

Abstract
Chronic traumatic encephalopathy (CTE) is a neurodegenerative tauopathy characterized by accumulation of hyperphosphorylated tau (p‐tau) in perivascular aggregates in neurons and glia at the depths of neocortical sulci and progresses to diffuse neocortical, allocortical and brainstem structures. The strongest risk factor is exposure to repetitive head impacts acquired most commonly through contact sports and military service. Given that CTE can only be definitively diagnosed after death, a better understanding of the cellular and molecular changes in CTE brains may lead to identification of mechanisms that could be used for novel biomarkers, monitoring progression or therapeutic development. Disruption of alternative pre‐mRNA splicing of tau mRNA plays a pathogenic role in tauopathy, with multiple characteristic patterns of isoform accumulation varying among tauopathies. Limited data are available on CTE, particularly at early stages. Using biochemical and histological approaches, we performed a detailed characterization of tau isoform signatures in post‐mortem human brain tissue from individuals with a range of CTE stages (n = 99). In immunoblot analyses, severity was associated with decreased total monomeric tau and increased total oligomeric tau. Immunoblot with isoform‐specific antisera revealed that oligomeric tau with three and four microtubule binding domain repeats (3R and 4R) also increased with CTE severity. Similarly, immunohistochemical studies revealed p‐tau accumulation consisting of both 3R and 4R in perivascular lesions. When the ratio of 4R:3R was analyzed, there was mixed expression throughout CTE stages, although 4R predominated in early CTE stages (I‐II), a 3R shift was observed in later stages (III‐IV). While neurons were found to contain both 3R and 4R, astrocytes only contained 4R. These 4R‐positive cells were exclusively neuronal at early stages. Overall, these findings demonstrate that CTE is a mixed 4R/3R tauopathy. Furthermore, histologic analysis reveals a progressive shift in tau isoforms that correlates with CTE stage and extent of neuronal pathology.
Keywords: chronic traumatic encephalopathy, gliofibrillary tangle, hyperphosphorylation, neurofibrillary tangle, tau isoform, tauopathy
Introduction
Tau is a microtubule‐associated protein enriched in neuronal axons (55). Under normal conditions, tau is a critical regulator of microtubule stability and supports axonal transport among other functions (32, 55). Through pathogenetic mechanisms, tau becomes abnormally hyperphosphorylated, dissociates from microtubules, aggregates into pathological oligomers and spreads in a prion‐like fashion (21). The presence of hyperphosphorylated tau (p‐tau) is a hallmark of several conditions collectively termed tauopathies. The clinical symptomatology, neuroanatomical pattern of disease progression, vulnerable cell types and tau filament ultrastructure vary among tauopathies. The most common is Alzheimer disease (AD), which arises secondarily to amyloid‐beta toxicity, but there are a number of primary tauopathies that include argyrophilic grain disease (AGD) (20), primary age‐related tauopathy (PART) (11) and aging‐related tau astrogliopathy (ARTAG) (26). A subset of these tauopathies are associated with alterations in the splicing of the tau pre‐mRNA, including frontotemporal lobar degeneration (FTLD) with tau mutation (52), progressive supranuclear palsy (PSP) (47), corticobasal degeneration (CBD) (43), globular glial tauopathy (GGT) (1). Chronic traumatic encephalopathy (CTE), which arises following mild yet repetitive traumatic brain injury (34, 37), may have differences in the structure of tau inclusions, with recent work using cryo‐electron microscopy demonstrating different conformational tau filament structures in AD, CTE and Pick disease (18, 19). Obtaining a better understanding of the differences in accumulation of tau isoforms in aggregates between the tauopathies is critical because of its potential to serve as a diagnostic biomarker and potential to reveal mechanisms that might serve as therapeutic targets. However, our understanding of the full complement and diversity of abnormal tau structural forms in CTE remains to be fully elucidated.
The microtubule‐associated protein tau gene (MAPT) produces predominantly six isoforms through alternative pre‐mRNA splicing of exons 2, 3 and 10 in the central nervous system (5). Inclusion of exon 10 results in isoforms with four microtubule‐binding domains repeats (4R) while exclusion produces isoforms with three (3R) (28). During human brain development, 3R predominates but the levels of 4R undergo an abrupt perinatal surge resulting in roughly equal levels of 3R and 4R in adulthood (23, 39). In tauopathies, imbalances in the normal isoform ratio occur, with diseases displaying predominance of 3R, 4R or both. PSP, CBD and AGD are 4R tauopathies while Pick’s disease displays 3R (6). AD, CTE and PART have aggregation of both 3R and 4R (11, 36, 53). While both 3R and 4R tau can exert pathologic effects, differences in the overt toxicity have been noted, where 4R tau is likely more pathogenic (45, 46).
Recent studies have advanced our ability to diagnose and stage CTE. The key pathologic feature that distinguishes and defines CTE as a unique neurodegenerative disease is a pathognomonic lesion that consists of perivascular accumulation of p‐tau in neurons, neurites and astrocytes at the depths of cortical sulci (34). Cross‐sectional studies have enabled delineation of the progression of tauopathy in the CTE brain, illustrating that the pathognomonic lesion is the earliest detectable p‐tau abnormality in CTE (37). The earliest stages (I and II) represent mild CTE with scattered foci of perivascular p‐tau throughout the cortex. In more advanced stages (III and IV), there is an increase in neocortical tau burden and p‐tau pathology involving the medial temporal lobe, basal ganglia and brainstem (37). Previous histological studies have identified accumulation of both 3R and 4R tau in CTE (36, 37) and a biochemical analysis of two severely affected boxers was consistent with this finding (44), however, it is unclear as to which tau isoforms accumulate in the earliest CTE stages and in which cell types.
Given the heterogeneity of tau isoforms in tauopathies and the need to better understand the cellular and molecular changes in CTE, we analyzed a series of post‐mortem human brains from the largest collection of individuals with neuropathologically confirmed CTE. Biochemical studies were performed to measure both monomeric and oligomeric p‐tau and tau isoforms. Morphologic assessment and multiplex immunofluorescence were used to examine the relative ratio of 4R and 3R tau‐containing astrocytes and neurons. Overall, we leveraged our emerging knowledge of CTE pathologic progression to pinpoint the spectrum of tau isoforms across disease and determine if unique expression patterns exist that in the future might improve neuropathological assessment and act as novel diagnostic biomarkers.
