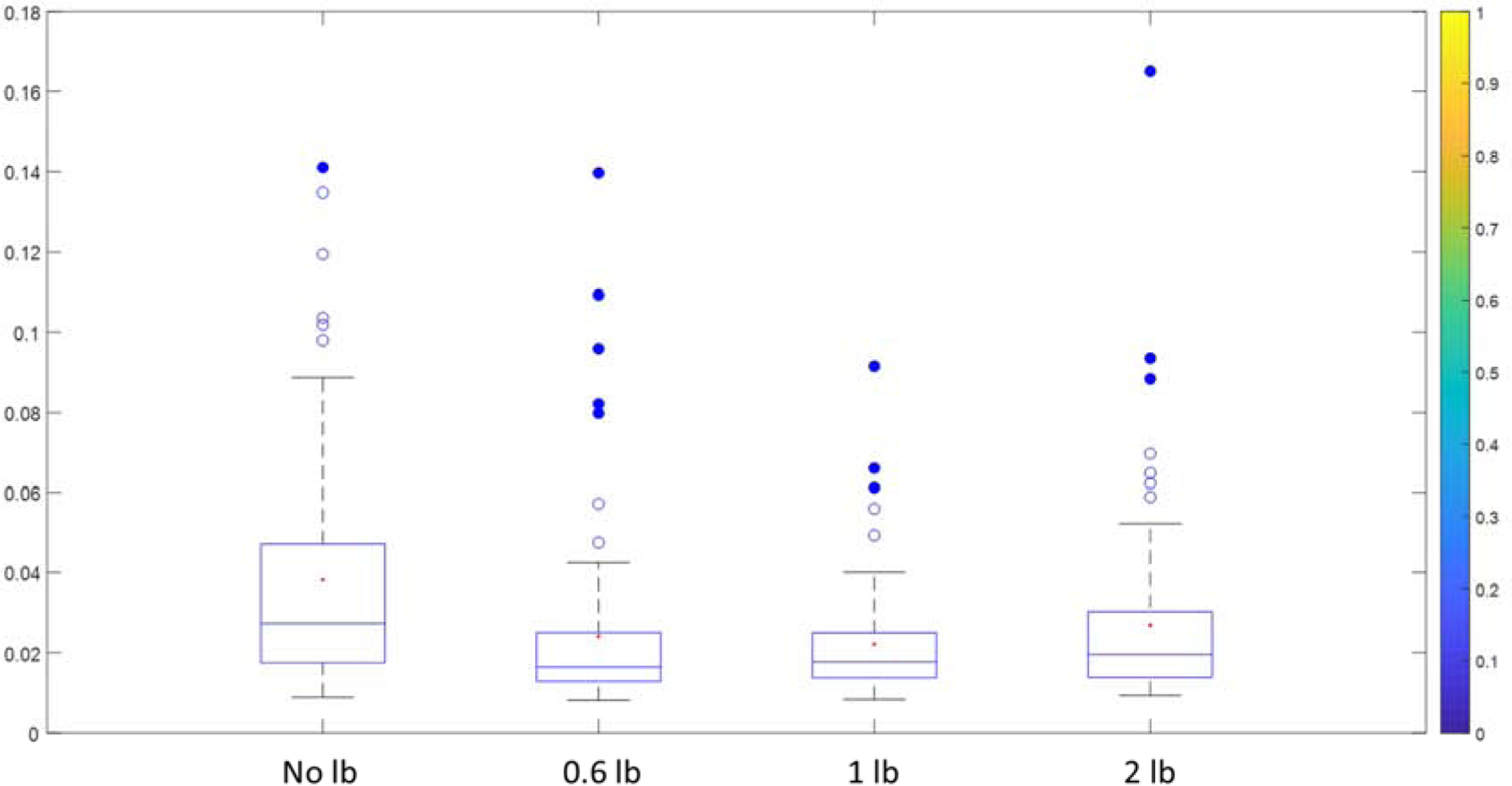
The box plot of the coefficient of variation (CV) for each line broadening method is normalized to a manual bucketing method. The CV was calculated for each metabolite among all the 12 samples. Each metabolite peak intensity was normalized to the same peak and the same sample but obtained by the manual bucketing method. For example, when the 1 lb method showed a low CV for glucose, it indicates that the glucose peak intensity obtained by the 1lb method is very close to the manual bucketing method for all samples.
5. Conclusion
In this study, the line broadening factors for the average spectrum of a fecal sample study was applied to NMR metabolomics spectra automatic bucketing. The boundary of peaks from the average spectrum after the applications of line broadening factors was applied as the size of the buckets to convert the NMR spectra to the data matrix. The bucket pattern could be easily visualized in Amix software where the bucketing can still be adjusted if needed. Our results showed that line broadening factor with the 1 lb method has the best performance in peak intensity accuracy and consistency between samples while both 0.6 and 2 lb methods showed acceptable performance. In summary, this study provides a convenient and efficient method for researchers to not only analyze NMR based metabolomics data automatically which is more efficient but also can generate visualized results that can help the end-users accurately convert instrument spectra to the data matrix.
