

SUMMARY
The development of fast Fourier transform (FFT) algorithms enabled the sampling of billions of complex conformations and thus revolutionized protein-protein docking. FFT based methods are now widely available and have been used in hundreds of thousands of docking calculations. Although the methods perform “soft” docking, which allows for some overlap of component proteins, the rigid body assumption clearly introduces limitations on accuracy and reliability. In addition, the method can work only with energy expressions represented by sums of correlation functions. In this paper we use a well-established protein-protein docking benchmark set to evaluate the results of these limitations by focusing on the performance of the docking server ClusPro, which implements one of the best rigid body methods. Furthermore, we explore the theoretical limits of accuracy when using established energy terms for scoring, provide comparison to flexible docking algorithms, and review the historical performance of servers in the CAPRI docking experiment.
Keywords: Structure prediction, fast Fourier transform, scoring function, ClusPro docking server, docking benchmark
Graphical Abstract
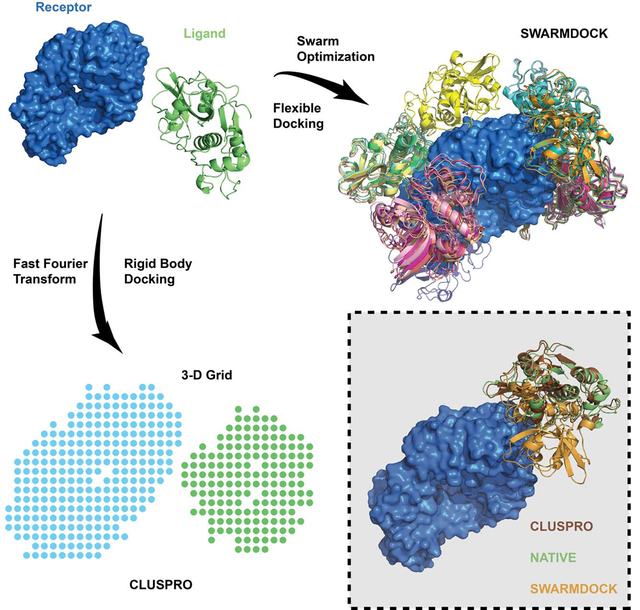
eTOC blurb
Rigid body docking methods, represented here by the ClusPro server, provide acceptable or better models for more complexes than flexible docking methods, but the later can yield higher accuracy for some targets. In spite of its shortcomings, ClusPro has over 15,000 registered users, and in 2019 performed 98,300 docking calculations.
INTRODUCTION
Protein-protein interactions (PPIs) are essential to the basic functioning of cells and larger biological systems in all living organisms. The golden standard of validating and understanding PPIs is X-ray crystallography. However, crystallizing protein complexes is sometimes very difficult, and the number of PPIs discovered is far outpacing the number of complex structures. Protein-protein docking is a computational tool that can potentially fill this gap by giving atomic-level details of the interactions between two proteins. In particular, docking can generate models that can be validated by simple tools such as crosslinking or site directed mutations.
It is widely recognized that one of the most important developments in protein-protein docking has been the introduction of fast Fourier transform (FFT) for energy evaluation (Katchalski-Katzir et al., 1992). In FFT based methods one of the proteins is placed at the origin of the coordinate system on a fixed grid, the second protein is placed on a movable grid, and the interaction energy is written as a sum of a few correlation functions. The numerical efficiency of the methods stems from the fact that such energy functions can be simultaneously evaluated for all translations using fast Fourier transforms, and only rotations need to be considered explicitly. This results in the ability of exhaustively sampling billions of the conformations of the two interacting proteins, obtaining energy values at each grid point. Thus, the FFT based algorithm enables global docking without any a priori information on the structure of the complex.
While the use of FFT yields impressive speed-up, it results in two major limitations. The first is the need for rigid body approximation. In all rigid body methods the shape complementarity term in the scoring function allows for some overlaps, and hence the methods are able to tolerate moderate differences between bound and unbound (separately crystallized) structures. However, the need for reducing sensitivity also reduces the specificity defined by the complementarity of the shape of the two proteins. In particular, the docked conformations that are close to the native structure do not necessarily have the lowest energies, whereas low energy conformations may occur far from the X-ray structures. Therefore rigid body methods must retain a large set of low energy docked structures for secondary processing that may include some type of refinement, hoping that the retained set includes at least some that are close to the native structure of the complex. The second limitation is that the energy expression should be written as a sum of correlation functions. The original work (Katchalski-Katzir et al., 1992) used a simple scoring function that accounted only for shape complementarity. However, subsequent methods based on the FFT correlation approach to docking introduced more complex and more accurate scoring functions that also included terms representing electrostatic interactions (Gabb et al., 1997; Mandell et al., 2001), or both electrostatic and desolvation terms (Chen and Weng, 2002; Kozakov et al., 2006). In fact, better models of the desolvation/nonpolar contributions to the binding free energy played a major role in making protein-protein docking useful for applications (Camacho et al., 2000; Camacho and Vajda, 2001), and remain critically important for the development of rigid body methods (Brenke et al., 2012; Chuang et al., 2008).
The goal of this paper is to rigorously evaluate the performance of rigid body protein-protein docking in view of the above limitations. The accuracy of docking methods has been continuously monitored since 2004 by the community-wide experiment called Critical Assessment of PRedicted Interactions (CAPRI) (Janin et al., 2003). More recently, another community-wide experiment focused on protein structure determination, Critical Assessment of Structure Prediction (CASP), joined with CAPRI. In CAPRI/CASP the challenge is predicting protein complex structures based on the sequences of the individual component proteins rather than their crystal structures, thus requiring the use of homology modeling tools. It is useful that CAPRI and CAPRI/CASP are blind prediction experiments and hence provide unbiased information on the accuracy of docking methods. However, the number, quality and diversity of these sporadic challenges lack the ability to give a full picture of a method’s strengths and weaknesses. In particular, in CAPRI/CASP the majority of new targets were homo-oligomers rather than complexes formed by two different proteins. In many cases such structures have homologous templates available in the Protein Data Bank (PDB), and the problems can be solved by homology modeling without the need for any docking (Porter et al., 2019a). In view of the limitations of the CAPRI and CAPRI/CASP experiments, evaluating docking methods on diverse and well-populated benchmark sets is still needed. Here we use version 5.0 of the well-established Protein Docking Benchmark which contains 230 protein pairs known to form complexes (Vreven et al., 2015). A key feature of the benchmark set is the availability of both the structure of each target complex and the unbound structures of the component proteins. The most recent version 5 of the Protein Docking Benchmark (BM5) includes 40 antibody-antigen, 88 enzyme-containing and 102 “other” type complexes. Based on the difference between bound and unbound conformations of the component proteins, these complexes are classified as 151 rigid-body (easy), 45 medium difficulty, and 34 difficult targets.
An important step in the evaluation of docking methods is calculating measures that describe the closeness of the models to experimentally determined crystal structures. CAPRI uses three related parameters for assessing a model: the fraction of native contacts, the backbone root mean square deviation of the ligand (LRMSD) from the reference ligand structure after superimposing the receptor structures, and the backbone RMSD of the interface residues (IRMSD). Based on these measures CAPRI defined four categories of accuracy, which are incorrect, acceptable, medium, and high accuracy (see Star Methods). More recently a continuous score called DockQ was developed that encapsulated the above three measures (Basu and Wallner, 2016). The DockQ values range from 0 to 1, where a value exceeding 0.80 implies high accuracy, between 0.80 and 0.49 medium accuracy, and between 0.49 and 0.23 acceptable accuracy. DockQ has been widely accepted and was even implemented in the official evaluations of the latest CAPRI round, and hence it is also used here.
While trying to reach general conclusions, here we discuss the results generated by the ClusPro server that performs rigid body docking (Kozakov et al., 2017). We focus on ClusPro results for three reasons. First, the use of a fully automated server makes the results reproducible. Second, ClusPro has consistently been one of the best docking servers as demonstrated in CAPRI rounds over the last 15 years. However, the other rigid body docking programs, including ZDOCK (Chen et al., 2003), GRAMM (Tovchigrechko and Vakser, 2005, 2006), pyDOCK (Cheng et al., 2007), and FTDock (Gabb et al., 1997) perform very similarly to ClusPro, and hence the latter can be used to explore the accuracy and limitations of rigid body methods. The third reason is that ClusPro has been developed in our lab, and hence we can also explore how variations in the parameters affect the docking results.
The main differences among FFT based docking methods are in the sampling density and in the scoring function used. The sampling density is defined by the step size of the translational grid, generally between 0.8 Å and 1.2 Å, and the number of rotations of the ligand protein, resulting in 5 to 12 degrees step size in terms of the Euler angles. For scoring, all methods use linear combinations of several energy terms that include attractive and repulsive van der Waals contributions describing shape complementarity, and one or more terms representing the electrostatic part of the binding energy. In spite of some differences, these energy terms are fairly standard. The scoring functions in most programs also include some structure-based energy expression for adding desolvation energy contributions, which is a key factor in determining the accuracy of docking results. In particular, ClusPro uses the pairwise potential DARS, based on decoys as the reference state (Chuang et al., 2008). As will be discussed, a major uncertainty is the relative weighting of these energy terms. Nevertheless, the best rigid body methods have similar success rates, and hence we assume that by analyzing the results provided by ClusPro we can assess the general limitations of the approach.
A protocol describing the use of ClusPro was published in 2017 (Kozakov et al., 2017). However, a detailed look at the advantages of each feature, strengths and weaknesses when it comes to different classes of proteins and rigidity of complexes has not been previously studied. This paper outlines the first systematic and rigorous evaluation of ClusPro on a well-accepted protein-protein docking benchmark. We mainly look at the performance across two dimensions: the conformational change upon complex formation, and the type of proteins in the target (antibody, enzyme, or “others”). As expected, results are better for more rigid complexes compared to complexes that have large conformational differences between unbound and bound states. As mentioned, an important question related to specific types of complexes is how to select the weights of energy terms to obtain the best results. Here we go one step further, and generate models using 105 different combinations of such coefficients. This will enable us to determine how the choice of weights affects docking accuracy and to find the best coefficient set for each particular complex to explore the theoretical limits of accuracy of rigid body docking with the given energy terms. Finally we compare ClusPro to some of the best flexible docking methods in terms of their success rates, considering both a well-studied subset of the benchmark set and a historical review of the CAPRI docking experiments.
