

Abstract
Changes in macrophage phenotype in injured muscle profoundly influence regeneration. In particular, the shift of macrophages from a pro-inflammatory (M1-biased) phenotype to a pro-regenerative (M2-biased) phenotype characterized by expression of CD206 and CD163 is essential for normal repair. According to the current canonical mechanism regulating for M1/M2 phenotype transition, signaling through PPARδ is necessary for obtaining the M2-biased phenotype. Our findings confirm that the murine myeloid cell targeted deletion of Ppard reduces expression in vitro of genes that are activated in M2-biased macrophages; however, the mutation in mice in vivo increased numbers of CD206+ M2-biased macrophages and did not reduce the expression of phenotypic markers of M2-biased macrophages in regenerating muscle. Nevertheless, the mutation impaired CCL2-mediated chemotaxis of macrophages and slowed revascularization of injured muscle. In contrast, null mutation of IL10 diminished M2-biased macrophages but produced no defects in muscle revascularization. Our results provide two significant findings. First, they illustrate that mechanisms that regulate macrophage phenotype transitions in vitro are not always predictive of mechanisms that are most important in vivo. Second, they show that mechanisms that regulate macrophage phenotype transitions differ in different in vivo environments.
Keywords: skeletal muscle, inflammation, acute muscle injury, macrophage, PPARδ, interleukin-10
Introduction
The inflammatory response to skeletal muscle injury or disease has important influences on the rate and extent of muscle repair. Although the initial population of myeloid cells that enter muscle following injury is dominated by neutrophils that can amplify muscle damage, their numbers rapidly decline and they are replaced by phenotypically-complex populations of macrophages that can remain at elevated concentrations in the regenerating muscle for weeks (1, 2, 3). Over that prolonged period of muscle regeneration, the predominant macrophage phenotypes shift from a population that expresses pro-inflammatory cytokines and increases proliferation of myogenic cells to a population that produces anti-inflammatory cytokines, promotes muscle fiber growth and increases connective tissue production (4, 5, 6, 7, 8, 9). The importance of the shifts in macrophage phenotypes on the course of muscle repair and regeneration has been well-established by investigations that have shown that disrupting the normal recruitment or phenotype specification of macrophages amplifies muscle damage and slows regeneration (10, 11, 12, 13, 14, 15).
The initial characterization of macrophage phenotypes as M1 (pro-inflammatory) or M2 (anti-inflammatory) was based upon changes in gene expression that occurred in macrophages in vitro following stimulation with Th1 cytokines (interferon-gamma [IFNγ]; tumor necrosis factor [TNF]) or Th2 cytokines (interleukin-4 [IL4]; IL13) (16, 17, 18). However, in the complex inflammatory environment in vivo, numerous other immunomodulatory molecules can influence macrophage activation to produce a broad and continuous spectrum of phenotypes that range between the M1 and M2 designations (19). Whether macrophage activation is biased along the spectrum toward the M1 or M2 phenotype is assessed by determining their expression of numerous phenotypic markers. Within injured and diseased muscles of mice, macrophages that are biased toward the M1 phenotype express inducible nitric oxide synthase (iNOS), CD68 and high levels of CD11b and Ly6C. Intramuscular macrophages biased toward the M2 phenotype express CD163, CD206, arginase-1 (Arg1) and low levels of CD11b and Ly6C (8, 10, 20, 21, 22).
Multiple signaling pathways contribute to activating macrophages to an M2-biased, pro-regenerative phenotype. For example, macrophage activation to a CD163+/CD206+, M2-biased phenotype in inflamed muscle is strongly influenced by IL10, transforming growth factor-β (TGFβ) and insulin-like growth factor-1 (IGF1) that are expressed by macrophages at the site of muscle damage (7, 8, 21, 23). Furthermore, diminishing expression of these regulators of M2-biased macrophage phenotype in injured or diseased muscle causes impaired muscle repair and retarded muscle growth (8, 21, 23), which emphasizes the functional importance of intramuscular macrophages biased to an M2-biased phenotype. However, recent discoveries also demonstrated that changes in the relative prevalence of lipid mediators that influence macrophage phenotype change over the course of muscle repair following injury (24). For example, lipid mediators that reduce inflammation (specialized, proresolving lipid mediators; SPMs) are more prevalent in Ly6Clo macrophages and intramuscular injection of one of the SPMs, resolvin D2, produced increases in Ly6Clo cell numbers in injured muscles and improved muscle function (24). Those findings suggest that SPM-induction of an M2-biased phenotype may also be able to improve muscle regeneration.
Although untested, the collective findings of numerous investigations support the hypothesis that lipid mediators may influence the shift of macrophages to an M2-biased phenotype in injured muscle by activating peroxisome proliferator-activated receptor-δ (PPARδ). PPARδ is a ligand activated nuclear receptor that is activated by polyunsaturated fatty acids and other lipid mediators that can be produced during breakdown of apoptotic cells that are engulfed by macrophages (25). Activated PPARδ then dimerizes with retinoic acid receptors (RXR), translocates to the nucleus and activates expression of M2-associated transcripts including Mrc1 (which encodes CD206), Arg1, Chi3l3, Retnla and Ppard (26, 27). The importance of PPARδ in activating macrophages to an M2-biased phenotype was demonstrated clearly in Kupffer cells in the liver and in adipose tissue macrophages (28, 29) in which the deletion of Ppard in LysM expressing cells or in bone marrow derived cells prevented activation of macrophages to an M2-biased phenotype. However, fatty acid ligation of PPARδ in macrophages can also cause an increase in IL10 expression that requires PPARδ (25) and induction of IL10 expression is increased by PPARδ in endothelial cells (30). Those findings show that IL10-mediated signaling and PPARδ-mediated signaling pathways can interact, suggesting the possibility that IL10 and lipid mediators may affect intramuscular macrophage phenotype through overlapping mechanisms.
In this investigation, we generated a mouse line in which there is a targeted deletion of Ppard in LysM expressing myeloid cells to test the hypothesis that activation of macrophages to an M2-biased phenotype in acutely-injured muscle is mediated by PPARδ. We also assayed whether Ppard mutation in macrophages influences macrophage function or affects muscle repair, growth and revascularization following injury. Finally, we assayed whether the same parameters of macrophage activation and muscle repair are similarly influenced by ablation of Il10 expression to address the question of whether IL10-mediated and PPARδ–mediated regulation of macrophage phenotype have similar effects on muscle inflammation, repair and regeneration, which would indicate a potential interaction between the two regulatory pathways in acutely injured muscle. The direct comparison of the effects of the two mutations in the same injury model enabled us to demonstrate the distinct roles of myeloid cell PPARδ and IL10 in regulating macrophage recruitment, phenotype and regeneration following acute muscle injury.
MATERIALS AND METHODS
Ethical approval and animals
All animals were handled according to guidelines provided by the Chancellor’s Animal Research Committee at the University of California, Los Angeles. C57BL/6, Ppardflox/flox (#005897), LysM-cre (#004781) and Il10−/− (#002250) mice were purchased from The Jackson Laboratory (Bar Harbor, ME) and bred in specific pathogen-free vivaria at the University of California, Los Angeles. Pparδfl/fl and LysM-cre mice were crossed to generate myeloid cell-specific Pparδ mutant mice. Knockdown of Pparδ was confirmed by quantitative real time polymerase chain reaction (QPCR) assays on bone marrow-derived macrophages (BMDMs) from Pparδfl/fl LysM-cre (Pparδ mutants) and Pparδfl/fl (Ppard WT) mice. Il10 mutation was confirmed by QPCR of skeletal muscle from C57BL6 and Il10−/− mice. Mice were housed on a 12/12 hour light/dark cycle and were provided food and water ad libitum. Following euthanasia by inhalation of isoflurane, muscles were collected, weighed and flash-frozen for histological or biochemical analyses.
Acute muscle injury
Mice were anesthetized with isoflurane inhalation in a chamber (4-5% isoflurane with 100% oxygen) then moved to a nose cone (1-2% isoflurane). Anesthesia was checked by testing mice for a positive reflex response to a hind foot pinch and by monitoring respiration. The lower limb was wiped with 70% ethanol before intramuscular injection. Sterile muscle injury was induced by the intramuscular injection of 50 μl of a 1.2% barium chloride (BaCl2) solution into the midbelly of tibialis anterior (TA) muscles of healthy, 4-6 months old male mice. Animals were monitored daily until they recovered.
RNA isolation and QPCR
RNA was isolated from cells and muscle homogenates and electrophoresed on agarose gels and its quality assessed by 28S and 18S ribosomal RNA integrity (31). RNA samples (2 μg) were reverse transcribed with Super Script Reverse Transcriptase II using oligo dTs to prime extension (Invitrogen, Carlsbad, CA, USA) to produce cDNA. Expression of selected transcripts was assayed using iTaq SYBR green qPCR Supermix according to the manufacturer’s protocol (BioRad, Hercules, CA, USA) and a QuantStudio 5 Real-time PCR System (Applied Biosystems, Foster City, CA, USA). Established guidelines for experimental design, data normalization and data analysis for QPCR were used to maximize the rigor of quantifying the relative levels of mRNA (32, 33). We empirically tested reference genes and identified those with the least variability between experimental groups. Based on that analysis, the relative expression of transcripts of interest were normalized to the reference genes Srp14 and Rnps1 for muscle regeneration, Srp14 and Tpt1 for Pparδ BMDMs, and Tbp and Rplo for Il10−/− BMDMs QPCR assays. The normalization factor for each sample was calculated by geometric averaging of the Ct values of reference genes. Expression for each gene in control samples was set to 1 and the other values were scaled to the control. Primers used for QPCR are listed in Table 1.