Methods
Subjects
Post‐mortem human brain tissue was obtained from 99 male former contact sport athletes following a comprehensive neuropathological workup (Tables 1 and S1). Assessments included the NINDS criteria for the pathological diagnosis of CTE and the McKee CTE staging of neuropathological severity (34, 37). Neuropathological criteria for CTE require at least one perivascular p‐tau lesion consisting of aggregates in neurons, astrocytes and cell processes around a small vessel; these pathognomonic lesions are most often distributed at the depths of the sulci in the cerebral cortex and are distinct from the lesions of ARTAG by the presence of neuronal tau pathology (35). Cases were also assessed for other neurodegenerative diseases using well‐established criteria for Alzheimer’s disease (AD) (24, 42), Lewy body disease (LBD) (38), frontotemporal lobar degeneration (FTLD) (8, 15, 30) and motor neuron disease (MND) (7, 29). Neuropathological evaluation occurred blinded to the clinical evaluation and was reviewed by four neuropathologists (VA, BH, TS, AM); discrepancies in the neuropathological diagnosis were resolved by consensus conference. In addition to diagnoses, the density of neuritic amyloid beta plaques (CERAD score) was measured semi‐quantitatively (0 to 3; 3 most severe). Next‐of‐kin provided written consent for participation and brain donation. IRB approval for brain donation was obtained through the Boston University Alzheimer's Disease Center (BU ADC) and CTE Center, Human Subjects Institutional Review Board of the Boston University School of Medicine, and Edith Nourse Rogers Memorial Veterans Hospital (Bedford, MA). Subjects were selected for inclusion into the study based on the presence of available tissue and a positive neuropathologic diagnosis of CTE. Cases that received a neuropathologic diagnosis of a neurodegenerative disease, including AD, PD, LBD, FTLD and MND were excluded from the study in order to observe CTE specific tau isoform changes. Demographics, educational attainment, athletic history (type of sports played, level, position, age of first exposure to sports and years playing contact sports), military history (branch, location of service and duration of combat exposure) and traumatic brain injury (TBI) history (including number of concussions) were queried during a telephone interview as detailed previously (50). All clinicians were blinded to neuropathology. Of the total 99 cases, 94 individuals played American football, two were boxers, two played hockey and one played Australian rules rugby.
Table 1.
Patient data.
| CTE stage | P | ||||
|---|---|---|---|---|---|
| I | II | III | IV | ||
| n | 9 | 23 | 42 | 25 | |
| Age at death (year) | 44.7 ± 16.4 | 48.3 ± 19.0 | 64.2 ± 13.1 | 73.2 ± 8.5 | <0.001 |
| Contact sports exposure (year) | 10.1 ± 5.7 | 13.7 ± 6.3 | 15.9 ± 5.3 | 20.0 ± 7.7 | <0.001 |
Data expressed as mean ± standard deviation. Age at death and years of exposure analyzed with a One‐way ANOVA.
Biochemistry
Fresh‐frozen brain tissue from the dorsolateral prefrontal cortex was homogenized with a glass‐Teflon homogenizer at 500 rpm in 10 volumes (wt/vol) of ice‐cold tissue homogenization buffer containing 20 mM Tris, pH 7.4, 250 mM sucrose, 1 mM EDTA, 1 mM EGTA and Halt protease and phosphatase inhibitor cocktail (Thermo Fisher Scientific). For each sample, 30 μg of protein were boiled in Laemmili sample buffer (Bio‐rad) for 5 minutes, run with tau protein ladder (rPeptide) on 10% PROTEAN TGX Precast SDS‐PAGE Gels (Bio‐Rad), blotted to nitrocellulose membranes and stained with anti‐Tau (HT7) (1:3000, Invitrogen), anti‐Tau (pSer202, pThr205, pS208 (31)) (AT8) (1:1000, Invitrogen), anti‐3R tau (1:2000, clone 8E6/C11, Sigma‐Aldrich) and anti‐4R tau (1:500, clone 1E1/A6, Sigma‐Aldrich). HRP‐labeled secondary anti‐mouse antibody (1:20 000, Vector Labs) was detected by Super Signal West Femto Maximum Sensitivity Substrate or Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific). To quantify and standardize protein levels without reliance on specific housekeeping proteins, total protein was detected with Amido Black as previously described (Sigma‐Aldrich) (2). Chemiluminescence was measured in an LAS‐4000 Intelligent Dark Box imager (Fuji Film), and relative optical densities were determined using Alpha EaseFC software, version 4.0.1 (Alpha Innotech), normalized to total protein loaded (Amido Black).
Immunoassay
Fresh‐frozen brain tissue was obtained from the dorsolateral prefrontal cortex was isolated, weighed, and placed on dry ice. Freshly prepared, ice‐cold 5M Guanidine Hydrochloride in Tris‐buffered saline (20 mM Tris‐HCl, 150 mM NaCl, pH 7.4 TBS) containing 1:100 Halt protease inhibitor cocktail (Thermo Scientific) and 1:100 Phosphatase inhibitor cocktail 2 & 3 (Sigma) was added to the brain tissue at 5:1 and homogenized with Qiagen Tissue Lyser LT, at 50 Hz for 5 minutes. The homogenate was then incubated while rocking overnight at room temperature. For Amyloid Beta (Aβ) 1‐42 protein levels, lysates were applied to an Aβ1‐42 immunoassay (MSD #K15200E‐2) according to the manufactures protocols and ran on MSD plate reader model 1250. Values were standardized to the amount of tissue used to generate lysates and expressed as pg/mg.
Immunohistochemistry
Brain tissue was taken from the dorsolateral frontal cortex (Broadmann area 9) (DLFC), embedded in paraffin and cut at 20 μm. Immunofluorescence staining was performed using Akoya Bioscience Opal Polaris 7 color manual IHC detection kit as per the manufacturer’s instructions. Briefly, the Opal multiplex method allows sequential staining of multiple antibodies irrespective of antibody species. A primary antibody, secondary antibody and Opal reactive fluorophore are added in sequential staining steps to the tissue. Microwave treatment is used to remove the primary antibody, secondary antibody, but not the Opal fluorophore. Additional primary, secondary and Opal fluorophore steps, followed by microwave treatment can then occur. Up to seven targets can be visualized. Sections were incubated with antibodies to RD3 (3R tau) (generous gift of Rohan de Silva, 1:6000, clone 8E6/C11), ET3 (4R tau) (generous gift from Peter Davis (17), 1:200), anti‐PHF‐tau (AT8) (Pierce Endogen, 1:2000) and DAPI. Section were visualized using an Akoya Bioscience Vectra Polaris Digital Slide Scanner. Automated deconvolution algorithms were created to remove background and spectral overlap using inForm (Akoya Bioscience). Images were analyzed in Halo (Indica Laboratory). The depth of the cortical sulcus (defined as the bottom third of two connecting gyri) was selected and highlighted. The white matter/gray matter boundary was used as the outer edge of the region of interest such that only gray matter was highlighted. The total amount of cells containing 3R and 4R tau was manually counted by a blinded observer. If multiple sulci were present on a section, each sulcus was counted and the results averaged. The number of 3R and 4R tau positive cells were standardized by the total area measured and expressed as the number of positive cells per mm2. To obtain 4R/3R positive cell ratios, the average amount of 4R cells were divided by the average amount of 3R cells for each case. To determine the overall extent of tau deposition in the DLFC at the depth of the cortical sulcus, immunochromogenic staining using the AT8 antibody and DAB visualization was performed and analyzed using previously published methods (9, 10).
Neuronal and glial quantification
To determine whether 3R or 4R tau was present in neurons or astrocytes, morphologic assessments were performed. Neurofibrillary tangles were morphologically defined as discreet and strong p‐tau positive pyramidal shaped staining compatible with a somatodendritic compartment often in association with large neuronal nuclei, whereas gliofibrillary tangles were defined as granular less defined cytoplasmic and glial process staining that was often radially oriented around a smaller nucleus. Morphologically ambiguous/indeterminant cells were not counted. Cell counts were restricted to a 0.5 mm2 circle around individual CTE lesions which captured most lesion‐associated cells while avoiding neighboring lesions. Lesions were identified in 6 cases of CTE stage I (all available cases), 10 stage II, 10, stage III and 10 stage IV. The number of neurons or astrocytes that contained 3R or 4R were counted for each lesion present in the DLFC and averaged together for each individual. The final counts of tau containing neurons or astrocytes were divided by the total number of tau containing cells to create a percentage of neurons and astrocytes around the CTE lesion. To verify the morphologic assessment, a subset of cases were stained with the previously mentioned 3R, 4R, AT8 tau, and DAPI, in addition to GFAP (Millipore, 1:2000), Iba1 (Wako, 1:1000) and MAP2 (Biolegends, 1:500) for preliminary cell type observations. Multispectral unmixing of the 7 color immunofluorescence images were performed using Phenochart and inForm (Akoya Bioscience). Images were compiled and viewed in Halo (Indica Laboratories).
