

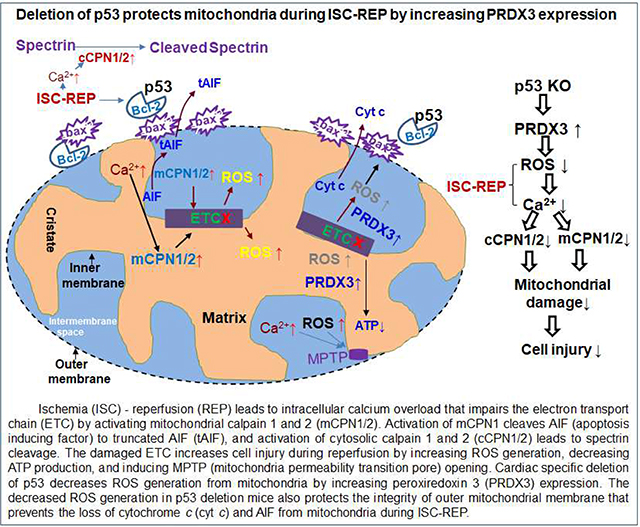
Abstract
p53 is a tumor suppressor protein with a very low content in the basal condition, but the content rapidly rises during stress conditions including ischemia-reperfusion. An increase in p53 content increases cardiac injury during ischemia-reperfusion. Since mitochondrial damage plays a key role in cardiac injury during ischemia-reperfusion, we asked if genetic ablation of p53 decreases cardiac injury by protecting mitochondria. Isolated, perfused hearts from cardiac specific p53 deletion or wild type underwent 25 min global ischemia at 37 oC and 60 min reperfusion. At the end of reperfusion, hearts were harvested for infarct size measurement. In separate groups, cardiac mitochondria were isolated at 30 min reperfusion. Time control hearts were buffer-perfused without ischemia. Compared to wild type, deletion of p53 improved cardiac functional recovery and decreased infarct size following ischemia-reperfusion. Oxidative phosphorylation was improved in p53 deletion mitochondria following ischemia-reperfusion compared to wild type. The net release of ROS generation from wild type but not in p53 deletion mitochondria was increased following ischemia-reperfusion. Peroxiredoxin 3 (PRDX 3) content was higher in p53 deletion than that in wild type, indicating that p53 deletion increases a key antioxidant. Ischemia-reperfusion led to increased spectrin cleavage (a marker of cytosolic calpain1 activation) in wild type but not in p53 deletion mice. Ischemia-reperfusion increased the truncation of mature AIF (apoptosis inducing factor, an indicator of mitochondrial calpain1 activation) in wild type but not in p53 deletion mice. The loss of cytochrome c from mitochondria was also decreased in p53 deletion following ischemia-reperfusion. Bcl-2 content was decreased in wild type but not in p53 deletion following reperfusion, suggesting that depletion of bcl-2 contributes to permeabilization of the mitochondrial outer membrane. Thus, deletion of p53 decreases cardiac injury by protecting mitochondria through attenuation of oxidative stress and calpain activation during ischemia-reperfusion.
Keywords: mitochondria, mitochondrial permeability transition pore, peroxiredoxins, reactive oxygen species
Graphical Abstract
1. Introduction
p53 is a tumor suppressor protein that regulates the cell cycle and DNA repair pathways [1]. The content of p53 is normally maintained at a very low level in cytosol through rapid ubiquitination and degradation [2]. However, p53 content is dramatically increased during stress conditions including ischemia and reperfusion due to slowed degradation [3]. Recent studies show that alteration of p53 content contributes to cardiac injury in pathological conditions. An increase in p53 content augments cell injury by interacting with death-related proteins [4]. p53 increases cell death by regulating the expression of a number of genes. For example, p53 negatively regulates apoptosis repressor with caspase recruitment domain to increase cardiac injury during ischemia-reperfusion [5]. Upregulation of p53 increases cardiac injury in rat hearts following ischemia-reperfusion by interacting with the mitochondrial E3 ubiquitin ligase 1 [6]. A reduced p53 expression decreases cardiac injury during ischemia-reperfusion [7–9]. p53 deletion affects gene expression in physiologic [10] and stress conditions [11–14]. p53 is also involved in maintaining cardiac homeostasis through regulation of microRNAs [11]. Administration of endogenous p53 antagonist with CHIP (carboxyl terminus of Hsp70-interacting protein) decreases apoptosis during ischemia-reperfusion by preventing poly-ADP-ribose-polymerase cleavage (PARP) cleavage [8]. Knockout of p53 maintains the integrity of outer mitochondrial membrane by increasing the ratio of bcl-2/bax through prevention of p53 translocation to the nucleus during triptolide-induced cardiac toxicity [12, 13]. Knockout of p53 also decreases cardiac injury during ischemia-reperfusion by promoting mitophagy [15] through inactivation of BNIP3 [9]. Genetic deletion of p53 also decreases cardiac injury during endoplasmic reticulum stress [16] and doxorubicin treatment [17, 18].
Mitochondrial damage contributes a central role in cardiac injury during ischemia-reperfusion [19, 20]. The damaged electron transport chain (ETC) leads to decreased energy production and increased ROS (reactive oxygen species) generation [21]. A burst of ROS generation during reperfusion favors mitochondrial permeability transition pore (MPTP) opening that leads to a release of mitochondrial intermembrane space proteins, including cytochrome c and apoptosis inducing factor (AIF), into cytosol leading to cell death [22]. In addition to MPTP, selective mitochondrial outer membrane permeation (MOMP) can occur [23, 24]. MOMP is mainly regulated by the bcl-2 family proteins including bcl-2 and bax located on the outer membrane [24]. A decrease in bcl-2 content or an increase in bax content increases MOMP [4]. An increase in p53 content increases MOMP by interacting with and inhibiting bcl-2 function [4].
The ETC is a key site of ROS generation in cardiac mitochondria [25]. A net release of ROS from mitochondria is balanced between the amount of ROS generated at the ETC and the capacity of mitochondrial antioxidants [26, 27]. During ischemia, complex I and complex III sustain damage leading to increased ROS generation and cell injury during reperfusion [25, 28, 29]. Mitochondria contain endogenous antioxidants including superoxide dismutase, catalase, and glutathione peroxidase. Knockout of p53 preserves Cu/Zn superoxide dismutase content in doxorubicin-treated hearts [18], suggesting that p53 contributes a role in regulation of antioxidants during stress conditions. Recent studies show that peroxiredoxins (PRDXs) are present within mitochondria [30]. PRDXs are a family of thiol specific antioxidant proteins. There are 6 PRDX isoforms in mammalian cells (PRDXs 1–6). PRDX 1, 2, 3, and 6 exist in the cytosol, and PRDX 4 is located in the endoplasmic reticulum [30]. PRDX 3 and 5 exist in the mitochondrial matrix [30]. PRDX 3 activity is inhibited in mouse heart mitochondria following ischemia-reperfusion [31]. Overexpression of PRDX3 decreases left ventricle remodeling and heart failure following myocardial infarction [32]. In addition, genetic knockout of PRDX 6 increases cell injury during ischemia-reperfusion [33], whereas administration of recombinant PRDX 6 decreases cardiac injury during oxidative stress [34]. These results support that PRDXs contribute to cardiac injury during ischemia-reperfusion. A recent study shows that alteration of an anti-aging gene, Klotho, affects p53 and PRDX 3 expression in the kidney [35]. We asked if genetic deletion of p53 in cardiac myocytes can alter PRDX 3 content in the heart.