RESULTS
Performance on the protein docking benchmark
For evaluating the ClusPro server we use the benchmark set 5.0 (BM5) (Vreven et al., 2015). The component proteins of all 230 target complexes were directly downloaded from BM5 and docked using ClusPro. For each complex, the 1000 lowest energy models were retained. The top 30 models (centers of the 30 largest clusters) were evaluated using the DockQ program. We note that some complexes may have fewer than 30 clusters and that a DockQ result of 0.23 and above (acceptable or better) will be termed as a good solution. In many cases we also list the success rates (i.e., the percentages of targets with acceptable, medium, or high accuracy models) in the top 1, top 5, top 10, and top 30 predictions, in some figures denoted as T1, T5, T10, and T30, respectively. These numbers are of interest for two reasons. First, as will be shown, docking methods rarely identify the best model as the top 1 prediction, but 5 or 10 models can be subjected to further refinement and rescoring. Second, recognizing this limitation, CAPRI generally allowed for the submission of 10 models for each target (Lensink et al., 2007; Lensink and Wodak, 2010, 2013; Mendez et al., 2003; Mendez et al., 2005), although in the more recent rounds ranking of the different groups was based on the top 5 rather than top 10 predictions (Lensink et al., 2019b; Lensink et al., 2018; Lensink et al., 2017).
Figures 1A, 1B, and Table 1 show the percentages of good models obtained by ClusPro in the top 10 and in the top 30 predictions. Unless otherwise noted, all figures of ClusPro’s results show with balanced coefficient set for enzymes, antibody mode for antibodies, and “others” mode for “other” type complexes (see Star Methods). The detailed results for each target in BM5, with the number of acceptable, medium and high accuracy predictions in T1, T5, T10 and T30, are shown in Data S1. According to Figure 1A and 1B, ClusPro performs best on enzyme containing targets. Indeed, 51 of the 88 such complexes (57.9%) had acceptable or better models in the top 10 predictions, compared to 19 of the 40 antibody-antigen complexes (50%) (note that only 38 of the 40 were evaluated successfully as DockQ failed to evaluate the predictions for 2 targets because of their large size), and 27 of the 102 “other” type complexes (26.7%) (only 101 of the 102 were evaluated successfully by DockQ). Increasing the number of predictions from top 10 to top 30, eight more enzyme containing complexes, five more antibody-antigen complexes, and nine more “other” type complexes had acceptable or better solutions. In total, 42.7% of the benchmark set had acceptable or better solutions in the top 10 predictions, which increases to 52.4% in the top 30. (Note that all percentages are based on the 227 successfully evaluated targets). That amounts to 97 and 119 good solutions in the top 10 and in the top 30, respectively.
Figure 1.
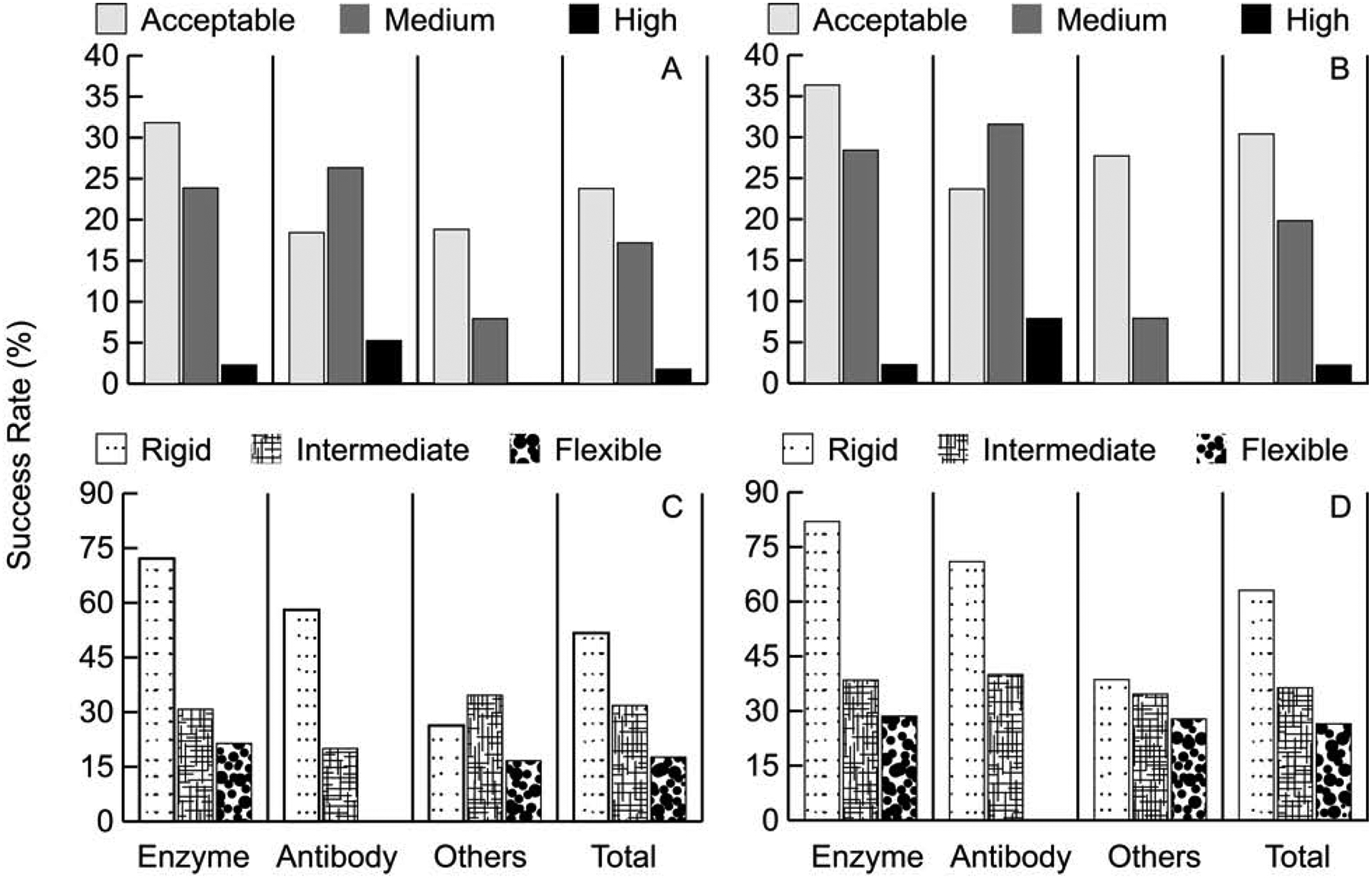
Performance on the ClusPro server on the protein docking benchmark. A. Percentage of targets with acceptable, medium, and high accuracy models in the top 10 (T10) predictions for different protein types. B. Same as in A, but in the top 30 (T30) predictions. C. Percentage of targets with acceptable, medium, and high accuracy models in the top 10 (T10) predictions, depending on the level of docking difficulty. D. Same as in C, but in the top 30 (T30) predictions.
Table 1.
ClusPro’s performance in top 10 and top 30 predictions for 227 targets in benchmark 5.0 (BM5)
| Target type | Number of targets testeda | Performance by target difficultyb | “Good” modelsd | |||
|---|---|---|---|---|---|---|
| Easyc | Intermediatec | Difficultc | Number | % | ||
| Enzyme | 88/61/13/14 | 44/2***/18** | 4/0***/2** | 3/0***/1** | 51 | 57.95% |
| 50/2***/21** | 5/0***/2** | 4/0***/2** | 59 | 67.04% | ||
| Unbound antibody | 26/20/5/1 | 11/0***/6** | 1/0***/0** | 0/0***/0** | 12 | 46.15% |
| 12/0***/6** | 2/0***/0** | 0/0***/0** | 14 | 53.85% | ||
| Bounde antibody | 12/11/0/1 | 7/2***/4** | N/A | 0/0***/0** | 7 | 58.33% |
| 10/3***/6** | N/A | 0/0***/0** | 10 | 83.33% | ||
| “Others” | 101/57/26/18 | 15/0***/5** | 9/0***/2** | 3/0***/1** | 27 | 26.73% |
| 22/0***/5** | 9/0***/2** | 5/0***/1** | 36 | 35.64% | ||
| Totalc | 227/149/44/34 | 87/4***/33** | 14/0***/4** | 6/0***/2** | 97 | 42.73% |
| 94/5***/38** | 16/0***/4** | 9/0***/3** | 119 | 52.42% | ||
| % “Good” predictions | 51.68% | 31.82% | 17.65% | |||
| 63.09% | 36.36% | 26.47% | ||||
Total/easy/medium/difficult targets
Number of targets with “good” (acceptable or better) predictions, among them predictions with high (***) and medium (**) accuracy.
Line 1: top 10 predictions, line 2: top 30 predictions
Targets with “good” (acceptable or better) predictions
Targets with the antibody structure from the complex
As expected, ClusPro performs best for rigid body protein complexes. As shown in Figure 1C, 1D, and Table 1, ClusPro solves 51.7% of rigid (easy) targets compared to 31.8% for intermediate and 17.6% for flexible (difficult) targets in the top 10 (T10) predictions. The overall success rate still reaches 42.7% because 151 of the 230 targets are classified as rigid body. The success rate increases to 63.1%, 36.36% and 26.5%, respectively, when considering the top 30 (T30) models. For enzymes, both in top 10 and top 30 models, more than twice the percentage of rigid targets were solved compared to intermediate, and over 3 times that of flexible ones. Similar trends are seen for antibodies. All high accuracy predictions were from the rigid category in both antibody and enzyme containing cases.
Energy-based versus cluster-based model selection
The set of the 1000 lowest energy models retained from PIPER is too large for post-processing and has to be reduced by selecting a number of predictions that are likely to be near-native. One of the major features of ClusPro is that it clusters the 1000 models using interface RMSD with a 9Å radius and ranks the clusters based on cluster population, retaining the top 1, top 5, top 10, and top 30 clusters. In Figure 2 we show the fraction of good models retained in this process, and compare energy-based and cluster-based model selection. Note that we consider only the top 30 clusters, and hence the performance of cluster-based selection does not improve beyond 30. However, up to that number selecting the centers of most populated clusters always provides substantially better performance than selecting the same number of lowest energy structures, and this is true for all types of protein complexes. Figures 2A–2C also show that PIPER generates good structures in the 1000 structures for 71.6%, 73.68%, and 51.2% of enzyme, antibody, and “other” type containing complexes, respectively. When retaining only the 100 best energy structures the success rates are about 10% lower. The top 30 cluster centers provide almost the same success rates as the top 100 energy ranked models for antibodies and “others”, showing only 2.6 and 3.9 percentage points loss. In enzymes, the top 30 cluster centers actually perform better than the top 100 energy ranked models. Thus, going from 1000 structures to 30 cluster centers does not lead to losing a very large fraction of good solutions (4.6%, 10.5%, and 15.8% for enzymes, antibodies and “others”, respectively). However, as shown in Figure 2, there is substantial drop from the top 30 cluster centers when considering only the top 10, top 5, and particularly the top 1 cluster.