Statistics
Statistical analysis was performed using SPSS (v.24; IBM, Inc. Armok, NY) and Prism (v.8, Graphpad Software, La Jolla, CA). To determine if increasing CTE stage correlated with the biochemical measures of tau and histologic counts of tau containing cells, ordinal regressions were used with CTE stage as the dependent variable in separate regressions. Analysis of variance (ANOVA) was also utilized to examine differences in the densities of 3R tau, 4R tau, and protein levels of Aβ1‐42 across CTE stages. As the data used for ANOVA analysis had a non‐normal distribution, Kruskal Wallis post‐tests were performed. For analysis of histologic counts of 4R/3R positive cell ratios, total AT8 p‐tau staining density and Aβ1‐42 protein levels were log transformed to normalize for regression analysis. Multiple linear regression analysis was used to examine if 4R/3R ratios correlated with AT8 p‐tau staining density independently of age at death and CERAD neuritic amyloid plaque score. For the tau containing cell type analysis, the average number of p‐tau containing neurons and astrocytes were divided by the total number of p‐tau containing cells to establish the percentage of the total population that each cell type comprised. Analysis of covariance (ANCOVA) was utilized to determine if age at death was implicated in the tau containing neuron vs. astrocyte percentage results. The percentage of neurons that contained tau was corrected for age at death and compared across CTE stages.
Results
Monomeric tau decreases while oligomeric and p‐tau increases with CTE severity
Given the progressive changes in the cellular and biochemical features of CTE and degenerative tauopathies in general, we first sought to characterize the quantitative burden of total normal and abnormal forms of tau in our case series biochemically. Immunoblot analysis of total brain homogenates using antisera recognizing total tau showed that low stage cases display predominantly low molecular weight tau monomers (Figure 1A). High molecular weight oligomeric tau assemblies, above 75 kDa, became progressively apparent as CTE stage increased. Unexpectedly, quantification revealed that total tau levels (combined monomeric and oligomeric) significantly decreased with increasing CTE stage (P = 0.018) (Figure 1B). Monomeric tau also significantly decreased with increasing CTE stage (P = 0.004) (Figure 1C), but oligomeric tau increased (P = 0.032) (Figure 1D). Phosphorylated tau (AT8) also significantly increased with CTE stage (P = 0.021) (Figure 1E,F).
Figure 1.
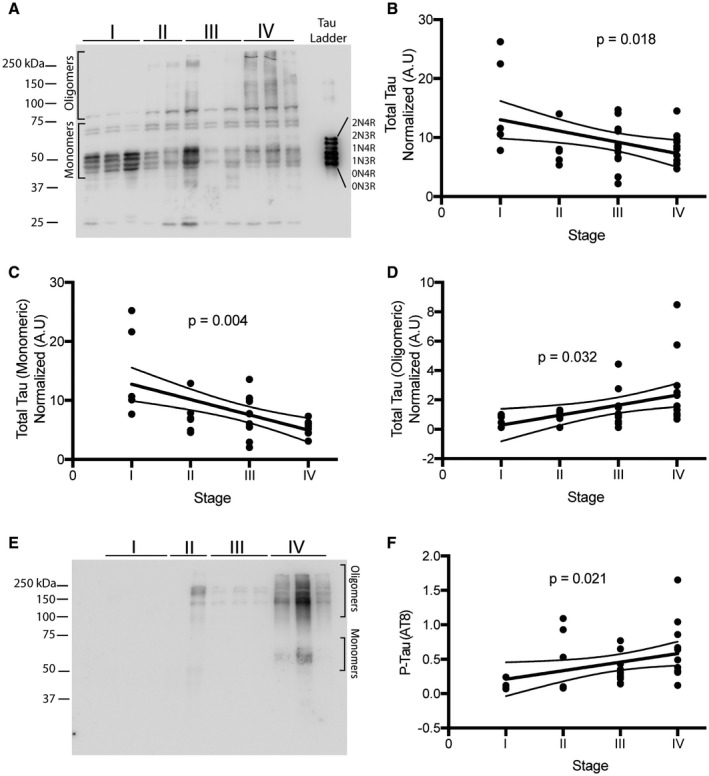
Biochemical analysis of total and hyperphosphorylated tau. A. Representative western blot images for total tau used to analyze 36 samples ranging from CTE stage I–IV. Oligomeric and monomeric species of tau are highlighted. B‐D. Densitometry analysis to determine relative levels of total tau (B), total monomeric tau (C) and oligomeric tau (D) was performed. E. Representative image of western blot for AT8 levels. F. Densitometry analysis to examine the amount of AT8 across CTE stage. P‐values were obtained using ordinal regression analysis. Lines on each graph depict linear regression analysis with 95% confidence intervals. Each dot is one case. For western blots, molecular ladder for each run is labeled on the right side.
Oligomeric levels of 3R and 4R increase with CTE severity
Next, we examined the levels of 3R and 4R tau using immunoblots probed with tau isoform specific antisera (anti‐3R tau (clone 8E6/C11) and anti‐4R tau (clone 1E1/A6), respectively), to determine whether either was preferentially altered in the monomeric or oligomeric fractions with advancing CTE stage (Figure 2A,B). With respect to 3R, there was an increase in the total levels (combined monomers and oligomers) (P = 0.036) and oligomeric levels (P = 0.004), but no change was observed in monomeric levels (P = 0.634) (Figure 2C–E). With respect to 4R, total (P = 0.868) and monomeric forms (P = 0.142) were unchanged, but oligomers were increased significantly (P = 0.025) in relation to stage (Figure 2F–H). There was no change in the ratio of total (P = 0.619), monomeric (P = 0.235) or oligomeric (P = 0.629) isoforms (Figure 2I–K).
Figure 2.
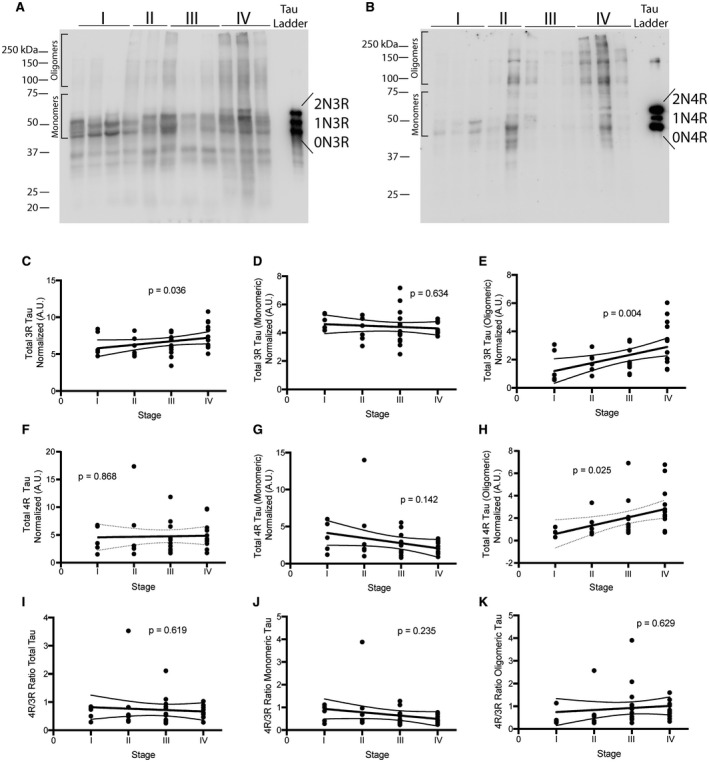
4R and 3R tau oligomers are increased in CTE. A. Representative image for 3R tau western blots using the antibody anti‐3R tau (clone 8E6/C11). B. Representative image for 4R tau western blot using the antibody anti‐4R tau (clone 1E1/A6). C‐E. Densitometry analysis to determine relative levels of total 3R tau (C), total 3R monomeric tau (D) and 3R oligomeric tau (E) was performed. F‐H. Densitometry analysis to determine relative levels of total 4R tau (F), total 4R monomeric tau (G) and 4R oligomeric tau (H) was performed. Ratio of 4R/3R was plotted for total tau (I), monomeric tau (J) and oligomeric tau (K). Ordinal regression was used to obtain P‐values. Lines on each graph depict linear regression analysis with 95% confidence intervals. Each dot is one case. For western blots, molecular ladder for each run is labeled on the right side. Oligomeric and monomeric species of tau are also highlighted on each blot.