Activation of calpains contributes to mitochondrial damage during ischemia-reperfusion [6, 36, 37]. Activation of mitochondrial calpain 1 cleaves AIF [36], subunits of complex I [37] and pyruvate dehydrogenase [38]. Activation of mitochondrial calpain 2 leads to the degradation of the complex I subunit ND6 (NADH dehydrogenase subunit 6) [39]. Activation of cytosolic calpains leads to the degradation of junctophilin-1 [40], junctophilin-2 [40, 41] and sarcolemmal Na+/K+-ATPase [42]. Interestingly, p53 is a potential substrate of calpain [43, 44]. In cancer cell line, activation of calpain increases apoptosis in a p53-dependent manner [45]. Genetic elimination of p53 may decrease cardiac injury during ischemia-reperfusion even despite the presence of activated calpains.
Specific deletion of p53 in cardiac myocytes decreases cell injury during endoplasmic reticulum stress [16]. In the present study, we asked if the cardiomyocyte specific deletion of p53 can decrease cardiac injury during ischemia-reperfusion. The focus of the current study is to investigate the contribution of cardiomyocyte deletion of p53 to decreased cardiac injury during ischemia-reperfusion in the isolated perfused mouse heart with a focus on the contribution of the protection of mitochondrial function due to p53 deletion. The role of cytosolic and mitochondrial calpain activation and decreased ROS generation, potentially via increased antioxidant capacity, in cardiac protection are studied.
2. METHODS
2.1. Preparation of mouse hearts for perfusion
The Animal Care and Use Committees of Virginia Commonwealth University and McGuire VA Medical Center approved this study. Floxed p53 mice were purchased from Jackson lab (Bar Harbor, ME). The floxed p53 mice were crossed with α-myosin heavy chain cre mice to generate cardiac specific p53 deletion mice [16]. p53 content was decreased in specific deletion mice compared to wild type (WT) (Figure 1A). Male p53 deletion or WT mice (2–3 months) were anesthetized with pentobarbital sodium (100 mg/kg, i.p.) and anticoagulated with heparin (1000 IU/kg, i.p.). The heart was isolated and mounted on a Langendorf apparatus and perfused retrograde via the aorta with modified Krebs-Henseleit buffer (115 mM NaCl, 4.0 mM KCl, 2.0 mM CaCl2, 26 mM NaHCO3, 1.1 mM MgSO4, 0.9 mM KH2PO4, and 5.5 mM glucose), gassed with 95% O2-5% CO2 to adjust pH to 7.35–7.45 [28]. Hearts were paced at 420 beats per minute during the periods of equilibration and reperfusion. The pacing was stopped during ischemia. The cardiac function was monitored with a balloon inserted into the left ventricle, and data were recorded digitally with Powerlab (AD Instruments, Colorado Springs, CO). After 15 min. equilibration perfusion, hearts underwent 25 min. global ischemia at 37°C and 30 min. reperfusion (mitochondrial study) or 60 min. reperfusion (infarct size measurement and functional assessment). In the time control group, hearts were only buffer perfused without ischemia.
Figure 1. Deletion of p53 decreases cardiac injury in mouse hearts following ischemia-reperfusion.

The content of p53 was decreased in cardiomyocyte specific deletion mice compared to wild type mice (WT) (Panel A). Mouse hearts were buffer perfused and exposed to 25 min. ischemia and 30 min. reperfusion. Cardiac contractility [reflected by left ventricular developed pressure (LVDP, mmHg)] was decreased following 60 min reperfusion (Panel B), whereas deletion of p53 improved contractile function compared to wild type mice (Panel B). Deletion of p53 also improved diastolic relaxation in hearts following 60 min reperfusion compared to wild type (Panel C). Representative pictures for infarct size measurement from wile type and p53 deletion mice are shown in Panel D. The infarct size was decreased in p53 deletion mice compared to wild type (Panel E). Mean ± SD. * p<0.05 vs. corresponding pre-ischemia. † p<0.05 vs. corresponding wild type hearts. n=7 in each group.
2.2. Determination of infarct size
Infarct size was measured at the end of 60 min. reperfusion in each group. The measurement of infarct size was previously described [46]. The heart was frozen at −20°C for 20 min., sectioned into 2 mm thick slices, incubated in 1% 2,3,5,-triphenyltetrazolium chloride (TTC) for 10 min. at 37 °C, and stored in 10% formalin followed by planimetry. Infarct size was expressed as percentage of the entire left ventricle.
2.3. Isolation of mouse heart mitochondria
The heart was removed from the perfusion column and placed in cold buffer A. The composition in mM was: 100 KCl, 50 MOPS [3-(N-morpholino)propanesulfonic acid], 1 EGTA, 5 MgSO4, and 1 mM ATP. The heart was blotted dry, weighed, and homogenized using a polytron tissue homogenizer at 10,000 rpm for 2.5 seconds in the presence of trypsin (5 mg/g tissue). The trypsin was used to increase the mitochondrial protein yield and remove potential cytosolic contamination. The homogenate was incubated for 15 min. at 4oC, then the same volume of buffer B [buffer A + 0.2% bovine serum albumin (BSA)] was added and the mixture was centrifuged at 500 × g for 10 min. The supernatant was again centrifuged at 3000 × g to pellet mitochondria. The mitochondrial pellet was first washed with buffer B, then re-suspended in KME (100 mM KCl, 50 mM MOPS, 0.5 mM EGTA), and centrifuged at 3000 × g to yield the final mitochondrial pellet. Mitochondria were re-suspended in KME for study [47].
2.4. Mitochondrial oxidative phosphorylation
Oxygen consumption in mitochondria was measured using a Clark-type oxygen electrode at 30°C as previously described [48]. Mitochondria were incubated in 80 mM KCl, 50 mM MOPS, 1 mM EGTA, 5 mM KH2PO4, and 1 mg defatted, dialyzed bovine serum albumin/ml at pH 7.4. Glutamate (20 mM) + Malate (10 mM) (complex I substrate), succinate (20 mM) plus 7.5 μM rotenone (complex II substrate), and TMPD (N,N,N’,N’ tetramethyl p-phenylenediamine, 1 mM)-ascorbate (10 mM, complex IV substrate) + rotenone were used.
2.5. Calcium retention capacity (CRC) in isolated mitochondria
CRC was used to assess calcium-induced mitochondrial permeability transition pore opening in isolated mitochondria [49]. CRC was evaluated in mitochondria (125 μg/ml) incubated in medium containing 150 mM sucrose, 50 mM KCl, 2 mM KH2PO4, 5 mM succinate in 20 mM Tris/HCl, pH 7.4 using sequential pulses of 5 nmol calcium. Extra-mitochondrial Ca2+ concentration was recorded with 0.5 μM Calcium Green-5N (Life Technologies) and fluorescence monitored with excitation and emission wavelengths set at 500 and 530 nm, respectively [49].
2.6. The production of H2O2 in isolated mitochondria
The net release of H2O2 from intact mitochondria was measured using the oxidation of the fluorogenic indicator amplex red in the presence of horseradish peroxidase (HRP)[25]. Glutamate (20 mM) + Malate (5 mM) or succinate (20 mM) + Rotenone (5 uM, rotenone was used to block reserve electron flow from complex II to complex I) were used as complex I or complex II substrates, respectively [25].
2.7. Immunoblotting
Proteins were separated using 12% or 4–15% Tris-glycine gels (Bio-Rad, Hercules, CA) and transferred to PVDF membrane (Millipore) using semi-dry transfer (Bio-Rad). The blots were incubated for 1 hr. at room temperature in 5% (w/v) non-fat dry milk (Bio-Rad) in TBST buffer (10 mM Tris pH 7.5, 150 mM NaCl, 0.1% Tween20) followed by the overnight incubation at 4 °C with primary antibody. After 1 hr. incubation at room temperature with a 1:10,000 dilution of HRP-conjugated anti-mouse or anti-rabbit IgG F(ab)2 (GE Healthcare Life Sciences, Piscataway, NJ), blots were developed using ECL Plus Western Blotting Detection Reagents (GE Healthcare Life Sciences) [37].