Table 1:
Primer sequences used for QPCR
| Gene | Forward | Reverse |
|---|---|---|
| Arg1 | CAATGAAGAGCTGGCTGGTGT | GTGTGAGCATCCACCCAAATG |
| Arg2 | GAAGTGGTTAGTAGAGCTGTGTC | GGTGAGAGGTGTATTAATGTCCG |
| Ccr2 | CCTGTAAATGCCATGCAAGTTC | GTATGCCGTGGATGAACTGAG |
| Cd68 | CAAAGCTTCTGCTGTGGAAAT | GACTGGTCACGGTTGCAAG |
| Cd163 | GCAAAAACTGGCAGTGGG | GTCAAAATCACAGACGGAG |
| Chil3 | GGGCATACCTTTATCCTGAG | CCACTGAAGTCATCCATGTC |
| Hif1a | GCTTACACACAGAAATGGCCC | CCTTCCACGTTGCTGACTTG |
| Ifng | GACAATCAGGCCATCAGCAAC | CGGATGAGCTCATTGAATGCTT |
| Il4 | GGATGTGCCAAACGTCCTC | GAGTTCTTCTTCAAGCATGGAG |
| Il10 | CAAGGAGCATTTGAATTCCC | GGCCTTGTAGACACCTTGGTC |
| Il13 | GTCCTGGCTCTTGCTTGC | CACTCCATACCATGCTGCC |
| Mrc1 | GGATTGTGGAGCAGATGGAAG | CTTGAATGGAAATGCACAGAC |
| Nos2 | CAGCACAGGAAATGTTTCAGC | TAGCCAGCGTACCGGATGA |
| Ppard | GAACACACGCTTCCTTCCAGC | CACCCGACATTCCATGTTGAG |
| Retnla | TCGTGGAGAATAAGGTCAAGG | GGAGGCCCATCTGTTCATAG |
| Rnsp1 | AGGCTCACCAGGAATGTGAC | CTTGGCCATCAATTTGTCCT |
| Rplp0 | GGACCCGAGAAGACCTCCTT | GCTGCCGTTGTCAAACACC |
| Srp14 | AGAGCGAGCAGTTCCTGAC | CGGTGCTGATCTTCCTTTTC |
| Tbp | TCCCCCTCTGCACTGAAATC | AGTGCCGCCCAAGTAGCA |
| Tpt1 | GGAGGGCAAGATGGTCAGTAG | CGGTGACTACTGTGCTTTCG |
| Tnf | CTTCTGTCTACTGAACTTCGGG | CACTTGGTGGTTTGCTACGAC |
Muscle fiber cross-sectional area
Frozen, cross-sections of TA muscles were sectioned at the midbelly and used for fiber cross-sectional area measurements. Sections were stained with hematoxylin and the cross-sectional areas of fibers were measured using a digital imaging system (Bioquant, Nashville, TN, USA). In muscle that had undergone acute injury, only centrally-nucleated, regenerating muscle fibers (34) were measured from the area of the central lesion, identified as the region that was least regenerated, as previously described (35). The classification of small and large fibers was determined by setting three standard deviations from the mean cross-sectional area for the Pparδ WT group at 15-dpi time point and quantifying the proportion of myofibers measured that fell within these ranges (adapted from reference 36). The threshold for small and large fibers at 15-dpi was determined to be less than 719 μm2 or more than 2304 μm2, respectively.
Production of Pax7 antibody
Pax7 hybridoma cells were purchased from Developmental Studies Hybridoma Bank anti-Pax7 (Developmental Studies Hybridoma, Iowa City, Iowa, USA; RRID: 2299243). Cells were cultured and antibody was isolated from the supernatant as previously described (37).
Immunohistochemistry
Muscles were dissected from euthanized mice and then rapidly frozen in liquid nitrogen-cooled isopentane. For identification of myogenic progenitor cells, cross-sections, 10 μm thick were taken from the mid-belly of muscles, air-dried for 30 minutes and then fixed in 4% paraformaldehyde for 10 minutes. Sections were then immersed in antigen retrieval buffer (10 mM sodium citrate, 0.05% Tween 20, pH 6.0) at 95–100° C for 40 minutes. Endogenous peroxidase activity in the tissue was quenched by immersion in 0.3% H2O2. Sections were then treated with blocking buffer from a mouse-on-mouse immunohistochemistry kit (M.O.M kit; Vector Labs, Burlingame, CA, USA) for 1 hour and immunolabeled with affinity purified mouse anti-Pax7 (1:500) antibody overnight at 4° C. Sections were washed with phosphate-buffered saline solution (PBS) and then incubated with biotin-conjugated anti-mouse IgG (1:250) for 30 minutes. Sections were subsequently washed with PBS and then incubated for 30 minutes with ABC reagents from the M.O.M kit. Staining was visualized with the peroxidase substrate 3-amino-9-ethylcarbazole (AEC; Vector), yielding a red reaction product.
For identification of macrophages and endothelial cells, cross-sections 10-μm thick were taken from the mid-belly of muscles and fixed in ice-cold acetone. Endogenous peroxidase activity in the sections was quenched by immersion in 0.3% H2O2. Sections were blocked for 1 hour with blocking buffer (3% bovine serum albumin (BSA), 2% gelatin and 0.05% Tween-20 in 50 mM Tris-HCl pH 7.6 containing 150 mM NaCl). Sections were then incubated with: rat anti-mouse F4/80 (1:100, overnight at 4° C, eBioscience #14-4801, San Diego, CA, USA; RRID: AB2314387), rat anti-mouse CD68 (1:100, 3 hours at room temperature (RT), Biorad #MCA1957, Hercules, CA, USA; RRID: AB322219), rabbit anti-mouse CD163 (1:100, 3 hours at RT, Santa Cruz Biotech #33560, Dallas, TX, USA; RRID: AB2074556), rat anti-mouse CD206 (1:50, 3 hours at RT, Biorad #MCA2235, Hercules, CA, USA; RRID: AB324622), and rat anti-CD31 (BD Pharmingen #01951D, San Diego, CA, USA). The sections were washed with PBS and probed with rabbit anti-rat IgG and goat anti-rabbit IgG (Vector Labs #BA-4001 and #BA-1000, Burlingame, CA, USA). Sections were then washed with PBS and incubated with avidin D-conjugated HRP (1:1000, 30 minutes at RT, Vector Labs, Burlingame, CA, USA). Staining was visualized with 3-amino-9-ethylcarbazole.
Stereology
The number of cells per volume of muscle was determined by measuring the total volume of each section using a stereological, point-counting technique to determine section area and then multiplying that value by the section thickness (10 μm) (38). The numbers of immunolabeled cells in each section were counted and expressed as the number of cells per unit volume of each section. In inflammatory lesions with high concentrations of macrophages, boundaries of individual cells were identified by using Nomarski optics (resolution ~400 nm) and changing plane of focus in the Z-axis.
Assay for ischemic muscle area
Ischemic areas of muscles were quantified by imaging the entire muscle section and merging the images using Image Composite Editor (Microsoft, Redmond, CA, USA). Bioquant digital imaging software (Nashville, TN, USA) was used to calculate the entire area of the muscle followed by measuring and subtracting areas of the tissue containing artifacts or imperfections (tears or folds). Areas of ischemia that were sparsely occupied by CD31+ endothelial cells were quantified by an investigator blinded to the identity of tissue sections. Data were expressed as the proportion of the ischemic area divided by the total muscle area.
Immunofluorescence
The immunofluorescence double-labeling protocol was similar to macrophage immunohistochemistry described above. Sections from the mid-belly of TA muscles were fixed with 4% PFA for 10 minutes and then blocked with PBS containing 10% horse serum and 0.1% Tween-20 for 60 minutes. Muscle sections were incubated with rat anti-mouse F4/80 (1:100) and goat anti-Ki67 (1:200; Santa Cruz Biotech #sc-7846, Dallas, TX, USA; RRID: AB2142374) or rabbit anti-CCR2 (1:50; Abcam #32144, Cambridge, UK; RRID:AB1603737) overnight at 4° C. Sections were subsequently incubated with a combination of donkey anti-rat IgG Alexa 488 (Abcam #ab102260, Cambridge, UK) and horse anti-goat IgG Dylight 594 (Vector #Dl-3094, Burlingame, CA, USA) or horse anti-rabbit IgG Dylight 594 (Vector #Dl-1094, Burlingame, CA, USA) and cover-slipped with Prolong Gold anti-fade reagent with DAPI (Invitrogen, Carlsbad, CA, USA).
Assay for muscle fiber damage
Damaged muscle fibers were identified in sections that were blocked in a 1% gelatin solution in PBS for 30 minutes and then labeled with horse anti-mouse IgG Dylight 488 (1:100; Vector Labs #Dl-2488, Burlingame, CA, USA) and cover slipped with Fluorogel (Electron Microscopy Sciences, Hatfield, PA, USA). Intracellular labeling of muscle fibers denoted the presence of the extracellular mouse IgG protein in the muscle fiber indicating damage to the muscle fiber membrane. The proportion of a muscle cross sections that contained damaged fibers was determined by using a sampling grid that was superimposed over the tissue after which the number of grid intercepts that overlaid IgG-positive fibers was counted and expressed as a percentage of the total number of intercepts (39).
Arginase activity assay
Arginase activity was measured in TA muscles at 7-dpi and in contralateral, noninjured TA muscles that were homogenized in 5 volumes of 25 mM Tris-HCl (pH 7.4) containing 5 mM MnCl2, 0.2% Triton X-100 and protease inhibitor cocktail (Sigma Aldrich, St. Louis, MO, USA). Lysates were centrifuged and the supernatant was heated to 56° C for 10 minutes to activate arginase. Substrate hydrolysis was performed by adding 0.5 M arginine, pH 9.7 to the tissue supernatant followed by 1-hour incubation at 37° C. The reaction was stopped by adding H2SO4 and H3PO4 in water (1:3:6 parts volume/volume). Samples were heated to 100° C for 45 minutes after adding 9% α-isonitrosopropriophenone. Urea content was then measured spectrophotometrically at 540 nm. Values were normalized to protein content of the tissue lysate. Six tissues were assayed at each per condition (adapted from 40).
Preparation of bone marrow-derived macrophages
For preparation of BMDMs, bone marrow cells were aseptically flushed from femurs and tibiae and differentiated using RPMI-1640, 20% heat-inactivated fetal bovine serum (FBS; Omega Scientific, Inc, Tarzana, CA, USA), 100 U/ml penicillin and 100 μg/ml streptomycin (1% P/S), and 10 ng/ml macrophage colony stimulating factor (M-CSF; Cell Applications Inc., #RP2008, San Diego, CA, USA) in vitro to BMDMs (31). BMDMs were stimulated for 24-hours with activation media consisting of Dulbecco’s Modified Eagle Medium (DMEM) with 0.25% heat-inactivated FBS, 1% P/S, 10 ng/ml M-CSF and recombinant IL4 (25 ng/ml) with IL13 (10 ng/ml) (BD Pharmingen #550067 and #554599, San Diego, CA, USA), recombinant TNFα (10 ng/ml) with IFNγ (10 ng/ml) (BD Pharmingen #554589 and #554587, San Diego, CA, USA) or recombinant IL10 (10 ng/ml) (BD Pharmingen #550070, San Diego, CA, USA).
Macrophage mobility and chemotaxis
Cell migration and chemotaxis was evaluated using a modified Boyden chemotaxis chamber according to the manufacturer’s protocol (Neuro Probe, Gaithersburg, MD, USA). Briefly, the lower chambers were filled with a volume of DMEM with 1% BSA or DMEM with 1% BSA and 100 ng/ml recombinant CCL2 (R&D Systems #479-JE-010, Minneapolis, MN, USA) resulting in a slight positive meniscus over the well. Next, a 5 μm porous membrane, silicone gasket and the upper plastic chambers were placed over top of the lower chambers. BMDMs were prepared as described above. Cells were dissociated from the plate and resuspended at a concentration of 2x106 cells/ml after which 105 cells were loaded into the upper chambers. The filled chambers were incubated for 2 hours in an incubator at 37° C in humidified air with 5% CO2. After 2 hours, the remaining cells were aspirated from the upper chamber, the chamber was disassembled and the cells on the non-migrated side of the membrane were cleared. The membrane was stained with hematoxylin, mounted to a slide and the number of cells that migrated across the membrane were quantified.