Supplementary Material
A convenient NMR spectra bucketing method was developed for metabolomics studies
Line broadening factors were applied to adjust the automatic generation of bucketing patterns
Bucketing patterns could be easily visualized and manually adjusted by end-users
The high accuracy bucketing method is comparable to a carefully designed manual pattern.
6. Acknowledgment
The project described was supported by the National Center for Advancing Translational Sciences (NCATS), National Institutes of Health, through Grant Award Number UL1TR002489 (AMD).
BW would like to thank the department of chemistry of North Carolina A&T State University for the startup funding and the support in the applications of the NMR facility. LJ would like to thank the financial support of the New College of Florida.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
7 Reference
- [1].Beger R, Schnackenberg L, Holland R, Li D, Dragan Y, Metabonomic models of human pancreatic cancer using 1D proton NMR spectra of lipids in plasma, Metabolomics, 2 (2006) 125–134. [Google Scholar]
- [2].Cuperlovic-Culf M, Ferguson D, Culf A, Morin P, Touaibia M, 1H NMR Metabolomics Analysis of Glioblastoma Subtypes CORRELATION BETWEEN METABOLOMICS AND GENE EXPRESSION CHARACTERISTICS, Journal of Biological Chemistry, 287 (2012) 20164–20175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Powers R, NMR metabolomics and drug discovery, Magnetic Resonance in Chemistry, 47 (2009) S2–S11. [DOI] [PubMed] [Google Scholar]
- [4].Ametaj B, Zebeli Q, Saleem F, Psychogios N, Lewis M, Dunn S, Xia J, Wishart D, Metabolomics reveals unhealthy alterations in rumen metabolism with increased proportion of cereal grain in the diet of dairy cows, Metabolomics, 6 (2010) 583–594. [Google Scholar]
- [5].Murovec B, Makuc D, Repinc SK, Prevorsek Z, Zavec D, Sket R, Pecnik K, Plavec J, Stres B, 1H NMR metabolomics of microbial metabolites in the four MW agricultural biogas plant reactors: A case study of inhibition mirroring the acute rumen acidosis symptoms, Journal of Environmental Management, 222 (2018) 428–435. [DOI] [PubMed] [Google Scholar]
- [6].Emwas AH, Saccenti E, Gao X, McKay RT, dos Santos V, Roy R, Wishart DS, Recommended strategies for spectral processing and post-processing of 1D H-1-NMR data of biofluids with a particular focus on urine, Metabolomics, 14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Vu TN, Laukens K, Getting your peaks in line: a review of alignment methods for NMR spectral data, Metabolites, 3 (2013) 259–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nielsen NPV, Carstensen JM, Smedsgaard J, Aligning of single and multiple wavelength chromatographic profiles for chemometric data analysis using correlation optimised warping, Journal of Chromatography A, 805 (1998) 17–35. [Google Scholar]
- [9].Kumar K, Chemometric assisted correlation optimized warping of chromatograms: optimizing the computational time for correcting the drifts in chromatographic peak positions, Analytical Methods, 10 (2018) 1006–1014. [Google Scholar]
- [10].Forshed J, Schuppe-Koistinen I, Jacobsson SP, Peak alignment of NMR signals by means of a genetic algorithm, Analytica Chimica Acta, 487 (2003) 189–199. [Google Scholar]
- [11].Savorani F, Tomasi G, Engelsen SB, icoshift: A versatile tool for the rapid alignment of 1D NMR spectra, Journal of Magnetic Resonance, 202 (2010) 190–202. [DOI] [PubMed] [Google Scholar]
- [12].Sousa SAA, Magalhaes A, Castro Ferreira MM, Optimized bucketing for NMR spectra: Three case studies, Chemometrics and Intelligent Laboratory Systems, 122 (2013) 93–102. [Google Scholar]
- [13].Craig A, Cloareo O, Holmes E, Nicholson J, Lindon J, Scaling and normalization effects in NMR spectroscopic metabonomic data sets, Analytical Chemistry, 78 (2006) 2262–2267. [DOI] [PubMed] [Google Scholar]
- [14].Anderson PE, Reo NV, DelRaso NJ, Doom TE, Raymer ML, Gaussian binning: a new kernelbased method for processing NMR spectroscopic data for metabolomics, Metabolomics, 4 (2008) 261–272. [Google Scholar]
- [15].De Meyer T, Sinnaeve D, Van Gasse B, Tsiporkova E, Rietzschel ER, De Buyzere ML, Gillebert TC, Bekaert S, Martins JC, Van Criekinge W, NMR-based characterization of metabolic alterations in hypertension using an adaptive, intelligent binning algorithm, Analytical Chemistry, 80 (2008) 3783–3790. [DOI] [PubMed] [Google Scholar]