2.8. Statistical analysis
Data are expressed as the mean ± standard error. For all analyses, differences between groups (≥ 3 groups) were compared by one-way ANOVA. When a significant F value was obtained, means were compared using the Student-Newman-Keuls test of multiple comparisons. Differences between two groups were compared by two tail unpaired Student t-test (Version 3.5, Systat Software, Inc., San Jose, CA). Statistical significance was defined as a value of p<0.05.
3. Results
3.1. Cardiomyocyte specific deletion of p53 decreases cardiac injury in buffer-perfused hearts during REP
In buffer-perfused wildtype mouse hearts, ischemia-reperfusion led to decreased left ventricle developed pressure (LVDP, mean ± SD: 15 ± 11 mmHg) (Figure 1B) and increased left ventricle end diastolic pressure (LVEDP, mean ± SD: 44 ± 6 mmHg) (Figure 1C) compared to pre-ischemia. Compared to wildtype, deletion of p53 improved the LVDP (mean ± SD: 30 ± 13 mmHg) and decreased LVEDP (mean ± SD: 37 ± 3 mmHg) during reperfusion (Figure 1B and C). The pre-ischemia LVDP in p53 deletion was slightly lower than that in WT (mean ± SD: WT, 77 ± 15 vs. p53 deletion, 63 ± 2 mmHg, p<0.05), whereas LVEDP was similar between wild type and p53 deletion before ischemia (mean ± SD: WT 9 ± 2 vs. 10 ± 1 mmHg, p=NS). The deletion of p53 decreased the myocardial infarct size compared to wildtype mice (Figure 1D). These results support that deletion of p53 decreases cardiac injury during ischemia-reperfusion.
3.2. Deletion of p53 improves oxidative phosphorylation in hearts following ischemia-reperfusion
There were no differences in the rate of oxidative phosphorylation in control mitochondria between wildtype and p53 deletion when glutamate + malate was used as complex I substrate (Table 1). Succinate oxidation was also not altered in p53 deletion mice compared to wildtype mice. The rate of oxidation of TMPD-ascorbate was slightly decreased in p53 deletion mice compared to wildtype (Table 1). Compared to time control, ischemia-reperfusion markedly decreased the rate of oxidative phosphorylation with complex I, II, and IV substrates in mitochondria from both wildtype and p53 deletion hearts (Table 1). Ischemia-reperfusion also decreased uncoupler (dinitrophenol, DNP) stimulated respiration in mitochondria from wild type using glutamate + malate as complex I substrates (mean ± SD: Time control, 253 ± 57 vs. ischemia-reperfusion, 141 ± 40 nAO/min/mg, p<0.05). However, DNP-stimulated respiration was not significantly altered in p53 deletion mice following ischemia-reperfusion (mean ± SD: Time control, 243 ± 64 vs. ischemia-reperfusion, 194 ± 57 nAO/min/mg, p=NS). These results indicate that deletion of p53 improves oxidative phosphorylation in mitochondria following ischemia-reperfusion.
Table 1.
Oxidative phosphorylation in mitochondria isolated from wild type and p53 deletion mice
| Wild type | p53 deletion | |||
|---|---|---|---|---|
| Time control (n=6) | IR (n=6) | Time control (n=8) | IR (n=8) | |
| Complex I substrate (Glutamate + malate) | ||||
| State 3 (nAO/min/mg) | 308 ± 89 | 139 ± 34* | 252 ± 47 | 186 ± 42*† |
| State 4 (nAO/min/mg) | 107 ± 19 | 74 ± 9* | 86 ± 14 | 84 ± 11 |
| RCR | 2.9 ± 0.5 | 1.9 ± 0.4* | 3.2 ± 0.6 | 2.2 ± 0.4* |
| 2 mM (nAO/min/mg) | 287 ± 59 | 137 ± 51* | 246 ± 62 | 192 ± 60* |
| Complex II substrate (Succinate + rotenone) | ||||
| State 3 (nAO/min/mg) | 726 ± 47 | 288 ± 48* | 670 ± 75 | 318 ± 39* |
| State 4 (nAO/min/mg) | 234 ± 23 | 165 ± 17* | 202 ± 28 | 168 ± 10* |
| RCR | 3.3 ± 0.5 | 1.8 ± 0.2* | 3.4 ± 0.3 | 1.9 ± 0.3* |
| 2 mM ADP (nAO/min/mg) | 703 ± 52 | 331 ± 66* | 614 ± 63† | 375 ± 48* |
| Complex IV substrate (TMPD + Ascorbate) | ||||
| 2 mM ADP (nAO/min/mg) | 1856 ± 204 | 991 ± 212* | 1633 ± 151† | 1104 ± 168* |
Mean ± SD.
p <0.05 vs. corresponding time control
p <0.05 vs. corresponding wild type
IR, ischemia-reperfusion.
3.4. Deletion of p53 decreased ROS generation from mitochondria following reperfusion
The net release of H2O2 from isolated mitochondria was measured. H2O2 generation was decreased using mitochondria from time control p53 deletion compared to wild type when glutamate + malate was used as complex I substrate (mean ± SD: wild type, 118 ± 17 vs. p53 deletion, 98 ± 16 μmol/min/mg, p<0.05) (Figure 2B). Ischemia-reperfusion increased H2O2 generation in both wildtype (mean ± SD: Time control, 118 ± 17 vs. ischemia-reperfusion, 162 ± 21 pmol/min/mg, p<0.05) and p53 deletion (mean ± SD: Time control, 98 ± 16 vs. ischemia-reperfusion, 147 ± 30 pmol/min/mg, p<0.05) mitochondria compared to time control oxidizing complex I substrate. H2O2 generation tended to be lower in p53 deletion vs. wild type, but it did not reach statistical significance (Figure 2B). Ischemia-reperfusion increased H2O2 generation in both wild type (mean ± SD: Time control, 481 ± 49 vs. ischemia-reperfusion, 756 ± 101 pmol/min/mg, p<0.05) and p53 deletion (mean ± SD: Time control, 423 ± 84 vs. ischemia-reperfusion, 551 ± 114 pmol/min/mg, p<0.05) compared to time control when succinate + rotenone was used as complex II substrate (Figure 2C). Compared to wildtype, deletion of p53 led to decreased ROS generation during reperfusion oxidizing complex II substrate (mean ± SD: wild type, 756 ± 101 vs. p53 deletion, 551 ± 114 pmol/min/mg, p<0.05) (Figure 2C). Compared to control, ischemia-reperfusion decreased the calcium retention capacity (CRC) in both wildtype (mean ± SD: Time control, 892 ± 109 vs. ischemia-reperfusion, 353 ± 59 pmol/min/mg, p<0.05) and p53 deletion (mean ± SD: Time control, 920 ± 142 vs. ischemia-reperfusion, 410 ± 56 pmol/min/mg, p<0.05) mitochondria, supporting that ischemia-reperfusion sensitized to MPTP opening (Figure 2D). Deletion of p53 did not markedly improve the CRC compared to wild type (Figure 2D), suggesting that p53 deletion has a limited effect on MPTP opening during reperfusion.
Figure 2. Deletion of p53 improved oxidative phosphorylation in mouse mitochondria following ischemia-reperfusion.