Results
Transcriptional regulation of macrophage activation by myeloid PPARδ in vitro
We generated myeloid specific Ppard mutant mice by crossing Ppardfl/fl mice (Ppard WT) with lysozyme Cre mice to assess the regulatory roles of PPARδ in muscle inflammation and repair following acute injury. First, we assayed the influence of Ppard mutation on myeloid cell phenotype and function in vitro by assaying changes in gene expression in BMDMs. Quantitative PCR (QPCR) analysis confirmed > 90% knockdown of Ppard expression in Ppard mutant macrophages (Figure 1A). The mutation also resulted in a 93% increase in the expression of the M1-biased transcript Nos2 while reducing the expression of M2-biased transcripts Arg1, Mrc1 and Chil3 by 61%, 30%, and 31%, respectively (Figure 1B). These findings generally agree with previous investigations showing that PPARδ signaling promotes the M2 phenotype in macrophages (28,29).
Figure 1:
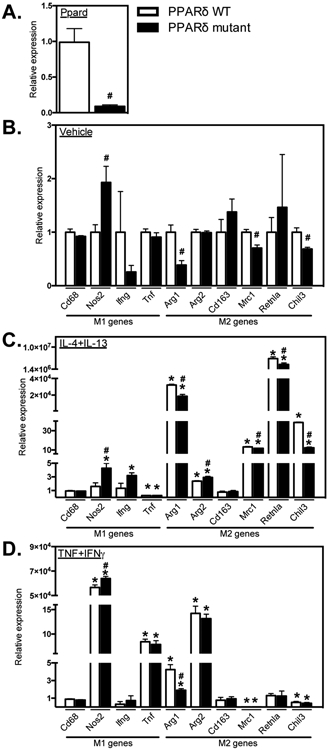
Ppard mutation reduced expression of M2-biased transcripts and promoted an M1-biased phenotype. A: QPCR data showing reduced Ppard expression in BMDMs from Ppard mutant mice compared to Ppard WT mice. B: Expression of the M1-biased transcript Nos2 was increased and M2-biased transcripts Arg1, Mrc1 and Chil3 were reduced in mutant BMDMs compared to WT in control media. C: With M2 activation (IL4 + IL13), Tnf expression was reduced and Arg1, Arg2, Mrc1, Retnla and Chil3 expression were increased compared to control media in Ppard WT BMDMs. Induction of M2-associated transcripts Arg1, Mrc1, Retnla and Chil3 was attenuated in mutant BMDMs. Nos2 and Arg2 expression were increased in M2-activated mutant BMDMs compared to WT. D: M1 activation (TNFα + IFNγ) increased the expression of Nos2, Tnf and had differential effects on M2-biased transcripts increasing Arg1, Arg2 and reducing Mrc1 and Chil3 expression in WT BMDMs. With M1 activation, Nos2 expression was increased and Arg1 expression was attenuated in mutant BMDMs compared to WT. Values in each data set were normalized to Ppard WT BMDMs in vehicle control media, set at 1. * indicates significant difference (P < 0.05) from control BMDMs within same genotype. # indicates significant difference (P < 0.05) versus treatment-matched Ppard WT BMDMs. P-values based on two-tailed t-test. N = 3-4 for each data set. Data are presented as mean ± SEM.
Because the activation state of macrophages in vitro along the M1/M2 spectrum of phenotypes is largely determined by the presence of Th1 or Th2 cytokines (17, 19), we assayed the influence of Ppard mutation on changes in gene expression that were induced by cytokines that may be modulated in muscle following acute injury or disease. Stimulation of wild-type (WT) BMDMs with the Th2 cytokines IL4 and IL13 showed the anticipated increases in expression of M2-biased genes (Arg1, Arg2, Mrc1, Retnla and Chil3), and Ppard mutation diminished the increase for each of those transcripts, except for Arg2 (Figure 1C). These observations are consistent with the previously-reported, Ppard-dependent increase in the expression of Mrc1, Retnla and Chil3 in BMDMs stimulated with IL4 only (29). Stimulation of WT BMDMs with the Th1 cytokines TNF and IFNγ produced expected increases in some M1-biased genes (Nos2 and Tnf) while reducing expression of some M2-biased genes (Mrc1, Chil3). Unexpectedly, TNF/IFNγ stimulation of WT BMDMs also increased expression of Arg1 and Arg2. Ppard mutation also caused a small increase in Nos2 expression in TNF/IFNγ stimulated BMDMs and diminished the increased expression of Arg1 that was caused by stimulation with the Th1 cytokines (Figure 1D). Those observations show that Ppard mutation somewhat amplifies the M1-biased phenotype in TNF/IFNγ activated cells. In addition, the findings show that the influence of Ppard mutation on the expression of some genes, such as the increase in Nos2 and reduction in Arg1, occurs regardless of the Th1 vs. Th2 cytokine environment. However, reductions in Mrc1 and Chil3 expression that were caused by Ppard mutation in unstimulated BMDMs or IL4/13 stimulated BMDMs, did not occur in TNF/IFNγ stimulated BMDMs. This suggests that the regulatory role of PPARδ signaling may vary with the stage of inflammation in injured tissue, as the cytokine environment changes.
Modulation of Th1 and Th2 cytokine gene expression following acute muscle injury
Because in vitro observations showed that the response of BMDMs to stimulation with either Th1 or Th2 cytokines in vitro was influenced by changes in Ppard expression, we assayed whether the expression of those cytokines changed over the time course of muscle repair following acute injury caused by BaCl2 injection. In addition, IFNγ, TNF and IL4 have all been shown previously to influence the muscle inflammation and repair following acute injury or disease (5, 8, 41, 42) Although changes in the expression of IL4 and IFNγ have been observed following muscle injury by snake toxin (5; 41), the inflammatory responses to muscle injury by snake toxin and BaCl2 differ (43). QPCR data showed that Tnf expression was greatly elevated at 3-days post-injury (dpi), remained elevated at 7-dpi and then declined to basal levels by 15-dpi (Figure 2A), which was consistent with a previous report that BaCl2–induced injury increases TNF expression in muscle (44). However, we also observed that Ifng, Il4 and Il13 expression were elevated at 7-dpi, with Il4 and Il13 returning to levels that did not differ from non-injured muscle by 15-dpi (Figures 2B-D). Notably, the increase in Il4 and Il13 expression at 7-dpi coincided to the time post-injury at which CD206+ macrophage populations are elevated (23, 45, 46). Because IL4 and IL13 can increase expression of Mrc1 (which encodes CD206) through a PPARδ-mediated mechanism (29), the observations support the expectation that elevations in IL4 and IL13 expression in muscle at 3- to 7-dpi could lead to increased activation of macrophages to the CD206+ M2-biased phenotype through a PPARδ–mediated process.
Figure 2.
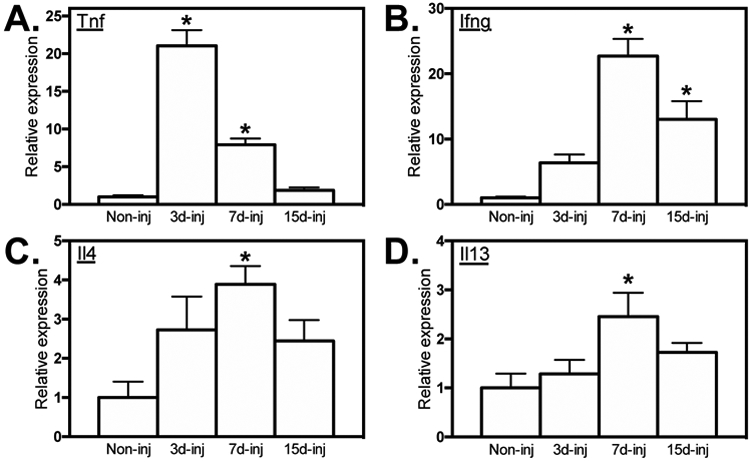
Elevated cytokine expression in skeletal muscle following acute injury. QPCR analysis showed elevated expression of Tnf, (A), Ifng (B), Il4 (C) and Il13 (D) in the TA muscles 3-, 7- and 15-dpi relative to non-injured muscle. All values were normalized to non-injured WT TA muscle and set at 1. * indicates significant difference (P < 0.05) from non-injured muscle. P-values based ANOVA with Tukey’s multiple comparison test. N = 4-5 for each data set. Data are presented as mean ± SEM.
Myeloid PPARδ affects resident macrophage phenotype in healthy muscle without affecting muscle mass
We tested whether Ppard mutation in myeloid cells affected their numbers or phenotype in healthy muscle in vivo. Using antibodies to the pan-macrophage marker F4/80, we found that deletion of Ppard in myeloid cells did not affect total numbers of macrophages in healthy, adult muscle (Figures 3A, B, I). However, the mutation yielded significant reductions of CD163+ (38%; Figures 3E, F, I) and CD206+ (55%; Figures 3G, H, I) M2-biased macrophages and we observed a strong trend (P = 0.06) for an increase in CD68+ macrophages per volume of muscle (Figure 3C, D, I). The reduction in CD206+ macrophages in Ppard mutant mice is consistent with our in vitro observations showing that the mutation reduced expression of Mrc1 (Figure 1). Despite the established role of M2-biased macrophages in promoting muscle growth following injury (10, 14, 22), we did not observe any influence of the reduction of intramuscular CD206+ M2-biased macrophages on the mass in healthy, adult TA muscles (Figures 3J-L).
Figure 3.

Ablation of Ppard in myeloid cells reduces M2-biased macrophages in healthy muscle without affecting muscle mass. A-H: Cross-sections of non-injured Ppard WT (A, C, E, G) and mutant (B, D, F, H) TA muscles were immunolabeled with anti-F4/80 (A, B), anti-CD68 (C, D), anti-CD163 (E, F) or anti-CD206 (G, H). Representative, positively labeled cells are indicated with arrows. Scale bars = 50 μm. I: Quantification of numbers of immunolabeled cells per sectioned muscle volume show that CD163+ and CD206+ cells were reduced in muscles of mutants. J-L: Myeloid Ppard deficiency did not affect body mass (J), TA muscle mass (K) or TA muscle mass-to-body mass ratio (L). # indicates significant difference (P < 0.05) from Ppard WT. P-values based on two-tailed t-test. N = 4-5 for each data set. Data are presented as mean ± SEM.