The CTE sulcal lesion undergoes a progressive shift in tau isoform ratio
Next, we performed a series of histological studies to examine tau isoform accumulation at different CTE stages on the cellular level using immunofluorescence microscopy. The antibody RD3 (clone 8E6/C11) was used to detect 3R tau and the ET3 antibody was used to detect 4R tau. In cortical regions most susceptible to early CTE pathology (ie, the depth of the sulcus in the neocortex), 3R and 4R tau immunopositivity were present within the perivascular CTE lesion in both mild CTE (stages I and II) and severe CTE (stage III and IV); Figure 3A). When quantifying the overall density of 3R or 4R tau positive cells at the depth of the cortical sulcus, similar to the biochemical analysis, an increase in 3R and 4R positive cells was observed during CTE disease progression (Figure 3B,C). Further, ordinal regression analysis demonstrated the levels of both 3R and 4R positive cells increased significantly with CTE stage (P = 0.029 for 3R, P = 0.021 for 4R). While the majority of cases showed more 4R cells compared to 3R cells at the depth of the cortical sulcus, 7.8% of stage III and 41.1% of stage IV cases showed greater 3R tau burden (Figure 3D). When the ratio of 4R/3R tau containing cells was plotted against the density of AT8 staining in the DLFC, a negative association between the 4R/3R ratio and AT8 density was observed (P = 0.0023, Figure 3E). Multiple linear regression was used to determine whether other variables contribute to the observed ratio change. Although individuals with a high burden of Aβ and a positive neuropathologic diagnosis of AD were excluded from the study, many CTE case still have Aβ pathology which could confound results (48). Age at death, CERAD neuritic amyloid beta plaque score and the AT8 staining density were used as covariates and demonstrated that AT8 staining density was significantly negatively correlated with the 4R/3R ratio (β = −0.231, P = 0.002) and that this was independent of age at death (β = 0.03, P = 0.364) or CERAD neuritic amyloid plaque score (β = −0.047, P = 0.697). Finally, to further rule out amyloid as a contributing factor toward the 4R/3R ratio, protein levels of the pathogenic oligomer Aβ1‐42 were analyzed via immunoassay (Figure 4). Overall Aβ1‐42 protein levels were not significantly changed across CTE stage (P = 0.768) (Figure 4A). Additionally, no significant correlation between Aβ1‐42 protein levels and the 4R/3R tau isoform ratio was observed (Figure 4B).
Figure 3.
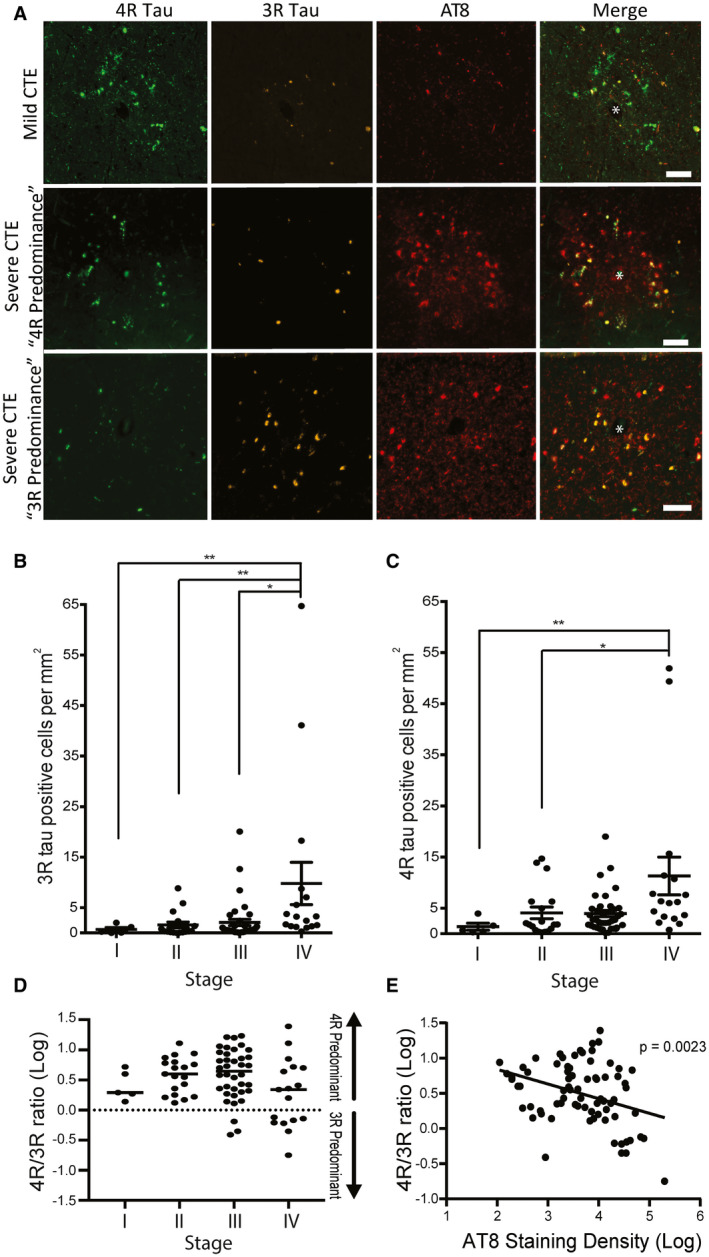
The p‐tau containing cells present in CTE are a mix of 3R and 4R tau. A. Representative images of CTE pathognomonic lesions detected using p‐tau antisera (AT8) in mild CTE (stage I&II) and severe CTE (stage III&IV). The cells in these lesions are variably positive for 3R, 4R, or both. The antibody RD3 (clone 8E6/C11) was used to detect 3R tau and the ET3 antibody was used to detect 4R tau. Representative images of 4R and 3R predominant lesion in severe CTE are included. * denotes a blood vessel where the lesions are centered around. Scale bar = 100 μm. B,C. Quantitative analysis of the overall density of 3R tau positive cells (B) and 4R tau positive cells (C) found at the depth of the cortical sulcus across the stages of CTE. ANOVA analysis with a Kruskal Wallis post‐tests were performed. Each dot represents one case. Errors bars represent standard error of the mean. D. Quantitative measurement of the relative ratios of 4R/3R tau present in the DLFC at the depth of the cortical sulcus across all stages of CTE. Data are log transformed to better visualize ratios with more 3R tau. Values over 0 contain more 4R tau while values under 0 contain more 3R tau. E. Linear analysis of the 4R/3R ratio compared to the AT8 p‐tau staining density in the DLFC. Linear regression analysis was used to obtain P‐values. Data are log transformed. Each dot represents one case. *P < 0.05, **P < 0.01.