Ischemia-reperfusion decreased the dinitrophenol (DNP)-uncoupled rate of respiration in mitochondria isolated from wildtype mice oxidizing complex I and II substrates. The baseline rate of oxidation in wildtype [Mean ± SD: complex I (253 ± 57) and complex II (606 ± 41)] and in p53 deletion [Mean ± SD, complex I (243 ± 64) and complex II (562 ± 45)] mice is given. The percentage decrease in respiration during reperfusion was attenuated in p53 deletion mice (Panel A). Compared to wild type mice, H2O2 generation was decreased in p53 deletion before ischemia with complex I substrate (Panel B). The generation of H2O2 was increased in mitochondria isolated from both wild type and p53 deletion following reperfusion using complex I substrate (Panel B). Deletion of p53 decreased H2O2 generation during reperfusion when succinate + rotenone was used as complex II substrate compared to wild type mice (Panel C). Ischemia-reperfusion decreased the calcium retention capacity (CRC) in mitochondria isolated from both wild type and p53 deletion mice compared to mitochondria from non-ischemic hearts. Deletion of p53 did not significantly improve the CRC in mitochondria isolated following ischemia and 30 min. reperfusion compared to wild type controls (Panel D). Mean ± SD.
* p<0.05 vs. corresponding pre-ischemia. † p<0.05 vs. corresponding wild type hearts. n=6–8 in each group.
3.5. Deletion of p53 decreased cytochrome c release and calpain activation during reperfusion
Compared to time control, the cytochrome c content in cytosol was increased in wild type mice following ischemia-reperfusion. Deletion of p53 significantly decreased the release of cytochrome c from mitochondria into cytosol during reperfusion (Figure 3A). Ischemia-reperfusion led to a decreased content of the mature AIF form (62 KD) in mitochondria isolated from wild type mice (Figure 3B), supporting that ischemia-reperfusion activates mitochondrial calpain 1. Deletion of p53 protected AIF content in mitochondria following reperfusion (Figure 3B). Compared to time control, the cleaved spectrin content was increased in wild type following ischemia-reperfusion (Figure 3C). In contrast, deletion of p53 did not increase the formation of cleaved spectrin during reperfusion (Figure 3C), indicating that deletion of p53 decreases cytosolic calpain activation. Ischemia-reperfusion led to decreased bcl-2 content in mitochondria isolated from wild type mice, whereas bcl-2 content was preserved in p53 deletion mice (Figure 3D).
Figure 3. Deletion of p53 decreased cytochrome c loss and calpain activation during reperfusion.

Immunoblotting showed that release of cytochrome c into the cytosol was increased in wild type mice following ischemia-reperfusion. Deletion of p53 led to decreased cytochrome c loss from mitochondria (Panel A). GAPDH was used as the cytosolic protein loading control (Panel A). The content of full-length AIF (62 KD) was decreased in wild type mitochondria following ischemia-reperfusion, whereas deletion of p53 protected AIF (62 KD) content during reperfusion (Panel B). VDAC was used as protein loading control (Panel B). The content of cleaved spectrin in cytosol was increased in wild type mice but not in p53 deletion mice with GAPDH as protein loading control (Panel C). bcl-2 content was decreased in mitochondria isolated from wild type hearts following ischemia-reperfusion. However, bcl-2 content was not decreased in mitochondria isolated from p53 deletion hearts following reperfusion (Panel D). Subunit 4 of cytochrome oxidase was used as a protein loading control. [Mean ± SD. *p <0.05 vs. TC (time control); n = 4 in each group)]
3.6. Deletion of p53 increased PRDX 3 but not PRDX 5 content
Compared to wild type mice, the content of PRDX 3 was increased in mitochondria from p53 deletion mice in the baseline condition (Figure 4A). The content of PRDX3 was still greater in p53 deletion mice following ischemia-reperfusion compared to wild type (Figure 4B). However, there were no differences in the content of PRDX 5 between wild type and p53 deletion mice (Figure 4C). These results indicate that deletion of p53 increased mitochondrial PRDX 3 content that may contribute to decreased ROS generation during reperfusion.
Figure 4. Deletion of p53 increased PRDX 3 content.

The contents of PRDX 3 and PRDX 5 were measured using immunoblotting. In mitochondria isolated from non-ischemic control mice, PRDX 3 content was increased in p53 deletion mitochondria compared to wild type mice in the baseline condition (Panel A). The PRDX3 content remained greater in p53 deletion mice following ischemia-reperfusion compared to wild type (Panel B). However, PRDX 5 content was not altered in p53 deletion mice compared to wild type mice (Panel C). Subunit 4 of cytochrome oxidase was used as a loading control. (Mean ± SD. *p <0.05 vs. wildtype; n = 4 in each group)
4. Discussion
In the present study, we show that cardiomyocyte specific deletion of p53 decreases cardiac injury during ischemia-reperfusion. Deletion of p53 decreases mitochondrial damage during reperfusion as shown by improved oxidative phosphorylation and decreased loss of cytochrome c and AIF from mitochondria. In addition, deletion of p53 decreases the activation of cytosolic calpains that contribute to cardiac injury during reperfusion. The deletion of p53 decreases the net release of ROS from mitochondria following reperfusion at least in part by increasing PRDX 3 content. The deletion of p53 preserves bcl-2 content that favors preserved integrity of the outer mitochondrial membrane. These results suggest that the deletion of p53 decreases cardiac injury during reperfusion by decreasing oxidative stress as well as the activation of cytosolic and mitochondrial calpains resulting in the protection of mitochondria.
4.1. Knockout of p53 protects hearts during ischemia-reperfusion
Modulation of p53 content alters cardiac injury during ischemia-reperfusion. An increase in p53 expression augments cardiac injury during ischemia-reperfusion [6]. Most studies show that inhibition of p53 decreases cardiac injury during reperfusion [7, 50], although negative results also exist in a recent study [51]. A reduced p53 expression decreases cardiac injury during ischemia-reperfusion [7–9]. Inhibition of p53 decreases cardiac injury by regulating gene expression including microRNA-30a [7], PARP [8], bcl-2, bax [12], p21 [14], and BNIP3 [9]. The current study shows that cardiac myocyte specific deletion of p53 decreases infarct size with improved cardiac function during reperfusion, supporting that p53 contributes to cardiac injury. In the present study, baseline contractility in p53 deletion mice is slightly lower than that observed in wild type. Deletion of p53 does not alter heart and body weight in the baseline condition [16]. Elimination of p53 leads to decreased cytochrome oxidase activity by reducing the contents of subunits 1 [52] and 2 [53]. Our current study finds a slight decrease in TMPD-ascorbate oxidation in p53 deletion mitochondria (Table 1). The mildly decreased cytochrome oxidase respiratory activity may contribute to a lower value of LVDP before ischemia in p53 deletion mice. A lower LVDP before ischemia may contribute to less injury during ischemia-reperfusion [37]. However, the substantial protection of mitochondrial function observed following ischemia-reperfusion in p53 deletion mice remains the likely effector of p53 mediated protection.
Activation of cytosolic calpains increases cardiac injury during reperfusion [54, 55]. Activation of cytosolic calpains leads to cleavage of spectrin that is used as a biomarker of cytosolic calpain activation [54, 55]. Ischemia-reperfusion increases the content of cleaved spectrin in wild type mice, indicating that cytosolic calpains are activated during reperfusion. Interestingly, a small amount of cleaved spectrin is shown in time control from p53 deletion mice, indicating that buffer-perfusion alone may start activating cytosolic calpains in p53 deletion mice. However, there were no differences in the cleaved spectrin content in time control and ischemia-reperfusion p53 deletion mice, suggesting that p53 deletion does not further increase cytosolic calpain activation, a mechanism of increased cardiac injury during reperfusion [37, 54]. Activation of calpain increases apoptotic cell death in a p53-dependent manner [45]. Therefore, p53 deletion may still decrease cardiac injury during ischemia-reperfusion.