The influence of myeloid cell deletion of Ppard differs between healthy and injured muscles
We tested whether PPARδ served similar regulatory roles for regulating macrophage phenotype in healthy and injured muscle by assaying numbers of macrophages in muscle that expressed CD68, CD163 or CD206 over the course of muscle inflammation following injury. Contrary to our observations on resident macrophages in healthy muscle, numbers of CD68+ macrophages were significantly reduced at 3-dpi, returning to levels present in non-mutant mice at 7-dpi (Figures 4A, B, C). Also contrary to the effect of the mutation in healthy muscle, numbers of CD163+ macrophages were not affected at any stage of inflammation following muscle injury (Figures 4D, E, F). However, in most distinct contrast to healthy muscle in which the mutation caused large reductions in CD206+ M2-biased macrophages, numbers of CD206+ cells were significantly elevated in mutant mice TAs at 7-dpi (Figures 4G, H, I).
Figure 4.

Ablation of Ppard in myeloid cells shifts macrophages to an M2-biased phenotype in injured muscle. A- I: Representative images and quantification of intramuscular macrophages after injury. Bars = 50 μm. A, B: CD68+ macrophages in PPARδ WT (A) and PPARδ mutant (B) muscles at 3-dpi. Representative CD68+ cells are indicated by arrows. C: Numbers of CD68+ cells per volume of sectioned tissue at 3-, 7- and 15-dpi. D, E: CD163+ macrophages in PPARδ WT (D) and PPARδ mutant (E) muscles at 7-dpi. Representative CD163+ cells are indicated by arrows. F: Quantification of numbers of CD163+ cells per volume of section tissue at 3-, 7- and 15-dpi. G, H: CD206+ macrophages in PPARδ WT (G) and PPARδ mutant (H) muscles at 7-dpi. Representative CD206+ cells are indicated by arrows. I: Numbers of CD206+ cells per volume of sectioned tissue at 3-, 7- and 15-dpi. J: QPCR analysis revealed increased expression of M2-biased transcript Arg1 in muscle of mutant mice after injury. K: Elevated Arg enzymatic activity was detected in Ppard WT and mutant TA muscles 7-dpi compared to non-injured contralateral control muscle. Arg activity was greater in mutant injured TA muscles compared to WT. L: QPCR analysis for expression of the M2-biased transcript Retnla in muscle of mutant mice after injury. All values for QPCR assays were normalized to non-injured Ppard WT TA muscle and set at 1. # indicates significant difference (P < 0.05) from WT within a time point. P-values based on two-tailed t-test. N = 4-6 for each data set. For Arg activity assay, * indicates significant difference (P < 0.05) from non-injured muscle of the same genotype. # indicates significant difference (P < 0.05) from PPARδ WT within a time point. P-values based ANOVA with Tukey’s multiple comparison test. N = 4-5 for each data set. Data are presented as mean ± SEM.
We also observed that the influence of Ppard deletion on the expression of transcripts related to the M2-biased phenotype differed greatly in macrophages in vitro compared to myeloid cells in injured muscles. For example, Ppard mutation in macrophages in vitro reduced Arg1 expression, regardless of the cytokine environment (Figure 1), but the mutation increased Arg1 expression in inflamed muscle at 7- and 15-dpi (Figure 4J) which was reflected by elevated Arg activity in the injured muscle (Figure 4K). Similarly, Retnla, a stereotypical indicator of the M2-biased phenotype, was significantly elevated at 15-dpi (Figure 4L). Thus, ablation of Ppard in macrophages within acutely injured muscle increased their expression of transcripts associated with activation of the M2-biased phenotype.
Ppard mutation in myeloid cells impairs macrophage accumulation in injured muscle and reduces chemotaxis
We assayed whether Ppard mutation in myeloid cells affected the kinetics of macrophage accumulation in injured muscle by assaying changes in total macrophage numbers, using antibodies to the pan macrophage marker, F4/80. The concentration of F4/80+ cells was substantially reduced in Ppard mutant mice at 3-dpi (Figures 5A-C). However, the number of F4/80+ cells at 7-dpi was significantly greater in injured muscles of mutant mice than in WT muscles and a strong trend (P = 0.08) for sustained elevations of F4/80+ cells in muscle persisted through 15-dpi (Figure 5C).
Figure 5.

Myeloid PPARδ mutation delayed macrophage accumulation in muscle after injury, regulated CCR2 expression and affected CCL2-mediated chemotaxis. A-C: Cross-sections of TA muscles 3-, 7- and 15-dpi were immunolabeled with anti-F4/80. Representative images of F4/80+ macrophages in PPARδ WT (A) and PPARδ mutant (B) muscles at 3-dpi. Representative F4/80+ cells are indicated by arrows. C: Numbers of F4/80+ cells per volume of sectioned tissue at 3-, 7- and 15-dpi. D: Cross-section of TA muscles 3-dpi from Ppard WT and mutant mice were immunolabeled with anti-F4/80 (green) and anti-Ki67 (red). Nuclei stained blue with DAPI. Arrow indicates an example of an F4/80+ cell with nuclear Ki67. E: Proportion of F4/80+ cells co-expressing nuclear Ki67 in PPARδ WT and PPARδ mutant muscles. F, G: Muscle sections were co-labeled with anti-F4/80 (green) and anti-CCR2 (red). Arrows indicate examples of F4/80+ cells that are CCR2+ at 3-dpi in PPARδ WT and PPARδ mutant muscles. Bars = 10 μm. H: Numbers of F4/80+ cells that were CCR2+ per volume of sectioned tissue at 3-dpi in PPARδ WT (F) and PPARδ mutant (G) muscles. H: Proportion of F4/80+ cells that were CCR2+ at 3-dpi in PPARδ WT and PPARδ mutant muscles. I: QPCR analysis of BMDMs demonstrated that Ppard mutation reduced Ccr2 expression. Values in each data set were normalized to Ppard WT BMDMs and set at 1. # indicates significant difference (P < 0.05) from Ppard WT within a time point. P-values based on two-tailed t-test. N = 4-5 for each data set. J: A chemotaxis assay was used to assess whether PPARδ affects macrophage mobility or chemotaxis. CCL2 increased chemotaxis but the Ppard mutation completely abrogated the chemotactic effect. Macrophage migration was unaffected by PPARδ in the absence of CCL2. * indicates significant difference (P < 0.05) from nontreated BMDMs within same genotype. # indicates significant difference (P < 0.05) from Ppard WT within a treatment group. P-values based ANOVA with Tukey’s multiple comparison test. N = 6 for each data set. Data are presented as mean ± SEM. K, L: QPCR analysis of non-injured or injured muscle at 7-dpi showed that Ppard mutation did not affect Ccl2 expression (K) but decreased Ccr2 expression in non-injured muscle and increased Ccr2 expression at 7-dpi (L). Values in each data set were normalized to Ppard WT BMDMs and set at 1. # indicates significant difference (P < 0.05) from Ppard WT with same treatment conditions. P-values based on two-tailed t-test. N = 5 for each data set.
Because previous investigators have shown that Ppard -deficient macrophages have a markedly reduced mitogenic response to IL4 stimulation in vitro (29), we tested whether the delayed accumulation of macrophages in injured muscles of Ppard mutants was attributable to impaired macrophage proliferation in vivo. However, the proportion of F4/80+ cells that co-expressed Ki67, a marker of cell proliferation, did not differ between injured muscles of control and mutant mice (Figures 5D, E), indicating that suppression of proliferation did not underlie the slowed kinetics of macrophage accumulation in injured muscles.
Alternatively, the delayed accumulation of macrophages in injured muscles of mutant mice could result from impaired recruitment because several previous investigations have shown that perturbations of CCL2/CCR2 signaling are particularly important in disrupting macrophage recruitment to injured muscle (6, 47-49). We observed that the proportion of F4/80+ cells that expressed detectible levels of CCR2 was significantly reduced in Ppard mutant muscles at 3-dpi (Figures 3F-H), suggesting that reductions in recruitment via CCR2 can contribute to the reduced numbers of total macrophages at this stage. In addition, we confirmed that Ppard mutation caused large reductions (41%) in Ccr2 expression by BMDMs in vitro (Figure 5I), consistent with previous reports (50). Furthermore, Ppard mutant BMDMs exhibited impaired chemotaxis towards CCL2, but did not affect cell mobility independent of the CCR2 ligand (Figure 5J). Finally, we also tested whether expression of either CCL2 or CCR2 differed in mutant and WT muscles at 7-dpi, when numbers of F4/80+ cells in mutant muscles increased, suggesting a recovery in a defect in CCR2-mediated chemotaxis. Although CCL2 expression did not differ between WT and mutants at 7-dpi (Figure 5K), CCR2 expression was significantly elevated and greater in mutant muscles at this stage (Figure 5L). Thus, our data indicate that myeloid cell PPARδ is important for influencing macrophage recruitment into muscle after acute injury, possibly by regulating CCR2 expression and reducing chemotaxis at early stages post-injury.
Ppard mutation in myeloid cells slows muscle repair and revascularization following acute injury Intramuscular macrophages can contribute to amplifying muscle damage or promoting muscle repair following injury or disease (3) which led us to examine the influence of Ppard mutation in myeloid cells on muscle injury and repair. We assayed muscle injury by measuring the proportion of total muscle cross-sections that contained injured fibers with large membrane lesions that allowed the unregulated influx of IgG (Figures 6A, B), which does not cross the intact cell membrane of healthy muscle fibers. The proportion of the muscle that contained injured fibers exceeded 73% at 3-dpi, and did not differ between WT and mutant mice at this stage (Figure 6C), indicating that PPARδ signaling is not an essential mediator of macrophage cytotoxicity in this injury model. Furthermore, because the numbers of macrophages in muscles at 3-dpi were greatly reduced (Figure 5C) although muscle injury did not differ between WT and mutant muscles at that stage (Figure 6C), the observations show that the magnitude of the inflammatory response is not an important determinant of the extent of muscle damage at this stage in this injury model. In addition, we observed that the regions of the most extensive muscle damage at 3-dpi contained few macrophages and regions of muscle fiber repair and regeneration were relatively enriched in macrophages (Figures 6D, E). Collectively, these observations show that neither the magnitude of macrophage accumulation, the localization of macrophages or their expression of PPARδ is an important determinant of the initial magnitude of muscle damage in this injury model. However, at 7-dpi, the area of injury in WT muscles returned almost to levels of non-injured muscle, although injury in muscles of mutant mice remained greatly elevated (Figure 6C), coinciding with the large elevation of macrophages in the muscle at this stage (Figure 5C). Repair of injury in muscles of mutant mice was complete by 15-dpi (Figure 6C), indicating that Ppard mutation slowed the rate of muscle repair.
Figure 6.