Figure 2.
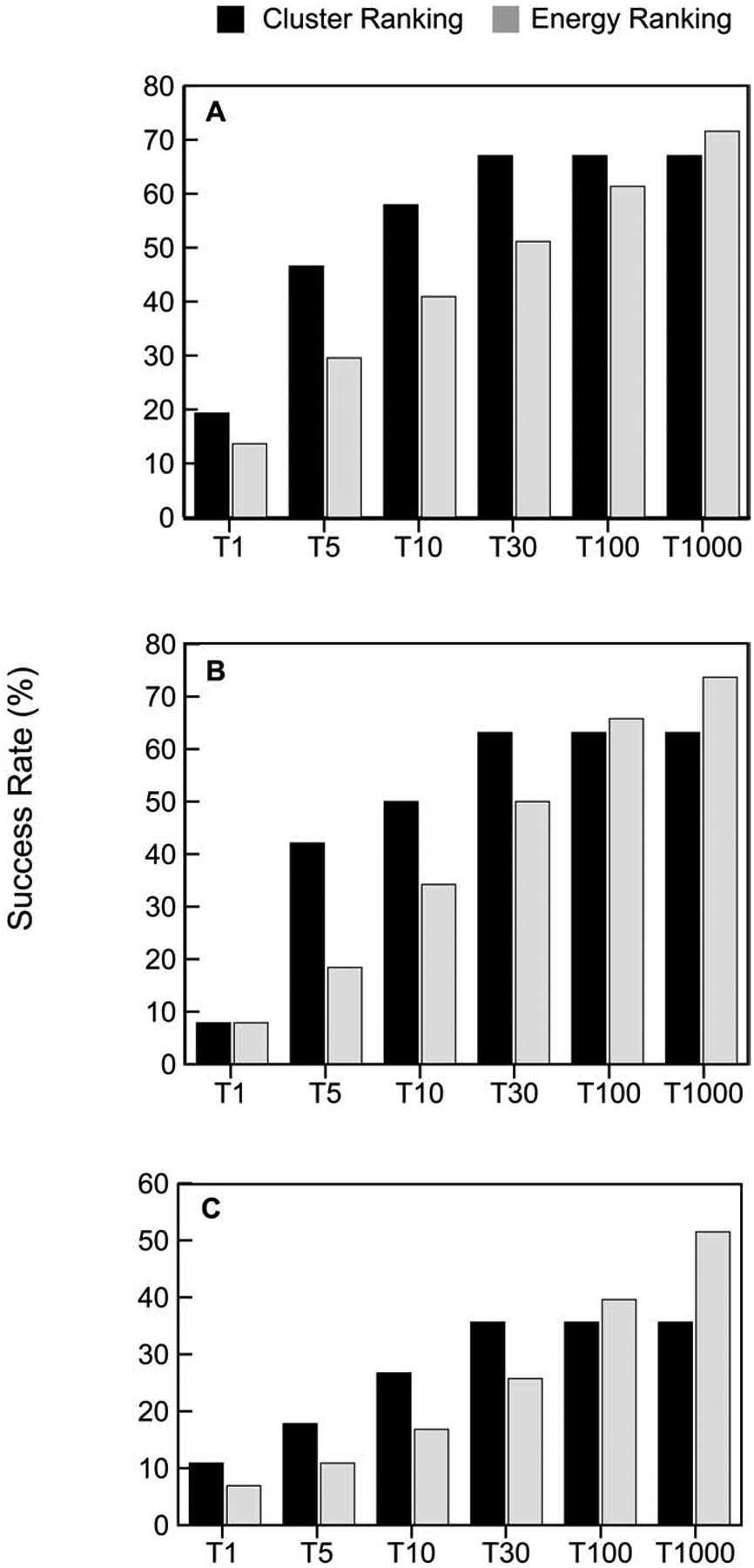
Cluster-based versus energy-based ranking of models. A. Percentage of “good” models in the top 1, top 5, top 10, top 30, and top 1000 predictions for enzyme containing complexes, based on either cluster size or energy value. We consider only up to 30 clusters, and hence the performance of cluster-based selection does not improve beyond 30. Note that energy-based selection of the top 1000 models means that all structures generated by ClusPro are retained. B. Same as A, but for antibody-antigen complexes. C. Same as A, but for “other” type complexes.
Antibody-Antigen Docking
Determining the structure of antibody-antigen complexes is an important application of protein docking, and results have been substantially improved by developing a structure based potential specific to these interactions (Brenke et al., 2012). Due to the limited number of antigen-antibody complex structures, the potential is defined for fewer atom types than the DARS potential developed for enzyme-inhibitor and “other” type complexes (Chuang et al., 2008). Antibody-antigen docking has two more special properties. The first is that the interactions are generally restricted to the Complementarity Determining Regions (CDRs) of antibodies. Thus, the docking can be restricted by “masking” the surface of the antibody except for the CDRs and a few additional surrounding residues. ClusPro includes both automated and manual options for masking. Automated masking recognizes known starting and ending motifs of CDR sequences, adds one to four residues on each end of the loops to allow for some flexibility in the docking, and masks the rest of the antibody surface (see Star Methods). Manual masking allows users to upload a masking file by themselves.
Figure 3A shows the percentage of antibody complexes solved without masking, with automated masking, and with a manually uploaded mask file. The manual masking in this study used the Chothia numbering system and boundaries for CDRs (Al-Lazikani et al., 1997). Masking generally increases the success rate, but the gain is moderate. Without masking ClusPro predicts good solutions in the top 30 for 63.2%% of antibody-antigen targets, compared to 68.4% with the automated masking option. The relative improvements are somewhat larger in the top 1, top 5 and top 10 predictions. Manual masking also shows only moderate improvements in T1, T5, and T10 over no masking. Thus, the interaction potential on its own is able to identify the CDR regions.
Figure 3.
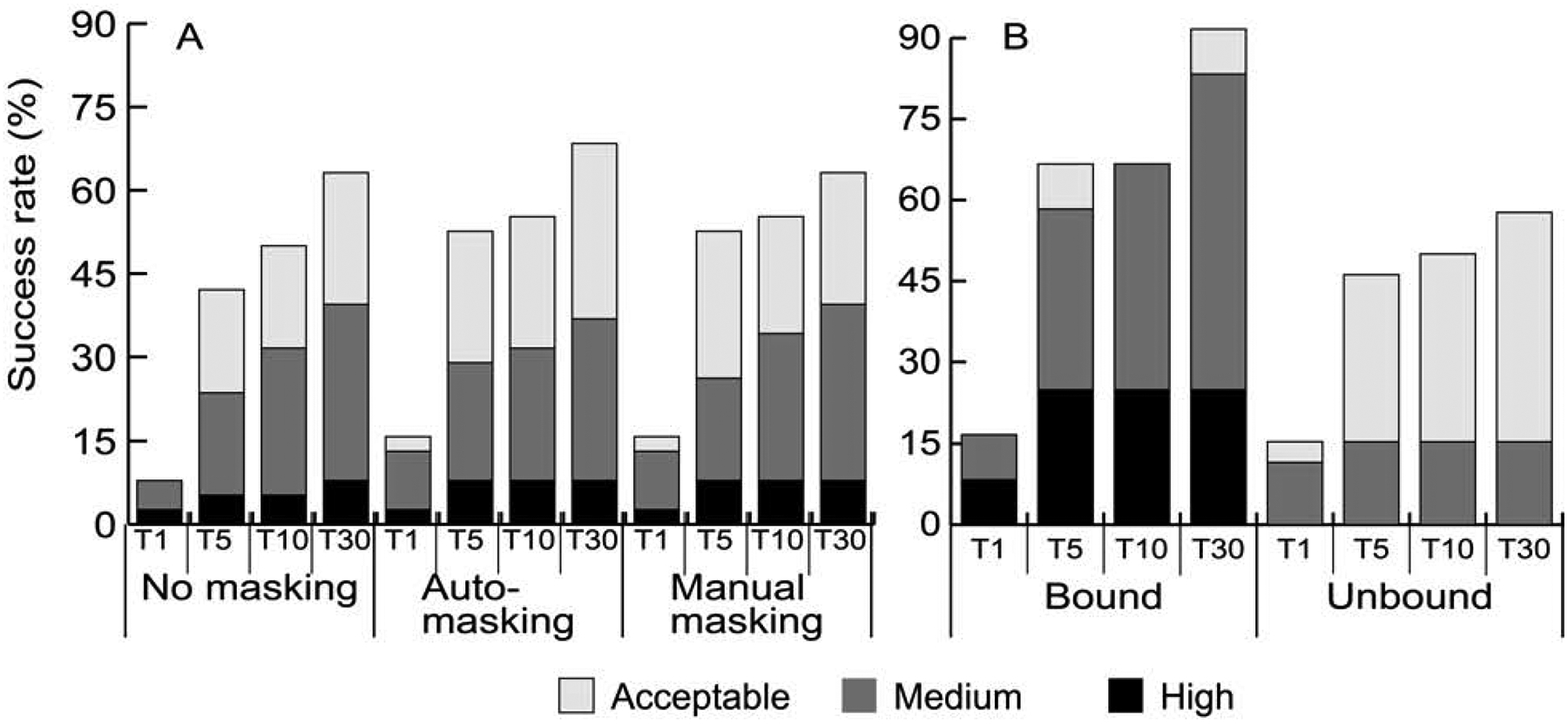
Results of antibody-antigen docking. A. Percentage of antibody-antigen targets with acceptable, medium, and high accuracy model in the top 1 (T1), top 5 (T5), top 10 (T10), and top 30 (T30) predictions, obtained using no masking, automated masking, and manual masking of non-CDR regions of the antibodies. B. Success rates for antibody-antigen targets with bound and unbound (separately crystallized) antibody structures with automated masking.