4.2. ROS generation and mitochondrial antioxidants
The ETC is a key source of ROS generation in cardiac mitochondria [25]. Complex III is a major site of ROS generation in ischemia-damaged rat heart mitochondria [25]. Superoxide generated at the complex III Qo site is released into the cytosol through VDAC [27] that contributes to cardiac injury [56]. In wild type mice, the net release of H2O2 is increased in mitochondria using succinate as complex II substrate in the presence of rotenone. When rotenone is used to block reverse electron flow [57], electron flow from succinate is forward from complex II to complex III, a major site of ROS generation in cardiac myocytes [25]. Deletion of p53 leads to the decreased net release of ROS from mitochondria following reperfusion with succinate + rotenone, suggesting that deletion of p53 decreases the net release of ROS generation from complex III. As discussed above, less damage to the ETC can lead to decreased ROS generation in p53 deletion mice. In addition, the amount of ROS generation from mitochondria is affected by the content of endogenous antioxidants. We find that p53 deletion increased PRDX 3 content in mitochondria in the basal condition and following ischemia-reperfusion. PRDX 3 and PRDX 5 are mitochondria localized PRDXs [30]. However, the content of PRDX 5 is not altered in p53 deletion mice. An increase in PRDX 3 content likely contributes to the decreased net release of ROS generation in the p53 deletion mouse.
4.3. Mitochondrial damage and cardiac injury during ischemia-reperfusion
Mitochondrial damage contributes to cardiac injury during ischemia-reperfusion [58, 59]. In the baseline condition, p53 deletion does not significantly alter mitochondrial function including oxidative phosphorylation and calcium retention capacity (CRC) [16]. In wild type mice, ischemia-reperfusion decreases oxidative phosphorylation using complex I, complex II, and complex IV substrates. In contrast, the rate of oxidative phosphorylation is improved in deletion mice, suggesting that the mitochondrial ETC suffers less damage in p53 deletion mice following ischemia-reperfusion. In addition, improved protection of the ETC decreases cardiac injury during reperfusion by reducing ROS generation [28, 58]. Protection of mitochondria also decreases cardiac injury by reducing the release of proteins located within mitochondria including cytochrome c and AIF as discussed below [58, 59]. Recent studies show that impaired mitophagy contributes to mitochondrial damage during ischemia-reperfusion [37, 60]. The mitophagy is improved in p53 knockout mice following in vivo ischemia-reperfusion by inhibiting BNIP3 [9]. In addition to impacts on mitochondria, p53 also alters metabolism by impairing glycolysis during hypoxia [61]. Knockout of p53 decreases cell injury during hypoxia by increasing glycolysis [61]. Thus, p53 deletion may decrease cardiac injury during ischemia-reperfusion in part by promoting glycolysis.
4.4. p53 deletion leads to decreased loss of cytochrome c and AIF during reperfusion
Cytochrome c shuttles electrons between cytochrome c1 in complex III and cytochrome oxidase under physiological conditions, whereas a release of cytochrome c from mitochondria into cytosol triggers caspase-dependent programmed cell death [19, 62]. In wild type mice, the content of cytochrome c in mitochondria following ischemia-reperfusion is decreased compared to control. Deletion of p53 preserves cytochrome c during reperfusion. Cytochrome c is located in the intermembrane space and permeation of the outer membrane is required for cytochrome c release into cytosol [63, 64].
AIF, a nuclear-encoded protein, is located in the mitochondrial intermembrane space and contributes an important role in stabilizing complex I [36, 65, 66]. The release of AIF induces caspase-independent apoptotic cell death [67]. Nascent AIF is transported into mitochondria to form mature AIF (62 KD) after the removal of a mitochondrial leader sequence [68]. Mature AIF (62 KD) is bound on the inner mitochondrial membrane [68]. A cleaved form of AIF (57 KD) is formed during pathological conditions including ischemia-reperfusion by activation of mitochondrial calpain 1 [36, 65]. The cleaved AIF (57 KD) is detached from the inner membrane and can be released into the cytosol when the outer mitochondrial membrane is permeabilized [36, 65]. The current study found that the content of mature AIF (62 KD) is decreased in wild type, supporting that ischemia-reperfusion activates mitochondrial calpain 1. Theoretically, a decrease in 62 KD AIF should lead to increased 57 KD AIF [36]. However, the content of AIF (57 KD) is not increased in mitochondria isolated from wild type mice following reperfusion, indicating that a portion of AIF (57 KD) has been released from mitochondria into cytosol. Activation of mitochondrial calpain1 also contributes to respiratory chain damage [37]. Cleavage of AIF by calpain 1 not only facilitates AIF release from mitochondria to trigger caspase-independent apoptosis [67], but also destabilizes complex I and leads to decreased complex I activity [69]. Activation of mitochondrial calpain 2 also contributes to complex I damage during reperfusion [39]. Thus, deletion of p53 may protect mitochondria by decreasing mitochondrial calpain activation.
4.5. p53 deletion decreases the permeability of the outer mitochondrial membrane during reperfusion
Cytochrome c and AIF are located in the mitochondrial inner membrane space. Permeation of the outer mitochondrial membrane is required for the release of these proteins from mitochondria into cytosol [58, 59]. The permeability of the outer membrane is increased during reperfusion [28, 36]. MPTP opening is a common mechanism to increase the permeability of the outer membrane during ischemia-reperfusion [22, 70]. MPTP opening mainly occurs during early reperfusion [71, 72]. The deletion of p53 does not improve the CRC in mitochondria following reperfusion, suggesting that p53 deletion has a limited effect on MPTP opening during reperfusion. Since in the present study CRC was measured in mitochondria at a later time, 30 min, of reperfusion, rather than in early reperfusion [72], the potential protection of p53 deletion on MPTP opening may have been underestimated. The increased retention of cytochrome c and AIF in mitochondria during reperfusion present in the p53 deletion mouse would support this notion.
In addition to MPTP, selective mitochondrial outer membrane permeation (MOMP) is a second mechanism to increase the permeability of the outer membrane. MOMP is regulated by bcl-2 family proteins [23, 24, 73]. A deletion of bcl-2 in ischemia-damaged mitochondria increases the release of cytochrome c from mitochondria into cytosol through MOMP [23, 24]. An increase in p53 translocation to the nucleus during triptolide-induced cardiac toxicity leads to decreased bcl-2 and increased bax expression that favors MOMP [12]. Knockout of p53 leads to decreased MOMP by restoring bcl-2 content during cardiac toxicity [12]. In the present study, p53 deletion preserves bcl-2 content in mitochondria following reperfusion, supporting that p53 activation contributes to decreased bcl-2 content during reperfusion. Thus, deletion of p53 likely decreases cytochrome c loss from mitochondria by decreasing MOMP through protection of bcl-2.
4.6. Limitations
Several limitations need to be considered in the current study. Although we find that p53 deletion improves mitochondrial function during ischemia-reperfusion, genes responsible for this protection were not identified, a substantial limitation. Since cardiac myocyte specific p53 deletion mice are generated in the current study, some residual p53 content is shown in cardiac homogenates of the p53 deletion hearts due to the presence of p53 in cells other than cardiac myocytes including endothelial cells [16]. A translocation of p53 from cytosol to mitochondria is a potential mechanism to induce mitochondrial dysfunction [4]. However, we were unable to detect an increased p53 content in mitochondria following reperfusion, consistent with previous publications [16, 51]. In the current study, mitochondria are purified by trypsin treatment that may potentially remove p53 attached to the mitochondrial outer membrane. Complex III is a key site of ROS generation [17, 27]. A direct measurement of complex III activity may provide further support that p53 deletion decreases ROS generation by improving complex III activity. Although we find the increased PRDX 3 content in p53 deletion mice, the mechanism by which deletion of p53 leads to increased PRDX 3 remains unknown. The relationship between p53 and Klotho gene expression warrants further study to address this potential mechanism [35].