Figure 4.
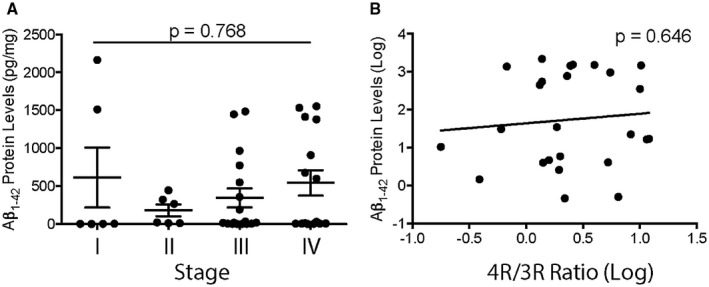
Oligomeric Amyloid Beta 1‐42 does not have an effect on the tau isoform ratio in CTE. A. Quantitative immunoassay measurement of the Aβ1‐42 protein levels across CTE stage. ANOVA was used to demonstrate no significant difference between groups. Each dot represents one case. Error bar represents standard error of the mean. B. Analysis of the Aβ1‐42 protein levels compared to the 4R/3R tau isoform ratio. Linear regression analysis was used to obtain P‐value. Data are log transformed. Each dot represents on case.
The early CTE perivascular lesion is composed of predominantly 3R and 4R containing neurons
To examine whether either astrocytes or neurons preferentially contain 3R or 4R tau, morphological analysis of cells around the perivascular lesion was performed. Overall, the primary cell type that contained p‐tau around the perivascular lesion were neurons (Figure 5A). In cells containing 4R tau in CTE stage I and II, 95.8% ± 10.2% and 96.1% ± 4.7% were neurons, respectively. For CTE stage III and IV, 65.6% ± 28.7% and 69.7% ± 26.0% of cells containing 4R were neurons, respectively. The remaining cells were morphologically identified as astrocytes. 3R was only detected in neurons. When the relative percentages of neurons and astrocytes were divided into decade of age at death, a trend toward increased astrocytes in the perivascular lesion was observed starting in individuals between 60–69 years old and increased in each subsequent decade (Figure 5B). This observation was confirmed using an ANCOVA analysis demonstrating that CTE stage was not significantly correlated with the percentage of 4R astrocytes around the lesion (P = 0.31) when including age at death as a covariate in the model (P = 0.004). To confirm the morphologic analysis and further explore the different cell types that express various tau isoforms, seven color multiplex immunofluorescent staining was utilized. In agreement with the morphologic assessment, in the sulcal lesions, 3R and 4R tau colocalized with MAP2+ neurons, and GFAP+ astrocytes only colocalized with 4R tau (Figure 5C,D). Neither 3R nor 4R tau colocalized with Iba1+ microglia.
Figure 5.
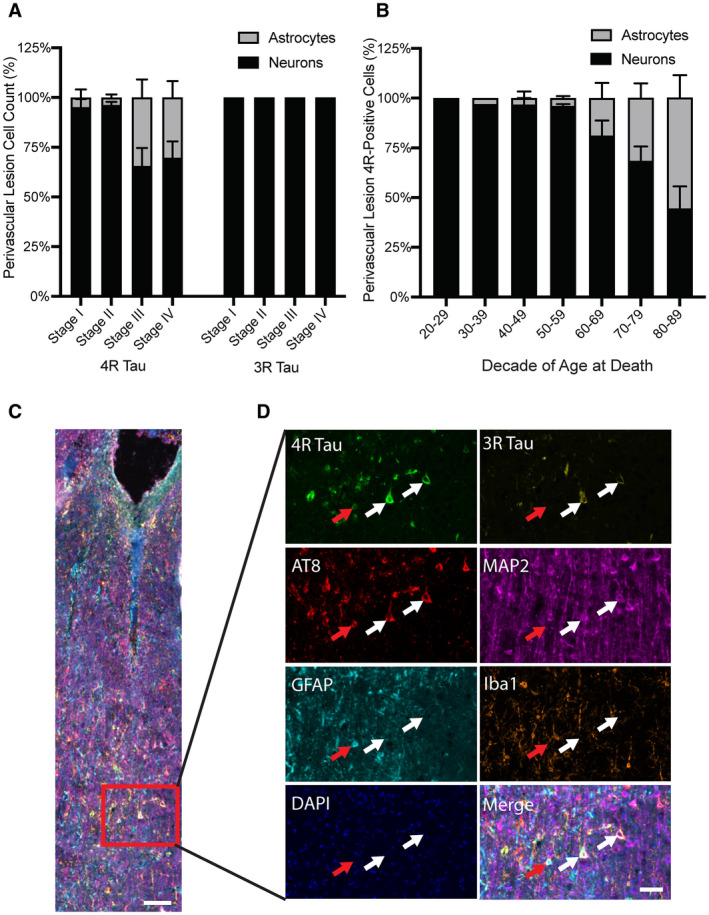
Neurons contain a mix of 3R and 4R tau while astrocytes contain 4R tau. A. Quantitative measurement of the relative percentages of neurons and astrocytes that contain tau found around a CTE lesion in the DLFC. Neurons contain both 3R and 4R while astrocytes only contain 4R tau. B. Relative percentages of neurons and astrocytes that contain 4R tau stratified by decade of age at death. Data are presented as mean ± SD. C. Overview image taken at the depth of the cortical sulcus from an individual with CTE. Scale bar represents 100 μm. A high powered representative images was obtained. D. Both 3R and 4R were observed in depth of sulcus deep gray matter. MAP2+ neurons colocalized with 3R and 4R tau. GFAP+ astrocytes colocalized with 4R tau. Iba1+ microglia did not colocalize with 3R or 4R. Scale bar represents 50 μm.
Discussion
Chronic traumatic encephalopathy (CTE) is a devastating, yet understudied, neurodegenerative disease. Like the other tauopathies, CTE is challenging to recognize clinically because of neuroanatomical vulnerability and resultant symptomatology that overlaps with other neurological and psychiatric conditions (12, 41). Thus, there is an urgent need to better understand the cellular and molecular degenerative changes in the CTE brain to pave the way toward better diagnostics and targeted therapeutics (27, 56). This study is the largest to date specifically measuring abnormal tau isoform accumulation biochemically in post‐mortem human CTE brain tissues, building on the previous limited work (36, 44), in the context of disease progression. Using a large cohort with a range of ages and severity, we provide quantitative data indicating that the proposed neuropathological staging system correlates with biochemical measures of hyperphosphorylated tau aggregate (p‐tau) burden and further demonstrate marked shifts in pools of tau with reduction of soluble total tau and tau monomers that coincides with increased abnormal insoluble p‐tau correlating with disease stage. Our findings reinforce previous studies detecting pathological p‐tau aggregates composed of both 3R and 4R, but further utilize multiplex immunofluorescence microscopy to spatially demonstrate early predominance of 4R tau in the perivascular regions in neurons, suggesting a potential mechanistic role for this isoform. Astrocyte pathology manifests after neuronal pathology and is exclusively 4R tau. Together, these findings indicate that abnormalities in 4R tau in perivascular neurons are among the earliest demonstrable changes in human CTE post‐mortem brain.