The second special property specific to antibody-antigen docking in the Protein Docking Benchmark is that 12 targets of the 40 antibody-antigen targets in BM5 include the structure of a separately crystallized antigen and the structure of the antibody extracted from the target complex. As shown in Figure 3B, the success rate for such complexes is much higher than for the separately crystallized antibodies. Calculated with masking of non-CDRs, 91.7% of targets with bound antibody structures had a good model in the top 30 predictions, compared to only 57.7% for targets with unbound antibody structures. In the top 1 prediction the difference is insignificant (16.7% versus 15.39%). High accuracy predictions were obtained only for targets with bound antibodies (Figure 3B). There is substantial difference in medium accuracy predictions, which was nearly 58% for targets with bound antibodies but only about 15% for the unbound cases in top 30. This is in line with the results shown for enzymes and rigid-body cases, demonstrating that ClusPro does well for targets with moderate conformational changes upon complex formation. Thus, as expected, the flexibility of the CDRs in antibodies makes docking much more challenging.
Weights of the Energy Terms
As described in Star Methods, unless the target is specified as an antibody-antigen or “other” type complex, ClusPro simultaneously generates four sets of models using the scoring schemes called balanced (denoted as 00), electrostatics-favored (02), hydrophobicity-favored (04), and van der Waals + electrostatics, i.e., no structure-based DARS energy term (06). Although all four predictions are available, for enzyme-inhibitor complexes we suggested using the results obtained by the balanced coefficient set (00). For “other” type complexes we have developed a special option that uses three different coefficient sets, generates 500 conformations for each, and clusters the resulting 1500 structures. These suggestions were based on a limited number of docking runs (Kozakov et al., 2013). However, application of ClusPro to the entire benchmark set enabled us to further investigate these choices. As shown in Figure 4A, for enzyme containing complexes the use of the balanced set (00) and of the electrostatics-favored set (02) yield about the same success rates, but the latter increases the number of targets with medium and high accuracy predictions, in agreement with the observation that many enzyme-inhibitor interactions are primarily driven by electrostatics. The coefficient sets 04 (hydrophobicity favored) and 06 (no Epair) yield substantially worse predictions (Figure S1, related to Figure 4A). These results were tested by five-fold cross-validation on 71 enzymes in Protein Docking Benchmark 4 (BM4) (Figure S2) and also tested on the BM5 additions (see Star Methods). According to the cross validation, the parameter set 00 generally yields slightly better results. However, this is not true for the test set (i.e., the added enzyme cases in BM5), as the parameter set 00 gave acceptable or better solutions for 7 targets in the top 10 predictions, and 10 targets in the top 30, whereas the parameter set 02 gave good solutions for 10 targets in the top 10 and 11 targets in the top 30. Thus, there is no clear preference, and either set 00 or 02 can be used. The coefficient set 04 was able to obtain good solutions only for 7 and 8 cases, while the coefficient 06 only gave 3 and 4 cases in T10 and T30 respectively, and thus these coefficient sets should not be used.
Figure 4.
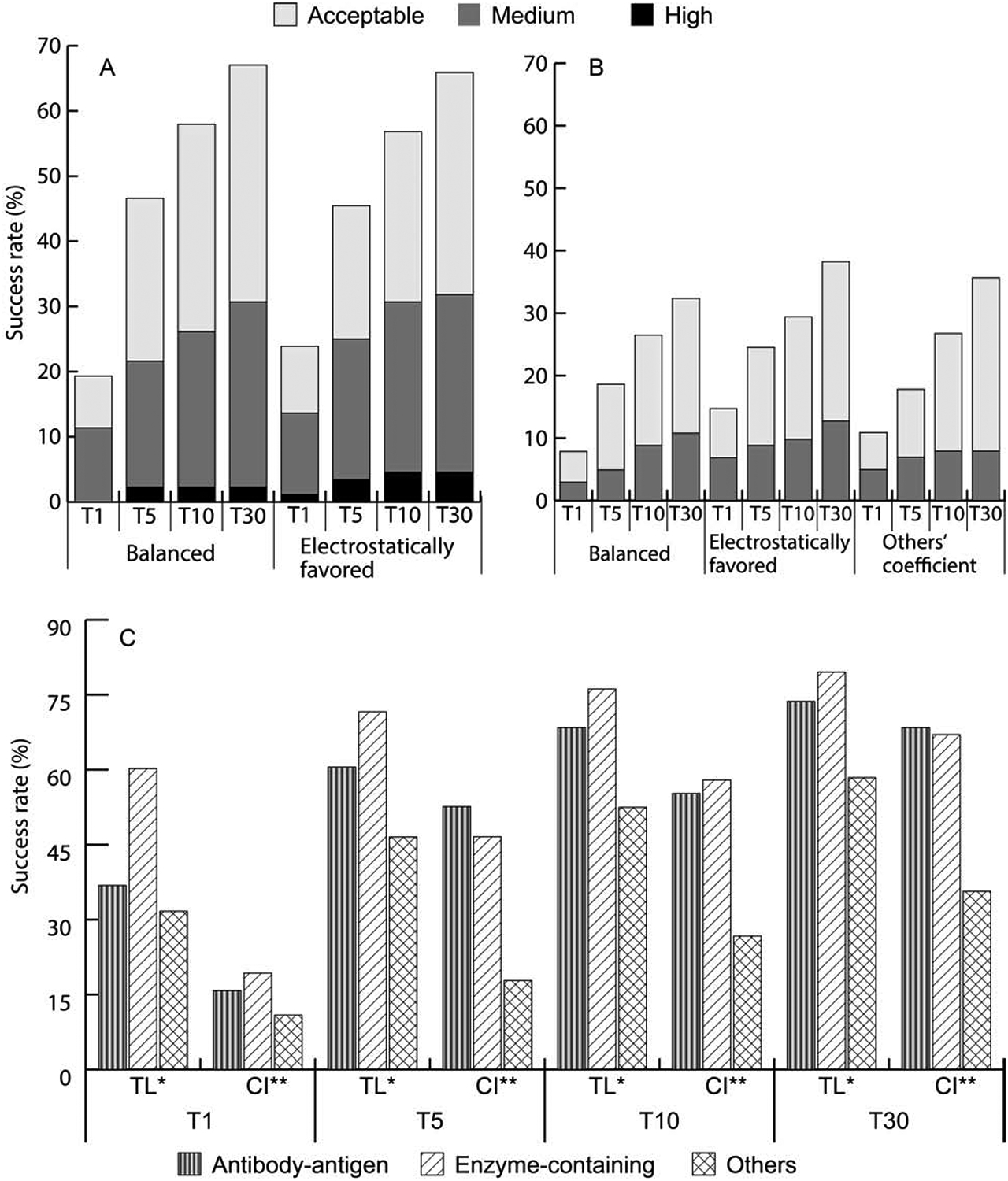
Impact of weighting the energy terms and exploring the limits of performance. A. Success rates for enzyme containing targets using the balanced (00) and electrostatics favored (02) weighting coefficient set in the ClusPro scoring function. B. Success rates for “other” type targets using the balanced (00), electrostatics favored (02), or special “other” (03) sets of weighting coefficients. For “other” type complexes, ClusPro employs 3 different sets of weighting coefficients, generates 500 conformations for each, and clusters the resulting 1500 structures. C. Success rates for the different types of targets obtained by using the best of 105 weighting coefficient sets (theoretical limit, TL), versus success rates obtained by using the weighting coefficient sets as currently implemented (CI) in ClusPro.
The reanalysis of the “other” type complexes were more successful, as it became clear that the electrostatic-favored set (02) yields the best results for these complexes, and there is no need for using the more complex algorithm of generating 1500 structures with three different parameter sets (Figure 4B). A five-fold cross validation was performed on 80 BM4 cases and in 4 of the 5 folds, the electrostatically driven coefficient set was found to be superior (Figure S3). Testing on BM5 additions, the coefficient set 02 obtained good solutions for 5 targets in the top 5 predictions and 6 targets in the top 10, and the coefficient set 00 gave 4 good predictions in T5 and 6 good predictions in T10. Both sets worked slightly better than the “others” option 03, which gave 3 good predictions in T5 and 5 good predictions in T10, and hence we recommend that users should use standard docking for complexes containing “others”. When all “others” targets in BM5 are evaluated, 4, 7, 3 and 4 more complexes were solved using the electrostatic-favored set (02) in the top 1, top 5, top 10 and top 30 predictions, respectively, than the currently preferred “others” mode. Figure 4B also shows that even the balanced coefficient set (00) has a slight advantage over the current “others” mode in top 5 and top 10 predictions.
The second interesting question concerning the coefficients of energy terms is the optimality of selected values, i.e., how much the results could be improved by selecting the individually best coefficient set for each target. To answer this question, we generated predictions using 105 different sets of coefficient values, and calculated the overall success rate considering the best result for each complex. Results are compared to the currently implemented coefficient sets in Figure 4C. For the antibody-antigen complexes the non-CDR regions were masked using the automatic feature available on the ClusPro server. However, 1KXQ and 2VIS did not have a light chain and so were docked with antibody mode but without masking. The “others” mode (03) for “others” and the balanced coefficient set (00) for enzymes were maintained for comparison. Figure 4C shows that taking the best of the 105 coefficients enables PIPER to find a good model in the top 1 prediction for 14 of the 38 antibody-antigen complexes compared to only 6 by the current implementation of ClusPro. The gap narrows significantly to only 2 complexes when considering the top 30 models. In the top 1 predictions for enzyme-containing complexes the best weight set yields a good solution for more than 60% of the cases as opposed to only 19% using the balanced parameter set. Even in the top 30 predictions, for enzymes the theoretical best parameter set yields 11 more cases than the current implementation of ClusPro (70 versus 59). The trend continues for the “other” type complexes. In the top 1 prediction there is nearly a three-fold increase in success rate from the current implementation (from 11 to 32). In top 30, there were 59 successful cases when using the best coefficient set compared to only 34 using the standard method. Overall, by using the best coefficient set, ClusPro could find good models for 76.1% of enzymes, 68.4% of antibodies and 52.5% of “others” in the top 10 predictions. This is compared to 57.9%, 55.3%, and 26.7%, respectively, for the currently selected coefficient sets. In addition to the number of cases solved, the accuracy of predictions also increases. Using the best of the 105 coefficient sets, almost 80% of the good solutions in T30 are medium accuracy or better, compared to only 50% with the currently implemented coefficient set. Although selecting the best coefficient for each protein significantly outperforms ClusPro using the standard coefficient sets, Figure S2, related to Figure 4C, shows that the latter still represents a reasonable choice. In particular, for antibodies the current set is better than three quarters of the sets available. In all cases, the selected coefficient set is better than the median.