4.7. Conclusion
The deletion of p53 decreases cardiac injury during endoplasmic reticulum stress [16, 51]. The current study shows that deletion of p53 decreases the cardiac injury during ischemia-reperfusion with mechanisms involving enhanced antioxidant expression via PRDX 3 and decreased cytosolic and mitochondrial calpain activation. These studies indicate that modulation of p53 expression is a potential strategy to decrease cardiac injury in pathological conditions.
Supplementary Material
Table 2.
Antibodies used
| Antibody name | Company | Catalog number | Concentration |
|---|---|---|---|
| AIF | Cell Signaling | 4642 | 1:1000 |
| Bcl-2 | Santa Cruz | sc-7382 | 1:500 |
| Calpain 1 | ThermoFisher Scientific | MA3-940 | 1:1000 |
| Complex IV (subunit 4) | Cell Signaling | 4844 | 1:1000 |
| Cytochrome c | ThermoFisher Scientific | 710627 | 1:2500 |
| GAPDH | Cell Signaling | 5174 | 1:1000 |
| p53 | Santa Cruz | sc-126 | 1:100 |
| PRDX3 | abcam | ab73349 | 1:500 |
| PRDX5 | ThermoFisher Scientific | LF-MA0002 | 1:1000 |
| Spectrin | Santa Cruz | sc-46696 | 1:100 |
| VDAC | Abcam | 14715 | 1:2500 |
Highlights.
Cardiomyocyte specific deletion of p53 decreases cardiac injury in mouse hearts following ischemia-reperfusion by protecting mitochondria.
Cardiomyocyte specific deletion of p53 maintains bcl-2 content during ischemia-reperfusion with enhanced retention of cytochrome c by mitochondria.
p53 knockout increased peroxiredoxin 3 content and decreased ROS generation from mitochondria.
Cardiomyocyte specific deletion of p53 decreased cytosolic and mitochondrial calpain 1 activation during ischemia-reperfusion.
ACKNOWLEDGMENTS
This work was supported by R21(AG054975-01) from NIA (QC), a Department of Veterans Affairs Merit Review Award (2IO1BX001355-01A2) (EJL, QC), the Pauley Heart Pilot grant (QC), and Virginia Commonwealth University (QC, EJL).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
5. References
- [1].Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T, Matoba S, Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart, Nat Commun 4 (2013) 2308. [DOI] [PubMed] [Google Scholar]
- [2].Perry ME, Mendrysa SM, Saucedo LJ, Tannous P, Holubar M, p76(MDM2) inhibits the ability of p90(MDM2) to destabilize p53, J Biol Chem 275 (2000) 5733–5738. [DOI] [PubMed] [Google Scholar]
- [3].Forini F, Kusmic C, Nicolini G, Mariani L, Zucchi R, Matteucci M, Iervasi G, Pitto L, Triiodothyronine prevents cardiac ischemia/reperfusion mitochondrial impairment and cell loss by regulating miR30a/p53 axis, Endocrinology 155 (2014) 4581–4590. [DOI] [PubMed] [Google Scholar]
- [4].Tsipis A, Athanassiadou AM, Athanassiadou P, Kavantzas N, Agrogiannis G, Patsouris E, Apoptosis-related factors p53, bcl-2 and the defects of force transmission in dilated cardiomyopathy, Pathol Res Pract 206 (2010) 625–630. [DOI] [PubMed] [Google Scholar]
- [5].Xu T, Ding W, Ao X, Chu X, Wan Q, Wang Y, Xiao D, Yu W, Li M, Yu F, Wang J, ARC regulates programmed necrosis and myocardial ischemia/reperfusion injury through the inhibition of mPTP opening, Redox Biol 20 (2019) 414–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Cao T, Fan S, Zheng D, Wang G, Yu Y, Chen R, Song LS, Fan GC, Zhang Z, Peng T, Increased calpain-1 in mitochondria induces dilated heart failure in mice: role of mitochondrial superoxide anion, Basic Res Cardiol 114 (2019) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang C, Liao P, Liang R, Zheng X, Jian J, Epigallocatechin gallate prevents mitochondrial impairment and cell apoptosis by regulating miR-30a/p53 axis, Phytomedicine 61 (2019) 152845. [DOI] [PubMed] [Google Scholar]
- [8].Naito AT, Okada S, Minamino T, Iwanaga K, Liu ML, Sumida T, Nomura S, Sahara N, Mizoroki T, Takashima A, Akazawa H, Nagai T, Shiojima I, Komuro I, Promotion of CHIP-mediated p53 degradation protects the heart from ischemic injury, Circ Res 106 (2010) 1692–1702. [DOI] [PubMed] [Google Scholar]
- [9].Hoshino A, Matoba S, Iwai-Kanai E, Nakamura H, Kimata M, Nakaoka M, Katamura M, Okawa Y, Ariyoshi M, Mita Y, Ikeda K, Ueyama T, Okigaki M, Matsubara H, p53-TIGAR axis attenuates mitophagy to exacerbate cardiac damage after ischemia, J Mol Cell Cardiol 52 (2012) 175–184. [DOI] [PubMed] [Google Scholar]
- [10].Mak TW, Hauck L, Grothe D, Billia F, p53 regulates the cardiac transcriptome, Proc Natl Acad Sci U S A 114 (2017) 2331–2336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Stanley-Hasnain S, Hauck L, Grothe D, Aschar-Sobbi R, Beca S, Butany J, Backx PH, Mak TW, Billia F, p53 and Mdm2 act synergistically to maintain cardiac homeostasis and mediate cardiomyocyte cell cycle arrest through a network of microRNAs, Cell Cycle 16 (2017) 1585–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Xi Y, Wang W, Wang L, Pan J, Cheng Y, Shen F, Huang Z, Triptolide induces p53-dependent cardiotoxicity through mitochondrial membrane permeabilization in cardiomyocytes, Toxicol Appl Pharmacol 355 (2018) 269–285. [DOI] [PubMed] [Google Scholar]
- [13].Regula KM, Kirshenbaum LA, p53 activates the mitochondrial death pathway and apoptosis of ventricular myocytes independent of de novo gene transcription, J Mol Cell Cardiol 33 (2001) 1435–1445. [DOI] [PubMed] [Google Scholar]
- [14].Li H, Zou T, Meng S, Peng YZ, Yang JF, p21 protects cardiomyocytes against ischemia-reperfusion injury by inhibiting oxidative stress, Mol Med Rep 17 (2018) 4665–4671. [DOI] [PubMed] [Google Scholar]
- [15].Liu CY, Zhang YH, Li RB, Zhou LY, An T, Zhang RC, Zhai M, Huang Y, Yan KW, Dong YH, Ponnusamy M, Shan C, Xu S, Wang Q, Zhang YH, Zhang J, Wang K, LncRNA CAIF inhibits autophagy and attenuates myocardial infarction by blocking p53-mediated myocardin transcription, Nat Commun 9 (2018) 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Chen Q, Thompson J, Hu Y, Das A, Lesnefsky EJ, Cardiac Specific Knockout of p53 Decreases ER Stress-Induced Mitochondrial Damage, Front Cardiovasc Med 6 (2019) 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Li J, Wang PY, Long NA, Zhuang J, Springer DA, Zou J, Lin Y, Bleck CKE, Park JH, Kang JG, Hwang PM, p53 prevents doxorubicin cardiotoxicity independently of its prototypical tumor suppressor activities, Proc Natl Acad Sci U S A 116 (2019) 19626–19634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shizukuda Y, Matoba S, Mian OY, Nguyen T, Hwang PM, Targeted disruption of p53 attenuates doxorubicin-induced cardiac toxicity in mice, Mol Cell Biochem 273 (2005) 25–32. [DOI] [PubMed] [Google Scholar]