Myeloid Ppard mutation impairs muscle repair and revascularization following injury. A, B: Representative cross-sections of TA muscles were immunolabeled with anti-mouse IgG (green) to assay for sarcolemma damage at 7-dpi in PPARδ WT and PPARδ mutant muscles. Arrows indicate examples of IgG+ muscle fibers. Bars = 100 μm. C: The volume fractions of TA muscles occupied by IgG+ fibers were quantified at 3-, 7- and 15-dpi. # indicates significant difference (P < 0.05) from WT within a time point. P-values based on two-tailed t-test. N = 4-6 for each data set. D-G: Muscle cross-sections at 7-dpi were co-labeled with anti-mouse IgG (green) and anti-CD68 (red) (D, E) or anti-mouse IgG (green) and anti-CD31 (red) (F, G). Bars = 15 μm. Both CD68+ and CD31+ cells were present in low concentrations in areas marked by muscle fiber damage (D, F) and in high concentrations in areas of regeneration containing centrally-nucleated muscle fibers (E, G). Arrows outline the surface of representative, IgG+, injured fibers (white arrows; D, F) or IgG-, central-nucleated, regenerative fibers (yellow arrows; E, G). H, I: Representative images of whole TA muscle cross-sections immunolabeled for CD31 in Ppard WT (H) and mutants (I) at 7-dpi. Arrow indicates ischemic region of muscle sparsely populated by CD31+ cells. J: Quantification of the volume fraction of the muscle sparsely occupied by CD31+ cells at 7-dpi. # indicates significant difference (P < 0.05) from Ppard WT. P-values based on two-tailed t-test. N = 5 for each data set. K: QPCR assays showed elevated expression of the hypoxia inducible transcript Hif1a in mutant muscle 3- and 7-dpi compared to WT. Values were normalized to non-injured Ppard WT TA muscle and set at 1. # indicates significant difference (P < 0.05) from Ppard WT within a time point. P-values based on two-tailed t-test. N = 4-6 for each data set. Data are presented as mean ± SEM.
Previous investigators demonstrated that treatments that reduce macrophage numbers in injured muscles can disrupt revascularization, which was associated with impaired muscle regeneration (6, 48, 51). We tested whether the myeloid cell targeted deletion of Ppard impaired revascularization, using immunohistochemistry for vascular endothelial cells that expressed CD31. We found that areas of muscle in which there was extensive muscle fiber damage contained few CD31+ cells, although areas in which regenerative muscle fiber displayed intact membranes were richly-vascularized (Figures 6F, G). We tested whether Ppard mutation affected the extent of ischemia in muscle during the repair phase following acute injury by assaying for the proportion of the muscle cross-section that was devoid of CD31+ vascular endothelial cells and found that mutant muscles showed over four-fold greater area of ischemia than WT at 7-dpi (Figures 6H-J). In addition, we assayed levels of expression of the hypoxia-inducible gene Hif1a as an independent index of ischemia and oxidative stress and found significantly elevated levels of Hif1α at each stage of muscle repair in Ppard mutants (Figure 6K).
Ppard mutation in myeloid cells slows myogenesis following acute injury
Macrophage-derived factors can influence the expansion of myogenic satellite cell populations and affect the growth of regenerative muscle fibers following muscle injury or disease (7, 23, 31). We tested whether PPARδ in myeloid cells played a significant role in these pro-myogenic roles of macrophages by assaying the effect of Ppard mutation on the numbers of Pax7+ myogenic cells and the size of regenerative muscle fibers over the course of muscle repair. Ppard mutants showed large reductions in the number of Pax7+ cells at 3-dpi which persisted in mutants at 7-dpi (Figures 7A-C). However, by 15-dpi, the numbers of Pax7+ cells in WT and mutant muscles were greatly reduced and did not differ between the two genotypes (Figure 7C). The negative influence of the Ppard mutation on myogenesis also manifests as a reduction in the cross-sectional area of central-nucleated, regenerative fibers (Figures 7D-F), which was 23% less in mutant mice at 15-dpi. In addition, the mutation produced a significant reduction in the proportion of large diameter muscle fibers (> 2304 μm2) and a nearly significant increase (p = 0.06) in small diameter fibers (Figure 7G).
Figure 7.

Myeloid Ppard mutation delayed expansion of satellite cell populations and reduced the size of regenerative fibers in injured muscle. A, B: Representative images of muscle cross-sections from PPARδ WT (A) and PPARδ mutant (B) muscles at 7-dpi immunolabeled with anti-Pax7. Arrows indicate examples of Pax7+ cells. Bars = 50 μm. C: Numbers of Pax7+ cells per volume of sectioned tissue at 3-, 7- and 15-dpi. # indicates significant difference (P < 0.05) from Ppard WT at a time point. P-values based on two-tailed t-test. N = 4-6 for each data set. Representative images of cross-sections of TA muscles from Ppard WT (D) and mutants (E) 15-dpi stained with hematoxylin to assay cross-sectional area of non-injured muscles and muscles 15-dpi. Bars = 100 μm. F: Cross-sectional areas of centrally-nucleated regenerating muscle fibers at 3-, 7- and 15-dpi. # indicates significant difference (P < 0.05) from Ppard WT within a time point. P-values based on two-tailed t-test. N = 5-6 for each data set. G: Frequency distribution of fiber cross-sectional areas for TA muscles from Ppard WT and mutants 15-dpi. N = 5-6 for each data set. # indicates a significant (P < 0.05) reduction in the proportion of large muscle fibers from mutant muscle 15-dpi compared to WT. Small (< 719 μm2) and large fibers (> 2304 μm2) were 3 standard deviations below and above the average cross-sectional area of Ppard WT mice 15-dpi. Data are presented as mean ± SEM.
IL10 is a greater effecter of macrophage activation to a CD163+/CD206+ M2-biased phenotype in injured muscle than PPARδ
Our unexpected finding that Ppard mutation did not diminish macrophage activation to a CD163+/CD206+ M2-biased phenotype in injured muscle led us to assay for other effector molecules that activate the CD163+/CD206+ phenotype in injured muscle. Our previous observation that ablation of Il10 in mice experiencing increased muscle use greatly reduced CD163+ M2-biased macrophages while increasing CD68+ macrophages (21) implicated IL10 as a significant modulator of intramuscular macrophage phenotype. In the present investigation, we showed that Il10 expression is greatly elevated in muscles following acute injury, reaching expression levels at 3-dpi that are more than 150-times the expression in uninjured muscle (Figure 8A). We also confirmed that IL10 is a strong activator of the M2-biased phenotype in BMDMs in vitro, causing large increases in the expression of Arg1, Arg2 and Mrc1 although Cd163 expression was not affected (Figure 8B).
Figure 8.
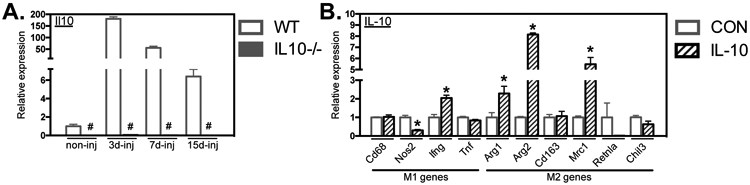
IL10 stimulation shifted BMDMs to an M2-biased phenotype in vitro. A: QPCR data showed induction of Il10 gene expression in WT muscle after injury. Il10 expression was ablated in non-injured and injured muscle 3-, 7- and 15-dpi from Il10−/− mice. Values were normalized to non-injured WT TA muscle. # indicates significant difference (P < 0.05) from WT at a time point. P-values based on two-tailed t-test. N = 4-5 for each data set. B: QPCR data showing that WT BMDMs stimulated with IL10 reduced iNOS and increased Ifng, Arg1, Arg2 and Mrc1 expression. All values were normalized to non-stimulated (CON) WT BMDMs and set at 1. # indicates significant difference (P < 0.05) from CON BMDMs. P-values based on two-tailed t-test. N = 3-4 for each data set. Data are presented as mean ± SEM.
In contrast to the effects of Ppard gene ablation in myeloid cells, Il10 mutation greatly influenced macrophage phenotype without affecting macrophage accumulation during repair of injured muscle. Assays for changes in F4/80+ total macrophages showed that Il10 mutation did not affect the numbers of macrophages or the time course of their accumulation in injured muscle (Figures 9A-C), which indicates that expansion and recruitment of macrophage populations were unaffected by the mutation. However, the mutation caused significant increases in the numbers of CD68+ macrophages at 15-dpi (Figures 9D-F) and caused large reductions in numbers of CD163+ cells at earlier stages of repair (Figures 9G-I). The most prominent treatment effect that we observed was the greatly reduced numbers of CD206+ macrophages that occurred at every stage of repair (Figure 9J-L), showing that IL10-mediated signaling rather than PPARδ signaling is an important regulator of the CD206+ M2-biased phenotype in injured muscle. Preventing expansion of CD206+ macrophages was not accompanied by a reduction in either Arg1 or Arg2 in injured muscles of Il10 mutant mice (Figures 9M, N), in contrast to the tremendous induction of Arg expression in IL10-stimulated BMDMs (Figure 8B).
Figure 9.

Il10 mutation attenuated the expansion of M2-biased macrophages in injured muscle affecting muscle injury and repair. A-L: Cross-sections of WT and Il10−/− TA muscles were immunolabeled with anti- F4/80 (A, B), anti-CD68 (D, E), anti-CD163 (G, H) and anti-CD206 (J, K). Representative images from WT (A, D, G, J) and mutant (B, E, H, K) muscles at either 7-dpi. Arrows indicate examples of antibody-labeled leukocytes. Bars = 50 μm. Numbers of F4/80+ (C), CD68+ (F), CD163+ (I) or CD206+ (L) cells per volume of sectioned tissue at 3-, 7- and 15-dpi are summarized in the histograms. M, N: QPCR data showed no change in the induction of Arg1 and Arg2 expression in WT and Il10−/− TA muscles after injury. O, P: Representative images of muscle cross-sections from WT (O) and Il10 mutant (P) muscles at 7-dpi immunolabeled with anti-Pax7. Arrows indicate examples of Pax7+ cells. Bars = 50 μm. Q: Numbers of Pax7+ cells per volume of sectioned tissue at 3-, 7- and 15-dpi. R, S: Representative cross-sections of TA muscles were immunolabeled with anti-mouse IgG (green) to assay for sarcolemma damage at 15-dpi in WT and Il10 mutant muscles. Bars = 100 μm. T: The volume fractions of TA muscles occupied by IgG+ fibers were quantified at 3-, 7- and 15-dpi. # indicates significant difference (P < 0.05) from WT within a time point. P-values based on two-tailed t-test. N = 4-6 for each data set. U, V: Representative images of whole TA muscle cross-sections at 7-dpi immunolabeled for CD31 in WT (U) and Il10 mutants (V). W: Quantification of the volume fraction of the muscle sparsely occupied by CD31+ cells at 7-dpi. X: QPCR analysis of Hif1a transcript expression in WT and Il10 mutant muscles at 3-, 7-, and 15-dpi. # indicates significant difference (P < 0.05) from WT within a time point. P-values based on two-tailed t-test. N = 4-5 for each data set. Data are presented as mean ± SEM.