Comparison to Flexible Docking
Four of the best docking servers have been tested on 55 complexes that were added to docking benchmark 4 (Hwang et al., 2010) to create version 5 of the benchmark (Vreven et al., 2015). These servers included two rigid body servers, ZDOCK (Pierce et al., 2014) and pyDock (Pons et al., 2010) that are similar to ClusPro, and the servers SwarmDock (Moal et al., 2018; Torchala et al., 2013), and HADDOCK (High Ambiguity Driven DOCKing) (de Vries et al., 2010; Vangone et al., 2017) that implement flexible docking algorithms. Overall, the success rates (at least one acceptable prediction for a benchmark case) ranged between 5% and 16% for the top 1 prediction and 20–38% for the top 10 predictions (Vreven et al., 2015). Overall the best results were obtained using SwarmDock, followed by ZDOCK (Vreven et al., 2015). We attempted to compare ClusPro to the four servers on the same 55 targets. However, the structures 1EXB, 4GXU, 4GAM and 4FQI all have more than 10,000 atoms in the PDB file, which could not be handled by the SWARMDOCK server, since it limits the number of atoms to 10,000. We also note that these four are multi-protein rather than binary complexes, and thus docking only two selected subunits is not an entirely valid test. Therefore, the testing was limited to the remaining 51 structures. Docking antibodies by ClusPro we selected the “antibody mode” with automated masking. For enzymes and the “other” type targets, the electrostatically driven coefficient (02) set was used. Success rates of the other four servers for the 51 remaining targets were adopted from the original publication (Vreven et al., 2015). Results in Figure S5 (related to Figure 5), show that ClusPro provides the highest number of good models, but SWARMDOCK produces slightly more models in the top 1 prediction, and HADDOCK yields more high accuracy models.
Figure 5.
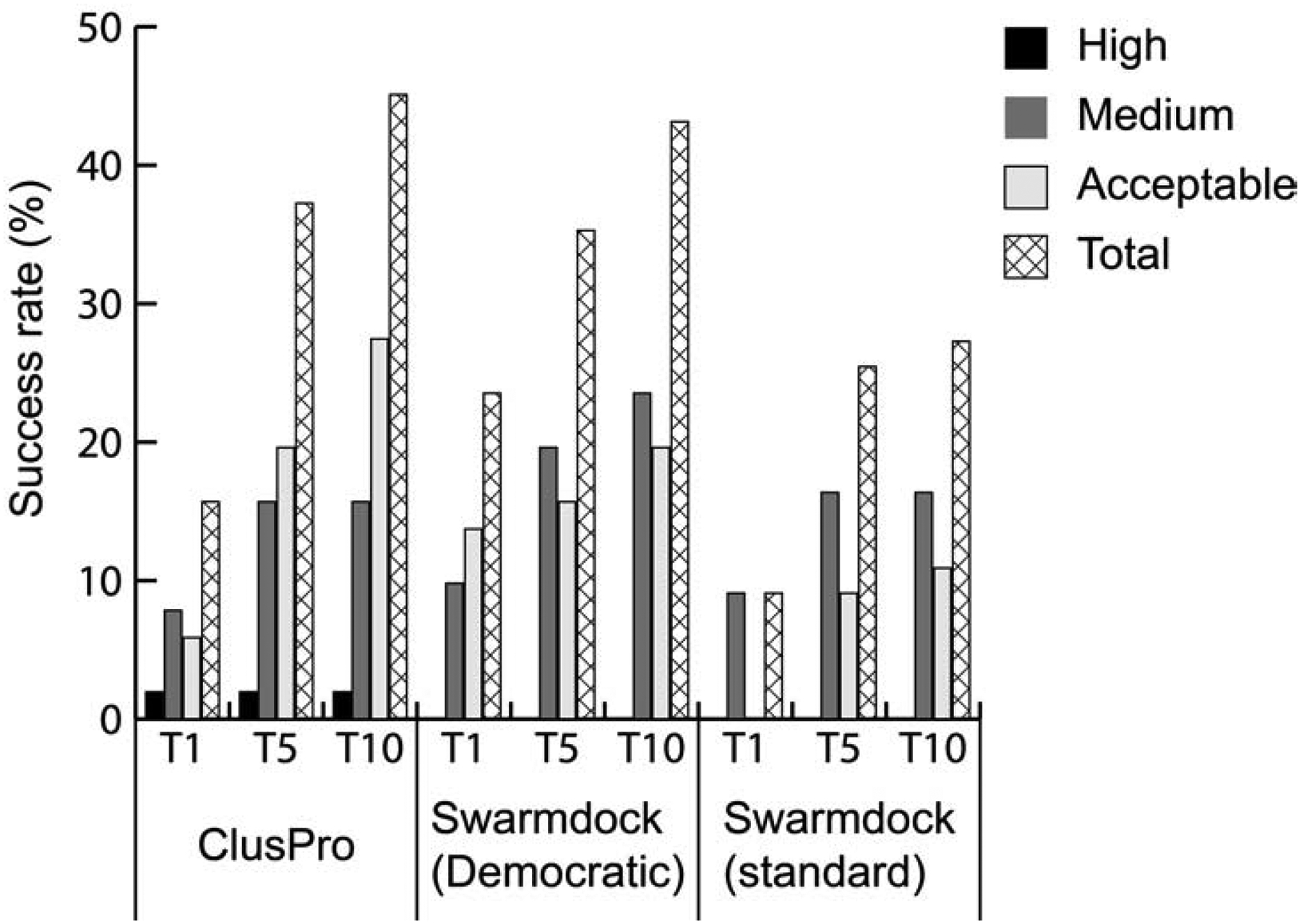
Rigid versus flexible docking. Success rates obtained by ClusPro, by the improved (“democratic”) version of the SwarmDock server, and by the standard versions of SwarmDock. All results are for 51 of the 55 new targets added to Benchmark version 4 (BM4) to form Benchmark 5.
Recently, the performance of SwarmDock has been substantially improved by the addition of a methodology of re-ranking models (Moal et al., 2017). The method employs an information-driven machine learning rescoring scheme called IRaPPA (Integrative Ranking of Protein-Protein Assemblies) (Moal et al., 2017). IRaPPA combines a large selection of metrics using support vector machines (R-SVMs) to obtain consensus ranking based on a voting method, resulting in so-called “democratic” modification of SwarmDock. Figure 5 shows success rates on the 51 targets for the standard SwarmDock, for the “democratic” versions of SwarmDock, and for ClusPro. The “democratic” ranking significantly improves the SwarmDock success rate, but ClusPro still performs somewhat better and obtains acceptable or better solutions for ~47% and ~41% compared to SWARMDOCK’s ~41% and ~35%, respectively, in the top 10 and in the top 5 predictions. However, the “democratic” SwarmDock predicts good models as the top 1 prediction for 23.5% of the 51 targets, whereas ClusPro obtains such result only for 15.69 % of the targets.
The CAPRI and CAPRI/CASP experiments
CAPRI is an ongoing protein docking experiment (Janin et al., 2003). Each round of CAPRI has a number of prediction targets, which are unpublished experimentally determined structures of protein-protein complexes. Given the atomic coordinates of the component proteins or of their homologs, the participants attempt to predict the structures of native complexes. Each group can submit up to ten predictions for each target, and the submitted models are evaluated by independent assessors. In 2014, CAPRI joined the protein structure prediction experiment CASP, adding target complexes to be predicted from the sequences of the individual component proteins (Lensink et al., 2016). The importance of these experiments is that the target structures are blinded, reducing any potential bias. ClusPro predictions have been submitted to CAPRI since 2004, but HADDOCK and SwarmDock started to participate only in 2009 and 2013, respectively, and hence we analyze only the past-2009 results. The predictions submitted by participating groups and servers have been evaluated at four CAPRI and three CAPRI/CASP meetings, namely at CAPRI 4 (Lensink and Wodak, 2010), CAPRI 5 (Lensink and Wodak, 2013), CAPRI 6 (Lensink et al., 2017), CAPRI 7 (Lensink et al., 2019b), CAPRI/CASP 11 (Lensink et al., 2016), CAPRI/CASP 12 (Lensink et al., 2018), and CAPRI/CASP 13 (Lensink et al., 2019a). While the limited number of targets in each round of CAPRI and CAPRI/CASP introduced substantial uncertainty into comparing the performances of the different methods, the reports on the above seven meetings present results for a total of 101 protein-protein targets. As shown in Table 2, based on all these results, ClusPro consistently produced more good predictions in the top 10 than HADDOCK, the second best performing server. SwarmDock did not participate in 2009, and hence its results cannot be directly compared to others. Assuming that SwarmDock would produce 4 acceptable or better predictions as ClusPro did, the latter still would be the best performer. However, Table 2 also shows that both HADDOCK and SwarmDock are better than ClusPro in terms of high accuracy predictions, although both are substantially worse in terms of medium accuracy ones. We note that other servers also had great performance in some CAPRI rounds, for example, the server LZerD had more good models than either SwarmDock or HADDOCK in both 2017 and 2018 (Peterson et al., 2017; Peterson et al., 2018). Nevertheless we focus on HADDOCK and SwarmDock in Table 2 because of their high overall success rates from 2009 through 2019.
Table 2.