- [19].Gustafsson AB, Gottlieb RA, Heart mitochondria: gates of life and death, Cardiovasc Res 77 (2008) 334–343. [DOI] [PubMed] [Google Scholar]
- [20].Lesnefsky EJ, Moghaddas S, Tandler B, Kerner J, Hoppel CL, Mitochondrial dysfunction in cardiac disease: ischemia-reperfusion, aging, and heart failure., J Mol Cell Cardiol 33 (2001) 1065–1089. [DOI] [PubMed] [Google Scholar]
- [21].Chen Q, Camara AK, Stowe DF, Hoppel CL, Lesnefsky EJ, Modulation of electron transport protects cardiac mitochondria and decreases myocardial injury during ischemia and reperfusion, Am J Physiol Cell Physiol 292 (2007) C137–147. [DOI] [PubMed] [Google Scholar]
- [22].Weiss JN, Korge P, Honda HM, Ping P, Role of the mitochondrial permeability transition in myocardial disease, Circ Res 93 (2003) 292–301. [DOI] [PubMed] [Google Scholar]
- [23].Chen Q, Xu H, Xu A, Ross T, Bowler E, Hu Y, Lesnefsky EJ, Inhibition of Bcl-2 sensitizes mitochondrial permeability transition pore (MPTP) opening in ischemia-damaged mitochondria, PLoS One 10 (2015) e0118834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Ishikita A, Matoba T, Ikeda G, Koga J, Mao Y, Nakano K, Takeuchi O, Sadoshima J, Egashira K, Nanoparticle-Mediated Delivery of Mitochondrial Division Inhibitor 1 to the Myocardium Protects the Heart From Ischemia-Reperfusion Injury Through Inhibition of Mitochondria Outer Membrane Permeabilization: A New Therapeutic Modality for Acute Myocardial Infarction, J Am Heart Assoc 5 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Chen Q, Vazquez EJ, Moghaddas S, Hoppel CL, Lesnefsky EJ, Production of reactive oxygen species by mitochondria: Central role of complex III, J Biol Chem 278 (2003) 36027–36031. [DOI] [PubMed] [Google Scholar]
- [26].Turrens JF, Mitochondrial formation of reactive oxygen species, J Physiol 552 (2003) 335–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Han D, Antunes F, Canali R, Rettori D, Cadenas E, Voltage-dependent anion channels control the release of the superoxide anion from mitochondria to cytosol, J Biol Chem 278 (2003) 5557–5563. [DOI] [PubMed] [Google Scholar]
- [28].Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ, Reversible blockade of electron transport during ischemia protects mitochondria and decreases myocardial injury following reperfusion, J Pharmacol Exp Ther 319 (2006) 1405–1412. [DOI] [PubMed] [Google Scholar]
- [29].Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ, Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria, Am J Physiol Cell Physiol 294 (2008) C460–466. [DOI] [PubMed] [Google Scholar]
- [30].Poynton RA, Hampton MB, Peroxiredoxins as biomarkers of oxidative stress, Biochim Biophys Acta 1840 (2014) 906–912. [DOI] [PubMed] [Google Scholar]
- [31].Kumar V, Kitaeff N, Hampton MB, Cannell MB, Winterbourn CC, Reversible oxidation of mitochondrial peroxiredoxin 3 in mouse heart subjected to ischemia and reperfusion, FEBS Lett 583 (2009) 997–1000. [DOI] [PubMed] [Google Scholar]
- [32].Matsushima S, Ide T, Yamato M, Matsusaka H, Hattori F, Ikeuchi M, Kubota T, Sunagawa K, Hasegawa Y, Kurihara T, Oikawa S, Kinugawa S, Tsutsui H, Overexpression of mitochondrial peroxiredoxin-3 prevents left ventricular remodeling and failure after myocardial infarction in mice, Circulation 113 (2006) 1779–1786. [DOI] [PubMed] [Google Scholar]
- [33].Eismann T, Huber N, Shin T, Kuboki S, Galloway E, Wyder M, Edwards MJ, Greis KD, Shertzer HG, Fisher AB, Lentsch AB, Peroxiredoxin-6 protects against mitochondrial dysfunction and liver injury during ischemia-reperfusion in mice, Am J Physiol Gastrointest Liver Physiol 296 (2009) G266–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Karaduleva EV, Mubarakshina EK, Sharapov MG, Volkova AE, Pimenov OY, Ravin VK, Kokoz YM, Novoselov VI, Cardioprotective Effect of Modified Peroxiredoxins in Retrograde Perfusion of Isolated Rat Heart under Conditions of Oxidative Stress, Bull Exp Biol Med 160 (2016) 639–642. [DOI] [PubMed] [Google Scholar]
- [35].Kimura T, Shiizaki K, Akimoto T, Shinzato T, Shimizu T, Kurosawa A, Kubo T, Nanmoku K, Kuro OM, Yagisawa T, The impact of preserved Klotho gene expression on antioxidative stress activity in healthy kidney, Am J Physiol Renal Physiol 315 (2018) F345–352. [DOI] [PubMed] [Google Scholar]
- [36].Chen Q, Paillard M, Gomez L, Ross T, Hu Y, Xu A, Lesnefsky EJ, Activation of mitochondrial mu-calpain increases AIF cleavage in cardiac mitochondria during ischemia-reperfusion, Biochem Biophys Res Commun 415 (2011) 533–538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chen Q, Thompson J, Hu Y, Dean J, Lesnefsky EJ, Inhibition of the ubiquitous calpains protects complex I activity and enables improved mitophagy in the heart following ischemia-reperfusion, Am J Physiol Cell Physiol 317 (2019) C910–921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Thompson J, Hu Y, Lesnefsky EJ, Chen Q, Activation of mitochondrial calpain and increased cardiac injury: beyond AIF release, Am J Physiol Heart Circ Physiol 310 (2016) H376–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Shintani-Ishida K, Yoshida K, Mitochondrial m-calpain opens the mitochondrial permeability transition pore in ischemia-reperfusion, Int J Cardiol 197 (2015) 26–32. [DOI] [PubMed] [Google Scholar]
- [40].Murphy RM, Dutka TL, Horvath D, Bell JR, Delbridge LM, Lamb GD, Ca2+-dependent proteolysis of junctophilin-1 and junctophilin-2 in skeletal and cardiac muscle, J Physiol 591 (2013) 719–729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Wu CY, Chen B, Jiang YP, Jia Z, Martin DW, Liu S, Entcheva E, Song LS, Lin RZ, Calpain-dependent cleavage of junctophilin-2 and T-tubule remodeling in a mouse model of reversible heart failure, J Am Heart Assoc 3 (2014) e000527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Singh RB, Dhalla NS, Ischemia-reperfusion-induced changes in sarcolemmal Na+/K+-ATPase are due to the activation of calpain in the heart, Can J Physiol Pharmacol 88 (2010) 388–397. [DOI] [PubMed] [Google Scholar]
- [43].Chen Z, Boor PJ, Finnerty CC, Herndon DN, Albrecht T, Calpain-mediated cleavage of p53 in human cytomegalovirus-infected lung fibroblasts, FASEB Bioadv 1 (2019) 151–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Chen Q, Lesnefsky EJ, Heart mitochondria and calpain 1: Location, function, and targets, Biochim Biophys Acta 1852 (2015) 2372–2378. [DOI] [PubMed] [Google Scholar]
- [45].Atencio IA, Ramachandra M, Shabram P, Demers GW, Calpain inhibitor 1 activates p53-dependent apoptosis in tumor cell lines, Cell Growth Differ 11 (2000) 247–253. [PubMed] [Google Scholar]