Because our observations of the effects of Ppard mutation on muscle repair showed PPARδ played a significant role in regulating the expansion of satellite cell numbers, reducing the extent of muscle damage and revascularization of muscle, we also assayed whether IL10 contributed to these features of muscle repair. Unlike the effects of Ppard mutation, Il10 mutation had no effect on the time course or magnitude of expansion of satellite cell populations following injury (Figures 9O-Q). Also unlike Ppard mutants in which the initial magnitude of muscle damage was unaffected by the mutation, the Il10 mutants experienced a small decrease in the proportion of the muscle that showed damage at 3-dpi (Figures 9R-T). However, at 15-dpi, Il10 mutants had more muscle damage than WT mice (Figure 9T). Finally, in further contrast to Ppard mutants, neither the extent of revascularization of muscle at 7-dpi (Figures 9U-W) or the levels of Hif1 expression, was affected by Il10 mutation (Figure 9X).
Discussion
Landmark discoveries by previous investigators demonstrated the importance of Ppard expression and activation in regulating the phenotype of resident macrophages in liver and adipose tissue in vivo and in BMDMs in vitro (28, 29). In particular, the reduced expression of markers of M2-biased macrophage activation (e.g., Arg1, Mrc1 (or CD206), Retnla, Chi3l3, Il10) in myeloid targeted Ppard mutants in these models (28, 29) served as the foundation for the broad view that PPARδ–mediated signaling provides a crucial control for the switch of macrophages from a pro-inflammatory, M1-biased phenotype to an anti-inflammatory, M2-biased phenotype (52). The present investigation shows that this regulatory function for PPARδ in myeloid cells does not exist in inflamed, regenerating muscle following acute injury. Conversely, we observed that the myeloid-cell-specific ablation of Ppard caused either increases or no change in the numbers of CD206+ macrophages over the course of muscle inflammation and caused either an increase or no change in the expression of phenotypic markers of M2-biased macrophages. These findings show that regulation of macrophage phenotype in vivo differs with the tissue environment in which phenotype switching occurs.
Our findings also confirm that regulatory controls of macrophage phenotype that are apparent in vitro are not necessarily predictive of controls that are most important in vivo, as emphasized previously by luminaries in the field of macrophage biology (19). Although our in vivo observations showed that myeloid cell targeted ablation of Ppard enhanced expression of markers of the M2-biased phenotype in injured muscle, our in vitro observations showed that the same mutation in BMDMs in vitro diminished their expression of M2-biased phenotypic markers. In addition, the mutation diminished the elevated expression of M2-biased phenotypic markers in BMDMs activated with the Th2 cytokines IL4 and IL13, as reported previously (29).
The distinct differences between the roles of PPARδ in controlling macrophage phenotype in vivo may reflect differences in the tissue in which activation occurred (i.e. fat or liver vs. skeletal muscle); however, our observations indicate that tissue type is not the most important variable for determining the influence of PPARδ on macrophage phenotype. Contrary to our findings in injured muscle, in which the myeloid-targeted mutation of Ppard increased CD206+ M2-biased macrophage number, the mutation reduced CD206+ resident macrophages in healthy, non-injured muscle. This effect of Ppard mutation on resident macrophages in healthy muscle resembles the influence of Ppard ablation in adipose tissue macrophages in healthy mice (29). Together, the observations suggest that differences between the regulatory roles of Ppard on macrophages in injured muscle vs. liver or adipose tissue may be attributable, in part, to differences in phenotype regulation in injured vs. non-injured tissue, rather than differences in tissue type. In contrast to the evidence that Ppard mutation does not prevent macrophage activation to an M2-biased phenotype in injured muscle, we observed that Il10 mutation produced large reductions in the numbers of CD206+ and CD163+ macrophages over the entire course of muscle regeneration that we studied. This is consistent with previous observations that IL10 plays a significant role in macrophage phenotype transitions in muscles of dystrophic mice and mice experiencing modified muscle loading (8, 21) and that treatment of infected muscles with exogenous IL10 increases the proportion of macrophages that express high levels of CD206 (53). We also observed that the rate of repair of the damaged muscle was impaired in Il10 mutant mice, which is consistent with previous findings that showed that diminishing macrophage phenotype transitions during muscle regeneration increased damage or slowed repair (7, 8, 21, 23). Thus, our findings collectively show that IL10 contributes significantly to the shift of macrophages to a CD206+, CD163+, M2-biased phenotype that is independent of PPARδ signaling and accelerates muscle repair.
Although our findings show that IL10 expression is mandatory for normal acquisition of a CD206+ M2-biased phenotype in acutely injured muscle and PPARδ is not required, it is feasible that PPARδ can contribute to acquisition of CD163+/CD206+ M2-biased phenotype in injured muscle, but that function is assumed by other molecules in mutant mice. Many other molecules could potentially serve that function, including other members of the PPAR family. For example, PPARγ has a well-established role in regulating macrophage function (54), although it is not essential for development of the macrophage lineage (55). Although numerous findings indicate that PPARγ-mediated signaling can promote an M2-biased phenotype in macrophages (56-60), other findings indicate that inhibition or loss of PPARγ can bias macrophages to a CD163+/CD206+ macrophage phenotype (61). These different outcomes may reflect differences in the environment in which macrophage activation occurred, and suggests that the role of PPARγ signaling in regulating macrophage phenotype may also vary in vivo according to the tissue and inflammatory context. However, there is no current evidence to show a role for PPARγ in regulating macrophage phenotype in injured muscle. Specifically, in skeletal muscle injured by snake toxin, ablation of PPARγ in myeloid cells did not affect the proportion of Ly6c+ macrophages (associated with an M1-biased phenotype) relative to Ly6c-macrophages (associated with an M2-biased phenotype) (62), in skeletal muscle injured by snake toxin.
Although mutation of Ppard in myeloid lineage cells did not prevent activation of macrophages to a CD206+ M2-biased phenotype in injured muscle, our data indicate that the mutation affected the kinetics of the inflammatory process. In particular, the amplification of F4/80+ macrophage numbers in injured muscles of mutant mice was delayed and was accompanied by reductions in the expression of CCR2 by intramuscular macrophages. Further, we observed an impaired chemotactic response of mutant macrophages to CCL2 in vitro. Because discoveries by other investigators showed that signaling via CCL2/CCR2 is tremendously important in promoting the traffic of macrophages into diseased or injured tissue (6, 47, 48, 63, 64), disrupting that signaling pathway by Ppard mutation would affect the time course and magnitude of inflammation. However, elevations in PPARδ signaling can also decrease CCL2/CCR2-mediated chemotaxis, at least in other tissue types and states of tissue damage. For example, activation of PPARδ by agonists suppresses atherogenic inflammation and atherosclerosis, which was attributed to the ability of PPARδ to repress the expression of CCL2 (65, 66) and thereby reduce transendothelial migration of monocytes, at least in vitro (65). These unpredictable relationships between changes in PPARδ signaling and signaling via CCL2/CCR2 and chemotaxis can be explained in part by opposing influences of PPARδ on CCL2 expression. Both pharmacological activation and genetic deletion of PPARδ can inhibit CCL2 promoter activity (66, 67), which indicates that any perturbation of Ppard expression or activity has the potential to reduce CCL2/CCR2-driven chemotaxis.
Notably, CCL2/CCR2 signaling does not only influence macrophage chemotaxis; it can also influence macrophage polarization. For example, disruption of CCL2/CCR2 signaling in human macrophages in vitro impairs their activation to a CD163+ M2-biased phenotype (68) and direct activation of human monocytes with CCL2 shifts them to a CD206+ M2-biased phenotype (69). However, in contrast with those observations, other investigators report that CCL2/CCR2 signaling in macrophages suppresses the M2 phenotype and promotes the M1 phenotype. For example, ablation of CCR2 in mice on a high-fat diet increased expression of M2 phenotypic markers in macrophages (70). Those observations suggest that the elevated numbers of CD206+ M2-biased macrophages that we observed in Ppard muscles at 7-dpi could reflect reductions of CCR2-mediated regulation of macrophage phenotype.
Because we found that Ppard mutation in myeloid cells produced either an increase or no change in numbers of macrophages that expressed markers of the M2-biased, pro-regenerative phenotype, we were surprised that the mutation greatly slowed muscle repair between 3 and 15-dpi. However, we observed that the mutation affected the distribution of macrophages in the injured muscles and that macrophages were more broadly distributed in injured WT muscles than in injured mutant muscles. In addition, macrophages were located in areas of regeneration and absent in areas of necrosis in both genotypes. These observations are consistent with a defect in macrophage recruitment to sites of muscle injury in the mutant mice, as a consequence of reduced expression of CCR2. Associated with the reduced dispersal of macrophages in the injured muscle of mutant mice, we found significantly larger areas of muscle lacking vascularization. Although we cannot definitively address the cause of this defect, the failure to recruit pro-regenerative macrophages to the sites of muscle damage may contribute. Previous investigators have established that macrophages play a significant role in affecting muscle revascularization following injury (71) and that reduction in numbers of macrophages in regenerative muscle is associated with impaired revascularization and slowed repair of injured muscle (6, 48, 51). This is similar to the outcomes that we observed. Interestingly, the systemic delivery of PPARδ agonists to experimental animals increases angiogenesis in synthetic implants in vivo (72) and in skeletal muscle (73). Because endothelial cells express PPARδ and signaling through PPARδ increases proliferation of endothelial cells (72, 74), the pro-vascular effects of PPARδ signaling were partially attributed to more rapid expansion of endothelial cell populations. However, our observations suggest the possibility that part of the increased angiogenesis in experimental animals treated with PPARδ agonists may result from an increase in PPARδ signaling in myeloid cells, in addition to endothelial cells.
Collectively, our findings indicate that PPARδ signaling in myeloid cells and IL10-mediated signaling both influence muscle inflammation and improve muscle repair following acute injury. However, their effects appear to be mediated by distinct influences on the inflammatory process, in which PPARδ signaling affects the chemotactic response of monocytes and macrophages to the injured muscle and IL10 signaling influences the shift in macrophages to an M2-biased phenotype, reflected by the numbers of cells that expressed the phenotypic markers CD206 and CD163. In addition to contributing to our understanding of processes that regulate muscle inflammation and repair following acute injury, these findings illustrate that the transcriptional control of macrophage phenotype switching in vivo varies with tissue type and is not always reliably predicted by in vitro findings.
Key points:
Macrophage deletion of PPARδ reduces inflammation/revascularization of injured muscle
PPARδ expression is not required for M2 macrophage activation in injured muscle
IL10 is required for M2 macrophage activation after sterile muscle injury
Acknowledgements
Confocal laser scanning microscopy was performed at the California NanoSystems Institute Advanced Light Microscopy/Spectroscopy Shared Resource Facility at UCLA. The Pax7 hybridoma developed by T.M. Jessell, Columbia University, was obtained from the Developmental Studies Hybridoma Bank, created by the NICHD of the NIH and maintained at The University of Iowa, Department of Biology, Iowa City, IA 52242, USA.