Identification of early pathological changes in CTE and other neurodegenerative diseases is a compelling approach to revealing causal mechanisms as the findings are less likely to be masked by secondary reactive changes, but this requires robust approaches to patient stratification and staging by disease severity for correlative studies. While recent questions as to whether neuropathological staging remains relevant have arisen (14), staging is essential for pinpointing early changes. The main challenge in post‐mortem neuropathological staging is that autopsy studies are not longitudinal by design. The fact that patients cannot be followed precludes confirmation that subjects with low pathology burden represent the presymptomatic disease phase rather than a resilience phenotype. But unlike other neurodegenerative diseases, there is little ambiguity as to the initiating event in CTE, which is mechanical brain injury, enabling better predictive power as to outcomes in our cohort which is a high‐risk population. Further, our unique cohort consists of a range of patients across the severity and aging spectrum, many being both relatively young and mildly affected, increasing our confidence in the findings. Further, with the exception of 2 Stage I CTE cases with elevated Aβ1‐42 protein levels, our early stage cases were mostly devoid of Aβ pathology, PART, and ARTAG, all common age‐associated comorbidities that serve as confounding factors. We found that the proposed staging system (34, 37) performed well in this study, correlating with biochemical measures of p‐tau burden, and enabling the observation of progressive decreases of soluble tau monomers and histological 4R predominance in perivascular lesions (see below). These findings are consistent with previous histological work demonstrating that the severity of p‐tau lesion burden in CTE positively correlates with the magnitude and duration of exposure to repetitive head injury (3, 10, 40) and rodent models of impact and blast neurotrauma showing that repetitive head injury leads to tau hyperphosphorylation, dissociation from microtubules, and aggregation into pathological oligomers (51). Together this work provides further support for a direct mechanistic link between repetitive head injury and tau pathology.
Direct histological examination of the pathognomonic perivascular lesion in early stage cases provides a window into the earliest cellular changes that have been proposed to be critical drivers of tau pathology in CTE, including damage to the blood brain barrier, activation of chronic neuroinflammation, and injury to neurons and synapses (9, 10, 36, 51). Our analysis revealed that the tau isoform signature in CTE might not be static, but rather evolving over the course of the disease with differences depending on cellular compartment. While the perivascular lesion was comprised of both 3R and 4R positive neurons, we noted that there was a higher burden of 4R compared to 3R, confirmed through quantitative analysis. We also observed a neuron‐specific shift at the perivascular lesion that may present an opportunity to monitor progression in the predominant isoform as stage increased with progressive enrichment of 3R tau. Shifts in isoform ratio at higher stages have been previously reported in AD (54). Notably, early pre‐tangles are enriched for 4R tau and extracellular end‐stage ghost tangles are preferentially labeled with 3R, which may provide a cellular context for this observation (22, 54). We also found that while neurons are the dominant p‐tau containing cell type, astrocytic p‐tau in perivascular lesion is predominantly 4R and the abundance is correlated with age. The nature of the astrocytes in CTE requires further clarification as to whether they are related to the thorn‐shaped or fuzzy astrocytes in ARTAG which will help unravel their potential mechanistic function during pathogenesis. Additionally, the present study chose to focus on the CTE pathognomonic lesion and perivascular astrocytes. It is still unknown how astrocytic pathology in other brain regions such as white matter, non‐perivascular regions of gray matter, or subpial surfaces evolves over the course of disease. Future work utilizing a diverse range of diseases and comorbid pathologies will be required. When comparing biochemical and immunofluorescence measurements, even though they are complementary approaches, certain discrepancies could be observed. Biochemical analysis is a global quantitative measure of total protein composition, while histology is less precise quantitatively yet allows assessment of incorporation within spatially defined cells in the perivascular lesion. Both methods also have inherent weaknesses, such as artifacts caused by fixation and masking of epitopes. Further, our histological analysis did not consider noncellular structures (eg, neuropil threads, grains, dot pathology, etc.). In summary, we interpret the histologic findings to indicate that CTE brain contains mixed levels of 3R and 4R isoforms and a shift to 3R positive cells (likely NFTs) occurs as the disease severity increases.
Evidence from patients with frontotemporal lobar degeneration with MAPT splice‐site mutations that cause increased inclusion of exon 10 and elevated 4R isoforms suggests that differences in the levels of tau isoforms may be neurotoxic in CTE as well. How this might occur remains unclear, but the isoform imbalance may promote microtubule instability and drive the formation of toxic p‐tau aggregates. The relative abundance of 4R isoforms we observed in the perivascular lesion in CTE may have a similar neurotoxic effect, being the primary cellular response to direct mechanical damage to neurons or secondary following vascular injury and breakdown of the blood brain barrier. Accumulation of p‐tau in astrocytes may be secondary, perhaps arising through prion‐like propagation, the phagocytic capabilities of astrocytes, or both. Another possibility is that astrocyte pathology is a primary result of injury, just taking longer to manifest. Differences in the expression of tau splicing factors, for example, RNA‐binding proteins such as TDP43, TIA1 or G3BP1 may play a role (33). Conversely, the resultant differences may occur independent of splicing changes, being the end result of differences in the relative abundance of cell populations following cell death and reactive changes, each with different inherent expression profiles. Finally, individuals with a predisposition to express high level of 3R tau, through perhaps genetic or environmental factors, may have a more severe CTE pathologic progression and are more likely to present as late stage disease. Genetic alterations have been shown to influence p‐tau deposition and neuroinflammation in CTE (9). However, if this were true, we would expect to find early CTE cases with 3R predominant expression. Additional studies are required to clarify these issues.
There are several limitations to this study in addition to those discussed above. The cohort that we examined represents a convenience sample, limited to voluntary brain donors and is neither population based nor prospective, which limits the ability to extrapolate to general populations. The immunoblots and immunofluorescence microscopy both rely on specificity of antisera and certain epitopes may be missed because specific protein conformation and structures can influence antibody binding. Notably, there is evidence that the 4R tau antisera we deployed recognizes an epitope that might become deaminated during AD (13, 16) and potentially obscure comparative results. Future studies looking at deamination of tau in CTE are warranted. Nevertheless, significant increases in 4R tau containing oligomers and tangles were observed with both the 4R antibodies which supports our interpretation. However, comparative analysis between each 4R antibody will be needed for further validation. Additional limitations were the subgroups of cases with biochemical and/or histological assessments did not completely overlap, but the sample characteristics were similar. Given that the CTE pathognomonic lesion is specific to CTE, a case only analysis was utilized for the present study. However, future work will be needed to determine if the observed changes in tau isoforms are similar or different to other tauopathies such as AD, PART, CBD, PSP, ARTAG or even normal aging.
In conclusion, we found that CTE is a mixed tauopathy consisting of both 3R and 4R tau and demonstrated that pathogenic oligomers and p‐tau positive cells for each isoform increases as CTE stage increases. Furthermore, a shift from 4R toward 3R tau isoforms was observed in the most severe cases suggesting that tau pathology evolves over time in CTE. While neurons within the perivascular lesion were the predominant p‐tau containing cell population and colocalized with both 4R and 3R tau, astrocytes only contained 4R tau and their abundance correlated with age of death as opposed to disease severity. Future work is still needed to determine the mechanistic implications of each isoform or how 3R predominant cases might differ clinically or neuropathologically from 4R predominate cases. Currently, p‐tau, among several other factors, is being used as a blood and cerebral spinal fluid‐based biomarker target to identify CTE in living individuals (4, 49). Next‐generation PET tracers and novel isoform seeding assays are beginning to be used to identify specific tau isoforms (25). Leveraging our findings may lead to novel biomarkers that can track progression of disease and help identify future preventative and therapeutic strategies.