Performance of three servers in CAPRI and CAPRI/CASP
| Namea | Yearb | ClusProc | HADDOCKc | SwarmDockc,d |
|---|---|---|---|---|
| CAPRI 4 | 2010 | 4/1***/3** | 3/1***/1** | N/A |
| CAPRI 5 | 2013 | 5/4** | 3/1*** | 3/1** |
| CAPRI/CASP 11 | 2016 | 16/8** | 16/9** | 11/4** |
| CAPRI 6 | 2017 | 5/2** | 2/2** | 2/2** |
| CAPRI/CASP 12 | 2018 | 7/3** | 6/1***/1** | 5/1***/1** |
| CAPRI 7 | 2019 | 4/3** | 4/1***/2** | 3/1***/1** |
| CAPRI/CASP 13 | 2019 | 12/10** | 9/3***/3** | 9/5***/4** |
| Total | 53/1***/33** | 43/7***/18** | 33/7***/13** |
Name of the meeting where the submissions were discussed
Year of the publication reporting the results
Number of targets with “good” (acceptable or better) predictions, among them predictions with high (***) and medium (**) accuracy, based on the top 10 models for each target
SwarmDock did not participate in CAPRI 4
DISCUSSION
As shown both by the benchmark-based evaluation and the overview of CAPRI and CAPRI/CASP experiments, the rigid body docking program ClusPro produces more good models in the top 5 and top 10 predictions than the best flexible docking methods. Since the performance of ClusPro heavily depends on the magnitude of conformational change between the separately crystallized component proteins and their bound forms, this result implies that the rigid body approximation is acceptable for a substantial fraction of interacting proteins. However, ClusPro has two shortcomings relative to methods such as SwarmDock and HADDOCK. First, the more advanced “democratic” version of SwarmDock produces more hits as the top 1 prediction. Second, both flexible methods generate more high accuracy models than ClusPro. These shortcomings are due to different but interrelated limitations on the scoring function available to a rigid body method. Indeed, some good solutions that are present in the top 5 or top 10 predictions are lost when restricting consideration to the top 1 model. This loss is particularly stark when it comes to protein types categorized as “other” type since the drop of success rate from top 1000 energy ranked models to the top 30 selected models was nearly 16% as noted in the results.
Optimal weighting of energy terms in ClusPro more than doubles the number of targets that have a good model as the top prediction. While this is a theoretical limit requiring the use of different weights for each target, the success of the “democratic” SwarmDock shows that the number of targets with a good model as the top 1 prediction can be improved by applying a higher accuracy scoring function. SwarmDock employs machine learning to optimally combine a very large number of energy terms from a variety of scoring functions (Moal et al., 2017). In addition to the DARS energy term used by ClusPro (Chuang et al., 2008), these include scoring functions such as the one implemented in the Rosetta program, which can be meaningfully evaluated only following energy minimization, thus allowing for flexibility (Gray et al., 2003). Based on these observations, we conclude that re-ranking of models generated by ClusPro after some flexible refinement is a reasonable and apparently necessary approach to increasing the number of good models as the top 1 prediction.
The need for flexible refinement is also emphasized by the relative scarcity of high accuracy models produced by ClusPro. Creating such models would require even more fundamental refinement, most likely by Monte Carlo minimization or by molecular dynamics simulation, adopting methods that currently achieved success in protein structure refinement (Heo et al., 2019; Terashi and Kihara, 2018). This can be done in a refinement stage following rigid body docking. Due to ongoing work it is likely that progress will be made to increase both the number of high accuracy models and the number of acceptable or better models in the top 1 prediction. The only disadvantage will be the increased computing time, which will be a problem for a heavily used public server such as ClusPro (see below).
A more important concern is that using any docking method, either rigid or flexible, the overall success rate in the top 10 predictions is still only around 43%, somewhat higher (~58%) for enzyme-containing complexes and much lower for complexes that are classified as “others” in the benchmark set (26.7%). The fraction of good solutions is also around 50% in the CAPRI experiments, with ClusPro producing 53 good predictions for the 101 targets since 2009. In spite of such low success rates, docking methods are heavily used. For example, ClusPro has almost 15,000 registered users (registration is not required), and has performed 98,300 docking calculations in 2019. The calculations have impact, as docked models generated by ClusPro have been reported in at least 600 publications. Thus, the question is, why are docking methods used at all with such a high level of uncertainty? The answer is that additional information is available in most applications, which can be used to restrain or validate the docking results, and thereby substantially reduce the uncertainty. Most frequently this information provides distance restraints that can be directly used in the process of docking. Accordingly, HADDOCK has been explicitly developed with these restraints in mind, and has included them as part of their scoring function. Since FFT based methods such as ClusPro perform a systematic search, restraints can be enforced simply by restricting considerations to the relevant regions of the search space, and the option to consider restraints has been added to the server (Xia et al., 2016). ClusPro also has the option of defining extra attraction or repulsion for selected residues (Kozakov et al., 2017).
The information to define restraints can come from a variety of sources. The most direct sources are experimental techniques, including cross-linking and site-directed mutagenesis, the latter paired with NMR, calorimetry, FRET, or surface plasmon resonance. Note that even the knowledge of a single interprotein residue-residue contact can remove a large number of false positive predictions. Accordingly, the majority of ClusPro users rely on some type of experimental information to validate docking results. Second, an increasing source of information is the availability of homologous complexes that can serve as templates for the structure to be determined. Such templates can be used for defining distance restraints to guide the docking. It is important to note that if a very good template complex with high sequence similarity is available, then accurate models can be obtained using homology modeling without any docking (Porter et al., 2019a; Porter et al., 2019b). However, docking with template-based restraints can be very useful if the available templates are less perfect. Finally, if neither experimental data nor structural templates are available, one can still predict inter-protein residue contacts by extracting co-evolutionary information. The analysis is based on the observed tendency for compensating mutations that occur at interacting residue positions in orthologous protein sequences across evolutionary lineages (Hopf et al., 2014; Marks et al., 2011; Ovchinnikov et al., 2014). The disadvantage of the method is that it usually requires sequences of the two component proteins in a very large number of organisms, and such information may not be available for eukaryotes. While acquiring additional information by any of the three methods is independent of the particular approach to docking, we emphasize that rigid body methods can efficiently account for the resulting distance restraints without altering the scoring function. \
STAR METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Sandor Vajda (vajda@bu.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
The published article includes all datasets generated during this study in the file Data S1. \
The ClusPro server is available free for academic and governmental use at https://cluspro.bu.edu. The PIPER docking engine of the server can be downloaded from the same site.
METHOD DETAILS
The Protein-Protein Docking Program PIPER
The program PIPER performs rigid body docking in the 6D space of rotations and translations. The center of mass of the receptor is fixed at the origin of the coordinate system, and the possible rotational and translational positions of the ligand are evaluated at the given level of discretization. The rotational space is sampled on a sphere-based grid that defines a subdivision of a spherical surface in which each pixel covers the same surface area as every other pixel (Yershova et al., 2010). The 70,000 rotations we consider correspond to about 5 degrees in terms of the Euler angles. The step size of the translational grid is 1 Å, and hence the program evaluates the energy for 109–1010 conformations.
The interaction energy between two proteins is described by the expression
where Erep and Eattr denote the repulsive and attractive contributions to the van der Waals interaction energy, and Eelec is an electrostatic energy term. Epair is a pairwise structure-based potential constructed by the Decoys as the Reference State (DARS) (Chuang et al., 2008) approach. As required for using FFT, all energy terms are written as correlation functions. Since Eelec is a truncated Columbic potential, it is already in this form. Erep and Eattr are written in terms of interacting atoms as correlation functions, whereas the pairwise potential Epair is converted by eigenvalue-eigenvector decomposition into the sum of a few correlation functions (Kozakov et al., 2006). The coefficients w1, w2, w3, and w4 define the weights of the corresponding terms, and are adjusted for different types of docking problems as will be discussed below.
The ClusPro Server
ClusPro is a web based server for docking of two interacting proteins. The server performs three computational steps as follows: (1) rigid body docking using PIPER, (2) clustering the 1000 lowest energy docked structures using pairwise IRMSD as the distance measure (Comeau et al., 2004; Kozakov et al., 2005), and (3) refinement of the predicted complex structures located at cluster centers by minimizing their energy. Unless specified otherwise, the server generates four sets of models using the scoring schemes called balanced (denoted as 00 in the server), electrostatics-favored (02), hydrophobicity-favored (04), and van der Waals + electrostatics only, i.e., no Epair term (06). In the balanced set 00, the weighting coefficients are selected to yield similar weights for the four different energy terms (Kozakov et al., 2017). The set was shown to generally provide good results for enzyme-inhibitor complexes. If it is known or assumed that the association of two proteins is mainly driven by their electrostatic interactions, then we select results obtained by the electrostatic-favored weights 02, in which the weight of the electrostatics is doubled relative to the balanced energy expression. In contrast, for complexes primarily stabilized by hydrophobic interactions, we use the hydrophobicity-favored potential 04, which thus doubles the weight of the Epair term. In the fourth option (van der Waals + electrostatics, the parameterization option 06 in the server) the pairwise potential Epair is not used. ClusPro has an additional option for complexes that are classified as “others” in the protein-protein docking benchmark (Vreven et al., 2015). The motivation for developing a special scoring function is the diverse nature and the generally limited shape and electrostatic complementarity in these complexes. In view of the assumed weaker shape complementarity and higher structural uncertainty, we reduce the coefficients of Eattr and Eelec and use three different values for the coefficient of Epair. Using each of the three sets of weighting coefficients, PIPER generates 500 structures and retains the resulting 1500 conformations. Finally, docking antibodies to antigens employs atom types and energy coefficients specifically designed for this type of target (Brenke et al., 2012).
As mentioned, the second step of ClusPro is clustering the lowest energy 1000 docked structures (or 1500 in the case of “other” type complexes) using pairwise IRMSD as the distance measure (Comeau et al., 2004; Kozakov et al., 2005). IRMSD values are calculated for each pair among the 1000 structures, and the structure that has the highest number of neighbors within 9Å IRMSD radius is identified. The selected structure will be defined as the center of the first cluster, and the structures within the 9Å IRMSD neighborhood of the center will constitute the first cluster. The members of this cluster are then removed, and we select the structure with the highest number of neighbors within the 9Å IRMSD radius among the remaining structures as the next cluster center. These neighbors will form the next cluster. Up to 30 clusters are generated in this manner. Finally, the energy of each cluster center structure is minimized with fixed backbone using only the van der Waals term of the Charmm potential (Brooks et al., 1983). The minimization removes steric overlaps but generally yields only very small conformational changes.
Antibody-Antigen Docking
ClusPro includes a special option to dock antigens to antibodies. First, we have developed a structure-based pairwise potential that is asymmetric, and thus a Phe residue of the antibody interacting with, say, a Leu of the antigen contributes more to the binding energy than a Phe residue of the antigen interacting with a Leu on the antibody. This asymmetric function represents the observed importance of some residues, such as Phe and Tyr, in the CDRs on the antibody side of the interface. The potential has been parameterized on antibody-antigen complexes, and since the number of such complexes is limited, the potential is defined for fewer atom types than the DARS potential developed for enzyme-inhibitor and “other” type complexes (Chuang et al., 2008).