This research was supported by the National Institute of Arthritis and Musculoskeletal and Skin Diseases and the National Institute on Aging of the National Institutes of Health under award numbers RO1AR066036 and RO1AG041147 (to JGT).
References
- 1.Belcastro AN, Arthur GD, Albisser TA, and Raj DA 1996. Heart, liver, and skeletal muscle myeloperoxidase activity during exercise. J. Appl. Physiol 80: 1331–1335. [DOI] [PubMed] [Google Scholar]
- 2.Fielding RA, Manfredi TJ, Ding W, Fiatarone MA, Evans WJ, and Cannon JG 1993. Acute phase response in exercise. III. Neutrophil and IL-1 beta accumulation in skeletal muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol 265: R166–R172. [DOI] [PubMed] [Google Scholar]
- 3.Tidball JG 2017. Regulation of muscle growth and regeneration by the immune system. Nature Rev. Immunol 17: 165–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bencze M, Negroni E, Vallese D, Yacoub-Youssef H, Chaouch S, Wolff A, Aamiri A, Di Santo JP, Chauzaud B, Butler-Browne G, Savino W, Mouly V, and Riederer I 2012. Proinflammatory macrophages enhance the regenerative capacity of human myoblasts by modifying their kinetics of proliferation and differentiation. Mol. Ther 20: 2168–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng M, Nguyen MH, Fantuzzi G, and Koh TJ 2008. Endogenous interferon-gamma is required for efficient skeletal muscle regeneration. Am. J. Physiol. Cell Physiol 294: C1183–C1191. [DOI] [PubMed] [Google Scholar]
- 6.Martinez CO, McHale MJ, Wells JT, Ochoa O, Michalek JE, McManus LM, and Shireman PK 2010. Regulation of skeletal muscle regeneration by CCR2-activating chemokines is directly related to macrophage recruitment. Am. J. Physiol. Regul. Integr. Comp. Physiol 299: R832–R842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vidal B, Serrano AL, Tjwa M, Suelves M, Ardite E, De Mori R, Baeza-Raja B, Martínez de Lagrán M, Lafuste P, Ruiz-Bonilla V, Jardi M, Gherardi R, Christov C, Dierssen M, Carmeliet P, Degen JL, Dewerchin M, and Muñoz- Cánoves P 2008. Fibrinogen drives dystrophic muscle fibrosis via a TGFbeta/alternative macrophage activation pathway. Genes Dev. 22: 1747–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Villalta SA, Deng B, Rinaldi C, Wehling-Henricks M, and Tidball JG 2011. IFN-γ promotes muscle damage in the mdx mouse model of Duchenne muscular dystrophy by suppressing M2 macrophage activation and inhibiting muscle cell proliferation. J. Immunol 187: 5419–5428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warren GL, O'Farrell L, Summan M, Hulderman T, Mishra D, Luster MI, Kuziel WA, and Simeonova PP 2004. Role of CC chemokines in skeletal muscle functional restoration after injury. Am. J. Physiol. Cell Physiol 286: C1031–C1036. [DOI] [PubMed] [Google Scholar]
- 10.Arnold L, Henry A, Poron F, Baba-Amer Y, van Rooijen N, Plonquet A, Gherardi RK, and Chazaud B 2007. Inflammatory monocytes recruited after skeletal muscle injury switch antiinflammatory macrophages to support myogenesis. J. Exp. Med 204: 1057–1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Perdiguero E, Sousa-Victor P, Ruiz-Bonilla V, Jardí M, Caelles C, Serrano AL, and Muñoz- Cánoves P 2011. p38/MKP-1-regulated AKT coordinates macrophage transitions and resolution of inflammation during tissue repair. J. Cell Biol 195: 307–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Segawa M, Fukada S, Yamamoto Y, Yahagi H, Kanematsu M, Sato M, Ito T, Uezumi A, Hayashi S, Miyagoe-Suzuki Y, Takeda S, Tsujikawa K, and Yamamoto H 2008. Suppression of macrophage functions impairs skeletal muscle regeneration with severe fibrosis. Exp. Cell Res. 314: 3232–3244. [DOI] [PubMed] [Google Scholar]
- 13.Summan M, Warren GL, Mercer RR, Chapman R, Hulderman T, Van Rooijen N, and Simeonova PP 2006. Macrophages and skeletal muscle regeneration: a clodronate-containing liposome depletion study. Am. J. Physiol. Regul. Integr. Comp. Physiol 290: R1488–R1495. [DOI] [PubMed] [Google Scholar]
- 14.Tidball JG, and Wehling-Henricks M 2007. Macrophages promote muscle membrane repair and muscle fibre growth and regeneration during modified muscle loading in mice in vivo. J. Physiol 578 327–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Warren GL, Hulderman T, Mishra D, Gao X, Millecchia L, O’Farrell L, Kuziel WA, and Simeonova PP 2005. Chemokine receptor CCR2 involvement in skeletal muscle regeneration. FASEB J. 19: 413–415. [DOI] [PubMed] [Google Scholar]
- 16.Locati M, Mantovani A, and Sica A 2013. Macrophage activation and polarization as an adaptive component of innate immunity. Adv. Immunol 120: 163–184. [DOI] [PubMed] [Google Scholar]
- 17.Mills CD, Kincaid K, Alt JM, Heilman MJ, and Hill AM 2000. M-1/M-2 macrophages and the TH1/TH2 paradigm. J. Immunol 164: 6166–6173.10843666 [Google Scholar]
- 18.Mills CD 2015. Anatomy of a discovery: M1 and M2 macrophages. Front. Immunol 6a: 212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, Gordon S, Hamilton JA, Ivashkiv LB, Lawrence T, Locati M, Mantovani A, Martinez FO, Mege JL, Mosser DM, Natoli G, Saeij JP, Schultze JL, Shirey KA, Sica A, Suttles J, Udalova I, van Ginderachter JA, Vogel SN, and Wynn TA 2014. Macrophage activation and polarization: nomenclature and experimental guidelines. Immunity 17: 14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Villalta SA, Nguyen HX, Deng B, Gotoh T, and Tidball JG 2009. Shifts in macrophage phenotypes and macrophage competition for arginine metabolism affect the severity of muscle pathology in muscular dystrophy. Hum. Mol. Genet 18: 482–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Deng B, Wehling-Henricks M, Villalta SA, Wang Y, and Tidball JG 2012. IL-10 triggers changes in macrophage phenotype that promote muscle growth and regeneration. J. Immunol 189: 3669–3680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mounier R, Théret M, Arnonld L, Cuvellier S, Bultot L, Göransson O, Sanz N, Ferry A, Sakamoto K, Foretz M, Viollet B, and Chazaud B 2013. AMPKα1 regulates macrophage skewing at the time of resolution of inflammation during skeletal muscle regeneration. Cell Metab. 18: 251–264. [DOI] [PubMed] [Google Scholar]
- 23.Tonkin J, Temmerman L, Sampson RD, Gallego-Colon E, Barberi L, Bilbao D, Schneider MD, Musarò A, and Rosenthal N 2015. Monocyte/macrophage-derived IGF-1 orchestrates murine skeletal muscle regeneration and modulates autocrine polarization. Mol. Ther 23: 1189–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Giannakis N, Sansbury BE, Patsalos A, Hays TT, Riley CO, Han X, Spite M, and Nagy L (2019) Dynamic changes to lipid mediators support transitions among macrophage subtypes during muscle regeneration. Nat. Immunol 20, 626–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mukundan L, Odegaard JI, Morel CR, Heredia JE, Mwangi JW, Ricardo-Gonzalez RR, Goh YP, Eagle AR, Dunn SE, Awakuni JU, Nguyen KD, Steinman L, Michie SA, and Chawla A 2009. PPAR-delta senses and orchestrates clearance of apoptotic cells to promote tolerance. Nat. Med 15: 1266–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bensinger SJ, and Tontonoz P 2008. Integration of metabolism and inflammation by lipid-activated nuclear receptors. Nature 454: 470–477. [DOI] [PubMed] [Google Scholar]
- 27.Odegaard JI, and Chawla A 2008. Mechanisms of macrophage activation in obesity-induced insulin resistance. Nat. Clin. Pract. Endocrinol. Metab 4: 619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kang K, Reilly SM, Karabacak V, Gangl MR, Fitzgerald K, Hatano B, and Lee C-H 2008. Adipocyte-derived Th2 cytokines and myeloid PPARdelta regulate macrophage polarization and insulin sensitivity. Cell Metab. 7: 485–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Odegaard JI, Ricardo-Gonzalez RR, Red Eagle A, Vats D, Morel CR, Goforth MH, Subramanian V, Mukundan L, Ferrante AW, and Chawla A 2008. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 7: 496–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hyun J, Romero L, Riveron R, Flores C, Kanagavelu S, Chung KD, Alonso A, Sotolongo J, Ruiz J, Manukyan A, Chun S, Singh G, Salas P, Targan SR, and Fukata M 2015. Human intestinal epithelial cells express interleukin-10 through Toll-like receptor 4-mediated epithelial-macrophage crosstalk. J. Innate Immun 7: 87–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wehling-Henricks M, Welc SS, Samengo G, Rinaldi C, Lindsay C, Wang Y, Lee J, Kuro-O M, and Tidball JG 2018. Macrophages escape Klotho gene silencing in the mdx mouse model of Duchenne muscular dystrophy and promote muscle growth and increase satellite cell numbers through a Klotho-mediated pathway. Hum. Mol. Genet 27: 14–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nolan T, Hands RE, and Bustin SA 2006. Quantification of mRNA using real-time RT-PCR. Nat. Protoc 1: 1559–1582. [DOI] [PubMed] [Google Scholar]
- 33.Bustin SA, Benes V, Garson JA, Hellemans J, Hugget J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, and Wittwer CT 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem 55: 611–622. [DOI] [PubMed] [Google Scholar]
- 34.Chargé SB, and Rudnicki MA 2004. Cellular and molecular regulation of muscle regeneration. Physiol. Rev 84: 209–238. [DOI] [PubMed] [Google Scholar]
- 35.Miller KJ, Thaloor D, Matteson S, and Pavlath GK 2000. Hepatocyte growth factor affects satellite cell activation and differentiation in regenerating skeletal muscle. Am J Physiol., Cell Physiol 278: C174–C181. [DOI] [PubMed] [Google Scholar]