- [16].Anderson PE, Mahle DA, Doom TE, Reo NV, DelRaso NJ, Raymer ML, Dynamic adaptive binning: an improved quantification technique for NMR spectroscopic data, Metabolomics, 7 (2011) 179–190. [Google Scholar]
- [17].Davis RA, Charlton AJ, Godward J, Jones SA, Harrison M, Wilson JC, Adaptive binning: An improved binning method for metabolomics data using the undecimated wavelet transform, Chemometrics and Intelligent Laboratory Systems, 85 (2007) 144–154. [Google Scholar]
- [18].Rodriguez-Martinez A, Ayala R, Posma JM, Harvey N, Jimenez B, Sonomura K, Sato T-A, Matsuda F, Zalloua P, Gauguier D, Nicholson JK, Dumas M-E, pJRES Binning Algorithm (JBA): a new method to facilitate the recovery of metabolic information from pJRES H-1 NMR spectra, Bioinformatics, 35 (2019) 1916–1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Shi Z, Mirza M, Wang B, Kennedy MA, Weber GF, Osteopontin-a alters glucose homeostasis in anchorage-independent breast cancer cells, Cancer letters, (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Wang B, Shi Z, Weber G, Kennedy M, Introduction of a new critical p value correction method for statistical significance analysis of metabonomics data, Analytical and Bioanalytical Chemistry, 405 (2013) 8419–8429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Soininen P, Kangas AJ, Wurtz P, Tukiainen T, Tynkkynen T, Laatikainen R, Jarvelin M-R, Kahonen M, Lehtimaki T, Viikari J, Raitakari OT, Savolainen MJ, Ala-Korpela M, High-throughput serum NMR metabonomics for cost-effective holistic studies on systemic metabolism, Analyst, 134 (2009) 1781–1785. [DOI] [PubMed] [Google Scholar]
- [22].Craigie JS, MacKinnon SL, Walter JA, Liquid seaweed extracts identified using H-1 NMR profiles, Journal of Applied Phycology, 20 (2008) 665–671. [Google Scholar]
- [23].Čuperlović-Culf M, 4 - Metabolomics NMR data preprocessing – analysis of individual spectrum, in: Čuperlović-Culf M (Ed.) NMR Metabolomics in Cancer Research, Woodhead Publishing2013, pp. 215–259. [Google Scholar]
- [24].Malz F, Jancke H, Validation of quantitative NMR, Journal of Pharmaceutical and Biomedical Analysis, 38 (2005) 813–823. [DOI] [PubMed] [Google Scholar]
- [25].Mora-Ortiz M, Ramos PN, Oregioni A, Claus SP, NMR metabolomics identifies over 60 biomarkers associated with Type II Diabetes impairment in db/db mice, Metabolomics, 15 (2019) 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ahmadi S, Razazan A, Nagpal R, Jain S, Wang B, Mishra SP, Wang S, Justice J, Ding J, McClain DA, Metformin reduces aging-related leaky gut and improves cognitive function by beneficially modulating gut microbiome/goblet cell/mucin axis, The Journals of Gerontology: Series A, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tiziani S, Kang Y, Choi JS, Roberts W, Paternostro G, Metabolomic high-content nuclear magnetic resonance-based drug screening of a kinase inhibitor library, Nature communications, 2 (2011) 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Romick-Rosendale L, Goodpaster A, Hanwright P, Patel N, Wheeler E, Chona D, Kennedy M, NMR-based metabonomics analysis of mouse urine and fecal extracts following oral treatment with the broad-spectrum antibiotic enrofloxacin (Baytril), Magnetic Resonance in Chemistry, 47 (2009) S36–S46. [DOI] [PubMed] [Google Scholar]
- [29].Lindon JC, Nicholson JK, Holmes E, Everett JR, Metabonomics: Metabolic processes studied by NMR spectroscopy of biofluids, Concepts in Magnetic Resonance, 12 (2000) 289–320. [Google Scholar]
- [30].Wang B, Goodpaster AM, Kennedy MA, Coefficient of variation, signal-to-noise ratio, and effects of normalization in validation of biomarkers from NMR-based metabonomics studies, Chemometrics and Intelligent Laboratory Systems, 128 (2013) 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kostidis S, Addie RD, Morreau H, Mayboroda OA, Giera M, Quantitative NMR analysis of intra- and extracellular metabolism of mammalian cells: A tutorial, Analytica chimica acta, 980 (2017) 1–24. [DOI] [PubMed] [Google Scholar]
- [32].Tynkkynen T, Wang Q, Ekholm J, Anufrieva O, Ohukainen P, Vepsäläinen J, Männikkö M, Keinänen-Kiukaanniemi S, Holmes MV, Goodwin M, Proof of concept for quantitative urine NMR metabolomics pipeline for large-scale epidemiology and genetics, International journal of epidemiology, 48 (2019) 978–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.