The second special factor in antibody-antigen docking is that the interactions are generally restricted to the Complementarity Determining Regions (CDRs) of antibodies. Thus, the docking can be restricted by “masking” the surface of the antibody except for the CDRs and a few additional surrounding residues. Accordingly, ClusPro includes both automated and manual options for masking. Manual masking allows users to upload a masking file by themselves. The automated masking that is currently implemented on ClusPro includes the following steps. (1) The residue sequences are obtained from the input PDB file of the antibody and the chains are separated. (2) Each chain is aligned to a set of known heavy and light chains using CLUSTAL (Madeira et al., 2019) and scored according to the alignment. The gap penalty was set to 10 and gap extension penalty was set as 0.1 with the GONNET identity matrix. (3) The location of the CDR termini for both the light and the heavy chains are determined using the Fab fragment from a mouse monoclonal antibody, NMC-4, from the crystal structure 1OAK where it is bound to the A1 domain of the von Willebrand factor as a reference (MacCallum et al., 1996). (4) After obtaining the indices of the start and end residues from 1OAK, the start and end indices for the aligned FASTA sequence of the target antibody are used to create a PDB file of the non-CDR region. This PDB format masking file is uploaded with the ligand and the antibody, and ClusPro essentially “ignores” the atoms in the masked regions during the docking.
QUANTIFICATION AND STATISTICAL ANALYSIS
Protein Docking Benchmark Version 5.0
Benchmark set 5.0 (BM5) is a nonredundant and fairly diverse set of protein complexes for testing protein-protein docking algorithms. For each complex the 3D structures of the complex and one or both unbound components are available. The set includes 40 antibody-antigen, 88 enzyme-containing and 102 “others” complexes. In terms of docking difficulty, the complexes are classified into the following groups: 151 rigid-body (easy), 45 medium difficulty and 34 difficult targets.
The CAPRI Criteria of Prediction Accuracy
CAPRI uses three related parameters for assessing a model: Fnat, iRMSD, and LRMSD(Lensink and Wodak, 2013). Fnat is the fraction of true interface contacts that were accurately predicted to non-native interface contacts in the predicted model where two residues are in contact if they are within 5Å of each other. Interface RMSD (iRMSD) is defined as the RMSD of all interface backbone atoms once superposed on the native structure (interface atoms are defined as those within 10Å of the other component of the complex) (Mendez et al., 2005). Finally, ligand RMSD (LRMSD) is calculated between the ligand backbone atoms, when the receptor of the predicted complex is superposed on that of the native. Four categories of prediction accuracy were defined by these three measures as follows. A prediction is considered of high accuracy if Fnat ≥ 0.5 and LRMSD ≤ 1.0 Å or iRMSD ≤ 1.0 Å. It is of medium accuracy if 0.5 > Fnat ≥ 0.3, and LRMSD ≤ 5.0 Å or iRMSD ≤ 2.0 Å, or if Fnat ≥ 0.5 but LRMSD > 1.0 Å and iRMSD > 1.0 Å. A prediction is acceptable if 0.3 > Fnat ≥ 0.1 and LRMS ≤ 10.0 Å or iRMSD ≤ 4.0 Å, or if Fnat ≥ 0.3 but LRMSD > 5.0 Å and iRMSD > 2.0 Å. Finally, the prediction is incorrect if none of these categories apply, i.e., if Fnat < 0.1 or LRMSD > 10 Å and iRMSD > 4.0 Å.
Evaluating Prediction Accuracy Using DOCKQ
To serve the docking community, a continuous score called DockQ was developed that encapsulated the three measures Fnat, iRMSD, and LRMSD (Basu and Wallner, 2016). The resulting combined measure adequately reproduced evaluation classes of CAPRI and is able to score predictions between 0 and 1 where ≥ 0.23 is acceptable, ≥ 0.49 is medium and ≥ 0.80 is high accuracy. Since DockQ recapitulates the CAPRI classification very well, it can be viewed as a higher resolution version of the classification scheme, making it possible to estimate model accuracy in a more quantitative way. Therefore DockQ has been widely accepted and was even implemented in the official evaluations of the latest CAPRI round. The program is available at http://github.com/bjornwallner/DockQ/.
Validation of energy weight coefficient sets for enzymes and “other” type complexes
As described, ClusPro currently generates results using four different sets of energy weights, which are the balanced set (denoted as 00), the electrostatics-favored set (02), hydrophobicity-favored set (04), and finally a set using only the van der Waals + electrostatics terms (06). In addition, special parameterization is used for “other” type complexes (03), and also for antibody-antigen targets. We reexamined the use of these coefficient sets for enzyme and “other” type complexes. In both cases we performed 5-fold cross-validation on the proteins in BM4, and also applied the selected parameters sets to the 55 targets in BM5 that were not part of BM4. The 71 enzyme-containing complexes in BM4 were shuffled randomly with a python script and divided into 5 sets. Each group was considered a test set, while the other four was considered the training sets for the balanced (00), the electrostatics-favored (02), the hydrophobicity-favored (04), and the van der Waals + electrostatics (06) sets. The best performing coefficient set on the other 4 groups was tested on the fifth group. The number of times each set would be considered the best and shown to be superior on the test set was used to determine the chosen coefficient set. Figure S2 shows the number of successful cases for each fold in the training set for better illustration. For enzyme containing complexes we concluded that the results obtained by using balanced (00) and electrostatics favored (02) do not significantly differ, and hence there is no preference using any of these two sets. The same validation protocol was applied to 80 “other”-containing complexes in Benchmark 4. Here we primarily compared the special protocol (03) suggested for this type of complexes to the use of the balanced (00) and electrostatics favored (02) sets. The 5-fold cross-validation revealed that the electrostatic-favored set (02) yields the best results, and there is no need for using the more complex algorithm of generating 1500 structures with three different parameter sets (Figure 4B).
Supplementary Material
Data S1.xlsx. Related to Table 1. ClusPro results for 227 targets in the BM5 benchmark set.
Figure S1. Related to Figure 4A. Success rates for enzyme containing complexes using different sets of weight in the scoring function of ClusPro.
Figure S2. Related to Figure 4A and Figure S1. Results of a five-fold cross validation on enzyme-containing BM4 cases.
Figure S3. Related to Figure 4B. Results of a five-fold cross validation on “others”-containing BM4 cases.
Figure S4. Related to Figure 4C. Comparing the performance of ClusPro with the currently implemented weight coefficients to the performance range with the 105 different coefficient sets,
Figure S5. Related to Figure 5. Comparing the success rate of ClusPro to those of four other servers.
KEY RESOURCES TABLE
| RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Software and algorithms | ||
| ClusPro | Kozakov et al., 2017 | https://cluspro.bu.edu/login.php |
| Protein Docking Benchmark | Vreven,et al., 2015 | https://zlab.umassmed.edu/benchmark/ |
| DockQ | Basu, S., and Wallner, B. (2016). | https://github.com/bjornwallner/DockQ |
| SwarmDock | Torchala et al., 2013 | https://bmm.crick.ac.uk/~svc-bmm-swarmdock/ |
| Antibody Informatics | MacCallum et al., 1996 | http://www.bioinf.org.uk/abs/ |
| PDB files | Protein Data Bank | https://www.rcsb.org/ |
Highlights.
Protein-protein docking by fast Fourier transform (FFT) samples billions of conformations.
FFT based methods need rigid body approximation and scoring by correlation functions.
Rigid methods yield more good models in the top 5 predictions than flexible docking.
Flexible methods yield higher accuracy models for some targets.
ACKNOWLEDGMENTS
This investigation was supported by grants DBI 1759277 and AF 1645512 from the National Science Foundation, and R35GM118078, R21GM127952, and RM1135136 from the National Institute of General Medical Sciences. We thank Dr. Paul Bates for his help with running the SwarmDock server to generate the results for the comparison described in this paper.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
Acpharis Inc. offers commercial licenses to PIPER. Dima Kozakov and Sandor Vajda occasionally consult for Acpharis, and own stock in the company. However, the PIPER program and the use of the ClusPro server are free for academic and governmental use.