- 36.White JP, Baltgalvis KA, Sato S, Wilson LB, and Carson JA 2009. Effect of nandrolone decanoate administration on recovery from bupivacaine-induced muscle injury. J. Appl. Physiol 107: 1420–1430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wehling-Henricks M, Li Z, Lindsey C, Wang Y, Welc SS, Ramos JN, Khanlou N, Kuro-O M, and Tidball JG 2016. Klotho gene silencing promotes pathology in the mdx mouse model of Duchenne muscular dystrophy. Hum. Molec. Genet 25: 2465–2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wehling M, Spencer MJ, and Tidball JG 2001. A nitric oxide synthase transgene ameliorates muscular dystrophy in mdx mice. J. Cell Biol 155: 123–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wehling-Henricks M, Jordan MC, Roos KP, Deng B, and Tidball JG 2005. Cardiomyopathy in dystrophin-deficient hearts is prevented by expression of a neuronal nitric oxide synthase transgene in the myocardium. Hum. Mol. Genet 14: 1921–1933. [DOI] [PubMed] [Google Scholar]
- 40.Corraliza IM, Campo ML, Soler G, and Modolell M 1994. Determination of arginase activity in macrophages: a micromethod. J. Immunol. Methods 174: 231–235. [DOI] [PubMed] [Google Scholar]
- 41.Heredia JE, Mukundan L, Chen FM, Mueller AA, Deo RC, Locksley RM, Rando TA, and Chawla A 2013. Type 2 innate signals stimulate fibro/adipogenic progenitors to facilitate muscle regeneration. Cell 153:376–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Warren GL, Hulderman T, Jensen N, McKinstry M, Mishra M, Luster MI, Simeonova PP 2002. Physiological role of tumor necrosis factor in traumatic muscle injury. FASEB J 16: 1630–1632. [DOI] [PubMed] [Google Scholar]
- 43.Hardy D, Besnard A, Latil M, Jouvion G, Briand D, Thépenier C, Pascal Q, Guguin A, Gayraud-Morel B, Cavaillon JM, Tajbakhsh S, Rocheteau P, and Chrétien F 2016. Comparative study of injury models for studying muscle regeneration in mice. PLoS One 11(1):e0147198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44., Baumann CW, and Otis JS 2015. 17-(allylamino)-17-demethoxygeldanamycin drives Hsp70 expression but fails to improve morphological or functional recovery in injured skeletal muscle. Clin. Exp. Pharmacol. Physiol 42:1308–16. [DOI] [PubMed] [Google Scholar]
- 45.Souza NHC, Mesquita -Ferrari RA, Rodrigues MFSD, da Silva DFT, Ribeiro BG, Alves AN, Garcia MP, Nunes FD, da Silva Junior EM, França CM, Bussadori SK, and Fernandes KPS 2018. Photobiomodulation and different macrophages phenotypes during muscle tissue repair. J. Cell. Mol. Med 22: 4922–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Welc SS, Wehling-Henricks M, Kuro-O M, Thomas KA, and Tidball JG 2020. Modulation of Klotho expression in injured muscle perturbs Wnt signalling and influences the rate of muscle growth. Exp. Physiol 105: 132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu H, Huang D, Saederup N, Charo IF, Ransohoff RM, and Zhou L 2011. Macrophages recruited via CCR2 produce insulin-like growth factor-1 to repair acute skeletal muscle injury. FASEB J. 25, 358–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shireman PK, Contreras-Shannon V, Ochoa O, Karia BP, Michalek JE, and McManus LM 2007. MCP-1 deficiency causes altered inflammation with impaired skeletal muscle regeneration. J. Leukoc. Biol 81: 775–785. [DOI] [PubMed] [Google Scholar]
- 49.Sun D, Martinez CO, Ochoa O, Ruiz-Willhite L, Bonilla JR, Centonze VE, Waite LL, Michalek JE, McManus LM, and Shireman PK 2009. Bone marrow-derived cell regulation of skeletal muscle regeneration. FASEB J. 23: 382–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Li G, Chen C, Laing SD, Ballard C, Biju KC, Reddick RL, Clark RA, and Li S 2016. Hematopoietic knockdown of PPARδ reduces atherosclerosis in LDLR−/− mice. Gene Therapy 23: 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ochoa O, Sun D, Reyes-Reyna SM, Waite LL, Michalek JE, McManus LM, and Shireman PK 2007. Delayed angiogenesis and VEGF production in CCR2−/− mice during impaired skeletal muscle regeneration. Am. J. Physiol. Regul. Integr. Comp. Physiol 293: R651–R661. [DOI] [PubMed] [Google Scholar]
- 52.Schug TT, and Li X 2009. PPARdelta-mediated macrophage activation: a matter of fat. Dis. Model. Mech 2: 421–422. [DOI] [PubMed] [Google Scholar]
- 53.Jin RM, Warunek J, and Wohlfert EA 2018. Therapeutic administration of IL-10 and amphiregulin alleviates chronic skeletal muscle inflammation and damage induced by infection. Immunohorizons 2: 142–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, and Evans RM 1998. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell 93: 241–252. [DOI] [PubMed] [Google Scholar]
- 55.Chawla A, Barak Y, Nagy L, Liao D, Tontonoz P, and Evans RM 2001. PPAR-gamma dependent and independent effects on macrophage-gene expression in lipid metabolism and inflammation. Nature Medicine 7:48–52. [DOI] [PubMed] [Google Scholar]
- 56.Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, and Glass CK 1999. Interleukin-4-dependent production of PPAR-γ ligands in macrophages by 12/15-lipoxygenase. Nature 400: 378–382. [DOI] [PubMed] [Google Scholar]
- 57.Odegaard JI, Ricardo-Gonzalez RR, Goforth MH, Morel CR, Subramanian V, Mukundan L, Red Eagle A, Vats D, Brombacher F, Ferrante AW, and Chawla A 2007. Macrophage-specific PPARγ controls alternative activation and improves insulin resistance. Nature 447: 1116–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dahten A, Mergemeier S, and Worm M 2007. PPARgamma expression profile and its cytokine driven regulation in atopic dermatitis. Allergy 62:926–933. [DOI] [PubMed] [Google Scholar]
- 59.Tikhanovich I, Zhao J, Olson J, Adams A, Taylor R, Bridges B, Marshall L, Roberts B, and Weinman SA 2017. Protein arginine methyltransferase 1 modulates innate immune responses through regulation of peroxisome proliferator-activated receptor γ-dependent macrophage differentiation. J. Biol. Chem 292:6882–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Luo W, Xu Q, Wang Q, Wu H, and Hua J 2017. Effect of modulation of PPAR-γ activity on Kupffer cells M1/M2 polarization in the development of non-alcoholic fatty liver disease. Sci. Rep 7:44612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Zizzo G, and Cohen PL 2015. The PPAR-γ antagonist GW9662 elicits differentiation of M2c-like cells and upregulation of the MerTK/Gas6 axis: a key role for PPAR-γ in human macrophage polarization. J. Inflamm 12: 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Varga T, Mounier R, Patsalos A, Gogolák P, Peloquin M, Horvath A, Pap A, Daniel B, Nagy G, Pintye E, Póliska S, Cuvellier S, Larbi SB, Sansbury BE, Spite M, Brown CW, Chazaud B, and Nagy L 2016. Macrophage PPARγ, a lipid activated transcription factor controls the growth factor GDF3 and skeletal muscle regeneration. Immunity 45: 1038–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Contreras-Shannon V, Ochoa O, Reyes-Reyna SM, Sun D, Michalek JE, Kuziel WA, McManus LM, and Shireman PK 2007. Fat accumulation with altered inflammation and regeneration in skeletal muscle of CCR2−/− mice following ischemic injury. Am. J. Physiol. Cell Physiol 292: C953–C967. [DOI] [PubMed] [Google Scholar]
- 64.Mojumdar K, Liang F, Giordano C, Lemaire C, Danialou G, Okazaki T, Bourdon J, Rafei M, Galipeau J, Divangahi M, and Petrof BJ 2014. Inflammatory monocytes promote progression of Duchenne muscular dystrophy and can be therapeutically targeted via CCR2. EMBO Mol. Med 6: 1476–1492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barish GD, Atkins AR, Downes M, Olson P, Chong LW, Nelson M, Zou Y, Hwang H, Kang H, Curtiss L, Evans RM, and Lee CH 2008. PPARdelta regulates multiple proinflammatory pathways to suppress atherosclerosis. Proc. Natl. Acad. Sci. USA 105: 4271–4276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Graham TL, Mookherjee C, Suckling KE, Palmer CN, and Patel L 2005. The PPARdelta agonist GW0742X reduces atherosclerosis in LDLR(−/−) mice. Atherosclerosis 181: 29–37. [DOI] [PubMed] [Google Scholar]
- 67.Lee CH, Chawla A, Urbiztondo N, Liao D, Boisvert WA, Evans RM, and Curtiss LK 2003. Transcriptional repression of atherogenic inflammation: modulation by PPARdelta. Science 302: 453–457. [DOI] [PubMed] [Google Scholar]
- 68.Sierra-Filardi E, Nieto C, Domínguez-Soto A, Barroso R, Sánchez-Mateos P, Puig-Kroger A, López-Bravo M, Joven J, Ardavín C, Rodríguez-Fernández JL, Sánchez-Torres C, Mellado M, and Corbí AL 2014. CCL2 shapes macrophage polarization by GM-CSF and M-CSF: identification of CCL2/CCR2-dependent gene expression profile. J. Immunol 192: 3858–3867. [DOI] [PubMed] [Google Scholar]
- 69.Roca H, Varsos ZS, Sud S, Craig MJ, Ying C, and Pienta KJ 2009. CCL2 and interleukin-6 promote survival of human CD11b+ peripheral blood mononuclear cells and induce M2-type macrophage polarization. J. Biol. Chem 284: 34342–34354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lumeng CN, Bodzin JL, and Saltiel AR 2007. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J. Clin. Invest 117: 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hsieh PL, Rybalko V, Baker AB, Suggs LJ, and Farrar RP 2018. Recruitment and therapeutic application of macrophages in skeletal muscles after hind limb ischemia. J. Vasc. Surg 67: 1908–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Piqueras L, Reynolds AR, Hodivala-Dilke KM, Alfranca A, Redondo JM, Hatae T, Tanabe T, Warner TD, and Bishop-Bailey D 2007. Activation of PPARbeta/delta induces endothelial cell proliferation and angiogenesis. Arterioscler. Thromb. Vasc. Biol 27: 63–69. [DOI] [PubMed] [Google Scholar]
- 73.Gaudel C, Schwartz C, Giordano C, Abumrad NA, and Grimaldi PA 2008. Pharmacological activation of PPAR beta promotes rapid and calcineurin-dependent fiber remodeling and angiogenesis in mouse skeletal muscle. Am. J. Physiol. Endocrinol. Metab 295: E297–E304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Stephen RL, Gustafsson MC, Jarvis M, Tatoud R, Marshall BR, Knight D, Ehrenborg E, Harris AL, Wolf CR, and Palmer CN 2004. Activation of peroxisome proliferator-activated receptor delta stimulates the proliferation of human breast and prostate cancer cell lines. Cancer Res. 64: 3162–3170. [DOI] [PubMed] [Google Scholar]