Conflict of Interest
The authors declare there are no conflicts of interest.
Data Availablity Statement
The data that supports the findings are available on request from the corresponding author. The data are not publicly available caused by privacy or ethical restrictions.
Supporting information
Table S1. Sample demographics broken down by experiment.
Acknowledgments
We would like to acknowledge all the donors and their families whose participation made this research possible. We also would like to acknowledge the support of the Edith Nourse Rogers Memorial Veterans Hospital (Bedford, MA, USA) Boston VA Healthcare system, Boston University School of Medicine, Boston University Alzheimer’s Disease Center and CTE program. We thank Nicole Saltiel, Hunter Kelly, Arsal Shah, Marcela Alvia, Rebecca Mathias, Kerry Cormier and Carol Kubilus for their assistance processing tissue. We also thank Madeline Uretsky and Evan Nair for their help obtaining samples. This work received funding through Alzheimer’s Association Research Fellowship (AARF‐17‐529888 to JDC), National Institutes of Health (U54NS115266, R01AG062348 to AM/JFC; K23NS102399 to MLA, and F32AG056098 to KF) (R01AG054008 and R01NS095252 to JFC), The United States Department of Defense (W81XWH‐14‐1‐0399 to AM/JFC), National Institute of Aging (RF1AG054156, R56AG057768, RF1AG057768 to TDS), Clinical Sciences Research and Development Merit Award (I01‐CX001038 to TDS), Rainwater Charitable Trust/Tau Consortium, the David Werber Family, the Nick and Lynn Buoniconti Foundation, the Concussion Legacy Foundation, the Andlinger Foundation and the WWE. The views, opinions and/or findings contained in this article are those of the authors and should not be construed as an official Veterans Affairs position, policy or decision, unless so designated by other official documentation.
Contributor Information
Ann C. McKee, Email: amckee@bu.edu.
John F. Crary, Email: john.crary@mountsinai.org.
References
- 1. Ahmed Z, Bigio EH, Budka H, Dickson DW, Ferrer I, Ghetti B et al (2013) Globular glial tauopathies (GGT): consensus recommendations. Acta Neuropathol 126:537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Aldridge GM, Podrebarac DM, Greenough WT, Weiler IJ (2008) The use of total protein stains as loading controls: an alternative to high‐abundance single‐protein controls in semi‐quantitative immunoblotting. J Neurosci Methods 172:250–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Alosco ML, Stein TD, Tripodis Y, Chua AS, Kowall NW, Huber BR et al (2019) Association of white matter rarefaction, arteriolosclerosis, and tau with dementia in chronic traumatic encephalopathy. JAMA Neurol 76:1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Alosco ML, Tripodis Y, Fritts NG, Heslegrave A, Baugh CM, Conneely S et al (2018) Cerebrospinal fluid tau, Abeta, and sTREM2 in Former National Football League Players: modeling the relationship between repetitive head impacts, microglial activation, and neurodegeneration. Alzheimer's Dement 14:1159–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Andreadis A (2005) Tau gene alternative splicing: expression patterns, regulation and modulation of function in normal brain and neurodegenerative diseases. Biochim Biophys Acta 1739:91–103. [DOI] [PubMed] [Google Scholar]
- 6. Arendt T, Stieler JT, Holzer M (2016) Tau and tauopathies. Brain Res Bull 126(Pt 3):238–292. [DOI] [PubMed] [Google Scholar]
- 7. Brownell B, Oppenheimer D, Hughes JT (1970) The central nervous system in motor neurone disease. J Neurol Neurosurg Psychiatr 33:338–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cairns NJ, Bigio EH, Mackenzie IR, Neumann M, Lee VM, Hatanpaa KJ et al (2007) Neuropathologic diagnostic and nosologic criteria for frontotemporal lobar degeneration: consensus of the Consortium for Frontotemporal Lobar Degeneration. Acta Neuropathol 114:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cherry JD, Mez J, Crary JF, Tripodis Y, Alvarez VE, Mahar I et al (2018) Variation in TMEM106B in chronic traumatic encephalopathy. Acta Neuropathol Commun 6:115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cherry JD, Tripodis Y, Alvarez VE, Huber B, Kiernan PT, Daneshvar DH et al (2016) Microglial neuroinflammation contributes to tau accumulation in chronic traumatic encephalopathy. Acta Neuropathol Commun 4:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Crary JF, Trojanowski JQ, Schneider JA, Abisambra JF, Abner EL, Alafuzoff I et al (2014) Primary age‐related tauopathy (PART): a common pathology associated with human aging. Acta Neuropathol 128:755–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. D'Ascanio S, Alosco ML, Stern RA (2018) Chronic traumatic encephalopathy: clinical presentation and in vivo diagnosis. Handb Clin Neurol 158:281–296. [DOI] [PubMed] [Google Scholar]
- 13. Dan A, Takahashi M, Masuda‐Suzukake M, Kametani F, Nonaka T, Kondo H et al (2013) Extensive deamidation at asparagine residue 279 accounts for weak immunoreactivity of tau with RD4 antibody in Alzheimer's disease brain. Acta Neuropathol Commun 1:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Del Tredici K, Braak H (2019) To stage, or not to stage. Curr Opin Neurobiol 61:10–22. [DOI] [PubMed] [Google Scholar]
- 15. Dickson DW (2009) Neuropathology of non‐Alzheimer degenerative disorders. Int J Clin Exp Pathol 3:1–23. [PMC free article] [PubMed] [Google Scholar]
- 16. Ebashi M, Toru S, Nakamura A, Kamei S, Yokota T, Hirokawa K, Uchihara T (2019) Detection of AD‐specific four repeat tau with deamidated asparagine residue 279‐specific fraction purified from 4R tau polyclonal antibody. Acta Neuropathol 138:163–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Espinoza M, de Silva R, Dickson DW, Davies P (2008) Differential incorporation of tau isoforms in Alzheimer's disease. J Alzheimers Dis 14:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Falcon B, Zhang W, Murzin AG, Murshudov G, Garringer HJ, Vidal R et al (2018) Structures of filaments from Pick's disease reveal a novel tau protein fold. Nature 561:137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Falcon B, Zivanov J, Zhang W, Murzin AG, Garringer HJ, Vidal R et al (2019) Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 568:420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ferrer I, Santpere G, van Leeuwen FW (2008) Argyrophilic grain disease. Brain 131(Pt 6):1416–1432. [DOI] [PubMed] [Google Scholar]
- 21. Goedert M, Eisenberg DS, Crowther RA (2017) Propagation of tau aggregates and neurodegeneration. Ann Rev Neurosci 40:189–210. [DOI] [PubMed] [Google Scholar]
- 22. Hara M, Hirokawa K, Kamei S, Uchihara T (2013) Isoform transition from four‐repeat to three‐repeat tau underlies dendrosomatic and regional progression of neurofibrillary pathology. Acta Neuropathol 125:565–579. [DOI] [PubMed] [Google Scholar]