- [46].Hedayati N, Schomisch SJ, Carino JL, Timothy Sherwood J, Lesnefsky EJ, Cmolik BL, Cardioprotection by St Thomas’ solution is mediated by protein kinase C and tyrosine kinase, J Surg Res 113 (2003) 121–127. [DOI] [PubMed] [Google Scholar]
- [47].Szczepanek K, Chen Q, Derecka M, Salloum FN, Zhang Q, Szelag M, Cichy J, Kukreja RC, Dulak J, Lesnefsky EJ, Larner AC, Mitochondrial-targeted Signal transducer and activator of transcription 3 (STAT3) protects against ischemia-induced changes in the electron transport chain and the generation of reactive oxygen species, J Biol Chem 286 (2011) 29610–29620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Lesnefsky EJ, Tandler B, Ye J, Slabe TJ, Turkaly J, Hoppel CL, Myocardial ischemia decreases oxidative phosphorylation through cytochrome oxidase in subsarcolemmal mitochondria, Am J Physiol 273 (1997) H1544–1554. [DOI] [PubMed] [Google Scholar]
- [49].Paillard M, Gomez L, Augeul L, Loufouat J, Lesnefsky EJ, Ovize M, Postconditioning inhibits mPTP opening independent of oxidative phosphorylation and membrane potential, J Mol Cell Cardiol 46 (2009) 902–909. [DOI] [PubMed] [Google Scholar]
- [50].Du JK, Cong BH, Yu Q, Wang H, Wang L, Wang CN, Tang XL, Lu JQ, Zhu XY, Ni X, Upregulation of microRNA-22 contributes to myocardial ischemia-reperfusion injury by interfering with the mitochondrial function, Free Radic Biol Med 96 (2016) 406–417. [DOI] [PubMed] [Google Scholar]
- [51].Yano T, Abe K, Tanno M, Miki T, Kuno A, Miura T, Steenbergen C, Does p53 Inhibition Suppress Myocardial Ischemia-Reperfusion Injury?, J Cardiovasc Pharmacol Ther 23 (2018) 350–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Flores ER, Lozano G, The p53 family grows old, Genes Dev 26 (2012) 1997–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Matoba S, Kang JG, Patino WD, Wragg A, Boehm M, Gavrilova O, Hurley PJ, Bunz F, Hwang PM, p53 regulates mitochondrial respiration, Science 312 (2006) 1650–1653. [DOI] [PubMed] [Google Scholar]
- [54].Hernando V, Inserte J, Sartorio CL, Parra VM, Poncelas-Nozal M, Garcia-Dorado D, Calpain translocation and activation as pharmacological targets during myocardial ischemia/reperfusion, J Mol Cell Cardiol 49 (2010) 271–279. [DOI] [PubMed] [Google Scholar]
- [55].Inserte J, Barba I, Hernando V, Abellan A, Ruiz-Meana M, Rodriguez-Sinovas A, Garcia-Dorado D, Effect of acidic reperfusion on prolongation of intracellular acidosis and myocardial salvage, Cardiovasc Res 77 (2008) 782–790. [DOI] [PubMed] [Google Scholar]
- [56].Becker LB, vanden Hoek TL, Shao ZH, Li CQ, Schumacker PT, Generation of superoxide in cardiomyocytes during ischemia before reperfusion, Am J Physiol 277 (1999) H2240–2246. [DOI] [PubMed] [Google Scholar]
- [57].Ross T, Szczepanek K, Bowler E, Hu Y, Larner A, Lesnefsky EJ, Chen Q, Reverse electron flow-mediated ROS generation in ischemia-damaged mitochondria: role of complex I inhibition vs. depolarization of inner mitochondrial membrane, Biochim Biophys Acta 1830 (2013) 4537–4542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Lesnefsky EJ, Chen Q, Hoppel CL, Mitochondrial Metabolism in Aging Heart, Circ Res 118 (2016) 1593–1611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Lesnefsky EJ, Chen Q, Tandler B, Hoppel CL, Mitochondrial Dysfunction and Myocardial Ischemia-Reperfusion: Implications for Novel Therapies, Annu Rev Pharmacol Toxicol 57 (2017) 535–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Kubli DA, Gustafsson AB, Mitochondria and mitophagy: the yin and yang of cell death control, Circ Res 111 (2012) 1208–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kimata M, Matoba S, Iwai-Kanai E, Nakamura H, Hoshino A, Nakaoka M, Katamura M, Okawa Y, Mita Y, Okigaki M, Ikeda K, Tatsumi T, Matsubara H, p53 and TIGAR regulate cardiac myocyte energy homeostasis under hypoxic stress, Am J Physiol Heart Circ Physiol 299 (2010) H1908–1916. [DOI] [PubMed] [Google Scholar]
- [62].Borutaite V, Brown GC, Mitochondria in apoptosis of ischemic heart, FEBS Lett 541 (2003) 1–5. [DOI] [PubMed] [Google Scholar]
- [63].Lesnefsky EJ, Chen Q, Moghaddas S, Hassan MO, Tandler B, Hoppel CL, Blockade of electron transport during Ischemia protects cardiac mitochondria, J Biol Chem 279 (2004) 47961–47967. [DOI] [PubMed] [Google Scholar]
- [64].Ott M, Robertson JD, Gogvadze V, Zhivotovsky B, Orrenius S, Cytochrome c release from mitochondria proceeds by a two-step process, Proc Natl Acad Sci U S A 99 (2002) 1259–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Ozaki T, Tomita H, Tamai M, Ishiguro S, Characteristics of mitochondrial calpains, J Biochem 142 (2007) 365–376. [DOI] [PubMed] [Google Scholar]
- [66].Hirst J, Carroll J, Fearnley IM, Shannon RJ, Walker JE, The nuclear encoded subunits of complex I from bovine heart mitochondria, Biochim Biophys Acta 1604 (2003) 135–150. [DOI] [PubMed] [Google Scholar]
- [67].Yu SW, Wang H, Poitras MF, Coombs C, Bowers WJ, Federoff HJ, Poirier GG, Dawson TM, Dawson VL, Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor, Science 297 (2002) 259–263. [DOI] [PubMed] [Google Scholar]
- [68].Sevrioukova IF, Apoptosis-inducing factor: structure, function, and redox regulation, Antioxid Redox Signal 14 (2011) 2545–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Vahsen N, Cande C, Briere JJ, Benit P, Joza N, Larochette N, Mastroberardino PG, Pequignot MO, Casares N, Lazar V, Feraud O, Debili N, Wissing S, Engelhardt S, Madeo F, Piacentini M, Penninger JM, Schagger H, Rustin P, Kroemer G, AIF deficiency compromises oxidative phosphorylation, Embo J 23 (2004) 4679–4689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Halestrap AP, What is the mitochondrial permeability transition pore?, J Mol Cell Cardiol 46 (2009) 821–831. [DOI] [PubMed] [Google Scholar]
- [71].Hausenloy DJ, Yellon DM, The mitochondrial permeability transition pore: its fundamental role in mediating cell death during ischaemia and reperfusion, J Mol Cell Cardiol 35 (2003) 339–341. [DOI] [PubMed] [Google Scholar]
- [72].Xu A, Szczepanek K, Maceyka MW, Ross T, Bowler E, Hu Y, Kenny B, Mehfoud C, Desai PN, Baumgarten CM, Chen Q, Lesnefsky EJ, Transient complex I inhibition at the onset of reperfusion by extracellular acidification decreases cardiac injury, Am J Physiol Cell Physiol 306 (2014) C1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Chen Q, Lesnefsky EJ, Blockade of electron transport during ischemia preserves bcl-2 and inhibits opening of the mitochondrial permeability transition pore, FEBS Lett 585 (2011) 921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.