REFERENCES
- Al-Lazikani B, Lesk AM, and Chothia C (1997). Standard conformations for the canonical structures of immunoglobulins. J. Mol. Biol 273, 927–948. [DOI] [PubMed] [Google Scholar]
- Basu S, and Wallner B (2016). DockQ: A Quality Measure for Protein-Protein Docking Models. PLoS One 11, e0161879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenke R, Hall DR, Chuang GY, Comeau SR, Bohnuud T, Beglov D, Schueler-Furman O, Vajda S, and Kozakov D (2012). Application of asymmetric statistical potentials to antibody-protein docking. Bioinformatics 28, 2608–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooks BR, Bruccoleri RE, Olafson BD, States DJ, Swaminathan S, and Karplus M (1983). Charmm - a Program for Macromolecular Energy, Minimization, and Dynamics Calculations. J. Comput. Chem 4, 187–217. [Google Scholar]
- Camacho CJ, Kimura SR, DeLisi C, and Vajda S (2000). Kinetics of desolvation-mediated protein-protein binding. Biophys. J 78, 1094–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camacho CJ, and Vajda S (2001). Protein docking along smooth association pathways. Proc. Natl. Acad. Sci. U S A 98, 10636–10641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Li L, and Weng Z (2003). ZDOCK: an initial-stage protein-docking algorithm. Proteins 52, 80–87. [DOI] [PubMed] [Google Scholar]
- Chen R, and Weng ZP (2002). Docking unbound proteins using shape complementarity, desolvation, and electrostatics. Proteins 47, 281–294. [DOI] [PubMed] [Google Scholar]
- Cheng TM, Blundell TL, and Fernandez-Recio J (2007). pyDock: electrostatics and desolvation for effective scoring of rigid-body protein-protein docking. Proteins 68, 503–515. [DOI] [PubMed] [Google Scholar]
- Chuang GY, Kozakov D, Brenke R, Comeau SR, and Vajda S (2008). DARS (Decoys As the Reference State) potentials for protein-protein docking. Biophys. J 95, 4217–4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comeau SR, Gatchell DW, Vajda S, and Camacho CJ (2004). ClusPro: an automated docking and discrimination method for the prediction of protein complexes. Bioinformatics 20, 45–50. [DOI] [PubMed] [Google Scholar]
- de Vries SJ, van Dijk M, and Bonvin AM (2010). The HADDOCK web server for data-driven biomolecular docking. Nat. Protoc 5, 883–897. [DOI] [PubMed] [Google Scholar]
- Gabb HA, Jackson RM, and Sternberg MJE (1997). Modelling protein docking using shape complementarity, electrostatics and biochemical information. J. Mol. Biol 272, 106–120. [DOI] [PubMed] [Google Scholar]
- Gray JJ, Moughon SE, Kortemme T, Schueler-Furman O, Misura KM, Morozov AV, and Baker D (2003). Protein-protein docking predictions for the CAPRI experiment. Proteins 52, 118–122. [DOI] [PubMed] [Google Scholar]
- Heo L, Arbour CF, and Feig M (2019). Driven to near-experimental accuracy by refinement via molecular dynamics simulations. Proteins 87, 1263–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopf TA, Scharfe CPI, Rodrigues JPGLM, Green AG, Kohlbacher O, Sander C, Bonvin AMJJ, and Marks DS (2014). Sequence co-evolution gives 3D contacts and structures of protein complexes. Elife 3 e03430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang H, Vreven T, Janin J, and Weng Z (2010). Protein-protein docking benchmark version 4.0. Proteins 78, 3111–3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janin J, Henrick K, Moult J, Eyck LT, Sternberg MJ, Vajda S, Vakser I, Wodak SJ, and Critical Assessment of PI (2003). CAPRI: a Critical Assessment of PRedicted Interactions. Proteins 52, 2–9. [DOI] [PubMed] [Google Scholar]
- Katchalski-Katzir E, Shariv I, Eisenstein M, Friesem AA, Aflalo C, and Vakser IA (1992). Molecular surface recognition: determination of geometric fit between proteins and their ligands by correlation techniques. Proc. Natl. Acad. Sci. U S A 89, 2195–2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D, Beglov D, Bohnuud T, Mottarella SE, Xia B, Hall DR, and Vajda S (2013). How good is automated protein docking? Proteins 81, 2159–2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D, Brenke R, Comeau SR, and Vajda S (2006). PIPER: an FFT-based protein docking program with pairwise potentials. Proteins 65, 392–406. [DOI] [PubMed] [Google Scholar]
- Kozakov D, Clodfelter KH, Vajda S, and Camacho CJ (2005). Optimal clustering for detecting near-native conformations in protein docking. Biophys. J 89, 867–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozakov D, Hall DR, Xia B, Porter KA, Padhorny D, Yueh C, Beglov D, and Vajda S (2017). The ClusPro web server for protein-protein docking. Nat. Protoc 12, 255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lensink MF, Brysbaert G, Nadzirin N, Velankar S, Chaleil RAG, Gerguri T, Bates PA, Laine E, Carbone A, Grudinin S, et al. (2019a). Blind prediction of homo- and heteroprotein complexes: The CASP13-CAPRI experiment. Proteins 87, 1200–1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lensink MF, Mendez R, and Wodak SJ (2007). Docking and scoring protein complexes: CAPRI 3rd Edition. Proteins 69, 704–718. [DOI] [PubMed] [Google Scholar]
- Lensink MF, Nadzirin N, Velankar S, and Wodak S (2019b). Modeling protein-protein, protein-peptide and protein-oligosaccharide complexes: CAPRI 7th edition. Proteins 10.1002/prot.25870 [DOI] [PubMed] [Google Scholar]
- Lensink MF, Velankar S, Baek M, Heo L, Seok C, and Wodak SJ (2018). The challenge of modeling protein assemblies: the CASP12-CAPRI experiment. Proteins 86 Suppl 1, 257–273. [DOI] [PubMed] [Google Scholar]
- Lensink MF, Velankar S, Kryshtafovych A, Huang SY, Schneidman-Duhovny D, Sali A, Segura J, Fernandez-Fuentes N, Viswanath S, and Elber R (2016). Prediction of homoprotein and heteroprotein complexes by protein docking and template-based modeling: A CASP-CAPRI experiment. Proteins 84, 323–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lensink MF, Velankar S, and Wodak SJ (2017). Modeling protein-protein and protein-peptide complexes: CAPRI 6th edition. Proteins 85, 359–377. [DOI] [PubMed] [Google Scholar]
- Lensink MF, and Wodak SJ (2010). Docking and scoring protein interactions: CAPRI 2009. Proteins 78, 3073–3084. [DOI] [PubMed] [Google Scholar]
- Lensink MF, and Wodak SJ (2013). Docking, scoring, and affinity prediction in CAPRI. Proteins 81, 2082–2095. [DOI] [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, et al. (2019). The EMBL-EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47, W636–W641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mandell JG, Roberts VA, Pique ME, Kotlovyi V, Mitchell JC, Nelson E, Tsigelny I, and Ten Eyck LF (2001). Protein docking using continuum electrostatics and geometric fit. Protein Eng 14, 105–113. [DOI] [PubMed] [Google Scholar]
- Marks DS, Colwell LJ, Sheridan R, Hopf TA, Pagnani A, Zecchina R, and Sander C (2011). Protein 3D structure computed from evolutionary sequence variation. PLoS One 6, e28766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez R, Leplae R, De Maria L, and Wodak SJ (2003). Assessment of blind predictions of protein-protein interactions: current status of docking methods. Proteins 52, 51–67. [DOI] [PubMed] [Google Scholar]
- Mendez R, Leplae R, Lensink MF, and Wodak SJ (2005). Assessment of CAPRI predictions in rounds 3–5 shows progress in docking procedures. Proteins 60, 150–169. [DOI] [PubMed] [Google Scholar]
- Moal IH, Barradas-Bautista D, Jimenez-Garcia B, Torchala M, van der Velde A, Vreven T, Weng Z, Bates PA, and Fernandez-Recio J (2017). IRaPPA: information retrieval based integration of biophysical models for protein assembly selection. Bioinformatics 33, 1806–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moal IH, Chaleil RAG, and Bates PA (2018). Flexible Protein-Protein Docking with SwarmDock. Methods Mol. Biol 1764, 413–428. [DOI] [PubMed] [Google Scholar]
- Ovchinnikov S, Kamisetty H, and Baker D (2014). Robust and accurate prediction of residue-residue interactions across protein interfaces using evolutionary information. Elife 3 e02030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LX, Kim H, Esquivel-Rodriguez J, Roy A, Han X, Shin WH, Zhang J, Terashi G, Lee M, and Kihara D (2017). Human and server docking prediction for CAPRI round 30–35 using LZerD with combined scoring functions. Proteins 85, 513–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson LX, Shin WH, Kim H, and Kihara D (2018). Improved performance in CAPRI round 37 using LZerD docking and template-based modeling with combined scoring functions. Proteins 86 Suppl 1, 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierce BG, Wiehe K, Hwang H, Kim BH, Vreven T, and Weng Z (2014). ZDOCK server: interactive docking prediction of protein-protein complexes and symmetric multimers. Bioinformatics 30, 1771–1773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pons C, Solernou A, Perez-Cano L, Grosdidier S, and Fernandez-Recio J (2010). Optimization of pyDock for the new CAPRI challenges: Docking of homology-based models, domain-domain assembly and protein-RNA binding. Proteins 78, 3182–3188. [DOI] [PubMed] [Google Scholar]
- Porter KA, Desta I, Kozakov D, and Vajda S (2019a). What method to use for protein-protein docking? Curr. Opin. Struct. Biol 55, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter KA, Padhorny D, Desta I, Ignatov M, Beglov D, Kotelnikov S, Sun Z, Alekseenko A, Anishchenko I, Cong Q, et al. (2019b). Template-based modeling by ClusPro in CASP13 and the potential for using co-evolutionary information in docking. Proteins 87, 1241–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terashi G, and Kihara D (2018). Protein structure model refinement in CASP12 using short and long molecular dynamics simulations in implicit solvent. Proteins 86 Suppl 1, 189–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torchala M, Moal IH, Chaleil RA, Fernandez-Recio J, and Bates PA (2013). SwarmDock: a server for flexible protein-protein docking. Bioinformatics 29, 807–809. [DOI] [PubMed] [Google Scholar]
- Tovchigrechko A, and Vakser IA (2005). Development and testing of an automated approach to protein docking. Proteins 60, 296–301. [DOI] [PubMed] [Google Scholar]
- Tovchigrechko A, and Vakser IA (2006). GRAMM-X public web server for protein-protein docking. Nucleic Acids Res 34, W310–W314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangone A, Rodrigues JP, Xue LC, van Zundert GC, Geng C, Kurkcuoglu Z, Nellen M, Narasimhan S, Karaca E, van Dijk M, et al. (2017). Sense and simplicity in HADDOCK scoring: Lessons from CASP-CAPRI round 1. Proteins 85, 417–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vreven T, Moal IH, Vangone A, Pierce BG, Kastritis PL, Torchala M, Chaleil R, Jimenez-Garcia B, Bates PA, Fernandez-Recio J, et al. (2015). Updates to the integrated protein-protein interaction benchmarks: Docking benchmark version 5 and affinity benchmark version 2. J. Mol. Biol 427, 3031–3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia B, Vajda S, and Kozakov D (2016). Accounting for pairwise distance restraints in FFT-based protein-protein docking. Bioinformatics 32, 3342–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yershova A, Jain S, LaValle SM, and Mitchell JC (2010). Generating uniform incremental grids on SO(3) using the Hopf fibration. Int. J. Robot. Res 29, 801–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1.xlsx. Related to Table 1. ClusPro results for 227 targets in the BM5 benchmark set.
Figure S1. Related to Figure 4A. Success rates for enzyme containing complexes using different sets of weight in the scoring function of ClusPro.
Figure S2. Related to Figure 4A and Figure S1. Results of a five-fold cross validation on enzyme-containing BM4 cases.
Figure S3. Related to Figure 4B. Results of a five-fold cross validation on “others”-containing BM4 cases.
Figure S4. Related to Figure 4C. Comparing the performance of ClusPro with the currently implemented weight coefficients to the performance range with the 105 different coefficient sets,
Figure S5. Related to Figure 5. Comparing the success rate of ClusPro to those of four other servers.
Data Availability Statement
The published article includes all datasets generated during this study in the file Data S1. \
The ClusPro server is available free for academic and governmental use at https://cluspro.bu.edu. The PIPER docking engine of the server can be downloaded from the same site.