- 23. Hefti MM, Farrell K, Kim S, Bowles KR, Fowkes ME, Raj T, Crary JF (2018) High‐resolution temporal and regional mapping of MAPT expression and splicing in human brain development. PLoS One 13:e0195771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hyman BT, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Carrillo MC et al (2012) National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease. Alzheimer's Dement 8:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Josephs KA, Whitwell JL, Tacik P, Duffy JR, Senjem ML, Tosakulwong N et al (2016) [18F]AV‐1451 tau‐PET uptake does correlate with quantitatively measured 4R‐tau burden in autopsy‐confirmed corticobasal degeneration. Acta Neuropathol 132:931–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kovacs GG, Ferrer I, Grinberg LT, Alafuzoff I, Attems J, Budka H et al (2016) Aging‐related tau astrogliopathy (ARTAG): harmonized evaluation strategy. Acta Neuropathol 131:87–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lin A, Charney M, Shenton ME, Koerte IK (2018) Chronic traumatic encephalopathy: neuroimaging biomarkers. Handb Clin Neurol 158:309–322. [DOI] [PubMed] [Google Scholar]
- 28. Liu F, Gong CX (2008) Tau exon 10 alternative splicing and tauopathies. Mol Neurodegener 3:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Love S, Louis D, Ellison DW (2008) Greenfield's Neuropathology, Vol. 2, 8 th edn. Abingdon, UK: Taylor & Francis. [Google Scholar]
- 30. Mackenzie IR, Neumann M, Bigio EH, Cairns NJ, Alafuzoff I, Kril J et al (2010) Nomenclature and nosology for neuropathologic subtypes of frontotemporal lobar degeneration: an update. Acta Neuropathol 119:1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Malia TJ, Teplyakov A, Ernst R, Wu SJ, Lacy ER, Liu X et al (2016) Epitope mapping and structural basis for the recognition of phosphorylated tau by the anti‐tau antibody AT8. Proteins 84:427–434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Mandelkow EM, Mandelkow E (2012) Biochemistry and cell biology of tau protein in neurofibrillary degeneration. Cold Spring Harb Perspect Med 2:a006247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Maziuk B, Ballance HI, Wolozin B (2017) Dysregulation of RNA binding protein aggregation in neurodegenerative disorders. Front Mol Neurosci 10:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Dirk Keene C, Litvan I et al (2015) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy. Acta Neuropathol 131:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. McKee AC, Cairns NJ, Dickson DW, Folkerth RD, Keene CD, Litvan I et al (2016) The first NINDS/NIBIB consensus meeting to define neuropathological criteria for the diagnosis of chronic traumatic encephalopathy . Acta Neuropathol 131:75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McKee AC, Stein TD, Kiernan PT, Alvarez VE (2015) The neuropathology of chronic traumatic encephalopathy. Brain Pathol 25:350–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McKee AC, Stern RA, Nowinski CJ, Stein TD, Alvarez VE, Daneshvar DH et al (2013) The spectrum of disease in chronic traumatic encephalopathy. Brain 136(Pt 1):43–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. McKeith IG (2006) Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the Consortium on DLB International Workshop. J Alzheimer's Dis 9(Suppl. 3):417–423. [DOI] [PubMed] [Google Scholar]
- 39. McMillan P, Korvatska E, Poorkaj P, Evstafjeva Z, Robinson L, Greenup L et al (2008) Tau isoform regulation is region‐ and cell‐specific in mouse brain. J Comp Neurol 511:788–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mez J, Daneshvar DH, Abdolmohammadi B, Chua AS, Alosco ML, Kiernan PT et al (2019) Duration of American football play and chronic traumatic encephalopathy. Ann Neurol 87:116–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Mez J, Daneshvar DH, Kiernan PT, Abdolmohammadi B, Alvarez VE, Huber BR et al (2017) Clinicopathological evaluation of chronic traumatic encephalopathy in players of American football. JAMA 318:360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Montine TJ, Phelps CH, Beach TG, Bigio EH, Cairns NJ, Dickson DW et al (2012) National Institute on Aging‐Alzheimer's Association guidelines for the neuropathologic assessment of Alzheimer's disease: a practical approach. Acta Neuropathol 123:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rebeiz JJ, Kolodny EH, Richardson EP, Jr. (1967) Corticodentatonigral degeneration with neuronal achromasia: a progressive disorder of late adult life. Trans Am Neurol Assoc 92:23–26. [PubMed] [Google Scholar]
- 44. Schmidt ML, Zhukareva V, Newell KL, Lee VM, Trojanowski JQ (2001) Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer's disease. Acta Neuropathol 101:518–524. [DOI] [PubMed] [Google Scholar]
- 45. Schoch KM, DeVos SL, Miller RL, Chun SJ, Norrbom M, Wozniak DF et al (2016) Increased 4R‐tau induces pathological changes in a human‐tau mouse model. Neuron 90:941–947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sealey MA, Vourkou E, Cowan CM, Bossing T, Quraishe S, Grammenoudi S et al (2017) Distinct phenotypes of three‐repeat and four‐repeat human tau in a transgenic model of tauopathy. Neurobiol Dis 105:74–83. [DOI] [PubMed] [Google Scholar]
- 47. Steele JC, Richardson JC, Olszewski J (1964) Progressive supranuclear palsy. A heterogeneous degeneration involving the brain stem, basal ganglia and cerebellum with vertical gaze and pseudobulbar palsy, nuchal dystonia and dementia. Arch Neurol 10:333–359. [DOI] [PubMed] [Google Scholar]
- 48. Stein TD, Montenigro PH, Alvarez VE, Xia W, Crary JF, Tripodis Y et al (2015) Beta‐amyloid deposition in chronic traumatic encephalopathy. Acta Neuropathologica 130:21–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Stern RA, Adler CH, Chen K, Navitsky M, Luo J, Dodick DW et al (2019) Tau positron‐emission tomography in former national football league players. N Eng J Med 380:1716–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Stern RA, Daneshvar DH, Baugh CM, Seichepine DR, Montenigro PH, Riley DO et al (2013) Clinical presentation of chronic traumatic encephalopathy. Neurology 81:1122–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tagge CA, Fisher AM, Minaeva OV, Gaudreau‐Balderrama A, Moncaster JA, Zhang XL et al (2018) Concussion, microvascular injury, and early tauopathy in young athletes after impact head injury and an impact concussion mouse model. Brain 141:422–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Thibodeau MP, Miller BL (2013) “Limits and current knowledge of Pick's disease: its differential diagnosis”. A translation of the 1957 Delay, Brion, Escourolle article. Neurocase 19:417–422. [DOI] [PubMed] [Google Scholar]
- 53. Togo T, Akiyama H, Iseki E, Uchikado H, Kondo H, Ikeda K et al (2004) Immunohistochemical study of tau accumulation in early stages of Alzheimer‐type neurofibrillary lesions. Acta Neuropathol 107:504–508. [DOI] [PubMed] [Google Scholar]
- 54. Uchihara T (2014) Pretangles and neurofibrillary changes: similarities and differences between AD and CBD based on molecular and morphological evolution. Neuropathology 34:571–577. [DOI] [PubMed] [Google Scholar]
- 55. Weingarten MD, Lockwood AH, Hwo SY, Kirschner MW (1975) A protein factor essential for microtubule assembly. Proc Natl Acad Sci U S A 72:1858–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Zetterberg H, Blennow K (2018) Chronic traumatic encephalopathy: fluid biomarkers. Handb Clin Neurol 158:323–333. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sample demographics broken down by experiment.